

Abstract
Background:
Rare genetic variants and genetic variation at loci in an enhancer in SRY-Box Transcription Factor 17 (SOX17) are identified in patients with idiopathic pulmonary arterial hypertension (PAH) and PAH with congenital heart disease. However, the exact role of genetic variants or mutation in SOX17 in PAH pathogenesis has not been reported.
Methods:
SOX17 expression was evaluated in the lungs and pulmonary endothelial cells (ECs) of idiopathic PAH patients. Mice with Tie2Cre mediated Sox17 knockdown and EC-specific Sox17 deletion were generated to determine the role of SOX17 deficiency in the pathogenesis of PAH. Human pulmonary ECs were cultured to understand the role of SOX17 deficiency. Single cell-RNA sequencing, RNA-sequencing analysis and luciferase assay were performed to understand the underlying molecular mechanisms of SOX17 deficiency-induced PAH. E2F1 inhibitor HLM006474 was used in EC-specific Sox17 mice.
Results:
SOX17 expression was downregulated in the lung and pulmonary ECs from IPAH patients. Mice with Tie2Cre mediated Sox17 knockdown and EC-specific Sox17 deletion induced spontaneously mild pulmonary hypertension (PH). Loss of endothelial Sox17 in EC exacerbated hypoxia-induced PH in mice. Loss of SOX17 in lung ECs induced endothelial dysfunctions including upregulation of cell cycle programming, proliferative and anti-apoptotic phenotypes, augmentation of paracrine effect on pulmonary arterial smooth muscle cells, impaired cellular junction, and BMP signaling. E2F Transcription Factor 1 (E2F1) signaling was shown to mediate the SOX17 deficiency-induced EC dysfunction. Pharmacological inhibition of E2F1 in Sox17 EC-deficient mice attenuated PH development.
Conclusions:
Our study demonstrated that endothelial SOX17 deficiency induces PH through E2F1. Thus, targeting E2F1 signaling represents a promising approach in PAH patients.
Keywords: pulmonary arterial hypertension, angiogenesis, vascular disease, proliferation, paracrine effect
Graphical Abstract
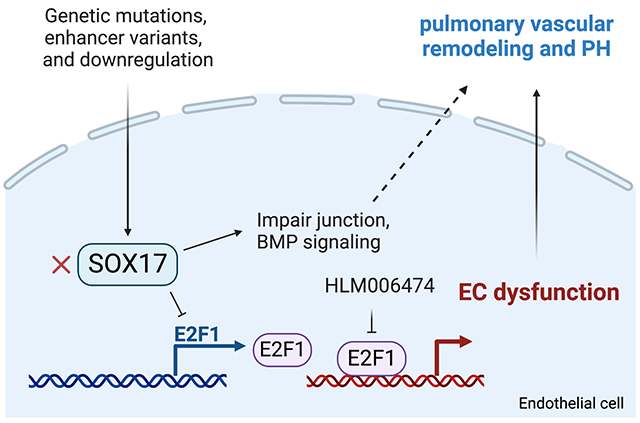
Introduction
Pulmonary hypertension (PH) is defined by a mean pulmonary arterial pressure more than 20 mmHg at rest. It is estimated that the PH prevalence is ~1% of the global population1. Heritable and idiopathic pulmonary arterial hypertension (IPAH) belong to group 1 PH. They are clinically identical progressive disorders charactered by elevation of pulmonary arterial pressure with pathologic remodeling in pulmonary arteries2. PH types with different etiologies share histopathologic features including eccentric and obliterative intima thickening and complex plexiform lesions. BMPR2, a gene encoding bone morphogenetic protein type 2 receptor (BMPR2), is mutated in 80% of familial PAH and approximately 20% of sporadic cases. Other mutations or pathogenic genes have been identified, including other TGF-β/BMP signaling members ACVRL1, ENG, SMAD1/4/9, and CAV1, KCNK3, and TBX43. Recent studies also identified a few rare sequence variations in the genes GDF2, ATP13A3, AQP1, and SOX174. However, the exact mechanisms by which these gene mutations or variants increase the susceptibility to PH remain elusive.
SOX17, a member of the Sry-related high mobility group domain family F (Sox F) transcription factors, is a critical regulator in the developmental stage of endothelial/hematopoietic lineages and maintenance of arterial identities5–7. In the developmental lung, SOX17 is selectively expressed in the pulmonary arteries and veins. Interestingly, SOX17 is only detected in the vasculature of the right ventricle in the developmental heart8. Deletion of Sox17 (Sox17Δ/Δ) at embryonic stage causes pulmonary vascular malformations, biventricular enlargement and postnatal lethality9, suggesting that endothelial SOX17 is critical to cardiopulmonary development. In the adult lung, Sox17 is required for endothelial regeneration following sepsis-induced vascular injury in mice10. Endothelial SOX17 also promotes tumor angiogenesis11. Rare genetic variants in SOX17 are identified in patients with IPAH and PAH with congenital heart disease12,13. Recent studies also identified genetic variation at loci in an enhancer near SOX17 is associated with human PAH14. Recent study showed that Sox17 deficiency promoted PH in mice via HGF/c-Met signaling or HIF-2α 15,16. CRISPR/Cas9-mediated Sox17 enhancer deletion in mice worsened PH17. Nevertheless, the exact role of genetic variants or mutation in SOX17 in the contribution of PH remain unclear. The goal of the present study is to understand the role of SOX17 in the pathogenesis of PH.
In our present studies, we determined the expression of SOX17 in pulmonary vascular endothelial cells (PVECs) isolated from IPAH patients. Using EC-specific deletion mouse model, we also evaluated the role of deficiency in Sox17 in ECs in contribution of pulmonary vascular remodeling and mild PH under normoxic and hypoxic condition. Moreover, we also investigated the underline mechanisms secondary to the loss of Sox17 in ECs in the development of PH.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. For expanded methods please see Supplemental Material.
Human samples
The use of archived human lung tissues and cells were granted by the University of Arizona Institutional Review Board. Human IPAH patients and failed donors (FD)’ PVECs were obtained from the Pulmonary Hypertension Breakthrough Initiative (PHBI).
Mice
cKO Sox17 mice were generated by breeding Sox17f/f mice with Tie2Cre mice18. ecKO Sox17 mice were generated by breeding Sox17f/f mice with EndoSCL-CreERT2 mice19. Both male and female mice were included for experiments. For HLM006474 treatment, ecKO Sox17 mice were treated with tamoxifen, followed by treatment with HLM006474 (HLM, 12.5 mg/kg) 3 times a week for 6 weeks. The protocol for animal care and studies was approved by the Institutional Animal Care and Use Committee of University of Arizona (#19-513).
Data availability
RNA-sequencing and single-cell RNA-sequencing (scRNA-seq) data have been deposited in the GEO database under accession number GSE192649 and GSE218398. Scripts used for single-cell RNA sequencing analysis and analyzed data in R objects are available in Figshare (https://figshare.com/s/37782988b8cac7cedcf9).
Statistical Analysis
Statistical determination was performed on Prism 9 (Graphpad Software Inc.). Two-group comparisons were compared by the unpaired 2-tailed Student t test for equal variance or the Welch t test for unequal variance. Multiple comparisons were performed by One Way ANOVA with a Tukey post hoc analysis that calculates corrected P values. P less than 0.05 indicated a statistically significant difference. All bar graphs represent mean±SD.
Results
SOX17 is downregulated in PVECs from PAH patients
SOX17 mutations and enhancer variants were found in patients with PAH. However, the expression pattern and levels of SOX17 in human PAH patients remain elusive. Leveraging the public single-cell RNA-sequencing dataset from healthy human lungs, we first analyzed the mRNA expression of SOX17. Our data demonstrated that SOX17 is highly expressed in the ECs and rarely expressed in other cell types in the adult lung (Figure 1A). To determine whether SOX17 is deficient in PVECs of PAH patients, we characterized the SOX17 expression in isolated PVECs from IPAH patients and failed donors (FD). We found that the SOX17 mRNA levels (Figure 1B) as well as the SOX17 protein levels (Figure 1C) were significantly downregulated in sub-confluent PVECs isolated from IPAH patients compared to that from FD subjects, suggesting that SOX17 deficiency is present in PAH patients. Our data is consistent the microarray analysis (GSE113439) of lung samples from IPAH patients and healthy donors, which showed that SOX17 mRNA level is decreased in IPAH patients 20 (Figure S1A). To determine the localization of SOX17, we performed immunofluorescent staining against SOX17 on human IPAH and FD lungs and our data showed that SOX17 is mainly located in the lung ECs. As shown in Figure 1D, 1E and Figure S1B, SOX17 is markedly downregulated in the ECs of less remodeled vessels and diminished in the occlusive vessels of IPAH patients. We also determine the levels of SOX17 in the lung of monocrotaline (MCT) induced PH rats, we found that there was a significant reduction of SOX17 in MCT-treated rats (Figure 1F).
Figure 1. Downregulation of endothelial SOX17 in the patients with PAH.
