

Abstract
Chemical probes are essential tools for understanding biological systems and for credentialing potential biomedical targets. Programmed cell death 2 (PDCD2) is a member of the B-cell lymphoma 2 (Bcl-2) family of proteins, which are critical regulators of apoptosis. Here we report the discovery and characterization of 10e, a first-in-class small molecule degrader of PDCD2. We discovered PDCD2 degrader by serendipity using a chemical proteomics approach in contrast to the conventional approach for making bivalent degraders starting from a known binding ligand targeting the protein of interest. Using 10e as a pharmacological probe, we demonstrate that PDCD2 functions as a critical regulator of cell growth by modulating the progression of the cell cycle in T lymphoblasts. Our work provides a useful pharmacological probe for investigating PDCD2 function and highlights using chemical proteomics to discover selective small molecule degraders of unanticipated targets.
Keywords: PDCD2, Degrader, Binder, Chemical probe, Proteomics
Graphical Abstract

A first-in-class Programmed cell death protein 2 (PDCD2) degrader 10e was developed by combination of mass spectrometry-based proteomics with a concise structure-degradation relationship campaign. Our work provides valuable tool compounds for PDCD2 to the scientific community and highlights that combination of proteomics technology with chemical optimization can be a powerful approach and a useful supplement for chemical probes discovery.
Elucidating the function of all human proteins represents one of the major challenges for biology.[1] Chemical probes represent powerful tools for illuminating biological processes and aid in the validation of emerging therapeutic targets while simultaneously providing starting points for the development of drugs. The ability of chemical probes to rapidly perturb biological function make them a complementary approach to nucleic-acid based approaches such as RNAi and CRISPR.[2] Despite the tremendous promise of chemical tools, small-molecule probes are currently available for less than 4% of the human proteome.[3] [4] Historically, the discovery of chemical probes has primarily followed a target-centric approach, and often relies on knowing an enzymatic function (e.g., kinase, protease) in order to screen for inhibitors. A complementary approach involves phenotypic screening which allows for identification of unexpected pharmacology but is often impeded by the difficulty of elucidating the target and mechanism of action of the identified compounds. [5] Over the last decades, a variety of innovative technologies have emerged to identify small-molecule binders of proteins, significantly expanding the ligand repertoire of the human proteome.[6] These technologies, which include fragment-based screening, DNA-encoded libraries, and chemical proteomic methods such as activity-based protein profiling (ABPP), have led to the discovery and development of valuable chemical probes for diverse arrays of proteins.[6] Nonetheless, the development of new methods to efficiently discover and characterize probes of poorly understood protein targets is urgently needed.
A popular approach is to make bivalent small molecule degraders called PROTACs by linking a small molecule capable of binding the target protein of interest (POI) with a ligand that can recruit an E3 ligase protein.[7] The PROTAC can stabilize a transient ternary complex between the POI and the ligase, which results in ligase-dependent ubiquitination of the POI and its destruction by the proteosome. PROTACs have been successfully developed for hundreds of proteins including proteins that were previously considered ‘undruggable’.[8] However, a limitation of the conventional PROTAC approach is the need for a known binder of POI which does not exist for many protein targets. Here we demonstrate that bivalent degraders can be identified using chemical proteomics to identify unanticipated degradation targets such as programmed cell death protein 2 (PDCD2).
