

Abstract
Most of the individuals who experience traumatic brain injury (TBI) develop neuropsychiatric and cognitive complications that negatively affect recovery and health span. Activation of multiple inflammatory pathways persists after TBI, but it is unclear how inflammation contributes to long-term behavioral and cognitive deficits. One outcome of TBI is microglial “priming” and subsequent “hyper-reactivity” to secondary stressors, injuries, or immune challenges that further augment complications. Additionally, microglia priming with aging contributes to exaggerated glial responses to TBI. One prominent inflammatory pathway, interferon (IFN) signaling, is increased after TBI and may contribute to microglial priming and subsequent reactivity. This review discusses the contributions microglia to inflammatory processes after TBI, as well as the influence of aging and interferons on microglia reactivity and chronic inflammation after TBI.
Keywords: Neurotrauma, Neurodegeneration, Microglia Priming, Neuroinflammation
Long-term Consequences of Traumatic Brain Injury
TBI is a leading cause of neurological disability and entails a significant risk of neurological and neuropsychiatric complications even after mild to moderate TBI [1]. For instance, brain-injured individuals are 5–10 times more likely to develop symptoms of depression compared to the general population [2, 3], and 15–30% have detectable cognitive decline over time [4–7]. In many individuals, depression and cognitive decline are comorbid [8, 9], and both may continue to progress with time after TBI [10]. Impulsivity is another complication of TBI that may increase risky behaviors like aggression [11, 12]. TBI also increases the risk of seizures and development of post-traumatic epilepsy [13–15]. TBI is also associated with increased risk for neurodegenerative diseases, including Chronic Traumatic Encephalopathy (CTE), Fronto-Temporal Dementia (FTD), and Alzheimer’s disease (AD) [16–23]. These clinical findings are indicative of persistent changes in the brain following TBI and raise the possibility of long-term brain inflammation that continues to disrupt brain homeostasis and confer vulnerability to neuropsychiatric complications.
Traumatic brain injury has been described previously as a “disease process” rather than a singular event [24]. This is because the response to TBI occurs in general stages; the initial impact causes what is known as “primary injury,” including immediate destruction or damage to cells. “Secondary injury” is used to describe the process of persistent inflammation, which contributes to chronic pathophysiological processes in the brain. While primary injury cannot be prevented without prevention of the initial hit, secondary injury represents a promising intervention target, and thus, substantial preclinical research is focused on defining features of inflammation that contribute to secondary injury and progressive neuropathology (the evolution of neuroinflammation after TBI has been reviewed elsewhere[25]). As such, microglia, the resident innate immune cells of the brain, have garnered significant attention in TBI research.
As long-lived cells with low turnover, microglia are susceptible to long-term effects of TBI and aging [26, 27]. In clinical studies, elevated metabolic activity in microglia, white matter abnormalities, and microglial activation are detectable long after TBI [28–34]. For instance, human brain imaging and post-mortem histology studies show that 17–18 years following injury, microglia have increased expression of activation markers CR3, CD68, CR3/43 (MHCII), and PK11195 in cases of single blunt-head trauma and repetitive hits (e.g., in former American football players, who may sustain hundreds of high-impact hits during their athletic careers) [26, 28, 31]. Experiments in animal studies have further characterized these CD68+/MHCII+ microglia as “primed.” Microglia priming represents an increase in the inflammatory profile of microglia with a corresponding enhanced readiness to respond to a secondary challenge [27]. In general, the primed microglia profile is characterized by increased expression of genes and proteins associated with antigen presentation (MHCII, CD11c, CD86), DAMPs/PAMPs (CD14, Toll-like receptors 2/4), complement components (C1q), and surface receptors (CD22, CD68, Trem2) [27, 35]. Notably, primed microglia exhibit an intermediate profile and do not actively produce cytokines and chemokines like they do acutely after TBI.
One potential mediator of microglia priming after TBI is interferon (IFN) signaling. Recent evidence indicates that TBI elicits a robust type I IFN response in the brain. While we will limit our discussions in this review to type I IFN responses, it should be noted that type II IFN response (mediated by T cells) may also be relevant in the context of TBI. Type I interferons bind to IFNAR1, and receptor activation drives STAT1, STAT2, and IRF9 signaling to control transcription of genes associated with anti-viral responses [36]. Type I IFNs (α and β) increase anti-viral properties of immune cells, preparing them to contest viral pathogens. As such, the IFN influence on myeloid cells is consistent with the idea of priming and is a potential mediator of TBI-induced microglia priming.
Preclinical researchers use several approaches to induce experimental TBIs of varying severity to illuminate pathophysiological processes, like IFN signaling, after injury [37]. In brief, animal models of TBI involve inducing diffuse, mixed, or focal/penetrating injuries. Mice, rats, and pigs are among the most prevalenty used species in TBI studies. Midline fluid percussion injury (mFPI), closed-head impact, and blast injuries represent approaches to model diffuse TBI. Of these, closed-head injuries are repeatable, allowing for the examination of multiple hits [38]. To induce focal TBI, controlled cortical impact (CCI) and ballistic injuries are used. Some models, including lateral FPI (lFPI), result in a mixed injury with aspects of both diffuse and focal injuries. Notably, focal injuries cause more tissue cavitation, cell death, and infiltration of peripheral monocytes to the lesion site compared to diffuse TBI (Fig.1). In all cases, “secondary” injury occurs, wherein there is progressive inflammation and tissue pathology (Fig. 1). These models are all relevant to a broad range of injuries that occur clinically.
Figure 1. Experimental models for the study of microglia activation and neurological complications following traumatic brain injury.
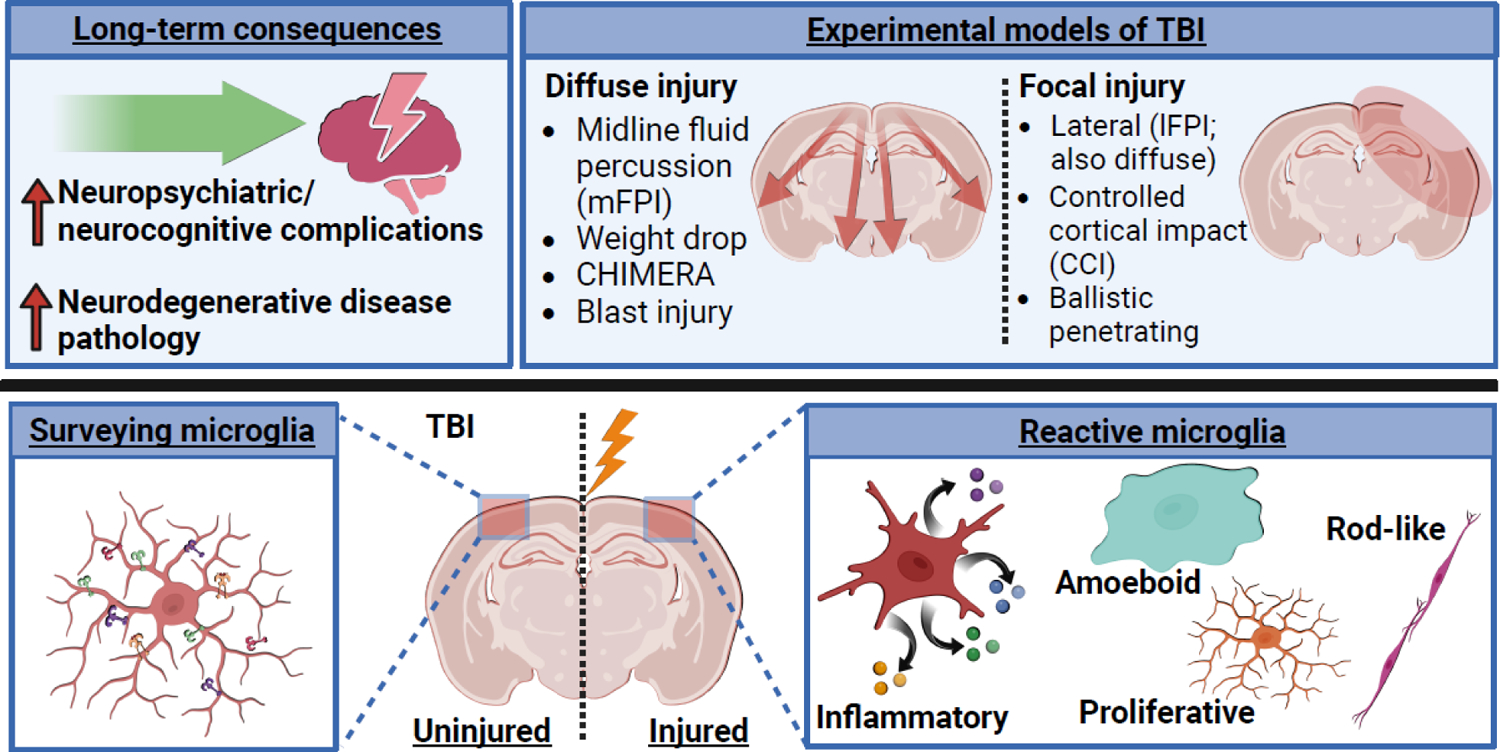
Traumatic brain injury often leads to the development and persistence of neurological and neuropsychiatric complications, including cognitive decline, depression, anxiety, epilepsy, aggression, and neurodegenerative diseases [1–20, 22, 23]. Preclinical researchers use several approaches to induce experimental TBIs to study the underlying pathophysiology. Microglia, the resident innate immune cells of the brain, are one well-documented cell type known to drive inflammation in the brain after TBI. Under steady-state conditions, microglia are actively surveying the micro-environment. Upon damage induced by TBI, microglia become reactive to drive several different processes. Microglia produce cytokines and chemokines (the signaling molecules of the immune system), become amoeboid and phagocytose debris, upregulate proliferation, and/or adopt rod-shaped morphology and align with damaged axons [39, 46]. Figure created using Biorender.com.
