

SUMMARY
Lung-resident memory B cells (lung-BRMs) differentiate into plasma cells after reinfection, providing enhanced pulmonary protection. Here we investigated the determinants of lung-BRM differentiation upon influenza infection. Kinetic analyses revealed that influenza nucleoprotein (NP)-specific BRMs preferentially differentiated early after infection and required Tfh cell help. BRM differentiation temporally coincided with transient IFN-γ production by Tfh cells. Depletion of IFN-γ in Tfh cells prevented lung-BRM differentiation and impaired protection against heterosubtypic infection. IFN-γ was required for expression of the transcription factor T-bet by germinal center (GC) B cells, which promoted differentiation of a CXCR3+ GC B cell subset that were precursors of lung-BRMs and CXCR3+ memory B cells in the mediastinal lymph node. Absence of IFN-γ signaling or T-bet in GC B cells prevented CXCR3+ pre-memory precursor development and hampered CXCR3+ memory B cell differentiation and subsequent lung-BRM responses. Thus, Tfh cell-derived IFN-γ is critical for lung-BRM development and pulmonary immunity, with implications for vaccination strategies targeting BRMs.
Keywords: BRMs, Tfh cells, T-bet, CXCR3, Tfh-1 cells, Influenza, pre-MEMs, memory B cells, Germinal centers, intranasal vaccination
Graphical Abstract
eTOC BLURB
Lung-resident memory B cells (lung-BRMs) provide superior protection against respiratory viruses. Arroyo-Díaz et al examine the requirements for lung-BRM development and reveal that IFN-γ production by Tfh cells is essential for the differentiation of memory B cell precursors that give rise to lung-BRMs and subsequent protection against heterosubtypic influenza infection.
INTRODUCTION
Lung-resident memory B cells (lung-BRMs) are a population of non-circulating memory B cells that develop in response to pulmonary challenges and permanently reside in the lungs 1–6. Phenotypically, lung-BRMs are class-switched, somatically hypermutated, and characteristically express CXCR3 1,5,7,8. The expression of CXCR3 is not required for the development nor the homing of lung-BRM cells into the lung 4,9. However, it allows lung-BRMs to rapidly localize to the infected areas of the lung after secondary infection, where they differentiate into plasma cells and secrete antibodies (Abs) that provide superior local protection against homologous and heterologous viruses 1–5,7,10. Highlighting the critical role of lung-BRMs, despite comparable titers of circulating Abs and similar systemic memory B cell responses, the lack of lung-BRMs substantially delays protection against secondary heterosubtypic influenza infection in systemically relative to intranasally vaccinated mice. 1,7. Thus, CXCR3+lung-BRMs have a critical role as first layer of defense against respiratory viruses.
CXCR3 is a transcriptional target of T-bet, a transcription factor that fine-tunes T-cell function and differentiation 11. T-bet can also contribute to the differentiation and function of memory B cells, as T-bet expression in germinal center (GC) B cells is required for the generation of antigen-experienced memory B cells with enhanced capacity to differentiate into antibody secreting cells (ASC) after rechallenge 12,13. Whether T-bet plays a role in lung-BRM development remains elusive.
Memory B cells can derive from the GCs or have a GC-independent origin 14–17. Correlating with a GC-dependent pathway, GC B cells poised to become memory B cells (pre-MEMs) have been identified at the edge of the light zone18–21. Influenza-specific BRMs are GC-derived memory B cells that initially arise from the GCs in the lung-draining lymph node and then home to the lung, establishing permanent residency therein 1,4,8,10. Most lung-BRMs seed the lungs within the first three weeks of infection 1,7. After this time, ablation of GCs 1 or blockade of lymphocyte egress 4 from the secondary lymphoid organs negligibly affects the accumulation of influenza-specific BRMs in the lung. Therefore, influenza-specific BRMs are GC-derived memory cells that are preferentially generated during the initial phase of the primary response.
GC formation depends on help provided by CD4+ T follicular helper (Tfh) cells 22, a population of CD4+ T cells that localizes in the B cell follicles and are characterized by expression of interleukin (IL)-21 and CD40L, which are required for the development and maintenance of the GCs 22,23. Tfh cells are plastic and can secrete various effector-like cytokines 21,24–29. As such, Tfh cells secrete IFN-γ during viral infections, which promotes IgG2c/a class switching 25,26,30. However, beyond promoting IgG2c/a class switching, the effects of IFN-γ+ Tfh cells in regulating GC fate decisions are unknown.
In this study, we investigated the mechanisms that control lung-BRM responses after intranasal influenza virus infection. We found that intrinsic IFN-γ-STAT1-T-bet signaling in GC B cells initiates a transcriptional program that is required for the differentiation of a population of CXCR3+ pre-MEMs, which are the precursors of CXCR3+ memory B cells in the lung-draining lymph node and ensuing lung-BRMs in the lungs. Without IFN-γ signaling, IfngR1−/−GCs failed to increase expression of T-bet, CXCR3+pre-MEMs did not differentiate, influenza-specific CXCR3+ memory B cells did not develop, and ensuing lung-BRM responses were compromised. IFN-γ was provided to GC B cells by IFN-γ-producing Tfh cells. Thus, in the absence of IFN-γ-producing Tfh cells, GCs normally differentiate, but influenza-specific lung-BRM responses are severely diminished. Consequently, BRM-mediated protection after heterologous re-challenge is impaired.
RESULTS
Lung-BRM responses to influenza require Tfh cell help early after infection
To determine the kinetics of the lung memory B cell response to influenza, we intranasally (i.n) infected C57BL/6 mice with A/PR8/34 (PR8) influenza virus and used fluorochrome-labeled recombinant influenza nucleoprotein (NP) tetramers to identify class-switched NP-specific memory B cells (CD38+IgD−IgM−NP+CD19+, Figure. 1A) in the lungs at different times after infection (Figure. 1B and C). We also enumerated NP-specific GC B cells (Figure. S1A and B) and class-switched NP-specific memory B cells in the lung-draining mediastinal lymph node (med-LN) (Figure. S1C and D). NP-specific GC B cells were readily identified in the med-LN on day 7 (Figure. S1A and B). At this time, class-switched NP-specific memory B cells were still undetectable in the lung (Figure. 1B and C). The frequency of NP-specific GC B cells increased after day 10, peaking on day 30 after infection (Figure. S1A and B). Correlating with the expansion of the GC B cell response, class-switched NP-specific memory B cells began to accrue in the lungs on day 10, reaching a peak between days 15 and 30 after infection. (Figure. 1B and C). As expected 1,5,7,8, most NP-specific memory B cells were CXCR3+ at all time points analyzed (Figure. 1A and Data not shown). Similar expansion kinetics were observed when we enumerated class-switched NP-specific memory B cells in the med-LN (Figure. S1C and D). However, whereas all the lung NP-specific memory B cells were CXCR3+ (Figure. 1A), NP-specific memory B cells in the med-LN separated into CXCR3+ and CXCR3− cells (Figure. S1E and F). As a control, the NP tetramer labeled class-switched memory B cells from influenza-infected mice but not from mice i.n challenged with an influenza-irrelevant protein (Figure. S1G).
Figure 1. Lung-BRM cells require Tfh help early after influenza infection.
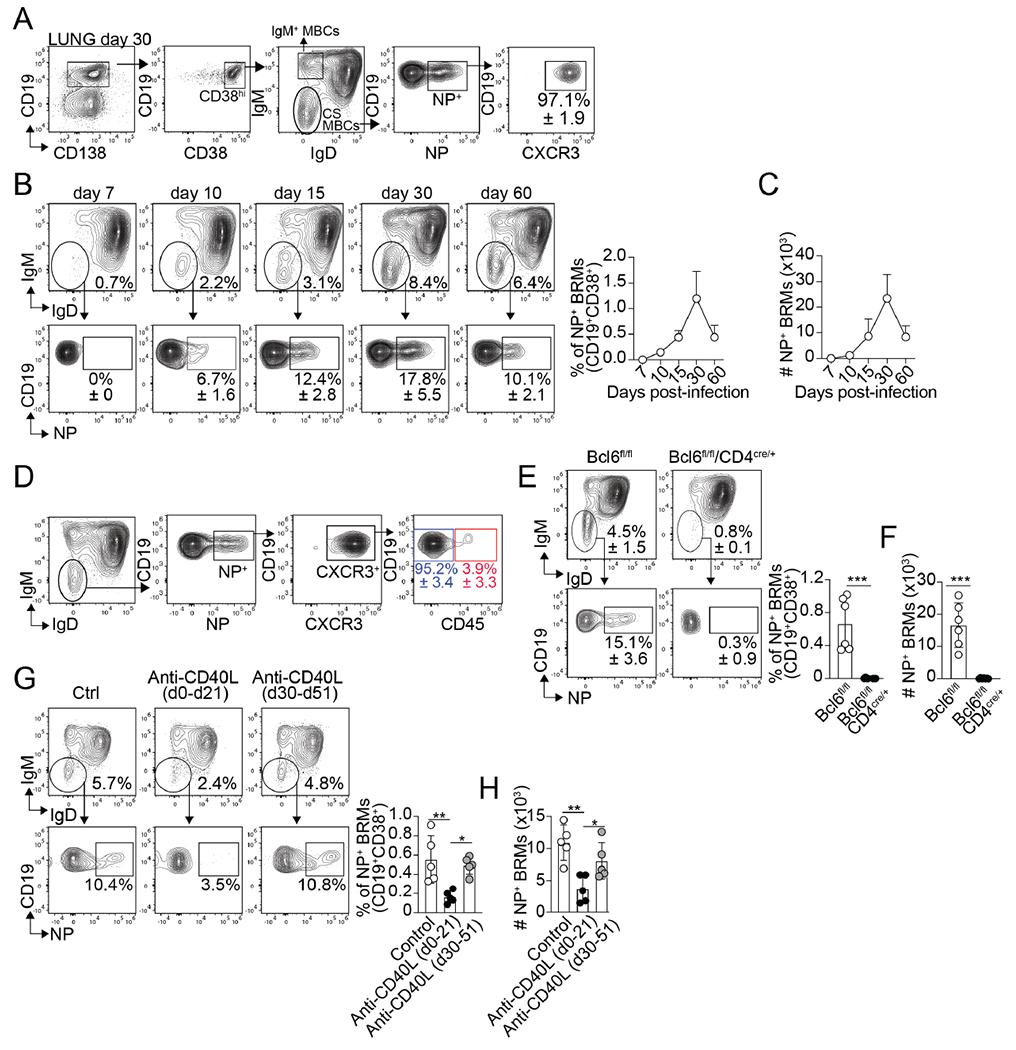
(A-C) B6 mice were infected with PR8. (A) Gating strategy for the identification of NP-specific BRMs in the lungs on day 30. Frequency (B) and number (C) of NP-specific BRMs in the lungs at the indicated time points. Representative plots gated on CD19+CD38+CD138− B cells are shown. Data are representative of four independent experiments (n=5 mice/time point). (D) PR8-infected B6 mice were treated with fluorochrome-labeled anti-CD45 antibody 5 minutes before euthanasia on day 30. Frequency of CD45+ and CD45− cells within the class-switched NP-specific lung-BRMs. Data are representative of four independent experiments (n=5 mice/time point). Frequency (E) and number (F) of NP-specific BRMs in the lung of PR8-infected control and Tfh−/− mice 30 days after infection. Data are representative of three independent experiments. (n=5 mice/time point) P value was determined using a two-tailed Student’s t-test. (G and H) PR8-infected mice were treated with 250 μg of anti-CD40L antibody between day 0 and day 21 or between day 30 and day 51. Frequency (G) and number (H) of NP-specific BRMs in the lung on day 55. Data are representative of three independent experiments (n=5 mice/time point). P values were determined by one-way ANOVA with a post-hoc Kruskal–Wallis comparison test. All data shown as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant.
To confirm that the NP-specific memory B cells were lung-BRMs, we treated influenza-infected mice with a fluorochrome-labeled anti-CD45 antibody 5 minutes before euthanasia to identify non-circulating (CD45−) class-switched BRMs in the lungs (Figure. 1D). As previously shown1,4, more than 95% of the NP-specific memory B cells were CD45− (Figure. 1D). Thus, in agreement with previous studies 1,4, the majority of the class-switched NP-specific lung memory B cells were non-circulating BRMs. Hereafter, we will refer to class-switched NP-specific memory B cells as NP-specific BRMs.
To test whether the differentiation of NP-specific BRMs required Tfh cell help, we infected control (B6.Bcl6fl/fl) and Tfh-deficient (B6.Bcl6fl/flCd4cre/+) mice with influenza and enumerated NP-specific BRMs in the lungs on day 30. As predicted, Tfh cells (Figure. S1H and I) and GC B cells (Figure. S1J and K) failed to accumulate in the Tfh-deficient compared to control mice. Correlating with the lack of GC B cells, NP-specific BRMs were virtually absent from the lungs of the Tfh-deficient mice (Figure. 1E and F). In contrast, no differences were detected in the number of lung IgM+ (CD38+IgD−IgM+) memory B cells (Figure. S1L and M). Thus, while dispensable for the differentiation of IgM+ lung memory B cells, Tfh cell help was required for class-switched NP-specific BRM responses to influenza.
