

Abstract
Postmating reproductive isolation can help maintain species boundaries when premating barriers to reproduction are incomplete. The strength and identity of postmating reproductive barriers are highly variable among diverging species, leading to questions about their genetic basis and evolutionary drivers. These questions have been tackled in model systems but are less often addressed with broader phylogenetic resolution. In this study we analyze patterns of genetic divergence alongside direct measures of postmating reproductive barriers in an overlooked group of sympatric species within the model monkeyflower genus, Mimulus. Within this Mimulus brevipes species group, we find substantial divergence among species, including a cryptic genetic lineage. However, rampant gene discordance and ancient signals of introgression suggest a complex history of divergence. In addition, we find multiple strong postmating barriers, including postmating prezygotic isolation, hybrid seed inviability, and hybrid male sterility. M. brevipes and M. fremontii have substantial but incomplete postmating isolation. For all other tested species pairs, we find essentially complete postmating isolation. Hybrid seed inviability appears linked to differences in seed size, providing a window into possible developmental mechanisms underlying this reproductive barrier. While geographic proximity and incomplete mating isolation may have allowed gene flow within this group in the distant past, strong postmating reproductive barriers today have likely played a key role in preventing ongoing introgression. By producing foundational information about reproductive isolation and genomic divergence in this understudied group, we add new diversity and phylogenetic resolution to our understanding of the mechanisms of plant speciation.
Keywords: postmating reproductive isolation, introgression, Mimulus, hybrid seed inviability, speciation
Graphical Abstract
INTRODUCTION
Evolutionary biologists have long been interested in understanding what patterns and processes lead to reproductive isolation among species (Coyne & Orr, 1997; Darwin, 1859; V. Grant, 1981). Reproductive isolation is often broken down into components that act at sequential stages of the life cycle – premating barriers such as ecogeographic isolation and pollinator-mediated isolation that prevent mating, postmating prezygotic barriers that reduce fertilization success after mating has occurred, and postmating postzygotic barriers that reduce the fitness of hybrid offspring relative to pure species (Ramsey et al., 2003; Sobel & Chen, 2014). In plants, premating barriers are typically thought to evolve more quickly than postmating barriers and to have a greater contribution to overall isolation (Christie et al., 2022). However, this general pattern masks a great deal of heterogeneity across species pairs: many systems are completely isolated by premating barriers, while others rely exclusively on strong postmating isolation; many have a mix of both. Postmating barriers are often the result of intrinsic genetic incompatibilities, making them potentially more stable over the long term than ecologically mediated premating barriers (Coughlan & Matute, 2020). Postmating barriers may also be a source of cryptic variation in groups without obvious morphological or ecological differences.
Species complexes, consisting of many groups at various stages of divergence and gene flow, have been useful tools for speciation research. By placing reproductive isolation and divergence in a phylogenomic context, species complexes allow us to make informed comparisons across groups, distinguishing patterns and trends while uncovering crucial differences. For example, the degree of reproductive isolation between two taxa tends to increase with genetic distance (Christie & Strauss, 2018; Coyne & Orr, 1989; Malone & Fontenot, 2008; Scopece et al., 2007), but careful work in irises and other species complexes has demonstrated substantial heterogeneity in this relationship (Moyle et al., 2004; Osmolovsky et al., 2022). In addition, the emergence speed of different categories of isolation (Christie & Strauss, 2018) or of specific reproductive barriers (e.g., sterility vs. inviability) (Coyne & Orr, 1989; Le Gac et al., 2007; Malone & Fontenot, 2008; Presgraves, 2002) may vary considerably.
The reasons behind these differences are a subject of ongoing investigation, but may include differences in genetic architecture (Guerrero et al., 2017; Moyle & Payseur, 2009) or the forms of selection acting on each barrier (Baack et al., 2015). For example, conflicting selection pressures between maternal and paternal genomes in the mammalian placenta or the flowering plant endosperm are thought to give rise to reproductive barriers as these conflicts are resolved differently in independent lineages (Crespi & Nosil, 2013; Haig & Westoby, 1991; Lafon-Placette & Köhler, 2016); the relative strength of this parental conflict could influence the rate of barrier evolution (Coughlan et al., 2020; Raunsgard et al., 2018). Different forms of isolation may also be genetically linked in colocalized QTL or chromosomal rearrangements, causing them to evolve non-independently (Charron et al., 2014; Ferris et al., 2017; Noor et al., 2001). The importance of this genomic colocalization in speciation, and the role of chance vs. selection in its appearance, are subjects of ongoing debate (Cruickshank & Hahn, 2014; Duranton et al., 2018; Fuller et al., 2018; Kirkpatrick & Barrett, 2015; Renaut et al., 2013).
An excellent system for building a phylogenetically informed understanding of reproductive isolation is the western North American radiation of monkeyflowers in the genus Mimulus (Lamiales: Phrymaceae). Multiple species complexes within Mimulus have already been studied extensively in a speciation context, including the Mimulus guttatus (e.g., Brandvain et al., 2014; Ferris et al., 2014; Lowry & Willis, 2010), M. aurantiacus (e.g., Stankowski et al., 2019; Streisfeld & Kohn, 2005), and M. lewisii (e.g., Nelson et al., 2021; Ramsey et al., 2003) complexes. A number of pre- and postmating reproductive barriers have been mapped to single genes or QTL (e.g., Fishman et al., 2014; Fishman & Willis, 2006; Streisfeld & Rausher, 2009; Sweigart et al., 2006; Yuan et al., 2013; Zuellig & Sweigart, 2018). Recently, hybrid seed inviability in particular has been identified as a strong barrier in multiple independent species pairs of the Mimulus guttatus species complex, with parental conflict repeatedly implicated as a common mechanism (Coughlan et al., 2020; Oneal et al., 2016; Sandstedt et al., 2020; Sandstedt & Sweigart, 2022). An expanding set of genetic and developmental resources across the genus allow for a holistic approach to understanding reproductive isolation from genic to phylogenetic scales (Yuan, 2019).
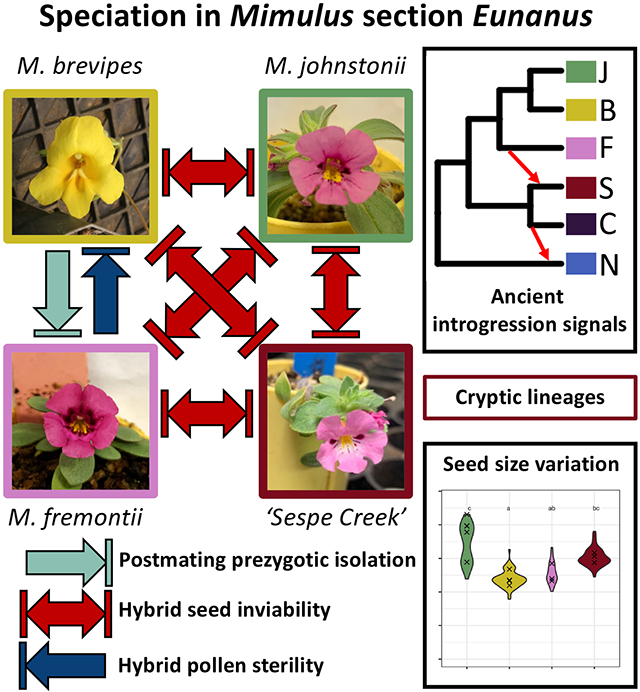
However, some Mimulus subgroups have received less attention, in part because they are less experimentally tractable or more difficult to access. Studies in these other groups would provide an important complement to existing knowledge, facilitating comparative analysis at the genus level while highlighting overlooked diversity in the genetic and ecological mechanisms of speciation. One such neglected group is Mimulus section Eunanus [synonym Diplacus section Eunanus, see (Barker et al., 2012; Lowry et al., 2019) for a discussion of nomenclatural issues]. Section Eunanus is a clade of ~23 species (Grant, 1924; Nesom, 2013) which prior to this publication has had no genome-scale sequencing and few assessments of reproductive isolation. The group includes both widespread and narrowly endemic species occupying a range of elevations and habitats in western North America. Here, we focus primarily on three species from this group: Mimulus brevipes Benth., Mimulus fremontii (Benth.) Gray, and Mimulus johnstonii Grant (Baldwin et al., 2012; Grant, 1924) (Figure 1A). These three morphologically defined species make up a well-supported clade according to a phylogenetic study of Mimulus based on three genetic markers (Beardsley et al., 2004). Their ranges are overlapping; M. johnstonii is restricted to higher elevations in the Transverse Ranges of southern California, while M. fremontii and M. brevipes are more widespread throughout the coastal ranges from Monterey into Baja California (Figure 1B).
Figure 1. Species and populations sampled in this study.

(A) Representative floral images for sampled species grown in growth chambers. (B) Map of occurrence records and sampled populations from the GBIF database for five species within California, USA and Baja California, Mexico. Smaller icons indicate GBIF records (GBIF.org, 2022) for each species, which include iNaturalist research grade observations and herbarium collections. Inset shows focal region of southern California where species ranges overlap. Larger named icons indicate sampled populations used in this study. The range of M. nanus extends into Nevada, Oregon, and Washington, but only occurrences for California are shown; population N01 (M. nanus) was collected from Washington state and is not shown. Population S25, ‘Sespe Creek’, is treated as a separate lineage despite being initially collected as M. johnstonii.
Studies of reproductive isolation in these three species are limited. Species distribution modeling between Mimulus johnstonii and M. brevipes identified strong but incomplete ecogeographical isolation (RI=0.65 and 0.82 reciprocally) (Sobel, 2014), and they can be found within hundreds of feet of each other (pers. obs.). No distribution models have been made with M. fremontii, but M. brevipes and M. fremontii are found at similar elevations, flower at similar times, and can be found growing within inches of each other (pers. obs.). Major differences in flower color, size, and shape between M. brevipes and its relatives might suggest some degree of pollinator-mediated isolation, though little is known about the identity of pollinators or levels of outcrossing; M. fremontii and M. johnstonii have similar floral characteristics and seem unlikely to have substantial pollinator isolation. Postmating barriers have not been studied, with the exception of one study showing low germination rates of F1 hybrid seeds between M. brevipes and M. johnstonii, potentially suggesting hybrid seed inviability (Sobel, 2010).
In this study, we present the first genome-scale analysis of divergence relationships in Mimulus section Eunanus, incorporating our three focal species as well as two more distant species, M. constrictus and M. nanus. In addition, we use controlled crosses to examine multiple postmating reproductive barriers between the focal species. We find clear genetic divergence and multiple strong postmating reproductive barriers between all tested species, but signals of historical introgression persist. Our results add to a growing body of evidence that intrinsic postmating barriers, especially hybrid seed inviability, are a key component in the maintenance of plant species.