Though chronic inflammation after TBI likely contributes to long-term vulnerability to neurological and neuropsychiatric deficits, the link between pathophysiology of TBI and these long-term consequences is not fully defined. Thus, determining the biological pathways that drive chronic inflammatory processes after TBI is critical to development of novel therapeutics to improve long-term recovery. This review article will discuss the role of microglia in propagating chronic inflammation after injury and how primed microglia confer vulnerability to secondary immune challenges. We will also discuss the role of aging on enhanced inflammatory responses to TBI and the potential role of interferons as a promising therapeutic to ameliorate chronic inflammation, microglia priming, and functional recovery.
Microglia Depletion and Forced Turn-over Studies Reveal Microglia Contributions to Pathology after TBI
Experimental animal-model studies of TBI (diffuse/focal/mixed) indicate that microglia mediate chronic inflammation and long-term functional deficits, including cognitive decline, depressive-like behaviors, and post-traumatic epilepsy [39–45] (Fig.2). For instance, microglia depletion using a CSF1R antagonist (Plexxikon 5622) with diffuse TBI (mFPI) in mice attenuates the inflammatory landscape of the cortex 1, 7, and 30 days post injury (dpi) [40, 46]. In these studies, depletion of microglia prior to mFPI reversed TBI-induced expression of several inflammation-associated genes in the cortex at each timepoint after TBI [40]. For instance, 33% of the differential gene expression between TBI and control mice 1dpi was prevented by microglia depletion [40]. By 7 and 30dpi, 70–80% of differential gene expression was prevented by microglia depletion. While NFkB-driven gene expression dominated the early response to TBI (8hpi), IFN signaling was a prominent mediator of inflammatory gene expression by 7dpi, and at 30dpi, expression of other innate immunity genes was still elevated [46]. Overall, inflammation evolves over time and is microglia dependent.
Figure 2. Microglia depletion and forced turn-over studies in animal models reveal stagespecific contributions of microglia in progression of pathology after TBI.
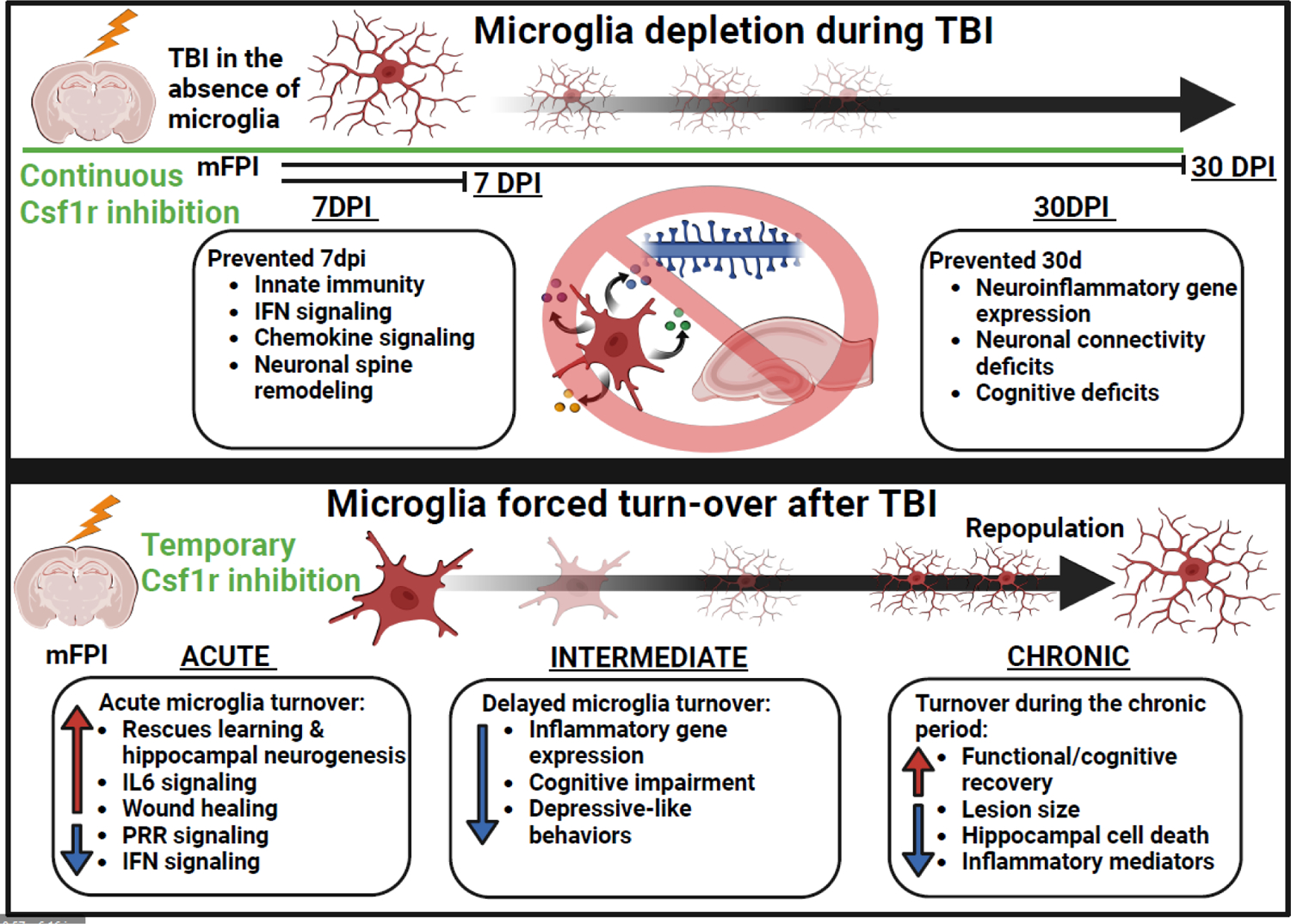
CSF1R antagonism results in rapid depopulation of microglia. Depletion of microglia throughout the time-course of TBI reverses inflammatory gene expression and neuronal spine remodeling, as well as cognitive deficits at a chronic timepoint, revealing the role of microglia in producing these effects after TBI [40, 46]. Forced turn-over or “repopulation” of microglia after TBI, by cessation of CSF1R antagonism, allows for assessment of applying microglia-specific intervention at later timepoints after injury. Acute (within 3 d), intermediate (5–14 d), or chronic (>30 d) repopulation of microglia after TBI generally improves inflammatory gene expression, cell death, cognitive impairment, depressive-like symptomology, and inflammatory responses to peripheral immune challenge[42, 48, 50, 51, 53]. These studies demonstrate that targeting microglia-mediated inflammation after TBI improves recovery. Figure created using Biorender.com.