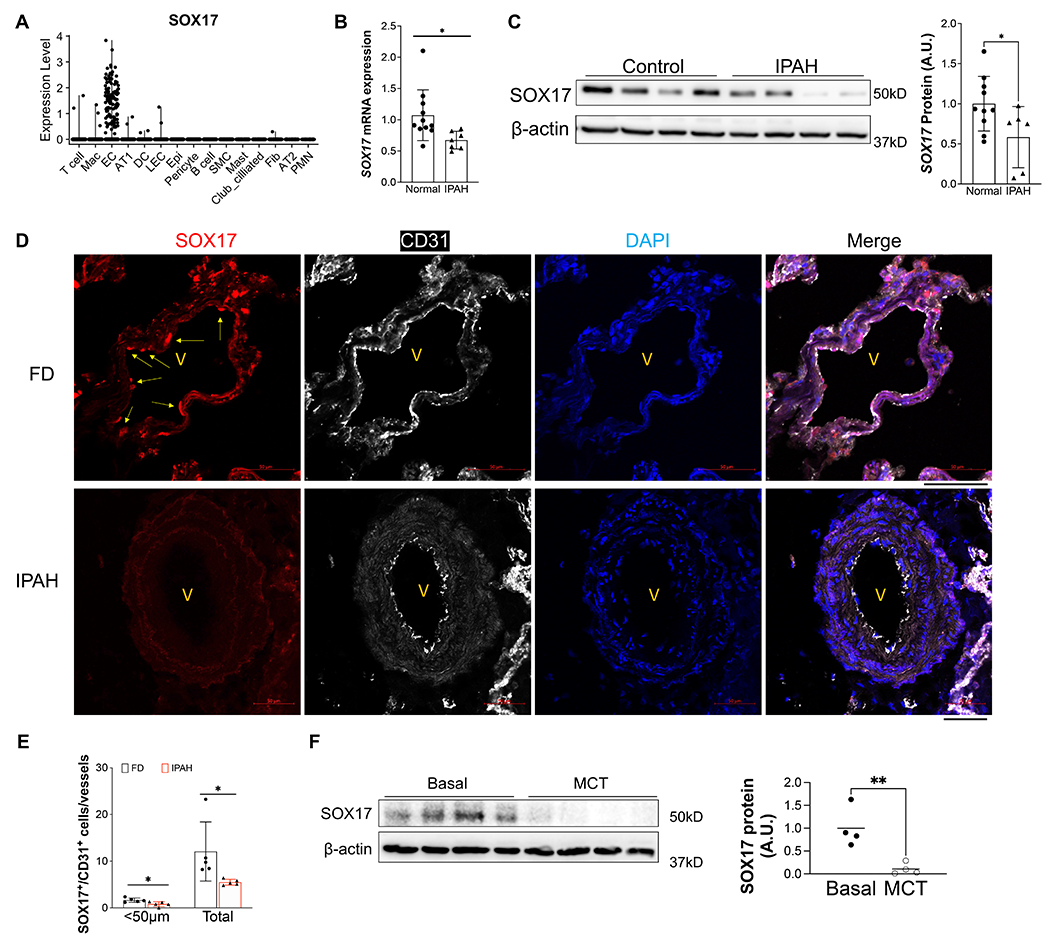
(A) A violin plot showing SOX17 is restricted in the ECs of human lungs via scRNA-seq. Mac=macrophage; DC= dendritic cell; LEC=lymphatic EC; Epi=epithelium; SMC= smooth muscle cell; Fib=fibroblast; AT1 or AT2 = alveolar type 1 or 2 epithelium; PMN=neutrophils. (B) qRT-PCR analysis showed that SOX17 mRNA levels were downregulated in the sub-confluent PVECs isolated from IPAH patients (n=11) and normal non-PAH donors (n=7). Each data point represents cells from one human subject including both male and female. (C) Western blotting demonstrated reduction of SOX17 protein expression in the IPAH PVECs (n=10) compared with normal non-PAH donors (n=6). Each data point represents cells from one human subject including both male and female. (D, E) Immunostaining against SOX17 showing diminished SOX17 expression in the ECs of remodeling lesions from IPAH patients. Arrows indicate SOX17 positive ECs in non-PAH failed donors (FD). SOX17+/CD31+ cell number was quantified and normalized by vessels number. Each dot represents one subject. (F) SOX17 is decreased in the lungs of established PH rats at 4 weeks post MCT (33mg/kg subcutaneously) treatment. Student t test (B, C, E, F). *, P< 0.05; **, P< 0.01. A.U. = arbitrary units; Scale bar, 50μm.
Loss of SOX17 in embryonic stage induces spontaneously mild PH and cardiac hypertrophy
To determine whether SOX17 deficiency is involved in the pathogenesis of PH in mice, we utilized EC specific Cre lines (Tie2Cre and EndoSCL-CreERT2) to delete Sox17 in the ECs. Constitute deletion of Sox17 mice (Sox17f/f;Tie2Cre) display vascular defect and embryonic lethal9, thus we generated Sox17f/+;Tie2Cre (KOEC/+, cKO) mice (Figure S2A and S2B). We then characterized the right ventricular (RV) hemodynamic and cardiac dissection of WT (Sox17f/f) and cKO mice. cKO mice at the basal developed mild PH by upregulation of right ventricle systolic pressure (RVSP), which is the indicator of pulmonary arterial pressure, when compared with WT mice in the similar age (Figure S2C). We also observe a significant increase in the weight ratio of the right ventricular free wall to left ventricle plus septum (RV/LV+S) and left ventricle weight vs body weight (LV/BW), indicative of right ventricular and left ventricular hypertrophy, in cKO mice. (Figure S2D and S2E), which is consistent with previous finding that embryonic deletion of Sox17 lead to enlargement of biventricles 9. To further determine whether Tie2Cre promoter mediated Sox17 knockdown regulates pulmonary vascular remodeling in mice, we performed Russell-Movat pentachrome staining and immunostaining of α-smooth muscle actin (SMA) and found that cKO mice exhibited increased of the thickness of pulmonary arterial wall, and muscularization of distal pulmonary arterioles (Figure S2H–S2I).
Loss of endothelial SOX17 in adult stage leads to spontaneously mild PH and exaggerated hypoxia-induced PH
Because Tie2Cre also induces gene deletion in hematopoietic stem cells besides ECs21, we then generated inducible deletion of EC Sox17 mice (Sox17f/f;EndoSCL-CreERT219,22, ecKO Sox17) by breeding Sox17 floxed mice with EndoSCL-CreERT219,22 (Figures S3A). Both Sox17f/f (WT) and ecKO Sox17 mice at the age of 7~8 weeks were treated with tamoxifen for 3 doses [100mg/kg, intraperitoneal injection (i.p.) daily] to induce SOX17 deletion only in ECs. Around 2 months post tamoxifen treatment, Immunostaining against SOX17 demonstrated that PVECs from ecKO Sox17 mice have significant decrease of SOX17 expression, suggest that SOX17 was selectively deleted in PVECs (Figures S3B). We then characterized the RV hemodynamic and cardiac dissection of WT and ecKO Sox17 mice. Our data showed that ecKO Sox17 mice showed a significant increase of RVSP when compared with WT mice (Figure 2A). However, we did not observe a significant change in RV/LV+S ratio and LV/BW ratio between WT and ecKO Sox17 mice (Figure 2B and Figure S3C). We also performed echocardiography measurement on these animals. We did not observe any significant alteration of cardiac size and function including heart rate, cardiac output, left ventricular fractional shorting and RV fraction area change in the ecKO Sox17 mice (Figure S3D–S3G). The difference cardiac phenotype between Sox17 cKO and ecKO mice might be due to the effect of constitute Sox17 deletion in the embryonic stage. Previous studies demonstrated that Sox17 is a HIF-1α target gene in the lung ECs 10. We did find that Sox17 is upregulated in the lung of chronic hypoxia incubated mice (Figure S4A). To further confirm if Sox17 deficiency in EC augments PH and RV remodeling in mice, we challenged both WT and ecKO Sox17 mice with hypoxia (10% O2) to assess the role of endothelial ecKO Sox17 in the hypoxia-induced PH in mice. 3 weeks post tamoxifen treatment, mice were incubated with hypoxia (10% O2) for 3 weeks or normoxia alone. Our data showed that ecKO Sox17 mice exposed to hypoxia exhibited a significantly elevated of RVSP when compared with WT mice (Figure 3A). ecKO Sox17 mice also showed a significantly increased weight ratio of RV/(LV+S), indicative of RV hypertrophy compared with WT mice (Figure 2B).
Figure 2. Endothelial SOX17 deficiency induced PH in mice.
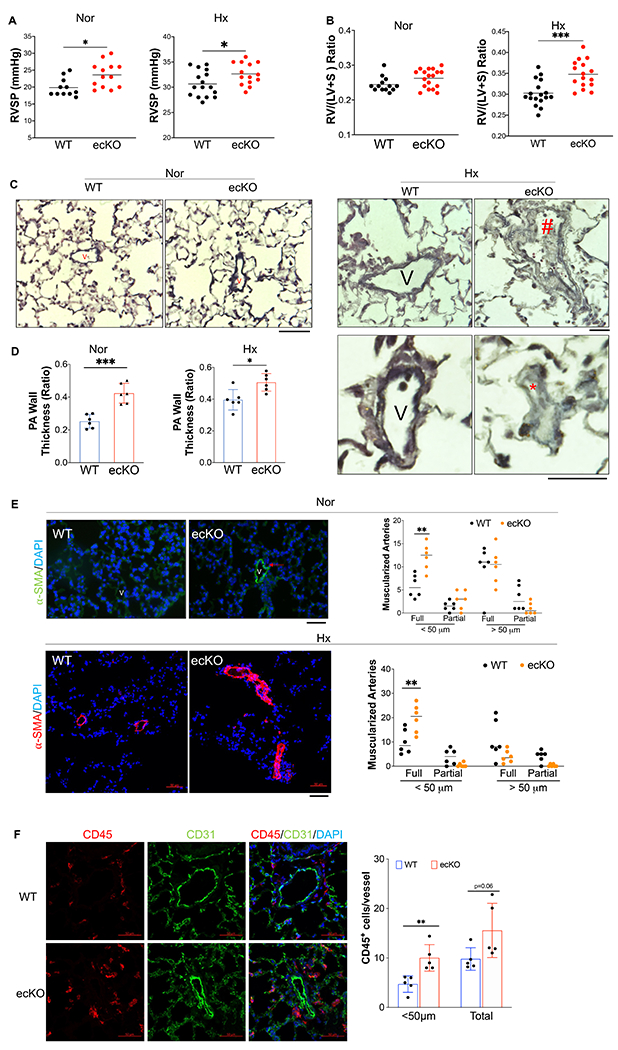
(A) ecKO Sox17 mice exhibited increase of RVSP at both normoxic (Nor) and hypoxic (Hx) condition. (B) ecKO Sox17 mice exhibited hypoxia-induced right heart hypertrophy compared with WT mice. (C) Representative micrographs of Russell-Movat pentachrome staining showing increased medial thickness in ecKO Sox17 mice compared with WT mice in normoxic and hypoxic condition. ecKO Sox17 mice also developed occlusive vascular lesion in response to hypoxia. V=vessel, # indicates narrower vessel, * indicates occlusive vessel. (D) Quantification of pulmonary artery wall thickness. Wall thickness was calculated by the distance between internal wall and external wall divided by the distance between external wall and the center of lumen. (E) Representative micrographs and quantification of muscularization of distal pulmonary vessels showed that markedly enhanced muscularization of distal pulmonary vessels in ecKO Sox17 mice compared with WT mice under normoxic and hypoxic condition. Lung sections were immunostained with anti–α-SMA (green). Red arrow indicates a-SMA+ distal pulmonary vessels. α-SMA+ vessels were quantified in 20 field at 10X magnification per mouse. (F) Immunostaining against CD45 (Red) demonstrated that there was upregulated accumulation of inflammatory cells in the perivascular bed of ecKO Sox17 mice. Student t test (A, B, D, E, F). *, P< 0.05; **, P< 0.01, ***, P< 0.001. Scale bar, 50μm.