Finally, we examined whether the blockade of Tfh cell help at different times after infection affected the lung-BRM response. To do this, we used a previously published strategy to test the temporarily of lung-BRM generation1. Briefly, we infected B6 mice with PR8 and treated them with control or anti-CD40L antibody (MR1) to block Tfh help either between day 0 and day 21 or between days 30 and 51. All mice were analyzed on day 55. In agreement with previous studies1, blockade of Tfh cell help significantly prevented the accumulation of NP-specific BRMs in mice treated between day 0 and day 21 (Figure. 1G and H). Blocking Tfh cell help after day 30, however, did not affect the NP-specific BRM response (Figure. 1G and H). As a control, treatment with anti-CD40L depleted GC B cells in all groups (Figure. S1N). Altogether, these results indicate that lung-BRM differentiation requires Tfh cell help during the first three weeks of the infection. These data are consistent with previous studies showing that long-lived, class-switched memory B cells differentiate through the GC-dependent pathway 14 and preferentially arise from the GCs early after immunization 31–33.
Tfh cells transiently produce IFNγ at the peak of the response
We next examined whether the preferential differentiation of lung-BRMs early after infection correlated with differences in the Tfh cell response at early relative to later time points. Tfh cells were identified as PD-1hiCXCR5hi Bcl6hiCD4+T cells (Figure. 2A). The frequency of Tfh cells rapidly increased after infection, peaked between days 10 and 15, and remained relatively constant until day 60 (Figure. 2B). Similar results were obtained when we used a fluorochrome-labeled MHC Class II tetramer to identify bona fide influenza NP-specific Tfh cells (Figure. S2A). Thus, Tfh cells were present throughout the entire duration of the GC B cell response.
Figure 2. Tfh cells transiently produce IFNγ after influenza infection.
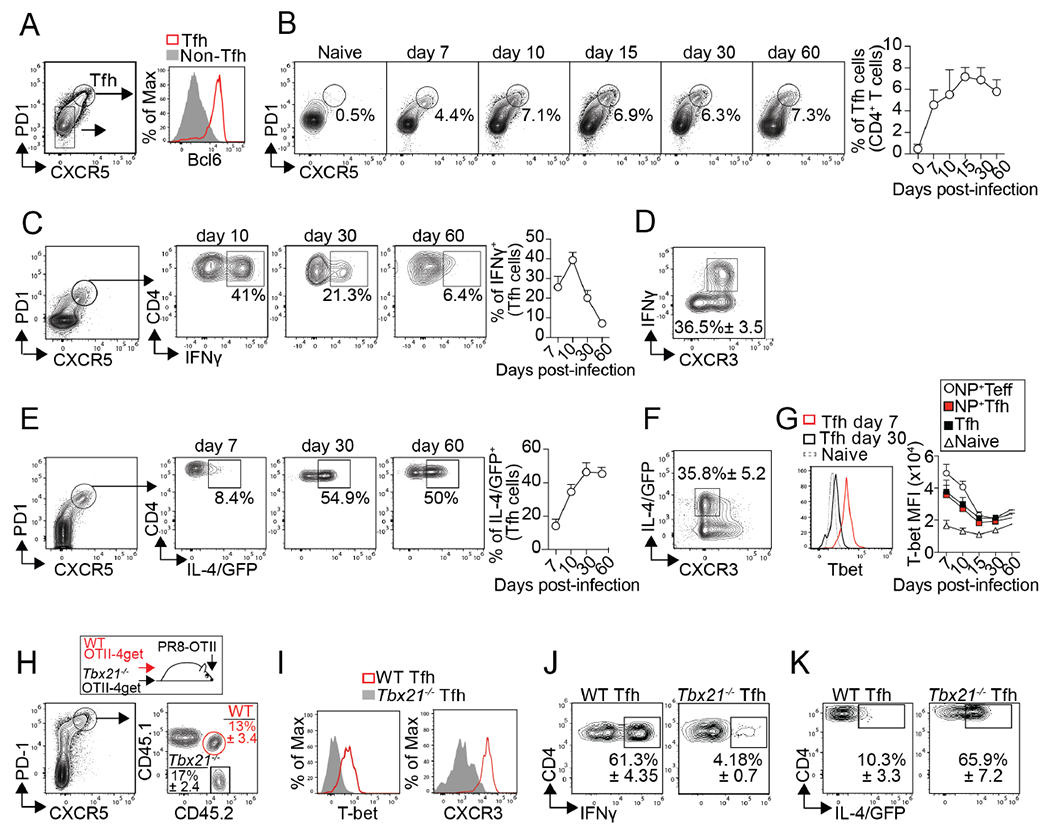
(A-D) Cells from the med-LN of PR8-infected B6 mice were analyzed (A) Tfh cells on day 15 after infection. (B) Frequency of Tfh cells at the indicated time points. Plots were gated on CD4+CD19− T cells. (C) Frequency of IFNγ+ cells within the Tfh cells after in vitro re-stimulation (D) Expression of CXCR3 and IFNγ in Tfh cells on day 15 after infection. Data are representative of three independent experiments (n=5 mice/time point). (E-F) B6.4get mice were infected with PR8. (E) Frequency of IL-4/GFP+ cells within the Tfh cells. (F) Expression of CXCR3 and IL-4/GFP in Tfh cells on day 15. Data are representative of three independent experiments (n=5 mice/time point). (G) T-bet expression in naïve CD4+ T cells, total Tfh cells, NP-specific Tfh cells, and NP-specific Teff cells. Data are representative of three independent experiments (n=5 mice/time point). (H-K) WT (CD45.1+CD45.2+) and Tbx21−/− (CD45.1−CD45.2+) OTII-4get cells were transferred into B6 mice (CD45.1+CD45.2−). Recipient mice were infected with PR8-OTII, and cells from the med-LN were analyzed on day 7. (H) Frequency of WT and Tbx21−/− donors with a Tfh phenotype. (I) T-bet and CXCR3 expression in WT and Tbx21−/−Tfh cells. Frequency of IFNγ+ (J) and IL-4/GFP+ cells (K) within the WT and Tbx21−/−Tfh cells. Data are representative of three independent experiments (n=4 mice).
Importantly, IFNγ-producing Tfh cells were easily identified on day 10 after infection (Figure. 2C). At this time, nearly 40% of the Tfh cells were IFNγ+ (Figure. 2C). However, the frequency of IFNγ+Tfh cells sharply declined thereafter (Figure. 2C). Phenotypically, IFNγ+Tfh cells characteristically expressed high levels of CXCR3 (Figure. 2D). Thus, IFNγ+Tfh cells were abundant at the peak of the infection but relatively rare at later time points.
The observation that Tfh cells produce IFN-γ early after influenza infection is at odds with studies showing that Tfh cells secrete IL-4 in the context of viral infections 10,26,30. To better characterize the Tfh cell response to influenza, we infected IL-4-reporter B6.4get (B6.4get) mice with influenza and enumerated GFP/IL-4+ Tfh cells at different times after infection (Figure. 2E). In agreement with previous work 26,30, we identified GFP/IL-4+ cells within the Tfh cell subset at all time points analyzed (Figure. 2E). However, contrary to IFNγ+ Tfh cells, the frequency of GFP/IL-4+ Tfh cells was relatively low at the beginning of the infection and then significantly increased at later time points (days 30 and 60) (Figure. 2E). Whereas the IFNγ+ Tfh cells were CXCR3hi (Figure. 2D), the GFP/IL-4+ Tfh cells were preferentially CXCR3low (Figure. 2F). Altogether, these results indicate that influenza-specific Tfh cells can be separated into IFN-γ+CXCR3hi and IL-4+CXCR3lo cells. The relative frequency of these subsets, however, changes over time as the immune response progresses.
Correlating with the kinetics of IFNγ production, Tfh cells expressed high levels of T-bet early after infection (Figure. 2G). Given that T-bet promotes IFNγ but inhibits IL-4 production 34,35, we next tested whether increased T-bet expression early after infection temporarily haltered the differentiation of IL-4+CXCR3lo Tfh cells. To test this possibility, we co-transferred congenically different WT-OTII-4get (CD45.1+CD45.2+) and Tbx21−/− OTII-4get (CD45.1−CD45.2+) cells into B6 (CD45.1+CD45.2) mice. One day later, the recipient mice were infected with a genetically modified strain of PR8 that expresses the OVA323–339 peptide (PR8-OTII) 36. Mice were analyzed on day 7 after infection, when IL-4+Tfh cells were normally scarce. WT and Tbx21−/− OTII-Tfh cells were easily distinguished from the host and each other based on the differential expression of CD45.1 and CD45.2 (Figure. 2H). As expected, WT-OTII Tfh cells were T-bethiCXCR3hi (Figure. 2I) and produced IFN-γ (Figure. 2J) but not IL-4 (Figure. 2K). In contrast, Tbx21−/−-OTII Tfh cells were T-betnegCXCR3lo (Figure. 2I) and produced IL-4 (Figure. 2K) but not IFN-γ (Figure. 2J). Collectively, these results indicate that increased T-bet expression early after infection temporarily favors the differentiation of IFN-γ+Tfh cells at the expense of IL-4+Tfh cells. However, as the immune response resolves, T-bet expression progressively decreases, and IFN-γ production subsequently declines. As a result, the Tfh cell response switches from secreting IFN-γ early after infection to mainly producing IL-4 at later time points.
IFN-γ production by Tfh cells is required for lung BRM responses
We next considered the possibility that the production of IFNγ by Tfh cells early after infection was required for the lung-BRM response to influenza. To test this hypothesis, we made bone marrow (BM) chimeras in which Tfh cells were selectively WT or Ifng−/− (Figure. 3A). Briefly, we reconstituted irradiated Tcrb−/−Tcrd−/− mice with a 50:50 mixture of Bcl6fl/flCD4cre and B6.Ifng−/− BM (Tfh-Ifng−/− chimeras). Because Tfh cells cannot differentiate from B6.Bcl6fl/flCD4cre precursors, all the Tfh cells in the Tfh-Ifng−/− chimeras developed from the Ifng−/− donors. In contrast, non-Tfh cells were a mix of WT and Ifng−/− cells. As controls, Tcrb−/−Tcrd−/− mice received a 50:50 mixture of B6.Bcl6fl/flCD4cre and B6 BM (Tfh-100%WT chimeras) or B6 and B6.Ifng−/− BM (Tfh-50%WT chimeras). Two months after reconstitution, we infected the chimeras with influenza and analyzed the Tfh cell response. The frequency and number of Tfh cells were comparable in all groups (Figure. 3B–C). As expected, however, Tfh cells failed to produce IFN-γ in the Tfh-Ifng−/− chimeras compared to Tfh-100%WT and Tfh-50%WT controls (Figure. 3D). In contrast, there were no differences in IFN-γ production within the non-Tfh cell compartment (Figure. 3E). We also found that the total NP-specific IgG titters were similar in all groups (Figure. S3A). However, corresponding with the lack of IFN-γ-producing Tfh cells, we observed a significant reduction in the NP-specific IgG2c Abs in the Tfh-Ifng−/− compared to the control chimeras (Figure. S3B). This result is consistent with a requirement for IFN-γ in IgG2c/a class switching. Collectively, these data show that Tfh cells normally accumulated but failed to produce IFN-γ in the Tfh-Ifng−/− compared to control chimeras.
Figure 3. IFNγ-producing Tfh cells are required for influenza-specific lung-BRM responses.