One of the microglia depletion studies mentioned above used single-cell RNA-sequencing (scRNA-seq) to examine the influence of microglia depletion on cellular profiles in the cortex post-TBI (mFPI). A cluster of trauma-associated microglia was identified 7dpi, which robustly expressed IFN-associated genes (Ifitm3, Isg15, Ifi27l2a, Irf7, Stat1) and genes associated with priming (Trem2, Apoe, Clec7a, H2-D1, H2-K1) [40]. The persistent inflammatory profile of microglia corresponded with global transcriptional suppression in neurons. For instance, scRNA-seq of cortical neurons 7dpi showed inhibition of long-term potentiation, dopamine signaling, calcium signaling, and synaptogenesis pathways [40]. The overall suppression of these pathways in neurons was associated with functional deficits 7 and 30dpi with impaired neuronal physiology (e.g., compound action potentials stimulated ex vivo) of corpus callosum axons and reduced cortical dendritic complexity [40]. TBI-induced neuronal connectivity deficits, neuronal restructuring, and cognitive impairment 30dpi were all microglia dependent [40]. These findings indicate that microglia significantly contribute to neuronal function and alterations in the synaptic landscape that persist chronically after diffuse TBI. Compared to diffuse TBI, focal injury induced by CCI creates a significant lesion, tissue loss, and cavitation with increased contribution from peripheral immune cells (including monocytes) [47]. In focal injury (CCI), microglia depletion prior to CCI did not rescue hippocampal-dependent memory deficits 12dpi and impaired hippocampal neurogenesis [48]. Lastly, a recent study showed that microglia depletion in mice prior to diffuse TBI transiently attenuated peripheral monocytes (CD115+/ Ly6Clo) in the blood (1 and 3dpi) and decreased microglia and neutrophils in the brain (7dpi) but elevated peripheral inflammatory monocytes in the blood at 7dpi [49]. The depletion of microglia prior to TBI provides important insights, but any benefits of the initial response of microglia to traumatic CNS injury is lost. Thus, experimental studies in animals sought to investigate the effects of delayed microglia forced turn-over and subsequent repopulation after injury.
Microglia “forced turnover” is accomplished by temporary administration of a CSF1R antagonist in animal models followed by a “repopulation” period. In diffuse TBI (mFPI), forced turnover (using PLX5622) starting at 7dpi with repopulation (14dpi-30dpi) reduced the cortical inflammatory profile 30dpi, prevented cognitive impairment, and attenuated depressive-like behavior in mice [42]. This strategy did not, however, prevent TBI-induced reductions in dendritic complexity [42]. In another study, depleting microglial acutely after CCI (0–5d) and allowing repopulation improved tissue loss at a chronic timepoint but did not prevent early neuropathology, hematoma development, or neurological severity scores [50]. Additionally, in focal TBI (CCI), microglia were repopulated within 3dpi in transgenic mice expressing CXCR3-DTR, rescuing CCI-induced deficits in learning and hippocampal neurogenesis [48]. In this study, the new microglia increased mediators of IL6 signaling and wound healing-associated gene expression while decreasing gene expression associated with pattern/damage recognition and interferon (IFN) signaling [48]. In another model of CCI, repopulation within 1mo after injury had multiple benefits. For instance, depletion for 1wk, 1mo after TBI reduced lesion size, hippocampal neuronal cell death, and inflammatory mediators NOX2 and NLRP3, while improving long-term motor and cognitive functional recovery [51]. Collectively, these studies indicate that there may be a critical window for application of this intervention after CCI. For instance, forced microglial turnover >4mo after CCI injury did not improve spatial learning or support production and survival of newborn neurons [48]. It is also important to consider the environment in which microglia are repopulated. For example, two studies found that forced turnover of microglia in aged mice (18–20mo) reduced the lipid-accumulating profile (lipofuscin/CD68+) of aged microglia but did not fully restore the young microglia profile [52, 53]. One caveat comes from a study in humans showing that minocycline (an antibiotic with microglia inhibiting properties) reduced neuroinflammation but increased a marker of neurodegeneration (neurofilament light chain, NFL) in plasma from patients 6mo after TBI [54]. These findings are difficult to interpret, however, because minocycline has many off-target effects. Nonetheless, one possibility, is that minocycline decreased microglia phagocytosis of damaged neurons and this increased NFL levels in blood. Overall, replacing trauma-associated microglia with nascent microglia may reduce inflammation and enhance intrinsic repair profiles if done within a critical timeframe after TBI.
Microglia Contribute to Amplified Neuroinflammation in the Aged Brain after TBI
Age at time of injury is a key factor in the recovery from TBI. Not surprisingly, elderly individuals have a significant risk of complications following TBI. For instance, individuals >75 years old have the highest rates of TBI-related deaths and hospitalizationsi and increased incidence of cognitive decline, psychiatric problems, and neurodegenerative disease pathology [21, 22]. Thus, it is important to determine the neurobiological mechanisms underlying these differences. As discussed in more detail below, recent studies of neuroinflammation after TBI in aging highlight several common themes, including enhanced inflammatory profiles of microglia with deficient anti-inflammatory mediators, dysregulated phagocytosis, increased infiltration of peripheral immune cells, increased oxidative stress, over activation of complement components with accompanying synapse loss, and amplified interferon-associated inflammation (Fig.3).
Figure 3. Primed microglia promote amplified immune responses and worsened functional recovery in the aged brain following TBI.
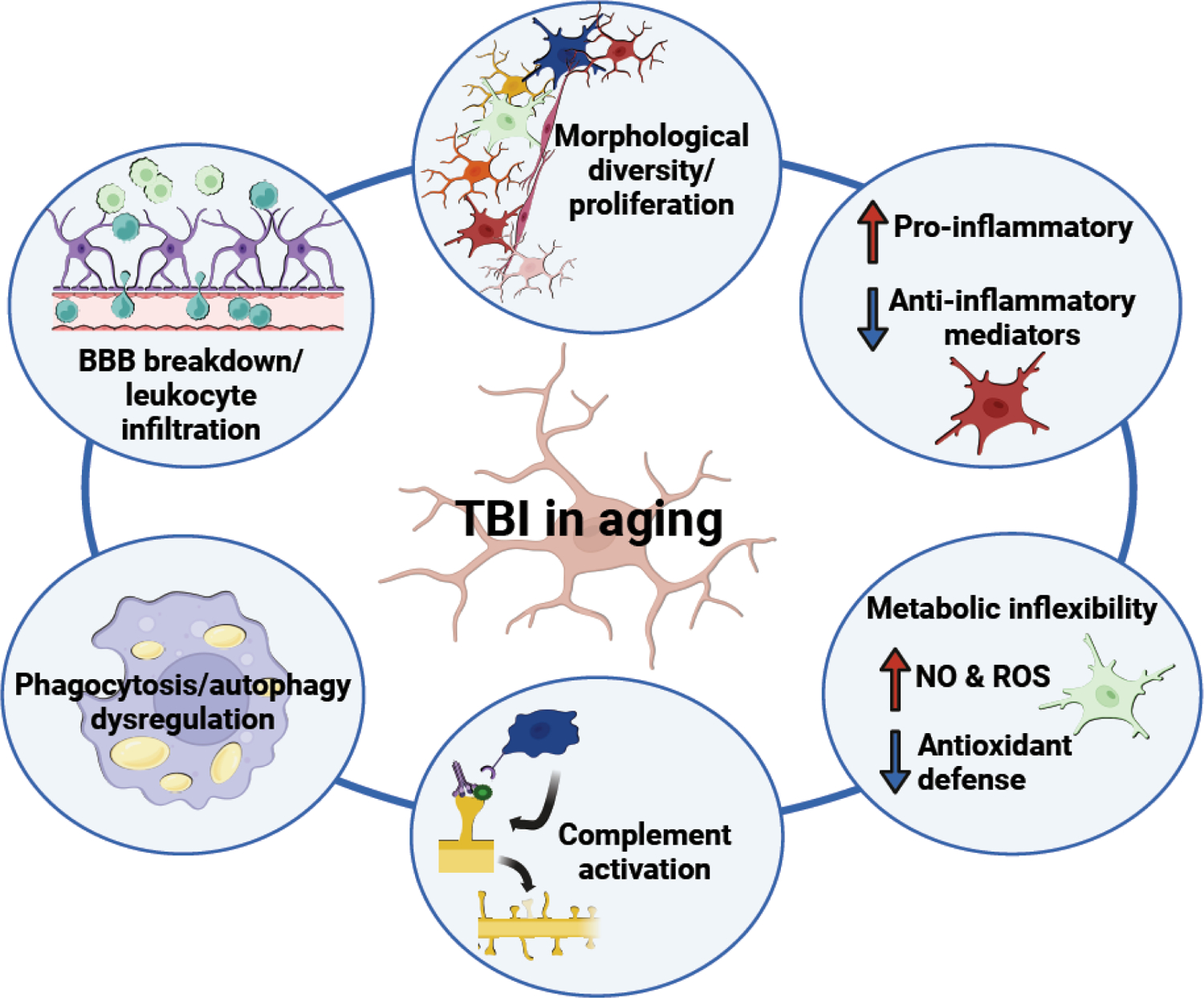
Aged individuals are particularly vulnerable to TBI; those > 75 yr old have the highest incidence rates of TBI, as well as the most TBI-related hospitalizations and deaths. Preclinical research has defined several biological themes that drive these effects in aging, including amplified morphological restructuring of microglia; enhanced production of inflammatory mediators accompanied by deficient anti-inflammatory signaling; increased production of damaging ROS and NO and decreased antioxidant defense; enhanced complement activation and synapse loss; dysregulated phagocytosis and autophagy; and exacerbated blood brain barrier breakdown with peripheral cell infiltration. Figure created using Biorender.com.