MATERIALS AND METHODS
Sample collections and growth conditions
Fruits were collected from wild plants of three focal species (M. brevipes, M. fremontii, and M. johnstonii) at nine locations in southern California in summer 2019 (Figure 1B, Table S1). A population of M. constrictus from California in 2019 and a population of M. nanus from Oregon in 2021 were collected as additional comparisons; these species are phylogenetically within section Eunanus but outside of the immediate focal group. To germinate, we treated seeds overnight with 1 mM gibberellic acid, then rinsed three times with distilled water and placed them individually in 2x2cm soil plugs. Soil was a 1:1:1 mix of sand, perlite, and peat with a thin layer of finely sifted soil mix on top. Germination trays were set in standing bottom water and kept moist with a squirt bottle as necessary. When germination rates were low, liquid smoke was added to standing water to simulate post-fire conditions and stimulate germination. After germinants developed one or two pairs of true leaves, we transplanted soil plugs into plastic conical pots filled with the same soil mix. The conical pots were maintained in standing bottom water in a growth chamber set to 23C days/18C nights, with low relative humidity and 16 hours of daylight. Germination and establishment was low overall and highly variable across species, populations, and trials, so germination and establishment rates were not used as a proxy for seed viability or hybrid fitness.
One population initially collected as M. johnstonii, S25 from Sespe Creek, showed morphological differences suggesting it may be a unique lineage, and is treated separately throughout this paper. Compared to M. johnstonii, Sespe Creek has inserted stigmas, a lighter purple corolla with a slight white wash at the mouth, and longer leaf trichomes.
Genomic sequencing
We collected leaf tissue from 33 individuals for genomic sequencing. All sequenced individuals were generated from outbred field-collected seeds and germinated in growth chambers. We flash-froze leaf tissue in liquid nitrogen and used a modified CTAB-based protocol to extract genomic DNA (Fishman, 2020). Briefly, tissue was ground to a powder and incubated in CTAB buffer with ß-mercaptoethanol. Supernatant was extracted with phenol-chloroform-isoamyl alcohol, then again with chloroform-isoamyl alcohol, followed by precipitation of carbohydrates with sodium chloride and polyethylene glycol. DNA was then precipitated with isopropanol, washed with ethanol, and resuspended in distilled water.
We used the Illumina Nextera XT DNA library prep kit (FC-131-1024), which uses a tagmentation enzyme to simultaneously fragment DNA and ligate adapters, to prepare genomic DNA for sequencing. The 33 individually barcoded samples were then sequenced with a read length of 2x150bp (paired end) in four batches at the Duke University Sequencing and Genomic Technologies Shared Resource with either an Illumina NextSeq 500 or an Illumina NovaSeq 6000 sequencer, in combined lanes with other barcoded projects (Table S2).
Sequence alignments and SNP calling
We combined our dataset of 33 samples with five previously published samples (Stankowski et al., 2019) from the Mimulus aurantiacus species complex as an outgroup for analysis. M. aurantiacus is outside of section Eunanus but is the closest group with previous genome-scale data, as well as the closest group for which a reference genome assembly is currently available. For all 38 samples, we trimmed raw sequencing reads using Trimmomatic v0.39 (Bolger et al., 2014) to remove adapters and low-quality ends. We mapped reads to the Mimulus aurantiacus reference genome (Stankowski et al., 2019) using bwa mem v0.7.17 (Li & Durbin, 2009), then sorted and indexed with samtools v1.10 (Danecek et al., 2021). We removed PCR duplicates using the MarkDuplicates tool in Picard v2.21.6 (Broad Institute, 2019), and removed reads with unmapped mate pairs or mapping quality <29 using samtools view (Danecek et al., 2021). Average read coverage per sample was 2.2-15.7 (mean 6.8) across 33 novel samples (details in Table S2).
We called SNPs in each sample using the GATK v4.1.6.0 haplotype caller, then used GATK GenotypeGVCFs to jointly call variant and invariant sites for the entire dataset in ‘all-sites’ mode (Van der Auwera & O’Connor, 2020). Only sites mapping to the ten major linkage groups of the reference genome, corresponding to ten nuclear chromosomes, were included. We split the resulting VCF into SNPs and invariant sites, and separately filtered each before recombining them into a joint dataset, following recommendations by Dmitri Kryvokhyza (Kryvokhyzha, 2022). We filtered both SNPs and invariant sites to remove sites with combined DP <83 (~1/3 of mean combined depth) or >1376 (two standard deviations above mean combined depth), QD<2, SOR>3, or MQ<40. We additionally filtered SNPs to keep only biallelic sites across the entire dataset, and to remove sites with QUAL<40, FS>60, MQRankSum<−12.5, ReadPosRankSum>12.5, or ReadPosRankSum<−12.5.
Starting with all sites that pass the above filters (14,814,282 biallelic SNPs and 96,871,941 invariant sites), we generated four datasets for analysis. For genome-wide phylogenetic and introgression analyses, we used a ‘complete’ dataset, which retained only sites where at least 31 of 38 (>80%) of samples had a called genotype, resulting in 7,728,322 biallelic SNPs and 32,658,171 invariant sites. When calculating genome-wide introgression metrics, uneven sampling of species or uneven coverage across individuals could produce a bias whereby variants in one taxon are called more frequently than in another. To reduce the effect of this bias, we created four independent iterations of ‘downsampled’ data with equalized sampling and coverage. For three iterations, we randomly chose two individuals from each species; for the fourth iteration, we chose the two most divergent individuals from each species (except for M. constrictus for which we only had one sample). We then randomly sampled 12 million raw read pairs from each chosen individual and used the same pipeline as above to generate SNPs and invariant sites. Again, we retained only sites where 11 of 13 samples (>80%) had called genotypes, resulting in between 3,253,505 and 3,778,609 SNPs.
For the calculation of diversity and divergence metrics, we made a ‘synonymous sites’ dataset. Starting with the ‘complete’ dataset, we used the M. aurantiacus gene annotation to select only sites in the 3rd codon position of coding sequences that were four-fold degenerate, i.e., any nucleotide change to that site in the reference background would not change the protein sequence, using a custom script by Tim Sackton (Sackton, 2014/2022). The ‘synonymous sites’ dataset included 967,470 SNPs and 1,654,798 invariant sites. We note that multiple mutation hits within a single codon are possible given the large proportion of variant sites across the dataset, which could change the synonymous nature of these sites, but we expect these events to be rare enough to not substantially affect our genome-wide results on average.
For gene-by-gene analyses, we used a ‘genic’ dataset that included all sites passing the initial filters that fell within each of the 22,421 genes in the M. aurantiacus genome annotation.
Phylogenetic tree building
To determine phylogenetic relationships among our samples, we built a maximum likelihood phylogenetic tree using the GTR+Gamma model in RAxML v8.2.12 (Stamatakis, 2014) with SNPs from the ‘complete’ dataset. SNPs were extracted into a concatenated fasta alignment using the ‘consensus’ function from bcftools v1.13 (Danecek et al., 2021) and invariant sites were excluded. Heterozygous sites were initially coded as IUPAC ambiguity codes, then randomly converted to a single allele with the ‘randbase’ function in seqtk v1.3 (Li, 2018). An ascertainment bias correction was included using the ‘Felsenstein’ method implemented in RAxML to account for the exclusion of invariant sites. We ran 4 separate iterations of RAxML with unique random seeds, then chose the iteration with the largest log-likelihood. We used the RAxML -b option to run 1000 rapid bootstrap trees. We also generated a neighbor-joining tree with 1000 bootstrap replicates from the same data, using the dist.ml, nj, and boostrap.phyDat functions in the R package phangorn (Schliep, 2011).
We generated gene trees to examine patterns of gene tree discordance on the phylogeny. We used SNPs from the ‘genic’ dataset in the M. aurantiacus genome annotation, following the same approach as above to generate a maximum-likelihood phylogenetic tree in RAxML for each gene. Genes with insufficient data to resolve a tree (any individual with no called sites, or any two individuals with identical sequence) were excluded, resulting in trees for 17,573 genes. We then input these trees into ASTRAL v5.6.1 (Rabiee et al., 2019) which uses a multispecies coalescent approach to calculate a consensus species tree and quartet support scores for each node.
Quantifying homozygosity, diversity, and divergence
Pairwise sequence differences were calculated from the ‘synonymous sites’ dataset using the ‘dxy’ and ‘pi’ functions in the program pixy v1.2.3 (Korunes & Samuk, 2021). For each comparison, sequence differences were calculated in 1Mb windows across the genome, then added together and divided by the total number of informative sites. We calculated pairwise diversity () for each pair of individuals within a species, pairwise divergence for each pair of individuals across species, and heterozygosity (, pairwise diversity within an individual) for each individual. Diversity, divergence, and heterozygosity values were averaged across individuals within each species, or across individual pairs within each species pair.
To estimate the divergence times between species, we used the following molecular clock formula: (divergence time in generations) = (synonymous divergence – synonymous ancestral diversity)/(2*mutation rate). We assumed ancestral diversity for any two species to be the average of within-species diversity for the two species, and took the mutation rate to be 1.5x10^−8 (Koch et al., 2000). These estimates are approximate, as they do not take into account the effect of changing effective population sizes, such as demographic bottlenecks, or the possibility that ancestral diversity was very different from current diversity.
Quantifying introgression
To explore historical introgression between species, we used three complementary approaches: ABBA-BABA tests and the related -statistics; gene tree discordance bias using TWISST; and the model-based TreeMix program.
We used Dsuite v0.4 (Malinsky et al., 2021) to calculate Patterson’s D, a measure of the relative frequency of ABBA and BABA sites in the genome, and the related f4 statistic, which uses allele frequencies to estimate the proportion of the genome resulting from introgression. We calculated D and f4 for all possible trios of species in our dataset, using the ‘complete’ dataset as well as four ‘downsampled’ datasets, with the M. aurantiacus complex as the outgroup. Z-scores and associated p-values were calculated for each trio using a block-jackknife approach with 100 blocks in Dsuite; to account for multiple tests, a Bonferroni correction was used to obtain a corrected p-value (pcorr). Because there are multiple trios available to estimate introgression for the same pair of species, we summarized results across trios using the Fbranch statistic in Dsuite for each species pair (Malinsky et al. 2018).
We used TWISST (github version d56cefb) (Martin & Van Belleghem, 2017) to obtain the proportion of gene trees supporting particular tree topologies. Gene trees produced for the ASTRAL analysis above, based on genes in the M. aurantiacus annotation, were used as input for TWISST, again excluding trees where any sample has 0 called sites or where any two samples have identical sequence. For each trio of species, we compared the two possible alternate (different from the consensus specie tree) tree topologies using a binomial test with expected ratio 1:1 to look for an excess of one topology over another. The M. aurantiacus complex was always used as the outgroup. This test is analogous to the SNP-based ABBA-BABA test, but uses gene trees rather than SNPs.