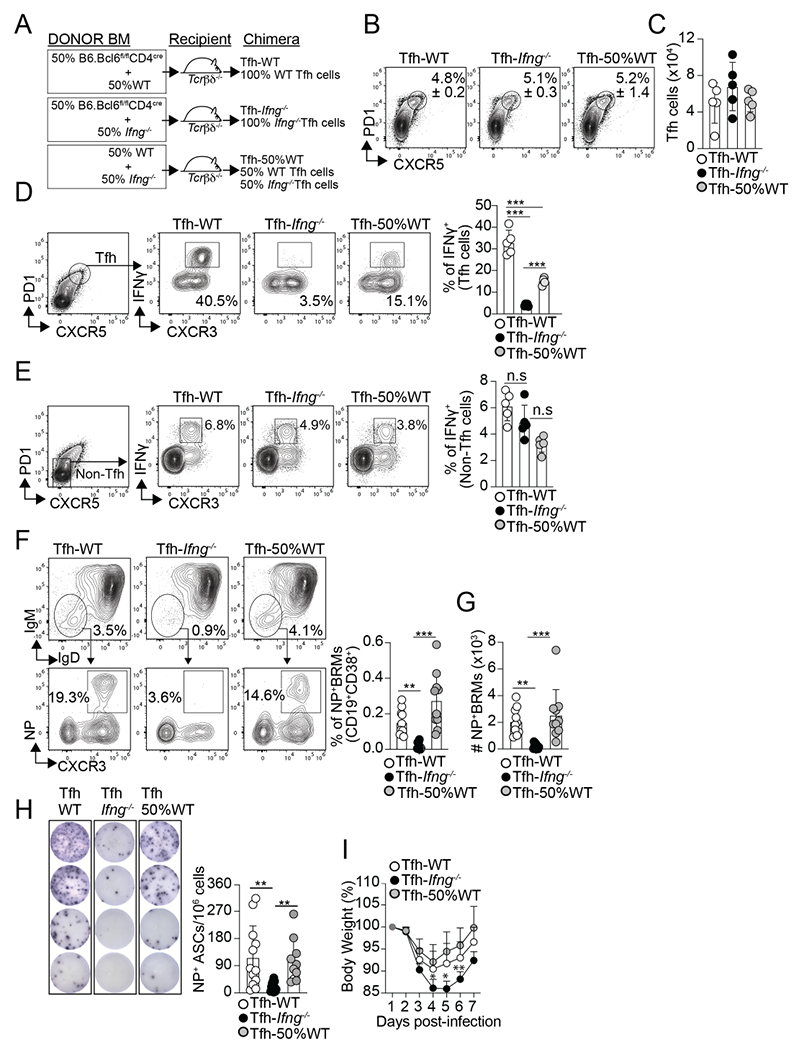
(A) Diagram showing the Tfh-WT, Tfh- Ifng−/−, and Tfh-50%WT BM chimeras design. Frequency (B) and number (C) of Tfh cells in the med-LN on day 12 after PR8 infection. Frequency of IFNγ+ cells within the Tfh cells (D) and non-Tfh cells (E) on day 12 after infection. Data are representative of three independent experiments (n=5 mice/group). Frequency (F) and number (G) of class-switched NP-specific BRMs in the lungs on day 60 after infection. Data were pooled from two independent experiments (n=10 mice/group). Data are representative of four independent experiments. (H and I) The chimeric mice were infected with PR8 and challenged with X31 on day 30. (H) IgG NP-specific ELISPOTs in the lungs on day 6 after rechallenge. Data were pooled from two independent experiments (n=10-12 mice/group). (I) Body weight loss after rechallenge (n=5-6 mice). Data are representative of two independent experiments. All P values were determined by one-way ANOVA with a post- hoc Kruskal–Wallis comparison test. Data shown as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant.
We next enumerated NP-specific BRMs in the lungs. The frequency (Figure. 3F) and number (Figure. 3G) of NP-specific BRMs were dramatically reduced in the lungs of the Tfh-Ifng−/− chimeras compared to the control chimeras. Importantly, the lack of NP-specific BRMs was not due to the absence of GC B cells in the Tfh Ifng−/− chimeras, as the frequency of NP-specific GC B cells in the med-LN (Figure. S3C and Data not shown) and lung (Figure. S3D and Data not shown) were similar in all groups at all time points analyzed. Thus, while dispensable for GC development, IFN-γ production by Tfh cells was required for the differentiation of class-switched influenza-specific BRMs.
Finally, we assessed the NP-specific memory B cell response in the med-LN (Figure. S3E–F). While virtually absent from the lungs (Figure. 3F), class-switched NP+ B memory cells were easily identified in the med-LN of the Tfh-Ifng−/− and control chimeras (Figure. S3E). However, when separated into CXCR3+ and CXCR3− cells, CXCR3+class-switched NP+ memory B failed to accumulate in the Tfh-Ifng−/− relative to the control chimeras (Figure. S3E and F). In contrast, CXCR3− class-switched NP+ memory B cell similarly accrued in all groups (Figure. S3E and F). Thus, similar to lung-BRMs, CXCR3+class-switched NP+ memory B cell responses in the med-LN required IFN-γ production by Tfh cells.
Lack of lung-BRMs in the absence of IFN-γ-producing Tfh cells compromises protection after reinfection.
Lung-BRMs rapidly proliferate and differentiate into antibody-secreting cells (ASCs) after rechallenge, producing Abs that are critical for protection after reinfection with heterologous influenza viruses 1–5,7,10. To test whether the lack of NP-specific BRMs in the lungs of the Tfh-Ifng−/− affected the pulmonary B cell response after recall, we rechallenge our chimeric mice with A/HK-X31 (X31). Whereas the PR8 virus expresses H1N1 proteins, X31 expresses H3N2. Thus, antibodies elicited by a previous PR8 infection do not neutralize X31, allowing a productive infection after X31 rechallenge. However, the NP protein is conserved between the two viruses, enabling NP-specific BRMs generated after infection with PR8 to respond after the X31 rechallenge 1,7. Hence, 30 days after PR8 infection, we challenged the chimeric mice with X31, monitored weight loss, and enumerated NP-specific ASCs by ELISPOT in the lungs on day 6 after rechallenge. Correlating with the lack NP-specific BRMs, the frequency of NP-specific ASCs in the lung was significantly decreased in the Tfh-IFN-γ−/− chimeras compared to the Tfh-100%WT and Tfh-50%WT controls (Figure. 3H). Furthermore, while all mice survive the infection, the Tfh-IFN-γ−/− chimeras lost more weight and showed a delayed recovery compared to the control groups (Figure. 3I). As a control, no differences in IFN-γ production within the CD4− cells were detected in the Tfh-Ifng−/− compared to the control chimeras (Figure. S3E), suggesting that global deficiency in IFN-γ production after rechallenge was not the likely cause of the lack of protection in the Tfh-Ifng−/− mice. Instead, our findings indicate that IFN-γ-producing Tfh cells are required to develop protective pulmonary BRM responses after heterosubtypic influenza infection.
Intrinsic IFNγ signaling is required for lung BRM differentiation
Our data suggest that IFN-γ is required for the generation of influenza-specific lung-BRMs. Thus, we next tested whether the requirement for IFN-γ signaling in lung-BRM development was B cell intrinsic. To do this, we made WT/IfngR1−/− mixed-BM chimeras, infected them with influenza, and enumerated NP-specific BRMs in the lung. This approach allowed us to study WT (CD45.1+) and IfngR1−/− (CD45.2+) B cells responding to influenza in the same animal. The frequency of total class-switched memory B cells (Figure. 4A) and NP-specific BRMs (Figure. 4B) was significantly diminished in the IfngR1−/− compared to the WT compartment at all time points analyzed. As a result, the WT to IfngR1−/− ratio of total and NP-specific class-switched BRMs was significantly increased compared to the ratio of naïve B cells (Figure. 4C). No differences were detected when we enumerated WT and IfngR1−/− class-switched GC B cells (Figure. S4A and B) and IgM+ memory B cells (Figure. S4C and D) in the lungs. However, similar to lung-BRMs, the frequency of CXCR3+class-switched NP+ memory B cells in the med-LN was significantly diminished in the IfngR1−/− compared to the WT compartment (Figure. S4E–F).
Figure 4. Intrinsic IFNγ/STAT1 signaling is required for the differentiation of lung-BRMs.
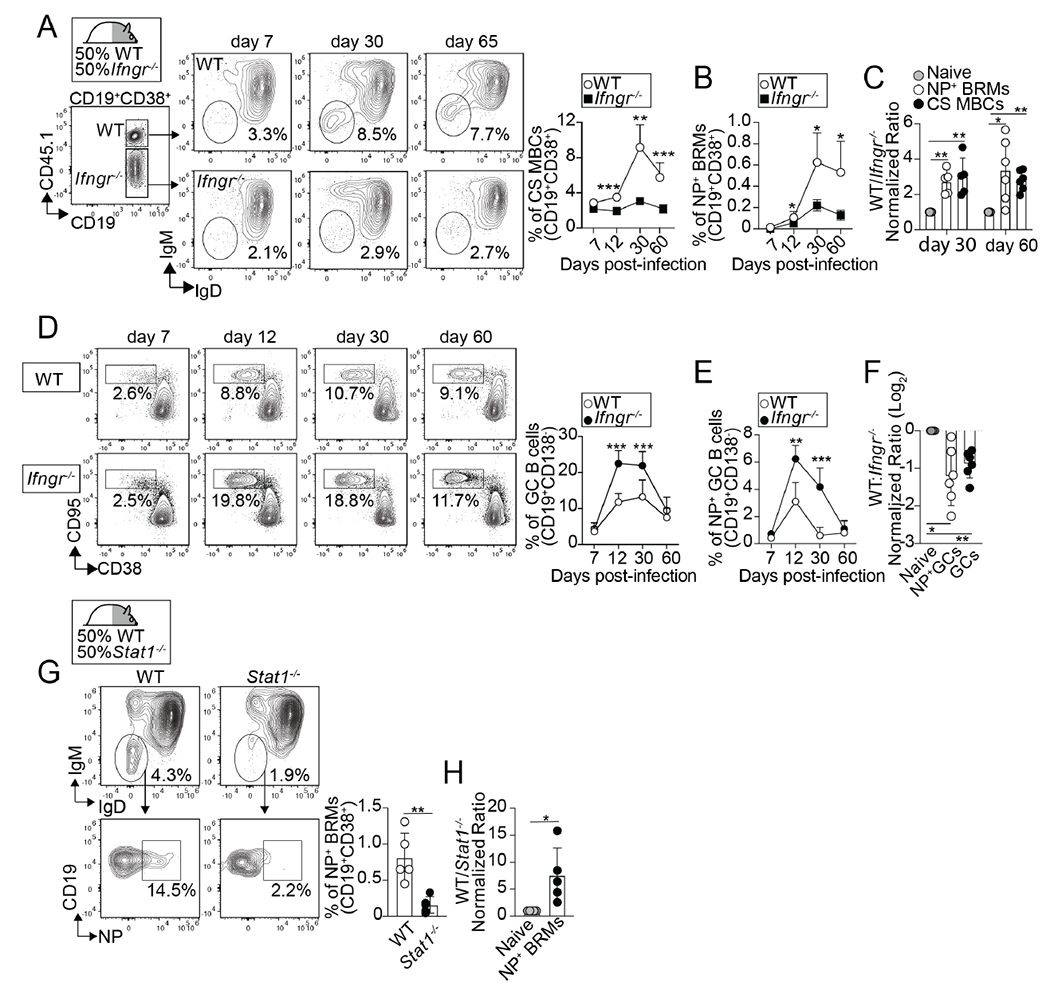
(A-F) WT/IfngR1−/− BM chimeras were infected with PR8. Frequency of class-switched memory B cells (A) and NP-specific lung BRMs (B) within the B6 and IfngR1−/− compartments in the lungs. P values were determined using a two-tailed Student´s t-test. (C) Ratio of B6 to IfngR1−/− naïve B cells (naïve), class-switched memory B cells (CS MBCs), and NP-specific BRMs. P values were determined by one-way ANOVA with a post-hoc Kruskal–Wallis comparison test. Frequency of total (D) and NP-specific GC B cells (E) within the B6 and IfngR1−/− compartments in the med-LN. P values were determined using a two-tailed Student´s t-test. (F) Ratio of B6 to IfngR1−/− naïve B cells, total GC B cells, and NP-specific GC B cells. P values were determined by one-way ANOVA with a post-hoc Kruskal–Wallis comparison test. Data are representative of three independent experiments (n=5-6 mice). (G-H) WT/ Stat1−/− BM chimeras were infected with PR8 and B cells from the lungs were analyzed on day 50.(G) Frequencies of NP-specific BRMs within the B6 and Stat1−/− compartments. Ratio of B6 to Stat1−/− naïve B cells and NP-specific BRMs. Data are representative of three independent experiments (n=5 mice). P values were determined using a two-tailed Student´s t-test.
We next examined the WT and IfngR1−/− GC B cell response in the med-LN of the WT/IfngR1−/− chimeras. We found a higher frequency of total (Figure. 4D) and NP-specific (Figure. 4E) GC B cells in the IfngR1−/− compared to the WT compartment at all time points analyzed. As a result, the WT to IfngR1−/− ratio of NP-specific GC B cells was significantly decreased compared to the ratio of naïve B cells (Figure. 4F). Thus, the scarcity of IfngR1−/− lung-BRMs was not due to the lack of GC B cells in the absence of IFNγ-STAT1 signaling. Altogether, these data indicate that, while dispensable for GC development, intrinsic IFNγ signaling in B cells was required for the differentiation of influenza-specific lung-BRMs after influenza virus infection.
STAT1 is required for optimal IFNγ signaling 37. To further confirm that IFNγ signaling was intrinsically required for lung-BRM development, we enumerated WT and Stat1−/− lung-BRMs in the lung of influenza-infected WT/Stat1−/− mixed-BM chimeras. Similar to IfngR1−/−, Stat1−/− NP-specific BRMs failed to accumulate compared to WT counterparts (Figure. 4G and H). Collectively, these results show that intrinsic IFNγ-STAT1 signaling in B cells is required for the development of influenza-specific lung-BRM responses after influenza virus infection.