In models of diffuse and focal TBI, age is an important contributor to the microglia response, and TBI-related structural changes in microglia were influenced by age in several studies. In focal TBI (CCI) in aged mice, hypertrophic and bushy microglia increased in 7dpi, while ramified “homeostatic” microglia decreased [55]. In diffuse TBI (mFPI), morphological restructuring was amplified by age in both the cortex and hippocampus 7dpi [56]. There were also more rod-shaped microglia in the cortex 30dpi in aged mice compared to adults [56]. While their function is unclear, rod-microglia localize to areas with damaged axons [46, 57, 58] and are detected in CNS infection, aging, and neurodegenerative disease [46, 59–63]. Thus, the enhanced prevalence of rod-microglia may be a response to greater neuronal damage after TBI in the aged. Overall, several aspects of TBI-induced microglia morphology appear to be influenced by age.
Consistent with enhanced morphological restructuring in the aged brain after TBI, animal model studies have also found age-dependent amplification of neuroinflammatory gene expression after diffuse and focal TBI. For instance, age enhanced expression of pro-inflammatory cytokine mRNA: Tnf and Il-1b [47, 55] and damage-associated transcripts Cd86, iNOS, Clec7a, Trem2, and Tyrobp [55, 56, 64] in the cortex of aged mice following injury. Moreover, mRNA levels of anti-inflammatory mediators (IL4Ra, SOCS3, TGFβ, IL10, and APOE) were inhibited or not induced to the same degree in aged mice after TBI [55, 56]. 7d after CCI, aged-TBI mice had the highest IL1-β and TNF microglia protein expression [64]. Furthermore, isolated myeloid cells from aged-TBI had decreased expression of anti-inflammatory mediators Ym1, CD206, TGFβ, and IL4RA [65]. Overall, TBI in aged mice was associated with exaggerated inflammation accompanied by reduced anti-inflammatory-related regulation.
Altered metabolic activity in the aged brain is associated with worse outcomes in both diffuse and focal TBI. For instance, Ingenuity Pathway Analysis of DEGs after diffuse TBI (mFPI) indicated production of NO and ROS in macrophages was significantly increased in aged mice [56]. After CCI, aged microglia had higher ROS production at baseline and acutely after injury, which lasted up to 16wks post-injury [64]. Moreover, focal TBI (CCI) induced higher mitochondrial activity, glucose uptake, and ATP production [64]. The cellular demand for glucose was higher in young-TBI than age-matched shams, even up to 16 weeks. Thus, TBI accelerated an aged-like metabolic profile of microglia [64]. Furthermore, aged-TBI microglia/macrophages had enhanced expression of NADPH oxidase mRNA accompanied by deficient induction of antioxidant defense enzyme mRNA for SOD1 and GPX1 [55]. Collectively, there were deficits in metabolic flexibility of aged microglia after TBI, which likely contributed to enhanced inflammation in the brain with age and TBI.
TBI in aged mice is also associated with over-activation of the complement cascade. The complement system helps shape the synaptic landscape during development and alters neuronal connections in neurodegenerative disease, brain injury, and aging [66–70]. In TBI, increased complement activation was detected both acutely and chronically. For instance, diffuse and focal TBI in aged mice increase complement components (including C1q(a-c) and C3) 7 and 30dpi and up to 16wks after injury [56, 64, 71]. Complement-mediated microglia phagocytosis of synapses was associated with cognitive impairment following focal TBI (CCI) [68]. In a focal TBI (CCI) study, Flow synaptocytometry revealed colocalization between individual synapses and C1q 30dpi that paired with reduced PSD95+ synapses in injured tissue [71]. Furthermore, hippocampal microglia in the aged brain after TBI (30dpi) engulfed more fluorescently-labeled synapse particles than sham [71]. These data indicate increases in phagocytic responses of aged microglia after TBI during the chronic phase [71]. In the same study, reducing complement activation in aged mice after TBI using C3 genetic knockout or anti-C1q antibody administration improved memory in aged mice 30dpi [71]. In summary, TBI in aging is associated with over-activation of the complement system with subsequent synapse loss and cognitive impairment.
Related to the discussion above, microglia phagocytosis was found to be dysregulated after TBI in the aged brain. Microglia and infiltrating macrophages 72h after CCI in mice had impaired phagocytic activity in a bead engulfment assay [72]. In an in vivo engulfment assay of fluorescently-labeled synapse particles, however, aged-TBI (CCI) microglia engulf more synapses than shams [71]. After diffuse TBI (mFPI), mRNA expression in the cortex 7dpi indicated age-enhanced phagosome formation [56]. Though there was evidence of phagosome formation after TBI, microglia/macrophages in the aged brain had impaired and delayed phagocytic capacity. Furthermore, aged microglia may be influenced by the overabundance of complement signaling to engulf synapses, rather than clear debris. One complicating factor is the apparent accumulation of lipids in aged microglia, which correlates with deficits in phagocytosis [73]. Moreover, aged microglia accumulate cellular “junk” called lipofuscin, composed of lipids, misfolded proteins, and metals [74]. Lipofuscin and lipid accumulation contribute to dysregulated phagocytosis and autophagy. Although lipid-droplet accumulating microglia (LDAMs) are characterized by decreased phagocytic capacity [73], microglia with high lipofuscin content in the aged brain have hyperphagocytic properties, showing increased intracellular detection of neuronal and myelin components [75]. Moreover, turnover of these microglia using pharmacological elimination and repopulation reduced lipofuscin content and restored phagocytosis[75], findings that corroborate previous work showing forced turn-over restored CD68+ lysosomal labeling in aged microglia [52]. Relatedly, treatment with autophagic enhancer trehalose improved functional recovery, cognition, and brain inflammation, while decreasing phagocytosis of neuronal particles after TBI in aged mice [64]. Collectively, dysregulated phagocytosis and debris clearance within microglia contributes to chronic inflammation, synapse loss, and cognitive impairment after TBI.
Another aspect of TBI in aging is increased recruitment of peripheral immune cells, especially in focal TBI induced by CCI. One important contributor to this is blood brain barrier (BBB) breakdown. BBB breakdown was evident in aged mice up to 1mo after focal TBI [71]. Compromised integrity of the BBB contributes to increased cellular infiltration from the periphery [76–78]. Penetrating CCI in aged mice resulted in amplified chemokine-driven recruitment of peripheral cells. For example, age increased the recruitment of CCR2+ and CD45hiCD11b+ cells to the brain after CCI 48h [64], and sub-acutely (4 and 7dpi) [65]. Furthermore, the enhanced effect of age on TBI-induced number of infiltrating myeloid cells remained at 16wks, indicating long-lasting changes in BBB integrity and chemokine signaling [64]. These infiltrating myeloid cells produced enhanced reactive oxygen species (ROS), a deleterious consequence [64]. Moreover, pharmacological, and genetic CCR2 inhibition improved TBI-induced neuroinflammation and cognitive deficits in aged mice [65, 79]. In diffuse TBI (mFPI), there was age-enhanced expression of genes associated with peripheral cell recruitment (Ccl2 and Ccl5) [56], although it is unclear if there was more recruitment. Previous studies in this model of diffuse injury have shown limited infiltration of monocytes from the periphery, but enhanced signaling in aged mice may promote increased cell recruitment [46]. Overall, penetrating injury in aged mice promotes enhanced recruitment of highly inflammatory peripheral immune cells that contribute to negative outcomes.
