

Summary
Demethylation of the Treg Specific Demethylation Region (TSDR) of the Foxp3 gene is the hallmark of Foxp3+ T regulatory cell (Treg) stability, but the cellular signaling that programs this epigenetic state remains undefined. Herein, we show that suppressed C3a and C5a receptor (C3ar1/C5ar1) signaling in murine Tregs plays an obligate role. Murine C3ar1−/−C5ar1−/− Foxp3+ cells showed increased SOCS1/2/3 expression, Vitamin C stabilization, and TET1/2/3 expression, all of which are linked to Treg stability. C3ar1−/−C5ar1−/− Foxp3+ cells additionally were devoid of BRD4 signaling that primes Th17 cell lineage commitment. Orally induced ova specific C3ar1−/−C5ar1−/− Foxp3+ OT-II Tregs transferred to ova-immunized WT recipients remained >90% Foxp3+ out to 4 months, whereas identically generated CD55−/− (DAF−/−) Foxp3+ OT-II Tregs (in which C3ar1/C5ar1 signaling is potentiated) lost >75% Foxp3 expression by 14 days. After 4 months in vivo, the C3ar1−/−C5ar1−/− Foxp3+ OT-II Tregs fully retained Foxp3 expression even with ova challenge and produced copious TGF-β and IL-10. Their TSDR was de-methylated comparably to that of thymic Tregs (tTregs). They exhibited nuclear translocation of NFAT and NF-ĸB reported to stabilize tTregs by inducing hairpin looping of the TSDR to the Foxp3 promoter. Thus, disabled CD4+ cell C3ar1/C5ar1 signaling triggers the sequential cellular events that lead to demethylation of the Foxp3 TSDR.
Introduction
Demethylation of the Foxp3 T regulatory cell (Treg) Specific Demethylation Region (TSDR) of the Foxp3 gene is the genomic signature of stable Tregs (1). This insight came from findings that the TSDR is demethylated in thymic Tregs (tTregs) which exhibit durable Foxp3 expression, whereas it is methylated in ex vivo TGF-β induced Tregs (iTregs) that rapidly lose Foxp3 expression. Characterizing the processes underlying TSDR demethylation is important immunologically for understanding T cell homeostasis, Treg control of T effector (Teff) responses, avoidance of autoimmunity, and tolerance.
Knowledge of the processes that give rise to TSDR demethylation is highly relevant therapeutically as durable Foxp3 expression can enable effective Treg biotherapy. This knowledge also is important for characterizing Treg dysfunction in disease. Because a demethylated TSDR is required for Treg durability, ex vivo expanded populations of thymic Tregs (tTregs) that possess this modification and peripherally generated Tregs (pTregs) that acquire it, have been employed. The tTreg and pTreg subsets, however, are restricted to their existing specificities, and can lack full suppressor functionality. De novo induction of polyclonal or antigen-specific iTregs containing a demethylated-TSDR and possessing robust suppressor activity, if achievable, could overcome these limitations in that they could function more potently. An iTreg induction protocol enabling the ex vivo generation of potent iTregs with demethylated TSDRs could facilitate the induction of both autologous and allogeneic immunosuppression.
While the signaling cascade in CD4+ cells that leads to TSDR demethylation and durable Tregs remains undefined, a number of processes connected with the acquisition of this state have been implicated. Among these processes are 1) upregulation of Suppressor Of Cytokine Signaling (SOCS1/2/3) expression, 2) suppression of phosphatidylinositol 3,4,5 trisphosphate [PtdIns (3,4,5)P3] assembly and AKT phosphorylation, 3) remodeling of genomic DNA, and 4) repression of Th17 gene transcription. A final process implicated in the stabilization is 5) hairpin looping of intron 1 Conserved Noncoding Sequence 2 (CNS2) (containing the TSDR) to the Foxp3 promoter (2). While these and other (see Discussion) reported processes seemingly appear unconnected, reversal of any one of them disrupts the induction of stable Tregs. Whether a shared signaling process underlies their collective induction has remained unknown.
In previous studies (3, 4), we found that interacting dendritic cells (DCs) and CD4+ T cells locally produce C3a and C5a from endogenously synthesized complement C3 and C5 proteins and the two anaphylatoxins establish autocrine C3ar1 and C5ar1 signaling loops in both partners. The C3ar1/C5ar1 G protein coupled receptor (GPCR) signaling is controlled by the cell associated regulator CD55 [aka decay accelerating factor (DAF)]. Potentiated C3ar1/C5ar1 signaling occurring as a result of downregulated CD55 promotes Th1 and Th17 Teff responses (4, 5). Conversely, repressed C3ar1/C5ar1 signaling occurring in conjunction with heightened CD55 expression programs T cells to endogenously synthesize TGF-β and IL-10 and become Tregs (6). The Tregs generated in this way show upregulated expression of C5a receptor 2 (C5ar2 aka C5L2), a decoy GPCR devoid of a G protein that scavenges C5a/C3a (7), preventing C3ar1/C5ar1 signaling into the Tregs. The C3ar1/C5ar1 repressed Tregs show disabled AKT phosphorylation (6), absent IL-6 production (6), and inhibited STAT3 activation (6). Functionally they exhibited 4–8-fold > suppressor activity than Tregs conventionally induced by incubating naïve CD4+ cells with exogenous TGF-β (6). They also possessed greater stability in short term (5 day) in vitro studies. Methylation assays, however, showed 88% TSDR methylation (6). We hypothesized that demethylation might require longer times and/or in vivo conditions.
In this study, we found that disrupted C3ar1/C5ar1 signaling elicits multiple processes that have been implicated in demethylation of the TSDR, including hairpin looping of the CNS2 region to the Foxp3 promoter that has been mechanistically connected with stabilizing Foxp3 transcription. We then tested whether disrupted C3ar1/C5ar1 transduction is the signaling condition required for the induction of stable Ag specific Tregs in vivo. We transferred sorted ova-specific Foxp3−OT-II CD4+ cells to naïve recipients and orally immunized the recipients by feeding them ova to induce de novo ova specific OT-II cell pTregs. We isolated the Foxp3+ cells and transferred the flow purified pTregs to naïve recipients. We then studied their properties over a 4-month period in the absence and presence of ova challenge. We found that pTregs in which CD55 restraint on C3ar1/C5ar1 signaling is lifted rapidly lost Foxp3 expression. In contrast, pTregs disabled in C3ar1/C5ar1 signaling survived > 4 months and retained full immunosuppressive capacity. Phenotypic analyses showed that the 4-month pTregs possessed a demethylated TSDR characteristic of stable tTregs. The analyses showed that the ova specific pTregs expressed high levels of TGF-β and IL-10 characteristic of robust Treg function..
Methods
Animals
Foxp3-GFP OT-II mice CD55−/−, WT and C3ar1−/−C5ar1−/− backgrounds were prepared in the Medof lab. Foxp3-GFP mice were kindly provided by Dr. V Kuckroo (Harvard Medical School). They were maintained in the Case Western Reserve Animal Facilities. All experiments were approved by CWRU Institutional Animal Care and Use Committee (IACUC).
Reagents and Antibodies
Ovalbumin (ova) was obtained from Sigma-Aldrich (St Louis, Mo). qPCR primers were designed by obtained Integrated DNA Technologies (IDT) (Coralville, IA). Vitamin C was obtained from Fisher Scientific. C3ar1-A 559410-10mg and C5ar1-A-234415-5mg were from Millipore 31908021. Fluorescein isothiocyanate–anti-CD4 (RM-4-5), phycoerythrin–indotricarbocyanine–anti-Thy-1.1 (53–2.1), allophycocyanin–anti-Thy-1.2 (53–2.1), phycoerythrin–anti-CD45.1 (A20) and allophycocyanin–eFluor 780–anti-CD45.2 (104) were from eBioscience.