IFNγ signaling skews GC B cells towards the BRM differentiation pathway
Studies indicate that lung-BRMs have a GC-dependent origin 1,4,8,10. Given that the lack of lung-BRMs in the absence of IFNγ cannot be explained by the concomitant absence of GC B cells (Figure. S3C and Figure. 4D–F), we next considered the possibility that IFNγ signaling promoted lung-BRM responses by skewing GC B cells towards the BRM differentiation pathway. To test this possibility, we first performed RNA-sequencing (RNA-seq) to compare the transcriptome of WT and IfngR1−/− GC B cells sorted from the med-LN of day 12 infected WT/IfngR1−/− BM chimeras. Principal component analysis (PCA) segregated WT and IfngR1−/− GC B cells into two separate clusters, showing that WT and IfngR1−/− GC B cells were transcriptionally different (Figure. 5A). In agreement, the expression of 140 genes were significantly increased in WT compared to IfngR1−/− GC B cells (Table S1). We will refer to this list of transcripts as the “GC IFNγ-induced program” (i.e., genes induced in GC B cells in response to IFNγ signaling).
Figure 5. Intrinsic IFNγ signaling initiates the lung-BRM differentiation program in GC B cells.
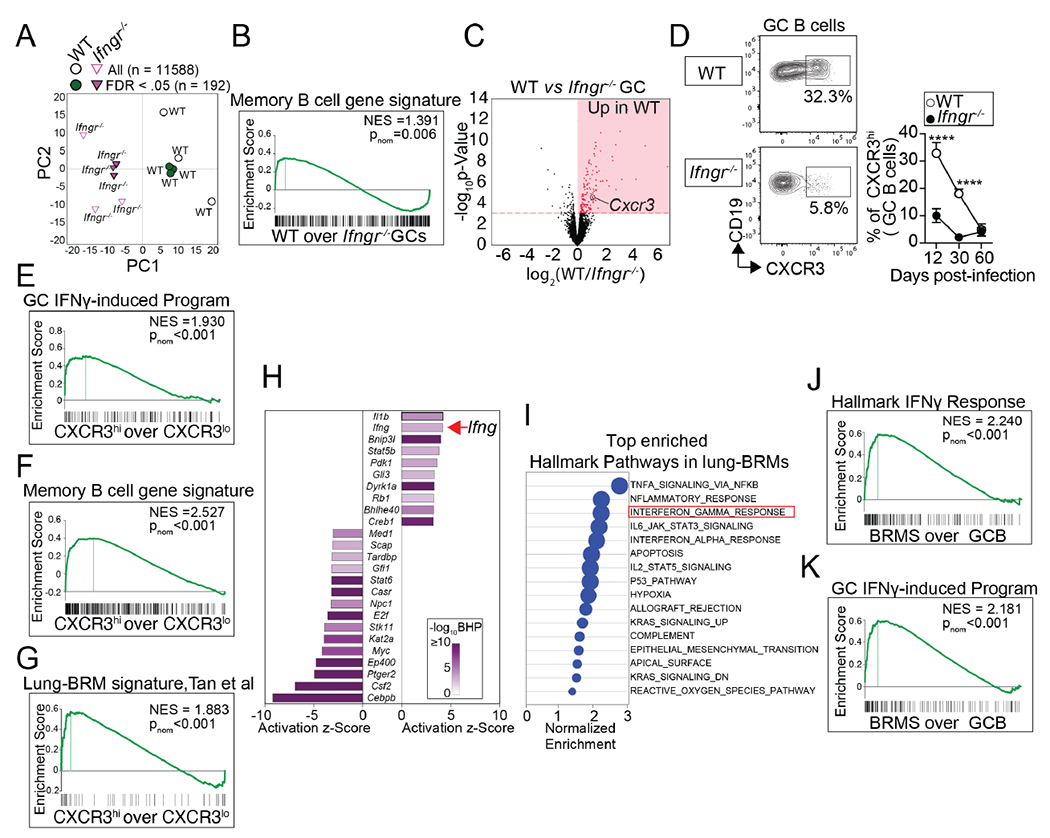
(A-C) WT and IfngR1−/− GC B cells were sorted from WT/IfngR1−/− BM chimeras on day 12 after PR8 infection, and RNA-seq was performed. (A) PCA of normalized gene expression in WT and IfngR1−/− GC B cells (B) GSEA for the memory B cell gene signature (Table S2) in WT vs. IfngR1−/− GC B cells. (C) Volcano plot highlighting genes differentially expressed in WT vs. IfngR1−/− GC B cells. Three replicates for each cell type were obtained from three independent experiments. (D) Frequency of CXCR3hi cells within WT and IfngR1−/− GC B cells from PR8-infected WT/IfngR1−/− BM chimeras on day 12 after infection. Data are representative of three independent experiments (n=5-6 mice/time point). P values were determined using a two-tailed Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant. (E-F) CXCR3hi and CXCR3hi GC B cells were sorted from the med-LN of day 12 PR8-infected B6 mice, and RNA-seq was performed. GSEA for the GC IFNγ-induced program (Table S1) (E), the memory B cell gene signature (Table S2) (F), and the lung-BRM gene signature 4 (G) in CXCR3hi vs CXCR3hi GC B cells. Three replicates for each cell type were obtained from three independent experiments. (H-K) Class-switched GC B cells from the med-LN of PR8-infected B6 mice and paired lung-BRMs (CXCR3+class-swicthed memory B cells) were sorted on day 30 and RNA-seq was performed. (H) IPA showing the activation Z-score for the top -ranked upstream regulators in BRM vs. GC B cells. (I) Top hallmark pathways significantly enriched in the BRM transcriptome. GSEA for the hallmark IFNγ signatures (J) and the IFNγ-induced program (Table S1) in BRM vs. GC B cells. Three replicates for each cell type were obtained from three independent experiments.
Next, to identify transcripts differentially expressed in lung-BRMs relative to GC B cells, we infected B6 mice with influenza. On day 30 after infection, we sorted class-switched lung-BRMs from the lungs and paired GC B cells from the med-LN, and performed RNA-seq. We identified a total of 399 genes that were significantly increased in the lung-BRMs compared to GC B cells (Table S2). We will refer to this list as the “memory B cell gene signature.” As expected, the expression of genes implicated in memory B cell differentiation and survival 17, such as Zbtb32, Klf2, Hhex, Ski, or Bcl2, were increased in lung-BRMs relative to GC B cells (Figure. S5A). On the contrary, the expression of transcripts characteristically expressed by GC B cells 17,38, such as Aicda or Bcl6, were decreased in lung-BRMs compared to GC B cells (Figure. S5A). As predicted, previously published gene expression signatures of memory B cells were significantly enriched in the transcriptome of lung-BRMs relative to GC B cells counterparts (Figure. S5B). Thus, lung-BRMs were bona fide memory B cells. Importantly, Gene Set Enrichment Analysis (GSEA) revealed that the “memory B cell gene signature” (Table S2) was significantly enriched in the WT relative to the IfngR1−/− GC B cell transcriptome (Figure. 5B). These results indicate that IFNγ signaling in GC B cells promotes the expression of a transcriptional program related to lung-BRMs.
To further confirm that the lung-BRM transcriptional program was initiated in GC B cells in response to IFNγ signaling, we sought to identify IFNγ-responding GC B cells. Additional analysis of the day 12 WT and IfngR1−/− GC B cell transcriptomes revealed that Cxcr3 was highly expressed in WT compared to IfngR1−/− GC B cells (Figure. 5C). In agreement with the transcriptional data, a fraction of the WT but not the IfngR1−/− GC B cells expressed CXCR3 early after infection (Figure. 5D). Similar results were obtained when we analyzed CXCR3 expression in WT and Stat1−/− GC B cells in the WT/IfngR1−/− chimeras (Figure. S5C). These results demonstrate that CXCR3 is expressed in GC B cells in response to IFNγ signaling. Thus, CXCR3 expression identifies a population of IFNγ-responding GC B cells.
To further characterize the CXCR3hiGC B cell subset, we sorted class-switched CXCR3hi and CXCR3lo GC B cells from day 12 infected B6 mice and performed RNA-seq. As predicted, the “GC IFNγ-induced program” (Table S1) was enriched in CXCR3hi compared to CXCR3loGC B cells (Figure. 5E), which is consistent with the notion that CXCR3hiGC B cells were IFNγ-responding GC B cells. Importantly, the expression of the “memory B cell gene signature” (Figure. 5F) and a previously published lung-BRM transcriptional signature4 were also significantly increased in CXCR3hi relative to CXCR3lo GC B cells (Figure. 5G). These data further support the conclusion that the lung-BRM transcriptional program is initiated in GC B cells in response to IFNγ signaling.
Finally, we used Ingenuity Pathway Analysis (IPA) to identify upstream regulators of the memory B cell gene signature. IPA ranked IFNγ as the second highest upstream regulator of the memory B cell gene signature (Figure. 5H). Corresponding with the IPA prediction, the IFNγ response signaling pathway 39 was one of the top three significantly enriched mSigDB Hallmark pathways in lung-BRM compared to the GC B cell transcriptome (Figure. 5I and J) 40. Similarly, the “GC IFNγ-induced program” (Table S1) was significantly enriched in the lung-BRMs compared to GC B cells counterparts (Figure. 5K). Collectively, these results indicate that IFNγ signaling skews the GC B cell transcriptional program towards the lung-BRM differentiation pathway.
IFNγ signaling is required for the differentiation of CXCR3+pre-MEMs
GC B cells poised to become memory express a characteristic pre-memory precursor (pre-MEM) transcriptional program 18–21. Previously published pre-MEM transcriptional signatures 18,21 were significantly increased in the WT relative to the IfngR1−/− GC B cells (Figure. 6A). We also found that the pre-MEM signatures were enriched in the IFNγ-responding CXCR3hi GC B cells compared to CXCR3lo GC B cells (Figure. 6B).
Figure 6. Intrinsic IFNγ signaling is required for the differentiation of pre-MEMs.
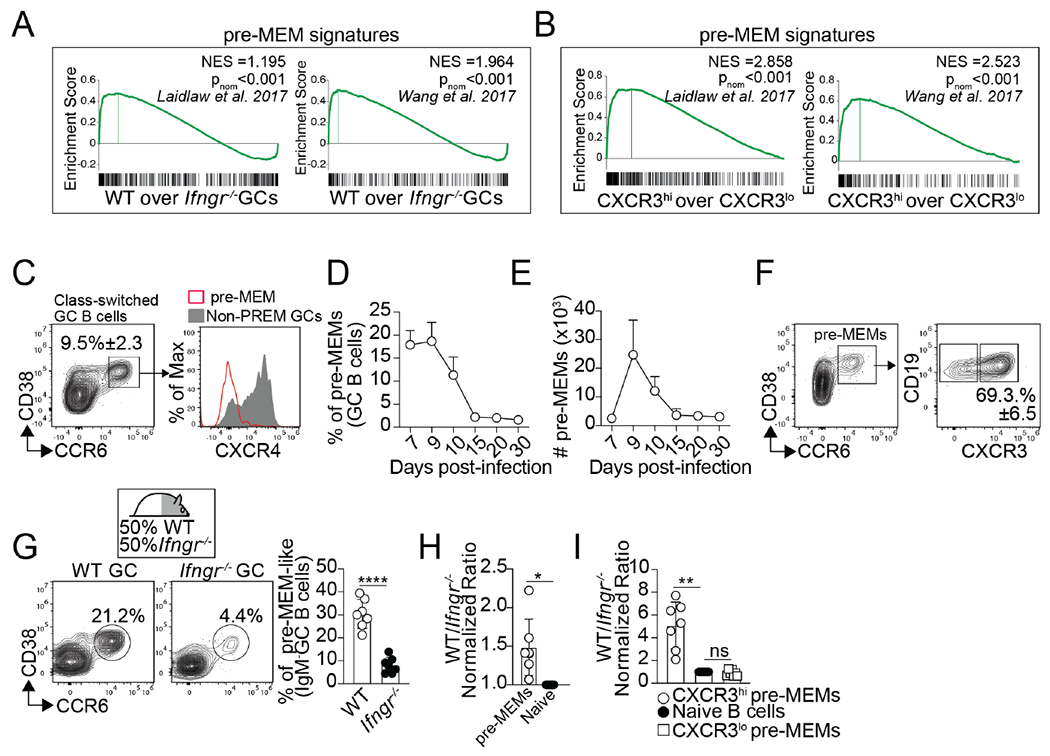
(A) WT and IfngR1−/− GC B cells were sorted from WT/IfngR1−/− BM chimeras on day 12 after PR8 infection and RNA-seq was performed. GSEA for the indicated pre-MEM gene signatures in WT vs. IfngR1−/− GC B cells. Three replicates for each cell type were obtained from three independent experiments. (B) CXCR3hi and CXCR3hi GC B cells were sorted from day 12 PR8-infected B6 mice and RNA-seq was performed. GSEA for the indicated pre-MEM gene signatures in CXCR3hi vs. CXCR3hi GC B cells. Three replicates for each cell type were obtained from three independent experiments. (C-F) B6 were infected with PR8. (C) Gating strategy for the identification of pre-MEMs. Frequency (D) and number (E) of pre-MEMs in the med-LN. (F) Expression of CXCR3 in pre-MEMs on day 10. Plots gated on CD19+CD138−IgD−IgM−FAS+ GC are shown. Data are representative of three independent experiments (n=5 mice/time point). (G) Frequency of pre-MEMs within B6 and IfngR1−/− GC B cells from the med-LN of day 10 PR8-infected WT/IfngR1−/− BM chimeras. (H) Ratio of B6 to IfngR1−/− pre-MEMs. (I) Ratio of B6 to IfngR1−/− CXCR3+ and CXCR3− pre-MEMs. Data are representative of three independent experiments (n=7 mice). P values were determined using a two-tailed Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant.