Collectively, microglia activation appears to be consistently amplified after TBI (diffuse and focal) in aged mice. This is associated with morphological, transcriptional, and protein changes indicative of chronic innate immune activation, increased production of damaging ROS, dysregulated phagocytosis and autophagy, and enhanced BBB breakdown leading to peripheral cell recruitment. These amplified responses with age are associated with long-lasting neuroinflammation and impaired functional recovery.
Mechanisms of Microglia Priming after TBI
One long-term consequence of TBI is “microglial priming.” Microglia priming is reported in models of aging, psychological stress, early life infection, and TBI [27, 35]. For example, CR3/43+ (MHCII) & CD68+ microglia were detected in 28% of human cases 1–18 years after a single, moderate to severe TBI [34]. In experimental models, disproportionate microglia responses lead to maladaptive behaviors (e.g., protracted sickness, cognitive deficits, depressive-like behaviors). Thus, priming after TBI is relevant because there is an increased risk of amplified maladaptive responses to secondary stressors, repeated head injuries, or immune challenges that further augment TBI-induced behavioral and cognitive complications.
Primed microglia were evident in experimental TBI studies. For example, there was increased gene expression of H2-eb1 (MHCII), Itgax (CD11c), toll-like receptor related (Tlr4,Tlr2, Cd14) and Cd68 in the cortex 7 and 30d after diffuse TBI (mFPI) in mice [40, 46]. These RNA increases were accompanied by increased MHCII protein expression in microglia 30dpi [80]. Structurally, these microglia had a morphological profile with more de-ramified processes and an enlarged soma compared to controls [80, 81]. Furthermore, scRNA-seq analysis of the cortex 7d after mFPI showed a novel cluster of “trauma-associated microglia” with higher expression of MHCII-related genes (H2-k1, H2-d1) and multiple interferon-related genes [40]. These microglia IFN-response RNA signatures at 7dpi were associated with microglia priming by 30dpi [40, 42]. Increased MHCII and CD68 labeling was also detected 14d after diffuse TBI (mFPI) in rats [60]. Furthermore, RNA analysis of the cortex after mixed diffuse/focal TBI (lFPI) showed higher expression of disease-associated/priming-related genes (C1q, Trem2, Tyrob, and Tlr2) 30dpi and enhanced morphological restructuring of microglia in the cortex [82]. These profiles were also present chronically (1yr) after focal TBI (CCI) with increased mRNA expression of MHCII, Cd68, and Nox2 [83]. Collectively, diffuse and focal TBI in animal models increase the inflammatory profiles of microglia, and components of this profile persist chronically.
Microglia priming may set the stage for an amplified response to a second hit, which might come in the form of an infection, a subsequent head injury, or a psychological stressor. Preclinical studies lend support to this notion. Microglia priming after TBI in mice was shown, for instance, to contribute to an exaggerated neuroinflammatory response to peripheral immune challenge 30dpi[80, 81]. Peripheral immune challenges, including lipopolysaccharide (LPS) administration, induce a transient inflammatory response that is directed by the immune system in communication with the brain to modulate physiology and behavior [84]. While this challenge causes transient immune and adaptive behavioral responses in control/uninjured mice, it elicits maladaptive responses in mice after TBI. For example, microglia from TBI mice (mFPI) had exaggerated levels of cytokine and chemokine RNA (Il-1β, Ccl2, and Tnf) after LPS (i.p.) challenge 30dpi.[80, 81]. Along with enhanced pro-inflammatory cytokine expression, there was pronounced morphological restructuring of microglia in TBI mice after LPS challenge at 30dpi [42, 80, 81]. This enhanced morphological restructuring supports the idea of amplified microglial activation. Moreover, the amplified and prolonged neuroinflammatory response to LPS challenge caused protracted sickness behavior (e.g., increased lethargy and decreased social exploration behavior) in TBI mice compared to controls at 24h after LPS administration. In addition, depressive-like behaviors were evident and cognitive deficits were amplified 72h after LPS challenge in TBI-mice [80, 81]. Conceptually, one can see parallels between these data and studies of microglia priming with age. For example, aged mice challenged with LPS had enhanced microglia expression of Il1b, Il1a, Tnf, and Ccl4, and increased intracellular IL-1β expression in MHCII+ microglia [85, 86]. Like TBI, the amplified neuroinflammatory response to LPS challenge in aged mice was associated with protracted sickness behavior, depressive-like behaviors, and cognitive impairment [87–89]. In addition, other stressors may amplify the microglial response post-TBI. For instance, the stress of sleep fragmentation after TBI in mice (lFPI) amplified gliosis, reduced neuronal activation (cfos labeling), and enhanced TBI-induced cortical inflammation through increased expression of interferon- and stress-associated genes, ultimately exacerbating cognitive deficits [82]. A recent study showed that forced turnover of microglia (using a CSFR1 antagonist) in mice during the subacute phase (7–14dpi) ameliorated TBI-induced microglia primed response to LPS 30dpi, including morphological restructuring, cortical inflammatory gene expression, and prolonged lethargy and social withdrawal [42]. Overall, a consequence of TBI is microglia priming that produces amplified responses to secondary innate immune challenge with subsequent neurocognitive and neuropsychiatric complications.
The Role of Interferons in Microglia Priming after TBI
The mechanisms that underlie priming of microglia after TBI are unclear. One possibility is enhanced interferon (IFN) signaling. Type I IFN responses are classically considered anti-viral and serve as an “alarm” system when a virus is present [90]. In the context of CNS infection, increased IFNs are beneficial for limiting viral spread [91, 92]. To our knowledge, no studies have yet directly investigated IFNs and microglia priming after TBI, but IFNs are generally known to increase mobilization, enhance antigen processing and presentation, and promote clearance of debris in macrophages, which is consistent with the primed profile [91]. IFNs also regulate the development of dendritic cells, professional antigen presenting cells, from monocyte precursors [93]. For example, monocytes exposed to GM-CSF with type I IFNs express MHC (I and II), co-stimulatory proteins (including CD86), and many TLRs [93]. Indeed, microglia have high relative expression of IFNAR1 mRNA compared to other cell types in the CNSii [94]. The IFN signature of microglia was also detected in murine AD models [95]. Collectively, the anti-viral activities induced by IFNs have a significant overlap with the type of genes that are chronically expressed in microglia after TBI, including MHCII-related, dendritic (Cdllc), and phagocytic (Cd68) (Fig.4). Though evolutionarily programmed to respond to viral infection, there is emerging evidence that TBI promotes activation of IFN.
Figure 4. Robust interferon activation primes microglia after TBI.