Foxp3+ T regulatory cell Induction
CD4+ T cells were isolated from spleens and LNs via CD4+ Negative Selection Cocktail Beads per the manufacturer’s instructions (Miltenyi, Biotec) and purified with an Automacs Pro. Foxp3− and Foxp3+ CD4+ T cells were purified by cell sorting gating on GFP+ and GFP− populations as described in (6) As described (6), iTregs were prepared by incubating Foxp3− (GFP−) CD4+ cells with anti-CD3/28 Dynabeads plus IL-2 (5 ng/ml) for 3–5 days after which Foxp3+ (GFP+) Tregs were purified by cell sorting. pTregs were prepared by adoptive transfer of Thy-1.2+ (CD45.1+) CD4+ OT-II cells (4 × 106) on each genotype via tail vein into ova-fed WT Thy-1.1+ mice followed by immunizing the recipients with ova (323–339) (100 ug) in IFA.
Flow Cytometry and Cell Sorting
All FACS analyses and studies involving cell sorting were performed on an LSR2 Flow Cytometer in the Flow Cytometry Core of the Comprehensive Cancer Center at Case Western Reserve University. Fluorescent labeled anti-Thy-1.2 and ova mAbs were obtained from Bioscience. Florescent labeled anti-human TGF-β and IL-10 mAbs were obtained from Bioscience. Staining and gating were performed as described in (4, 6).
Amnis Cytometry
Assays for nuclear NFAT and NF-kB were carried out on an Amnis Cytometer (Beckman-Coulter San Diego, CA) (Dept of Pathology, Case Western Reserve University) using fluorescent labeled anti-NFAT and NF-kB mAbs (Cell Signaling Inc. Danvers MA). A gate was placed on the nuclear stain and NFAT and NF-kB signals in and outside of the gate were assessed.
TSDR Methylation Analysis
Measurements of TSDR methylation was performed by bisulfite sequencing as described in (8, 9) in the Shevach lab (NIAID, NIH, Bethesda, MD).
Statistics.
Statistical significance was determined by Student’s t-test (unpaired, two-tailed) with Microsoft Excel or GraphPad Prism 5.
Animal Studies
All mouse breeding and adoptive transfers, long-term follow-up and harvesting of spleen and lymph node cells were approved by the Case Western Reserve Institutional Animal Care and Use Committee (IACUC).
Results
Disrupted C3ar1/C5ar1 signaling in Tregs induces expression of SOCS1, 2, and 3
Prior work by others on Treg stability found that STAT3 activation in Tregs leads to their conversion to “exTregs” or Th17 cells (10, 11). Signaling studies showed that SOCS1 and SOCS3 which inactivate STAT3 participate in inhibiting this conversion and thereby promote Treg stability (12). Our previous studies of Treg induction (6) linked disabled CD4+ cell C3ar1/C5ar1 signaling with inhibited STAT3 activation but did not provide a mechanism for the inhibition.
To determine if absent C3ar1/C5ar1 signaling induces the expression of the STAT3 phosphatases SOCS1 and/or 3 in CD4+ cells, we sorted Foxp3+ CD25+ cells deriving from Treg induction cultures of Foxp3−CD55−/−, WT, and C3ar1−/−C5ar1−/− Foxp3− CD4+ cells and quantitated SOCS 1,2 and 3 mRNA levels in induced Foxp3+ CD25+ cells Levels of SOCS1 and 3 were 200–300% higher in Foxp3+ C3ar1−/−C5ar1−/− CD4+ cells than in Foxp3+ WT CD4+ cells, whereas they were >500% lower in Foxp3+ CD55−/− CD4+ cells (Fig 1A). Similar results were observed for SOCS2 which inhibits IL-4 induction of STAT6 activation (Fig 1A).
Figure 1: Metabolic pathways connected with absent C3ar1/C5ar1 signaling in activated CD4+ cells.
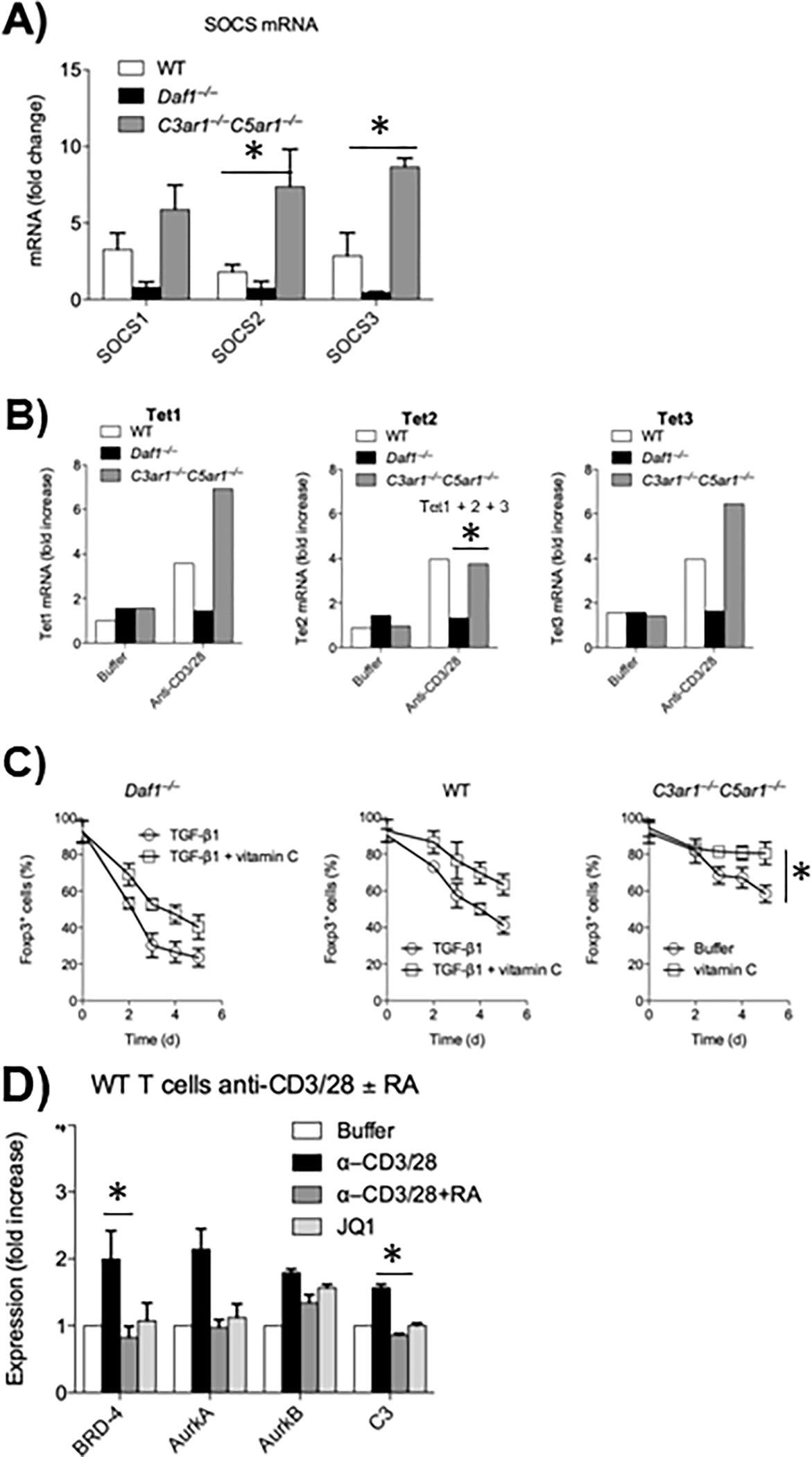
A) Sorted CD55−/−, WT, and C3ar1−/−C5ar1−/− CD4+ cells were incubated with anti-CD3/28 Dynabeads plus IL-2 for 5 days after which Foxp3+ cells were sorted. A) SOCS1, 3, and 4 mRNA levels. Induction in C3ar1−/−C5ar1−/− vs WT cells and B) TET1, TET2, and TET3 mRNA levels were quantitated by qPCR. Representative of 2 assays. Induction in C3ar1−/−C5ar1−/− vs CD55−/− cells. C) Sorted Foxp3− CD55−/−, WT, and C3ar1−/−C5ar1−/− CD4+ cells were incubated with anti-CD3/28 Dynabeads and IL-2 plus TGF-β in the absence and presence of Vitamin C and percent Foxp3+ cells quantitated daily. Induction in C3ar1−/−C5ar1−/− vs WT− cells. D) WT CD4+ cells were incubated for 1 h with anti-CD3/28 Dynabeads without or with C3ar1/C5ar1 receptor antagonists (RA) or the BRD inhibitor JQ1 after which BRD4, AuraA, AuraB and C3 mRNA levels were assayed by qPCR. Induction in C3ar1−/−C5ar1−/− vs WT− cells. All n=2. * = p<.05.