Studies show that pre-MEMs are CD38+CCR6+CXCR4lo GC B cells 18–21. Class-switched GC B cells with a pre-MEM-like phenotype were easily identified in B6 mice early after infection (Figure. 6C). The frequency (Figure. 6D) and number (Figure. 6E) of pre-MEMs progressively declined after day 10. Further phenotypic characterization revealed that pre-MEM cell could be separated into CXCR3lo and CXCR3hi cells, being the majority (70%) CXCR3hipre-MEMs (Figure. 6F). Importantly, in a reverse analysis, we found that, when gated on CXCR3lo and CXCR3hiGC B cells, only 5% of the CXCR3loGC B cells had a pre-MEM phenotype (Figure. S6A). In contrast, nearly 60% of the IFNγ-responding CXCR3hiGC B cells were pre-MEMs, thereby explaining the enrichment of the pre-MEM signatures in the CXCR3hi relative to the CXCR3lo GC B cell transcriptome (Figure. 6B). These results indicate that pre-MEMs were easily identified after influenza infection and could be divided into CXCR3lo and CXCR3hi cells.
To investigate whether IFN-γ signaling affected the differentiation of pre-MEMs, we enumerated pre-MEM cells in our WT/ IfngR1−/− BM chimeras. The frequency of pre-MEMs was significantly reduced in the IfngR1−/− compared to the WT GC B cell compartment (Figure. 6G). As a result, the WT to IfngR1−/− ratio of pre-MEMs was significantly increased compared to the ratio of naïve B cells (Figure. 6H). Similar results were obtained when we enumerated NP-specific pre-MEMs (Figure. S6B). Importantly, the lack pre-MEMs in the IfngR1−/− GC B cell compartment was due to the lack of CXCR3hipre-MEMs, as WT and IfngR1−/− CXCR3lo pre-MEMs similarly accumulated (Figure. 6I). Similar results were obtained when we enumerated WT and Stat1−/− pre-MEM-like cells in our WT/Stat1−/− BM chimeras (Figure. S6C–E). These data suggest that CXCR3hiexpression identifies a population of pre-MEM cells that are generated in response to IFNγ-STAT1 signaling, likely representing a population of GC-derived lung-BRM precursors.
T-bet expression in response to IFNγ is required for CXCR3+pre-MEMs and lung-BRM differentiation
Further analysis of the WT and IfngR1−/− GC B cell transcriptomes revealed that Tbx21 (the gene encoding T-bet) was highly expressed in WT compared to IfngR1−/− GC B cells (Figure. 7A). In agreement with the transcriptional differences, IfngR1−/− GC B cells failed to express T-bet compared to WT GC B cells (Figure.7B). These results indicate that T-bet was expressed in GC B cells in response to IFNγ at the peak of the infection. However, as the immune response progresses, T-bet expression progressively decreased (Figure. 7C).
Figure 7. T-bet expression in response to IFNγ signaling is required for pre-MEM and lung-BRM differentiation after influenza infection.
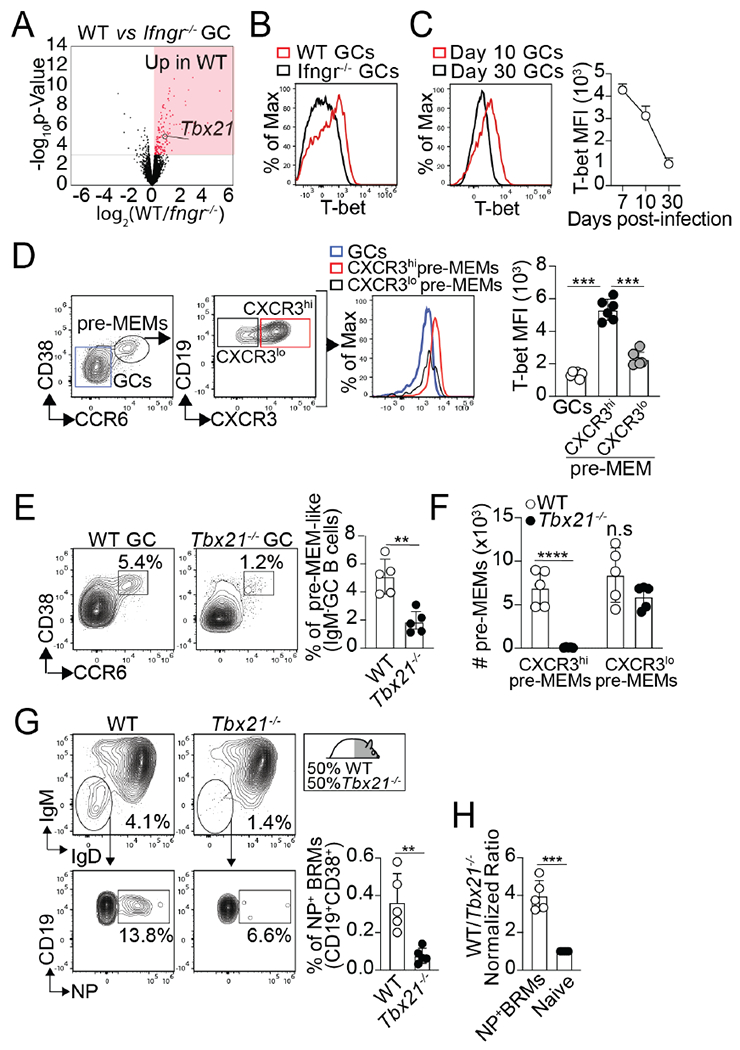
(A) Volcano plot highlighting DEGs in WT vs. IfngR1−/− GC B cells from day 12 PR8-infected WT/IfngR1−/− BM chimeras. Three replicates for each cell type were obtained from three independent experiments. (B) T-bet expression in WT and IfngR1−/− GC B cells from day 10 PR8-infected WT/IfngR1−/− BM chimeras. Data are representative of three independent experiments. (C) T-bet expression in GC B cells from PR8-infected B6 mice at the indicated time points. Data are representative of three independent experiments (n=5-7 mice). (D) T-bet expression in CXCR3hi and CXCR3lo pre-MEMs from day 10 PR8-infected B6 mice. Data are representative of three independent experiments (n=5-6 mice). P values were determined by one-way ANOVA with a post-hoc Kruskal–Wallis comparison test. (E-F) B6 and Tbx21−/− mice were infected with PR8 and GC B cells from the med-LN were analyzed on day 10. (E) Frequency of pre-MEMs. (F) Number of CXCR3+ and CXCR3− pre-MEMs. Data are representative of two independent experiments (n=5mice). P values were determined using a two-tailed Student’s t-test. (G and H) WT/Tbx21−/− BM chimeras were infected with PR8. (G) Frequency of NP-specific BRMs within the B6 and Tbx21−/− compartments in the lungs on day 30. (H) Ratio of B6 to Tbx21−/− naïve B cells and NP-specific BRMs. Data are representative of three independent experiments (n=5-6 mice). P values were determined using a two-tailed Student’s t-test. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant.
We next examined T-bet expression in pre-MEMs. We found that CXCR3hipre-MEMs expressed high levels of T-bet compared to CXCR3lopre-MEMs and conventional GC B cells (Figure. 7D). To test whether T-bet was required for the differentiation of pre-MEMs, we infected WT and Tbx21−/− mice with influenza, and enumerated pre-MEMs on day 10 after infection (Figure. 7E and F). The frequency (Figure. S7A) and number (Figure. S7B) of total GC B cells were similar in both groups. However, the frequency of GC B cells with a pre-MEM phenotype was significantly reduced in the Tbx21−/− compared to control mice (Figure. 7E). Importantly, when separated into CXCR3lo and CXCR3hi pre-MEMs, we found no differences in the number of CXCR3lopre-MEMs (Figure. 7F). In contrast, CXCR3hipre-MEMs failed to accumulate in Tbx21−/− compared to WT mice (Figure. 7F). These results indicate that T-bet expression was required for the normal CXCR3hi pre-MEM responses.
Finally, to test whether the lack of CXCR3hipre-MEMs in Tbx21−/− GC B cells correlated with defects in the influenza-specific lung-BRM response, we enumerated NP-specific BRMs in the lung of influenza-infected WT/Tbx21−/− BM chimeras. The frequency of NP-specific BRMs was significantly decreased in the Tbx21−/− compared to the WT compartment at all time points after infection (Figure. 7G and Data not shown). Thus, the WT to Tbx21−/− ratio of total and NP-specific class-switched BRMs was significantly increased compared to the ratio of naïve B cells (Figure. 7H). Altogether, these results indicate that T-bet expression in GC B cells in response to IFNγ signaling is required for the differentiation of CXCR3hipre-MEMs and the subsequent development of influenza-specific lung-BRM cell responses.
DISCUSSION
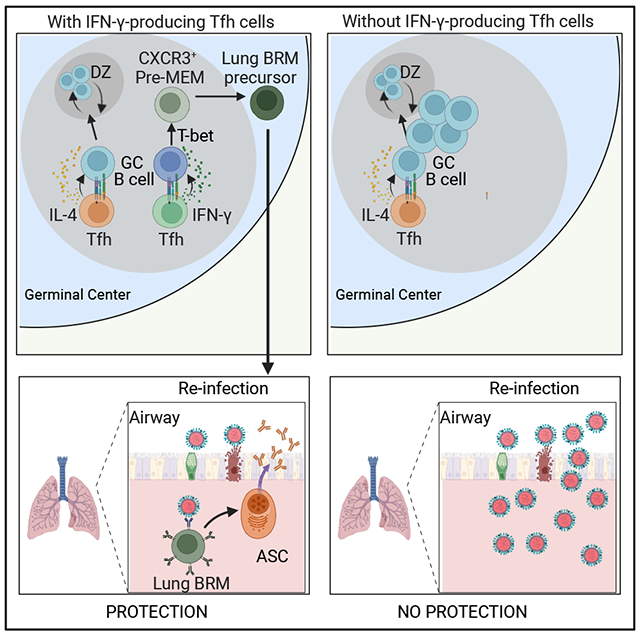
Studies indicate that lung-BRMs derive from the GC developing in the secondary lymphoid organs 1,4,8,10. However, the mechanisms that control lung-BRM responses are unknown. We show here that intrinsic IFN-γ signaling in GC B cells was required for the development of lung-BRMs after influenza virus infection. As such, when B cells were unable to respond to IFN-γ, the influenza-specific BRM response was significantly reduced. IFN-γ was provided to GC B cells by Tfh cells. Correspondently, in the absence of IFN-γ-producing Tfh cells, lung-BRMs were not generated, and lung-BRM-mediated protection after heterologous re-challenge was compromised. Thus, our results identify IFN-γ as a critical regulator of lung-BRM differentiation and demonstrate that IFN-γ-producing Tfh cells are essential for generating protective lung-BRM responses after influenza virus infection.
The lack of lung-BRMs cannot be attributed to the lack of GC B cells, as GC B cells largely accrue in the med-LN without IFN-γ signaling. Instead, our findings suggest a model where the generation of lung-BRMs is impaired in the absence of IFN-γ signaling due to the lack CXCR3+pre-MEMs in the med-LN. Mechanistically, intrinsic IFN-γ-STAT1 signaling in GC B cells promotes T-bet expression, which is necessary for the differentiation of CXCR3+pre-MEMs. Subsequently, CXCR3+pre-MEMs differentiate into CXCR3+memory B cells, which egress the med-LN and home to the lung to become lung-BRMs. In this proposed model, CXCR3+ memory B cells in the med-LN are the precursors of lung-BRMs. However, it is also plausible that CXCR3+ memory B cells and lung-BRMs represent two distinct subsets that originate from CXCR3+ pre-MEM precursors independently of each other. Regardless of the developmental relationship between CXCR3+ memory B cells and lung-BRMs, our data indicate that the absence of CXCR3+ pre-MEMs is the fundamental cause underlying the impaired lung-BRM response observed in the absence of IFN-γ signaling.