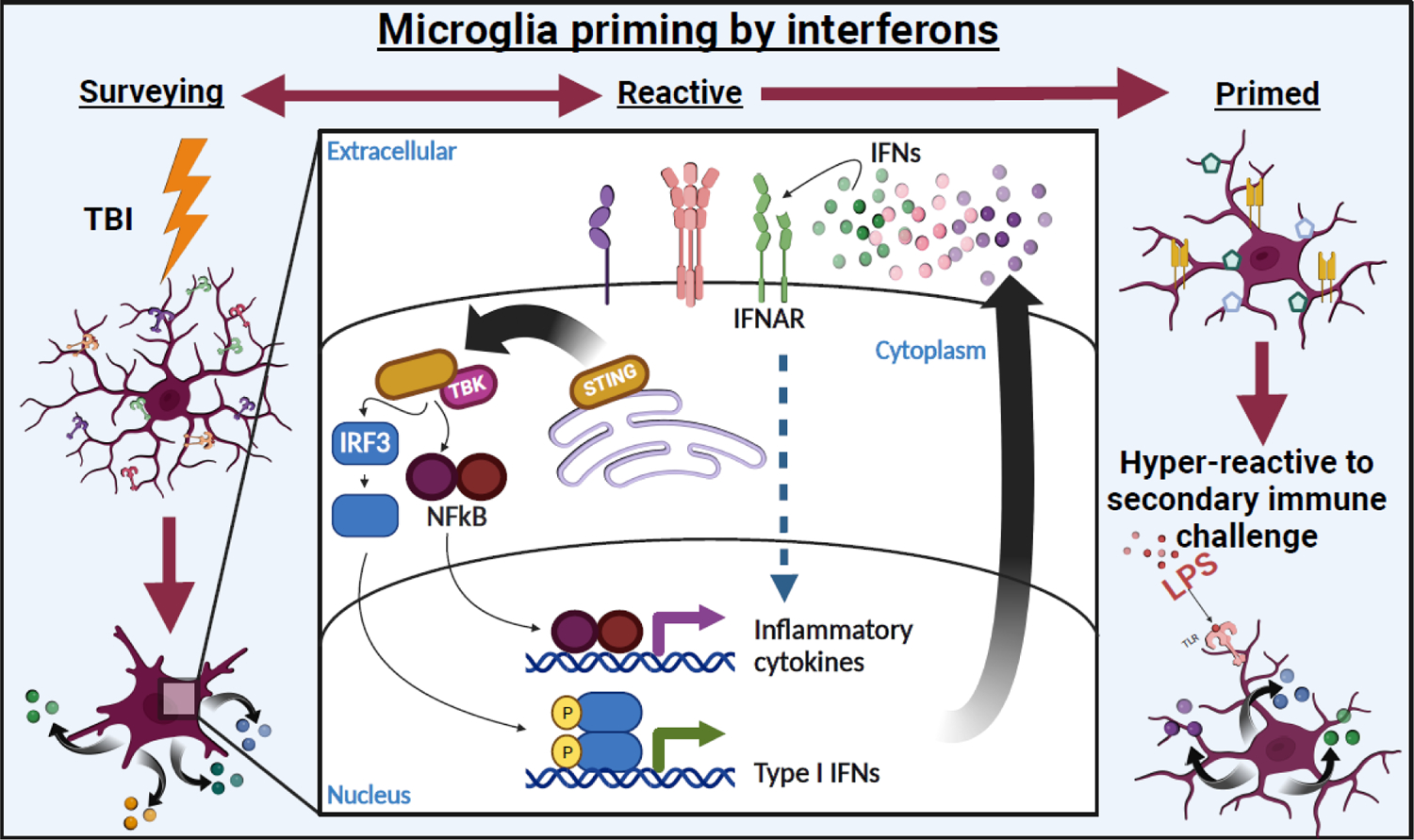
Microglia remain activated chronically after injury. Furthermore, while some microglia return to basal status, a subset of microglia adopt a “primed” profile characterized by increased reactivity to secondary stimuli (e.g., infection, stress, etc.). Microglia priming has been associated with increased inflammation and development of chronic cognitive and psychiatric complications following TBI. IFN activation has been associated with priming cells to respond to viral infection by increasing phagocytic and antigen presentation machinery similar to the profile of primed microglia. Figure created using Biorender.com.
IFN responses are activated by cellular distress and damage that are evident with TBI in humans and rodents. For example, IFNB mRNA levels are significantly higher in individuals who died shortly after TBI [96]. One important mediator of IFN responses in TBI is stimulator of IFN genes (STING). STING is a stress-responsive ER protein that coordinates with cGAS to detect tissue damage, extracellular DNA, and mitochondrial dysfunction to promote downstream type I IFN gene expression [97]. In the context of TBI, transcription of inflammatory cytokines and interferon stimulated genes (ISGs) is triggered by binding of DAMPs and PAMPs to Pattern Recognition Receptors, and cGAS-STING-mediated sensing of self-DNA from damaged cells [98–100]. Moreover, increased STING was detected in human brain samples late post-injury [101]. STING activates the transcription factors IRF3 and NFkB, upon detection of foreign and self-DNA [102, 103] to drive a wide array of IFN- and NFKB-mediated responses.
It is evident that IFN is activated by TBI, but the extent to which different cell types contribute to IFN production and responses remains unclear. As IFN signaling represents a conserved anti-viral program, all cell types respond to IFNs [104]. One study using lFPI showed that both microglia and astrocyte transcriptomes were enriched for type I IFN response 7dpi [105]. Furthermore, scRNA-sequencing after diffuse TBI (mFPI) revealed unique clusters of trauma-associated microglia expressing IFN-response transcripts 7dpi (Ifitm3, H2-d1, Stat1, Irf7, H2d, 1fi27i2a) [40]. Studies also showed that STING and cGAS were localized to microglia in the brain [98, 101]. Thus, microglia are one source and responder to IFNs after TBI. IBA-1+ meningeal macrophages are another potential source of IFN and have increased expression of type I IFN genes after mild head injury [106]. Bone marrow-derived-cells are another possible source of IFNs, especially in models of focal TBI with exacerbated peripheral cell infiltration [96]. Damaged neurons are another possible source of IFN production and/or response. Collectively, IFNs are produced by several different cells and IFN responses are likely driven by autocrine and paracrine signaling or intracellular activation of cytosolic pattern recognition receptors, like cGAS-STING. More studies are needed to determine the contributions of intercellular and intracellular IFN signal propagation after TBI.
Inhibiting IFN type I responses with a variety of methods in focal, diffuse, and mixed TBI reduced inflammation and improved recovery in rodent experimental studies. Differential gene expression (nCounter NanoString) analysis in cortex showed that TBI-induced IFN responses 7dpi were ablated by microglia depletion in mice [40, 46]. These data indicate that microglia-targeted interventions can reduce IFN signaling and subsequent inflammation. Moreover, several preclinical TBI studies demonstrate the benefits of attenuating IFN activation (Fig.5). Studies using focal injury (CCI) in mice showed increased expression of type I IFNs (IFNα and IFNβ) within 24 hours [96]. IFNAR1 knockout attenuated lesion volume and expression of proinflammatory genes Il1b and Il6 at acute time points after CCI (<24h) [96]. Furthermore, administration of an anti-IFNAR1 blocking antibody after TBI reduced gliosis and stride/locomotor deficits [96]. In another study, cortical expression of interferon-associated genes (Irf1, Irf7, Stat1) and pro-inflammatory genes (Tnf, Nox2, Ccl5, Cxcl10) was attenuated in Ifnb knock-out mice 72h after CCI [107]. In addition, the lesion induced by CCI 24h post-TBI and mRNA levels of autophagy-related genes were attenuated in STING knock-out mice compared to TBI-WT mice [98].
Figure 5. Inhibiting interferon (IFN) signaling in animal models improves inflammation and functional recovery after TBI.
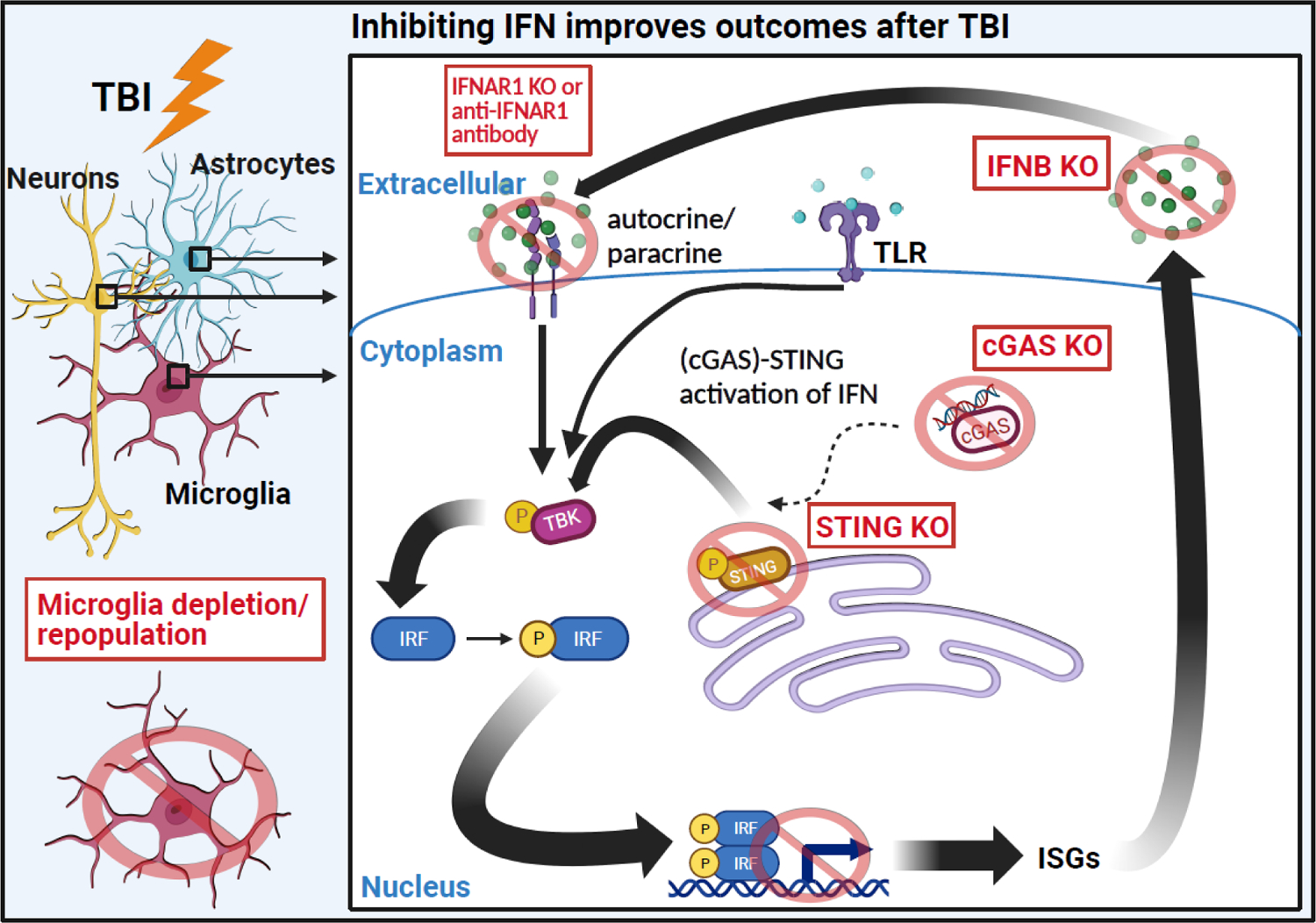
IFN gene expression is strongly induced by both focal and diffuse TBI in multiple cell types. Blocking the IFN response to TBI through various methods is generally beneficial to neuroinflammation and functional recovery. For instance, IFNAR (receptor) knockout or antibody inhibition decreases proinflammatory gene expression and lesion volume [96]. Direct knock-out of Ifnb also successfully ameliorates inflammatory gene expression [109]. Furthermore, knock-out of cGAS or STING, proteins that mediate the IFN response to TBI, improves neuronal pathology, inflammation, and functional recovery [98]. Figure created using Biorender.com.
