

SUMMARY
Circadian disruptions impact nearly all people with Alzheimer’s Disease (AD), emphasizing both, its potential role in pathology and the critical need to investigate the therapeutic potential of circadian-modulating interventions. Here we show that time-restricted feeding (TRF) without caloric restriction improved key disease components including behavioral timing, disease pathology, hippocampal transcription, and memory in two transgenic mouse models of AD. We found that TRF had the remarkable capability of simultaneously reducing amyloid deposition, increasing Aβ42 clearance, improving sleep and memory, and normalizing daily transcription patterns of multiple genes, including those associated with AD and neuroinflammation. Thus, our study unveils for the first time the pleiotropic nature of timed feeding on AD, which has far-reaching effects beyond metabolism, ameliorating neurodegeneration and the misalignment of circadian rhythmicity. Since TRF can substantially modify disease trajectory, this intervention has immediate translational potential, addressing the urgent demand for accessible approaches to reduce or halt AD progression.
Keywords: Alzheimer’s disease, circadian rhythms, feed/fast cycle, time-restricted feeding, rhythmic transcription, amyloid plaque deposition, neuroinflammation, APP23, APP-KI, hippocampal transcriptome, cognition, memory, Alzheimer’s disease-associated genes
Graphical Abstract
eTOC blurb
Circadian alterations are prevalent in patients with Alzheimer’s disease (AD). Whittaker et al. report circadian disruptions in AD mouse models, that emerge at early pathology stages. The application of a time-restricted feeding strategy efficiently restored brain transcription, slowed amyloid deposition, and improved memory deficits in AD mice.
INTRODUCTION
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder affecting the lives of more than 6 million afflicted Americans and their families, and it is the biggest forthcoming health challenge in the United States 1. Beyond accumulation of beta-amyloid and phosphorylated Tau proteins in the brain, disturbance of circadian rhythmicity is a common complaint for more than 80% of patients with AD, evidenced by altered sleep/wake cycles and behaviors, such as increased cognitive impairment and confusion in the evening (a.k.a. sundowning), as well as difficulty in falling and staying asleep 2,3. These factors are the leading cause of nursing home placement and are also associated with decreased survival 4. Treatments delaying AD progression remain elusive; thus, approaches that prolong patient independence and daily functioning are likely to have a great impact in clinical care.
Emerging evidence, including studies from our group, suggests that circadian alterations in AD occur earlier in disease progression than previously estimated and may directly aggravate pathology 5,6. Circadian rest-activity patterns are fragmented in preclinical stages of AD, and weakened circadian activity patterns increase the risk of dementia and precede cognitive impairment by several years 7,8. However, the pathways that mediate circadian dysregulation in AD and how they are modulated are not yet well defined.
Circadian (ca 24 h) clocks coordinate the daily temporal organization of physiology and behavior through tightly regulated transcriptional programs. In addition, clock transcription factors can modulate downstream targets outside the core clock, thus imposing rhythmicity in up to 50% of the transcriptome 9. Cell-autonomous clocks reside in various brain regions, including those severely affected by AD like hippocampus and frontal cortex 10,11. Clock misalignment is associated with poor health and AD risk factors including diabetes, cardiovascular diseases, inflammation, and sleep disorders 12. Deletion of core circadian clock genes Bmal1 and Per1 in the mouse brain triggers synaptic degeneration, impaired cortical connectivity, oxidative damage, behavioral abnormalities, and memory impairment, highlighting the impact of circadian alterations on cognitive function and neuronal viability 13,14.
Modulation of the circadian clock as a strategy to improve health outcomes is gaining momentum. Particularly, the daily feed/fast cycle provides a strong stimulus for the synchronization of metabolic and behavioral functions even in the absence of a functional central circadian clock 15–18. We recently demonstrated that time-restricted feeding (TRF; 6 h feed, 18 h fast cycles) can improve sleep/wake cycles, motor performance, and inflammation in mouse models of Huntington’s disease 19,20. Furthermore, human studies applying analogous “Time Restricted Eating” paradigms showed changes in diurnal patterns of cortisol, and in the expression of circadian clock genes and autophagy markers in blood 21.
The mechanisms through which TRF conveys the observed benefits are not well understood and are likely pleiotropic. The alterations brought about by TRF may result from changes in metabolite interactions, bioenergetic pathway responses, modulation of circadian rhythm timing and strength, epigenetic modifications, and effects on food-anticipatory activity and reward circuits 22–28.
In this study, we identified progressive circadian disruptions in the APP23 transgenic (TG) mouse model of AD, including excessive wakefulness, altered behavioral circadian rhythms, hyperactivity, and severe deregulation of daily expression patterns of many genes associated with AD pathology and neuroinflammation in the hippocampus. We evaluated whether modulation of the circadian clock at early-disease stages mitigates behavioral and transcriptional alterations and ameliorates pathology. We report that a TRF protocol limiting food access to 6 h – aligned to the active phase – improved diurnal locomotor activity patterns and behavioral circadian rhythms and increased total sleep, without caloric restriction. TRF normalized the transcription patterns of genes associated with AD, neuroinflammation, lipid processing, and autophagy in the hippocampus of APP23 TG mice. Critically, TRF had a major impact on neuropathology, with treated APP23 TG mice displaying a significant reduction in plaque burden, amyloid deposition, and a reversal of circulating biomarkers of AD. Furthermore, this TRF protocol was effective in reducing amyloid burden and improving memory in the APP-KI mouse model, which exhibits a more aggressive disease progression. Overall, our results demonstrate that the application of TRF can forestall behavioral, cognitive, and molecular disruptions that contribute to AD pathology in mouse models.
RESULTS
AD mice exhibit disrupted sleep, cage activity, and circadian regulation
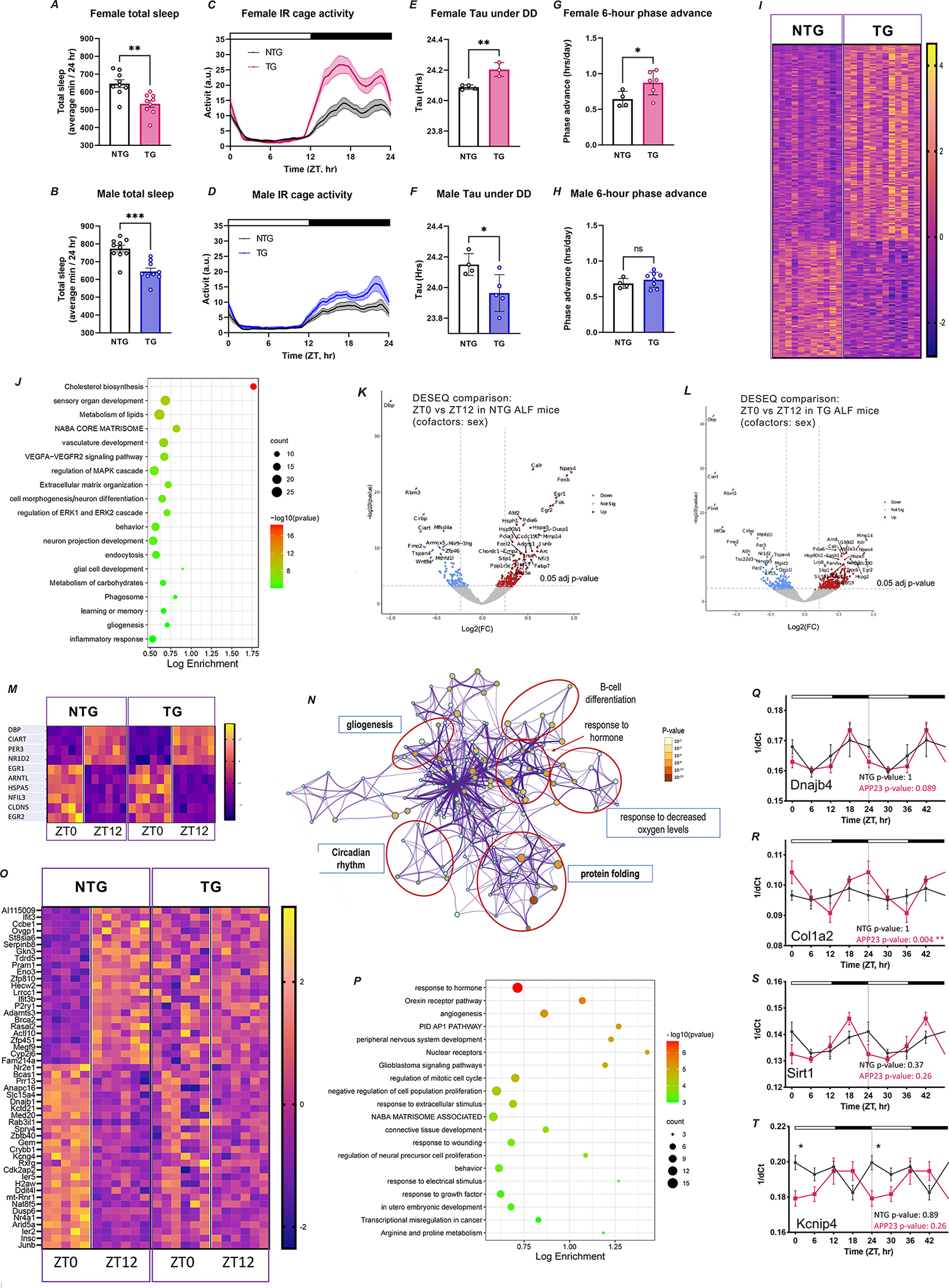
APP23 TG mice develop progressive pathology starting with sparse amyloid plaques in the cortex and hippocampus around six months of age 29. Importantly, TG mice at this age already display increased fragmentation of sleep during the light phase and some cognitive alterations 30. Sleep disruptions continue to worsen with age and pathology, and we observed that 11-month-old TG mice showed decreased mean total sleep and significant hyposomnia during the active phase compared with non-transgenic (NTG) controls (Figure 1A–B; Figure S1a–b) and altered activity rhythms with excessive cage activity during the dark phase (Figure 1C–D; Figure S1c–d).
FIGURE 1. AD mice show alterations in sleep, activity, circadian rhythms and brain transcription.
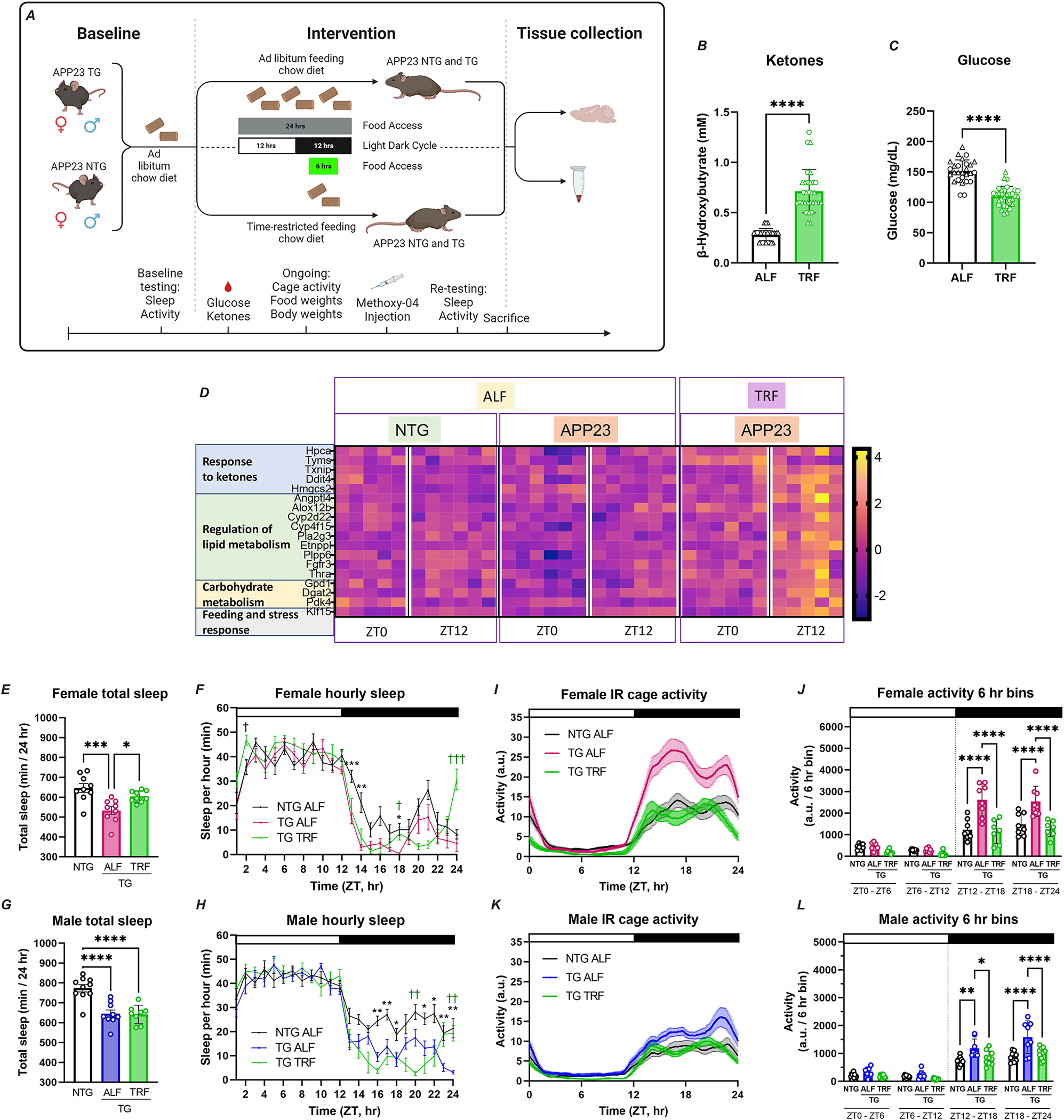
A-B. Total sleep is represented as average minutes per 24 h from data collected over two 24 h sleep-wake cycles. C-D. Activity recordings reported in 3-min bins averaged from 7–10 days of activity. E-F. Tau (circadian period length) under DD was calculated from activity onset times and is presented in hour units. G-H. Phase shift was calculated as [(Δh “activity onset” to “entrained onset”)/days to entrainment] and is presented as the shift in h/day. Top bar in waveforms represents the light-dark cycle. Bar graphs represent individual data plots with standard error of the mean. Statistical significance represents the comparison between TG and NTG mice as per unpaired Students’ t-test *p≤0.05; **p≤0.01; ***p≤0.001. I. Heatmap of significant DEGs in APP23 TG vs NTG mice based on RNA-Seq analysis of hippocampus tissue using DESEQ2; threshold adj.p≤0.2. Each row is one gene and expression is represented by z-score. J. Gene ontology terms for genes differentially expressed in TG mice using Metascape. K-L. Transcriptional profiling by RNA-Seq shows time-of-day expression in both APP23 TG (K; n=12) and NTG mice (L; n=13) hippocampus. Depicted as volcano plots at adj.p≤0.05; red denotes increased and blue decreased at ZT0 in comparison to ZT12. M. Expression of circadian clock genes in NTG and TG mice, presented as heatmap sorted by phase of gene expression in z-scores. N. Pathway enrichment analysis of rhythmic genes using Metascape and showing enrichment in functions associated with neurodegeneration and circadian clock. O. Time-of-day expression patterns are obliterated in some genes in APP23 TG mouse hippocampus, as shown by heatmap sorted by phase of expression in NTG and represented by z-scores. P. GO analysis of the genes that lost rhythmicity in TG mice, showing top enriched functions in Metascape, including pathways associated with transcription regulation, sleep, and behavior. Q-T. Alterations in the rhythmic patterns of expression of selected AD-associated core clock were detected by qPCR using samples taken every 6 hr. Transcript abundance at each time point is expressed as inverse delta Ct. The periodicity p-value is denoted on each plot for NTG and TG. Values are double plotted. Asterisks in the waveform show statistical significance representing the comparison between TG and NTG mice at single timepoints as per unpaired Students’ t-test, *p≤0.05; **p≤0.01.
Evaluation of endogenous circadian activity rhythms in constant darkness conditions (DD) showed some alterations in circadian period, with TG females showing longer and males exhibiting shorter periods than NTG controls (Figure 1E–F; Figure S2i–l). TG mice also showed increased fragmentation of activity under DD, indicated by reduced activity bout lengths and a trend towards increased activity bout number, with no changes in total activity in LD vs DD (Figure S2a–f). Furthermore, female TG mice re-entrained significantly faster than NTG controls in response to a 6 h phase advance (a behavior not observed in males; Figure 1G–H; Figure S2m–p), and both female and male TG mice showed significant activity suppression in response to negative light masking, similar to NTG animals (Figure S2g–h). Taken together, these observations demonstrate that TG mice present disruptions in sleep, cage activity, and circadian regulation (without exhibiting impairments in their response to light) at early disease stages that precede substantial amyloid pathology.