Disrupted C3ar1/C5ar1 signaling in Tregs augments upregulation of the ten-eleven translocation enzymes (TET1/TET3) and enables Vitamin C enhancement of their upregulatory effect on Foxp3 stability
TET enzymes [which oxidize 5-methylcytosine (5mC) to 5-hydroxymethylcytosine and other oxidized methylcytosines] participate in demethylation of genomic DNA (13, 14). There are three TET genes. A previous Treg study (14) connected upregulation of TET2 and TET3 with demethylation of the TSDR. Knock-out of these genes led to loss of Foxp3 expression comparably to knockout of the CNS2 region itself (14). Another Treg study (13) found that Vitamin C participates in demethylation of the CNS2 region via promoting TET2 upregulation.
To determine if CD4+ cell expression of CD55 and its suppression of autocrine C3ar1/C5ar1 signaling are involved in TET gene upregulation and Vitamin C augmentation of Foxp3 associated TET activity, we performed two experiments. In the first, we assayed TET mRNA expression in sorted Foxp3+ cells derived from Treg induction cultures of flow sorted WT, CD55−/−, or C3ar1−/−C5ar1−/− Foxp3− CD4+ cells. Sorted WT Foxp3+ CD4+ cells showed ~4-fold increased mRNA levels of all three TET enzymes, whereas sorted Foxp3+ CD55−/− CD4+ cells showed no changes in any of the TETs. In contrast, sorted Foxp3+ C3ar1−/−C5ar1−/− cells showed ~ 9-fold increases in TET1 and TET3 mRNAs (Fig 1B). Previous studies showed that TET mRNA levels closely correlate with TET protein levels and TET function (14). In the second experiment, we repeated the above iTreg induction protocol in the absence and presence of Vitamin C. Kinetic analyses of the sorted Foxp3+ cells in cultures showed that the inclusion of Vitamin C augmented Treg stability most profoundly in C3ar1−/−C5ar1−/− cells and least profoundly in CD55−/− cells (Fig 1C). While Vitamin C reversed the loss of Foxp3+ in the CD55−/− cells from 70% to 60% (a ~ 14% difference), it decreased the loss from ~40% to 10% in the C3ar1−/−C5ar1−/− cells (a 75% increase). Thus, TET gene upregulation is essential for Treg induction as well as stability while Vitamin C is Foxp3 modulatory as it enhances TET effects.
Disrupted C3ar1/C5ar1 signaling in Tregs represses expression of BRD4 needed for Th17 gene expression.
Bromodomain and Extra-Terminal (BET) family proteins enable the productive transcription of genes that drive cell growth and enable proinflammatory processes (15, 16). They do so by binding to acetylated lysines on histones and TFs and in so doing de-repress transcriptional elongation. There are four BET protein family members BRD2, BRD3, BRD4 and BRDT (testes specific). Recent studies (17) showed that BRD4 plays an obligatory role in Th17 lineage commitment. In line with this, the BET protein inhibitor JQ1 totally abolishes Th17 cell generation (17). Motivated by findings that Treg instability, i.e. the production of “exTregs”, involves the conversion of Tregs into Th17 cells, we examined whether BRD4 expression and C3ar1/C5ar1 signaling are interconnected. Sorted WT Foxp3− CD4 cells activated with anti-CD3/28 under Teff conditions upregulated BRD4. The activated CD4+ cells concurrently upregulated the endogenous production of C3 and the aurora kinases aura1 and aura2 which participate in mitosis. The inclusion of C3ar1/C5ar1 antagonists inhibited all of the upregulations. The pharmaceutical C3ar1/C5ar1 blockade inhibited BRD4 mRNA expression similarly to JQ1 (Fig 1D). Collectively, the findings argue that absent C3ar1/C5ar1 signaling in CD4+ cells not only functions endogenously to confer the signature phenotypic markers of Tregs, but at the same time endogenously represses a process that would promote the conversion of Tregs to Th17+ “ex Tregs”.
Repressed C3ar1/C5ar1 signaling in developing Tregs evokes nuclear translocation of NFAT and NF-kB.
While the above studies showed that disabled C3ar1/C5ar1 signaling in Tregs induces several of the reported processes that lead to a demethylated TSDR and long term pTreg stability, they did not provide a molecular mechanism.
A previous study (2) linked nuclear translocation of NFAT to the TSDR-containing CNS2 region to hairpin looping of the CNS2 intronic region to the Foxp3 promoter. Other studies linked concomitant nuclear translocation of NF-kB to the Foxp3 promoter (18, 19). To determine if these translocations are interconnected with CD55 restraint of C3ar1/C5ar1 signaling in CD4+ cells, we activated sorted Foxp3− WT, CD55−/−, and C3ar1−/−C5ar1−/− CD4+ cells with anti-CD3/28 Dynabeads and IL-2 plus TGF-β (Treg conditions). After 3 d, we assayed sorted Foxp3+ cells induced from sorted Foxp3− cells of each genotype for nuclear NFAT and NF-kB by Amnis Cytometry. Both nuclear NFAT and NF-kB were upregulated in Foxp3+ cells generated from sorted Foxp3−C3ar1−/−C5ar1−/− cells, but not in Foxp3+ cells induced from sorted Foxp3− WT or CD55−/− CD4+ cells (Fig 2). These data link upregulated TET expression with their targeting to the CNS2 region in Foxp3+ cells that develop on a background disabled in C3ar1 and C5ar1 signaling.
Figure 2: Nuclear translocation of NFAT and NF-kB in Tregs devoid of C3ar1/C5ar1 signaling.
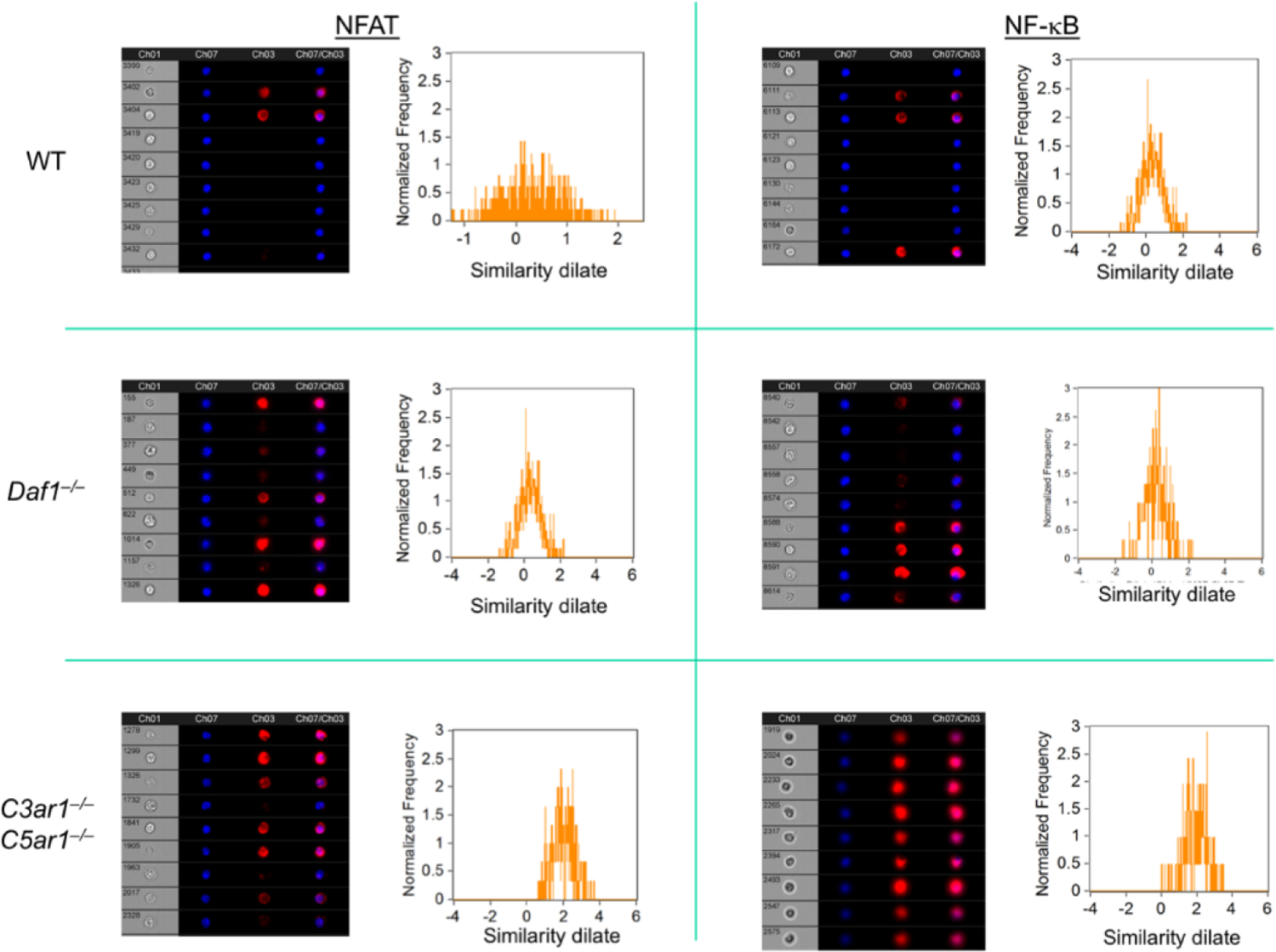
Sorted induced Foxp3+ cells on each genotype were assayed for nuclear NFAT and NF-kB by Amnis Cytometry. The flow plots show colocalization of NFAT and NF-kB with the nuclear stain. Representative of 2 assays. The red lines are the anti-TGF-β and anti-IL-10 stains. The black lines are nonrelevant controls.