Figure 3. Loss of SOX17 induced EC proliferation.
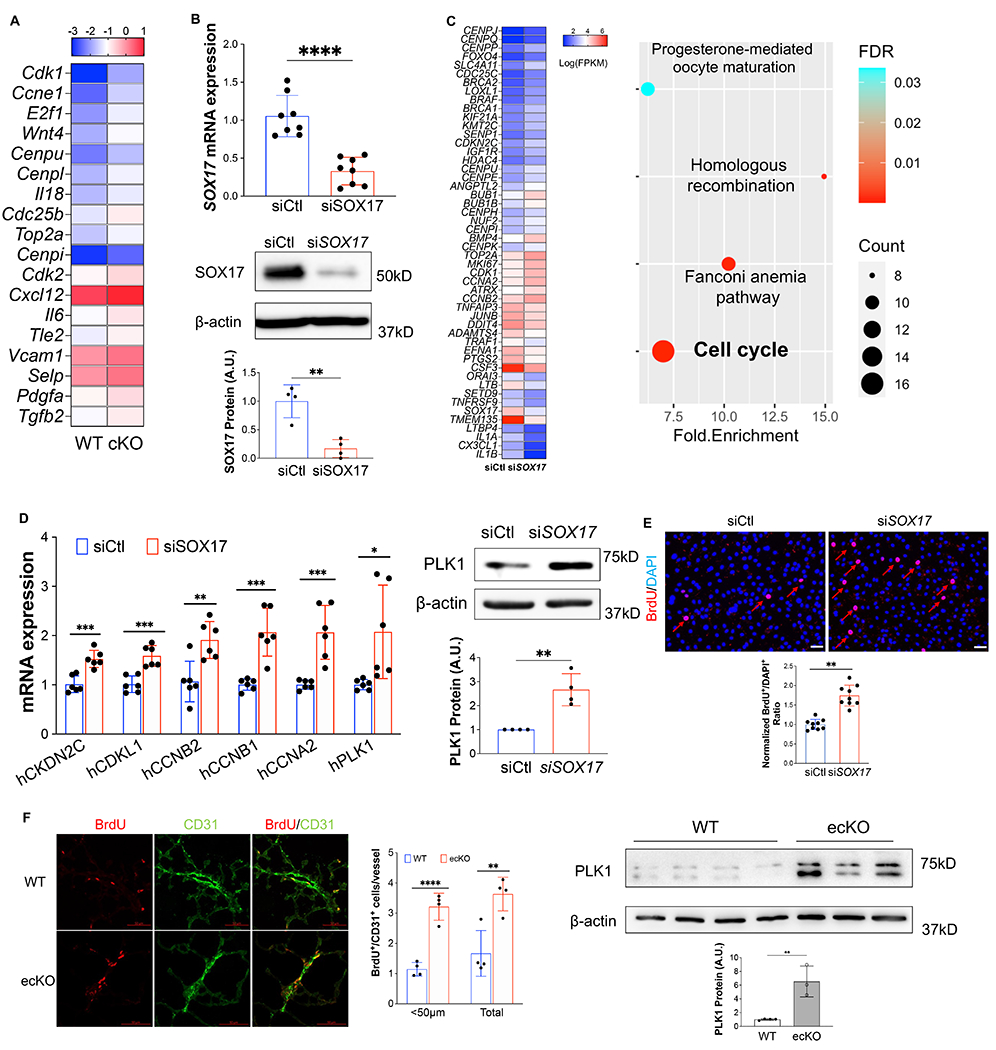
(A) scRNA transcriptomics showed that Sox17 deficiency ECs expressed higher levels of proliferation genes compared to WT ECs. scRNA-seq analysis was performed on the whole lung of WT and cKO mice. Lung ECs transcriptomics were analyzed. (B) qRT-PCR analysis showing efficient knockdown of SOX17 via siRNA against SOX17 in HPVECs assessed by QPCR and western blot. (C)A representative heatmap of RNA-sequencing analysis of SOX17 knockdown in HPVECs. HPVECs were transfected with control siRNA (siCtl) or SOX17 siRNA for 48 hours. Equal amount of RNA from three replicates per group were pooled for RNA-seq. KEGG pathway enrichment analysis of upregulated genes in SOX17 deficient lung ECs demonstrating that cell cycle pathway is the top upregulated signaling induced by loss of SOX17. (D) qRT-PCR analysis confirmed the upregulation of cell proliferation related genes including CKDN2C, CDKL1, CCNB2, CCNB1, CCNA2, and PLK1. Western Blotting analysis demonstrated induction of PLK1 protein expression by SOX17 deficiency. (E) BrdU incorporation assay demonstrated increased of EC proliferation in SOX17 deficient HPVECs. At 48 hours post-transfection, HPVECs were starved in serum/growth factors free medium for 12 hours. BrdU was added in the medium at 4 hours prior to cells harvest. BrdU was stained with anti-BrdU antibodies. Red indicated BrdU positive cells. Nucleus were co-stained with DAPI. (F) In vivo BrdU incorporation assay showed upregulation of lung ECs proliferation in ecKO Sox17 mice during hypoxia condition. WT and ecKO Sox17 mice were incubated in hypoxia (10% O2) for 10 days. BrdU (25 mg/kg) was injected i.p. between day 7 to day 9. Lung sections were stained with anti-BrdU and anti-CD31. BrdU+/CD31+ cells were quantified. Augmentation of cell proliferation marker PLK1 expression in the lung of ecKO Sox17 (ecKO) mice compared to WT mice. β-actin level was used as an internal control. Student t test (B, D, E,F). *, P< 0.05; **, P< 0.01. ***, P< 0.001. Scale bar, 50μm.
To further determine whether endothelial Sox17 deficiency regulates pulmonary vascular remodeling in basal and hypoxia incubated mice, we then performed Russell-Movat pentachrome staining (Figure 2C and 2D) and immunostaining of α-SMA (Figure 2E). Examination of lung pathology showed that ecKO Sox17 mice exhibited a marked increase of pulmonary wall thickness and distal pulmonary arterial muscularization at both basal and hypoxic condition. Notably, we found the narrower pulmonary vessel lumen and thicker wall in the big vessels of ecKO Sox17 mice, as well as occasional occlusion in the small vessels of ecKO Sox17 mice under hypoxic condition (Figures 2C and 2E), demonstrating loss of endothelial SOX17 aggravates pulmonary vascular remodeling in mice. As PAH is associated with upregulation of accumulation of perivascular inflammatory, we found that ecKO Sox17 mice exhibited increased CD45+cells accumulation in the vascular bed compared to WT mice (Figure 2F). Taken together, our data demonstrated that Sox17 deficiency induces spontaneous PH in mice and augmented hypoxia-induced pulmonary vascular remodeling and vasoconstriction.
SOX17 deficiency induces endothelial cell proliferation
To validate the impact of Sox17 deletion in vivo, we applied scRNA-seq analysis on cKO mice and WT mice (Figure S5A). scRNA-seq data revealed an increase of EC proportion in cKO mice compared with WT mice (Figure S5B). Transcriptomic analysis demonstrated that the lung ECs from cKO mice exhibited increased expression of genes related to cell proliferation, including Cdk1, E2f1, Top2a, etc (Figure 3A). To understand the direct impact of SOX17 deficiency in pulmonary EC in vitro, we also performed whole transcriptome RNA-sequencing in HPVECs with SOX17 knockdown. Short interfering RNA (siRNA) against SOX17 efficiently reduced SOX17 mRNA level and proteins expression (Figures 3B). RNA-seq analysis and pathway enrichment analysis showed that there was an alteration of many genes (i.e., CENPP, BRCA2, CDKN2C, CCNB2) and pathway (i.e., cell cycle) related to cell proliferation (Figures 3B). QRT-PCR analysis confirmed that SOX17 knockdown significantly induced expression of genes related to cell proliferation including PLK1, CCNA2, CCNB1, CCNB2, CDKL1, and CKDN2C (Figure 3D). Western Blotting confirmed upregulation of PLK1 protein expression by SOX17 knockdown (Figure 3D). As SOX17 deficiency in EC induces cell cycle program, we hypothesize that SOX17 deficiency might lead to endothelial hyperproliferation during the development of PH. We employed siRNA to knockdown SOX17 in cultured HPVECs and evaluated cell proliferation. Cell proliferation, assessed by 5-bromo-2’-deoxyuridine (BrdU) incorporation assay, in SOX17-deficient cells was markedly augmented compared to control siRNA-transfected HPVECs (Figure 3E). We also evaluated in vivo proliferation via injecting BrdU into WT and ecKO mice. We found that BrdU incorporation in CD31+ cells were markedly increased in ecKO mice (Figure 3F). The expression levels of a cell proliferation marker, proliferating cell nuclear antigen (PCNA), and polo-like kinase 1 (PLK1) were upregulated in the lung from ecKO Sox17 mice compared to WT mice (Figure 3F). These data suggest that SOX17 deficiency induces EC proliferation in vitro and in vivo.