We used TreeMix (Pickrell & Pritchard, 2012) to create phylogenetic network models with or without migration edges as a further test of introgression. We used the ‘synonymous sites’ dataset, including only variant SNPs with no missing data, and grouped individuals by species. The dataset was pruned to select SNPs not in linkage disequilibrium using a custom script by Joana Meier (https://github.com/joanam/scripts/raw/master/ldPruning.sh). We ran models with zero to eight migration edges, choosing the best of 10 replicate runs for each model type, then used likelihood ratio tests to determine whether each additional edge significantly improved the model fit. The program OptM (Fitak, 2021) was used as an alternative measure of the best fitting TreeMix migration model.
Identifying candidate windows with historical introgression
To identify candidate genomic regions with signatures of historical introgression, we used Dsuite (Malinsky et al., 2021) to calculate the window-based df statistic (Pfeifer and Kapan, 2019) for windows of 100 informative SNPs, each overlapping the previous by 50 SNPs. Low diversity is a potential confounding factor when using D- and f-statistics, particularly for small genomic windows, so we used pixy as before to calculate ‘pi’ for each species in 10kb regions, using the ‘complete’ dataset of SNPs and invariant sites (the ‘synonymous sites’ dataset did not provide good coverage of some areas at 10kb resolution). We assigned a π value to each 100-SNP df window based on the 10kb region containing the midpoint of that 100-SNP window.
We calculated df across the genome for two focal trios with genome-wide support for introgression, using the M. aurantiacus complex as the outgroup. We then excluded any df window whose midpoint lies within a 10kb region with π=0 for any of the three focal species for that trio. To test the relationship between π and df, we binned all df windows by their associated π value for each species into 10 deciles of increasing π, and calculated mean df for each decile. Finally, to identify the most extreme outliers for df across the genome, we selected the 100 windows (out of 4732 or 4141, depending on the quartet) with the most extreme (positive or negative) values as potential introgression outliers, with positive values set to match the direction of the genome-wide Fbranch signal.
Measuring reproductive barriers
We assessed three different barriers to reproduction: postmating prezygotic isolation, F1 hybrid seed inviability, and F1 hybrid sterility. We conducted hand pollinations of greenhouse-grown plants within and among species by first removing anthers from the maternal flower to prevent self-pollination, then using forceps to place pollen from the paternal flower on the maternal stigma surface. Stigma lobes in these species close quickly in response to touch, so hand-pollination is not always successful. In a few cases, supplemental pollen from the same paternal plant was added the following day once the stigma had re-opened. We collected mature, browned fruits, categorized according to cross type. Cross type throughout this paper refers to the combination of maternal species and paternal species, with maternal species always listed first; the 16 cross types are JxJ, JxB, BxJ, JxF, FxJ, JxS, SxJ, BxB, BxF, FxB, BxS, SxB, FxF, FxS, SxF, SxS where J=M. johnstonii, B=M. brevipes, F=M. fremontii, S=Sespe creek population. For the cross type BxF, we also germinated and grew F1 hybrids from four independent crosses in the same conditions as parental plants (Table S3). These F1 hybrids were crossed to themselves and reciprocally with M. fremontii and M. brevipes as above (cross types HxB, BxH, HxH, HxF, FxH, where H=F1 hybrid). F1 hybrids between other species pairs were not grown because of low crossing success or low seed viability.
To measure postmating prezygotic isolation, we quantified crossing success (probability that a pollination produces at least one seed) and seed production (number of seeds per fruit) after hand-pollination; both viable and inviable seeds were included but unfertilized ovules were not. To test for an association between cross type and crossing success, we ran a penalized Firth regression with a binomial family function using the R package ‘brglm’. A Firth regression was chosen to account for complete separation (all successes or all failures) of some groups. To test the dominance relationships of this barrier, we compared the crossing success of M. brevipes x M. fremontii F1 hybrids to their parental species. We ran a generalized linear mixed-effects model (GLMM) with a binomial family and logit link function using the R package ‘lme4’ on cross types BxB, BxH, BxF, HxB, HxH, HxF, FxB, FxH, and FxF, where H indicates F1 hybrid offspring from a BxF cross. For this model, maternal population was included as a random effect, treating each independent F1 cross as its own population.
To assess seed viability in each cross, we used a combination of visual inspection and chemical treatment. All seeds were inspected under a dissecting scope and scored as inviable or viable. Viable seeds were plump and generally ovate, although they were allowed to be slightly misshapen as long as they appeared to be full. Inviable seeds included any seed that was severely shriveled, concave, empty, much smaller than the typical within-species seed, or with a shape very different from the typical ovate shape. To confirm that seeds visually scored as inviable were in fact inviable, we treated a subset of seeds with tetrazolium chloride, which stains viable seeds dark red. For this treatment, seeds were scarified using 1:5 bleach:distilled water with 0.83 uL/mL Triton-X for 15 minutes, washed twice with distilled water, and placed in 1% (w/v) tetrazolium chloride, then incubated in the dark at room temperature for ~48 hours. To test for an association between cross type and the relative counts of viable vs. inviable seeds, we ran a penalized Firth regression using the R package ‘brglm’ with a binomial family and logit link function. A Firth regression was chosen to account for complete separation (all viable or all inviable) of some groups.
Parental conflict is a potential driver of hybrid seed inviability, and is associated with asymmetries in hybrid seed traits (Haig & Westoby, 1991). To investigate seed size and shape in parental vs. hybrid seeds, we imaged a random subset of seeds from 49 representative fruits under a dissecting scope and used imageJ to manually measure seed length and width. To avoid conflating viability with seed measurements within a cross, we measured only viable seeds for majority-viable cross types, and only inviable seeds for majority-inviable cross types. We ran a linear mixed-effects model using the R package ‘lme4’ to test for differences in seed length between the four intraspecific cross types (JxJ, BxB, FxF, and SxS), using fruit as a random effect. We also ran six independent LMMs, one for each pair of species, to directly compare intra- and interspecific seeds. For example, we tested the M. johnstonii vs. M. brevipes pair by comparing cross types JxJ, JxB, BxJ, BxB. For each species-pair, we ran one LMM to test for an association between cross type and seed length, and a second to test for an association between cross type and seed length/width ratio.
To assess hybrid male sterility of M. brevipes-M. fremontii F1 plants compared with their parental species, we scored pollen number and pollen viability using an aniline blue stain. For each individual, all anthers from a single flower were collected in 50 μL 0.25% aniline blue in lactophenol solution. Blue stained pollen grains were scored as viable, while unstained clear pollen grains were counted as inviable. Pollen was counted in full 1mm2 (0.1 μL) squares of a hemocytometer until 9 squares or at least 100 total pollen grains were counted. For each individual, we estimated pollen viability by determining the proportion of viable pollen grains out of the total counted (for some individuals, pollen viability was measured from multiple flowers and the average was used). We used an LMM to test for an association between species ID and the total count of pollen grains scored per hemocytometer square, with population as a random effect. Similarly, we used a GLM with a binomial family and logit link function to test for an association between species ID (M. brevipes, M. fremontii, or F1 hybrid) and the relative counts of viable vs. inviable pollen grains, adding population as a random effect (each hybrid family was treated as its own population).
To assess M. brevipes-M. fremontii F1 female fertility, we hand-pollinated F1 hybrids with pollen from either parent and measured seed production. To determine whether F1 hybrids produce fewer seeds than within-species crosses, we used a linear model to test for an association between cross type and the number of seeds produced per fruit, comparing cross types BxB, BxH, HxF, and FxF and only counting fruits that produced at least one seed.
For every statistical model above, we ran Tukey post-hoc tests implemented in the package ‘multcomp’ to test for pairwise differences between each cross type or group.
We followed the methods of Sobel and Chen (2014) to calculate standardized measures of reproductive isolation for each of three reproductive barriers, as well as total postmating reproductive isolation, between each species. All measures used the equation
where H=heterospecific fitness and C=conspecific fitness. In this framework, RI ranges from −1 to 1 where −1 is complete heterosis, 0 is random assortment, and 1 is complete isolation. We calculated total measured reproductive isolation sequentially with the equation
where RIprevious is the total RI from all previous barriers and RInew is the next barrier. Barriers that could not be measured due to complete or near-complete previous RI were left blank.
RESULTS
Genome-wide variation in the M. brevipes group defines clear species but also reveals a complex history of introgression.
A maximum-likelihood phylogeny generated from whole-genome SNP data resolves each of the three focal species (M. brevipes, M. johnstonii, and M. fremontii) as monophyletic with 100% bootstrap support (Figure 2A). Within these species, each population is also recovered as monophyletic, with the exception of one sample from a M. brevipes population (Figure 2A: one individual from population B11 clusters with B19). M. johnstonii is supported as sister to M. brevipes, in contrast to (Beardsley et al., 2004) which found M. brevipes and M. fremontii as sister based on just three loci. Genome-wide estimates of pairwise nucleotide divergence at synonymous sites also support strong divergence among the three focal species, with pairwise divergence values (, 4.8-6.0%) consistently well outside the range of nucleotide diversity within species (, range 1.6-3.7%) (Figure 2B, Table S4). Heterozygosity is similar to pairwise diversity (, range 1.1-3.1%), supporting a primarily outcrossing strategy in these species (Figure 2B, Table S4). Speciation times were estimated at >1 million generations for all species pairs (Figure 2A).
Figure 2. Phylogenetic relationships and divergence between sampled species in Mimulus section Eunanus.

(A) Maximum likelihood phylogeny inferred using RAxML from the ‘complete’ dataset of genome-wide SNPs, rooted by the M. aurantiacus complex. All nodes except those marked with open circles have 100% bootstrap support. Species divergence times were estimated separately using synonymous divergence relative to synonymous diversity in Pixy. All sampled populations are monophyletic with strong support, with the exception of one sample from B11 that clusters with B19 (marked with an asterisk). (B) Nucleotide heterozygosity , pairwise diversity , and divergence at genome-wide synonymous sites, indicating substantially higher between-species divergence than within-species diversity. Bars represent species or comparison means, while points represent individual pairwise sample comparisons. Diversity includes both within- and between-population comparison where possible. Sespe creek is substantially diverged from its closest sampled relative, M. constrictus (SxC ds=6.0%). J = M. johnstonii, B = M. brevipes, F = M. fremontii, S = Sespe creek population, C = M. constrictus, N = M. nanus, AUR = M. aurantiacus complex.