The differentiation of IFN-γ-producing Tfh cells depended on T-bet, which promoted IFN-γ production while hindering IL-4 secretion. However, T-bet expression in Tfh cells was transient. Thus, as the inflammation subsided, T-bet expression gradually diminished, and Tfh cells progressively switched from secreting IFN-γ to secreting IL-4. Previous studies 1,4 and our research demonstrate that lung-BRMs are preferentially generated within the initial three weeks of infection. The transient nature of the IFN-γ-producing Tfh cell response provides a plausible explanation for the temporality of lung-BRM differentiation. We propose that the interaction between GC B cells and IFN-γ-producing Tfh cells transiently skews the responding GC B cells toward the CXCR3+pre-MEM differentiation pathway, thus facilitating lung-BRM formation early after infection. However, as Tfh cells cease IFN-γ production and begin to secrete IL-4, GC B cells fail to initiate the CXCR3+pre-MEM program, resulting in a gradual decline of lung-BRM progenitors and the stabilization of the GC fate. In this model, T-bet is a critical regulator that controls a time-dependent switch in Tfh cell functionality and fine-tunes the output of the GC response throughout the infection.
CXCR3 is required for the migration of NK cells and T cells to the lung 41. Since CXCR3 is expressed in response to IFN-γ, one possible explanation for the impaired lung-BRM responses in the absence of IFN-γ signaling could be the inability of lung-BRM precursors to migrate to the lung due to the lack of CXCR3. However, previous studies demonstrate that CXCR3 is dispensable for lung-BRM responses 4,9. Hence, the absence of CXCR3 expression in lung-BRM precursors is unlikely to be the primary mechanism responsible for the lack of lung-BRMs when IFN-γ signaling is absent. Instead, our data suggest that CXCR3 identifies a transcriptionally distinct population of pre-MEMs that are generated in response to IFN-γ and are the precursors of lung-BRMs.
T-bet physically associates with Bcl6 leading to the formation of T-bet-Bcl6 complexes that direct Bcl6 to a different set of promoters in T cells, resulting in different transcriptional patterns depending on the relative abundance of these two transcription factors 42. The molecular mechanism by which the IFN-γ-STAT1-T-bet axis promotes lung-BRM responses remains to be elucidated. One possibility is that, similar to T cells, the induction of T-bet expression in GC B cells in response to IFN-γ alters the transcriptional output of Bcl6, thereby influencing GC B cell fate at multiple levels. For example, positioning of memory B cell precursors in the periphery of the light zone, away from the follicular dendritic cell network, contributes to memory B cell differentiation by limiting the amount of Tfh cell help received, a prerequisite for Bcl6 downregulation and memory formation 18–20,43. The location of GC B cells at the GC border depends on the expression of sphingosine 1-phosphate receptor 1 (S1PR1), which is also required for LN egress 44. Bcl6 inhibits S1PR1 expression and promotes the expression of S1PR2, restricting the positioning of GC B cells at the GC border 45. In contrast, STAT1 is a positive regulator of S1PR146. Thus, by antagonizing Bcl6-mediated inhibition of S1PR1 expression, the IFN-γ-STAT1-T-bet axis may license the positioning of CXCR3+pre-MENs in specialized LN microenvironments prone to memory B cell differentiation and LN egress. Additionally, STAT1 may further contribute to biasing the GC B cell response towards the memory differentiation pathway by promoting cell cycle arrest, which is necessary for memory B cell formation 47. All these possibilities are consistent with the idea that limiting Bcl6 activity is a prerequisite for memory formation 20,43.
In contrast to IFN-γ, IL-4 signaling supports Bcl6 expression and GC maintenance 48, while inhibiting the differentiation of pre-MEMs 49. Thus, an additional non-mutually excluding possibility is that IFN-γ-STAT1 signaling contributes to lung-BRM differentiation by counteracting the inhibitory effects of IL-4 signaling in memory B cell formation. We propose a division of labor model in which, while IFN-γ signaling promotes memory B cell development at the expense of GC maintenance, IL-4 favors the continuation of the GC fate, hindering memory B cell formation. Consequently, in the absence of IFN-γ signaling, GC B cells continue their GC fate and accumulate in the med-LN. However, these possibilities are speculative and further studies are required to elucidate the precise role of IFN-γ-STAT1-T-bet signaling in the differentiation of memory B cell precursors and the development of lung-BRMs.
IL-9 is required for the development of pre-MEMs and can activate STAT121. It will be important to determine whether, in addition to IFN-γ, IL-9 and other STAT1-activating cytokines influence lung-BRM responses. Given that different pathogens, adjuvants, or allergens are likely to prime diverse populations of cytokine-producing Tfh cells, understanding the relative contribution and importance of varying STAT1-activating cytokines in the generation of lung-BRM responses will require further research using different models of infection or immunization.
In summary, our data evidence a critical role for IFN-γ-producing Tfh cells in generating lung-BRM responses and provide new insights into the mechanisms that fine-tune GC B cell fate decisions after influenza virus infection. This knowledge is essential for designing new vaccine strategies tailored to elicit potent lung-BRM responses, which have the potential to generate enhanced cross-protection to escape variants.
LIMITATIONS OF THE STUDY
In this study, we show that IFN-γ-production by Tfh cells is critically required for the differentiation of lung-BRM cells after influenza infection. Collectively our data demonstrate an essential function of Tfh cells in establishing pulmonary immunity and identifies IFN-γ as a critical regulator of lung-BRM differentiation. However, the precise molecular mechanism by which the IFN-γ-STAT1-T-bet axes regulated lung-BRM differentiation remains undefined. Future work will also be needed to determine whether IFN-γ signaling is also required for differentiating BRM residing in tissues other than the lungs. Similarly, further studies are necessary to better characterize the IFN-γ-producing Tfh cells. For example, given that IFN-γ-producing Tfh cells express high levels of CXCR3, they are likely to localize in different GC microenvironments compared to CXCR3lo Tfh cells. Finally, in this study, we primarily focused on the role of IFN-γ-producing Tfh cells in regulating lung-BRM differentiation. Thus, the exact role of IL-4-producing Tfh was not determined.
STAR ★METHODS
RESOURCE AVAILABILITY
Lead contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Andre Ballesteros-Tato (andreballesterostato@uabmc.edu).
Materials availability
Unique reagents and non-commercially available mice generated in this study will be made available upon request, subject to availability and completion of a materials transfer agreement.
Data and code availability.
The datasets generated for this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE208322. Additional information and scripts for data processing and analysis of RNA sequencing reported in this paper are available on GitHub (https://github.com/afr-uab/Arroyo2023).
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice.
C57BL/6 (B6), B6.SJL-Ptprca Pepcb/BoyJ (B6.CD45.1), B6.129P2-Tcrβtm1MomTcrδtm1Mom (Tcrb−/−Tcrd−/−),C57BL/6-Tg(TcraTcrb)425Cbn/J (OTII), B6.129S6-Tbx21tm1Glm/J (B6. Tbx21−/−), B6.129S7-Ifngr1tm1Agt/J (Ifngr1−/−), B6.129P2-Cxcr3tm1Dgen/J, B6.129S7-Ifngtm1Ts/J (Ifng−/−), B6.129S(Cg)-Stat1tm1Dlv/J (Stat1−/−), and Tg(Cd4-cre)1Cwi/BfluJ (Cd4-cre) were originally purchased obtained from The Jackson Laboratories. B6.Bcl6fl/fl mice 50 were originally obtained from Dr. Changchun Xiao, Scripps Research Institute and crossed to CD4-cre mice to generate Tfh-deficient (B6.Bcl6fl/fl/Cd4cre/+) mice. B6.4get mice were originally obtained from Dr. M. Mohrs (Trudeau Institute). OTII mice were crossed to B6. Tbx21−/− and B6.4get to obtain Tbx21−/− OTII-4get mice (CD45.1−CD45.2+). OTII mice were crossed to B6.CD45.1and B6.4get mice to generate WT-OTII-4get (CD45.1+CD45.2+) mice. All mice were bred in the University of Alabama at Birmingham (UAB) animal facility. Experiments were equally performed with eight-week-old male and female mice. All experimental procedures involving animals were approved by the UAB Institutional Animal Care and Use Committee and were performed according to guidelines outlined by the National Research Council.
METHOD DETAILS
Infections
Primary influenza virus infections were performed intranasally (i.n) with 9,750 viral focal units (VFU) of A/PR8/34 (PR8) 51administered in 100μl of PBS or with 500 VFU of PR8-OTII 52 influenza virus in 100μl of PBS. Secondary infections were performed with 125,000VFU of X31 influenza virus administered in 100μl of PBS 51.
BM chimeras
BM chimeric mice were generated by irradiating the indicated recipient mice with 950 Rads from an X-ray source delivered in two equal doses of 475 Rads administered 4-5 hours apart. Following irradiation, mice were intravenously injected with 5 × 106 cells of the indicated mix of BM cells. Mice were allowed to reconstitute for 8-10 weeks before infection. The normalized ratios of the indicated target populations were calculated by dividing the CD45.1+ to CD45.2+ ratio of the target cells by the CD45.1+ to CD45.2+ ratio of naïve B cells.
Adoptive transfers and in vivo treatments
In some experiments, mice were intraperitoneally treated with 250 μg anti-CD154 (MR-1) obtained from BioXcell. For the adoptive transfer experiments, CD4+ OTII cells were purified from the spleens of naïve WT-OTII-4get (CD45.1+CD45.2+) and Tbx21−/− OTII-4get mice (CD45.1−CD45.2+) by positive selection with the EasySep™ CD4+ positive selection kit (STEMCELL Technologies). Purified OTII cells (2.5× 104) were transferred i.v into B6.CD45.1+ mice. One day later, the recipient mice were infected with PR8-OTII influenza virus.
Cell preparation and flow cytometry.