Endothelial SOX17 deficiency induces PASMCs proliferation
The muscularization of distal pulmonary arterials and neointima formation seen in ecKO mice are likely due to increased proliferation of pulmonary arterials smooth muscle cell (PASMCs). PAECs from PAH patients produce pro-proliferative signaling through secreting many growth factors such as PDGF-B, ET-1, CXCL12, and MIF, and promote perivascular cells such as PASMCs proliferation23. We then seeded SOX17 deficient HPVECs on the top chamber and co-cultured with PASMCs, and found that SOX17 knockdown promoted PASMCs proliferation (Figure 4A). In vivo BrdU assay also showed the increased of BrdU+/α-SMA+ cells (indicating PASMCs) proliferation in the Sox17 ecKO mice compared to WT mice (Figure 4B). These data suggest that SOX17 deficiency induces paracrine effect and enhances PASMCs proliferation. To identify the potential factors derived from SOX17 deficiency ECs, we leveraged the scRNA-seq dataset and predicted the potential ligand and receptor pairs between ECs and SMCs using CellChat24. CellChat prediction showed that there were increased ligand-receptor pairs such as Pdgfb-Pdgfra, Edn1-Ednra from ECs to PASMCs (Figure 4C). Transcriptomes analysis showed that lung ECs from CKO mice showed an increase of multiple paracrine factors including Cxcl12, Edn1, Pdgfb, and Pdgfd (Figure 4C), suggesting that SOX17 deficiency in ECs induces paracrine effect on PASMCs.
Figure 4. Loss of endothelial SOX17 promoted EC dysfunction.
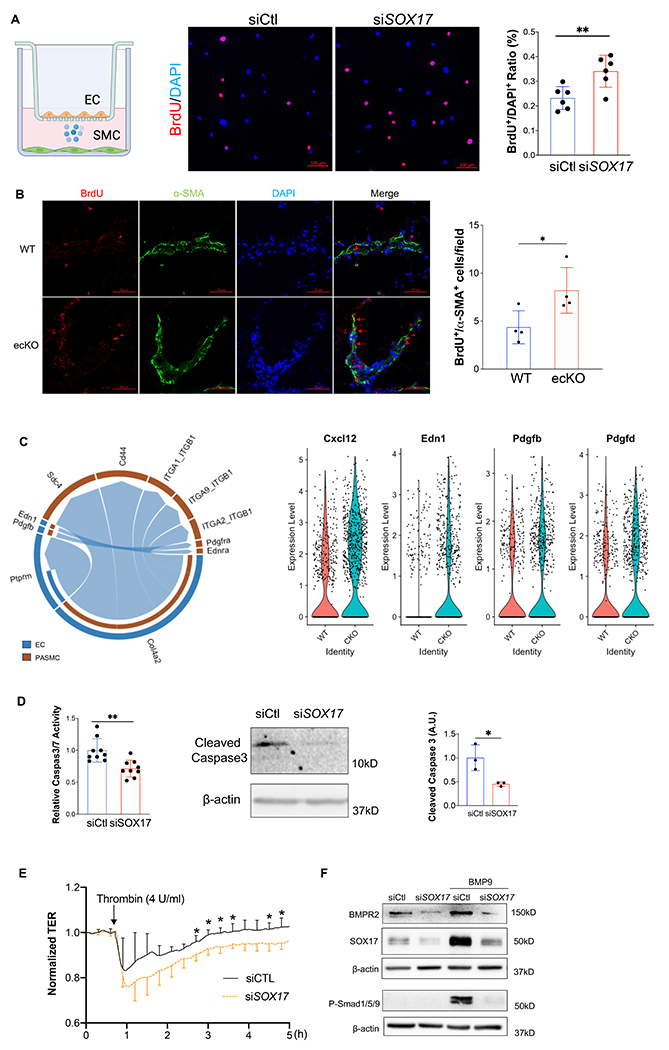
(A) SOX17 deficiency in lung ECs promoted PASMCs proliferation assessed by Transwell co-culture and BrdU assay. PASMCs were seeded on the cover slides on the lower chamber. SOX17 deficiency or control HPVECs were seeded on the top chamber for 48 hours. PASMCs were starved overnight, then co-cultured with HPVECs. BrdU was added in the lower chamber at 8 hours prior to cells harvest. BrdU was stained with anti-BrdU antibodies. Red indicated BrdU positive cells. Nucleus were co-stained with DAPI. (B) In vivo BrdU incorporation assay showed upregulation of PASMCs proliferation in ecKO Sox17 mice during hypoxia condition. WT and ecKO Sox17 mice were incubated in hypoxia (10% O2) for 10 days. BrdU (25 mg/kg) was injected i.p. between day 7 to day 9. Lung sections were stained with anti-BrdU and anti-α-SMA. BrdU+/α-SMA+ cells were quantified. (C) CellChat prediction using scRNA-seq dataset showed the upregulation of ligand and receptor pairs (Pdgfb-Pdgfra, Edn1-Ednra) in cKO mice. ScRNA-seq analysis showed the increase of EC derived cytokines including Cxcl12, Edn1, Pdgfb, Pdgfd. (D) SOX17 deficiency promoted anti-apoptotic phenotype of HPVECs during starvation assessed by Caspase 3/7 activities. (J) Western blotting analysis demonstrated reduction of cleaved Caspase 3 in SOX17 deficient HPVECs. (E) Impairment of endothelial barrier function in SOX17 deficient HPVECs. At 60 hours post-transfection, TER was monitored for up to 5 hours. Thrombin (4U/ml) was added to disrupt the cellular junction. (n=4). (F) Sox17 deficiency reduced BMPR2 expression and impaired BMPR2 activity via assessing P-Smad1/5/9 expression. Student t test (A, B, D, E). *, P< 0.05; **, P< 0.01. Scale bar, 50μm.
SOX17 deficiency induces endothelial dysfunction
EC hyperproliferation and upregulation of glycolysis are hallmarks of PAH EC25,26, we then measure the Extracellular Acidification Rate (ECAR) level and found that SOX17 deficient HPVECs enhanced glycolysis compared to control. (Figure S6). Since anti-apoptotic and hyperproliferative features are hallmarks of PAH ECs, we also evaluated cell apoptosis after SOX17 knockdown. After starvation for 24 hours, HPVECs with SOX17 knockdown exhibited a significant reduction in Caspase 3/7 activity and cleaved Caspase 3 expression, suggesting that SOX17 deficiency promotes anti-apoptotic phenotype of HPVECs (Figures 4D). Endothelial junction integrity is important to maintain vascular homeostasis. We then measured the EC junction via ECIS system in the presence of Thrombin. Junction integrity is significantly impaired in SOX17 deficient ECs (Figure 4E). BMPR2 deficiency is evident in patients with PAH. Our data also demonstrated that SOX17 knockdown reduced BMPR2 expression and BMP9-induced phosphorylation of Smad1/5/9 (Figure 4F). These data suggest that SOX17 deficiency induces EC dysfunction including hyperproliferation, enhanced paracrine effect and glycolysis, anti-apoptosis, and impaired junction integrity and BMPR2 signaling leading to EC dysfunction.
E2F1 mediated SOX17 deficiency-induced EC dysfunction
To further determine what regulators or transcriptional factors that mediate the upregulation of the proliferative gene program induced by loss of SOX17, we performed transcription factor prediction using iRegulon27. iRegulon prediction showed that E2F family member E2F1 is the top transcription factor governing the proliferative program induced by SOX17 deficiency (Figure 5A). Western blotting analysis confirmed that SOX17 knockdown markedly induced E2F1 expression in HPVECs (Figure 5A). We also observed that E2F1 was significantly upregulated in the lung of ecKO Sox17 mice (Figure 5A). To determine whether E2F1 activation mediates the effect of SOX17 deficiency-induced HPVECs proliferation and survival, we performed siRNA-mediated knockdown of E2F1 in SOX17 deficient HPVECs. siRNA against E2F1 significantly reduced E2F1 mRNA and protein expression (Figures 5B). We found that E2F1 inhibition via siRNA blocked the expression of cell proliferation genes including PLK1, CCNB1, and CCNB2, as well as HPVECs proliferation assessed by BrdU incorporation assay (Figures 5C). Finally, E2F1 knockdown significantly inhibited SOX17 deficiency-induced cell survival (Figure. 5D).
Figure 5. E2F1 mediated SOX17 deficiency-induced dysfunction.