Programmed cell death (PDCD) is a class of proteins that function in the regulation of cell survival, cell death and regulation of the immune system.[9] The PDCD family comprises at least 12 members and exhibits widespread expression across human tissues and cell lines. Furthermore, these proteins are highly conserved across evolution underscoring their functional importance in diverse biological contexts.[9] The remarkable success of immune checkpoint inhibitors targeting programmed death protein 1 (PD1) for activating an anti-tumor immune response, which is encoded by the PDCD1 gene, has drawn considerable interest to the broader PDCD gene family.[10] Among them, PDCD2 is a zinc finger MYND domain‑containing protein that exhibits widespread expression, with particular abundance in T lymphocytes.[11] Originally identified in thymocytes undergoing programmed cell death, PDCD2 has been implicated in apoptotic regulation.[12], [13], [14] Recent studies suggest that PDCD2 may also regulate the expression of host cell factor 1 (HCF-1), potentially modulating GA-binding protein (GABP)-mediated production of the critical cytokine IL-2.[15] Moreover, the observed correlation between PDCD2 expression and CD4+/ CD8+ cell percentages implies a direct involvement of PDCD2 in immune responses.[16]
Additionally, PDCD2 was reported to control hematopoietic stem cell (HSC) differentiation during development.[17] It also functions as a ribosomal protein chaperone that facilitates ribosome assembly.[18] Studies using inducible PDCD2 knockout mice have revealed a potential role for PDCD2 in regulating the cell cycle, as loss of PDCD2 led to activation of p53 and its downstream target genes.[19] Moreover, PDCD2 has been found to interact with a variety of regulatory proteins, such as ERBB receptor feedback inhibitor 1 (ERRFI1),[20] ubiquitin protein ligase parkin,[21] indicating its involvement in a broader range of cellular processes. Dysregulation of PDCD2 has been associated with several diseases, including cancer. For example, PDCD2 is overexpressed in human acute leukemia cells and genetic knockdown of PDCD2 impairs cell proliferation,[22] suggesting PDCD2 as a potential target for new anticancer therapies. However, its functions in carcinogenesis are debatable, individual studies have reported that PDCD2 was downregulated and might function as a tumor suppressor in osteosarcoma and gastric cancer.[16],[23]. Additionally, PDCD2 is dysregulated in drug‑resistant breast cancer cells, indicating that PDCD2 may be involved in multidrug resistance.[24] Nonetheless, the physiological functions of PDCD2 have not been fully elucidated. The potential for therapeutic targeting of PDCD2 is currently unknown, as there are no potent or selective chemical probes and small molecule binders available to date. PDCD2 lacks intrinsic enzymatic activity and a well-defined ligand binding pocket. It functions via protein-protein interactions (PPIs),[18] creating unique challenges to develop a conventional antagonist. Herein, we described the serendipitous discovery and characterization of the first-in-class PDCD2 degrader based on quantitative proteomics, providing a potentially valuable chemical probe for validating PDCD2-related biological pathways and a good starting point for further optimization.
Mass spectrometry-based proteomics assays have emerged as indispensable tools in drug discovery, specifically in the domains of target identification, selectivity profiling, and characterization of biological input materials.[25] The application of proteomics has played a pivotal role in advancing the field of protein degradation. [26] [27] [28] For example, proteomics has been used to characterize bivalent kinase degraders to annotate the degradable kinome, providing a valuable resource to expedite kinase degrader development.[29] Global proteomics not only enables the comprehensive evaluation of degrader molecule selectivity across the proteome, but also presents a unique opportunity for the discovery of novel degradable targets. We designed compound XJZ-25 (Compound 1), as a bivalent compound linking a reported transcription enhancing activation domain (TEAD) palmitoylation inhibitor to thalidomide via a piperidine-containing linker (Figure 1a). We had anticipated that the compound would degrade TEAD or another protein with a similar lipid binding pocket, but surprisingly, quantitative MS proteomics identified PDCD2 as the sole downregulated protein following a 6-hour treatment of compound XJZ-25 in MOLT-4 (acute lymphoblastic leukemia) cells. (Figure 1b). To validate the degradation of PDCD2, we treated MOLT-4 cells with XJZ-25 at three different concentrations (0.01, 0.1, and 1 μM). As illustrated by Figure 1c, our findings demonstrate a dose-dependent degradation of PDCD2. After 24 hours of treatment, western blotting analysis revealed a significant reduction in PDCD2 protein levels, with a remarkable 73% decrease observed at a concentration of 1 μM.
Figure 1.