AD mice show dysregulation of gene expression in the hippocampus
To investigate early transcriptional alterations associated with AD pathology in the hippocampus of APP23 TG mice, we applied RNA-Seq and performed differential gene expression analysis in comparison to NTG animals. We identified 258 differentially expressed genes (DEGs) in TG mice, including 28 AD-genes overlapping with AMP-AD and DisGENET Databases (Figure 1I–J; Table S1). Gene ontology (GO) analysis revealed involvement of DEGs in cholesterol biosynthesis, core matrisome and extracellular matrix (ECM) organization, vasculature development, neuron and glial cell functions, memory, inflammation, endocytosis and phagocytosis; with significant enrichment for astrocyte markers. Upstream regulator analysis predicted activation of MAP2K5 kinase (z-score=4.2; p-value of the overlap=1.68E-30) and SREBF2 (z-score=3.8; p-value of the overlap=1.58E-24), both implicated in cholesterol synthesis and insulin metabolism. Among the top inhibited regulators, we identified the group of β-adrenergic receptors (ADRB; z-score=−3.3; p-value of the overlap=6.12E-9), which mediate norepinephrine signaling facilitating synaptic plasticity and memory formation and that have been extensively investigated as therapeutic targets for AD 31.
Impaired daily diurnal variations of transcription in the hippocampus of AD mice
To evaluate potential alterations in time-of-day gene expression in the hippocampus of APP23 TG mice, we analyzed RNA-Seq data from brain samples obtained at ZT0 and ZT12 (n=3/4 mice per time point per genotype). We identified 248 genes in NTG mice and 623 genes in TG mice that were differentially expressed between ZT0 and ZT12 (Figure 1K–L; Table S2A), with 127 DEGs overlapping between NTG and TG mice. This group included the core clock genes Dbp, Nr1d2, Per3, and Arntl, as well as clock-controlled genes Ciart, Egr1, Hspa5, Nfil3, Cldn5, and Egr2 (Figure 1M). Comparison of genes showing time-of-day expression with a dataset of rhythmic genes in the mouse hippocampus from Debski et al 32 showed that 93 (38%) transcripts identified in NTG mice and 145 (23%) observed in TG mice indeed have oscillatory expression, validating our findings (Table S2A). The observation of a fraction of genes that gained time-of-day gene expression in TG mice is in agreement with previous observations in AD and other disease models, although the mechanisms that mediate this effect are unknown 33. Pathway analysis of hippocampal genes with diurnal variation showed enrichment in protein folding, response to hormones and growth factors, and circadian rhythms (Figure 1N). Notably, we identified 121 genes that lost time-of-day-specific expression in TG mice (Figure 1O; Table S2A), and were functionally associated with response to hormones, angiogenesis, ECM, and orexin signaling, and enriched for oligodendrocyte cell markers (Figure 1P). Evaluation of core clock gene expression using RNAseq data including additional tissue collected at ZT6 and ZT18 and JTK-Cycle analysis showed very modest alterations in the expression of core clock genes in the hippocampus of TG mice (Figure S4; Table S2B). Using this extended time point sample set, we also tested the rhythmic expression of selected AD-associated genes that ranked as top DEGs in the RNA-seq analysis. Quantitative real-time PCR followed by JTK analysis showed oscillatory patterns of transcript abundance in NTG mice which were altered in TG mice. We observed earlier time of peak for Kcnip4 and Sirt1, and increased amplitude for Col1a2 and Sirt1 (Figure 1Q–T; Table S2B). Overall, these results demonstrate that a large fraction of genes perturbed in AD exhibit time-of-day-specific expression, underscoring the impact of circadian dysfunction on disrupting the hippocampal transcriptome at early disease stages.
TRF effectively modulates metabolic markers and ketone-responsive genes in AD mice
Having detected disruptions in activity rhythms, sleep, and brain transcription that potentially contribute to pathology in APP23 TG mice, we evaluated whether a circadian and metabolic intervention could ameliorate AD phenotypes. TG and NTG littermate mice were randomly assigned to either ad libitum feeding (ALF) or time-restricted access to food (TRF) with a 6 h feeding/18 h fasting regimen, and the feeding window aligned to the middle of the active period (ZT15-ZT21; Figure 2A).
FIGURE 2. Time-restricted feeding induces metabolic changes, modulates brain transcription, and rescues behavior and sleep in AD mice.
A. Schematic representation of TRF intervention indicating feeding models and assay/evaluations performed. B-C. Significant changes in β-hydroxybutyrate and glucose in TRF-treated animals (n=31) in comparison to ALF mice (n=27), as detected in blood and presented as individual values with standard error of the mean. Statistical significance as per unpaired Students’ t-test. ◯ female; △ male. D. The expression of a set of metabolism-associated genes was modulated by TRF in the hippocampus of APP23 TG mice in a phase-dependent fashion, as shown by heatmap based on z-score of normalized reads and sorted by phase of expression in NTG mice. E-H. Total sleep is represented as average minutes per 24 h and sleep as average sleep per 1 h bin. Data was analyzed averaged over two 24 h sleep-wake cycles. Statistical significance represents one-way ANOVA with Tukey’s multiple comparisons test (E and G), or two-way ANOVA with Tukey’s multiple comparisons test (F and H) I-L. Activity recordings reported in 3-min and 6 h bins averaged from 7–10 days of activity. Statistical significance represents one-way ANOVA followed by Šídák’s multiple comparisons test (J and L). Top bar in waveforms represents the light-dark cycle. Bar graphs represent individual data plots with standard error of the mean. *p≤0.05; **p≤0.01; *** p≤0.001; **** p≤0.0001.
The metabolic effects of TRF are well described, including modulation of glucose and ketone pathways 21,34,35. In the present intervention, mice under TRF showed significantly increased β-Hydroxybutyrate and reduced glucose levels in blood at ZT14 in comparison to mice under ALF conditions (Figure 2B–C). Importantly, mice in all groups consumed equivalent volumes of food and showed no significant differences in body weight (Figure S3a–d), establishing that any observed effects were not mediated by caloric restriction. In addition, ketone-response genes and transcripts associated with lipid and carbohydrate metabolism were differentially expressed in the brains of TG mice under TRF, as shown by RNA-Seq data (Figure 2D). Importantly, TRF imposed time-of-day-specific changes in the expression of these genes, with the largest variations occurring at ZT12, after 15 h of fasting. Among these genes, we detected the transcription factor Klf15, which binds hippocampal glucocorticoid receptors and whose diurnal rhythmicity is known to be directly regulated by feeding 36. These findings confirm that TRF was effective in modulating the appropriate blood metabolites, and, importantly, that these peripheral changes were able to induce specific and temporal transcriptional responses in the brain. Moreover, these data demonstrate that despite ongoing brain pathology, TG mice can respond to this TRF intervention.
TRF improves sleep and restores activity rhythms in AD mice
We next evaluated the impact of TRF on behavior in APP23 mice. Temporal restriction of food intake for three months improved different aspects of sleep in females and males. Female TG mice on TRF showed increased total sleep, reaching the levels observed in NTG animals (Figure 2E–F). While no changes in total sleep were observed in TG males under TRF, these animals still showed increased mid-day wakefulness (Figure 2G–H). Importantly, both TG females and males under TRF showed improvements in the onset of sleep at ZT24, the beginning of the sleep phase (Figure 2F and H).
Markedly, TRF rescued specific behavioral abnormalities in both female and male TG mice, which no longer exhibited active phase hyperactivity, and whose activity patterns became indistinguishable from those observed in NTG mice (Figure 2I–L).
TRF modulates hippocampal transcription and pathways related to AD and inflammation
TRF effects have been extensively investigated in the liver, a metabolic hub with strong circadian rhythmicity. However, the potential effects of TRF on the brain are unknown. We first sought to understand the impact of feeding entrainment on brain transcription pathways associated with neurodegeneration by using the NanoString® focused panels for AD and neuroinflammation 37–39. Comparison of APP23 TG mice under TRF or ALF regimens showed differential expression of 86 AD genes and 100 neuroinflammation genes functionally associated with myelination, transmitter synthesis and storage, autophagy, cytokine remodeling, and adaptive immune response (Figure 3A–J; Table S3). These results indicate that TRF can affect the brain transcriptome, but importantly it can modulate pathways directly involved in AD pathology.
FIGURE 3. Effects of TRF on hippocampus transcription and diurnal rhythms of gene expression.
A-J. Differential expression of genes associated with Alzheimer’s disease (A-E) and neuroinflammation (F-J) in APP123 TG mice under TRF (n=10) vs ALF regimen (n=9) and as detected by NanoString® panels. Heatmaps sorted by fold-change after normalization and analysis in Rosalind. Comparison based on treatment, controlling by genotype, sex, and time of collection. Examples of gene expression are shown by box plots for the top changed genes in each group (B-E and G-J) and are based on normalized counts and presented as individual values with standard error of the mean. Statistical significance as per unpaired Students’ t-test *p≤0.05; **p≤0.01. ◯ female; △ male; white symbols for samples taken at ZT0; black symbols for samples taken at ZT12. K. Heatmap of significant DEGs in APP23 TG mice in TRF (n=18) vs ALF (n=20) conditions, based on RNA-Seq analysis of hippocampus tissue using DESEQ2; threshold adj.p≤0.05. Each row is one gene and normalized expression is represented by z-score. L. Gene ontology terms for the top significantly enriched functions for genes modulated by TRF in TG mice. M. Heatmap of genes showing changes in rhythmic expression in response to TRF in TG mice hippocampus, as defined by Metacycle analysis using JTK and LS statistics on normalized transcript levels detected by RNAseq across 4-time points. N. Gene ontology terms for genes that gained or lost rhythmicity under TRF. O-P. Volcano plot representation of DEGs between TG TRF and ALF at the maximum fasting time ZT12 (O) or after re-feeding at ZT18 (P). DESEQ2 analysis of RNA-Seq data, red denotes increased and blue decreased expression at adj.p<0.05. Q-R. Gene ontology terms for genes found to be differentially expressed at ZT12 (Q) or ZT18 (R) in TG mice under TRF.
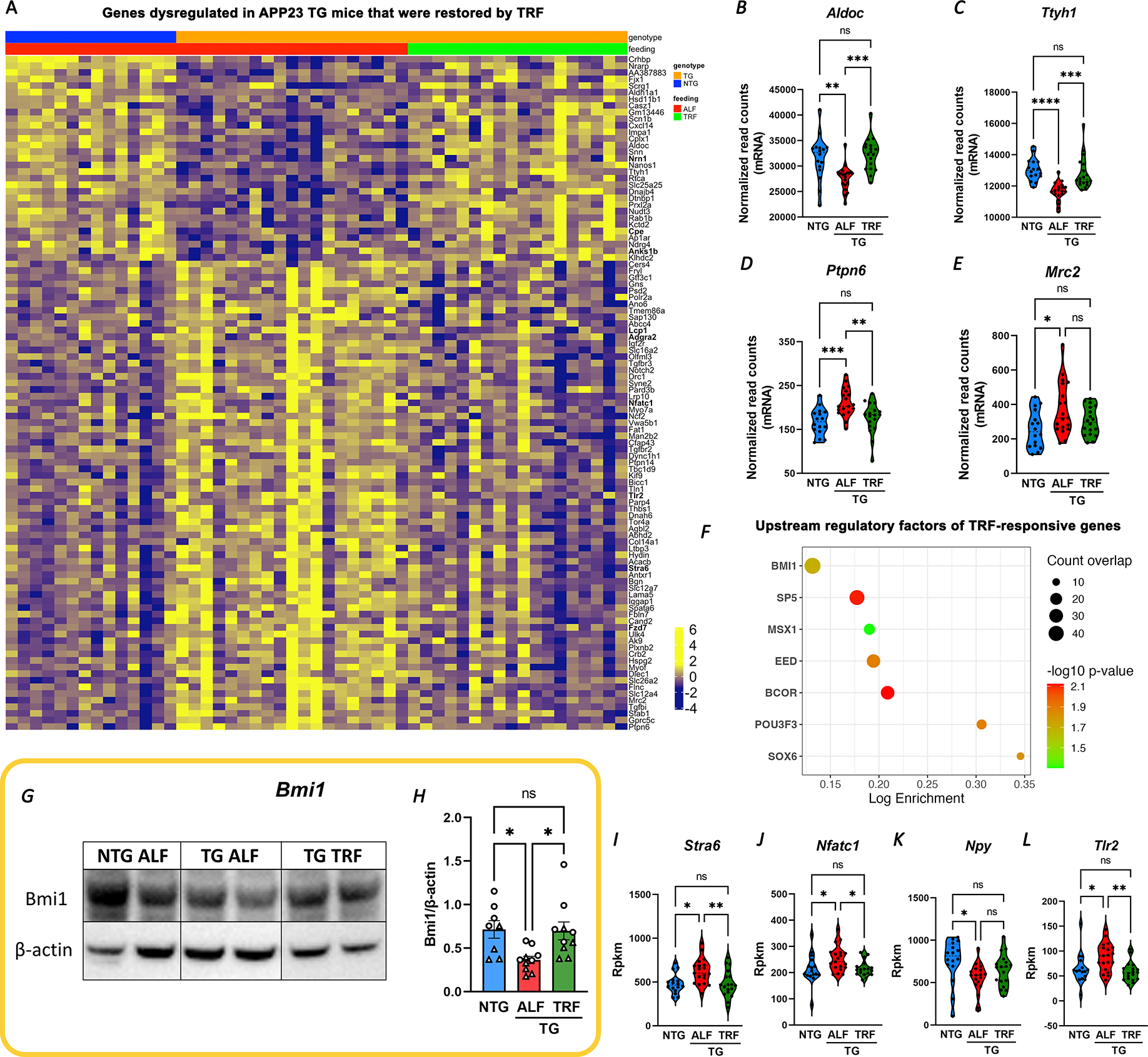
We next applied unbiased transcriptome-wide analysis using RNA-Seq for a comprehensive evaluation of the effects of TRF on the brain. For this analysis we included brain samples from TG mice collected at four time points for increased power. We analyzed the changes induced by TRF in TG mice from two dimensions: a) transcripts showing differential abundance in response to TRF in cross-sectional analysis, and b) genes that showed changes in rhythmicity upon TRF treatment, either gaining or loosing cyclic expression. In the former category, we identified 415 genes differentially expressed in TRF-TG mice in comparison to ALF (adj. pvalue<0.05, Figure 3K, Table S4). Notably, 41 of these genes are associated with AD (Table S4). Functionally, this group of genes was enriched in pathways that converge into AD pathology, including myelination, vasculature, microglia differentiation, cerebrospinal fluid circulation, and integrin signaling (Figure 3L). Analysis of upstream factors that may co-regulate these TRF-responsive genes showed enrichment for Bmi1 (regulating 45 genes in the set; FDR qvalue=2.7 E-7); Gsk3β (regulating 34 genes in the set; FDR qvalue=3.9 E-6), and Id2 (regulating 32 genes in the set; FDR qvalue=6.5 E-6.
On the other hand, Metacycle analysis using JTK and LS statistics identified 142 genes, including 7 AD-associated transcripts, showing differential oscillatory patterns in response to TRF, with 59 genes gaining rhythmicity (adj. pvalue <0.05 in TRF and nominal pvalue >0.2 in ALF) and 83 genes losing rhythmic expression under TRF (nominal pvalue >0.2 in TRF and adj. pvalue <0.05 in ALF, Figure 3M, Table S5A). Gene ontology analysis of this set showed enrichment for VEGFA/VEGFR2 signaling, vasculature development, and adherens junction, potentially implicating effects in the brain-blood barrier, alongside chromatin organization and histone modifications, cholesterol metabolism, fatty acids transport and autophagy (Figure 3N). Among the transcription factors enriched in this set of rhythmic genes modulated by TRF we identified Phf2 (top enrichment, targeting 23 genes in the set; FDR qvalue=2.1 E-6), a histone demethylase that increases excitatory postsynaptic potential in hippocampal neurons contributing to memory consolidation 40, and Id2 (targeting 38 genes in the set; FDR qvalue=7.2 E-10), a transcriptional repressor that can abolish gene activation by Bmal1:Clock dimers, and whose deficiency in mice is associated with disrupted locomotor activity rhythms and enhanced photoentrainment 41.
Lastly, we evaluated changes in gene expression at specific time-points that mark distinct metabolic states, as an approach to partially dissect the contribution of metabolic signaling to the observed effects of TRF, and including ZT12, which represents the maximum duration of fasting, and ZT18, which corresponds to re-feeding. We identified 30 genes differentially expressed in TRF-treated TG mice at ZT12 (adj. pvalue <0.05), and functionally associated with the response to nutrients, mTOR signaling, metabolism of lipids, myelination and PPAR signaling. Since the PPAR pathway is activated in response to fasting and integrates energy metabolism with the circadian clock, our observations are in alignment with other studies characterizing the metabolic changes activated by fasting 22. On the other hand, 151 genes were modulated by TRF at ZT18, and these were functionally enriched for gliogenesis, chemotaxis, apoptosis, integrin signaling, fatty acid oxidation, and indicating that the effects of circadian-aligned fasting are still present during the feeding state (Figure 3O–R; Table S5B).