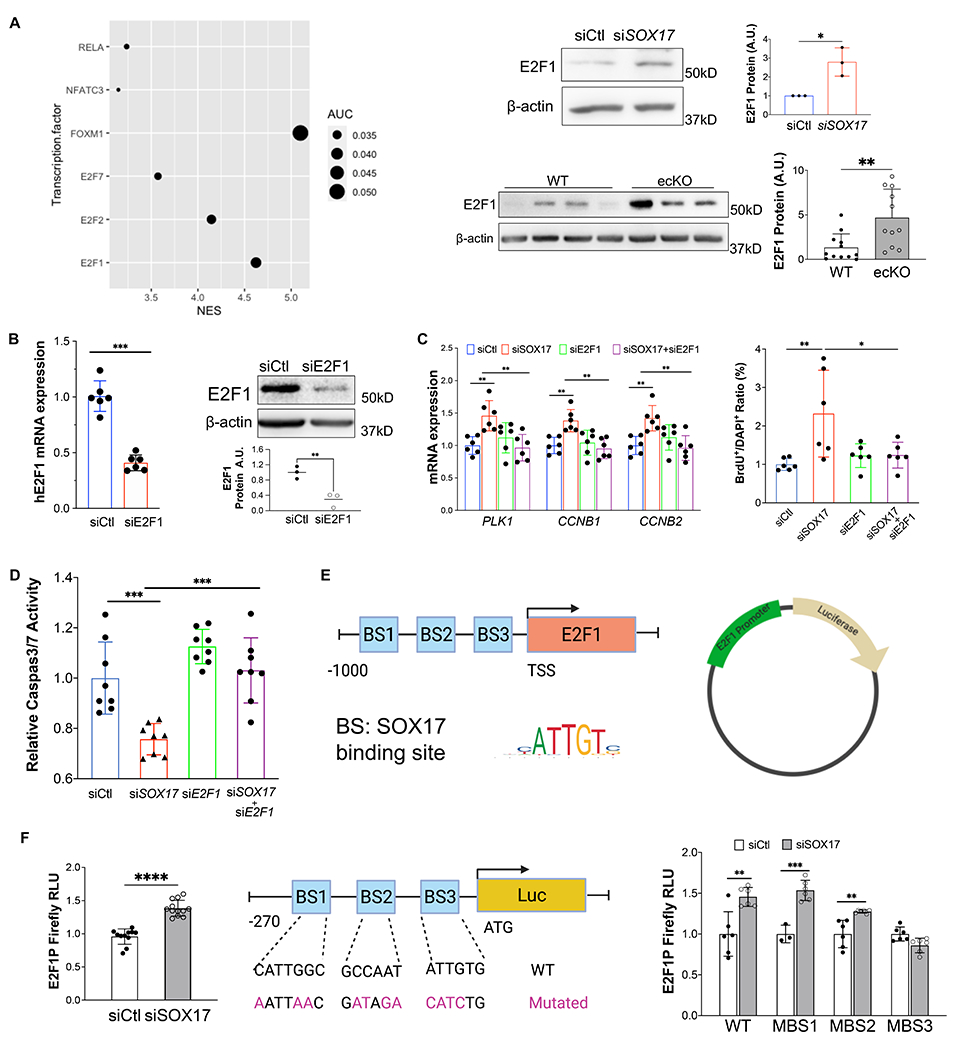
(A) iRegulon analysis demonstrated that E2F1 is the top enriched transcriptional factors potentially governing cell cycle programming in SOX17 deficient HPVECs. Upregulation of E2F1 protein expression by SOX17 knockdown. Increased of E2F1 expression in the lung of ecKO Sox17 mice compared to WT mice. (B) E2F1 siRNA markedly reduced E2F1 mRNA and protein expression. (C) QRT-PCR analysis demonstrated that E2F1 knockdown blocked the genes associated with proliferation including PLK1, CCNB1, and CCNB2 in the presence of SOX17 deficiency. BrdU incorporation assay demonstrated that E2F1 knockdown normalized cell proliferation induced by loss of SOX17. (D) E2F1 knockdown restored EC apoptosis which was inhibited by SOX17 deficiency. Studies were repeated at least 3 times. (E) A diagram shows that there are 3 putative SOX17 binding sites in the proximal promoter region of human E2F1 gene and a representative map for pLV-E2F1P/Luc plasmid. (F) Loss of SOX17 increased E2F1 promoter activities assessed by luciferase assay. HPVECs were transfected with control of SOX17 siRNA for 12 hours, followed by infected with pLV-E2F1P/luc lentivirus for 48 hours. A diagram showing that the SOX17 putative binding sites in E2F1 promoter/luciferase constructs were mutated. Purple highlight letters indicate mutated DNA sequences of the SOX17 putative binding sites in the E2F1 promoter. Binding site 3 mutation blocked SOX17 deficiency-induced E2F1 promoter activation. MBS1/2/3 indicate mutated binding site 1/2/3. HPVECs were transfected with control of SOX17 siRNA for 12 hours, followed by infected with WT or mutated pLV-E2F1P/luc lentiviruses for 48 hours. Studies were repeated at least 3 times. One-way ANOVA with Tukey post hoc analysis (C and D). Student t test (A, B, F). *, P< 0.05; **, P< 0.01, ***, P< 0.001, ****, P< 0.0001.
Transcriptional upregulation of E2F1 promoter is activated by SOX17 deficiency
To characterize whether E2F1 is a direct transcriptional binding target of SOX17 in HPVECs, we did in silico promoter analysis (Eukaryotic Promoter Database)28 of the human E2F1 promoter and found that there are 3 putative SOX17 binding sites in the human E2F1 proximal promoter (−200bp to +1bp of transcription start site) (Figure 5E). We then cloned the E2F1 promoter into the upstream of luciferase gene (Figure 5E). Knockdown of SOX17 significantly upregulated the promoter activity of E2F1 assessed by luciferase assay (Figures 5F), suggesting that SOX17 might repress E2F1 through binding to SOX17 binding sites in the promoter of E2F1. To determine which putative binding sites in the E2F1 promoter are response for E2F1 suppression by SOX17, we mutated individual binding (MBS) site and co-transfected with SOX17 siRNA (Figure 5F). Our data showed that binding site 3 mutation inhibited SOX17 deficiency induced E2F1 promoter activation, suggesting that binding side 3 is likely the binding region of SOX17 in E2F1 promoter in lung ECs (Figures 5F).
E2F1 signaling inhibition rescued SOX17 deficiency-induced PH in mice
To determine whether E2F1 is involved in SOX17 deficiency-induced EC dysfunction, E2F1 inhibitor (HLM) was added to in HPVECs for 6 hours. BrdU assay and qRT-PCR and Western Blot analysis showed that E2F1 inhibition significantly impeded cell proliferation and the levels of the genes (PLK1, CDKN2C, CCNA2) related to cell proliferation. (Figures 6A). We also found that E2F1 inhibition rescued the anti-apoptotic phenotype and paracrine effect of SOX17 deficient HPVECs. (Figure 6B). To further determine the therapeutic potential of targeting E2F1 signaling, we treated ecKO Sox17 mice with HLM or vehicle (Figure 6C). We found that HLM treatment almost completely rescued the PH phenotype, as RVSP levels was significantly reduced by HLM treatment compared to vehicle (Figure 6D). The RV/LV+S ratio was not changed by the treatment of HLM (Figure 6D). Further examination of pulmonary pathology showed that the muscularization of distal pulmonary arteries and pulmonary wall thickness were markedly attenuated by HLM treatment (Figures 6E). Collectively, our studies suggest that E2F1 signaling mediates SOX17 deficiency-induced PH in mice and targeting E2F1 represents a novel therapeutic approach for the treatment of PH with SOX17 deficiency.
Figure 6. Pharmacological inhibition of E2F1 reduced EC dysfunction and PH development in ecKO Sox17 mice.
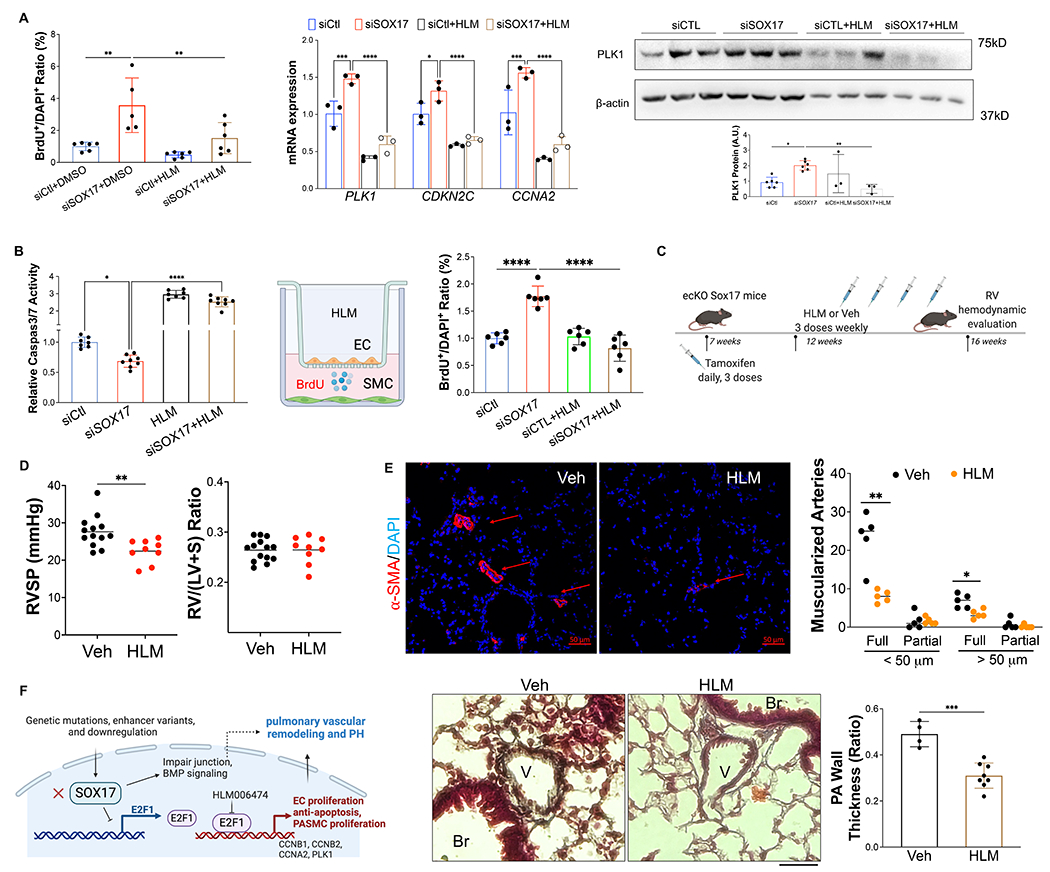
(A) E2F1 inhibition reduced EC proliferation measured by BrdU incorporation assay. At 48 hours post-transfection of siRNA against SOX17 or control siRNA, HPVECs were treated with DMSO or HLM (40 μM) for 12 hours in serum/growth factors free medium. 2.5% FBS and BrdU were added in the medium at 4 hours prior to cells harvest. qRT-PCR analysis demonstrated normalization of the expression of genes related to cell proliferation after E2F1 inhibition in HPVECs. At 48 hours post-transfection, HPVECs were treated with DMSO or HLM for 12 hours in serum/growth factors free medium. 2.5% FBS were added in the medium at 4 hours prior to RNA isolation. E2F1 inhibition reduced cell proliferation marker PLK1 expression in SOX17 deficiency in HPVECs. (B) Pharmacological inhibition of E2F1 increased EC apoptosis in SOX17 deficient HPVECs. At 48 hours post-transfection, HPVECs were treated with DMSO or HLM for 12 hours in serum/growth factors free medium, followed by measurement of Caspase 3/7 activities. (C) A diagram showing the strategy of E2F1 inhibition in ecKO Sox17 mice. (D) RVSP was attenuated by E2F1 inhibition in ecKO Sox17 mice. RV hypertrophy was not altered by E2F1 inhibition. (E) Muscularization of distal pulmonary arteries were reduced by E2F1 inhibition in ecKO Sox17 mice compared to vehicle. α-SMA+ vessels were quantified in 20 field at 10X magnification per mouse. Pentachrome staining showed that E2F1 inhibition by HLM attenuated pulmonary wall thickness. Wall thickness was calculated by the distance between internal wall and external wall divided by the distance between external wall and the center of lumen. Studies were repeated at least 3 times (A and B). One-way ANOVA with Tukey post hoc analysis (A and B) and Student t test (D and E). *, P< 0.05; **, P< 0.01, ***, P< 0.001, ****, P< 0.0001. Scale bar, 50μm.