a) Chemical structure of compound 1 (XZJ-25). b) Quantitative proteomics of compound 1 showing the relative abundance of proteins in MOLT-4 cells treated for 6 h at 1 μM. c) Western blotting analysis of PDCD2 in MOLT-4 cells treated with indicated concentrations of compound 1. The data are expressed as the mean values ± SD (n = 3) and are representative of three separate experiments. All uncropped blots were presented in Figure S6.
To gain insight into the structure–degradation relationship (SDR) of PDCD2 degraders, a series of analogues of compound 1 were synthesized (Table 1). The general synthetic route of compounds 10a – 10h is illustrated in Scheme 1. Briefly, Palladium-catalyzed Suzuki coupling reaction between the commercially available 5-bromo-2-naphthoic acid (or 8-bromoquinoline-3-carboxylic acid) and phenylboronic acid derivatives thereof led to the formation of corresponding intermediate 3 which was subject to a HATU-mediated amide coupling with (S)-1-(6-bromopyridin-2-yl)ethan-1-amine 4 to give 2-bromopyridine derivative 5. Buchwald-Hartwig C-N coupling reaction between bromopyridine intermediate 5 with various substituted amines afforded key intermediate 7. Removal of the Boc protection group under acid conditions yielded corresponding amines 8. The final products 10a – 10h were synthesized following HATU-mediated coupling of the amine 9 with 2-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-4-yl)oxy)acetic acid.
Table 1.
Chemical structure of compounds 10a-10h.

| |||
|---|---|---|---|
|
| |||
| Compound |

|
X | R |
|
| |||
| 10a |
|
C |

|
| 10b |
|
C |

|
| 10c |
|
C |
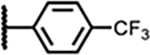
|
| 10d |
|
C |

|
| 10e |

|
C |

|
| 10f |

|
N |

|
| 10g |

|
C |

|
| 10h |

|
C |

|
Scheme 1.

Synthesis of analogues of compound 1 (XJZ-25).
Next, we evaluated the degradation activities of these analogues in MOLT-4 cells. Western blotting analysis of PDCD2 levels after 24 hours treatment with 1 μM of each compound was shown in Figure 2a. Replacement of piperidine moiety in compound 1 with all carbon linkers resulted in decreased degradation activity (compound 10a, 10b). Compound 10c, 10d with either polyethylene glycol (PEG)1 or PEG2 in the middle induced moderate degradation of PDCD2. Notably, Compound 10e which is one carbon shorter than compound 1 displayed a significant improvement in effect with over 95% reduction of PDCD2 protein levels after 24 h treatment with 10e. The switch of naphthalene core to quinoline resulted in compound 10f slightly less potent than 10e. Trifluoromethyl group is crucial to the degradation activity, as removal of CF3 in 10e resulted in a significant decrease in potency. Further truncation of trifluoromethylphenyl as shown in compound 10h resulted in a loss of activity. Among analogues, compound 10e proved to be our most potent degrader for PDCD2 and global proteomics demonstrated high proteome-wide selectivity of compound 10e (Figure S1).
Figure 2.