Evaluation of the effects of TRF on the expression of core clock genes in the hippocampus of TG mice showed very mild effects of treatment (Figure S4; Table S2B), suggesting that the broad remodeling of the hippocampal transcriptome elicited by TRF, may be transduced by regulation of clock-controlled genes.
Altogether, these findings indicate that TRF has a profound impact on the brain transcriptome in APP23 TG mice. Not only did we detect gene expression remodeling in response to fasting, we also identified a substantial proportion of TRF-responsive genes that show rhythmic expression, and are regulated by factors like PPAR, ID2, and GSK3β that directly interact with clock genes 41–44. Thus, these data support the hypothesis that the pleiotropic effects of TRF in the brain are mediated by a complex cross-talk between the circadian clock and homeostatic systems that respond to temporal metabolic cues.
Bmi1 is a potential mediator of TRF-induced transcriptional changes in APP23 TG mice hippocampus
One salient finding of the above analysis is the extent of TRF-induced changes in the AD-associated transcriptome. We observed that 40% of the genes aberrantly expressed in the hippocampus of APP23 TG mice (103/258, from Fig. 1 I and Supp. Table1) were restored by TRF treatment, with their expression levels approaching those from NTG ALF mice (Figure 4A–E, Table S6). The rescued genes included 18 AD-transcripts (Tgfbr2, Gns, Tlr2, Col1a2, Cd74, Nrn1, Iqgap1 and others, Table S6) and were functionally enriched for gliogenesis, synaptic transmission, glutamatergic synapse, and ECM-receptor interaction.
FIGURE 4. TRF rescue of hippocampal transcription is partially mediated by Bmi1.
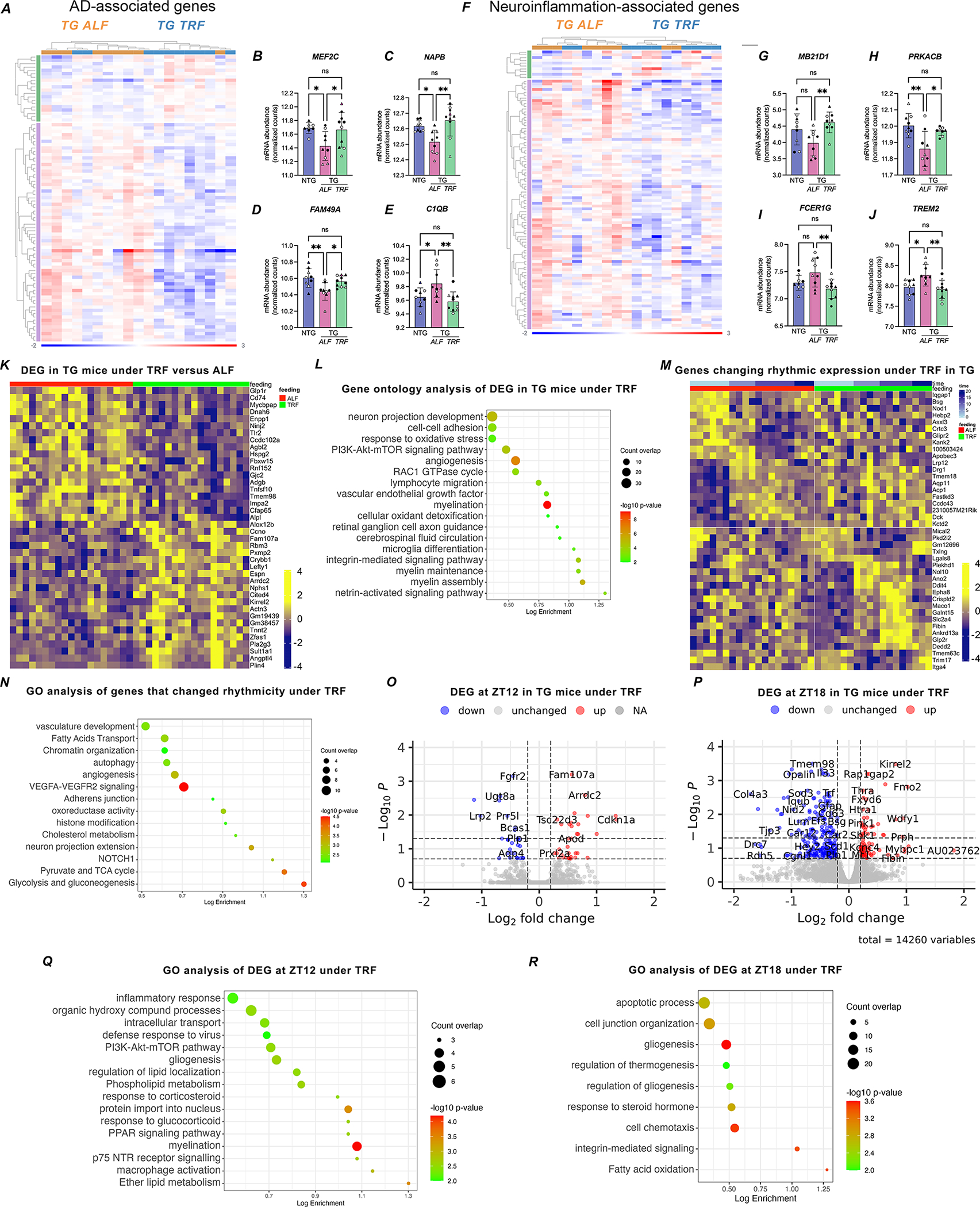
A-E. Heatmap representing changes in expression of genes deregulated in TG mice that were rescued by TRF. DESEQ2 analysis of RNA-Seq data at adj. pvalue<0.05 (A). Examples of gene expression changes are shown by violin plots for the top changed genes in each group (B-E) based on normalized counts. F. Top upstream regulatory factors enriched in TRF-responsive genes using GSEA; FDR q-value= 7.67E-07. G-H. Analysis of Bmi1 protein levels in brain lysates from NTG (n=8), TG ALF (n=10), and TG TRF (n=10) mice western blot (G) and quantification of Bmi1 abundance by densitometry, normalized to β-actin levels, and represented as individual values with standard error of the mean (H). I-L. Violin plots of selected Bmi1 downstream targets restored under TRF, based on normalized counts from RNAseq data. Statistical significance represents one-way ANOVA with Tukey’s multiple comparisons test *p≤0.05; **p≤0.01; ***p≤0.001 (B-E; H and I-L). ◯ female; △ male.
We applied in silico analysis using GSEA to identify potential upstream regulators of TRF-responsive genes, as these factors may shed light on the mechanisms that mediate the effects of TRF onto the AD transcriptome. Among the candidate molecules returned by this analysis, we focused on Bmi1 (a.k.a. Pcg4, Polycomb Group RING Finger Protein 4), the top enriched transcription factor modulating 47/415 TRF-responsive genes (FDR q-value= 7.67E-07, Figure 4F). This regulatory protein is abundantly expressed in the brain and involved in DNA damage response and chromatin remodeling, and importantly is specifically decreased in AD brains. Bmi1-deficient mice recapitulate late-onset AD pathology and show increased amyloid plaques and phosphorylated tau (pTau) 45–47. We found that Bmi1 regulates 25 genes disrupted in APP23 TG mice, and almost half of them are rescued by TRF (Table S6). In alignment with the role of Bmi1 in AD, we detected lower levels of Bmi1 protein in TG mice in ALF, in comparison to NTG animals, while TRF normalized Bmi1 expression in TG brains (Figure 4 G–H). Moreover, we validated that several downstream targets of Bmi1 were restored by TRF, including some highly relevant for AD pathology, like the retinol transporter Stra6 48, the neuromodulator Npy 49, and Tlr2, which acts as a receptor for amyloid-β triggering microglia inflammatory responses 50 (Figure 4 I–L). Taken in all, these results support a role for Bmi1 in partially mediating the beneficial effects of TRF on APP23 TG mice.
TRF reduces AD pathology in APP23 TG mice
In view of the effects of TRF in modulating pathways strongly associated with AD pathology, we next assessed the impact of this intervention on disease trajectory. Neuropathological evaluation by immunostaining and quantification of NeuN+ nuclei revealed significant, albeit modest, neuronal loss in the hippocampus of TG animals, with a trend towards reduced neuronal counts in the cortex. These deficiencies were partially restored by TRF (Figure S5a–n). Similarly, a trend towards increased Iba1+ microglia abundance was apparent in TG mice both in cortex and hippocampus, although TRF treatment did not significantly affected Iba1+ microglial cell number (Figure S5o–bb). Further, we observed increased astrogliosis in the cortex of TG mice, which was also reduced by TRF treatment, whereas no significant changes were detected in the hippocampus (Figure S5cc–pp).
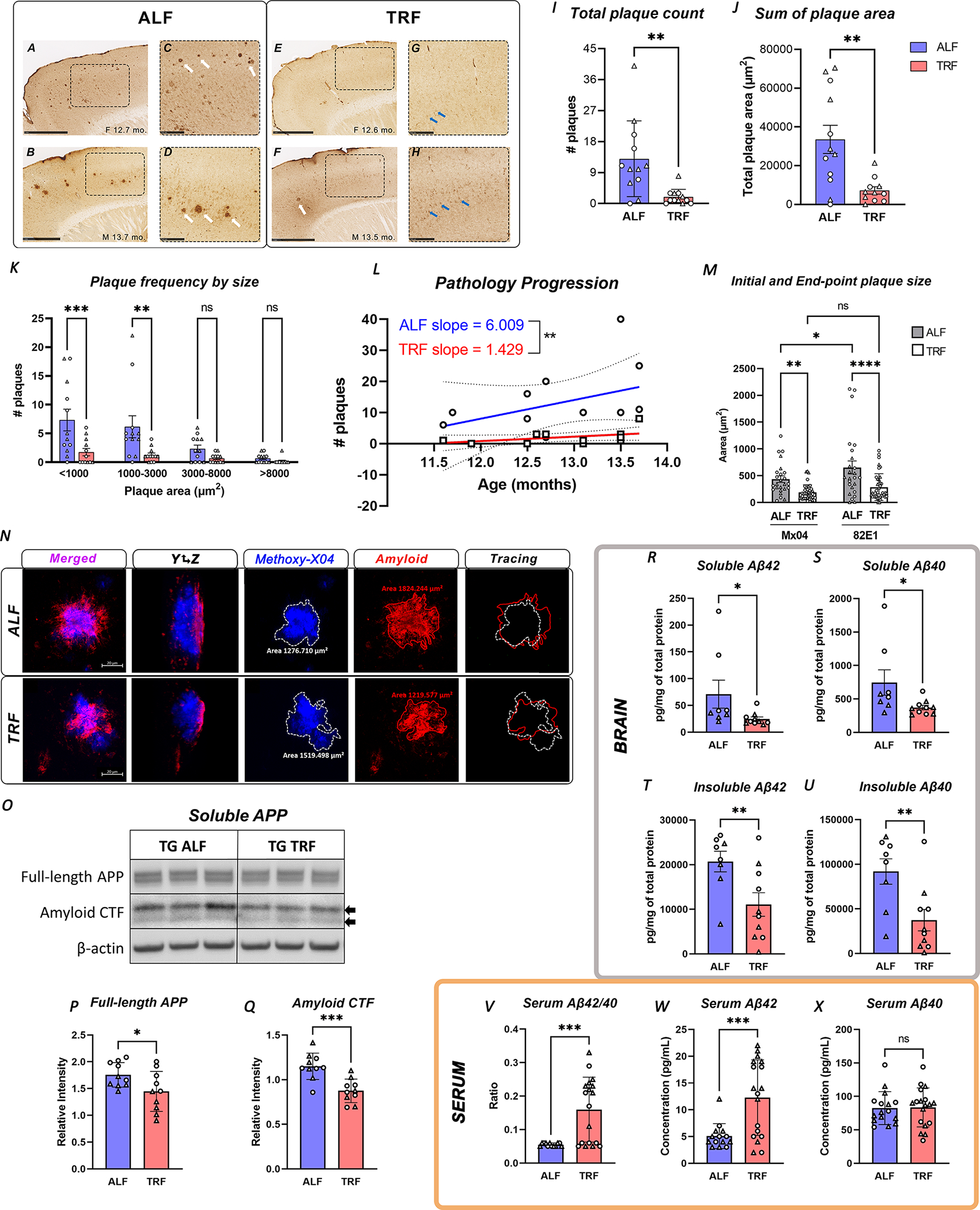
Immunostaining of amyloid-β showed that TRF-treated mice presented sparse, small core (dense) plaques, and intracellular accumulation of amyloid-β (Figure 5A–H), which may represent a pool for the formation of extracellular aggregates later in disease 51. The total area occupied by plaques, and the number of plaques per mouse were both significantly reduced in TG mice under TRF (Figure 5I–J). Plaque-size analysis showed fewer plaques of any size under TRF, with more pronounced and significant changes in smaller plaques (under 3,000 μm2), potentially indicating changes in the rate of amyloid deposition (Figure 5K). Moreover, linear regression analysis showed the expected positive correlation between plaque load and age for APP23 TG mice in all groups, but there was a significant difference in the elevations of the curves (p=0.0021), with TG mice in TRF showing a 42.8% reduction in the slope (1.429) in comparison to the ALF group (6.009) and demonstrating that overall disease progression was significantly attenuated by treatment (Figure 5L).
FIGURE 5. TRF significantly ameliorates amyloid pathology and disease progression in AD mice.
A-H. APP23 TG mice show progressive amyloid pathology, with plaque accumulation in the frontal and medial cortex. Pathology is substantially reduced for APP23 TG mice in TRF, as sex- and age-matched animals show only sparse plaques (white arrows) and intracellular accumulation of amyloid-β (blue arrows) in higher magnification sections (C, D, G and H). Lower magnification views with outlines indicating the magnified sections (A, B, E and F). I-K. Quantification of amyloid plaque counts (I), area occupied by plaques (J), and comparison of plaque number after binning by size (K) in APP23 TG mice in TRF (n=11) vs ALF (n=12). L. TRF slows down disease progression as seen by comparing the slopes obtained by simple linear regression of plotting total plaque number per mouse as a function of age in APP23 TG mice in TRF and ALF. M-N. In vivo analysis of plaque growth in APP23 TG mice. We labeled the initial time-stamp plaques that were present 7 weeks into the treatment by i.p. injection of Methoxy-X04 (Mx04). End point plaques were detected by immunostaining of sagittal sections with anti-amyloid antibody 82E1. Orthogonal projections of z-stacks obtained by confocal microscopy (80x) were used to trace and calculate the plaque area using ZEN Blue digital imaging software. Plot showing plaque sizes at initial (Mx04) and end point (82E1) times in APP23 TG mice under ALF or TRF from 12 mice/condition (M). Panels show representative images from single plaques analyzed in one TG TRF and one TG ALF (N; scale bar = 20 μm). O-Q. Detection of full length APP and amyloid C-terminal fractions in soluble brain lysates from TG mice on TRF (n=10) or ALF (n=10). Representative western blot image (O) and quantification of protein abundance normalized to β-actin (P-Q). R-U. TRF decreased soluble and insoluble Aβ40 and Aβ42 in brain lysates in APP23 TG mice comparing TRF (n=10) vs ALF (n=8) as quantified by ELISA (Mesoscale). V-X. TRF modulates clinically relevant blood biomarkers in treated mice. Total serum levels of amyloid-β fragments 40 and 42 were quantified by ELISA (Mesoscale) in APP23 TG mice in TRF (n=18) vs ALF (n=12) and presented individually or as ratio. Bar graphs represent individual data plots with standard error of the mean. ◯ female; △ male. Statistical significance represents the comparison between TG TRF vs ALF mice as per unpaired Students’ t-test (I-J and P-X) or one-way ANOVA with multiple comparison (K-M) * p≤0.05; **p≤0.01; *** p≤0.001; **** p≤0.0001.
To understand whether the slowed pathology progression was mediated by decreased amyloid-β deposition or increased clearance, we examined plaque dynamics with a multi-color timestamp approach using probes that lack competitive displacement, as previously reported 52. After 7 weeks of treatment, we performed an initial time-stamp-labeling via i.p. injection of Methoxy-X04 (Mx04). At the end of TRF, we followed with post-mortem immunohistochemical labeling of newly accumulated amyloid using the anti-amyloid 82E1 antibody. We quantified the longitudinal change in the size of plaques that occur between weeks 7 and 18 of treatment using orthogonal projections of z-stacked images from Mx04-labelled plaques (blue channel) and 82E1+ plaques (red channel, Figure 5N). Strikingly, at 7 weeks of treatment plaque size already appears significantly reduced in TRF mice in comparison to ALF animals (Mx04 plaques). Moreover, new plaque accumulation was also decreased under TRF, as evidenced by 82E1 staining (Figure 5M–N). These data support a role for TRF in both, inducing the clearance of pre-existing plaques and reducing the rate of new amyloid deposition.