The in vivo stability of Tregs depends on CD55 restraint of autocrine C3ar1/C5ar1 signaling in pTregs.
To determine the extent to which restraint of C3ar1/C5ar1 signaling in pTregs impacts the stability of Foxp3 expression and consequent durability of Tregs in vivo, we performed in vivo studies with ovalbumin (ova) specific Tregs generated from ova specific OT-II CD4+ cells. We adoptively transferred 4 × 106 flow sorted Foxp3− (GFP−) WT, CD55−/−, or C3ar1−/−C5ar1−/− OT-II cells (all Thy-1.2) to Thy-1.1 recipients. We added 20 mg/ml of ova to the drinking water for 5 d, after which we immunized the mice with 100 ug of ova in IFA and followed the mice thereafter without further manipulation. After 8 months without any other intervention, we harvested Thy-1.2 cells on each genetic background. Following flow sorting of the Thy-1.2+ Foxp3+ (GFP+) CD4+ cells, we examined the TSDR in the sorted cells. While the recovered Thy-1.2+ Foxp3+ CD55−/− and WT OT-II cells contained methylated TSDRs, the recovered Thy-1.2+ Foxp3+ C3ar1−/−C5ar1−/− OT-II cells contained a de-methylated TSDR (Fig 3A).
Figure 3: Long-term in vivo stability of Tregs induced in the absence of C3ar1/c5ar1 signaling.
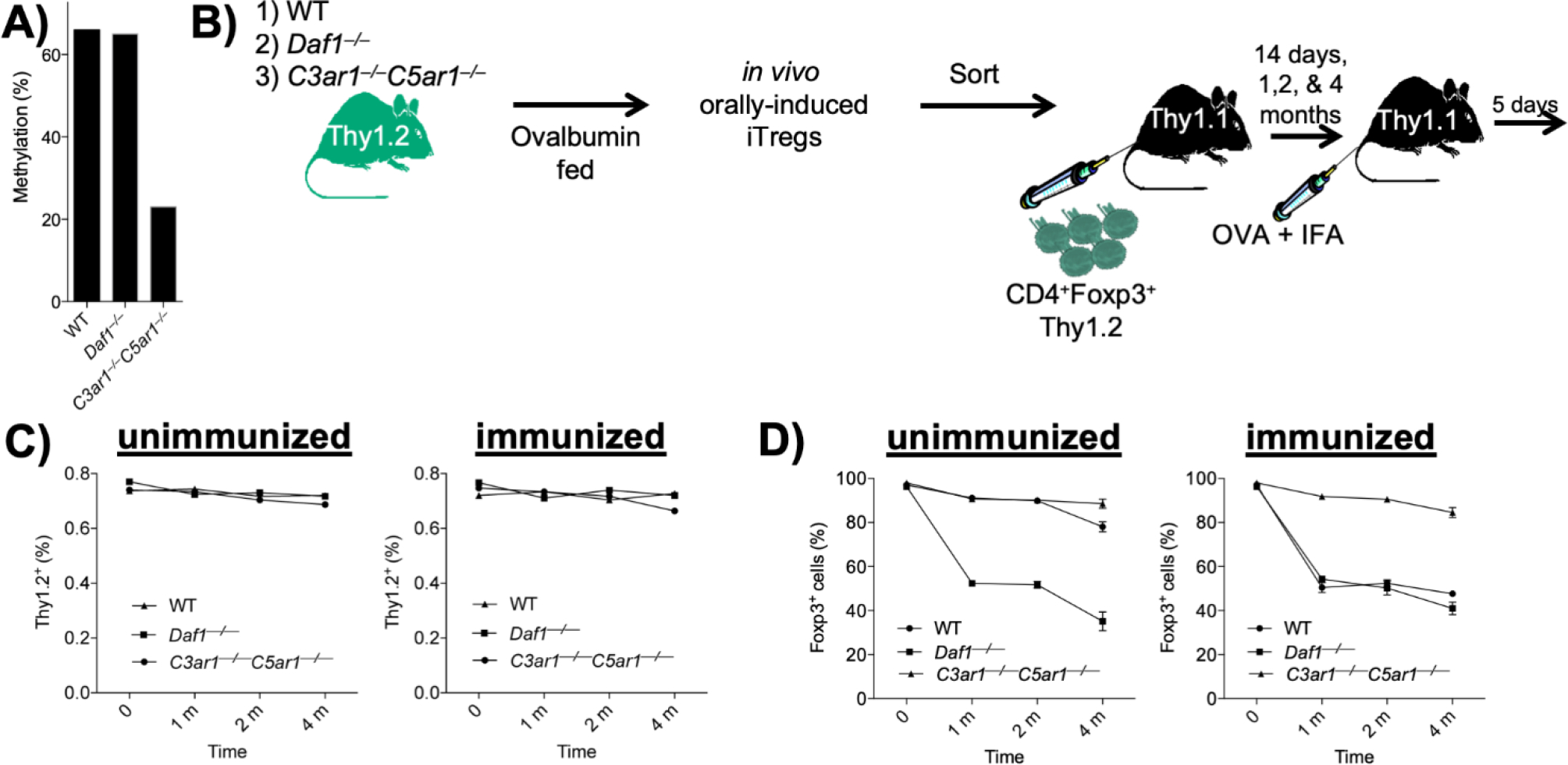
A) Thy-1.2 flow sorted Foxp3− (GFP−) WT, CD55−/−, or C3ar1−/−C5ar1−/− OT-II cells were adoptively transferred to Thy-1.1 recipients. Ova (20 mg/ml) was added to the drinking water of the recipients for 5 d after which the recipients were immunized with 100 ug/ml of ova in IFA. Eighty d later Thy-1.2 Foxp3+ cells in the inguinal LNs and spleen of the recipients were harvested and methylation status of the TSDR assayed. Values representative of 2 assays. B) Orally induced Thy-1.2 Foxp3+ OT-II cells on the CD55−/−, WT, and C3ar1−/−C5ar1−/− backgrounds were prepared as in A) and sorted Foxp3+ cells on each background were transferred to a second set of naïve Thy-1.1 recipients. The recipients were immunized with ova in IFA or IFA alone, and Thy-1.2 CD4+ cells in the inguinal LNs and spleens of the recipients were assayed at 1, 2, and 4 months for retention of Foxp3 expression. The cartoon diagrams the experimental protocol. C) The number of Thy-1.2 CD4+ on each background recovered at each time point. D) The percentage of Thy-1.2 CD4+ cells on each genetic background retaining Foxp3 (GFP+) expression at each time point.