a) Western blotting analysis of PDCD2 in MOLT-4 cells treated with compounds 10a –10h for 24 h at 1uM. b) Western blotting analysis of PDCD2 in MOLT-4 cells treated with indicated concentrations of compound 10e. c) Western blotting analysis of PDCD2 in MOLT-4 treated with 1 μM of compound 10e at the indicated time points. The data are expressed as the mean values ± SD (n = 2) and are representative of two separate experiments. All uncropped blots were presented in Figure S6.
To further characterize compound 10e, we treated MOLT-4 cells with the compound at different concentrations for 24 h. Compound 10e efficiently induced PDCD2 degradation in a dose-dependent manner, with a DC50 value of 2 nM and a Dmax value of 96.7%. No “hook effect” was observed for compound 10e at concentrations up to 10 μM (Figure 2b and S2). To determine the kinetics of PDCD2 degradation, we carried out time-course experiments (Figure 2c). Degradation of PDCD2 was observable as early as 4 hours after treatment and continued until 24 hours after treatment.
We next determined the mechanism of action (MOA) for 10e-induced PDCD2 degradation. First, to investigate whether the observed PDCD2 degradation is E3 ubiquitin ligase CRBN-dependent, we synthesized the negative control compound 10e-Neg in which a methyl group was placed on the N of the glutarimide moiety of pomalidomide, which is known to abrogate the ability to bind CRBN (Figure 3a). 10e-Neg fails to reduce the levels of PDCD2 protein in the MOLT-4 cells. As a further control, we evaluated the parental inhibitor 7e which also did not induce PDCD2 degradation (Figure 3b). To further demonstrate the observed PDCD2 degradation is dependent on the ubiquitin-proteasome system, we performed a set of rescue experiments (Figure 3c). Pretreatment with a NEDD8 activating E1 enzyme inhibitor (MLN-4924),[30] which inhibits the process of ubiquitination and proteasomal degradation rescued PDCD2 degradation induced by 10e. A similar rescue effect was observed with either pretreatment with lenalidomide (a CRBN ligand)[31] or compound 7e. Furthermore, the PDCD2 degradation induced by 10e was completely abrogated in cells lacking CRBN, compared with wild-type controls (Figure 3d). Taken together, these data demonstrated that 10e degrades PDCD2 in a CRBN-dependent manner.
Figure 3.

a) Chemical structures of negative control compound (10e-Neg) and 7e. b) Western blotting analysis of PDCD2 in MOLT-4 cells treated with 10e, 10e-Neg, or 7e for 24 h. c) Western blotting analysis of PDCD2 in MOLT-4 cells pretreated for 2 h with MLN-4924, lenalidomide and 7e and then treated with 10e for additional 6 h. d ) Western blotting analysis of PDCD2 in MOLT-4 and CRBN-knockout MOLT-4 cells treated with 10e for 24 h. All uncropped blots were presented in Figure S6.
The observation that pretreatment of compound 7e rescues PDCD2 degradation induced by 10e suggests that 7e might directly bind to PDCD2. To investigate this, a biotinylated 7e derivative (7e-biotin) was synthesized by chemically attaching biotin to the piperidine of 7e (Figure 4a). MOLT-4 cell lysates incubated with biotinylated 7e followed by streptavidin beads demonstrated robust enrichment for PDCD2 in a dose-dependent manner (Figure 4b). In addition, pretreatment of cells with 7e followed by biotinylated 7e incubation showed that enrichment of PDCD2 could be outcompeted by 7e (Figure 4c). To further investigate cellular target engagement, we employed a cellular thermal shift assay (CETSA). After treating MOLT-4 cells with 10 μM of 7e, the thermal stability of PDCD2 was markedly increased (Figure 4d). While not achieving the identical level of enhancement as observed with the 7e treatment, it has been verified that applying 10e to MOLT-4 cell lysates also moderately improves PDCD2’s thermal stability (Figure S3). The reduced thermal stabilizing potency of the 10e compared to the 7e could potentially result from alteration in binding affinity due to structural modifications. These data support a model that posits that 7e and 10e might directly bind to PDCD2. To explore whether there is a potential binding pocket in PDCD2, we harnessed an AlphaFold predicted PDCD2 structure and employed Sitemap.[32] The top ranked site was chosen for docking of compound 7e, followed by molecular dynamics simulations to assess the binding stability. We uncovered a putative binding pocket capable of accommodating compound 7e (Figure S4). Validation of this potential binding pocket will require future structural biology efforts.
Figure 4.