We next evaluated the expression of 70 enzymes involved in APP metabolism (mSigDB GO) using our RNA-Seq data. TG mice (in ALF) presented only slight changes in the expression of Neprylisin, an enzyme implicated in the catabolism of Aβ peptides (fold change 0.86, pvalue=0.034), and which may correlate with amyloid deposition in TG mice. On the other hand, TRF only induced a slight decrease in the transcription of Bace1, which cleaves APP generating C-terminal amyloid fragments (CTFs; fold change 0.93, pvalue=0.033). We found the levels of full-length APP and the levels of C-terminal amyloid fragments to be reduced by TRF in TG mice (Figure 5O–Q). In addition, detection of amyloid fragments in the soluble and insoluble fractions using an anti-human amyloid 6E10 multispot assay showed a reduction of Aβ40 and Aβ42 levels in the brains of TRF-treated mice (Figure 5R–U). While we cannot rule out changes in protein levels or activity of APP metabolizing enzymes, the observed reductions in CTFs and Aβ40 and Aβ42 in the brain highly suggest that TRF treatment may facilitate amyloid clearance by multiple mechanisms 53–55. In alignment with these observations, detection of Aβ40 and Aβ42 levels in serum showed a significant elevation of the Aβ42/40 ratio in TRF-treated TG mice, driven by increased Aβ42 concentrations (Figure 5V–X), and further validating the increase in Aβ clearance induced by TRF.
TRF reduces pathology in a new rapidly progressing AD mouse model
The effectiveness of treatments initiated after the onset of amyloid deposition, and their capacity to restore cognitive impairment are fundamental questions for AD therapeutic development. Since our primary goal was to identify the effects of TRF on the alterations in clock-controlled transcription that appear early in disease progression, the TRF intervention in APP23 mice was started when amyloid pathology was negligible and cognitive deficits were not yet apparent in these animals. We therefore conducted a preliminary study utilizing the novel humanized APP knock-in mouse (APP-KI) harboring three familial mutations in the APP human gene, and which presents a much faster disease progression than APP23 mice, including amyloid deposition starting at three months of age and also accumulation of phospho-Tau in dystrophic neurites surrounding plaques 56. We validated the reported pathology progression in our mouse cohort and corroborated that 6-month-old AAP-KI mice present advanced amyloid accumulation, some microgliosis and more abundant astrogliosis (Figure S6). Based on these observations we started a 3-month-long TRF regimen on APP-KI mice at 4.5 months of age (Figure 6A). Similar to our observations in APP23, APP-KI mice under TRF showed significantly increased β-Hydroxybutyrate and reduced glucose levels in blood at ZT14 in comparison to mice under ALF (Figure 6B–C). Strikingly, despite the advanced pathology that APP-KI mice presented at the end point of treatment, quantification of amyloid plaques showed that TRF reduced amyloid load in these animals, both in plaque counts and area, compared to ALF mice (Figure 6D–E). Plaque-size analysis also showed a reduction in plaques of any size under TRF in APP-KI mice, with more pronounced and significant changes observed for small plaques (Figure 6F). Importantly, we detected a reduction of pTau puncta associated with plaques (Figure 6G–H), although the proportion of plaques that were positive for pTau (as % of total plaques present) did not change between TRF and ALF conditions (Figure 6I). While these results may be driven by the overall reduction in plaques, the possible implications for TRF inducing a reduction in the total burden of Tau aggregates in dystrophic neurites surrounding Aβ plaques (NP Tau), one of the major hallmarks of AD 6, is intriguing and warrants further investigation in Tau models.
FIGURE 6. Time-restricted feeding rescues cognitive behavior and reduces disease progression in the APP-KI AD mouse model.
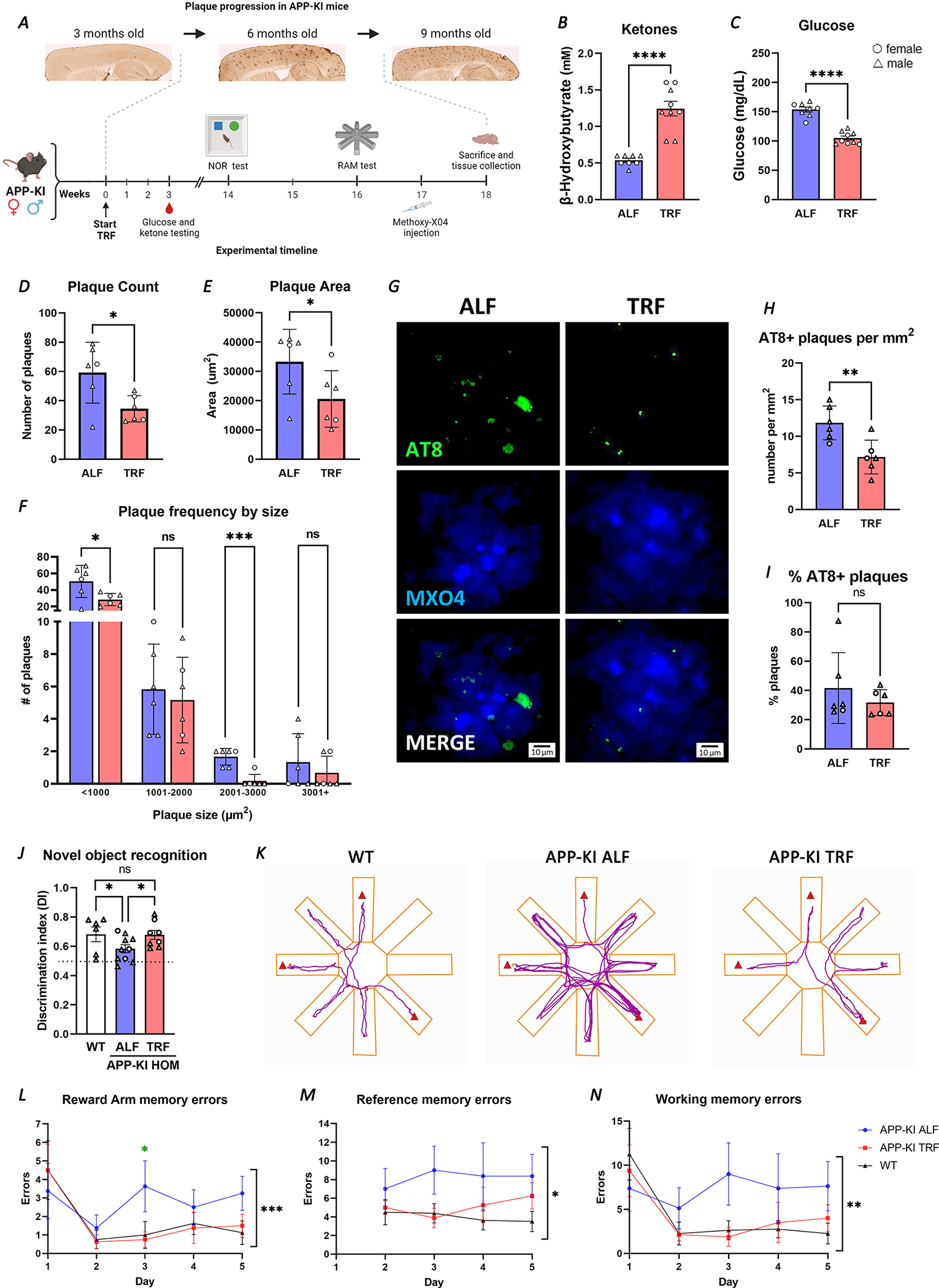
A. Schematic representation of the TRF intervention in APP-KI indicating evaluations performed with relation to timescale of pathology progression. B-C. Significant changes in β-hydroxybutyrate (B) and glucose (C) in APP-KI TRF treated mice (n=9) in comparison to APP-KI ALF mice (n=8), as detected in blood and presented as individual values. D-F. Plaque counts (D) and total plaque area (E) are reduced, and frequency by size (F) shows fewer plaques at any size under TRF. Plaques assessed in Methoxy-X04 injected APP-KI under ALF and TRF at 8.5-months (n=6 per treatment). 20 μm sagittal brain section images were analyzed using ImageJ. G-I. AT8+ puncta co-occurring with plaques was assessed in Methoxy-X04 injected APP-KI under TRF and ALF. Representative images show plaque staining with MX04, P-Tau detected by AT8 antibody and merged images (G). Number of AT8+ plaques per mm2 (H), and the percentage of assessed plaques that are AT8+ (I). J. Learning and memory was tested using the Novel Object Recognition test (n=6 WT; n=10 ALF; n=8 TRF). Values above the dotted line represent greater novel object exploration. K-N. Hippocampal dependent memory deficits were assessed using the Radial Arm Maze (n=8 per condition). Representative trace plots of animal center point for the duration of Radial Arm test on day 3. Red triangles indicate reward arms (K). 5 days of Radial Arm Maze testing showed Reward arm, and total Reference and Working memory errors (L-N). All graphs plotted with standard error of the mean. ◯ female; △ male. Statistical significance as per unpaired Students’ t-test (B-E and H-I), repeat measures two-way ANOVA (F), one-way ANOVA with multiple comparisons (J), multiple unpaired t-tests for APP-KI ALF vs TRF (L), and two-way ANOVA (L-N). *p≤0.05; **p≤0.01; ***p≤0.001; **** p<0.0001.
TRF improves cognition in APP-KI mice
Finally, we evaluated whether the TRF-induced improvements in AD phenotypes extend to cognitive function, specifically testing hippocampal-dependent memory, using the Novel Object Recognition test and the Radial Arm Maze (n=8 per condition). The APP-KI mice displayed a deficit in learning and memory in the Novel Object Recognition test that was improved by TRF, as treated mice spent more time exploring the novel object than mice in ALF (Figure 6J). Furthermore, TRF improved both short- and long-term memory in APP-KI mice in the Radial Arm Maze test, where the impairments in recalling recently visited (working memory) and reward (reference memory) locations displayed by mice on ALF were rescued by treatment (Figure 6K). The working and reference memory impairments in APP-KI mice were evident from day 3, with mice on ALF displaying deficits in short-term, reward arm and whole maze working memory (Figure 6L and N), and in long-term reference memory (Figure 6M). Notably, TRF restored cognitive performance in all of these domains in APP-KI mice to near wild type levels.
DISCUSSION
Disruptions in sleep and daily activity patterns are highly prevalent among people with AD, and emerging data indicate that these changes occur early in disease progression, potentially representing causal factors rather than consequences of neurodegeneration.
Here, we demonstrate the efficacy of a circadian intervention based on time-restricting feeding in rescuing pathology and behavior in two mouse models of AD. We provide ample evidence of the pleiotropic effects of TRF treatment in modulating behavior and sleep, in normalizing hippocampal gene expression in specific pathways associated with AD and neuroinflammation, and in improving memory deficits. Importantly, our results show that TRF can alter disease trajectory by slowing the progression of amyloid pathology, as evidenced by reduced plaque load, slower rate of amyloid deposition and increased Aβ42 clearance. While the effects of TRF in synchronizing and strengthening circadian rhythmicity in metabolic hubs like the liver are very well known, our study shows for the first time that circadian modulation through time-restricted feeding in AD models has the capacity of modulating crucial pathways that trigger neurodegeneration.
In addition to increasing total sleep and resolving sundowning-like hyperactivity, TRF specifically improved sleep at the beginning of the daily sleep period. Among the most common sleep disturbances in patients with AD are excessive daytime sleepiness, agitated behavior after sundown, and sleep disruptions, including difficulty in falling asleep and difficulty staying asleep (sleep fragmentation), all associated with declining cognitive performance and reduced white matter volumes 57. These alterations are viewed as disruptions in the circadian organization of sleep and wakefulness, which have been reported to increase the accumulation of Aβ. At the same time, Aβ accumulation and the resulting neuronal and synaptic damage can drive sleep disturbances and dysfunction of circadian clocks during the preclinical phase of AD, thus fueling a vicious cycle between AD pathology and circadian disruptions 6. Here we show that APP23 TG mice lose diurnal oscillation in genes regulated by the orexin signaling pathway, which may partly explain the hyperexcitability we observed in the mice and their reduced sleep. Orexin is a rhythmic neurotransmitter expressed in the hippocampus that modulates sleep, excitability, reward, and motivated behaviors. Notably, orexin and its receptors are diminished in patients with AD, contributing to sleep disturbances 58–60.
Impairments in the light input pathway have been reported in several AD mouse models 61,62, but responses to light were intact in APP23 TG mice in the current study, suggesting that the observed circadian impairments were not a result of light input deficits. In line with TRF acting as a synchronizing stimulus (zeitgeber), we saw rhythmic behavioral and transcriptomic changes in response to TRF. This may have significance for patients with AD, where pathology in the retina and light input pathways have been reported, and which may contribute to light response impairment and circadian rhythm dysfunction 63. Whereas light therapy may help some patients with AD presenting the most severe rest-activity disruptions 64, TRF may benefit a larger patient population, representing an efficacious approach to ameliorating circadian rhythm dysfunction.
Entrainment of circadian clocks by timed feeding and arousal are well studied 65, yet the benefits of the TRF protocol presented in this study extended beyond transient arousal at the time of feeding in the dark phase. Rather, the impact of TRF was sufficient to drive transcriptional changes in several interconnected pathways, including ECM and vascular remodeling, glycosylation, lipid and cholesterol dynamics, vesicle trafficking, autophagy, protein degradation, Aβ clearance, neuroglial functions, and inflammation, which collectively restored systems disrupted by AD pathology. Further, although there were only mild effects of TRF on the expression of core-clock genes, TRF restored diurnal oscillation in a significant number of genes, including in AD-associated transcripts. Cholesterol pathways were strongly disrupted in APP23 TG mice, representing a primary transcriptional target for the action of TRF. This is particularly relevant to AD, as abnormalities in cholesterol-related gene transcription play a role in altered amyloidogenic processing of APP and disease severity 66,67. The extent of TRF-driven changes in hippocampal gene expression, especially as they relate to pathways that impact AD pathogenesis and circadian disruption, may underlie the breadth of benefits observed in this study.
Shedding light on potential mechanisms that may mediate effects of TRF, we identified multiple upstream regulators for TRF-responsive genes. The finding of Bmi1 as a major regulator of genes modulated by TRF in APP23 TG mice is highly significant in relation to AD pathology. Haplodeficiency of Bmi1 in aged mice is sufficient to recapitulate late-onset AD pathology, including increased amyloid plaques and pTau, and the neuronal deficiency of Bmi1 appears to be highly specific for AD and not found in related dementias 47. In contrast to some previous studies finding no changes in Bmi1 expression in APP-overexpressing mice, we observed a significant reduction of Bmi1 protein expression in the cortex of APP23 mice, which was restored by TRF treatment. The effects of TRF on Bmi1 may also partially explain the improvements in hippocampal-dependent memory that we observed in APP-KI mice, as Bmi1 was associated with cognitive performance and CSF Aβ42 levels in patients with AD in a large ADNI study, which identified a specific SNP in Bmi1 that confers protection against AD 46. Bmi1 activity includes regulation of histone H2A mono-ubiquitination, suggesting the engagement of epigenetic factors in the observed remodeling of hippocampal transcription.
While future studies will be required to delineate the precise mechanisms by which TRF modulates Bmi1 activity, we observed that TRF-increased Bmi1 levels resulted in repression of downstream targets Nfatc1, Stra6, and Tlr2, and activation of Npy. Stra6 is a receptor for the binding of retinol and mediates signaling pathways that inhibit insulin. The increased Stra6 protein levels reported in AD brains and multiple animal models may be associated with dysregulation of metabolism in AD, which may be normalized by TRF 48. The Toll-like receptor (Tlr) family is involved in innate immunity and in the response to danger-associated molecular patterns arising from tissue damage. Tlr2 is constitutively expressed by microglia and mediates the activation of the NF-κB pro-inflammatory cascade induced by amyloid-β. While some studies point at Tlr2 functions as protective, with genetic ablation of Tlr2 in APP/PS1 mice worsening pathology and cognition, other studies indicate that suppression of Tlr2 mitigates amyloid pathology. The observation of increased Tlr2 expression in APP23 mice in our study, which was normalized by TRF treatment, may be a reflection of changes in microglial expression and neuroinflammation that were attenuated by TRF 50. Lastly, Npy is a neuromodulator with protective functions in the CNS, and transcript levels of Npy are decreased in plasma from patients with AD and in the cortex and hippocampus of transgenic AD mouse models. Importantly, supplementation of Npy reduces amyloid toxicity in vivo and in vitro, rescues glutamate excitotoxicity, and suppresses neuroinflammation 49. Taken together, our results on Bmi1 engagement in response to TRF and the changes observed in downstream targets directly associated with AD pathology implicate these pathways as plausible mechanisms mediating at least some of the pleiotropic beneficial effects of TRF.