Lungs were harvested and cut into small fragments. The ensuing tissue was digested for 40 min at 37°C with 0.6 mg/ml collagenase A (Sigma) and 30 μg/ml DNAse I (Sigma) in RPMI-1640 medium (GIBCO). Digested lungs, LNs, and spleens were mechanically disrupted to obtained single cell suspensions. Lung red blood cells were lysed with 150 mM NH4Cl, 10 mM KHCO3 and 0.1 mM EDTA for 5 minutes. Cells were washed and then filtered through a 70 μm nylon strainer. Cells were resuspended and stained with fluorochrome-conjugated antibodies diluted in PBS with 2% donor calf serum. Biotin-conjugated primary antibodies were detected with fluorochrome-labeled streptavidin from BD Biosciences. Dead cell exclusion was performed using 7-AAD (Calbiochem). Intracellular staining for cytokines was performed using BD Cytofix/Cytoperm™ Fixation and Permeabilization Solution (BD Biosciences) following the manufacturer’s instructions. Intracellular staining for transcription factors was performed using the mouse regulatory T cell staining kit (eBioscience). Flow cytometry was performed using an Attune NxT Flow Cytometer (ThermoFischer Scientific). The NP-specific B cell tetramers were prepared as previously described 1,53. The IA bNP311-325 MHC class II tetramer was obtained from the NIH Tetramer Core Facility. The specific fluorochrome-conjugated antibodies used for this work are listed in the key resource table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD19 APC Cy7, Clone 1D3 | BD Biosciences | Cat# 557655, RRID:AB_396770 |
| Anti-CD45.1 APC, Clone A20 | BD Biosciences | Cat# 558701, RRID:AB_1645214 |
| Anti-CD45.1 APC R700, Clone A20 | BD Biosciences | Cat# 565813, RRID:AB_2744397 |
| Anti-CD45.1 BV510, Clone A20 | BD Biosciences | Cat# 565278, RRID:AB_2739150 |
| Anti-CD45.1 PECy7, Clone A20 | BD Biosciences | Cat# 560578, RRID:AB_1727488 |
| Anti-CD45.2 PECy7, Clone 104 | BD Biosciences | Cat# 560696, RRID:AB_1727494 |
| Anti-CD45.2 PE, Clone 104 | BD Biosciences | Cat# 560695, RRID:AB_1727493 |
| Anti-CD45.2 BV711, Clone 104 | BD Biosciences | Cat# 563685, RRID:AB_2738374 |
| Anti-Bcl6 PE, Clone K112-91 | BD Biosciences | Cat# 561522, RRID:AB_10717126 |
| Anti-CD138 BV510, Clone 281-2 | BD Biosciences | Cat# 558626, RRID:AB_1645216 |
| Anti-CD38 APC, Clone 90/CD38 | BD Biosciences | Cat# 102711, RRID:AB_312932 |
| Anti-CD4 BV711, Clone RM4-5 | BD Biosciences | Cat# 563726, RRID:AB_2738389 |
| Anti-CD4 PE, Clone RM4-5 | BD Biosciences | Cat# 553049, RRID:AB_394585 |
| Anti-CD95 BV421, Clone Jo2 | BD Biosciences | Cat# 562633, RRID:AB_2737690 |
| Anti-CD95 FITC, Clone Jo2 | BD Biosciences | Cat# 554257, RRID:AB_395329 |
| Anti-CD95 APC R700, Clone Jo2 | BD Biosciences | Cat# 565130, RRID:AB_2739078 |
| Anti-CXCR3 BV421, Clone CXCR3-173 | BioLegend | Cat# 126522, RRID:AB_2562205 |
| Anti-CXCR3 PECy7, Clone CXCR3-173 | BioLegend | Cat# 126516, RRID:AB_2245493 |
| Anti-CXCR5 Biotin, Clone 2G8 | BD Biosciences | Cat# 551960, RRID:AB_394301 |
| Anti-CXCR5 Biotin, Clone SPRCL5 | eBioscience | Cat# 13-7185-82, RRID:AB_2572800 |
| Anti-IgD FITC, Clone 11-26c.2a | BD Biosciences | Cat# 562022, RRID:AB_10894208 |
| Anti-IgM PECy7, Clone ll / 41 | eBioscience | Cat# 25-5790-82, RRID:AB_469655 |
| Anti-IFNγ PECy7, Clone XMG1.2 | BD Biosciences | Cat# 557649, RRID:AB_396766 |
| Anti-PD1 FITC, Clone J43 | eBioscience | Cat# 11-9985-85, RRID:AB_465473 |
| Anti-PD1 eFluorTM 450, Clone J43 | eBioscience | Cat# 48-9985-82, RRID:AB_2574139 |
| Anti-T-bet PECy7, Clone 4B10 | BioLegend | Cat# 644823, RRID:AB_2561760 |
| Anti-T-bet APC, Clone 4B10 | BioLegend | Cat# 644814, RRID:AB_10901173 |
| Anti-CD184 (CXCR4) PE, Clone 2B11 | BD Biosciences | 551966, RRID:AB_394305 |
| Anti-CD196 (CCR6) Alexa Fluor 647, Clone140706 | BD Biosciences | 557976, RRID:AB_2228793 |
| Anti-CD196 (CCR6) BV510, Clone140706 | BD Biosciences | Cat# 747832, RRID:AB_2872295 |
| anti-CD154, Clone MR-1 | BioXCell | Cat# BE0017-1, RRID:AB_1107601 |
| Streptavidin, V500 | BD Biosciences | Cat# 561419, RRID:AB_10611863 |
| HRP-conjugated goat Anti-Mouse IgG | Southern Biotech | Cat# 1030-05, RRID:AB_2619742 |
| HRP-conjugated Goat Anti-Mouse IgG2c | Southern Biotech | 1079-05, RRID:AB_2794466 |
| Bacterial and Virus Strains | ||
| PR8-OTII | Lee et al., 200551 | N/A |
| X31 | Lee et al., 200551 | N/A |
| A/PR8/34 (PR8) | Thomas et al., 201652 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PMA | Sigma-Aldrich | Cat# P8139-1MG |
| Calcimycin | Life Technologies | Cat# A1493 |
| Brefeldin-A | Sigma-Aldrich | Cat# B7651-5MG |
| DNAse I | Qiagen | Cat# 79254 |
| 7-AAD | Calbiochem | Cat# 12-993-51 |
| Recombinant NP tetramer | Allie et al., 2019 1 | N/A |
| Alkaline phosphatase substrate BCIP/NBT | Moss Substrates | Cat#NBIM-1000 |
| Critical Commercial Assays | ||
| EasySep™ CD4+ positive selection kit | STEMCELL Technologies | Cat# 18952 |
| BD Cytofix/Cytoperm™ Fixation and Permeabilization Solution | BD Biosciences | Cat# 554722 |
| Norgen Single Cell RNA Purification Kit | Norgen Biotek | Cat# 51800 |
| Transcription factor staining buffer set | eBioscience | Cat# 00-5523-00 |
| Deposited Data | ||
| RNA-seq data | This paper | GSE208322. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE208322 |
| Public pre-MEM signature | Laidlaw et al., 2017 18 | GSE89897 |
| Public pre-MEM signature | Wang et al., 2017 21 | GSE85018. |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 (B6) | The Jackson Laboratories | Strain #:000664 RRID:IMSR_JAX:000664 |
| Mouse: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratories | Strain #:002014 RRID:IMSR_JAX:002014 |
| Mouse: B6.129P2-Tcrβtm1MomTcrδtm1Mom | The Jackson Laboratories | N/A |
| Mouse: C57BL/6-Tg(TcraTcrb)425Cbn/J | The Jackson Laboratories | Strain #:004194 RRID:IMSR_JAX:004194 |
| Mouse: B6.129S6-Tbx21tm1Glm/J | The Jackson Laboratories | Strain #:004648 RRID:IMSR_JAX:004648 |
| Mouse: B6.129S7-Ifngr1tm1Agt/J | The Jackson Laboratories | Strain #:003288 RRID:IMSR_JAX:003288 |
| Mouse: B6.129P2-Cxcr3tm1Dgen/J | The Jackson Laboratories | Strain #:005796 RRID:IMSR_JAX:005796 |
| Mouse: B6.129S7-Ifngtm1Ts/J | The Jackson Laboratories | Strain #:002287 RRID:IMSR_JAX:002287 |
| Mouse: B6.129S(Cg)-Stat1tm1Dlv/J | The Jackson Laboratories | Strain #:012606 RRID:IMSR_JAX:012606 |
| Mouse: Tg(Cd4-cre)1Cwi/BfluJ | The Jackson Laboratories | Strain #:017336 RRID:IMSR_JAX:017336 |
| Mouse: B6.Bcl6fl/fl | Laboratory of Dr. Changchun Xiao (Scripps Research Institute) 50 | N/A |
| Mouse: B6.129-Il4tm1Lky/J (B6.4get IL-4 reporter mice) | Laboratory of Dr. M. Mohrs (Trudeau Institute) | N/A |
| Software and Algorithms | ||
| Prism V9 | GraphPad | https://www.graphpad.com/ |
| FlowJo V10.8.0 | TreeStar | https://www.flowjo.com |
| javaGSEA V3.0 | Broad Institute | http://www.gsea-msigdb.org/gsea/index.jsp |
| MATLAB | The Mathworks Inc. | https://www.mathworks.com/products/matlab/whatsnew.html |
| fastQC | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| Trim Galore V0.4.4. | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore |
| STAR aligner V2.5.2a | Ilumina | https://support.illumina.com/sequencing/sequencing_software/igenome.html |
| Ingenuity Pathway Analysis | Qiagen | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| R package edgeR | Robinson et al., 2019 56 | https://www.bioconductor.org |
ELISPOT
For ELISPOT assays, multiscreen 96 well plates (MAHAS4510, Millipore) were coated with 10ug of purified NP antigen in PBS overnight at 4 °C. Plates were washed with PBS and blocked with complete medium (RPMI supplemented with 10% fetal bovine serum (FBS), 0.5% Penicillin (100X), 0.5% streptomycin (100X), 1% glutamine (200 mM), 1% sodium pyruvate (100 mM), 1% HEPES pH7.4 (1 M), 0.15% sodium bicarbonate, 1.2% amino acids (50X ), 1.2% non-essential amino acids (100X ), 1.2% vitamins (100X ), 0.7% glucose, 0.1% 2-mercaptoethanol (1000X, 55 mM). Lung single-cell suspensions prepared as described above were washed, diluted in complete medium and cultured on coated plates. After 5 hours, the wells were washed with PBS containing 0.5% BSA and 0.05% Tween 20, and IgG was detected using alkaline phosphatase-conjugated goat anti-mouse IgG (Jackson immunoresearch). Plates were washed with 0.2% Tween 20 in PBS and developed with BCIP/NBT (Moss Substrates). Spots were recorded using a CTL Immunospot S6 Macroplate Imager Reader (New Life Scientific) and counted manually.
ELISAs.
96-well plates (Corning Clear Polystyrene 96-Well Microplates) were coated overnight with recombinant NP protein at 1 μg/ml in 0.05 M Na2CO3 pH 9.6. Coated plates were then blocked for 1 h with 1% BSA in PBS. Serum from infected mice was collected and serially diluted (threefold) in PBS with 10 mg/ml BSA and 0.1% Tween 20 before incubation on coated plates. After washing, bound antibody was detected with HRP-conjugated goat Anti-Mouse IgG and IgG2c (Southern Biotech) and quantified by spectrophotometry at 405 nm (OD).
RNA Sequencing
Lung-BRMs (CD38+IgD−IgM−CXCR3+CD19+CD138− FAS−) from the lungs and class-switched GC B cells (CD38−IgD−IgM− CD19+CD138− FAS+) from the med-LN were sorted from day 30 infected B6 after positive selection with anti-CD19 MACS beads (Miltenyi Biotec). WT and IfngR1−/− GC were sorted from the med-LN of day 12 infected WT/IfngR1−/− mixed-bone marrow chimeras. CXCR3hi (IgD−IgM−CD19+CD138−FAS+CXCR3+) and CXCR3lo (IgD−IgM− CD19+CD138− FAS+CXCR3−) GC B cells were sorted from the med-LN of day 12 infected B6 mice. All sorting experiments were performed using a FACSAria™ (BD Biosciences) sorter in the University of Alabama at Birmingham Flow Cytometry core. All sorted populations were more than 95% pure as determined by flow cytometry. RNA was isolated from the sorted cells using the Norgen Single Cell RNA Purification Kit (Norgen Biotek). Three replicates from three independent experiments for each condition were analyzed with RNA-seq.
Primary Analysis
Library preparation and RNA sequencing was conducted through Genewiz. Libraries were sequenced using a 1x50bp single end rapid run on the HiSeq2500 platform. The quality of raw sequence fastq-formatted files was assessed using fastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Sequences were trimmed using Trim Galore using phred33 scores (version 0.4.4, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore), the paired setting and the nextera adapter option. Trimmed sequences were aligned with STAR aligner 54 (version 2.5.2a) using mouse GRCm39 genome (https://support.illumina.com/sequencing/sequencing_software/igenome.html) and default settings. The STAR genome was constructed using the GRCm39 annotation file and sjdbOverhang = 100 as recommended in the documentation. Aligned reads were counted with HTseq-count (version 0.6.1p1) 55 set for un-stranded and using the GRCm39 genes.gtf annotation file.
Downstream Analysis
The R package edgeR 56 was used to assess differential expression between pairs of groups and to generate gene-by-sample matrices for both RPKM and counts per million (CPM). Genes were considered for further analysis if their CPM were above 1 for at least three samples. Because sorted pared populations were derived from the same mouse per replicate, we used a paired model for the indicated comparisons. Additional downstream analysis and visualization (principal components, clustered heat maps, volcano plots) was performed using custom Matlab (The Mathworks Inc., Natick MA, USA) scripts (https://github.com/afr-uab/Jenkins2021). Comparison of our data to published data sets was accomplished using Gene Set Enrichment Analysis (GSEA) 40. Identification of enriched functions based on differentially expressed genes was performed using Ingenuity Pathway Analysis (Ingenuity Pathway Analysis, Qiagen, Redwood City CA, USA) 57. Genes for GSEA were ranked by −log10(p-value) times the sign of the fold change for the indicated comparisons.
QUANTIFICATION AND STATISTICAL ANALYSIS
All plots and histograms were plotted in FlowJo v.9 software (Treestar). GraphPad Prism (Version 10) was used for data analysis. The statistical significance of differences in mean values was determined using two-tailed Student’s t test or one-way ANOVA with post-hoc Tukey’s multiple comparison test. P values of less than 0.05 were considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary Material
Table S1. Genes induced in GC B cells in response to IFNγ signaling, related to Figure 5.
Table S2. Differentially expressed genes in memory B cells compared to GC B cells, related to Figure 5.
HIGHLIGHTS
B6.Bcl6fl/flCd4cre/+ mice lack lung-resident memory B cells (BRMs)
Lung-BRMs fail to differentiate in bone marrow chimeras in which Tfh cells are Ifng−/
IfngR1−/−, Tbx21−/− and Stat1−/− B cells fail to differentiate into lung-BRMs
T-bet expression in GC B cells is required for the development of lung-BRM precursors
ACKNOWLEDGEMENTS.
The authors would like to thank Thomas S. Simpler, Kelsey Browning, and Rebecca Burnham for animal husbandry. This work was supported by The University of Alabama at Birmingham (UAB) and National Institutes of Health grants 1R01 AI162698 and 1R01 AI150664 to A.B.-T, and 2R01AI116584 to B.L. N.M.A.D was supported by the National Institutes of Health (NIH) Basic Immunology and Immunological Diseases training grant T32AI007051.The X-RAD 320 unit was purchased using a Research Facility Improvement Grant, 1 G20RR022807-01, from the National Center for Research Resources, National Institutes of Health.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare that they have no competing interests.