Discussion
The present study has demonstrated that genetic disruption of Sox17 in ECs induces mild PH as evident by increased RVSP and pulmonary vascular remodeling. We also observed that SOX17 expression is significantly downregulated in isolated PAECs from IPAH patients and is diminished in the occlusive vessels of IPAH lungs. In addition, we found the increased cell proliferation, survival and paracrine effect, impairment of cellular junction and BMP signaling in SOX17 deficient PAECs. We then demonstrated that E2F1 is induced by loss of SOX17 and mediates the cell dysfunctions induced by SOX17 deficiency. Pharmacological inhibition of E2F1 attenuated PH in ecKO Sox17 mice. These findings raise the exciting possibility that inhibition of E2F1 signaling could treat PAH patients with SOX17 deficiency (Figure 6F).
Endothelial dysfunction is believed to be the initial event during the development of PAH29. Single-cell transcriptomics analysis showed that expression of SOX17 is preferentially expressed in the lung ECs compared to other cell types. However, SOX17 expression is markedly downregulated in the lung ECs isolated from IPAH patients and the lung of MCT-induced PH models, suggesting that EC SOX17 deficiency mediates the development of PAH in patients.
Endothelial dysfunctions including hyperproliferation and anti-apoptosis are hallmark of PAH 25,26,30. Increased cell proliferation and apoptosis-resistance were evident in the SOX17-deficienct ECs. SOX17 deficiency also led to upregulation of glycolysis, one of important mechanisms mediating EC dysfunction in PAH31. We also observed that loss of SOX17 resulted in impairment of cellular junction integrity and BMP signaling, important features of lung vasculature in maintaining lung hemostasis. Loss of SOX17 in ECs also enhanced the paracrine effect such as promotion of PASMCs proliferation. Our scRNA-seq analysis also indicated there might be deficiency of lung arterial EC differentiation in Sox17 deficiency lung (Figure S7), as SOX17 is critical for maintaining arterial identity7, which is consistence with recent study showing Notch1 deficiency due to Sox17 loss in mice15. Using tamoxifen-inducible EC-specific Sox17 deletion in adult mice, our work demonstrated the causal role of SOX17 deficiency in inducing endothelial dysfunction, pulmonary vascular remodeling and the development of PH. This observation is consistent with the finding that SOX17 mutations were present in patients with IPAH and congenital heart disease associated PAH12,13. The effects and role of SOX17 in PH have been recently reported. Park et al showed that Sox17 deficiency and hypoxia activated HGF/c-Met signaling in lung ECs15. Walters et al demonstrated that SOX17 enhancer variants lead to downregulation of SOX17 and result in disturbed lung ECs function and PAH through different binding of HOXA5 and ROR-α15. Sangam et al reported that SOX17 deficiency inhibits mitochondrial bioenergetics and induces PAH partly via HIF-2α signaling16. Zou et al identified SOX17-associated exosomes including miR-224-5p and miR-361-3p improves endothelial function32. Taken together, these studies highlight the critical role of SOX17 in maintaining endothelial function and repressing PH development.
In addition to PAH patients, SOX17 expression is downregulated in many forms of cancer, including colorectal cancer33, breast cancer34, endometrial cancer35, and cholangiocarcinoma36, due to DNA hypermethylation at SOX17 promoter loci. Mechanistically, SOX17 serves as a tumor suppressor through the suppression of tumor cell proliferation and migration via modulation of Wnt signaling36–38. Reduced SOX17 expression is also present in the intracerebral arteries of intracerebral aneurysm patients39. Deficiency of SOX17 in ECs induces intracerebral aneurysm 39. Other studies demonstrated that EC-specific inactivation of Sox17 in mice leads to brain microcirculation leakage due to loss of Wnt/β-catenin signaling40. It seems that β-catenin is not involved in the pro-proliferation and anti-apoptosis phenotypes of SOX17 deficient HPVECs, as β-catenin knockdown did not block the pro-proliferation effect induced by loss of SOX17 (Figure S8).
Using unbiased analysis of the single-cell and bulk transcriptomes altered by SOX17 deficiency, we identified cell proliferation and paracrine effect program (including Pdgfb, Edn1, Cxcl12) is upregulated by loss of SOX17 in vitro and in vivo. Other study also showed that combined Sox17 deficiency with hypoxia induced HGF signaling and endothelial proliferation in vivo15. We then predicted and validated that E2F1 is the central governor controlling the EC dysfunction by SOX17 deficiency. E2F1 belongs to a subclass of the E2F transcription factor family and is thought to act as a transcriptional activator, mediating cell proliferation and apoptosis41,42. E2F1 is critical for the expression of various genes regulating G1 to S transition and S phase, including cyclin E, PCNA, Ki67, BUB1, Cyclin A2, Cyclin B1, Cyclin B2, etc43,44. Loss of E2F1 was shown to mediate TNF-α-induced cell cycle arrest in proliferating bovine aortic ECs45. Restoration of E2F activities via adenovirus-mediated E2F1 overexpression promoted EC cell cycle progress and rescued TNF-α-induced apoptosis45. Our studies demonstrated that E2F1 expression and promoter activities are upregulated by SOX17 deficiency in HPVECs likely due to absence of suppression of SOX17 in the proximal region of E2F1 promoter. Moreover, E2F1 has been shown to mediate sodium–hydrogen exchanger 1 (NHE1) induced PASMCs proliferation, hypertrophy and migration in vitro46. E2F1 expression is also significantly increased in the lung of other PH models such as monocrotaline-exposed rats47 and Egln1Tie2Cre mice18,48 (Figures S9A and S9B). Overexpression of E2F1 suppressed BMPR2 expression in the HPVECs (Figure S9C). Taken together, E2F1 activation is likely the common mechanisms mediating pulmonary vascular remodeling and PH development.
The present study has demonstrated that targeting E2F1 signaling with HLM effectively inhibited Sox17 deficiency-induced PH development in mice. Pharmacological inhibition of E2F1 reduced HPVECs pro-proliferation, anti-apoptotic phenotypes and paracrine effect due to SOX17 deficiency and pulmonary vascular remodeling and PH in ecKO Sox17 mice. It is possible that E2F1 inhibition also reduced PASMCs proliferation in ecKO mice. Other studies showed that inhibition of E2F1 signaling prevented occlusive thickening of the vessel wall in venous bypass grafts49. Future studies are warranted to investigate whether or not E2F1 inhibition could attenuate PH development and right heart dysfunction in more severe PH models such as MCT-exposed rat, Sugen5416/hypoxia exposed-rats, or Egln1Tie2Cre mice.
There are several limitations in the current studies. Firstly, it remains unknown whether any of the samples from patients with PAH exhibit mutations in SOX17 or other genes, or if SOX17 deficiency is also present in other subgroups of PAH or PH groups. Secondly, there is a higher prevalence of SOX17 mutations in PAH patients with congenital heart defects compared to other subgroups of PAH. Additionally, PAH patients with SOX17 mutations tend to be younger than those with other mutations. However, it is worth noting that Sox17 ecKO mice did not exhibit any obvious cardiac abnormalities, suggesting that earlier deletion of Sox17 may be necessary to observe cardiac abnormalities in mice with Sox17 deficiency. Lastly, HLM006474 is an inhibitor that targets the E2F family, including E2F1 and other E2F proteins. It has been found to inhibit DNA binding to E2F1, E2F2, and E2F4 in A375 melanoma cells, indicating that there might be some off-target effects of HLM006474 treatment in mice with pulmonary hypertension.
In summary, our studies demonstrate a pathogenic role of endothelial SOX17 deficiency in mediating lung EC dysfunction and pulmonary vascular remodeling, and provide clear evidence of E2F1 activation in the pathogenesis of PH. We also show that pharmacologic inhibition of E2F1 attenuated PH development in ecKO Sox17 mice. These studies suggest that E2F1 inhibition could be a promising approach for the treatment of PAH patients with loss of SOX17 or E2F1 activation.
Perspective
Our studies identified a novel mechanism of SOX17 deficiency in ECs in inducing the development of PH via activation of transcriptional factor E2F1. Future studies are warranty to determine whether E2F1 activation is the common mechanism for PAH development. Genetic deletion and pharmacological inhibition of E2F1 approaches might be used to confirm the role of E2F1 in PH mice or rat models. A clinical work is required for evaluating whether E2F1 is a good therapeutic target for PAH patients.
Supplementary Material
Pathophysiological Novelty and Relevance.
What is new?
In this study, we identified the novel mechanism of SOX17 deficiency in inducing endothelial dysfunction via activation of transcriptional factor E2F1. Pharmacological inhibition of E2F1 attenuated PH development in mice.
What is Relevant?
SOX17 mutation has been identified in patients with PAH. This study found that SOX17 expression is downregulated in the pulmonary endothelial cells of PAH patients. Genetic deletion of Sox17 in endothelial cells leads to PH development in mice, demonstrating that SOX17 deficiency in endothelial cells might induce PAH development in human.
Clinical/Pathophysiological Implications?
This study provides direct evidence that genetic loss of SOX17 in endothelial cells induces PH in mice, which is consistent with the observation that endothelial SOX17 is downregulated in lungs of PAH patients. We validated that inhibition of E2F1 could be a novel approach to treat patients with PAH.
Acknowledgements:
We thank Dr. Marlene Rabinovitch (Stanford University) for her advice on the experimental design and data interpretation and Dr. Indrapal Singh (University of Arizona)’s help on Seahorse experiment. The authors the Pulmonary Hypertension Breakthrough Initiative for providing the Data/tissue samples. Funding for the Pulmonary Hypertension Breakthrough Initiative is provided under an NHLBI R24 grant (R24HL123767) and by the Cardiovascular Medical Research and Education Fund.