a) Chemical structure of 7e-biotin. b) MOLT-4 cell lysates incubated with 7e-biotin followed by streptavidin biotin pull-down and western blotting analysis. c) MOLT-4 cells pretreated with 7e, followed by cell lysis, incubation with 7e-biotin, and streptavidin biotin pull-down and western blotting analysis. d) Cellular thermal shift assay (CETSA) of 7e in MOLT-4 cells. All uncropped blots were presented in Figure S6.
Several studies have noted cell-type dependent degradation profiles for degrader molecules [8, 33], and therefore, it is critical to characterize the degradation profile of the compound 10e in different cell lines. As shown in Figure 5, robust degradation of PDCD2 was observed across Jurkat, MOLT-3, HepG2, and K-562 cells treated with 10e for 24 h, though the potency varies in these cell lines, which may be due to differential expression levels of CRBN E3 ligase or different resynthesis rates of PDCD2 in these cell lines. A putative hook effect was observed in HepG2 and K-562 cell at 20 μM treatment.[34] These results demonstrate 10e could be a useful chemical knockdown tool in different cellular contexts.
Figure 5.

Western blotting analysis of PDCD2 in Jurkat, MOLT-3, HepG2, and K-562 cells treated with indicated concentration of compound 10e for 24 h. All uncropped blots were presented in Figure S6.
Lastly, we investigated the impact of PDCD2 depletion on cell viability. To do so, we treated MOLT-4 cells with 10e for 72 h. Intriguingly, although 10e displayed a significant reduction of PDCD2 in MOLT-4 cells, it does not significantly impede cell viability as measured using CellTiter-Glo® (Figure 6a). Instead, it notably resulted in a deceleration of cell growth (Figure S5a). To further explore the mechanism, we investigated the impact of compound 10e on the cell cycle of MOLT-4 cells. Remarkably, our results demonstrated that compound 10e induced a concentration-dependent G2/M phase arrest, effectively inhibiting the cell cycle progression in MOLT-4 cells (Figure 6b-c). To validate these findings, we employed siRNA-mediated knockdown of PDCD2 in MOLT-4 cells (Figure S5b). Consistently, it led to a slowdown in cell growth (Figure S5c), mirroring the phenotypic outcomes observed with compound 10e treatment. Collectively, the findings indicate that PDCD2 is not essential for cell viability in MOLT-4 cells, but rather plays a role in regulating cell growth through modulation of the cell cycle progression. These results also align with previous research demonstrating that genetic knockdown of PDCD2 has limited impact on the viability of leukemia cells.[22]
Figure 6.