The above data prompted two subsequent antigen challenge experiments directed at Treg durability. In the first, we added 20 mg/ml of ova to the drinking water of WT, CD55−/−, and C3ar1−/−C5ar1−/− mice all on the OT-II-Foxp3-GFP background. After 5 d, we immunized each mouse group with 100 ug of ova in IFA as above. Seven d later, we harvested CD4+ cells from the mLNs. We then transferred 4 × 106 sorted Foxp3+ (GFP+) OT-II cells (Thy-1.2) from each genotype to 3 sets (18 mice each) of naïve Thy-1.1 recipients for analyses at 1, 2, and 4 m. At each time point thereafter, we immunized half of the recipient mice with ova in IFA and half with IFA alone as control. Five d later, we assayed (Thy-1.2) CD4+ cells in the inguinal LNs and in the spleen for Foxp3+ cells (diagramed in Fig 3B). This monitoring of Foxp3+ OT-II cells over time showed that the number of transferred (Thy-1.2) OT-II cells remained stable for all groups over the full 4 m period (Fig 3C left and right). In recipients that received IFA alone, Foxp3 expression on CD55−/− (Thy-1.2) Foxp3+ OT-II cells progressively declined, and that on WT (Thy-1.2) Foxp3+ OT-II cells began to decline at 4 m. In contrast, Foxp3 expression on C3ar1−/−C5ar1−/− (Thy-1.2) Foxp3+ OT-II cells remained uniformly stable out to 4 m (Fig 3D left). Moreover, in recipients that were immunized with ova, only C3ar1−/−C5ar1−/− OT-II Foxp3+ cells retained Foxp3+ expression (Fig 2D right) supporting the proposition that disabled C3ar1/C5ar1 signaling sustains Foxp3 expression even following activation with specific antigen.
Orally induced pTregs in the absence of C3ar1/C5ar1 signaling show durable Foxp3 expression, de-methylated TSDRs and robust TGF-β and IL-10 expression.
Because Foxp3+ OT-II cells recovered from the recipients in the above study could have contained tTregs, in a second study we adoptively transferred sorted (Thy-1.2) Foxp3− OT-II cells on each genetic background to Thy1.1 recipients. We fed the recipients 20 mg/ml of ova and transferred sorted Thy-1.2 Foxp3+ OT-II that were orally induced to a second set of naïve recipients (diagrammed in Fig 4A). We then studied the stability of Foxp3 expression on the WT, CD55−/− and C3ar1−/−C5ar1−/− OT-II Tregs at 14 days, 1 month, and 2 months as described in the prior experiment. Analyses of the recovered Thy1.2 CD4+ cells at each time point yielded similar results (Fig 4B). We isolated Foxp3+ WT, CD55−/−, and C3ar1−/−C5ar1−/− (Thy-1.2) OT-II cells from the LNs and spleen and expanded the cells in cultures with anti-CD3/28 Dynabeads and IL-2. Analyses of the TSDR of the Foxp3 gene of each genotype at 2 months showed that it was ~80% methylated in CD55−/− or WT OT-II Foxp3+ cells similar to conventional T cells. In contrast, the level of demethylation in C3ar1−/−C5ar1−/− OT-II Foxp3+ cells was similar to that of tTregs (Fig 4C). A replicate analysis of the TSDR gave the same result. This finding of sustained TSDR de-methylation only in C3ar1−/−C5ar1−/− OT-II Foxp3+ cells despite TCR stimulation with ova further validates the interpretation that disabled C3ar1/C5ar1 signaling into Tregs is a requisite condition that sustains antigen specific Treg stability. In line with this interpretation, an earlier experiment without the use the Foxp3-GFP transgene in which we used adoptively transferred CD25+ cells from the parental WT, CD55−/−, or C3ar1−/−C5ar1−/− OT-II mice and analyzed Foxp3+ expression on recovered Thy-1.2 cells by intracellular staining showed stability of Foxp3 expression only in Foxp3+ C3ar1−/−C5ar1−/− OT-II cells (not shown).
Figure 4: Demethylation of the TSDR in Tregs devoid of C3ar1/C5ar1 signaling.
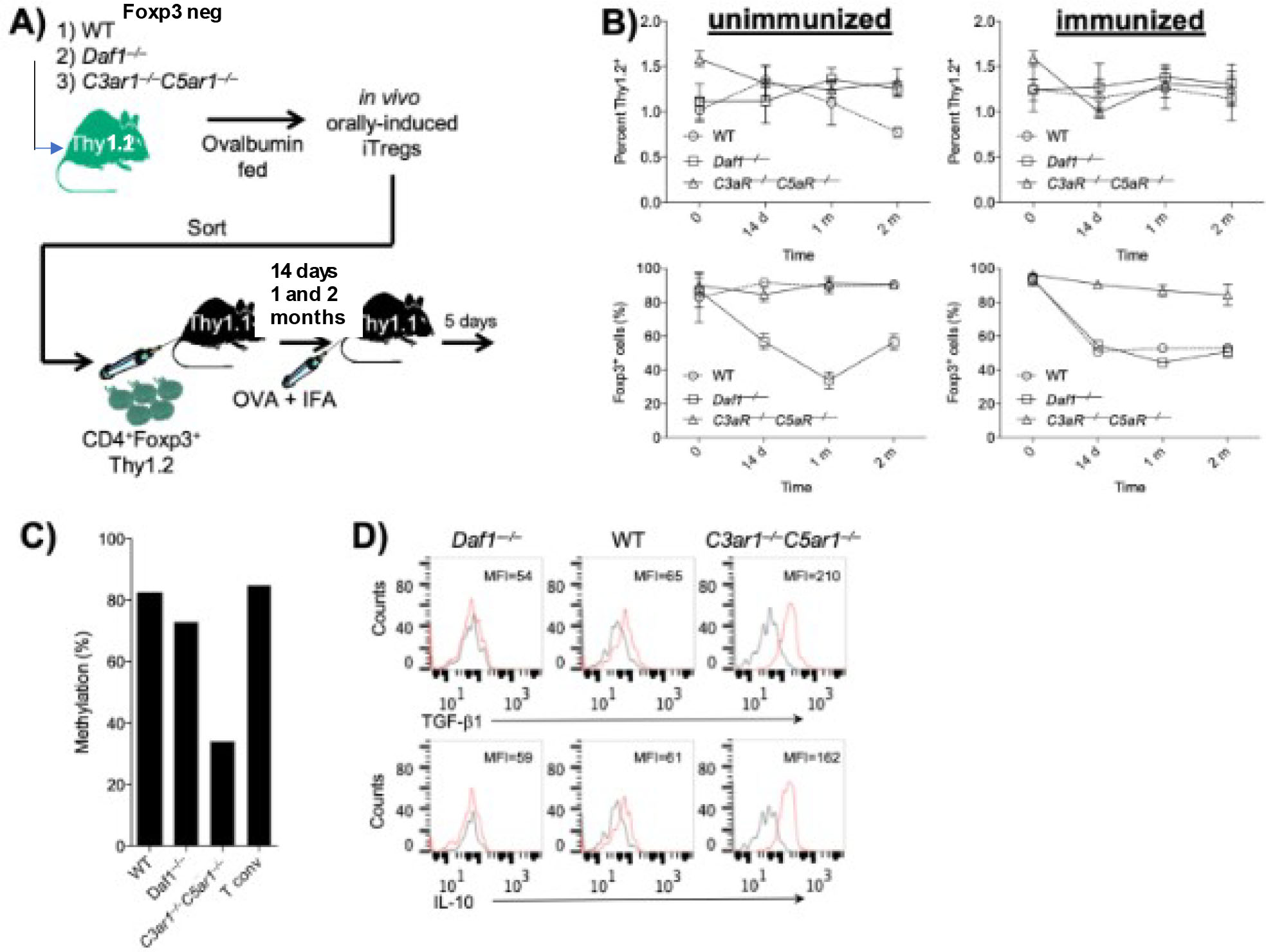
A) Sorted Foxp3− CD55−/−, WT, and C3ar1−/−C5ar1−/− OT-II cells (all Thy-1.2) were adoptively transferred to naïve Thy-1.1 recipients. After feeding the recipients 20 mg/ml of ova and immunizing them with 100 ug of ova in IFA as above, sorted Thy-1.2 Foxp3+ OT-II cells on each genotype in the spleens and inguinal LNs of the recipients were transferred to a second set of naïve Thy-1.1 recipients. Thy-1.2 OT-II cells on each genotype were recovered from the spleens and inguinal LNs of the second set of recipients at 1, 2 or 4 months and analyzed as in Fig 2. A) Cartoon of the experimental protocol. B) numbers (Upper) and Foxp3 (GFP+) expression (Lower) of Thy1.2 OT-II cells on each genotype recovered at each timepoint. C) The methylation status of TSDR in the sorted Foxp3+ OT-II cells on each genotype was assayed. N=2. D) Intracellular TGF-β and IL-10 in the sorted Foxp3+ OT-II cells on each genotype were assayed by intracellular FACS.