Furthermore, the functional enrichment of PPARA-regulated pathways among TRF-responsive genes also links the beneficial effects of this intervention with improved circadian regulation. Members of the ligand-regulated nuclear receptor family (PPARs) are rhythmically expressed in mouse tissues, with PPARα and PPARγ directly modulating core clock genes Bmal1 and Rev-erbα, linking circadian rhythms and metabolism 43. PPAR-α regulates genes associated with glutamatergic, dopaminergic, and cholinergic signaling, and modulates the activity of α-secretase and BACE1, thus directly impacting APP processing and degradation. Importantly, PPAR-α is decreased in the brains of patients with AD, driving inflammation, oxidative stress, and alterations in lipid metabolism, all pathways that we showed here to be dysregulated in APP23 TG mice 68. Interestingly, Bmi1 and PPARa are currently being investigated as therapeutic targets for AD. Our findings showing the potential of TRF to engage these pivotal factors offer a new promise for non-pharmacological interventions in the treatment of AD.
The peripheral benefits of various feeding regimens have been explored in previous studies, with a recent paper elegantly demonstrating how a circadian-aligned 30% caloric-restricted feeding regimen leads to complex genome-wide reprogramming of circadian gene expression in the liver, including amelioration of age-related changes and protective effects on lifespan 69. In numerous preclinical animal models and human studies, restricting feeding to particular time windows, even without reducing caloric intake, attenuated metabolic diseases like obesity, glucose intolerance, dyslipidemia, and age-related decline in cardiac function 34,70. Similarly, a fast-mimicking diet was found to promote regeneration, including rejuvenating immune cells, promoting hippocampal neurogenesis, and increasing lifespan in mice, whereas a companion trial in humans improved regenerative markers and reduced disease risk factor biomarkers 71. Specifically for AD, recent work has shown that 40% caloric restriction in APP mice reduced amyloid load in the hippocampus 72. Intriguingly, a ketogenic diet was shown to reduce Aβ42 and Aβ40 in APP mice 73, and recently nutritional ketosis has been shown to improve the function of aquaporin 4 while reducing astrogliosis 74. Whereas this body of work shows a variety of benefits from various feeding approaches, the circadian-aligned TRF without caloric restriction in the current study not only normalizes AD pathology-related and daily patterns of gene expression in the hippocampus, but also ameliorates AD-related behaviors, cognition, and pathology.
Critically, administration of TRF in APP23 TG mice resulted in a significant reduction in amyloid load, with increased clearance of Aβ42, resulting in higher Aβ42/40 plasma ratios. In sporadic AD, clearance of Aβ42 into cerebrospinal fluid is reduced as early as 10–20 years before the onset of clinical symptoms and shows an inverse correlation with cortical Aβ burden 75–77. This observation has immediate translational value, as these clinical biomarkers could be used to monitor the efficacy of a time-restricted eating regimen in patients with AD.
Two important questions with clinical relevance are whether this intervention is still effective if initiated after the onset of amyloid plaque and Tau accumulation, at the stage when AD clinical diagnoses are made, and whether TRF is effective in mitigating cognitive decline as the cardinal, debilitating feature of the disease. Our results using APP-KI mice showing the rescue of short- and long-term memory by TRF are highly encouraging, and further support the potential translational application of TRF.
Restriction of the time window of eating has emerged as an important treatment option with whole-system effects. Here we demonstrate that these interventions can also modulate key disease pathways in the brain. Drug-based strategies targeting amyloid are still not widely accessible for patients, hence there is an urgent need for novel approaches to reduce or halt disease progression. Our findings support future studies exploring the therapeutic potential of time-restricted eating as a powerful circadian modulator that could significantly modify disease trajectory in patients with AD and which is eminently feasible for integration into clinical care.
LIMITATIONS OF STUDY
Our study has limitations associated with the AD models, the nature of circadian regulation, and the frequency of sampling. APP23 TG mice and APP-KI mice are AD models based on amyloidosis, and despite some accumulation of Tau being detectable, they don’t present neurofibrillary tangle formation or neuronal loss. Further, healthy mice accrue 25% of their sleep in the active phase, which was lost in the TG mice due to hyperexcitability. This differs from the typical sundowning hyperexcitability and the deterioration of rest phase sleep and active phase wakefulness observed in patients with AD and may represent a limitation to the generalizability of the study’s findings. The intricate cross-talk between metabolism and the circadian clock makes it quite difficult to disentangle their individual contributions to the observed phenotype rescue, and future studies are needed to dissect specific mechanisms, including manipulations of potential common regulators like Bmi1.
STAR METHODS
Resource availability
Lead contact
Further information and requests for Data and resources should be directed to and will be fulfilled by the lead contact, Dr. Paula Desplats (pdesplat@health.ucsd.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-Seq data have been deposited at the Gene Expression Omnibus (GEO) and are publicly accessible as of the date of publication. Accession numbers are listed in the key resources table.
All the values used to create graphs in the paper can be found in the Data S1 file in a single Excel spreadsheet.
The raw uncropped western blots can be found in the Data S1 file in a single PDF document.
Additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
This paper does not report original code.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Iba1 Rabbit antibody | FUJIFILM Wako Shibayagi | Cat# 019-19741; RRID:AB_839504 |
| Anti-NeuN Mouse antibody | Millipore Corp. | Cat# MAB377; RRID:AB_2298772 |
| Amyloid β (N) (82E1) Anti-Human Mouse IgG MoAb | Immuno-biological Laboratories | Cat# 10323; RRID:AB_10707424 |
| Anti-GFAP Rabbit antibody | Thermo Fisher Scientific Inc. | Cat# 18-0063; RRID:AB_138737 |
| Goat Anti-Rabbit IgG Antibody (H+L), Biotinylated | Vector Laboratories | Cat# BA-1000; RRID:AB_2313606 |
| Horse Anti-Mouse IgG Antibody (H+L), Biotinylated | Vector Laboratories | Cat# BA-2000; RRID:AB_2313581 |
| Methoxy-X04 | Tocris Bioscience | Cat. No. 4920 |
| Chicken anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 594 | Thermo Fisher Scientific Inc. | Cat# A-21201; RRID:AB_2535787 |
| Phospho-Tau (Ser202; Thr205) Monoclonal Antibody (AT8) | Thermo Fisher Scientific Inc. | Cat# MN1020; RRID:AB_223647 |
| Chicken anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 | Thermo Fisher Scientific Inc. | Cat# A-21200; RRID:AB_2535786 |
| Anti-Amyloid Precursor Protein; C-Terminal antibody produced in rabbit | MilliporeSigma | RRID:AB_258409 |
| Bmi1 (D42B3) Rabbit mAb | Cell Signaling Technology | Cat# 5856; RRID:AB_10838137 |
| Goat Anti-Rabbit IgG (H L)-HRP Conjugate | Bio-Rad | RRID:AB_11125143 |
| Horse Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076; RRID:AB_330924 |
| Anti-β-Actin Antibody (C4) | Santa Cruz Biotechnology | Cat# sc-47778; RRID:AB_626632 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals; peptides; and recombinant proteins | ||
| Critical commercial assays | ||
| Applied Biosystems™ High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific Inc. | Cat. No. 43-688-14 |
| RNeasy Lipid Tissue Mini kit | QIAGEN | Cat. No. 74804 |
| DAB Substrate Kit; Peroxidase (HRP); with Nickel; (3;3’-diaminobenzidine) | Vector Laboratories; Inc. | Cat. No. SK-4100 |
| VECTASTAIN Elite ABC HRP Kit (Peroxidase; Standard) Kit | Vector Laboratories; Inc. | Cat. No. PK-6100 |
| V-PLEX Aβ Peptide Panel 1 (6E10) Kit | https://www.mesoscale.com | Cat. No. K15200E |
| Deposited data | ||
| Raw and analyzed RNA-Seq data | This Paper | GEO: GSE236731 |
| Data S1. Uncropped western blots and data underlying graphs | This Paper | N/A |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| APP23 mice; B6.Cg-Tg(Thy1-APP)3Somm/J | Jackson Laboratory | RRID:IMSR_JAX:030504 |
| APP-SAA KI mice; B6.Cg-Apptm1.1Dnli/J | Jackson Laboratory | RRID:IMSR_JAX:034711 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| ImageJ (v1.53t) | NIH | RRID:SCR_023336 |
| Clocklab Analysis 6 (v6.1.12) | ActiMetrics | RRID:SCR_014309 |
| VitalView | Starr Life Sciences Corp. | RRID:SCR_014497 |
| ANY-maze (v4.99) | Stoelting Co. | RRID:SCR_014289 |
| El Temps program (v.313) | A. Diez-Nogura; Barcelona; Spain | http://www.el-temps.com/principal.html |
| Cran-package fpc (v2.2-10) | https://cran.r-project.org/ | https://cran.r-project.org/web/packages/fpc/index.html |
| DESeq2 | https://bioconductor.org/ | RRID:SCR_015687 |
| HISAT2 | http://ccb.jhu.edu/software/hisat2/index.shtml | RRID:SCR_015530 |
| JTK_CYCLE | https://openwetware.org/wiki/HughesLab:JTK_Cycle | RRID:SCR_017962 |
| Ingenuity Pathway Analysis | QIAGEN | RRID:SCR_008653 |
| Metascape | Metascape.org | RRID:SCR_016620 |
| Enrichr | https://maayanlab.cloud/Enrichr/ | RRID:SCR_001575 |
| DisGeNET | https://www.disgenet.org/ | RRID:SCR_006178 |
| Rosalind (v3.35.10.0) | https://www.rosalind.bio/ | RRID:SCR_006233 |
| geNORM | http://www.bioconductor.org | RRID:SCR_006763 |
| NormqPCR | http://www.bioconductor.org | RRID:SCR_003388 |
| Other | ||
| nCounter Analysis System | NanoString Technologies; Inc. | RRID:SCR_021712 |
| RNAlater Stabilization Solution | Thermo Fisher Scientific Inc. | Cat. No. AM7021 |
| AMP-AD Knowledge Portal | https://adknowledgeportal.synapse.org/ | RRID:SCR_016316 |
| NovaSeq 6000 Sequencing System | Illumina; Inc. | RRID:SCR_016387 |
| NanoZoomer S60 Digital slide scanner | https://www.hamamatsu.com/ | RRID:SCR_022537 |
| ZEN Digital Imaging for Light Microscopy | https://www.zeiss.com/ | RRID:SCR_013672 |
| OlyVIA (V3.4.1) | https://www.olympus-lifescience.com | RRID:SCR_016167 |
| DISCOVERY WORKBENCH | https://www.mesoscale.com | RRID:SCR_019192 |
| Prism 9 | https://www.graphpad.com/ | RRID:SCR_002798 |
| MESO QuickPlex SQ 120 | https://www.mesoscale.com | RRID:SCR_020304 |
| Precision Xtra Blood Glucose and Ketone Monitoring System | Abbott Laboratories; https://abbottstore.com | Cat. No. 9881465 |
| Agilent 4200 TapeStation System | Agilent Technologies; Inc. | RRID:SCR_019398 |
| TaqMan custom probes | Thermo Fisher Scientific Inc. | https://www.thermofisher.com/us/en/home/life-science/oligonucleotides-primers-probes-genes/applied-biosystems-custom-primers-probes.html |
| TaqMan™ Fast Advanced Master Mix | Thermo Fisher Scientific Inc. | Cat. No. 44-449-64 |
| LSM 800 confocal microscope | https://www.zeiss.com/ | RRID:SCR_015963 |
| SLIDEVIEW VS200 Slide Scanner | Olympus Life Science | https://www.olympus-lifescience.com/en/solutions-based-systems/vs200/ |
| iBright™ CL1500 Imaging System | Thermo Fisher Scientific Inc | Cat. No. A44240CFR |
Experimental model details
APP23 transgenic (TG) mice (B6.Cg-Tg(Thy1-APP)3Somm/J; RRID:IMSR_JAX:030504) and non-transgenic (NTG) littermates control mice were housed in light-tight enclosures at the University of California, Los Angeles in the Laboratory of Circadian and Sleep Medicine. APP-SAA KI (B6.Cg-Apptm1.1Dnli/J; RRID:IMSR_JAX:034711) were obtained from Jackson Laboratory as heterozygotes and bred to develop homozygote APP-SAA KI mice (APP-KI) and wild type (WT) controls. APP-KI were housed in light-tight enclosures at the University of California, San Diego. The mice were given ad libitum food (Teklad rodent diet 8604; Envigo, Indianapolis, IN) and water access until they were placed in the time-restricted feeding (TRF) or ad libitum feeding (ALF) cohorts. This study used a total of 110 APP23 mice almost equally distributed across sex/genotype/treatment throughout the study, and further grouped by ZT collection times. Further, an additional 26 mice consisting of APP-KI and WT controls were used. The work presented in this study followed all guidelines and regulations of the UCSD and UCLA Division of Animal Medicine that are consistent with the Animal Welfare Policy Statements and the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association.
Method details
Experimental design
Prior to any measurements or beginning any experiments, mice were habituated to a 12:12 light-dark (LD 12:12) cycle and single housing conditions in custom light-tight cabinets. We then measured diurnal rhythms in activity and sleep behavior from the mice. To assess circadian impairments in the mice, some animals were placed in constant darkness (DD), followed by light masking and 6 h phase advance protocols. Feeding manipulations were then performed on mice habituated to LD 12:12. Animals within each sex and genotype were randomly divided into two groups with one group continuing ALF and the other placed on TRF, with food available during 6 h in the middle of the mouse active phase, during zeitgeber time (ZT) 15 to ZT21. By definition, ZT0 is the time when lights go on and ZT12 is the time when the lights go off when the mice are in a 12:12 LD cycle. After habituating to the feeding protocol, metabolites were measured in the mice to ensure the treatment was inducing the expected metabolic effects. The mice remained on TRF or ALF food access until sacrificed for tissue collection. After all tests were completed and the mice had re-entrained to the LD cycle, mice within each genotype and treatment group were randomly assigned for sacrifice at one of four time points: ZT0, ZT6, ZT12, or ZT18. ZT0 and ZT6 tissues were collected in the light; ZT12 and ZT18 were collected in the dark. Brain tissue and blood serum were collected for analysis.
Time-restricted feeding
Experimental mice were housed in standard cages with either normal ad libitum food or with a custom-made programmable food hopper that could temporally control access to food and prevent food consumption during restricted times. Feeders were open for 6 h in the middle of the mouse active phase (ZT15 to ZT21). Food consumption was initially measured twice per week to ensure that all mice acclimated to the new feeding regimen. Mice are coprophagic and were moved to new cages twice per week, as determined empirically, to diminish coprophagic behavior during the fasting period.
β-hydroxybutyrate and glucose measurements
Tail blood sampling was performed at ZT14 under dim red-light conditions (3 lux). Blood was tested for β-hydroxybutyrate (BOHB, 1.5 μL sample) and glucose (0.6 μL sample) using a commercially available glucose/ketone meter (Precision Xtra Blood Glucose and Ketone Monitoring System, Abbott Laboratories, Chicago, IL).
Monitoring of cage locomotor activity
Experimental mice were singly housed in cages with IR motion sensors (Honeywell, Charlotte, NC) and activity data were analyzed using the El Temps programs (A. Diez-Nogura, Barcelona, Spain; http://www.el-temps.com/principal.html) and ClockLab Analysis 6 (Actimetrics, Wilmette, IL). The data were recorded and analyzed as previously described 78,79. Mice were initially entrained to a 12:12 LD cycle. Locomotor activity was recorded using Mini Mitter (Bend, OR) data loggers in 3-min bins, and 7 to 10 days of data were averaged for analysis. Activity data were collected for a month prior to placement in total darkness or any feeding treatments. The amount of cage activity over a 24 h period was averaged and reported in arbitrary units (a.u.)/h. The number of activity bouts and the average length of bouts were determined, with a new activity bout defined after a gap of either 21 min (maximum gap: 21 min; threshold: 3 counts/min) or 1 min (maximum gap: 1 min; threshold: 3 counts/min). Circadian period length (Tau) was determined in DD, and re-entraining phase shifts were measured after a 6 h advance in the light/dark cycle (see below). Tau in DD was obtained from the slope of a line fitted to activity onset times. Phase shifts were calculated as: [(Δh “activity onset” to “entrained onset”)/days to entrainment] and presented as the shift in h/day.
Monitoring of immobility-defined sleep behavior
Immobility-defined sleep was determined as previously described 78,79. Mice were housed in standard see-through plastic cages containing bedding (without the addition of nesting material). A side-on view of each cage was obtained, with minimal occlusion by the food bin or water bottle, both of which were top-mounted. Cages were top lit using 850nm IR LED lights (LLH-850nm-60; LEDLightinghut, Shenzhen City, China). Video capture was accomplished using surveillance cameras with visible light filters (Gadspot Inc., City of Industry, CA) connected to a video-capture card (Adlink Technology Inc., Irvine, CA) on a custom-built computer system. ANY-maze software (Stoelting Co., Wood Dale, IL) was used for automated acquisition of mouse immobility.