Enhanced IFN signaling in the brain after TBI is observed in aged mice as well. CCI in aged mice amplified the expression of type I IFN genes Ifnb, Irf7, Ifi204, and Isg15 in the cortex 24h after TBI [108]. Furthermore, cGAS and pSTAT1 protein levels were increased by age after injury [108]. Adult and aged mice had increased gene expression associated with DNA damage and neuronal death, which may augment IFN signaling. Additionally, diffuse brain injury increased various master regulators in the cortex 7dpi associated with IFN signaling [56]. Furthermore, enhanced STING signaling in adult mice after diffuse TBI recapitulated gene expression associated with aged TBI [56]. Overall, enhanced type I IFN signaling contributes to detrimental neuroinflammation in adult and aged after TBI and may contribute to long-term microglia priming.
Concluding Remarks & Future Perspectives
This review aimed to discuss microglia priming and reactivity after TBI in the context of neuroinflammation, aging, and interferon signaling. As discussed in earlier sections, preclinical and clinical data show that neuroinflammation persists after focal and diffuse TBI and is associated with chronic microglial responses. Moreover, aging is a major factor that contributes to exaggerated inflammatory responses to TBI. Complicating matters is the potential for “microglial priming” and subsequent “reactivity” to secondary stressors, injuries, or immune challenges that further augment neuroinflammation and functional complications. Several aspects of these processes remain to be elucidated in future work (see Outstanding Questions). Preclinical studies also illustrate various strategies used to inhibit, eliminate, or force the turnover of microglia. In addition to serving as proof of concept, these studies highlight certain pathways as potential therapeutic targets. One potential pathway is interferon (IFN) signaling. IFN-driven gene expression is increased after TBI, and recent data indicate that this may set the stage for chronic microglial reactivity. Whether targeting interferon-driven inflammation may to ameliorate neuroinflammation and attenuate chronic microglia reactivity and priming in clinical settings remains to examined.
Outstanding Questions.
Are there clinical interventions that could be applied immediately after TBI to prevent microgliadriven neuroinflammation and long-term changes in glial profiles and brain function?
Are there clinical interventions that could be applied at later timepoints after TBI that would still benefit long-term recovery and chronic inflammation?
To what extent does age-related microglia priming worsen the aged brain’s response to TBI?
To what extent does activation of interferon signaling after TBI contribute to chronic microglia reactivity and primed profiles?
Which cell types are producing interferons and what is the contribution of damaged self-DNA to the activation and production of IFN responses?
Could interferon or other inflammatory-related interventions be applied after TBI to prevent microglia primed profiles and neuropsychiatric complications?
Highlights.
Traumatic brain injury (TBI) often entails chronic neuroinflammation, which contributes to the neuropsychiatric and cognitive complications often observed after TBI.
Following TBI, microglia promote chronic neuroinflammation that is associated with impaired functional recovery and sensitization to secondary insults.
In animal models, forced turn-over of microglia ameliorates TBI-induced neuroinflammation and associated deficits.
Chronic microglia activation and dysregulation in the aged brain promotes amplified neuroinflammation associated with impaired recovery and development of neurodegenerative disease pathology after TBI.
IFN- and STING-driven responses are potential regulators of microglial priming and could reveal therapeutic targets to improve outcomes after TBI.
Acknowledgements
This work was supported by National Institute of Health grant R01-NS118037 to JPG. LMW was supported by OSU Distinguished University Fellowship. In addition, the authors thank Chronic Brain Injury Program, Discovery Themes Initiative at Ohio State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests in relation to this work.
References
- 1.Perry DC, et al. , Association of traumatic brain injury with subsequent neurological and psychiatric disease: a meta-analysis. J Neurosurg, 2016. 124(2): p. 511–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gualtieri T and Cox DR, The delayed neurobehavioural sequelae of traumatic brain injury. Brain Injury, 1991. 5(3): p. 219–232. [DOI] [PubMed] [Google Scholar]
- 3.Bodnar CN, Morganti JM, and Bachstetter AD, Depression following a traumatic brain injury: uncovering cytokine dysregulation as a pathogenic mechanism. Neural Regen Res, 2018. 13(10): p. 1693–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Millis SR, et al. , Long-term neuropsychological outcome after traumatic brain injury. J Head Trauma Rehabil, 2001. 16(4): p. 343–55. [DOI] [PubMed] [Google Scholar]
- 5.Salmond CH, et al. , Changes over time in cognitive and structural profiles of head injury survivors. Neuropsychologia, 2006. 44(10): p. 1995–1998. [DOI] [PubMed] [Google Scholar]
- 6.Himanen L, et al. , Longitudinal cognitive changes in traumatic brain injury: a 30-year follow-up study. Neurology, 2006. 66(2): p. 187–92. [DOI] [PubMed] [Google Scholar]
- 7.Till C, et al. , Postrecovery cognitive decline in adults with traumatic brain injury. Arch Phys Med Rehabil, 2008. 89(12 Suppl): p. S25–34. [DOI] [PubMed] [Google Scholar]
- 8.Silver JM, McAllister TW, and Arciniegas DB, Depression and cognitive complaints following mild traumatic brain injury. Am J Psychiatry, 2009. 166(6): p. 653–61. [DOI] [PubMed] [Google Scholar]
- 9.Jackson JC, et al. , The association between delirium and cognitive decline: a review of the empirical literature. Neuropsychol Rev, 2004. 14(2): p. 87–98. [DOI] [PubMed] [Google Scholar]
- 10.Fleminger S, Long-term psychiatric disorders after traumatic brain injury. Eur J Anaesthesiol Suppl, 2008. 42: p. 123–30. [DOI] [PubMed] [Google Scholar]
- 11.Jorge RE, et al. , Major depression following traumatic brain injury. Arch Gen Psychiatry, 2004. 61(1): p. 42–50. [DOI] [PubMed] [Google Scholar]
- 12.Rao V, et al. , Aggression after traumatic brain injury: prevalence and correlates. J Neuropsychiatry Clin Neurosci, 2009. 21(4): p. 420–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamar Cory D., M.D.,, et al. , Post-Traumatic Epilepsy: Review of Risks, Pathophysiology, and Potential Biomarkers. The Journal of Neuropsychiatry and Clinical Neurosciences, 2014. 26(2): p. iv–113. [DOI] [PubMed] [Google Scholar]
- 14.Sharma S, et al. , Neuropathophysiological Mechanisms and Treatment Strategies for Posttraumatic Epilepsy. Front Mol Neurosci, 2021. 14: p. 612073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fordington S and Manford M, A review of seizures and epilepsy following traumatic brain injury. J Neurol, 2020. 267(10): p. 3105–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Asken BM, et al. , Research Gaps and Controversies in Chronic Traumatic Encephalopathy: A Review. JAMA Neurol, 2017. 74(10): p. 1255–1262. [DOI] [PubMed] [Google Scholar]
- 17.Daneshvar DH, et al. , Post-traumatic neurodegeneration and chronic traumatic encephalopathy. Mol Cell Neurosci, 2015. 66(Pt B): p. 81–90. [DOI] [PubMed] [Google Scholar]
- 18.Fleminger S, et al. , Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry, 2003. 74(7): p. 857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardner RC, et al. , Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol, 2014. 71(12): p. 1490–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardner RC, et al. , Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study. Neurology, 2018. 90(20): p. e1771–e1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gardner RC and Yaffe K, Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramos-Cejudo J, et al. , Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine, 2018. 28: p. 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang HK, et al. , Population based study on patients with traumatic brain injury suggests increased risk of dementia. J Neurol Neurosurg Psychiatry, 2012. 83(11): p. 1080–5. [DOI] [PubMed] [Google Scholar]
- 24.Masel BE and DeWitt DS, Traumatic brain injury: a disease process, not an event. J Neurotrauma, 2010. 27(8): p. 1529–40. [DOI] [PubMed] [Google Scholar]