Sources of Funding:
This work was supported in part by NIH grant R01HL169509, R01HL170096, R00HL138278, R01HL158596, R01HL62794, AHA Career Development Award 20CDA35310084, The Cardiovascular Research and Education Foundation, Arizona Biomedical Research Centre funding (RFGA2022-01-06), and University of Arizona startup funding to Z.D.
Nonstandard Abbreviations and Acronyms
- PH
Pulmonary hypertension
- IPAH
idiopathic pulmonary arterial hypertension
- BMPR2
bone morphogenetic protein type 2 receptor
- HPVECs
human pulmonary vascular endothelial cells
- PHBI
Pulmonary Hypertension Breakthrough Initiative
- FD
failed donors
- HLM
HLM006474
- scRNA-seq
single-cell RNA-sequencing
- RVSP
right ventricle systolic pressure
- RV/LV+S
the weight ratio of the right ventricular free wall to left ventricle plus septum
- LV/BW
the weight ratio of left ventricle weight vs body weight
- cKO
Sox17f/+;Tie2Cre
- SMA
smooth muscle actin
- ecKO
Sox17f/f;EndoSCL-CreERT2
- WT
Sox17f/f
- BrdU
5-bromo-2′-deoxyuridine
- PLK1
polo-like kinase 1
- PASMCs
pulmonary arterials smooth muscle cell
- siRNA
short interfering RNA
- MBS
mutated binding site
Footnotes
Disclosure: None.
References
- 1.Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, Carlsen J, Coats AJS, Escribano-Subias P, Ferrari P, Ferreira DS, Ghofrani HA, Giannakoulas G, Kiely DG, Mayer E, Meszaros G, Nagavci B, Olsson KM, Pepke-Zaba J, Quint JK, Rådegran G, Simonneau G, Sitbon O, Tonia T, Toshner M, Vachiery JL, Vonk Noordegraaf A, Delcroix M, Rosenkranz S, ESC/ERS Scientific Document Group, Schwerzmann M, Dinh-Xuan AT, Bush A, Abdelhamid M, Aboyans V, Arbustini E, Asteggiano R, Barberà JA, Beghetti M, Čelutkienė J, Cikes M, Condliffe R, De Man F, Falk V, Fauchier L, Gaine S, Galié N, Gin-Sing W, Granton J, Grünig E, Hassoun PM, Hellemons M, Jaarsma T, Kjellström B, Klok FA, Konradi A, Koskinas KC, Kotecha D, Lang I, Lewis BS, Linhart A, Lip GYH, Løchen ML, Mathioudakis AG, Mindham R, Moledina S, Naeije R, Nielsen JC, Olschewski H, Opitz I, Petersen SE, Prescott E, Rakisheva A, Reis A, Ristić AD, Roche N, Rodrigues R, Selton-Suty C, Souza R, Swift AJ, Touyz RM, Ulrich S, Wilkins MR, Wort SJ. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. European Heart Journal. 2022;43:3618–3731. [DOI] [PubMed] [Google Scholar]
- 2.Austin ED, Newman JH, Loyd JE, Phillips JA. Heritable and Idiopathic Forms of Pulmonary Arterial Hypertension. In: Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics. Elsevier; 2020. p. 439–462. [Google Scholar]
- 3.Evans JDW, Girerd B, Montani D, Wang X-J, Galiè N, Austin ED, Elliott G, Asano K, Grünig E, Yan Y, Jing Z-C, Manes A, Palazzini M, Wheeler LA, Nakayama I, Satoh T, Eichstaedt C, Hinderhofer K, Wolf M, Rosenzweig EB, Chung WK, Soubrier F, Simonneau G, Sitbon O, Gräf S, Kaptoge S, Di Angelantonio E, Humbert M, Morrell NW. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. The Lancet Respiratory Medicine. 2016;4:129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Southgate L, Machado RD, Gräf S, Morrell NW. Molecular genetic framework underlying pulmonary arterial hypertension. Nature Reviews Cardiology. 2020;17:85–95. [DOI] [PubMed] [Google Scholar]
- 5.Clarke RL, Yzaguirre AD, Yashiro-Ohtani Y, Bondue A, Blanpain C, Pear WS, Speck NA, Keller G. The expression of Sox17 identifies and regulates haemogenic endothelium. Nature Cell Biology. 2013;15:502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim I, Saunders TL, Morrison SJ. Sox17 Dependence Distinguishes the Transcriptional Regulation of Fetal from Adult Hematopoietic Stem Cells. Cell. 2007;130:470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corada M, Orsenigo F, Morini MF, Pitulescu ME, Bhat G, Nyqvist D, Breviario F, Conti V, Briot A, Iruela-Arispe ML, Adams RH, Dejana E. Sox17 is indispensable for acquisition and maintenance of arterial identity. Nature Communications. 2013;4:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engert S, Burtscher I, Kalali B, Gerhard M, Lickert H. The Sox17CreERT2 knock-in mouse line displays spatiotemporal activation of Cre recombinase in distinct Sox17 lineage progenitors. Genesis. 2013;51:793–802. [DOI] [PubMed] [Google Scholar]
- 9.Lange AW, Haitchi HM, LeCras TD, Sridharan A, Xu Y, Wert SE, James J, Udell N, Thurner PJ, Whitsett JA. Sox17 is required for normal pulmonary vascular morphogenesis. Developmental Biology. 2014;387:109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu M, Zhang L, Marsboom G, Jambusaria A, Xiong S, Toth PT, Benevolenskaya EV, Rehman J, Malik AB. Sox17 is required for endothelial regeneration following inflammation-induced vascular injury. Nature Communications. 2019;10:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang H, Lee S, Lee S, Kim K, Yang Y, Kim JH, Adams RH, Wells JM, Morrison SJ, Koh GY, Kim I. Sox17 promotes tumor angiogenesis and destabilizes tumor vessels in mice. Journal of Clinical Investigation. 2013;123:418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gräf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W, Hodgson J, Liu B, Salmon RM, Southwood M, Machado RD, Martin JM, Treacy CM, Yates K, Daugherty LC, Shamardina O, Whitehorn D, Holden S, Aldred M, Bogaard HJ, Church C, Coghlan G, Condliffe R, Corris PA, Danesino C, Eyries M, Gall H, Ghio S, Ghofrani HA, Gibbs JSR, Girerd B, Houweling AC, Howard L, Humbert M, Kiely DG, Kovacs G, MacKenzie Ross RV, Moledina S, Montani D, Newnham M, Olschewski A, Olschewski H, Peacock AJ, Pepke-Zaba J, Prokopenko I, Rhodes CJ, Scelsi L, Seeger W, Soubrier F, Stein DF, Suntharalingam J, Swietlik EM, Toshner MR, Van Heel DA, Vonk Noordegraaf A, Waisfisz Q, Wharton J, Wort SJ, Ouwehand WH, Soranzo N, Lawrie A, Upton PD, Wilkins MR, Trembath RC, Morrell NW. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nature Communications. 2018;9:1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu N, Welch CL, Wang J, Allen PM, Gonzaga-Jauregui C, Ma L, King AK, Krishnan U, Rosenzweig EB, Ivy DD, Austin ED, Hamid R, Pauciulo MW, Lutz KA, Nichols WC, Reid JG, Overton JD, Baras A, Dewey FE, Shen Y, Chung WK. Rare variants in SOX17 are associated with pulmonary arterial hypertension with congenital heart disease. Genome Medicine. 2018;10:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rhodes CJ, Batai K, Bleda M, Haimel M, Southgate L, Germain M, Pauciulo MW, Hadinnapola C, Aman J, Girerd B, Arora A, Knight J, Hanscombe KB, Karnes JH, Kaakinen M, Gall H, Ulrich A, Harbaum L, Cebola I, Ferrer J, Lutz K, Swietlik EM, Ahmad F, Amouyel P, Archer SL, Argula R, Austin ED, Badesch D, Bakshi S, Barnett C, Benza R, Bhatt N, Bogaard HJ, Burger CD, Chakinala M, Church C, Coghlan JG, Condliffe R, Corris PA, Danesino C, Debette S, Elliott CG, Elwing J, Eyries M, Fortin T, Franke A, Frantz RP, Frost A, Garcia JGN, Ghio S, Ghofrani HA, Gibbs JSR, Harley J, He H, Hill NS, Hirsch R, Houweling AC, Howard LS, Ivy D, Kiely DG, Klinger J, Kovacs G, Lahm T, Laudes M, Machado RD, MacKenzie Ross RV, Marsolo K, Martin LJ, Moledina S, Montani D, Nathan SD, Newnham M, Olschewski A, Olschewski H, Oudiz RJ, Ouwehand WH, Peacock AJ, Pepke-Zaba J, Rehman Z, Robbins I, Roden DM, Rosenzweig EB, Saydain G, Scelsi L, Schilz R, Seeger W, Shaffer CM, Simms RW, Simon M, Sitbon O, Suntharalingam J, Tang H, Tchourbanov AY, Thenappan T, Torres F, Toshner MR, Treacy CM, Vonk Noordegraaf A, et al. Genetic determinants of risk in pulmonary arterial hypertension: international genome-wide association studies and meta-analysis. The Lancet Respiratory Medicine. 2019;7:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soon Park C, Hyun Kim S, Young Yang H, Kim J-H, Theo Schermuly R, Seul Cho Y, Kang H, Park J-H, Lee E, Park H, Myung Yang J, Wook Noh T, Lee S, Sik Bae S, Han J, Seok Ju Y, Park J, Kim I. Sox17 Deficiency Promotes Pulmonary Arterial Hypertension via HGF (Hepatocyte Grow Factor)/c-Met Signaling. Circulation research. 2022;101161CIRCRESAHA122320845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sangam S, Sun X, Schwantes-An T-H, Yegambaram M, Lu Q, Shi Y, Cook T, Fisher A, Frump AL, Coleman A, Sun Y, Liang S, Crawford H, Lutz KA, Maun AD, Pauciulo MW, Karnes JH, Chaudhary KR, Stewart DJ, Langlais PR, Jain M, Alotaibi M, Lahm T, Jin Y, Gu H, Tang H, Nichols WC, Black SM, Desai AA. SOX17 Deficiency Mediates Pulmonary Hypertension: At the Crossroads of Sex, Metabolism, and Genetics. Am J Respir Crit Care Med [Internet]. 2023. [cited 2023 Mar 29]; Available from: 10.1164/rccm.202203-0450OC [DOI] [PMC free article] [PubMed]