a) MOLT-4 cells were treated with 10e for 72 h and the cell proliferation was measured using CellTiter-Glo assay. b) MOLT-4 cells were treated with 10e at the concentrations specified for 24 h. After incubation with RNase A and propidium iodide (PI), the distribution of cells in each cell cycle phase was determined using flow cytometry. c) Distribution of cells in each cell cycle phase was calculated and graphed using GraphPad Prism 9.5.1. The data are expressed as the mean values ± SD (n = 2) and are representative of two separate experiments.
The PDCD (programmed cell death) family of proteins plays a crucial role in governing cellular survival, cell death, and the regulation of immune responses.[9] However, the investigation of the physiological functions of PDCD2 and the assessment of its therapeutic potential are hindered by the lack of useful chemical probes. PDCD2 operates through protein-protein interactions (PPIs) and lacks intrinsic enzymatic activity, posing significant obstacles in the development of conventional antagonists.[18] Here we reported the identification and characterization of a promising PDCD2 degrader, uncovered through proteomics-based approach, followed by a concise structure-degradation relationship (SDR) campaign. Compound 10e was identified as the most potent candidate among this class of PDCD2 degraders. 10e degrades PDCD2 in a concentration- and time-dependent manner, with a DC50 value of 2 nM and a Dmax value of 96.7%. Mechanistic studies validated that 10e induced PDCD2 degradation is mediated by CRBN E3 ligase. Employing this compound, we investigated the essentiality of PDCD2 in T lymphoblast cells. Our study revealed that PDCD2 functions as a critical regulator of cell growth by modulating the progression of the cell cycle in T lymphoblasts. We anticipate 10e may serve as a useful small molecule degrader of PDCD2 to study PDCD2-related biological pathways. Additionally, by performing biotin pull-down and cellular thermal shift assay we provide evidence that 7e may directly bind to PDCD2. Computational approach revealed a possible binding site of 7e in PDCD2. Validation of this potential binding pocket will require future structural biology efforts. With the rapid emerging proximity-inducing paradigms, such as DUBTAC,[35] PHIC,[36] and DEPTAC,[37] we envisage that 7e could be deployed as PDCD2 recruitment handle to modulate PDCD2-related cellular function by inducing or eliminating its post-translational modifications. Further studies to understand the cell biology of PDCD2 with these tool compounds are underway and will be the topic of future publications. Collectively, our work demonstrates that proteomics profiling of E3 ligase-recruiting compounds combined with chemical optimization can represent a powerful and efficient strategy to provide novel chemical tool molecules to the scientific community.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (NIH) grant R01CA262188 (to E.S.F.) R01CA218278 (to E.S.F. and N.S.G.) and K08CA230220 (to S.M.C).
Footnotes
Conflict of interest
The authors declare the following competing financial interest(s): N.S.G. is a founder, science advisory board (SAB) member, and equity holder in Syros, Lighthorse, Inception, C4, Voronoi, Larkspur (board member), Shenandoah (board member) and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Arbella, Epiphanes, Deerfield, and Sanofi. E.S.F. is a founder, SAB member, and equity holder in Civetta Therapeutics, Proximity Therapeutics, and Neomorph Inc. (board member), SAB member and equity holder in Photys Therapeutics and Avilar Therapeutics, an equity holder in Lighthorse, and a consultant to Novartis, Sanofi, EcoR1 Capital, Ajax Therapeutics and Deerfield. The Fischer lab receives or has received research funding from Astellas, Novartis, Voronoi, Interline, Ajax, and Deerfield. T.Z. is a scientific founder, equity holder and consultant of Matchpoint, equity holder of Shenandoah, consultant of Lighthorse. K.A.D is a consultant to Kronos Bio and Neomorph Inc. J.C is a founder of matchpoint, and consultant and equity holder of Matchpoint, Allorion, and Soltego.
Supporting information for this article is given via a link at the end of the document.
References
- [1].Müller S, Ackloo S, Al Chawaf A, Al-Lazikani B, Antolin A, Baell JB, Beck H, Beedie S, Betz UA, Bezerra GA, RSC Med.Chem. 2022, 13, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schreiber SL, Kotz JD, Li M, Aubé J, Austin CP, Reed JC, Rosen H, White EL, Sklar LA, Lindsley CW, Cell 2015, 161, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Antolin AA, Tym JE, Komianou A, Collins I, Workman P, Al-Lazikani B, Cell Chem. Biol. 2018, 25, 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Licciardello MP, Workman P, RSC Med. Chem. 2022, 13, 1446–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moffat JG, Rudolph J, Bailey D, Nat. Rev. Drug Discov. 2014, 13, 588–602. [DOI] [PubMed] [Google Scholar]