In a subsequent experiment, we examined the properties of the recovered WT, CD55−/−, or C3ar1−/−C5ar1−/− OT-II Tregs that were orally induced in vivo. To do this, we used an identical protocol of feeding ovalbumin to Thy-1.1 recipients of sorted Thy-1.2 Foxp3− OT-II cells on the three genotypes. We then harvested the Thy-1.2 Foxp3+ OT II cells in the mLNs after 2 months. Intracellular staining of C3ar1−/−C5ar1−/− Foxp3+ OT-II cells showed more TGF-β1 and IL-10 than in WT Foxp3+ OT-II cells and little of either immunosuppressive cytokine in CD55−/− Foxp3+ OT-II cells (Fig 4D).
Summarizing the in vivo data, the experiment in Fig 3A analyzed ova specific CD55−/−, WT, and C3ar1−/−C5ar1−/− Tregs generated in vivo out to 8 months at which time the TSDR was demethylated only in the C3ar1−/−C5ar1−/− Tregs. The experiment in Fig 3B examined CD55−/−, WT, and C3ar1−/−C5ar1−/− Tregs at 1, 2 and 4 months and found that CD55−/− and WT Tregs markedly lost Foxp3 expression with immunization (activation of their TCR), whereas C3ar1−/−C5ar1−/− mice fully retained Foxp3 expression. The experiment in Fig 4 used the same protocol, except the mice received adoptively transferred Tregs orally generated in a prior set of mice. At 2 months, only the C3ar1−/−C5ar1−/− cells again were stable and uniformly demethylated, A fourth experiment employing the same protocol with non Foxp3-GFP cells staining intracellularly for Foxp3 showed the same full stability uniquely in the C3ar1−/−C5ar1−/− Tregs. A fifth experiment used the same protocol and isolated the Tregs at 2 months. Only the C3ar1−/−C5ar1−/− cells retained robust production of the prototypical Treg cytokines TGF-β and IL-10. The five experiments taken together combined with the in vitro data thus argue that absent C3ar1/C5ar1 signaling in Tregs enables TSDR demethylation, Treg stability, and promotes Treg immunosuppressive function.
Discussion
The experiments in this study show that disrupted C3ar1/C5ar1 signaling in naïve CD4+ cells triggers multiple processes connected in the literature with TSDR demethylation in antigen specific CD4+ cells. The processes ultimately give rise to Tregs that exhibit long-term stability and robust Foxp3 expression when activated in vivo with specific antigen. Specifically, they show that disabled C3ar1/C5ar1 signaling is connected with upregulation of the TET demethylases which together with Smad2 activation documented in our previous study (6) empower activation of the TET proteins shown by others (20–22) to target the Foxp3 locus. In conjunction with the TET upregulation, repressed C3ar1/C5ar1 signaling is connected with Vitamin C augmentation of Foxp3 transcriptional stabilization mediated via enhanced TET demethylation (13, 14, 23). Disabled Treg C3ar1/C5ar1 signaling inactivates BRD4 that otherwise would favor Th17 cell differentiation (17, 24). The experiments additionally show that SOCS1/2/3 mRNA expression levels are upregulated 5-fold sustaining stable Foxp3 expression, since one process linked to decay of Foxp3 expression is STAT3/4 signaling (25–27).
Despite the connection of TSDR demethylation with Treg stability 14 years ago (28), how this epigenetic state confers Treg stability has remained incompletely clarified. The studies by Li et.al. in 2014 (2) implicating hairpin looping of the TSDR-containing CNS2 region to the Foxp3 promoter triggered by nuclear recruitment of NFAT to the former and NF-kB to the latter provided a critical insight into the mechanics of how the transcriptional stabilizing effect occurs. The Treg signaling that enables this Foxp3 promoter alteration, however, remained unclarified. The experiments herein are in line with the Li et al findings. They document by Amnis cytometry that nuclear translocation of both NF-kB and NFAT occurs when C3ar1/C5ar1 signals are absent but not when they are present. Taken together with the above findings, they thus for the first time provide a coherent signaling connection that is directly tied to TSDR demethylation and stabilized Foxp3 transcription.
The finding that CNS2 looping to the Foxp3 promoter is associated with nuclear translocation of NF-kB as well as NFAT seemingly differs from recent findings by Mikami et.al (29) that NF-kB is suppressed in connection with TSDR demethylation occurring when Tregs are generated under conditions of CD28 deficiency. We are unable to explain the difference. However, our previous findings (4, 6) that C3a/C5a production is induced by CD28 costimulation argue that CD28−/− CD4+ cells are likely disabled in C3ar1/C5ar1 signaling. As referenced in that communication, many past studies (18, 19, 30–34) provided evidence that strong CD28 signaling and consequent NF-kB activation is needed for tTreg and pTreg TSDR demethylation and stability. Another point is that while the MikamI et al study focused on TSDR demethylation, it did not investigate long-term Treg stability. Further studies will be needed to precisely determine the role of CD28 costimulation and NF-kB in programming i-, p-, and tTreg induction, function, and stability.
Other studies have connected additional Foxp3 Treg processes with repression of C3ar1/C5ar1 signaling. Relevant to tolerance and to checkpoint therapy in cancer, studies of oral and ocular tolerance (35) showed that suppression of C3ar1/C5ar1 signaling upregulates GARP that delivers latent TGF-β to TGFβR1/2. The consequent autocrine TFG-β signaling upregulates ICOS, PD1 and CTLA-4 (35) which transmit coinhibitory signals into CD4+ cells and reciprocally downregulates CD28, CD40 ligand expression needed for Teff responses. Repression of C3ar1/C5ar1 signaling concomitantly upregulates CD37, CD73 and adenosine that inhibits Teff. Repressed C3ar1/C5ar1 signaling additionally upregulates interferon regulatory factor-8 (IRF-8) that enables the expansion of Tregs (36) and aldehyde dehydrogenase 1 family member A2 (Aldh1a2) that generates retinoic acid in the GI tract. It additionally upregulates CX3CR1, CCR7, and CCR9 which enable CD4+ cell transit to LNs and the lamina propria (35). Studies of vascular endothelial cell (EC) homeostasis (35) (Feng-Qi A, et.al. under review) showed that absent C3ar1/C5ar1 signaling enables the recruitment of CREB binding protein (CBP) or p300, a co-activator tied to p-CREB co-transcriptional activity in the Foxp3 promoter (37). Studies of growth factor signaling (38) showed that disruption of C3ar1/C5ar1 signaling derepresses PTEN activity which dephosphorylates PtdIns (3,4,5)P3 and thereby suppresses AKT activation, a hallmark of Tregs. The repressed PtdIns (3,4,5)P3 and p-AKT prevent Th1/Th17 differentiation but amplify autocrine TGF-β production. The growth factor studies additionally showed that repressed PI-3Kɣ resulting from disabled C3ar1/C5ar1 signaling evokes PHLLP phosphatase activity that inactivates any preexisting p-AKT and prevents activation of CK2 and Fyn (38) that otherwise would introduce phosphorylations in PTEN that inactivate its phosphatase activity. They additionally showed that the absence of C3ar1/C5ar1 signaling disables autocrine IL-6 production and IL-6R-gp130 signaling (38, 39) and consequently impairs the transition of Tregs to Th17 “ex-Tregs”. The absence of C3ar1/C5ar1 signaling is thus connected with many cellular processes tied to Treg induction and TSDR demethylation. Because of the collective immunoinhibitory effects of these multiple signaling alterations, C3ar1−/−C5ar1−/− Tregs possess more robust suppressor activity than Tregs conventionally induced by TGF-β (6).