The Sespe Creek population (S25) is clearly a distinct lineage well outside of the M. brevipes trio (Figure 2A). Although S25 is most closely related to M. constrictus, high genetic divergence between these lineages suggest that S25 is a distinct species (Figure 2A-B, Table S4). Other species in this clade are not sampled and could be closer relatives to Sespe Creek, but none are known to occur in that geographic area. M. nanus is recovered as an outgroup to the other Eunanus section species as expected, with the M. aurantiacus complex set as the ultimate outgroup (Figure 2B). Species-level relationships from the maximum-likelihood tree were corroborated by a neighbor-joining tree and ASTRAL consensus tree (Figure S1).
Despite clear divergence between these Mimulus species, we also discovered a complex history of introgression. Using ABBA-BABA tests (Tables S5-S6) and associated F statistics (Figures 3A and S2), TWISST (Figure S3), and TreeMix (Figure 3B), we detected two signals of gene flow that were consistent across all methods, as well as additional signals that were more ambiguous (summarized in Figure S4). First, we found a strong signal of directional introgression from the M. brevipes-M. fremontii-M. johnstonii clade into the Sespe Creek population (Table S5: D = 0.186-0.199; Figure 3A: Fbranch = 0.072-0.093; Figure S3: TWISST quartet scores 41.1/41.8/17.0, Figure 3B: TreeMix m=0.193). Second, we detected a signal of gene flow between M. nanus and the M. constrictus – Sespe Creek clade (Table S5: D = 0.091-0.131, Figure 3A: Fbranch = 0.032, Figure S3: TWISST quartet scores 73.5/15.2/11.3, Figure 3B: TreeMix m=0.296).
Figure 3. Signatures of historical introgression between species in Mimulus section Eunanus.

(A) Genome-wide Fbranch statistics, an estimate of the proportion of the genome affected by introgression based on multiple f4-ratio statistics, calculated from the ‘complete’ SNP dataset. Seven bold-outlined boxes indicate comparisons with significant gene discordance bias supporting introgression from TWISST gene tree summaries (pcorr<0.01). Full TWISST summaries are provided in Figure S3. (B) TreeMix network analysis results for four migration edges. Migration weights are provided next to each edge. The four-edge model was a significant improvement over the three-edge or smaller models (likelihood ratio test p=0.048, llik=197.423); a five-edge model did not significantly improve the model. The optimizer program OptM selected a single-edge model as the model that maximized the change in log-likelihood; this model had an edge of weight 0.249 from M. constrictus into M. nanus.
Introgression from Sespe Creek back into M. fremontii was supported by TWISST (Figure S3: quartet scores 70.3/15.7/14.0) and by ABBA-BABA tests for the ‘complete’ dataset (Table S5: D = 0.080; Figure 3A: Fbranch = 0.016) but only some downsampled iterations of -statistics (Table S6: median D = 0.044, Figure S2: median Fbranch = 0.007) and not by TreeMix (Figure 3B). Introgression from Sespe Creek into M. johnstonii was supported by ABBA-BABA tests for the ‘complete’ dataset (Table S5: D = 0.077; Figure 3A: Fbranch = 0.023) and by TreeMix (Figure 3B: m=0.193), but not by TWISST. Results from TWISST were in the opposite direction of D- and f-statistics for the trio (M. fremontii, M. brevipes, M. johnstonii) (Figure 3A), while downsampled datasets produced a wide range of Fbranch values from 0 to 0.033 (Figure S2), indicating that this signal was unstable depending on which individuals were included in the dataset. We note that Fbranch and TWISST results cannot explicitly test for the directionality of introgression, though they may show asymmetry depending on the direction and chosen taxa; we use our TreeMix results to infer the directionality of each introgression case when possible.
For the two strongest cases of species-level introgression, we chose the trio with the strongest signal and scanned the genome for signatures of introgression using the statistic df. For both comparisons, df windows were asymmetric, with both mean and median df greater than 0 and more positive outliers (signals of introgression in the same direction as the genome-wide Fbranch pattern) than negative outliers of the same magnitude (Figure 4). Positive outliers were widely distributed throughout the genome, present on all ten linkage groups for both trios and with no large blocks of uninterrupted introgression, indicating that gene flow in this group is ancient rather than ongoing in these comparisons (Figure 4). Higher df values were generally associated with lower-diversity regions: top outliers were found in lower-π regions than the genome-wide average, and df decreased across deciles of increasing π (Table S7). However, df remained greater than 0 across all deciles of π for all species and for both quartets, suggesting that elevated df is not driven only by low diversity (Table S7).
Figure 4. Introgression outlier signals are distributed throughout the genome.

(A) Position along ten linkage groups of extreme df values in 100-SNP windows for the quartet (M. constrictus, Sespe Creek; M. fremontii, M. aurantiacus group). The top 100 windows with the largest absolute value of df are highlighted: positive outliers (red) indicate allele sharing consistent with introgression between Sespe Creek and M. fremontii, while negative outliers (black) indicate excess allele sharing between M. constrictus and M. fremontii. The blue line shows a rolling mean of 51 windows, indicating that the pattern of elevated df is distributed across the genome rather than focused in a few blocks. (B) Position along ten linkage groups of extreme df values in 100-SNP windows for the quartet (M. brevipes, M. constrictus; M. nanus, M. aurantiacus group). The top 100 windows with the largest absolute value of df are highlighted: positive outliers (red) indicate allele sharing consistent with introgression between M. constrictus and M. nanus, while negative outliers (black) indicate excess allele sharing between M. brevipes and M. nanus. The blue line shows a rolling mean of 21 windows. Dotted grey lines indicate the most extreme negative outlier and its equivalent positive value, and dashed black lines indicate df=0 (no excess allele sharing). Total tested windows after removal of values in regions where π=0 for any species in the tested trio: (A) 4732, (B) 4141.
Multiple strong postmating reproductive barriers between species in the M. brevipes group.
Most crosses between four focal species (M. johnstonii, M. brevipes, M. fremontii, and the Sespe Creek population) showed little to no reduction in crossing success (probability of producing a seed, or seeds produced per fruit) compared to within-species crosses (Figures 5 and S5, Table S8). In one case (FxB), there is evidence of strong unidirectional postmating prezygotic isolation: only one in 58 crosses between maternal M. fremontii and paternal M. brevipes produced any seeds, a significantly lower proportion than the within-species or reciprocal cross types (Figure 4, Table S8). This pattern appears to be additive on both the maternal and paternal sides in M. brevipes x M. fremontii F1 hybrids, with intermediate crossing success in both HxB and FxH crosses (Figure S6).
Figure 5. Postmating prezygotic isolation between at least one species pair.

Crossing success (probability of producing at least one seed from a cross) for intra- and interspecific cross types. Shown are least-square means and asymptotic confidence intervals from a Firth binomial model. Letters indicate significance in post-hoc Tukey tests; cross types sharing a letter are not significantly different. One interspecific cross type, FxB, was significantly reduced compared to both intraspecific parental crosses, indicating an asymmetric barrier to fruit set. One cross, BxJ, was significantly reduced compared to the maternal (BxB) parent but not the paternal (JxJ) parent. Two other crosses, FxS and SxF, had low fruit set possibly indicating a reproductive barrier, but were not significant in the model due to low sample sizes; e.g., 0 out of 4 SxF crosses resulted in seeds. J = M. johnstonii, B = M. brevipes, F = M. fremontii, S = Sespe creek population. Cross types are listed as (maternal x paternal).
A few other interspecific cross types also had low or zero seed production (e.g., reciprocal crosses of M. fremontii and Sespe Creek), which could indicate a postmating prezygotic barrier, but these crosses had low sample sizes and did not reach significance compared to within-species crosses (Figure 5). In addition, cross type BxJ had significantly lower fruit success than BxB, possibly indicating premating postzygotic isolation – however, JxJ crosses also had relatively low fruit success, making it difficult to rule out poor performance of M. johnstonii pollen overall. We note that these species are difficult to keep happy in growth chamber conditions, and some cases of fruit failure (such as low intraspecific cross success in M. johnstonii) may be a byproduct of less-than-perfect growth conditions rather than the particulars of a given cross type.
For crosses that did produce seeds, we found strong F1 hybrid seed inviability in nine interspecific crosses: 0% viability for five cross types and <2% viability for four others (Figure 6A-B, Table S8). The exception was species pair M. fremontii and M. brevipes, which produced viable seeds in both directions (Figure 6A-B, Table S8). We could not score seed viability for cross type SxF because the only potential seeds produced were not distinguishable from unfertilized ovules. Seed viability results from the tetrazolium staining assay were qualitatively similar to results from scoring seeds by eye (Table S9).
Figure 6. Strong hybrid seed inviability between multiple species pairs.

(A) Representative seed images from intra- and interspecific laboratory crosses between four species. All images are adjusted to the same scale. For the SxF cross, marked with an asterisk, the only ‘seeds’ produced were not distinguishable from unfertilized ovules and were not counted as seeds for crossing success or viability scoring, but it is possible they represent early seed abortion. (B) Proportion of viable seeds produced by intra- and interspecific laboratory crosses. Shown are least-square means and asymptotic confidence intervals from a Firth binomial model, with letters indicating significance in post-hoc Tukey tests. Intraspecific, BxF, and FxB crosses are mostly viable, but all other interspecific cross types have almost complete inviability. Cross types are listed as (maternal x paternal). (C) Seed lengths for intraspecific crosses. Violin plots show distribution of data for individual seeds, with x’s marking the means of individual fruits. Letters indicate significance from post-hoc Tukey tests of an LMM.
M. johnstonii intraspecific seeds were significantly larger than M. brevipes or M. fremontii seeds, with Sespe Creek seeds intermediate in size (Figure 6C, Table S10). Inviable seeds overall showed no evidence of overgrowth or undergrowth relative to parents, but instead tended to match the seed length of the maternal parent. However, F1 hybrids with Sespe Creek as maternal parent did seem to have slightly smaller seeds than either parent (this trend was only significant for SxF) (Figures 6A and S7A, Table S10). Crosses with M. johnstonii, the largest-seeded species, as the maternal parent produced inviable seeds that were collapsed and thin, as shown by a significant increase in hybrid seed length/width ratios (three cross types) compared to both parents and to the reciprocal cross type (Figures 6A and S7B, Table S10). In the reciprocal direction, hybrid seeds were typically shriveled, as were hybrid seeds in both directions of Sespe Creek crosses (Figures 6A and S7B).
Although in most cases, strong seed inviability prevented us from quantifying later-acting barriers, we were able to assess F1 hybrid sterility between M. brevipes and M. fremontii. Pollen number per flower was intermediate for hybrids compared to the two parental species (Figure 7A, Table S10). However, only ~20% of hybrid pollen grains were fertile, a sharp reduction from either parent, indicating strong F1 male sterility (Figure 7B, Table S11). We find no evidence of hybrid female sterility in M. brevipes x M. fremontii F1s; seed production from pollinated hybrids was intermediate compared to pure species (Figure 7C).