- [6].Schreiber SL, Isr. J. Chem. 2019, 59, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li K, Crews CM, Chemical Society Reviews 2022, 51, 5214–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Békés M, Langley DR, Crews CM, Nat. Rev. Drug Discov. 2022, 21, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guan X, Lu J, Sun F, Li Q, Pang Y, Front. Immunol. 2019, 10, 1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Durvalumab N, Nat. Rev. Drug Discov. 2020, 19, 163–164. [DOI] [PubMed] [Google Scholar]
- [11].Baron BW, Anastasi J, Thirman MJ, Furukawa Y, Fears S, Kim DC, Simone F, Birkenbach M, Montag A, Sadhu A, Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2860–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Owens GP, Hahn W, Cohen J, Mol. Cell. Biol. 1991, 11, 4177–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Baron BW, Zeleznik-Le N, Baron MJ, Theisler C, Huo D, Krasowski MD, Thirman MJ, Baron RM, Baron JM, Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7449–7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Baron BW, Hyjek E, Gladstone B, Thirman MJ, Baron JM, Blood Cells Mol. Dis. 2010, 45, 169–175. [DOI] [PubMed] [Google Scholar]
- [15].Scarr RB, Sharp PA, Oncogene 2002, 21, 5245–5254. [DOI] [PubMed] [Google Scholar]
- [16].Yang Y, Jin Y, Du W, Int. J. Clin. Exp. Pathol 2015, 8, 10894–10900. [PMC free article] [PubMed] [Google Scholar]
- [17].Li Y, Liu S, Hongzhu Q, Yang Y, Ding N, Ruan X, Stamatoyannopoulos G, Fang X, Blood 2014, 124, 1331.24916509 [Google Scholar]
- [18].Landry-Voyer A-M, Bergeron D, Yague-Sanz C, Baker B, Bachand F, Nucleic Acids Res. 2020, 48, 12900–12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Granier CJ, Wang W, Tsang T, Steward R, Sabaawy HE, Bhaumik M, Rabson AB, Biol. Open 2014, 3, 821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cui M, Liu D, Xiong W, Wang Y, Mi J, Cell Death Discov. 2021, 7, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fukae J, Sato S, Shiba K, Sato K.-i, Mori H, Sharp PA, Mizuno Y, Hattori N, FEBS lett. 2009, 583, 521–525. [DOI] [PubMed] [Google Scholar]
- [22].Barboza N, Minakhina S, Medina DJ, Balsara B, Greenwood S, Huzzy L, Rabson AB, Steward R, Schaar DG, Cancer Biol. Ther. 2013, 14, 546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang J, Wei W, Jin H-C, Ying R-C, Zhu A-K, Zhang F-J, Oncol. Rep. 2015, 33, 103–110. [DOI] [PubMed] [Google Scholar]
- [24].Kars MD, Işeri ÖD, Gündüz U, Eur.J. Pharmacol. 2011, 657, 4–9. [DOI] [PubMed] [Google Scholar]
- [25].Meissner F, Geddes-McAlister J, Mann M, Bantscheff M, Nat. Rev. Drug Discov. 2022, 21, 637–654. [DOI] [PubMed] [Google Scholar]
- [26].Zhang AX, Cassidy K, Dahl G, Moreau K, Pachl F, Zuhl AM, SLAS Discovery 2021, 26, 518–523. [DOI] [PubMed] [Google Scholar]
- [27].Xiong Y, Donovan KA, Eleuteri NA, Kirmani N, Yue H, Razov A, Krupnick NM, Nowak RP, Fischer ES, Cell Chem. Biol. 2021, 28, 1514–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Spradlin JN, Zhang E, Nomura DK, Acc. Chem. Res. 2021, 54, 1801–1813. [DOI] [PubMed] [Google Scholar]
- [29].Donovan KA, Ferguson FM, Bushman JW, Eleuteri NA, Bhunia D, Ryu S, Tan L, Shi K, Yue H, Liu X, Cell 2020, 183, 1714–1731.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, Nature 2009, 458, 732–736. [DOI] [PubMed] [Google Scholar]
- [31].Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, Nature 2014, 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Halgren TA, J. Chem. Inf. Model. 2009, 49, 377–389. [DOI] [PubMed] [Google Scholar]
- [33].Huang H-T, Dobrovolsky D, Paulk J, Yang G, Weisberg EL, Doctor ZM, Buckley DL, Cho J-H, Ko E, Jang J, Cell Chem. Biol. 2018, 25, 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang X, Dixit VM, Cell research 2016, 26, 484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Henning NJ, Boike L, Spradlin JN, Ward CC, Liu G, Zhang E, Belcher BP, Brittain SM, Hesse MJ, Dovala D, Nat. Chem. Biol. 2022, 18, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shoba VM, Munkanatta Godage DN, Chaudhary SK, Deb A, Siriwardena SU, Choudhary A, Angew. Chem. 2022, 134, e202202770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hu Z, Chen P-H, Li W, Douglas T, Hines J, Liu Y, Crews CM, J. Am. Chem. Soc. 2023,145, 4045–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.