- 17.Walters R, Vasilaki E, Aman J, Chen C-N, Wu Y, Liang OD, Ashek A, Dubois O, Zhao L, Sabrin F, Cebola I, Ferrer J, Morrell NW, Klinger JR, Wilkins MR, Zhao L, Rhodes CJ. SOX17 Enhancer Variants Disrupt Transcription Factor Binding And Enhancer Inactivity Drives Pulmonary Hypertension. Circulation. 2023;CIRCULATIONAHA.122.061940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai Z, Li M, Wharton J, Zhu MM, Zhao YY. Prolyl-4 Hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2α. Circulation. 2016;133:2447–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran KA, Zhang X, Predescu D, Huang X, Machado RF, Göthert JR, Malik AB, Valyi-Nagy T, Zhao Y-Y. Endothelial β-Catenin Signaling Is Required for Maintaining Adult Blood–Brain Barrier Integrity and Central Nervous System Homeostasis. Circulation. 2016;133:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mura M, Cecchini MJ, Joseph M, Granton JT. Osteopontin lung gene expression is a marker of disease severity in pulmonary arterial hypertension. Respirology. 2019;24:1104–1110. [DOI] [PubMed] [Google Scholar]
- 21.Lee CH, Taketo T. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2Cre mouse line. Genesis. 2001;30:36–44. [DOI] [PubMed] [Google Scholar]
- 22.Göthert JR, Gustin SE, Van Eekelen JAM, Schmidt U, Hall MA, Jane SM, Green AR, Göttgens B, Izon DJ, Begley CG. Genetically tagging endothelial cells in vivo: Bone marrow-derived cells do not contribute to tumor endothelium. Blood. 2004;104:1769–1777. [DOI] [PubMed] [Google Scholar]
- 23.Dai Z, Zhu MM, Peng Y, Jin H, Machireddy N, Qian Z, Zhang X, Zhao YY. Endothelial and smooth muscle cell interaction via FoxM1 signaling mediates vascular remodeling and pulmonary hypertension. American Journal of Respiratory and Critical Care Medicine. 2018;198:788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, Nie Q. Inference and analysis of cell-cell communication using CellChat. Nature Communications. 2021;12:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dabral S, Tian X, Kojonazarov B, Savai R, Ghofrani HA, Weissmann N, Florio M, Sun J, Jonigk D, Maegel L, Grimminger F, Seeger W, Pullamsetti SS, Schermuly RT. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. European Respiratory Journal. 2016;48:1137–1149. [DOI] [PubMed] [Google Scholar]
- 26.Caruso P, Dunmore BJ, Schlosser K, Schoors S, Dos Santos C, Perez-Iratxeta C, Lavoie JR, Zhang H, Long L, Flockton AR, Frid MG, Upton PD, D’Alessandro A, Hadinnapola C, Kiskin FN, Taha M, Hurst LA, Ormiston ML, Hata A, Stenmark KR, Carmeliet P, Stewart DJ, Morrell NW. Identification of MicroRNA-124 as a Major Regulator of Enhanced Endothelial Cell Glycolysis in Pulmonary Arterial Hypertension via PTBP1 (Polypyrimidine Tract Binding Protein) and Pyruvate Kinase M2. Circulation. 2017;136:2451–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Janky R, Verfaillie A, Imrichová H, van de Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval Sanchez M, Potier D, Svetlichnyy D, Kalender Atak Z, Fiers M, Marine JC, Aerts S. iRegulon: From a Gene List to a Gene Regulatory Network Using Large Motif and Track Collections. PLoS Computational Biology. 2014;10:e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dreos R, Ambrosini G, Périer RC, Bucher P. The Eukaryotic Promoter Database: Expansion of EPDNew and new promoter analysis tools. Nucleic Acids Research. 2015;43:D92–D96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483–5495. [DOI] [PubMed] [Google Scholar]
- 30.Xu W, Erzurum SC. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Comprehensive Physiology. 2011;1:357–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan SY, Rubin LJ. Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. European Respiratory Review [Internet]. 2017;26. Available from: 10.1183/16000617.0094-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou X, Liu T, Huang Z, Zhou W, Yuan M, Zhao H, Pan Z, Chen P, Shao Y, Hu X, Zhang S, Zheng S, Zhang Y, Huang P. SOX17 is a Critical Factor in Maintaining Endothelial Function in Pulmonary Hypertension by an Exosome-Mediated Autocrine Manner. Advanced Science. 2023;n/a:2206139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang W, Glöckner SC, Guo M, Machida EO, Wang DH, Easwaran H, Van Neste L, Herman JG, Schuebel KE, Watkins DN, Ahuja N, Baylin SB. Epigenetic inactivation of the canonical Wnt antagonist SRY-box containing gene 17 in colorectal cancer. Cancer Research. 2008;68:2764–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu DY, Wang ZM, Li-Chen, Wang BL, Shen ZZ, Huang W, Shao ZM. Sox17, the canonical Wnt antagonist, is epigenetically inactivated by promoter methylation in human breast cancer. Breast Cancer Research and Treatment. 2010;119:601–612. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Bao W, Wang K, Lu W, Wang H, Tong H, Wan X. SOX17 is a tumor suppressor in endometrial cancer. Oncotarget. 2016;7:76036–76046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merino-Azpitarte M, Lozano E, Perugorria MJ, Esparza-Baquer A, Erice O, Santos-Laso Á, O’Rourke CJ, Andersen JB, Jiménez-Agüero R, Lacasta A, D’Amato M, Briz Ó, Jalan-Sakrikar N, Huebert RC, Thelen KM, Gradilone SA, Aransay AM, Lavín JL, Fernández-Barrena MG, Matheu A, Marzioni M, Gores GJ, Bujanda L, Marin JJG, Banales JM. SOX17 regulates cholangiocyte differentiation and acts as a tumor suppressor in cholangiocarcinoma. Journal of Hepatology. 2017;67:72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hopman ANH, Moshi JM, Hoogduin KJ, Ummelen M, Henfling MER, van Engeland M, Wouters KAD, Stoop H, Looijenga LHJ, Ramaekers FCS. SOX17 expression and its downregulation by promoter methylation in cervical adenocarcinoma in situ and adenocarcinoma. Histopathology. 2020;76:383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tan DS, Holzner M, Weng M, Srivastava Y, Jauch R. SOX17 in cellular reprogramming and cancer. Seminars in Cancer Biology. 2019;0–1. [DOI] [PubMed] [Google Scholar]
- 39.Lee S, Kim IK, Ahn JS, Woo DC, Kim ST, Song S, Koh GY, Kim HS, Jeon BH, Kim I. Deficiency of endothelium-specific transcription factor Sox17 induces intracranial aneurysm. Circulation. 2015;131:995–1005. [DOI] [PubMed] [Google Scholar]
- 40.Corada M, Orsenigo F, Bhat GP, Conze LL, Breviario F, Cunha SI, Claesson-Welsh L, Beznoussenko GV, Mironov AA, Bacigaluppi M, Martino G, Pitulescu ME, Adams RH, Magnusson P, Dejana E. Fine-Tuning of Sox17 and Canonical Wnt Coordinates the Permeability Properties of the Blood-Brain Barrier. Circulation research. 2019;124:511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buccitelli C, Salgueiro L, Rowald K, Sotillo R, Mardin BR, Korbel JO. Pan-cancer analysis distinguishes transcriptional changes of aneuploidy from proliferation. Genome Research. 2017;27:501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitfield ML, George LK, Grant GD, Perou CM. Common markers of proliferation. Nature Reviews Cancer. 2006;6:99–106. [DOI] [PubMed] [Google Scholar]
- 43.Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR. Role for E2F in Control of Both DNA Replication and Mitotic Functions as Revealed from DNA Microarray Analysis. Molecular and Cellular Biology. 2001;21:4684–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9309–9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spyridopoulos I, Principe N, Krasinski KL, Xu SH, Kearney M, Magner M, Isner JM, Losordo DW. Restoration of E2F expression rescues vascular endothelial cells from tumor necrosis factor-α-induced apoptosis. Circulation. 1998;98:2883–2890. [DOI] [PubMed] [Google Scholar]
- 46.Yu L, Hales CA. Silencing of sodium-hydrogen exchanger 1 attenuates the proliferation, hypertrophy, and migration of pulmonary artery smooth muscle cells via E2F1. American Journal of Respiratory Cell and Molecular Biology. 2011;45:923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai Z, Zhu MM, Peng Y, Machireddy N, Evans CE, Machado R, Zhang X, Zhao YY. Therapeutic targeting of vascular remodeling and right heart failure in pulmonary arterial hypertension with a HIF-2a inhibitor. American Journal of Respiratory and Critical Care Medicine. 2018;198:1423–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu B, Peng Y, Yi D, Machireddy N, Dong D, Ramirez K, Dai J, Vanderpool R, Zhu MM, Dai Z, Zhao Y-Y. Endothelial PHD2 deficiency induces nitrative stress via suppression of caveolin-1 in pulmonary hypertension. The European respiratory journal. 2022;33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mann MJ, Whittemore AD, Donaldson MC, Belkin M, Conte MS, Polak JF, Orav EJ, Ehsan A, Dell’Acqua G, Dzau VJ. Ex-vivo gene therapy of human vascular bypass grafts with E2F decoy: The PREVENT single-centre, randomised, controlled trial. Lancet. 1999;354:1493–1498. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing and single-cell RNA-sequencing (scRNA-seq) data have been deposited in the GEO database under accession number GSE192649 and GSE218398. Scripts used for single-cell RNA sequencing analysis and analyzed data in R objects are available in Figshare (https://figshare.com/s/37782988b8cac7cedcf9).