REFERENCES
- 1.Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, Lund FE, and Randall TD (2019). The establishment of resident memory B cells in the lung requires local antigen encounter. Nat Immunol 20, 97–108. 10.1038/s41590-018-0260-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Onodera T, Takahashi Y, Yokoi Y, Ato M, Kodama Y, Hachimura S, Kurosaki T, and Kobayashi K (2012). Memory B cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc Natl Acad Sci U S A 109, 2485–2490. 10.1073/pnas.1115369109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adachi Y, Onodera T, Yamada Y, Daio R, Tsuiji M, Inoue T, Kobayashi K, Kurosaki T, Ato M, and Takahashi Y (2015). Distinct germinal center selection at local sites shapes memory B cell response to viral escape. J Exp Med 212, 1709–1723. 10.1084/jem.20142284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan HX, Juno JA, Esterbauer R, Kelly HG, Wragg KM, Konstandopoulos P, Alcantara S, Alvarado C, Jones R, Starkey G, et al. (2022). Lung-resident memory B cells established after pulmonary influenza infection display distinct transcriptional and phenotypic profiles. Sci Immunol 7, eabf5314. 10.1126/sciimmunol.abf5314. [DOI] [PubMed] [Google Scholar]
- 5.MacLean AJ, Richmond N, Koneva L, Attar M, Medina CAP, Thornton EE, Gomes AC, El-Turabi A, Bachmann MF, Rijal P, et al. (2022). Secondary influenza challenge triggers resident memory B cell migration and rapid relocation to boost antibody secretion at infected sites. Immunity 55, 718–733 e718. 10.1016/j.immuni.2022.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allie SR, and Randall TD (2020). Resident Memory B Cells. Viral Immunol 33, 282–293. 10.1089/vim.2019.0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oh JE, Song E, Moriyama M, Wong P, Zhang S, Jiang R, Strohmeier S, Kleinstein SH, Krammer F, and Iwasaki A (2021). Intranasal priming induces local lung-resident B cell populations that secrete protective mucosal antiviral IgA. Sci Immunol 6, eabj5129. 10.1126/sciimmunol.abj5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathew NR, Jayanthan JK, Smirnov IV, Robinson JL, Axelsson H, Nakka SS, Emmanouilidi A, Czarnewski P, Yewdell WT, Schon K, et al. (2021). Single-cell BCR and transcriptome analysis after influenza infection reveals spatiotemporal dynamics of antigen-specific B cells. Cell Rep 35, 109286. 10.1016/j.celrep.2021.109286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Denton AE, Innocentin S, Carr EJ, Bradford BM, Lafouresse F, Mabbott NA, Morbe U, Ludewig B, Groom JR, Good-Jacobson KL, and Linterman MA (2019). Type I interferon induces CXCL13 to support ectopic germinal center formation. J Exp Med 216, 621–637. 10.1084/jem.20181216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gregoire C, Spinelli L, Villazala-Merino S, Gil L, Holgado MP, Moussa M, Dong C, Zarubica A, Fallet M, Navarro JM, et al. (2022). Viral infection engenders bona fide and bystander subsets of lung-resident memory B cells through a permissive mechanism. Immunity 55, 1216–1233 e1219. 10.1016/j.immuni.2022.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beima KM, Miazgowicz MM, Lewis MD, Yan PS, Huang TH, and Weinmann AS (2006). T-bet binding to newly identified target gene promoters is cell type-independent but results in variable context-dependent functional effects. J Biol Chem 281, 11992–12000. 10.1074/jbc.M513613200. [DOI] [PubMed] [Google Scholar]
- 12.Knox JJ, Myles A, and Cancro MP (2019). T-bet(+) memory B cells: Generation, function, and fate. Immunol Rev 288, 149–160. 10.1111/imr.12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stone SL, Peel JN, Scharer CD, Risley CA, Chisolm DA, Schultz MD, Yu B, Ballesteros-Tato A, Wojciechowski W, Mousseau B, et al. (2019). T-bet Transcription Factor Promotes Antibody-Secreting Cell Differentiation by Limiting the Inflammatory Effects of IFN-gamma on B Cells. Immunity 50, 1172–1187 e1177. 10.1016/j.immuni.2019.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor JJ, Pape KA, and Jenkins MK (2012). A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med 209, 597–606. 10.1084/jem.20111696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Viant C, Wirthmiller T, ElTanbouly MA, Chen ST, Cipolla M, Ramos V, Oliveira TY, Stamatatos L, and Nussenzweig MC (2021). Germinal center-dependent and -independent memory B cells produced throughout the immune response. J Exp Med 218. 10.1084/jem.20202489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toyama H, Okada S, Hatano M, Takahashi Y, Takeda N, Ichii H, Takemori T, Kuroda Y, and Tokuhisa T (2002). Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity 17, 329–339. 10.1016/s1074-7613(02)00387-4. [DOI] [PubMed] [Google Scholar]
- 17.Laidlaw BJ, and Cyster JG (2021). Transcriptional regulation of memory B cell differentiation. Nat Rev Immunol 21, 209–220. 10.1038/s41577-020-00446-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laidlaw BJ, Schmidt TH, Green JA, Allen CD, Okada T, and Cyster JG (2017). The Eph-related tyrosine kinase ligand Ephrin-B1 marks germinal center and memory precursor B cells. J Exp Med 214, 639–649. 10.1084/jem.20161461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suan D, Krautler NJ, Maag JLV, Butt D, Bourne K, Hermes JR, Avery DT, Young C, Statham A, Elliott M, et al. (2017). CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immunity 47, 1142–1153 e1144. 10.1016/j.immuni.2017.11.022. [DOI] [PubMed] [Google Scholar]
- 20.Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, and Kurosaki T (2016). Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 17, 861–869. 10.1038/ni.3460. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Shi J, Yan J, Xiao Z, Hou X, Lu P, Hou S, Mao T, Liu W, Ma Y, et al. (2017). Germinal-center development of memory B cells driven by IL-9 from follicular helper T cells. Nat Immunol 18, 921–930. 10.1038/ni.3788. [DOI] [PubMed] [Google Scholar]
- 22.Crotty S (2019). T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 50, 1132–1148. 10.1016/j.immuni.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bryant VL, Ma CS, Avery DT, Li Y, Good KL, Corcoran LM, de Waal Malefyt R, and Tangye SG (2007). Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol 179, 8180–8190. 10.4049/jimmunol.179.12.8180. [DOI] [PubMed] [Google Scholar]
- 24.Mountz JD, Hsu HC, and Ballesteros-Tato A (2019). Dysregulation of T Follicular Helper Cells in Lupus. J Immunol 202, 1649–1658. 10.4049/jimmunol.1801150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinstein JS, Laidlaw BJ, Lu Y, Wang JK, Schulz VP, Li N, Herman EI, Kaech SM, Gallagher PG, and Craft J (2018). STAT4 and T-bet control follicular helper T cell development in viral infections. J Exp Med 215, 337–355. 10.1084/jem.20170457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM, and Nutt SL (2012). The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat Immunol 13, 491–498. 10.1038/ni.2261. [DOI] [PubMed] [Google Scholar]
- 27.Ballesteros-Tato A, Randall TD, Lund FE, Spolski R, Leonard WJ, and Leon B (2016). T Follicular Helper Cell Plasticity Shapes Pathogenic T Helper 2 Cell-Mediated Immunity to Inhaled House Dust Mite. Immunity 44, 259–273. 10.1016/j.immuni.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gowthaman U, Chen JS, Zhang B, Flynn WF, Lu Y, Song W, Joseph J, Gertie JA, Xu L, Collet MA, et al. (2019). Identification of a T follicular helper cell subset that drives anaphylactic IgE. Science 365. 10.1126/science.aaw6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papillion A, Powell MD, Chisolm DA, Bachus H, Fuller MJ, Weinmann AS, Villarino A, O’Shea JJ, Leon B, Oestreich KJ, and Ballesteros-Tato A (2019). Inhibition of IL-2 responsiveness by IL-6 is required for the generation of GC-TFH cells. Sci Immunol 4. 10.1126/sciimmunol.aaw7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yusuf I, Kageyama R, Monticelli L, Johnston RJ, Ditoro D, Hansen K, Barnett B, and Crotty S (2010). Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150). J Immunol 185, 190–202. 10.4049/jimmunol.0903505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith KG, Light A, Nossal GJ, and Tarlinton DM (1997). The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J 16, 2996–3006. 10.1093/emboj/16.11.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dal Porto JM, Haberman AM, Kelsoe G, and Shlomchik MJ (2002). Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J Exp Med 195, 1215–1221. 10.1084/jem.20011550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weisel FJ, Zuccarino-Catania GV, Chikina M, and Shlomchik MJ (2016). A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity 44, 116–130. 10.1016/j.immuni.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, and Ansel KM (2007). Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol 8, 145–153. 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 35.Naoe Y, Setoguchi R, Akiyama K, Muroi S, Kuroda M, Hatam F, Littman DR, and Taniuchi I (2007). Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. J Exp Med 204, 1749–1755. 10.1084/jem.20062456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas PG, Brown SA, Morris MY, Yue W, So J, Reynolds C, Webby RJ, and Doherty PC (2010). Physiological numbers of CD4+ T cells generate weak recall responses following influenza virus challenge. J Immunol 184, 1721–1727. 10.4049/jimmunol.0901427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu X, and Ivashkiv LB (2009). Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity 31, 539–550. 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Victora GD, and Nussenzweig MC (2022). Germinal Centers. Annu Rev Immunol 40, 413–442. 10.1146/annurev-immunol-120419-022408. [DOI] [PubMed] [Google Scholar]
- 39.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kohlmeier JE, Cookenham T, Miller SC, Roberts AD, Christensen JP, Thomsen AR, and Woodland DL (2009). CXCR3 directs antigen-specific effector CD4+ T cell migration to the lung during parainfluenza virus infection. J Immunol 183, 4378–4384. 10.4049/jimmunol.0902022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oestreich KJ, Mohn SE, and Weinmann AS (2012). Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat Immunol 13, 405–411. 10.1038/ni.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inoue T, Shinnakasu R, Kawai C, Ise W, Kawakami E, Sax N, Oki T, Kitamura T, Yamashita K, Fukuyama H, and Kurosaki T (2021). Exit from germinal center to become quiescent memory B cells depends on metabolic reprograming and provision of a survival signal. J Exp Med 218. 10.1084/jem.20200866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cyster JG, and Schwab SR (2012). Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol 30, 69–94. 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 45.Huang C, Gonzalez DG, Cote CM, Jiang Y, Hatzi K, Teater M, Dai K, Hla T, Haberman AM, and Melnick A (2014). The BCL6 RD2 domain governs commitment of activated B cells to form germinal centers. Cell Rep 8, 1497–1508. 10.1016/j.celrep.2014.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xin Q, Cheng G, Kong F, Ji Q, Li H, Jiang W, Wang J, Luan Y, Sun C, Chen X, et al. (2020). STAT1 transcriptionally regulates the expression of S1PR1 by binding its promoter region. Gene 736, 144417. 10.1016/j.gene.2020.144417. [DOI] [PubMed] [Google Scholar]
- 47.Avalle L, Pensa S, Regis G, Novelli F, and Poli V (2012). STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT 1, 65–72. 10.4161/jkst.20045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chevrier S, Kratina T, Emslie D, Tarlinton DM, and Corcoran LM (2017). IL4 and IL21 cooperate to induce the high Bcl6 protein level required for germinal center formation. Immunol Cell Biol 95, 925–932. 10.1038/icb.2017.71. [DOI] [PubMed] [Google Scholar]
- 49.Duan L, Liu D, Chen H, Mintz MA, Chou MY, Kotov DI, Xu Y, An J, Laidlaw BJ, and Cyster JG (2021). Follicular dendritic cells restrict interleukin-4 availability in germinal centers and foster memory B cell generation. Immunity 54, 2256–2272 e2256. 10.1016/j.immuni.2021.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang Z, Kang SG, Li Y, Zak J, Shaabani N, Deng K, Shepherd J, Bhargava R, Teijaro JR, and Xiao C (2021). IFNAR1 signaling in NK cells promotes persistent virus infection. Sci Adv 7. 10.1126/sciadv.abb8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee BO, Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Makris M, Sprague F, Lund FE, and Randall TD (2005). CD4 T cell-independent antibody response promotes resolution of primary influenza infection and helps to prevent reinfection. J Immunol 175, 5827–5838. 10.4049/jimmunol.175.9.5827. [DOI] [PubMed] [Google Scholar]
- 52.Thomas PG, Brown SA, Yue W, So J, Webby RJ, and Doherty PC (2006). An unexpected antibody response to an engineered influenza virus modifies CD8+ T cell responses. Proc Natl Acad Sci U S A 103, 2764–2769. 10.1073/pnas.0511185103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, and Randall TD (2012). Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity 36, 847–856. 10.1016/j.immuni.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kramer A, Green J, Pollard J Jr., and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes induced in GC B cells in response to IFNγ signaling, related to Figure 5.
Table S2. Differentially expressed genes in memory B cells compared to GC B cells, related to Figure 5.
Data Availability Statement
The datasets generated for this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession numbers GSE208322. Additional information and scripts for data processing and analysis of RNA sequencing reported in this paper are available on GitHub (https://github.com/afr-uab/Arroyo2023).