Immobility was registered when 95% of the area of the animal remained immobile for more than 40 sec, which was previously determined to have 99% correlation with simultaneous EEG/EMG defined sleep 80,81. Continuous tracking of the mice was performed for a minimum of 5 sleep-wake cycles, with time-randomized visits (1 time/day) by the experimenter to confirm mouse health and video recording. Two sleep-wake cycles over 48 h were averaged for further analysis. Immobility-defined sleep data were exported in 1-min bins, and total sleep time was determined by summing the immobility durations in the rest phase (ZT0 to ZT12) and active phase (ZT12 to ZT24). An average waveform of hourly immobile-sleep over the two sleep-wake cycles was produced per sex, genotype, and treatment for graphical display.
Assessments of circadian function
To assess the endogenous circadian period in the mice we measured free-running behavior in constant darkness for 10–14 days. To assess circadian response to external cues, mice were exposed to a single activity-augmenting dark pulse in the light phase (positive masking: 1 h dark pulse during the light phase at ZT3), then a single activity-suppressing light pulse in the dark phase (negative masking: 1 h light pulse in the dark phase at ZT15). Further, a 6 h phase advance was performed to assess circadian entrainment. Mice were permitted to recover for at least a week between light cycle manipulations, and entrainment was verified before each test was performed 82. These tests were not performed under TRF treatment, as TRF is itself a synchronizing stimulus (zeitgeber).
Assessments of cognitive function
Novel Object recognition test (NOR) was carried out in the mouse active phase over 5 consecutive days under dim red light (5 lux) as previously described 83. Each testing day, mice were acclimated to the testing room for 10 minutes before trials and testing. Each mouse was first habituated to the testing arena (35 cm × 35 cm × 35 cm) in a 10 min period on two consecutive days. Next, mice performed object familiarization trials on the following two consecutive days where two identical objects were placed equidistant from the walls on opposite sides of the arena and mice were allowed to explore for 10 min. On the day of testing for NOR, one familiar object was replaced with a novel object with a different shape and made of a different material. The testing trial was 5 min in duration. Arenas and objects were wiped between animals with 70% ethanol and dried using paper towels. ANY-maze software was used for acquisition using overhead CCTV cameras. The time spent was operationally defined as occurring when an animal directed its nose to the object at a distance <2.0 cm. Discrimination index was calculated as time spent with the novel object (Tnovel) divided by the sum of time spent with both objects (Tnovel + Tfamiliar).
8-Arm Radial Arm Maze (RAM) was carried out under dim light (5 lux) and over 5 consecutive days as previously described 84. For a week prior to starting RAM testing and each day after completion of the daily testing, all mice received 2.5 grams of food each day. Three random, nonconsecutive maze arms contained a highly palatable food reward consisting of a 1 kcal (250 uL) droplet of condensed milk. Spatial cues were placed above the terminal ends of reward arms which were unchanged for the duration of the test. The maze was cleaned by blotting up urine and picking up feces with kimwipes then wiping with 70% ethanol between each test and was completely washed and randomly rotated each day. To randomize the direction the mice were facing, mice were placed inside an opaque cylinder at the center of the maze for 15-sec before initiation of each test. An overhead CCTV camera fitted with infrared lighting connected to ANY-maze software was used for acquisition and scoring. On the first day of testing, each mouse was free to explore the maze and to discover the food rewards in the baited arms. Training and day 1 sessions were terminated when the mouse had explored all maze arms. On day 1, working memory errors were assessed. On days 2 to 5, tests were stopped when the mice had visited all three reward arms, after which working and reference memory were assessed. Returning to a previously visited arm is scored as a working memory error. Visiting an arm without a reward is scored as a reference memory error.
Tissue Collection
Mice were euthanized with isoflurane, either in the dark (ZT12 and ZT18) or in the light (ZT0 and ZT6). Brain hemispheres were collected and placed in either RNAlater Stabilization Solution (Thermo Fisher, Waltham, MA) or 4% paraformaldehyde. Serum was collected from blood incubated on ice for 30 min, centrifuged at 2000 g for 10 min at 4°C, and then stored until used at −80°C.
RNA isolation and RNA-Seq analysis
Total RNA was isolated from hippocampus tissue dissected from one hemibrain per animal using RNeasy Lipid Tissue Mini kit (Qiagen, Hilden, Germany) as indicated by the manufacturer. Quality of the extracted RNA was assessed using TapeStation (Agilent Technologies, Inc., Santa Clara, CA). All samples showed RIN ≥8.
RNA-Seq library preparation was performed with poly-A enrichment using poly-T oligo-attached magnetic beads employing the PE150 sequencing strategy performed on a NovaSeq 6000 (Illumina, Inc., San Diego, CA) by Novogene Inc. (Sacramento, CA). Raw reads (>20M per sample) were imported to Galaxy platform for analysis. Reads were first cleaned by removing adapter sequences, trimming low-quality ends, and filtering reads with low quality using Trimmomatic. Sequence alignment of the resulting high-quality reads to the mouse reference genome (build GRCm38) was done using HISAT2 and quantification of gene-level expression was performed using featureCounts. DESEQ2 was used to generate normalized read counts and to determine differentially expressed genes. Genes with an average expression of more than 2 counts per minute (CPM) across all samples were considered as expressed. Pathway enrichment analysis was performed using Ingenuity Pathway Analysis (IPA) software (Qiagen), Metascape [http://metascape.org], and Enrichr [https://maayanlab.cloud/Enrichr/] web-based tools.
Quantification of mRNA by Real time PCR
Total RNA (1.0 μg) was used for reverse transcription to cDNA using a High-Capacity cDNA Reverse Transcription Kit (Cat. No. 43-688-14; Applied Biosystems, Waltham, MA). Quantitative real-time PCR (qPCR) was performed using TaqMan Fast Advanced Master Mix and mouse specific TaqMan probes (Cat. No. 44-449-64; Thermo Fisher, Waltham, MA): Sirt1 (Mm01168521); Col1a2 (Mm00483888); Dnajb4 (Mm00508908); Kcnip4 (Mm00518835). qPCR reactions were performed in duplicate. Relative quantification of gene expression was calculated using β-actin (Actb (Mm00607939)) as an internal control and expressed as the inverse ratio to threshold cycle (1/dCt). AD-related genes were selected based on three sources: AMP-AD, DisGENET, and IPA AD-related molecules.
NanoString AD and Neuroinflammation Panels
Total RNA (1.0 μg) from hippocampus was used to hybridize either the nCounter© Mouse AD Consortium Panel or the nCounter© Mouse Neuroinflammation Panel (NanoString Technologies, Inc.). Raw data were exported into ROSALIND (version 3.35.10.0, https://rosalind.bio/) for analysis, including QC steps and differential gene expression. Normalization, fold changes and p-values were calculated using criteria provided by NanoString. ROSALIND® follows the nCounter® Advanced Analysis protocol of dividing counts within a lane by the geometric mean of the normalizer probes from the same lane. Housekeeping probes to be used for normalization are selected based on the geNorm algorithm as implemented in the NormqPCR R library (Perkins, JR. et al. BMC Genomics 13, 286 2012). Clustering of genes for the final heat-map of differentially expressed genes was done using the PAM (Partitioning Around Medoids) method using the fpc R library that takes into consideration the direction and type of all signals on a pathway, the position, role, and type of every gene, among other parameters (Hennig, C. Cran-package fpc. https://cran.r-project.org/web/packages/fpc/index.html).
Immunostaining
For immunohistochemistry (IHC), mice were sacrificed at the end of treatment and hemi-brains were extracted and fixed by 4% paraformaldehyde. Fixed APP23 brains were sectioned sagitally at 40 μm using a Leica VT1000S vibratome. Sections were washed three times in PBS, pre-treated with 1% Triton X-100, 10% H2O2 in PBS for 20 min at room temperature, washed again, and incubated for 1 h at room temperature in 10% serum according to secondary antibody species. The sections were incubated with primary antibodies to microglia marker Iba1 (1:500, FUJIFILM Wako Chemicals U.S.A. Corporation, code number 019-19741), neuronal marker NeuN (1:200, Millipore Corp., Cat. No. MAB377), amyloid marker 82E1 (1:500, Immuno-biological Laboratories, Cat. No. 10323), and astrocyte marker GFAP (1:200, Invitrogen 180063) at 4°C overnight. Sections were washed three times, incubated in 1:100 biotinylated secondary antibody (goat anti-rabbit, Vector Laboratories, Cat. No. BA-1000; or horse anti-mouse Vector Laboratories, Cat. No. BA-2000) for 30 min at room temperature, washed again, incubated in biotinylated HRP and avidin (ABC, Vector Laboratories, Cat. No. PK-6100) for 1 h in the dark at room temperature and then treated with diaminobenzidine (DAB) Substrate Kit, Peroxidase (Vector Laboratories, Cat. No. SK-4100) for coloration. 20X images were collected using a Nanozoomer slide scanner (Humamatsu, Japan).
Quantification of each cell type was done in ImageJ using the “analyze particles” tool or determining the % area covered by the signal in hippocampus and cortex. Plaque count and area were manually acquired by blinded researchers using 82E1 DAB-stained sections exported into ImageJ.
Longitudinal labeling of plaques
To determine the rate of amyloid deposition in APP23 mice, amyloid plaques were labelled in vivo on a subset of n=16 TG mice 1 mo after TRF protocol was started, by i.p. administration of 10 mg/kg Methoxy-X04 (Tocris Bioscience, Cat. No. 4920; 25 mg/mL in DMSO and diluted 1:10 in PBS) as previously reported 52,85. Fixed hemibrains collected after sacrificing the mice were later stained with anti-human amyloid beta antibody 82E1 (IBL 10323; 1:500 in PBST) using chicken anti-mouse Alexa Fluor 594. All Methoxy-X04 stained plaques were imaged on a Zeiss LSM 800 confocal microscope. Three-channel 12-bit images were sequentially acquired in 1 μM z-steps spanning the entire thickness of the plaque (Zeiss 63xplain apochromatic oil immersion lens, NA:1.4). Uniform pinhole AUM=1 was applied to all sections, with laser power ranging between 10 and 23% for the red (561 nm) and blue (405 nm) channels; digital gain ranging between 0.3 and 3.4 (red) and 0.3 and 2 (blue) and detector offset ranging for 1 to 2 in both channels. Settings were defined as to obtain the same exposure level based on pixel intensity in the blue channel (Methoxy-X04).
Plaques were manually quantified in ZEN Blue (Carl Zeiss AG) by blinded investigators who determined the areas detected in the red (82E1) and blue (Methoxy-X04) channels in orthogonal projection images generated for each plaque analyzed. A total of 16 APP23 TG mice and two sections per animal were analyzed (n=7 ALF; n=9 TRF; 0–7 plaque image/section/mouse).
Methoxy-X04 plaques in APP-KI were assessed in sagittal brain sections (n=6 per treatment) collected at 20 μm thickness on a Leica VT1000S vibratome. Two sections per animal were scanned on a SLIDEVIEW VS200 Slide Scanner (Olympus Life Science, USA). Fluorescent images were exported using OlyVia software (V3.4.1, Olympus Life Science, USA) and analyzed using ImageJ. Additionally, phospho-tau per plaque was assessed by immunofluorescence in three cortical regions (posterior, medial and anterior; 0.36 mm × 1.0 mm) per sagittal section, resulting in the inclusion of 16 to 49 plaques per animal (n=6 mice per condition). The sections were washed three times in PBST for 10 min followed by 10 min in 0.1% Triton-X in PBST and blocking for 1 h in 10% chicken serum in PBST all at room temperature. Sections were incubated with primary antibody to phospho-tau (Ser202, Thr205) marker AT8 (1:100, Invitrogen Cat. No. MN1020) at 4°C overnight. Following three 10 min washes in PBST the sections were incubated in secondary antibody (1:200, Invitrogen Cat. No. A21200), washed three times in PBST for 10 min and once in H2O, all at room temperature. Mounted sections were scanned, with images exported and analyzed as above.
Soluble and insoluble Aβ extraction from brain
Soluble and insoluble Aβ fractions from APP23 brain lysates were prepared as previously described 86. Brain tissue was briefly homogenized in Dounce then a PowerGen 125 (Thermo Fisher, Waltham, MA) homogenizers in 0.2 μm filtered 1% CHAPSO (Cat. No. C3649, Sigma-Aldrich, St. Louis, MO) in PBS with cOmplete™, Mini Protease Inhibitor Cocktail (Cat. No. 11836153001, Roche, Germany) for 3 × 10 sec, followed by 30 min incubation at 4°C with shaking. Homogenates were ultra-centrifuged (Beckman TLA 100.4 Rotor) at 46k rpm (100k g) at 4°C for 1 hour then supernatants were collected as “soluble” fraction. 1 mL 70% formic acid was added to each pellet and homogenized for 3 × 10 sec on ice then ultra-centrifuged at 46k rpm (100k g) at 4°C for 1 hour. The clean layer between the lipid layer and pellet was collected and then diluted with a 10x volume of 2M Tris base pH 11–11.5 to achieve a final pH of 8–8.5. Protease Inhibitor was added (Cat. No. P8340, Sigma-Aldrich, St. Louis, MO) followed by centrifugation at 3K rpm for 10 min and collecting samples as “insoluble” fraction. Samples were stored at −80°C until used.
Aβ40 and Aβ42 Quantification
In APP23 mice, levels of Aβ40 and Aβ42 were determined in soluble and insoluble brain lysate fractions (n=18) and in peripheral serum (n=38) using the Meso Scale V-PLEX Aβ Peptide Panel 1 (6E10) Kit (Cat. No. K15200E) processed on a MESO QuickPlex SQ 120 as indicated by the manufacturer, with final incubation at 4°C overnight. NTG mice assayed had no detectable levels of human Aβ40 or Aβ42 (not shown). Analysis was performed in the Discovery Workbench (Meso Scale Diagnostics, Rockville, MD).
Western Blots
Western blotting detection of target proteins was performed as previously described 5. Protein concentrations were measured by Pierce™ BCA Protein Assay Kit (Cat. No. 23225, Thermo Fisher, Waltham, MA). After dilution, 20 μg of protein per sample were loaded on NuPAGE 4–12% Bis-Tris gels (Cat. No. NP03022BOX, Thermo Fisher, Waltham, MA) and subsequently transferred using the iBlot 2 Dry Blotting System onto iBlot 2 Transfer Stack nitrocellulose (Cat. No. IB23001, Thermo Fisher, Waltham, MA). After blocking, membranes were incubated with primary antibodies overnight at 4°C with gentle rocking with: Anti-Amyloid Precursor Protein, C-Terminal (1:1,000, Cat. No. A8717, MilliporeSigma, Burlington, MA), Bmi1 (D42B3) (1:500, Cat. No. 5856, Cell Signaling Technologies, Danvers, MA). After washing, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies, either anti-rabbit (1:3000, Cat. No. 1721019, Bio-Rad, Hercules, CA) or anti-mouse (1:3000, Cat. No. 7076S, Cell Signaling Technologies, Danvers, MA), for 1 h at room temperature. Digital imaging was performed on an iBright 1500 (Thermo Fisher, Waltham, MA) using Clarity Western ECL (Cat. No. 1705060, Bio-Rad, Hercules, CA). Blots were subsequently stripped with 30% hydrogen peroxide for 15 min at 37° C, followed by washing, blocking, and then probing with β-actin (C4) (1:1,000, Cat. No. sc-47778, Santa Cruz Biotechnology, Dallas, TX). Protein abundance was assessed by densitometry in ImageJ and normalized to β-actin.
Quantification and statistical analysis
Data were analyzed in GraphPad Prism 9 (GraphPad Software, Inc., San Diego, CA) unless indicated otherwise. Figure error bars represent the standard error of the mean (SEM) unless stated otherwise. For comparisons, t-tests were used to determine significant differences between means of two group values, either paired or unpaired as appropriate. To analyze sleep waveforms, we performed an analysis of variance (ANOVA) followed by multiple unpaired t-tests corrected for multiple comparisons using the Holm-Šídák method. Significance is indicated as follows: * = p≤0.05, ** = p≤0.01, *** = p≤0.001, **** = p≤0.0001. NanoString data for gene expression was analyzed using the Rosalind suite with P-value adjustment using the Benjamini-Hochberg method of estimating false discovery rates (FDR). Differential gene expression from RNA-Seq data was determined by DESEQ2 using the negative binomial distribution. Differential expression was considered when the median of groups had an adjusted pvalue <0.05 while a relaxed significance threshold of adj.p≤0.2 was used for discovery and pathway analysis. We explored the impact of sex on gene expression by Principal Component Analysis and we found this factor uncorrelated with overall transcriptional changes. Sex was still used as a variable controlled for in DESEQ2 analysis. The periodicity of gene expression data was evaluated using LS and JTK_Cycle algorithm within the MetaCycle R package suite.