Figure 7. Strong hybrid male sterility in hybrids between M. brevipes and M. fremontii.

(A) Pollen number (counts per mm2 on a hemocytometer) and (B) pollen viability (proportion of pollen grains scored as viable in aniline blue stain). Letters indicate significance in post-hoc Tukey tests from a poisson (abundance) or binomial (viability) GLMM with population as a random variable. Each point represents the average across 1-3 flowers from a single individual. For each panel, groups sharing a letter are not significantly different. (C) Seed production from hybrid vs. parental fruits as a test of hybrid female sterility, including only fruits that produced at least one seed. Letters indicate significance in post-hoc Tukey tests from a linear model. B=M. brevipes, F=M. fremontii, H=M. brevipes x M. fremontii F1 hybrid. Cross types are listed as (maternal x paternal).
Total postmating reproductive isolation was near-complete (RI>=0.95) for all tested species pairs except for M. brevipes vs. M. fremontii, primarily driven by strong hybrid seed inviability (Table 1). The exception, M. brevipes vs. M. fremontii, had incomplete but still substantial total postmating RI from a combination of postmating prezygotic isolation and F1 male sterility (B -> F: RI=0.91; F -> B: RI=0.66).
Table 1.
Summary of Postmating Reproductive Isolation (RI).
| Pair a, b | Crossing success RI | Seed viability RI | F1 viability RI | Total measured RI | ||||
|---|---|---|---|---|---|---|---|---|
| a->b | b->a | a->b | b->a | a->b | b->a | a->b | b->a | |
| J, B | 0.30 | 0.10 | 0.99 | 1.00 | -- | -- | 0.99 | 1.00 |
| J, F | −0.13 | −0.05 | 0.97 | 1.00 | -- | -- | 0.97 | 1.00 |
| B, F | 0.91 | 0.13 | −0.03 | 0.09 | -- | 0.57 | 0.91 | 0.66 |
| J, S | 0.35 | −0.07 | 1.00 | 0.97 | -- | -- | 1.00 | 0.96 |
| B, S | 0.05 | 0.16 | 1.00 | 0.99 | -- | -- | 1.00 | 0.99 |
| F, S | 1.00 | 0.68 | -- | 1.00 | -- | -- | 1.00 | 1.00 |
a->b indicates reproductive isolation preventing gene flow from species a into species b, calculated by comparing the fitness of b x a crosses relative to b x b crosses, with b>a indicating the reciprocal direction using the fitness of a x b crosses relative to a x a crosses, following (Sobel & Chen, 2014). Values range from −1 (complete heterosis) to 0 (free mating) to 1 (complete reproductive isolation).
Bolded values include contributions from barriers with significant model results.
DISCUSSION
A major goal of speciation research is to understand similarities and differences in how reproductive isolation emerges as groups of species diverge. We find that Mimulus brevipes, Mimulus johnstonii, and Mimulus fremontii are clearly delineated species both genetically and by intrinsic reproductive barriers. We find strong hybrid seed inviability between multiple species pairs, as well as a postmating prezygotic barrier and severe hybrid male sterility between one species pair. While premating barriers have not yet been thoroughly quantified in this group, the combination of close geographic proximity (within centimeters in some cases) and similar floral morphology (excluding M. brevipes) makes it unlikely that premating barriers alone can eliminate hybridization. These strong postmating barriers are therefore likely to be important in preventing contemporary gene flow.
While many studies have shown that reproductive isolation increases with divergence on average, individual species pairs are often idiosyncratic (Coyne & Orr, 1989; Malone & Fontenot, 2008; Matute & Cooper, 2021; Moyle et al., 2004). Our results highlight this heterogeneity – while reproductive isolation and divergence are both strong overall, the species pair with the least postmating reproductive isolation in this group (M. brevipes and M. fremontii) is not the most recently diverged pair (M. brevipes and M. johnstonii). Paying greater attention to the exceptions may help us better understand the factors that drive reproductive isolation.
Gene discordance and introgression
We find evidence of extensive phylogenetic discordance across the genome, including signatures of historical introgression between multiple species in this group. Our strongest signals of introgression appear between groups with essentially complete postmating barriers and without stark differences in floral morphology. These signals are likely ancient, and may have predated the postmating barriers we see today. Without postmating reproductive barriers, even small amounts of introgression can lead to species collapse in sympatry, especially if premating barriers are weak or are disrupted by environmental change (Behm et al., 2010; Kleindorfer et al., 2014; Xiong & Mallet, 2022). Alternatively, species can persist despite ongoing introgression, depending on the circumstances (Kay et al., 2018; Kenney & Sweigart, 2016; Servedio & Hermisson, 2020). Given the lack of obvious prezygotic barriers between these species, it is possible that postmating reproductive barriers have played an important and ongoing role in reducing gene flow as divergence increased in section Eunanus, suggesting they are not just ‘after-the-fact’ byproducts with little influence on speciation trajectories. More work on prezygotic barriers in section Eunanus would help to confirm or refute this hypothesis.
Without further sampling of populations and species in this section, attributing these historical signals of introgression to precise events on the phylogeny is difficult. In particular, gene flow involving unsampled lineages can result in incorrectly attributed ‘ghost’ introgression (Beerli, 2004; Slatkin, 2005; Tricou et al., 2022), and complex demographic histories may produce false positive signals of hybridization. Taken as a whole, however, these signals demonstrate a high degree of complexity in the divergence process, consistent with reticulate evolutionary histories of other groups within Mimulus (e.g., Brandvain et al., 2014; Nelson et al., 2021; Stankowski et al., 2019), in plants more broadly (e.g., Curtu et al., 2009; Goulet et al., 2017; Hamlin et al., 2020; Kay et al., 2018; Scascitelli et al., 2010), and across the tree of life (e.g., D’Angiolo et al., 2020; Green et al., 2010; Kleindorfer et al., 2014; Mallet et al., 2007; Schumer et al., 2018).
Cryptic diversity in Mimulus section Eunanus
Genetic data allow us to uncover cryptic variation in the form of distinct genetic lineages that are not readily distinguished by morphological features in the field (Bickford et al., 2007; Struck et al., 2018). Our population from Sespe Creek does not appear to match any of the likely species from the area, and we find that it is a genetically distinct lineage with a history of introgression. In addition, it appears to be reproductively isolated from other members of the group by strong hybrid seed inviability. We do not yet know the exact nature of its origin, or how widely distributed this lineage is; more sampling from the area and genetic data from more species in the group will be necessary to fully resolve these questions. Studies of speciation in plants often focus on ecologically mediated premating reproductive isolation, but cryptic diversity may be better explained by postmating barriers (Coughlan & Matute, 2020). Mimulus section Eunanus is home to many more understudied, small, pink-flowered taxa; if our study is any indication, strong postmating reproductive isolation may play an outsized role in generating diversity in this group.
Parallel evolution of hybrid seed inviability in Mimulus
Hybrid seed inviability has been found repeatedly in species pairs from the Mimulus guttatus species complex (section Simiolus) (Coughlan et al., 2020; Oneal et al., 2016; Sandstedt & Sweigart, 2022). By demonstrating hybrid seed inviability in a distant section of Mimulus, we show that this pattern is widespread and important, both across Mimulus and likely in plants more broadly. In the M. guttatus complex and other systems, hybrid seed inviability has been tied to parental conflict in resource allocation, a process mediated by the endosperm (Coughlan et al., 2020; Haig & Westoby, 1991; Lafon-Placette et al., 2017; Oneal et al., 2016; Rebernig et al., 2015; Roth et al., 2019; Sandstedt & Sweigart, 2022). We do not find clear evidence of parental conflict in our system -- hybrid seed sizes tended to track maternal seed sizes, without the telltale overgrowth or undergrowth phenotypes. However, hybrid seed size is not always a good predictor of parental conflict (Sandstedt & Sweigart, 2022), so future work to characterize differences in seed development would be required to rule out a conflict hypothesis. Alternatively, differences in parental seed sizes between our species could result in Dobhzansky-Muller-like incompatibilities during hybrid seed development, without invoking parental conflict as a driver. Seed size is an important life history trait tied to ecological strategies; larger seeds tend to have more success establishing under a variety of hazardous conditions, from nutrient deprivation to low soil moisture to deep burial (Leishman et al., 2000). Larger, heavier seeds may also prevent long-range dispersal and keep offspring in maternal habitats for which they are locally adapted, as is the case in dune-adapted plants (Bowers, 1982; Schwarzbach et al., 2001). Both M. johnstonii and the Sespe Creek population are found on unstable scree slopes, which may pose a particular challenge to seed establishment or favor local dispersal to maintain local adaptation. This habitat preference is therefore a possible candidate for a selective force, which could have acted in parallel in M. johnstonii and Sespe Creek to drive seed size differences leading to hybrid inviability.
Postmating prezygotic isolation
Crossing failure (postmating prezygotic isolation) can be caused by a failure of pollen tube germination, pollen tube growth, or fertilization (Wheeler et al., 2001); it may be a passive incompatibility between pollen and pistil, or an active rejection mechanism to prevent maladaptive hybridization (Hogenboom et al., 1975; Roda & Hopkins, 2019; Rushworth et al., 2022). Most studies on the mechanisms of postmating prezygotic isolation come from systems with self-incompatibility, where they are thought to be related to self-incompatibility mechanisms, e.g. Solanum (Bernacchi & Tanksley, 1997; Tovar-Méndez et al., 2014), Nicotiana (Kuboyama et al., 1994), and Lilium (Ascher & Drewlow, 1975), though a mechanism unrelated to self-incompatibility has been described in Brassica (Fujii et al., 2019). Since Mimulus lacks self-incompatibility mechanisms, this group provides an opportunity to investigate how interspecific incompatibility may arise on its own. In other systems with differences in style length, germinating pollen tubes can under- or over-shoot the ovules (Gore et al., 1990; Williams & Rouse, 1988). Style length could be important in our system, since M. brevipes styles are substantially longer than M. fremontii styles, though our case is unusual in that pollen from the long-styled parent fails while pollen from the short-styled parent is successful. Pollen competition driving differential coevolution of the pollen and pistil is another possible source of incompatibilities (Brandvain & Haig, 2005; Skogsmyr & Lankinen, 2002), a scenario which has been documented in the M. guttatus complex (Aagaard et al., 2013; Fishman et al., 2008) and Arabidopsis (Takeuchi & Higashiyama, 2012).
Hybrid male sterility
Hybrid male sterility has been characterized multiple times in Mimulus sections Simiolus and Erythranthe, with a variety of genetic causes, including a nuclear DMI interaction (Sweigart et al., 2006), a cytoplasmic-nuclear interaction (Fishman & Willis, 2006), or underdominant chromosomal rearrangements (Stathos & Fishman, 2014), but the relative frequency of these mechanisms across plants is unknown. M. brevipes and M. fremontii are more genetically divergent than most pairs for which male sterility has been studied, in Mimulus or elsewhere. Since they still produce viable hybrids and sterility is not 100% complete, genetic mapping could be used in the future to determine the mechanisms underlying sterility in this system, and the degree of parallelism with other cases in Mimulus.