Although the full signaling cascade(s) underlying the linkage of absent C3ar1/C5ar1 signaling in Tregs with TSDR de-methylation remains to be characterized, the findings herein that circumvention of C3ar1/C5ar1 signaling into CD4+ cells activates TET1/2/3, shown by others to target the Foxp3 gene (20–22) provides a mechanism for demethylating the TSDR and thereby enabling its NFAT uptake. Our prior findings (6) that C3a/C5a activate STAT3 dependent Th1/Th17 responses and repress SOCS1/3/4 activation (this study) provide one mechanistic insight into the unwanted outcome of the collapse of Tregs into “ex Tregs” in Treg biotherapy (28, 40).
In support of the physiological connection of repression of C3ar1/C5ar1 signaling with TSDR demethylation and durable Foxp3 transcription, the in vivo experiments in this study showed that disabled CD4+ cell C3ar1/C5ar1 signaling in Tregs is an essential condition for long term Foxp3 expression and Treg stability. The experiments with (ova-specific) orally induced pTregs showed that suppressed C3ar1/C5ar1 signaling prevented the loss of Foxp3 expression for 8 months. The experiments in which we adoptively transferred Thy-1.2 WT, CD55−/−, and C3ar1−/−C5ar1−/− Foxp3− OT-II pTregs prepared in vivo in Thy-1.1 recipients to a second set of naïve Thy-1.1 recipients revealed that Foxp3 expression of CD55−/− Foxp3+ OT-II cells was unstable at 14 days even in the absence of ova immunization. They conversely showed that Foxp3 expression in transferred C3ar1−/−C5ar1−/− Foxp3+ OT-II cells was stable at 4 months even with TCR stimulation (i.e. ova immunization). Consistent with Foxp3 stability occurring on the C3ar1−/−C5ar1−/− Foxp3+ background, the TSDR in the 4-month C3ar1−/−C5ar1−/− Foxp3+ OT-II cells was de-methylated. In contrast, the TSDR in CD55−/− and WT Foxp3+ OT-II cells was methylated directly tying repressed C3ar1/C5ar1 signaling to TSDR de-methylation. In further support of this relationship, the analysis of the C3ar1−/−C5ar1−/− Foxp3+ OT-II cells at 8 months without adoptive transfer showed that Foxp3 stability in C3ar1−/−C5ar1−/− Tregs likewise was associated with TSDR demethylation. Our findings are similar to those of Van der Touw et al (38) who found that alloantigen-induced C3ar1−/−C5ar1−/− iTreg were more stable than WT iTreg upon transfer to Rag−/− recipients after 5 days (41). Our findings of TSDR demethylation and-long term stability of ova specific Tregs generated in vivo however differ. One possible explanation for the difference is that the Van der Touw studies examined iTregs induced in vitro by allo-stimulation.
While these results differ from our previous 5 day in vitro studies (6), they argue that either more time, an in vivo process, or both are required for stability of the TSDR demethylated state. Consistent with the interpretation that Foxp3 stability did not derive from the outgrowth of another contaminating cell type, a short term (14 day) study in which Treg “fate mapping” mice (42) crossed with C3ar1−/−C5ar1−/− or WT mice were treated with CpG showed Foxp3 stability on pTregs in the C3ar1−/−C5ar1−/− mice but not the WT mice. Thus, absent CD4+ cell C3ar1/C5ar1 signaling gives rise to processes that both enable pTreg generation and prevent pTreg instability. As examples, the inductive processes include upregulation of CD55 which downregulates C3ar1/C5ar1 signaling (3), interference with C3ar1/C5ar1 signaling by upregulation of non-G-protein coupled C5ar2 (6) which competes for C5a, upregulation of CTLA-4, PD1 and PD-L1 which transmit coinhibitory signals into CD4+ cells (35), expression of SOCS1/4/3 which inactivate STAT1/2/3 [this paper] and repression of MyD88 signaling which inactivates TLR signaling (43). While the inherent Foxp3 instability of ex vivo TGF-β prepared Tregs has limited the effectiveness of biotherapy connected with their use, it remains to be determined how the stability of Tregs prepared by C3ar1/C5ar1 antagonism will be affected by inflammation. This will require more experiments as studies by others of antigen (MOG35–55) specific pTregs in experimental autoimmune encephalitis (EAE) have shown loss of Foxp3 expression and function (44). However, our past studies of MOG35–55 specific Tregs generated in C3ar1−/−C5ar1−/− CD4+ cells or prepared by C3ar1/C5ar1 antagonism in EAE have shown retained Foxp3 (GFP) expression, robust immune suppression, and resolution of disease (6).
The findings herein that suppression of C3ar1/C5ar1 signaling in pTregs induces stabilized Foxp3 transcription and immunosuppressive TGF-β and IL-10 expression by iTregs argue that it may be possible to achieve durable immune suppression to an exogenous antigen in a recipient if Tregs can be generated in a way that corresponds to the C3ar1−/−C5ar1−/− background in in vivo conditions, i.e. disabled C3ar1/C5ar1, but retained C5ar2 function. This formulation could have important clinical relevance for immune-mediated conditions, e.g. autoimmunity, transplantation, allergic airway disease, or other disorders in which suppression of T effector responses would be beneficial.
Key Points.
Foxp3 TSDR demethylation is governed by autocrine C3ar1/C5ar1 signaling in Tregs.
Treg generation under in vivo conditions is required for TSDR demethylation
Acknowledgements
The authors thank Dawn Smith for help with cells and John Denker for help with genotyping Ophthalmology Core Facility (EY11373) Director Irena Pikuleva (Case Western Reserve University). We also thank Mike Sramkoski Flow Cytometry Core (Case Comprehensive Cancer Center) for help with cell sorting.
The work in this study was supported by NIH grants AR067182 and HL109561 (MEM) and the Division of Intramural Research, National Institute of Allergy and Infectious Diseases.