Supplementary Material
Data S1. Unprocessed data underlying the display items in the manuscript, related to Figures 1–6 and Figure S1–5
Table S1. DEGs between APP23 TG and NTG mice, Related to Figure 1
Table S2A. Genes showing time-of-day transcription changes in APP23 TG and NTG mice, Related to Figure 1.
Table S2B. Analysis of rhythmicity in selected Core Clock and AD-associated genes, Related to Figure 1 and Figure S4
Table S3. Nanostring DEGs in APP23 TG under TRF vs ALF treatment, Related to Figure 3
Table S4. DEGs in APP23 TG under TRF vs ALF treatment, Related to Figure 3
Table S5A. Analysis of rhythmic expression changes in APP23 TG mice under TRF, Related to Figure 3.
Table S5B. Analysis of genes modulated by TRF in APP23 TG mice at ZT12 and ZT18, Related to Figure 3
Table S6. Genes rescued by TRF treatment in APP23 mice, Related to Figure 4
Highlights.
AD mice present circadian deregulation and aberrant time-of-day brain transcription.
TRF modulates hippocampal gene expression and pathways related to AD and inflammation.
Increased Aβ clearance and reduced amyloid deposition mediate TRF benefits.
Time-restricted feeding recovers sleep and activity rhythms and improves cognition.
ACKNOWLEDGEMENTS
This study was supported by NIA grant #AG061831 to PD and DKW and by a training fellowship to DSW from NIA grant #5T32AG066596-02. The UCSD Microscopy Core is supported by NINDS grant #P30NS047101. We thank Charisse Winston-Gray, PhD, and Floyd Sarsoza for their technical assistance with the MSD MULTI-SPOT Assay System. We thank Janna Wu for her technical assistance with microglial IHC quantification. We would also like to thank Diego Martinez, Noora El-sherif, Deap Bhandal, Sophia Anne Marie Villanueva, Kevin O’Donnell and Karan Singh for vigilantly undertaking the vivarium work, activity record monitoring, and overseeing TRF.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research.
We worked to ensure sex balance in the selection of non-human subjects.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Rajan KB, Weuve J, Barnes LL, McAninch EA, Wilson RS, and Evans DA (2021). Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimers Dement. 17, 1966–1975. 10.1002/alz.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colwell CS (2021). Defining circadian disruption in neurodegenerative disorders. J. Clin. Invest. 131. 10.1172/JCI148288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fifel K, and Videnovic A (2021). Circadian and Sleep Dysfunctions in Neurodegenerative Disorders—An Update. Front. Neurosci. 0. 10.3389/fnins.2020.627330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holth JK, Patel TK, and Holtzman DM (2016). Sleep in Alzheimer’s Disease–Beyond Amyloid. Neurobiol. Sleep Circadian Rhythms 2, 4–14. 10.1016/j.nbscr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cronin P, McCarthy MJ, Lim ASP, Salmon DP, Galasko D, Masliah E, De Jager PL, Bennett DA, and Desplats P (2017). Circadian alterations during early stages of Alzheimer’s disease are associated with aberrant cycles of DNA methylation in BMAL1. Alzheimers Dement. 13, 689–700. 10.1016/j.jalz.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang C, and Holtzman DM (2020). Bidirectional relationship between sleep and Alzheimer’s disease: role of amyloid, tau, and other factors. Neuropsychopharmacology 45, 104–120. 10.1038/s41386-019-0478-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li P, Gao L, Gaba A, Yu L, Cui L, Fan W, Lim ASP, Bennett DA, Buchman AS, and Hu K (2020). Circadian disturbances in Alzheimer’s disease progression: a prospective observational cohort study of community-based older adults. Lancet Healthy Longev. 1, e96–e105. 10.1016/s2666-7568(20)30015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musiek ES, Bhimasani M, Zangrilli MA, Morris JC, Holtzman DM, and Ju Y-ES (2018). Circadian Rest-Activity Pattern Changes in Aging and Preclinical Alzheimer Disease. JAMA Neurol. 75, 582–590. 10.1001/jamaneurol.2017.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruben MD, Wu G, Smith DF, Schmidt RE, Francey LJ, Lee YY, Anafi RC, and Hogenesch JB (2018). A database of tissue-specific rhythmically expressed human genes has potential applications in circadian medicine. Sci. Transl. Med. 10, eaat8806. 10.1126/scitranslmed.aat8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoyt KR, and Obrietan K (2022). Circadian clocks, cognition, and Alzheimer’s disease: synaptic mechanisms, signaling effectors, and chronotherapeutics. Mol. Neurodegener. 17, 35. 10.1186/s13024-022-00537-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welsh DK, Takahashi JS, and Kay SA (2010). Suprachiasmatic Nucleus: Cell Autonomy and Network Properties. Annu. Rev. Physiol. 72, 551–577. 10.1146/annurev-physiol-021909-135919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koronowski KB, and Sassone-Corsi P (2021). Communicating Clocks Shape Circadian Homeostasis. Science 371. 10.1126/science.abd0951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haque SN, Booreddy SR, and Welsh DK (2019). Effects of BMAL1 Manipulation on the Brain’s Master Circadian Clock and Behavior. Yale J. Biol. Med. 92, 251–258. [PMC free article] [PubMed] [Google Scholar]
- 14.Kwapis JL, Alaghband Y, Kramár EA, López AJ, Vogel Ciernia A, White AO, Shu G, Rhee D, Michael CM, Montellier E, et al. (2018). Epigenetic regulation of the circadian gene Per1 contributes to age-related changes in hippocampal memory. Nat. Commun. 9, 1–14. 10.1038/s41467-018-05868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Acosta-Galvan G, Yi C-X, van der Vliet J, Jhamandas JH, Panula P, Angeles-Castellanos M, del Carmen Basualdo M, Escobar C, and Buijs RM (2011). Interaction between hypothalamic dorsomedial nucleus and the suprachiasmatic nucleus determines intensity of food anticipatory behavior. Proc. Natl. Acad. Sci. U. S. A. 108, 5813–5818. 10.1073/pnas.1015551108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bass J, and Takahashi JS (2010). Circadian Integration of Metabolism and Energetics. Science 330, 1349–1354. 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephan FK (2002). The “Other” Circadian System: Food as a Zeitgeber. J. Biol. Rhythms 17, 284–292. 10.1177/074873040201700402. [DOI] [PubMed] [Google Scholar]
- 18.Tahara Y, and Shibata S (2013). Chronobiology and nutrition. Neuroscience 253, 78–88. 10.1016/j.neuroscience.2013.08.049. [DOI] [PubMed] [Google Scholar]
- 19.Wang H-B, Loh DH, Whittaker DS, Cutler T, Howland D, and Colwell CS (2018). Time-Restricted Feeding Improves Circadian Dysfunction as well as Motor Symptoms in the Q175 Mouse Model of Huntington’s Disease. eNeuro 5. 10.1523/ENEURO.0431-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whittaker DS, Loh DH, Wang H-B, Tahara Y, Kuljis D, Cutler T, Ghiani CA, Shibata S, Block GD, and Colwell CS (2018). Circadian-based Treatment Strategy Effective in the BACHD Mouse Model of Huntington’s Disease: J. Biol. Rhythms 33, 535–554. 10.1177/0748730418790401. [DOI] [PubMed] [Google Scholar]
- 21.Jamshed H, Beyl RA, Della Manna DL, Yang ES, Ravussin E, and Peterson CM (2019). Early Time-Restricted Feeding Improves 24-Hour Glucose Levels and Affects Markers of the Circadian Clock, Aging, and Autophagy in Humans. Nutrients 11, 1234. 10.3390/nu11061234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anton SD, Moehl K, Donahoo WT, Marosi K, Lee S, Mainous AG, Leeuwenburgh C, and Mattson MP (2018). Flipping the metabolic switch: Understanding and applying health benefits of fasting. Obes. Silver Spring Md 26, 254–268. 10.1002/oby.22065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colwell CS (2015). Circadian medicine (John Wiley & Sons Inc; ). [Google Scholar]
- 24.Lee Y, and Wisor JP (2021). Multi-Modal Regulation of Circadian Physiology by Interactive Features of Biological Clocks. Biology 11, 21. 10.3390/biology11010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lundell LS, Parr EB, Devlin BL, Ingerslev LR, Altıntaş A, Sato S, Sassone-Corsi P, Barrès R, Zierath JR, and Hawley JA (2020). Time-restricted feeding alters lipid and amino acid metabolite rhythmicity without perturbing clock gene expression. Nat. Commun. 11. 10.1038/s41467-020-18412-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruan H-B, and Crawford PA (2018). Ketone bodies as epigenetic modifiers. Curr. Opin. Clin. Nutr. Metab. Care 21, 260–266. 10.1097/MCO.0000000000000475. [DOI] [PubMed] [Google Scholar]
- 27.Tognini P, Samad M, Kinouchi K, Liu Y, Helbling J-C, Moisan M-P, Eckel-Mahan KL, Baldi P, and Sassone-Corsi P (2020). Reshaping circadian metabolism in the suprachiasmatic nucleus and prefrontal cortex by nutritional challenge. Proc. Natl. Acad. Sci. 117, 29904–29913. 10.1073/pnas.2016589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Webb IC, Baltazar RM, Lehman MN, and Coolen LM (2009). Bidirectional interactions between the circadian and reward systems: is restricted food access a unique zeitgeber? Eur. J. Neurosci. 30, 1739–1748. 10.1111/j.1460-9568.2009.06966.x. [DOI] [PubMed] [Google Scholar]
- 29.Van Dam D, Vloeberghs E, Abramowski D, Staufenbiel M, and De Deyn PP (2005). APP23 Mice as a Model of Alzheimer’s Disease: An Example of a Transgenic Approach to Modeling a CNS Disorder. CNS Spectr. 10, 207–222. 10.1017/S1092852900010051. [DOI] [PubMed] [Google Scholar]
- 30.Van Erum J, Van Dam D, Sheorajpanday R, and De Deyn PP (2019). Sleep architecture changes in the APP23 mouse model manifest at onset of cognitive deficits. Behav. Brain Res. 373, 112089. 10.1016/j.bbr.2019.112089. [DOI] [PubMed] [Google Scholar]
- 31.Goodman AM, Langner BM, Jackson N, Alex C, and McMahon LL (2021). Heightened Hippocampal β-Adrenergic Receptor Function Drives Synaptic Potentiation and Supports Learning and Memory in the TgF344-AD Rat Model during Prodromal Alzheimer’s Disease. J. Neurosci. 41, 5747–5761. 10.1523/JNEUROSCI.0119-21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Debski KJ, Ceglia N, Ghestem A, Ivanov AI, Brancati GE, Bröer S, Bot AM, Müller JA, Schoch S, Becker A, et al. (2020). The circadian dynamics of the hippocampal transcriptome and proteome is altered in experimental temporal lobe epilepsy. Sci. Adv. 6, eaat5979. 10.1126/sciadv.aat5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carter B, Justin HS, Gulick D, and Gamsby JJ (2021). The Molecular Clock and Neurodegenerative Disease: A Stressful Time. Front. Mol. Biosci. 8, 644747. 10.3389/fmolb.2021.644747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaix A, Zarrinpar A, Miu P, and Panda S (2014). Time-Restricted Feeding Is a Preventative and Therapeutic Intervention against Diverse Nutritional Challenges. Cell Metab. 20, 991–1005. 10.1016/j.cmet.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, Leblanc M, Chaix A, Joens M, Fitzpatrick JAJ, et al. (2012). Time restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high fat diet. Cell Metab. 15, 848–860. 10.1016/j.cmet.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mifsud KR, Kennedy CLM, Salatino S, Sharma E, Price EM, Haque SN, Gialeli A, Goss HM, Panchenko PE, Broxholme J, et al. (2021). Distinct regulation of hippocampal neuroplasticity and ciliary genes by corticosteroid receptors. Nat. Commun. 12, 4737. 10.1038/s41467-021-24967-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danaher P, Warren S, Dennis L, D’Amico L, White A, Disis ML, Geller MA, Odunsi K, Beechem J, and Fling SP (2017). Gene expression markers of Tumor Infiltrating Leukocytes. J. Immunother. Cancer 5, 18. 10.1186/s40425-017-0215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Preuss C, Pandey R, Piazza E, Fine A, Uyar A, Perumal T, Garceau D, Kotredes KP, Williams H, Mangravite LM, et al. (2020). A novel systems biology approach to evaluate mouse models of late-onset Alzheimer’s disease. Mol. Neurodegener. 15, 67. 10.1186/s13024-020-00412-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan Y-W, Al-Ouran R, Mangleburg CG, Perumal TM, Lee TV, Allison K, Swarup V, Funk CC, Gaiteri C, Allen M, et al. (2020). Meta-Analysis of the Alzheimer’s Disease Human Brain Transcriptome and Functional Dissection in Mouse Models. Cell Rep. 32, 107908. 10.1016/j.celrep.2020.107908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim H-J, Hur SW, Park JB, Seo J, Shin JJ, Kim S-Y, Kim M-H, Han DH, Park J-W, Park JM, et al. (2019). Histone demethylase PHF2 activates CREB and promotes memory consolidation. EMBO Rep. 20, e45907. 10.15252/embr.201845907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward SM, Fernando SJ, Hou TY, and Duffield GE (2010). The Transcriptional Repressor ID2 Can Interact with the Canonical Clock Components CLOCK and BMAL1 and Mediate Inhibitory Effects on mPer1 Expression *. J. Biol. Chem. 285, 38987–39000. 10.1074/jbc.M110.175182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bozek K, Relógio A, Kielbasa SM, Heine M, Dame C, Kramer A, and Herzel H (2009). Regulation of clock-controlled genes in mammals. PloS One 4, e4882. 10.1371/journal.pone.0004882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen L, and Yang G (2014). PPARs Integrate the Mammalian Clock and Energy Metabolism. PPAR Res. 2014, 653017. 10.1155/2014/653017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Besing RC, Rogers CO, Paul JR, Hablitz LM, Johnson RL, McMahon LL, and Gamble KL (2017). GSK3 activity regulates rhythms in hippocampal clock gene expression and synaptic plasticity. Hippocampus 27, 890–898. 10.1002/hipo.22739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abdouh M, Chatoo W, Hajjar JE, David J, Ferreira J, and Bernier G (2012). Bmi1 Is Down-Regulated in the Aging Brain and Displays Antioxidant and Protective Activities in Neurons. PLOS ONE 7, e31870. 10.1371/journal.pone.0031870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim JP, Kim B-H, Bice PJ, Seo SW, Bennett DA, Saykin AJ, and Nho K (2021). BMI1 is associated with CSF amyloid-β and rates of cognitive decline in Alzheimer’s disease. Alzheimers Res. Ther. 13, 164. 10.1186/s13195-021-00906-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flamier A, El Hajjar J, Adjaye J, Fernandes KJ, Abdouh M, and Bernier G (2018). Modeling Late-Onset Sporadic Alzheimer’s Disease through BMI1 Deficiency. Cell Rep. 23, 2653–2666. 10.1016/j.celrep.2018.04.097. [DOI] [PubMed] [Google Scholar]
- 48.Khatib T, Chisholm DR, Whiting A, Platt B, and McCaffery P Decay in Retinoic Acid Signaling in Varied Models of Alzheimer’s Disease and In-Vitro Test of Novel Retinoic Acid Receptor Ligands (RAR-Ms) to Regulate Protective Genes. J. Alzheimers Dis. 73, 935–954. 10.3233/JAD-190931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li C, Wu X, Liu S, Zhao Y, Zhu J, and Liu K (2019). Roles of Neuropeptide Y in Neurodegenerative and Neuroimmune Diseases. Front. Neurosci. 13, 869. 10.3389/fnins.2019.00869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dallas ML, and Widera D (2021). TLR2 and TLR4-mediated inflammation in Alzheimer’s disease: self-defense or sabotage? Neural Regen. Res. 16, 1552–1553. 10.4103/1673-5374.303016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.LaFerla FM, Green KN, and Oddo S (2007). Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509. 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 52.Condello C, Schain A, and Grutzendler J (2011). Multicolor time-stamp reveals the dynamics and toxicity of amyloid deposition. Sci. Rep. 1, 19. 10.1038/srep00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cacquevel M, Aeschbach L, Houacine J, and Fraering PC (2012). Alzheimer’s Disease-Linked Mutations in Presenilin-1 Result in a Drastic Loss of Activity in Purified γ-Secretase Complexes. PLoS ONE 7, e35133. 10.1371/journal.pone.0035133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chakraborty R, Vepuri V, Mhatre SD, Paddock BE, Miller S, Michelson SJ, Delvadia R, Desai A, Vinokur M, Melicharek DJ, et al. (2011). Characterization of a Drosophila Alzheimer’s Disease Model: Pharmacological Rescue of Cognitive Defects. PLoS ONE 6, e20799. 10.1371/journal.pone.0020799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abramowski D, Wiederhold K-H, Furrer U, Jaton A-L, Neuenschwander A, Runser M-J, Danner S, Reichwald J, Ammaturo D, Staab D, et al. (2008). Dynamics of Aβ Turnover and Deposition in Different β-Amyloid Precursor Protein Transgenic Mouse Models Following γ-Secretase Inhibition. J. Pharmacol. Exp. Ther. 327, 411–424. 10.1124/jpet.108.140327. [DOI] [PubMed] [Google Scholar]