Future directions
As the first in-depth genomic and phenotypic investigation of speciation within Mimulus section Eunanus, this study fills an important phylogenetic gap within the speciation literature, helping to position the genus Mimulus as a leading model for broad phylogenetically informed comparisons of how species form and diverge. Our study system highlights both universal and idiosyncratic patterns in the speciation process; we add to a growing body of evidence that hybrid seed inviability and hybrid male sterility are important barriers to gene flow in Mimulus and across plant taxa; and we demonstrate the complexity of gene discordance and historical introgression among species despite substantial overall divergence. In addition, we lay the groundwork for fruitful avenues of future mechanistic study: seed size evolution and hybrid seed inviability; style length and postmating prezygotic isolation; and highly divergent hybrid male sterility are all worthy of future exploration in this group. Out study also provides the starting point for a more complete phylogenetic sampling of section Eunanus, which would enhance our understanding of cryptic divergence and the interplay between ecological, geographic, and genetic forces driving the diversification of species.
Supplementary Material
Acknowledgments
We thank James Sobel for seed collections, for advice on germination and growth conditions, and for helpful discussions. Hagar Soliman contributed a photo of M. constrictus and an extracted DNA sample from this species. Samuel Mantel, Makenzie Whitener, Natalie Gonzalez, and Emma Chandler provided comments that improved the manuscript. Kelly Dyer, Jill Anderson, Casey Bergman, Molly Schumer, John Willis, Jenn Coughlan, Elen Oneal, and Robert Franks also contributed to useful discussions. This work is supported by the National Science Foundation DEB1856180; the National Institutes of Health Predoctoral Training Grant 5T32GM007103, the Society for the Study of Evolution Lewontin Award, and a UGA Plant Center Doctoral Dissertation Improvement Grant.
Footnotes
Competing interests
The authors declare no competing interests.
Data accessibility
All newly generated whole genome sequence data are deposited at the NCBI Sequence Read Archive (accession PRJNA922914). The complete genotype call dataset, phylogenetic tree files, and raw crossing data, including seed counts, seed measurements, and pollen counts, are archived in the DRYAD digital data repository at https://doi.org/10.5061/dryad.7wm37pvzc. Code used in these analyses is archived at https://github.com/mfarnitano/Eunanus_popgen.
REFERENCES
- Aagaard JE, George RD, Fishman L, MacCoss MJ, & Swanson WJ (2013). Selection on Plant Male Function Genes Identifies Candidates for Reproductive Isolation of Yellow Monkeyflowers. PLoS Genetics, 9(12). 10.1371/journal.pgen.1003965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascher PD, & Drewlow LW (1975). The effect of prepollination injection with stigmatic exudate on interspecific pollen tube growth in Lilium longiflorum thunb. Styles. Plant Science Letters, 4(6), 401–405. 10.1016/0304-4211(75)90269-2 [DOI] [Google Scholar]
- Baack E, Melo MC, Rieseberg LH, & Ortiz-Barrientos D (2015). The origins of reproductive isolation in plants. New Phytologist, 207(4), 968–984. 10.1111/nph.13424 [DOI] [PubMed] [Google Scholar]
- Baldwin BG, Goldman D, Keil DJ, Patterson R, Rosatti TJ, & Wilken D (Eds.). (2012). The Jepson Manual: Vascular Plants of California, Thoroughly Revised and Expanded (2nd ed.). [Google Scholar]
- Barker W. R.(Bill) , Nesom GL, Beardsley PM, & Fraga NS (2012). A taxonomic conspectus of phrymaceae: A narrowed circumscription for. Phytoneuron, May, 1–60. [Google Scholar]
- Beardsley PM, Schoenig SE, Whittall JB, & Olmstead RG (2004). Patterns of evolution in western North American Mimulus (Phrymaceae). American Journal of Botany, 91(3), 474–489. 10.3732/ajb.91.3.474 [DOI] [PubMed] [Google Scholar]
- Beerli P (2004). Effect of unsampled populations on the estimation of population sizes and migration rates between sampled populations. Molecular Ecology, 13(4), 827–836. 10.1111/j.1365-294X.2004.02101.x [DOI] [PubMed] [Google Scholar]
- Behm JE, Ives AR, & Boughman JW (2010). Breakdown in postmating isolation and the collapse of a species pair through hybridization. The American Naturalist, 175(1), 11–26. 10.1086/648559 [DOI] [PubMed] [Google Scholar]
- Bernacchi D, & Tanksley SD (1997). An Interspecific Backcross of Lycopersicon esculentum × L. hirsutum: Linkage Analysis and a QTL Study of Sexual Compatibility Factors and Floral Traits. Genetics, 147(2), 861–877. 10.1093/genetics/147.2.861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, Ingram KK, & Das I (2007). Cryptic species as a window on diversity and conservation. Trends in Ecology & Evolution, 22(3), 148–155. 10.1016/j.tree.2006.11.004 [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, & Usadel B (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers JE (1982). The plant ecology of inland dunes in western North America. Journal of Arid Environments, 5(3), 199–220. 10.1016/S0140-1963(18)31444-7 [DOI] [Google Scholar]
- Brandvain Y, & Haig D (2005). Divergent Mating Systems and Parental Conflict as a Barrier to Hybridization in Flowering Plants. The American Naturalist, 166(3), 330–338. 10.1086/432036 [DOI] [PubMed] [Google Scholar]
- Brandvain Y, Kenney AM, Flagel L, Coop G, & Sweigart AL (2014). Speciation and Introgression between Mimulus nasutus and Mimulus guttatus. PLoS Genetics, 10(6), e1004410. 10.1371/journal.pgen.1004410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute. (2019). Picard Toolkit, GitHub repository [Computer software]. https://broadinstitute.github.io/picard/
- Charron G, Leducq JB, & Landry CR (2014). Chromosomal variation segregates within incipient species and correlates with reproductive isolation. Molecular Ecology, 23(17), 4362–4372. 10.1111/mec.12864 [DOI] [PubMed] [Google Scholar]
- Christie K, Fraser LS, & Lowry DB (2022). The strength of reproductive isolating barriers in seed plants: Insights from studies quantifying premating and postmating reproductive barriers over the past 15 years. Evolution, evo.14565. 10.1111/evo.14565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie K, & Strauss SY (2018). Along the speciation continuum: Quantifying intrinsic and extrinsic isolating barriers across five million years of evolutionary divergence in California jewelflowers. Evolution, 72(5), 1063–1079. 10.1111/evo.13477 [DOI] [PubMed] [Google Scholar]
- Coughlan JM, & Matute DR (2020). The importance of intrinsic postzygotic barriers throughout the speciation process: Intrinsic barriers throughout speciation. Philosophical Transactions of the Royal Society B: Biological Sciences, 375(1806). 10.1098/rstb.2019.0533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan JM, Wilson Brown M, & Willis JH (2020). Patterns of Hybrid Seed Inviability in the Mimulus guttatus sp. Complex Reveal a Potential Role of Parental Conflict in Reproductive Isolation. Current Biology, 30(1), 83–93.e5. 10.1016/j.cub.2019.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA, & Orr HA (1989). Patterns of Speciation in Drosophila. Evolution, 43(2), 362–381. [DOI] [PubMed] [Google Scholar]
- Coyne JA, & Orr HA (1997). Patterns of Speciation in Drosophila Revisited. Evolution, 51(1), 295. 10.2307/2410984 [DOI] [PubMed] [Google Scholar]
- Crespi B, & Nosil P (2013). Conflictual speciation: Species formation via genomic conflict. Trends in Ecology & Evolution, 28(1), 48–57. 10.1016/j.tree.2012.08.015 [DOI] [PubMed] [Google Scholar]
- Cruickshank TE, & Hahn MW (2014). Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Molecular Ecology, 23(13), 3133–3157. 10.1111/mec.12796 [DOI] [PubMed] [Google Scholar]
- Curtu AL, Gailing O, & Finkeldey R (2009). Patterns of contemporary hybridization inferred from paternity analysis in a four-oak-species forest. BMC Evolutionary Biology, 9(1), 284. 10.1186/1471-2148-9-284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, & Li H (2021). Twelve years of SAMtools and BCFtools. GigaScience, 10(2), giab008. 10.1093/gigascience/giab008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolo M, De Chiara M, Yue J-X, Irizar A, Stenberg S, Persson K, Llored A, Barré B, Schacherer J, Marangoni R, Gilson E, Warringer J, & Liti G (2020). A yeast living ancestor reveals the origin of genomic introgressions. Nature, 587(7834), Article 7834. 10.1038/s41586-020-2889-1 [DOI] [PubMed] [Google Scholar]
- Darwin C (1859). On the origin of species by means of natural selection. John Murray. http://darwin-online.org.uk/converted/pdf/1861_OriginNY_F382.pdf [Google Scholar]
- Duranton M, Allal F, Fraïsse C, Bierne N, Bonhomme F, & Gagnaire PA (2018). The origin and remolding of genomic islands of differentiation in the European sea bass. Nature Communications, 9(1), 1–11. 10.1038/s41467-018-04963-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris KG, Barnett LL, Blackman BK, & Willis JH (2017). The genetic architecture of local adaptation and reproductive isolation in sympatry within the Mimulus guttatus species complex. Molecular Ecology, 26(1), 208–224. 10.1111/mec.13763 [DOI] [PubMed] [Google Scholar]
- Ferris KG, Sexton JP, & Willis JH (2014). Speciation on a local geographic scale: The evolution of a rare rock outcrop specialist in Mimulus. Philosophical Transactions of the Royal Society B: Biological Sciences, 369(1648), 20140001. 10.1098/rstb.2014.0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman L (2020). 96-well CTAB-chloroform DNA extraction. protocols.io. 10.17504/protocols.io.bgv6jw9e [DOI] [Google Scholar]
- Fishman L, Aagaard J, & Tuthill JC (2008). Toward the Evolutionary Genomics of Gametophytic Divergence: Patterns of Transmission Ratio Distortion in Monkeyflower (mimulus) Hybrids Reveal a Complex Genetic Basis for Conspecific Pollen Precedence. Evolution, 62(12), 2958–2970. 10.1111/j.1558-5646.2008.00475.x [DOI] [PubMed] [Google Scholar]