Footnotes
The authors have declared that no conflict of interest exists
References
- 1.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, and Rudensky AY. 2010. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463: 808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li X, Liang Y, LeBlanc M, Benner C, and Zheng Y. 2014. Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell 158: 734–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heeger PS, Lalli PN, Lin F, Valujskikh A, Liu J, Muqim N, Xu Y, and Medof ME. 2005. Decay-accelerating factor modulates induction of T cell immunity. The Journal of experimental medicine 201: 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, Shapiro VS, Dubyak GR, Heeger PS, and Medof ME. 2008. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 28: 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, Lin F, Strainic MG, An F, Miller RH, Altuntas CZ, Heeger PS, Tuohy VK, and Medof ME. 2008. IFN-gamma and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. Journal of immunology 180: 5882–5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strainic MG, Shevach EM, An F, Lin F, and Medof ME. 2013. Absence of signaling into CD4(+) cells via C3aR and C5aR enables autoinductive TGF-beta1 signaling and induction of Foxp3(+) regulatory T cells. Nature immunology 14: 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bamberg CE, Mackay CR, Lee H, Zahra D, Jackson J, Lim YS, Whitfeld PL, Craig S, Corsini E, Lu B, Gerard C, and Gerard NP. 2010. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. The Journal of biological chemistry 285: 7633–7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sebastian M, Lopez-Ocasio M, Metidji A, Rieder SA, Shevach EM, and Thornton AM. 2016. Helios Controls a Limited Subset of Regulatory T Cell Functions. Journal of immunology 196: 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thornton AM, Lu J, Korty PE, Kim YC, Martens C, Sun PD, and Shevach EM. 2019. Helios(+) and Helios(−) Treg subpopulations are phenotypically and functionally distinct and express dissimilar TCR repertoires. European journal of immunology 49: 398–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, and Rudensky AY. 2014. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 158: 749–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Liang Y, LeBlanc M, Benner C, and Zheng Y. 2014. Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell 158: 734–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, Pear WS, Hatton RD, and Weaver CT. 2015. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nature immunology 16: 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasidharan Nair V, Song MH, and Oh KI. 2016. Vitamin C Facilitates Demethylation of the Foxp3 Enhancer in a Tet-Dependent Manner. Journal of immunology 196: 2119–2131. [DOI] [PubMed] [Google Scholar]
- 14.Yue X, Trifari S, Aijo T, Tsagaratou A, Pastor WA, Zepeda-Martinez JA, Lio CW, Li X, Huang Y, Vijayanand P, Lahdesmaki H, and Rao A. 2016. Control of Foxp3 stability through modulation of TET activity. The Journal of experimental medicine 213: 377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shu S, and Polyak K. 2016. BET Bromodomain Proteins as Cancer Therapeutic Targets. Cold Spring Harb Symp Quant Biol 81: 123–129. [DOI] [PubMed] [Google Scholar]
- 16.Sahni JM, and Keri RA. 2017. Targeting bromodomain and extraterminal proteins in breast cancer. Pharmacological research : the official journal of the Italian Pharmacological Society. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheung KL, Zhang F, Jaganathan A, Sharma R, Zhang Q, Konuma T, Shen T, Lee JY, Ren C, Chen CH, Lu G, Olson MR, Zhang W, Kaplan MH, Littman DR, Walsh MJ, Xiong H, Zeng L, and Zhou MM. 2017. Distinct Roles of Brd2 and Brd4 in Potentiating the Transcriptional Program for Th17 Cell Differentiation. Molecular cell 65: 1068–1080 e1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, and Chen YH. 2009. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 31: 932–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Long M, Park SG, Strickland I, Hayden MS, and Ghosh S. 2009. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 31: 921–931. [DOI] [PubMed] [Google Scholar]
- 20.Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, Gao P, Helmin KA, Abdala-Valencia H, Sena LA, Schumacker PT, Turka LA, and Chandel NS. 2019. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565: 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nair VS, Song MH, Ko M, and Oh KI. 2016. DNA Demethylation of the Foxp3 Enhancer Is Maintained through Modulation of Ten-Eleven-Translocation and DNA Methyltransferases. Mol Cells 39: 888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song MH, Nair VS, and Oh KI. 2017. Vitamin C enhances the expression of IL17 in a Jmjd2-dependent manner. BMB Rep 50: 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kasahara H, Kondo T, Nakatsukasa H, Chikuma S, Ito M, Ando M, Kurebayashi Y, Sekiya T, Yamada T, Okamoto S, and Yoshimura A. 2017. Generation of allo-antigen-specific induced Treg stabilized by vitamin C treatment and its application for prevention of acute graft versus host disease model. International immunology 29: 457–469. [DOI] [PubMed] [Google Scholar]
- 24.Mele DA, Salmeron A, Ghosh S, Huang HR, Bryant BM, and Lora JM. 2013. BET bromodomain inhibition suppresses TH17-mediated pathology. The Journal of experimental medicine 210: 2181–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C, Wang D, Zhao H, Wang J, Liu N, Shi H, Tian J, Wang X, and Zhang Z. 2021. Traffic-related PM2.5 and diverse constituents disturb the balance of Th17/Treg cells by STAT3/RORgammat-STAT5/Foxp3 signaling pathway in a rat model of asthma. Int Immunopharmacol 96: 107788. [DOI] [PubMed] [Google Scholar]
- 26.Hua Y, Liu R, Lu M, Guan X, Zhuang S, Tian Y, Zhang Z, and Cui L. 2021. Juglone regulates gut microbiota and Th17/Treg balance in DSS-induced ulcerative colitis. Int Immunopharmacol 97: 107683. [DOI] [PubMed] [Google Scholar]
- 27.Hosseini A, Gharibi T, Mohammadzadeh A, Ebrahimi-Kalan A, Jadidi-Niaragh F, Babaloo Z, Shanehbandi D, Baghbani E, and Baradaran B. 2021. Ruxolitinib attenuates experimental autoimmune encephalomyelitis (EAE) development as animal models of multiple sclerosis (MS). Life sciences 276: 119395. [DOI] [PubMed] [Google Scholar]
- 28.Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, Olek S, Hamann A, von Boehmer H, and Huehn J. 2008. DNA methylation controls Foxp3 gene expression. European journal of immunology 38: 1654–1663. [DOI] [PubMed] [Google Scholar]
- 29.Mikami N, Kawakami R, Chen KY, Sugimoto A, Ohkura N, and Sakaguchi S. 2020. Epigenetic conversion of conventional T cells into regulatory T cells by CD28 signal deprivation. Proceedings of the National Academy of Sciences of the United States of America 117: 12258–12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahmud SA, Manlove LS, Schmitz HM, Xing Y, Wang Y, Owen DL, Schenkel JM, Boomer JS, Green JM, Yagita H, Chi H, Hogquist KA, and Farrar MA. 2014. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nature immunology 15: 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, Banerjee A, Proietto A, Gugasyan R, Wu L, McNally A, Steptoe RJ, Thomas R, Shannon MF, and Gerondakis S. 2009. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. The Journal of experimental medicine 206: 3001–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo F, Iclozan C, Suh WK, Anasetti C, and Yu XZ. 2008. CD28 controls differentiation of regulatory T cells from naive CD4 T cells. Journal of immunology 181: 2285–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tai X, Cowan M, Feigenbaum L, and Singer A. 2005. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nature immunology 6: 152–162. [DOI] [PubMed] [Google Scholar]
- 34.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, and Bluestone JA. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12: 431–440. [DOI] [PubMed] [Google Scholar]
- 35.Strainic MG, Liu J, An F, Bailey E, Esposito A, Hamann J, Heeger PS, and Medof ME. 2019. CD55 Is Essential for CD103(+) Dendritic Cell Tolerogenic Responses that Protect against Autoimmunity. The American journal of pathology 189: 1386–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valanparambil RM, Tam M, Gros PP, Auger JP, Segura M, Gros P, Jardim A, Geary TG, Ozato K, and Stevenson MM. 2017. IRF-8 regulates expansion of myeloid-derived suppressor cells and Foxp3+ regulatory T cells and modulates Th2 immune responses to gastrointestinal nematode infection. PLoS Pathog 13: e1006647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Wang L, Han R, Beier UH, Akimova T, Bhatti T, Xiao H, Cole PA, Brindle PK, and Hancock WW. 2014. Two histone/protein acetyltransferases, CBP and p300, are indispensable for Foxp3+ T-regulatory cell development and function. Molecular and cellular biology 34: 3993–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strainic MG, Pohlmann E, Valley CC, Sammeta A, Hussain W, Lidke DS, and Medof ME. 2020. RTK signaling requires C3ar1/C5ar1 and IL-6R joint signaling to repress dominant PTEN, SOCS1/3 and PHLPP restraint. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 34: 2105–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hwang MS, Strainic MG, Pohlmann E, Kim H, Pluskota E, Ramirez-Bergeron DL, Plow EF, and Medof ME. 2019. VEGFR2 survival and mitotic signaling depends on joint activation of associated C3ar1/C5ar1 and IL-6R-gp130. J Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Q, Kim YC, Laurence A, Punkosdy GA, and Shevach EM. 2011. IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. Journal of immunology 186: 6329–6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Touw W, Cravedi P, Kwan WH, Paz-Artal E, Merad M, and Heeger PS. 2013. Cutting edge: Receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. Journal of immunology 190: 5921–5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, and Rudensky AY. 2010. Stability of the regulatory T cell lineage in vivo. Science 329: 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheen JH, Strainic MG, Liu J, Zhang W, Yi Z, Medof ME, and Heeger PS. 2017. TLR-Induced Murine Dendritic Cell (DC) Activation Requires DC-Intrinsic Complement. Journal of immunology 199: 278–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, Fehling HJ, and Bluestone JA. 2013. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity 39: 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]