- 56.Xia D, Lianoglou S, Sandmann T, Calvert M, Suh JH, Thomsen E, Dugas J, Pizzo ME, DeVos SL, Earr TK, et al. (2022). Novel App knock-in mouse model shows key features of amyloid pathology and reveals profound metabolic dysregulation of microglia. Mol. Neurodegener. 17, 41. 10.1186/s13024-022-00547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yaffe K, Falvey CM, and Hoang T (2014). Connections between sleep and cognition in older adults. Lancet Neurol. 13, 1017–1028. 10.1016/S1474-4422(14)70172-3. [DOI] [PubMed] [Google Scholar]
- 58.Gao F, Liu T, Tuo M, and Chi S (2021). The role of orexin in Alzheimer disease: From sleep-wake disturbance to therapeutic target. Neurosci. Lett. 765, 136247. 10.1016/j.neulet.2021.136247. [DOI] [PubMed] [Google Scholar]
- 59.Li S-B, Damonte VM, Chen C, Wang GX, Kebschull JM, Yamaguchi H, Bian W-J, Purmann C, Pattni R, Urban AE, et al. (2022). Hyperexcitable arousal circuits drive sleep instability during aging. Science 375, eabh3021. 10.1126/science.abh3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma Z, Jiang W, and Zhang EE (2016). Orexin signaling regulates both the hippocampal clock and the circadian oscillation of Alzheimer’s disease-risk genes. Sci. Rep. 6, 36035. 10.1038/srep36035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Georgevsky D, Retsas S, Raoufi N, Shimoni O, and Golzan SM (2019). A longitudinal assessment of retinal function and structure in the APP/PS1 transgenic mouse model of Alzheimer’s disease. Transl. Neurodegener. 8, 30. 10.1186/s40035-019-0170-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lim JKH, Li Q-X, He Z, Vingrys AJ, Chinnery HR, Mullen J, Bui BV, and Nguyen CTO (2020). Retinal Functional and Structural Changes in the 5xFAD Mouse Model of Alzheimer’s Disease. Front. Neurosci. 14. 10.3389/fnins.2020.00862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.La Morgia C, Ross-Cisneros FN, Sadun AA, and Carelli V (2017). Retinal Ganglion Cells and Circadian Rhythms in Alzheimer’s Disease, Parkinson’s Disease, and Beyond. Front. Neurol. 8. 10.3389/fneur.2017.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dowling GA, Hubbard EM, Mastick J, Luxenberg JS, Burr RL, and Van Someren EJW (2005). Effect of morning bright light treatment for rest–activity disruption in institutionalized patients with severe Alzheimer’s disease. Int. Psychogeriatr. IPA 17, 221–236. 10.1017/S1041610205001584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mistlberger RE, and Antle MC (2011). Entrainment of circadian clocks in mammals by arousal and food. Essays Biochem. 49, 119–136. 10.1042/bse0490119. [DOI] [PubMed] [Google Scholar]
- 66.Cho YY, Kwon O-H, and Chung S (2020). Preferred Endocytosis of Amyloid Precursor Protein from Cholesterol-Enriched Lipid Raft Microdomains. Molecules 25, 5490. 10.3390/molecules25235490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, and Mattson MP (2004). Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 101, 2070–2075. 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wójtowicz S, Strosznajder AK, Jeżyna M, and Strosznajder JB (2020). The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 45, 972–988. 10.1007/s11064-020-02993-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Acosta-Rodríguez V, Rijo-Ferreira F, Izumo M, Xu P, Wight-Carter M, Green CB, and Takahashi JS (2022). Circadian alignment of early onset caloric restriction promotes longevity in male C57BL/6J mice. Science 376, 1192–1202. 10.1126/science.abk0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Manoogian ENC, Chow LS, Taub PR, Laferrère B, and Panda S (2022). Time-restricted Eating for the Prevention and Management of Metabolic Diseases. Endocr. Rev. 43, 405–436. 10.1210/endrev/bnab027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, Dubeau L, Yap LP, Park R, Vinciguerra M, et al. (2015). A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 22, 86–99. 10.1016/j.cmet.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gregosa A, Vinuesa Á, Todero MF, Pomilio C, Rossi SP, Bentivegna M, Presa J, Wenker S, Saravia F, and Beauquis J (2019). Periodic dietary restriction ameliorates amyloid pathology and cognitive impairment in PDAPP-J20 mice: Potential implication of glial autophagy. Neurobiol. Dis, 104542. 10.1016/j.nbd.2019.104542. [DOI] [PubMed] [Google Scholar]
- 73.Van der Auwera I, Wera S, Van Leuven F, and Henderson ST (2005). A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2, 28. 10.1186/1743-7075-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morris G, Maes M, Berk M, Carvalho AF, and Puri BK (2020). Nutritional ketosis as an intervention to relieve astrogliosis: Possible therapeutic applications in the treatment of neurodegenerative and neuroprogressive disorders. Eur. Psychiatry 63, e8. 10.1192/j.eurpsy.2019.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doecke JD, Pérez-Grijalba V, Fandos N, Fowler C, Villemagne VL, Masters CL, Pesini P, Sarasa M, and Group, for the A.R. (2020). Total Aβ42/Aβ40 ratio in plasma predicts amyloid-PET status, independent of clinical AD diagnosis. Neurology 94, e1580–e1591. 10.1212/WNL.0000000000009240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fandos N, Pérez-Grijalba V, Pesini P, Olmos S, Bossa M, Villemagne VL, Doecke J, Fowler C, Masters CL, and Sarasa M (2017). Plasma amyloid β 42/40 ratios as biomarkers for amyloid β cerebral deposition in cognitively normal individuals. Alzheimers Dement. Diagn. Assess. Dis. Monit. 8, 179–187. 10.1016/j.dadm.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Janelidze S, Stomrud E, Palmqvist S, Zetterberg H, van Westen D, Jeromin A, Song L, Hanlon D, Tan Hehir CA, Baker D, et al. (2016). Plasma β-amyloid in Alzheimer’s disease and vascular disease. Sci. Rep. 6, 26801. 10.1038/srep26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loh DH, Kudo T, Truong D, Wu Y, and Colwell CS (2013). The Q175 mouse model of Huntington’s disease shows gene dosage- and age-related decline in circadian rhythms of activity and sleep. PLoS ONE 8. 10.1371/journal.pone.0069993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang H-B, Whittaker DS, Truong D, Mulji AK, Ghiani CA, Loh DH, and Colwell CS (2017). Blue light therapy improves circadian dysfunction as well as motor symptoms in two mouse models of Huntington’s disease. Neurobiol. Sleep Circadian Rhythms 2, 39–52. 10.1016/j.nbscr.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fisher SP, Godinho SIH, Pothecary CA, Hankins MW, Foster RG, and Peirson SN (2012). Rapid assessment of sleep/wake behaviour in mice. J. Biol. Rhythms 27, 48–58. 10.1177/0748730411431550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pack AI, Galante RJ, Maislin G, Cater J, Metaxas D, Lu S, Zhang L, Smith RV, Kay T, Lian J, et al. (2007). Novel method for high-throughput phenotyping of sleep in mice. Physiol. Genomics 28, 232–238. 10.1152/physiolgenomics.00139.2006. [DOI] [PubMed] [Google Scholar]
- 82.Redlin U, Hattar S, and Mrosovsky N (2005). The circadian Clock mutant mouse: impaired masking response to light. J. Comp. Physiol. A 191, 51–59. 10.1007/s00359-004-0570-z. [DOI] [PubMed] [Google Scholar]
- 83.Lueptow LM (2017). Novel Object Recognition Test for the Investigation of Learning and Memory in Mice. J. Vis. Exp. JoVE, 55718. 10.3791/55718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wenk GL (2001). Assessment of Spatial Memory. In Current Protocols in Toxicology (John Wiley & Sons, Inc.). [DOI] [PubMed] [Google Scholar]
- 85.Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, and Hyman BT (2002). Imaging Aβ Plaques in Living Transgenic Mice with Multiphoton Microscopy and Methoxy-X04, a Systemically Administered Congo Red Derivative. J. Neuropathol. Exp. Neurol. 61, 797–805. 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- 86.Casali BT, and Landreth GE (2016). Aβ Extraction from Murine Brain Homogenates. Bio-Protoc. 6, e1787. 10.21769/BioProtoc.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Unprocessed data underlying the display items in the manuscript, related to Figures 1–6 and Figure S1–5
Table S1. DEGs between APP23 TG and NTG mice, Related to Figure 1
Table S2A. Genes showing time-of-day transcription changes in APP23 TG and NTG mice, Related to Figure 1.
Table S2B. Analysis of rhythmicity in selected Core Clock and AD-associated genes, Related to Figure 1 and Figure S4
Table S3. Nanostring DEGs in APP23 TG under TRF vs ALF treatment, Related to Figure 3
Table S4. DEGs in APP23 TG under TRF vs ALF treatment, Related to Figure 3
Table S5A. Analysis of rhythmic expression changes in APP23 TG mice under TRF, Related to Figure 3.
Table S5B. Analysis of genes modulated by TRF in APP23 TG mice at ZT12 and ZT18, Related to Figure 3
Table S6. Genes rescued by TRF treatment in APP23 mice, Related to Figure 4
Data Availability Statement
RNA-Seq data have been deposited at the Gene Expression Omnibus (GEO) and are publicly accessible as of the date of publication. Accession numbers are listed in the key resources table.
All the values used to create graphs in the paper can be found in the Data S1 file in a single Excel spreadsheet.
The raw uncropped western blots can be found in the Data S1 file in a single PDF document.
Additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
This paper does not report original code.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Iba1 Rabbit antibody | FUJIFILM Wako Shibayagi | Cat# 019-19741; RRID:AB_839504 |
| Anti-NeuN Mouse antibody | Millipore Corp. | Cat# MAB377; RRID:AB_2298772 |
| Amyloid β (N) (82E1) Anti-Human Mouse IgG MoAb | Immuno-biological Laboratories | Cat# 10323; RRID:AB_10707424 |
| Anti-GFAP Rabbit antibody | Thermo Fisher Scientific Inc. | Cat# 18-0063; RRID:AB_138737 |
| Goat Anti-Rabbit IgG Antibody (H+L), Biotinylated | Vector Laboratories | Cat# BA-1000; RRID:AB_2313606 |
| Horse Anti-Mouse IgG Antibody (H+L), Biotinylated | Vector Laboratories | Cat# BA-2000; RRID:AB_2313581 |
| Methoxy-X04 | Tocris Bioscience | Cat. No. 4920 |
| Chicken anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 594 | Thermo Fisher Scientific Inc. | Cat# A-21201; RRID:AB_2535787 |
| Phospho-Tau (Ser202; Thr205) Monoclonal Antibody (AT8) | Thermo Fisher Scientific Inc. | Cat# MN1020; RRID:AB_223647 |
| Chicken anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 | Thermo Fisher Scientific Inc. | Cat# A-21200; RRID:AB_2535786 |
| Anti-Amyloid Precursor Protein; C-Terminal antibody produced in rabbit | MilliporeSigma | RRID:AB_258409 |
| Bmi1 (D42B3) Rabbit mAb | Cell Signaling Technology | Cat# 5856; RRID:AB_10838137 |
| Goat Anti-Rabbit IgG (H L)-HRP Conjugate | Bio-Rad | RRID:AB_11125143 |
| Horse Anti-mouse IgG, HRP-linked Antibody | Cell Signaling Technology | Cat# 7076; RRID:AB_330924 |
| Anti-β-Actin Antibody (C4) | Santa Cruz Biotechnology | Cat# sc-47778; RRID:AB_626632 |
| Bacterial and virus strains | ||
| Biological samples | ||
| Chemicals; peptides; and recombinant proteins | ||
| Critical commercial assays | ||
| Applied Biosystems™ High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific Inc. | Cat. No. 43-688-14 |
| RNeasy Lipid Tissue Mini kit | QIAGEN | Cat. No. 74804 |
| DAB Substrate Kit; Peroxidase (HRP); with Nickel; (3;3’-diaminobenzidine) | Vector Laboratories; Inc. | Cat. No. SK-4100 |
| VECTASTAIN Elite ABC HRP Kit (Peroxidase; Standard) Kit | Vector Laboratories; Inc. | Cat. No. PK-6100 |
| V-PLEX Aβ Peptide Panel 1 (6E10) Kit | https://www.mesoscale.com | Cat. No. K15200E |
| Deposited data | ||
| Raw and analyzed RNA-Seq data | This Paper | GEO: GSE236731 |
| Data S1. Uncropped western blots and data underlying graphs | This Paper | N/A |
| Experimental models: Cell lines | ||
| Experimental models: Organisms/strains | ||
| APP23 mice; B6.Cg-Tg(Thy1-APP)3Somm/J | Jackson Laboratory | RRID:IMSR_JAX:030504 |
| APP-SAA KI mice; B6.Cg-Apptm1.1Dnli/J | Jackson Laboratory | RRID:IMSR_JAX:034711 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and algorithms | ||
| ImageJ (v1.53t) | NIH | RRID:SCR_023336 |
| Clocklab Analysis 6 (v6.1.12) | ActiMetrics | RRID:SCR_014309 |
| VitalView | Starr Life Sciences Corp. | RRID:SCR_014497 |
| ANY-maze (v4.99) | Stoelting Co. | RRID:SCR_014289 |
| El Temps program (v.313) | A. Diez-Nogura; Barcelona; Spain | http://www.el-temps.com/principal.html |
| Cran-package fpc (v2.2-10) | https://cran.r-project.org/ | https://cran.r-project.org/web/packages/fpc/index.html |
| DESeq2 | https://bioconductor.org/ | RRID:SCR_015687 |
| HISAT2 | http://ccb.jhu.edu/software/hisat2/index.shtml | RRID:SCR_015530 |
| JTK_CYCLE | https://openwetware.org/wiki/HughesLab:JTK_Cycle | RRID:SCR_017962 |
| Ingenuity Pathway Analysis | QIAGEN | RRID:SCR_008653 |
| Metascape | Metascape.org | RRID:SCR_016620 |
| Enrichr | https://maayanlab.cloud/Enrichr/ | RRID:SCR_001575 |
| DisGeNET | https://www.disgenet.org/ | RRID:SCR_006178 |
| Rosalind (v3.35.10.0) | https://www.rosalind.bio/ | RRID:SCR_006233 |
| geNORM | http://www.bioconductor.org | RRID:SCR_006763 |
| NormqPCR | http://www.bioconductor.org | RRID:SCR_003388 |
| Other | ||
| nCounter Analysis System | NanoString Technologies; Inc. | RRID:SCR_021712 |
| RNAlater Stabilization Solution | Thermo Fisher Scientific Inc. | Cat. No. AM7021 |
| AMP-AD Knowledge Portal | https://adknowledgeportal.synapse.org/ | RRID:SCR_016316 |
| NovaSeq 6000 Sequencing System | Illumina; Inc. | RRID:SCR_016387 |
| NanoZoomer S60 Digital slide scanner | https://www.hamamatsu.com/ | RRID:SCR_022537 |
| ZEN Digital Imaging for Light Microscopy | https://www.zeiss.com/ | RRID:SCR_013672 |
| OlyVIA (V3.4.1) | https://www.olympus-lifescience.com | RRID:SCR_016167 |
| DISCOVERY WORKBENCH | https://www.mesoscale.com | RRID:SCR_019192 |
| Prism 9 | https://www.graphpad.com/ | RRID:SCR_002798 |
| MESO QuickPlex SQ 120 | https://www.mesoscale.com | RRID:SCR_020304 |
| Precision Xtra Blood Glucose and Ketone Monitoring System | Abbott Laboratories; https://abbottstore.com | Cat. No. 9881465 |
| Agilent 4200 TapeStation System | Agilent Technologies; Inc. | RRID:SCR_019398 |
| TaqMan custom probes | Thermo Fisher Scientific Inc. | https://www.thermofisher.com/us/en/home/life-science/oligonucleotides-primers-probes-genes/applied-biosystems-custom-primers-probes.html |
| TaqMan™ Fast Advanced Master Mix | Thermo Fisher Scientific Inc. | Cat. No. 44-449-64 |
| LSM 800 confocal microscope | https://www.zeiss.com/ | RRID:SCR_015963 |
| SLIDEVIEW VS200 Slide Scanner | Olympus Life Science | https://www.olympus-lifescience.com/en/solutions-based-systems/vs200/ |
| iBright™ CL1500 Imaging System | Thermo Fisher Scientific Inc | Cat. No. A44240CFR |