- Fishman L, Sweigart AL, Kenney AM, & Campbell S (2014). Major quantitative trait loci control divergence in critical photoperiod for flowering between selfing and outcrossing species of monkeyflower (Mimulus). New Phytologist, 201(4), 1498–1507. 10.1111/nph.12618 [DOI] [PubMed] [Google Scholar]
- Fishman L, & Willis JH (2006). A cytonuclear incompatibility causes anther sterility in Mimulus hybrids. Evolution, 60(7), 1372–1381. 10.1111/j.0014-3820.2006.tb01216.x [DOI] [PubMed] [Google Scholar]
- Fitak RR (2021). OptM: Estimating the optimal number of migration edges on population trees using Treemix. Biology Methods and Protocols, 6(1), bpab017. 10.1093/biomethods/bpab017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Tsuchimatsu T, Kimura Y, Ishida S, Tangpranomkorn S, Shimosato-Asano H, Iwano M, Furukawa S, Itoyama W, Wada Y, Shimizu KK, & Takayama S (2019). A stigmatic gene confers interspecies incompatibility in the Brassicaceae. Nature Plants, 5(7), Article 7. 10.1038/s41477-019-0444-6 [DOI] [PubMed] [Google Scholar]
- Fuller ZL, Leonard CJ, Young RE, Schaeffer SW, & Phadnis N (2018). Ancestral polymorphisms explain the role of chromosomal inversions in speciation. PLOS Genetics, 14(7), e1007526. 10.1371/journal.pgen.1007526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GBIF.org. (2022). GBIF Occurrence Download (12 September 2022). 10.15468/dl.j8dxeq [DOI] [Google Scholar]
- Gore P, Potts B, Volker P, & Megalos J (1990). Unilateral Cross-Incompatibility in Eucalyptus: The Case of Hybridisation Between E. globulus and E. nitens. Australian Journal of Botany, 38(4), 383. 10.1071/BT9900383 [DOI] [Google Scholar]
- Goulet BE, Roda F, & Hopkins R (2017). Hybridization in plants: Old ideas, new techniques. Plant Physiology, 173(1), 65–78. 10.1104/pp.16.01340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant AL (1924). A Monograph of the Genus Mimulus. Annals of the Missouri Botanical Garden, 11(2/3), 99–388. 10.2307/2394024 [DOI] [Google Scholar]
- Grant V (1981). Plant Speciation. Columbia University Press. [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH-Y, Hansen NF, Durand EY, Malaspinas A-S, Jensen JD, Marques-Bonet T, Alkan C, Prüfer K, Meyer M, Burbano HA, … Pääbo S (2010). A draft sequence of the Neandertal genome. Science (New York, N.Y.), 328(5979), 710–722. 10.1126/science.1188021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero RF, Muir CD, Josway S, & Moyle LC (2017). Pervasive antagonistic interactions among hybrid incompatibility loci. PLOS Genetics, 13(6), e1006817. 10.1371/journal.pgen.1006817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haig D, & Westoby M (1991). Genomic imprinting in endosperm: Its effect on seed development in crosses between species, and between different ploidies of the same species, and its implications for the evolution of apomixis. Philosophical Transactions - Royal Society of London, B, 333, 1–13. [Google Scholar]
- Hamlin JAP, Hibbins MS, & Moyle LC (2020). Assessing biological factors affecting postspeciation introgression. Evolution Letters, 4(2), 137–154. 10.1002/evl3.159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogenboom NG, Mather K, Heslop-Harrison J, & Lewis D (1975). Incompatibility and incongruity: Two different mechanisms for the non-functioning of intimate partner relationships. Proceedings of the Royal Society of London. Series B. Biological Sciences, 188(1092), 361–375. 10.1098/rspb.1975.0025 [DOI] [Google Scholar]
- Kay KM, Woolhouse S, Smith BA, Pope NS, & Rajakaruna N (2018). Sympatric serpentine endemic Monardella (Lamiaceae) species maintain habitat differences despite hybridization. Molecular Ecology, 27(9), 2302–2316. 10.1111/mec.14582 [DOI] [PubMed] [Google Scholar]
- Kenney AM, & Sweigart AL (2016). Reproductive isolation and introgression between sympatric Mimulus species. Molecular Ecology, 25(11), 2499–2517. 10.1111/mec.13630 [DOI] [PubMed] [Google Scholar]
- Kirkpatrick M, & Barrett B (2015). Chromosome inversions, adaptive cassettes and the evolution of species’ ranges. Molecular Ecology, 24(9), 2046–2055. 10.1111/mec.13074 [DOI] [PubMed] [Google Scholar]
- Kleindorfer S, O’Connor JA, Dudaniec RY, Myers SA, Robertson J, & Sulloway FJ (2014). Species collapse via hybridization in Darwin’s tree finches. The American Naturalist, 183(3), 325–341. 10.1086/674899 [DOI] [PubMed] [Google Scholar]
- Koch MA, Haubold B, & Mitchell-Olds T (2000). Comparative Evolutionary Analysis of Chalcone Synthase and Alcohol Dehydrogenase Loci in Arabidopsis, Arabis, and Related Genera (Brassicaceae). Molecular Biology and Evolution, 17(10), 1483–1498. 10.1093/oxfordjournals.molbev.a026248 [DOI] [PubMed] [Google Scholar]
- Korunes KL, & Samuk K (2021). pixy: Unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Molecular Ecology Resources, 21(4), 1359–1368. 10.1111/1755-0998.13326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryvokhyzha D (n.d.). GATK: The best practice for genotype calling in a non-model organism. Dmytro Kryvokhyzha - Bioinformatics & Genomics Scientist. Retrieved August 8, 2022, from https://evodify.com/gatk-in-non-model-organism/ [Google Scholar]
- Kuboyama T, Chung CS, & Takeda G (1994). The diversity of interspecific pollen-pistil incongruity in Nicotiana. Sexual Plant Reproduction, 7(4), 250–258. 10.1007/BF00232744 [DOI] [Google Scholar]
- Lafon-Placette C, Johannessen IM, Hornslien KS, Ali MF, Bjerkan KN, Bramsiepe J, Glöckle BM, Rebernig CA, Brysting AK, Grini PE, & Köhler C (2017). Endosperm-based hybridization barriers explain the pattern of gene flow between Arabidopsis lyrata and Arabidopsis arenosa in Central Europe. Proceedings of the National Academy of Sciences, 114(6), E1027–E1035. 10.1073/pnas.1615123114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon-Placette C, & Köhler C (2016). Endosperm-based postzygotic hybridization barriers: Developmental mechanisms and evolutionary drivers. Molecular Ecology, 25(11), 2620–2629. 10.1111/mec.13552 [DOI] [PubMed] [Google Scholar]
- Le Gac M, Hood ME, & Giraud T (2007). Evolution of Reproductive Isolation Within a Parasitic Fungal Species Complex. Evolution, 61(7), 1781–1787. 10.1111/j.1558-5646.2007.00144.x [DOI] [PubMed] [Google Scholar]
- Leishman M, Wright I, Moles A, & Westoby M (2000). The Evolutionary Ecology of Seed Size. Seeds: The Ecology of Regeneration in Plant Communities, 2. 10.1079/9780851994321.0031 [DOI] [Google Scholar]
- Li H (2018). Seqtk (1.3) [Computer software]. https://github.com/lh3/seqtk [Google Scholar]
- Li H, & Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry DB, Sobel JM, Angert AL, Ashman T-L, Baker RL, Blackman BK, Brandvain Y, Byers KJRP, Cooley AM, Coughlan JM, Dudash MR, Fenster CB, Ferris KG, Fishman L, Friedman J, Grossenbacher DL, Holeski LM, Ivey CT, Kay KM, … Yuan Y-W (2019). The case for the continued use of the genus name Mimulus for all monkeyflowers. TAXON, 68(4), 617–623. 10.1002/tax.12122 [DOI] [Google Scholar]
- Lowry DB, & Willis JH (2010). A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biology, 8(9). 10.1371/journal.pbio.1000500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinsky M, Svardal H, Tyers AM et al. Whole-genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nat Ecol Evol 2, 1940–1955 (2018). 10.1038/s41559-018-0717-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinsky M, Matschiner M, & Svardal H (2021). Dsuite—Fast D-statistics and related admixture evidence from VCF files. Molecular Ecology Resources, 21(2), 584–595. 10.1111/1755-0998.13265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J, Beltrán M, Neukirchen W, & Linares M (2007). Natural hybridization in heliconiine butterflies: The species boundary as a continuum. BMC Evolutionary Biology, 7(1), 28. 10.1186/1471-2148-7-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone JH, & Fontenot BE (2008). Patterns of Reproductive Isolation in Toads. PLoS ONE, 3(12), e3900. 10.1371/journal.pone.0003900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SH, & Van Belleghem SM (2017). Exploring Evolutionary Relationships Across the Genome Using Topology Weighting. Genetics, 206(1), 429–438. 10.1534/genetics.116.194720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute DR, & Cooper BS (2021). Comparative studies on speciation: 30 years since Coyne and Orr. Evolution, 1989, 1–15. 10.1111/evo.14181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle LC, Olson MS, & Tiffin P (2004). Patterns of reproductive isolation in three angiosperm genera. Evolution, 58(6), 1195–1208. 10.1111/j.0014-3820.2004.tb01700.x [DOI] [PubMed] [Google Scholar]
- Moyle LC, & Payseur BA (2009). Reproductive isolation grows on trees. Trends in Ecology & Evolution, 24(11), 591–598. 10.1016/j.tree.2009.05.010 [DOI] [PubMed] [Google Scholar]
- Nelson TC, Stathos AM, Vanderpool DD, Finseth FR, Yuan Y, & Fishman L (2021). Ancient and recent introgression shape the evolutionary history of pollinator adaptation and speciation in a model monkeyflower radiation (Mimulus section Erythranthe). PLoS Genetics, 17(2), e1009095. 10.1371/journal.pgen.1009095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesom GL (2013). Taxonomic notes on Diplacus (Phrymaceae). Phytoneuron, 8. [Google Scholar]
- Noor MAF, Grams KL, Bertucci LA, & Reiland J (2001). Chromosomal inversions and the reproductive isolation of species. Proceedings of the National Academy of Sciences, 98(21), 12084–12088. 10.1073/pnas.221274498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oneal E, Willis JH, & Franks RG (2016). Disruption of endosperm development is a major cause of hybrid seed inviability between Mimulus guttatus and Mimulus nudatus. New Phytologist, 210(3), 1107–1120. 10.1111/nph.13842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmolovsky I, Shifrin M, Gamliel I, Belmaker J, & Sapir Y (2022). Eco-Geography and Phenology Are the Major Drivers of Reproductive Isolation in the Royal Irises, a Species Complex in the Course of Speciation. Plants, 11(23), Article 23. 10.3390/plants11233306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer B, & Kapan DD (2019). Estimates of introgression as a function of pairwise distances. BMC Bioinformatics, 20(1), 207. 10.1186/s12859-019-2747-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, & Pritchard JK (2012). Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLOS Genetics, 8(11), e1002967. 10.1371/journal.pgen.1002967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presgraves DC (2002). Patterns of Postzygotic Isolation in Lepidoptera. Evolution, 56(6), 1168–1183. 10.1111/j.0014-3820.2002.tb01430.x [DOI] [PubMed] [Google Scholar]
- Rabiee M, Sayyari E, & Mirarab S (2019). Multi-allele species reconstruction using ASTRAL. Molecular Phylogenetics and Evolution, 130, 286–296. 10.1016/j.ympev.2018.10.033 [DOI] [PubMed] [Google Scholar]
- Ramsey J, Bradshaw HD, & Schemske DW (2003). Components of reproductive isolation between the monkeyflowers Mimulus lewisii and M. cardinalis (Phrymaceae). Evolution, 57(7), 1520–1534. 10.1111/j.0014-3820.2003.tb00360.x [DOI] [PubMed] [Google Scholar]
- Raunsgard A, Opedal ØH, Ekrem RK, Wright J, Bolstad GH, Armbruster WS, & Pélabon C (2018). Intersexual conflict over seed size is stronger in more outcrossed populations of a mixed-mating plant. Proceedings of the National Academy of Sciences, 115(45), 11561–11566. 10.1073/pnas.1810979115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebernig CA, Lafon-Placette C, Hatorangan MR, Slotte T, & Köhler C (2015). Non-reciprocal Interspecies Hybridization Barriers in the Capsella Genus Are Established in the Endosperm. PLOS Genetics, 11(6), e1005295. 10.1371/journal.pgen.1005295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaut S, Grassa CJ, Yeaman S, Moyers BT, Lai Z, Kane NC, Bowers JE, Burke JM, & Rieseberg LH (2013). Genomic islands of divergence are not affected by geography of speciation in sunflowers. Nature Communications, 4(1), 1827. 10.1038/ncomms2833 [DOI] [PubMed] [Google Scholar]
- Roda F, & Hopkins R (2019). Correlated evolution of self and interspecific incompatibility across the range of a Texas wildflower. New Phytologist, 221(1), 553–564. 10.1111/nph.15340 [DOI] [PubMed] [Google Scholar]
- Roth M, Florez-Rueda AM, & Städler T (2019). Differences in Effective Ploidy Drive Genome-Wide Endosperm Expression Polarization and Seed Failure in Wild Tomato Hybrids. Genetics, 212(1), 141–152. 10.1534/genetics.119.302056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushworth CA, Wardlaw AM, Ross-Ibarra J, & Brandvain Y (2022). Conflict over fertilization underlies the transient evolution of reinforcement. PLOS Biology, 20(10), e3001814. 10.1371/journal.pbio.3001814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackton T (2022). Natural selection constrains neutral diversity across a wide range of species [R]. https://github.com/tsackton/linked-selection/blob/da62043544dce9c9342f0a393f41c63bec292fcf/misc_scripts/Identify_4D_Sites.pl (Original work published 2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstedt GD, & Sweigart AL (2022). Developmental evidence for parental conflict in driving Mimulus species barriers. New Phytologist, 236(4), 1545–1557. 10.1111/nph.18438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstedt GD, Wu CA, & Sweigart AL (2020). Evolution of multiple postzygotic barriers between species of the Mimulus tilingii complex. Evolution, 75, 600–613. 10.1111/evo.14105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scascitelli M, Whitney KD, Randell RA, King M, Buerkle CA, & Rieseberg LH (2010). Genome scan of hybridizing sunflowers from Texas ( Helianthus annuus and H. debilis ) reveals asymmetric patterns of introgression and small islands of genomic differentiation. Molecular Ecology, 19(3), 521–541. 10.1111/j.1365-294X.2009.04504.x [DOI] [PubMed] [Google Scholar]
- Schliep KP (2011). phangorn: Phylogenetic analysis in R. Bioinformatics, 27(4), 592–593. 10.1093/bioinformatics/btq706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M, Xu C, Powell DL, Durvasula A, Skov L, Holland C, Blazier JC, Sankararaman S, Andolfatto P, Gil, †, Rosenthal G, & Przeworski M (2018). Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science, 360(6389), 656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbach AE, Donovan LA, & Rieseberg LH (2001). Transgressive character expression in a hybrid sunflower species. American Journal of Botany, 88(2), 270–277. 10.2307/2657018 [DOI] [PubMed] [Google Scholar]
- Scopece G, Musacchio A, Widmer A, & Cozzolino S (2007). Patterns of reproductive isolation in Mediterranean deceptive orchids. Evolution, 61(11), 2623–2642. 10.1111/j.1558-5646.2007.00231.x [DOI] [PubMed] [Google Scholar]
- Servedio MR, & Hermisson J (2020). The evolution of partial reproductive isolation as an adaptive optimum. Evolution, 74(1), 4–14. 10.1111/evo.13880 [DOI] [PubMed] [Google Scholar]
- Skogsmyr I, & Lankinen Å (2002). Sexual selection: An evolutionary force in plants? Biological Reviews, 77(4), 537–562. 10.1017/S1464793102005973 [DOI] [PubMed] [Google Scholar]
- Slatkin M (2005). Seeing ghosts: The effect of unsampled populations on migration rates estimated for sampled populations. Molecular Ecology, 14(1), 67–73. 10.1111/j.1365-294X.2004.02393.x [DOI] [PubMed] [Google Scholar]
- Sobel JM (2010). Speciation in the western North American wildflower genus Mimulus [Ph.D., Michigan State University; ]. https://www.proquest.com/docview/873378456/abstract/950327CDE46D4690PQ/1 [Google Scholar]
- Sobel JM (2014). Ecogeographic Isolation and Speciation in the Genus Mimulus. The American Naturalist, 184(5), 565–579. 10.1086/678235 [DOI] [PubMed] [Google Scholar]
- Sobel JM, & Chen GF (2014). Unification of methods for estimating the strength of reproductive isolation. Evolution, 68(5), 1511–1522. 10.1111/evo.12362 [DOI] [PubMed] [Google Scholar]
- Stamatakis A (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30(9), 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankowski S, Chase MA, Fuiten AM, Rodrigues MF, Ralph PL, & Streisfeld MA (2019). Widespread selection and gene flow shape the genomic landscape during a radiation of monkeyflowers. PLOS Biology, 17(7), e3000391. 10.1371/journal.pbio.3000391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathos A, & Fishman L (2014). Chromosomal rearrangements directly cause underdominant F1 pollen sterility in Mimulus lewisii–Mimulus cardinalis hybrids. Evolution, 68(11), 3109–3119. 10.1111/evo.12503 [DOI] [PubMed] [Google Scholar]
- Streisfeld MA, & Kohn JR (2005). Contrasting patterns of floral and molecular variation across a cline in Mimulus aurantiacus. Evolution, 59(12), 2548–2559. 10.1111/j.0014-3820.2005.tb00968.x [DOI] [PubMed] [Google Scholar]
- Streisfeld MA, & Rausher MD (2009). Altered trans-Regulatory Control of Gene Expression in Multiple Anthocyanin Genes Contributes to Adaptive Flower Color Evolution in Mimulus aurantiacus. Molecular Biology and Evolution, 26(2), 433–444. 10.1093/molbev/msn268 [DOI] [PubMed] [Google Scholar]
- Struck TH, Feder JL, Bendiksby M, Birkeland S, Cerca J, Gusarov VI, Kistenich S, Larsson K-H, Liow LH, Nowak MD, Stedje B, Bachmann L, & Dimitrov D (2018). Finding Evolutionary Processes Hidden in Cryptic Species. Trends in Ecology & Evolution, 33(3), 153–163. 10.1016/j.tree.2017.11.007 [DOI] [PubMed] [Google Scholar]
- Sweigart AL, Fishman L, & Willis JH (2006). A simple genetic incompatibility causes hybrid male sterility in mimulus. Genetics, 172(4), 2465–2479. 10.1534/genetics.105.053686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, & Higashiyama T (2012). A Species-Specific Cluster of Defensin-Like Genes Encodes Diffusible Pollen Tube Attractants in Arabidopsis. PLOS Biology, 10(12), e1001449. 10.1371/journal.pbio.1001449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar-Méndez A, Kumar A, Kondo K, Ashford A, Baek YS, Welch L, Bedinger PA, & McClure BA (2014). Restoring pistil-side self-incompatibility factors recapitulates an interspecific reproductive barrier between tomato species. Plant Journal, 77(5), 727–736. 10.1111/tpj.12424 [DOI] [PubMed] [Google Scholar]
- Tricou T, Tannier E, & de Vienne DM (2022). Ghost Lineages Highly Influence the Interpretation of Introgression Tests. Systematic Biology, 71(5), 1147–1158. 10.1093/sysbio/syac011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera G, & O’Connor B (2020). Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (1st Edition). O’Reilly Media. [Google Scholar]
- Wheeler MJ, Franklin-Tong VE, & Franklin FCH (2001). The molecular and genetic basis of pollen–pistil interactions. New Phytologist, 151(3), 565–584. 10.1046/j.0028-646x.2001.00229.x [DOI] [PubMed] [Google Scholar]
- Williams E, & Rouse J (1988). Disparate Style Lengths Contribute to Isolation of Species in Rhododendron. Australian Journal of Botany, 36(2), 183. 10.1071/BT9880183 [DOI] [Google Scholar]
- Xiong T, & Mallet J (2022). On the impermanence of species: The collapse of genetic incompatibilities in hybridizing populations. Evolution, 76(11), 2498–2512. 10.1111/evo.14626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y-W (2019). Monkeyflowers (Mimulus): New model for plant developmental genetics and evo-devo. New Phytologist, 222(2), 694–700. 10.1111/nph.15560 [DOI] [PubMed] [Google Scholar]
- Yuan Y-W, Sagawa JM, Young RC, Christensen BJ, & Bradshaw HD Jr. (2013). Genetic Dissection of a Major Anthocyanin QTL Contributing to Pollinator-Mediated Reproductive Isolation Between Sister Species of Mimulus. Genetics, 194(1), 255–263. 10.1534/genetics.112.146852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuellig MP, & Sweigart AL (2018). A two-locus hybrid incompatibility is widespread, polymorphic, and active in natural populations of Mimulus. Evolution, 72(11), 2394–2405. 10.1111/evo.13596 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All newly generated whole genome sequence data are deposited at the NCBI Sequence Read Archive (accession PRJNA922914). The complete genotype call dataset, phylogenetic tree files, and raw crossing data, including seed counts, seed measurements, and pollen counts, are archived in the DRYAD digital data repository at https://doi.org/10.5061/dryad.7wm37pvzc. Code used in these analyses is archived at https://github.com/mfarnitano/Eunanus_popgen.