- 25.Simon DW, et al. , The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol, 2017. 13(3): p. 171–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DiSabato DJ, Quan N, and Godbout JP, Neuroinflammation: the devil is in the details. J Neurochem, 2016. 139 Suppl 2(Suppl 2): p. 136–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Witcher KG, Eiferman DS, and Godbout JP, Priming the inflammatory pump of the CNS after traumatic brain injury. Trends Neurosci, 2015. 38(10): p. 609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks WM, et al. , Metabolic and cognitive response to human traumatic brain injury: a quantitative proton magnetic resonance study. J Neurotrauma, 2000. 17(8): p. 629–40. [DOI] [PubMed] [Google Scholar]
- 29.Ramlackhansingh AF, et al. , Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol, 2011. 70(3): p. 374–83. [DOI] [PubMed] [Google Scholar]
- 30.Johnson VE, et al. , Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain, 2013. 136(Pt 1): p. 28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gentleman SM, et al. , Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int, 2004. 146(2–3): p. 97–104. [DOI] [PubMed] [Google Scholar]
- 32.Coughlin JM, et al. , Imaging of Glial Cell Activation and White Matter Integrity in Brains of Active and Recently Retired National Football League Players. JAMA Neurol, 2017. 74(1): p. 6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coughlin JM, et al. , Neuroinflammation and brain atrophy in former NFL players: An in vivo multimodal imaging pilot study. Neurobiol Dis, 2015. 74: p. 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith C, et al. , The neuroinflammatory response in humans after traumatic brain injury. Neuropathol Appl Neurobiol, 2013. 39(6): p. 654–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niraula A, Sheridan JF, and Godbout JP, Microglia Priming with Aging and Stress. Neuropsychopharmacology, 2017. 42(1): p. 318–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee AJ and Ashkar AA, The Dual Nature of Type I and Type II Interferons. Frontiers in Immunology, 2018. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiong Y, Mahmood A, and Chopp M, Animal models of traumatic brain injury. Nature Reviews Neuroscience, 2013. 14(2): p. 128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shitaka Y, et al. , Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol, 2011. 70(7): p. 55167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Donat CK, et al. , Microglial Activation in Traumatic Brain Injury. Front Aging Neurosci, 2017. 9: p. 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Witcher KG, et al. , Traumatic Brain Injury Causes Chronic Cortical Inflammation and Neuronal Dysfunction Mediated by Microglia. J Neurosci, 2021. 41(7): p. 1597–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber MD, et al. , The Influence of Microglial Elimination and Repopulation on Stress Sensitization Induced by Repeated Social Defeat. Biological Psychiatry, 2019. 85(8): p. 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bray CE, et al. , Chronic Cortical Inflammation, Cognitive Impairment, and Immune Reactivity Associated with Diffuse Brain Injury Are Ameliorated by Forced Turnover of Microglia. The Journal of Neuroscience, 2022. 42(20): p. 4215–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spangenberg E, et al. , Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun, 2019. 10(1): p. 3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mouzon BC, et al. , Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol, 2014. 75(2): p. 241–54. [DOI] [PubMed] [Google Scholar]
- 45.Mukherjee S, et al. , Neuroinflammatory mechanisms of post-traumatic epilepsy. J Neuroinflammation, 2020. 17(1): p. 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witcher KG, et al. , Traumatic brain injury-induced neuronal damage in the somatosensory cortex causes formation of rod-shaped microglia that promote astrogliosis and persistent neuroinflammation. Glia, 2018. 66(12): p. 2719–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morganti JM, et al. , CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci, 2015. 35(2): p. 748–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Willis EF, et al. , Repopulating Microglia Promote Brain Repair in an IL-6-Dependent Manner. Cell, 2020. 180(5): p. 833–846.e16. [DOI] [PubMed] [Google Scholar]
- 49.Giordano KR, et al. , Colony-Stimulating Factor-1 Receptor Inhibition Transiently Attenuated the Peripheral Immune Response to Experimental Traumatic Brain Injury. Neurotrauma Rep, 2023. 4(1): p. 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, et al. , Early posttraumatic CSF1R inhibition via PLX3397 leads to time- and sexdependent effects on inflammation and neuronal maintenance after traumatic brain injury in mice. Brain Behav Immun, 2022. 106: p. 49–66. [DOI] [PubMed] [Google Scholar]
- 51.Henry RJ, et al. , Microglial Depletion with CSF1R Inhibitor During Chronic Phase of Experimental Traumatic Brain Injury Reduces Neurodegeneration and Neurological Deficits. J Neurosci, 2020. 40(14): p. 2960–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Neil SM, et al. , Forced turnover of aged microglia induces an intermediate phenotype but does not rebalance CNS environmental cues driving priming to immune challenge. Acta Neuropathol Commun, 2018. 6(1): p. 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ritzel RM, et al. , Brain injury accelerates the onset of a reversible age-related microglial phenotype associated with inflammatory neurodegeneration. Sci Adv, 2023. 9(10): p. eadd1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scott G, et al. , Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain, 2018. 141(2): p. 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar A, et al. , Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging, 2013. 34(5): p. 1397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wangler LM, et al. , Amplified Gliosis and Interferon-Associated Inflammation in the Aging Brain following Diffuse Traumatic Brain Injury. The Journal of Neuroscience, 2022. 42(48): p. 9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Greer JE, McGinn MJ, and Povlishock JT, Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. J Neurosci, 2011. 31(13): p. 5089–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lifshitz J and Lisembee AM, Neurodegeneration in the somatosensory cortex after experimental diffuse brain injury. Brain Struct Funct, 2012. 217(1): p. 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor SE, et al. , Rod microglia: a morphological definition. PLoS One, 2014. 9(5): p. e97096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ziebell JM, et al. , Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J Neuroinflammation, 2012. 9: p. 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bachstetter AD, et al. , Rod-shaped microglia morphology is associated with aging in 2 human autopsy series. Neurobiol Aging, 2017. 52: p. 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ji P, et al. , Peripheral viral infection induced microglial sensome genes and enhanced microglial cell activity in the hippocampus of neonatal piglets. Brain Behav Immun, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wierzba-Bobrowicz T, et al. , Morphological analysis of active microglia--rod and ramified microglia in human brains affected by some neurological diseases (SSPE, Alzheimer’s disease and Wilson’s disease). Folia Neuropathol, 2002. 40(3): p. 125–31. [PubMed] [Google Scholar]
- 64.Ritzel RM, et al. , Functional and transcriptional profiling of microglial activation during the chronic phase of TBI identifies an age-related driver of poor outcome in old mice. Geroscience, 2022. 44(3): p. 1407–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chou A, et al. , Persistent Infiltration and Impaired Response of Peripherally-Derived Monocytes after Traumatic Brain Injury in the Aged Brain. Int J Mol Sci, 2018. 19(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stevens B, et al. , The classical complement cascade mediates CNS synapse elimination. Cell, 2007. 131(6): p. 1164–78. [DOI] [PubMed] [Google Scholar]
- 67.Hong S, et al. , Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 2016. 352(6286): p. 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mallah K, et al. , Complement mediates neuroinflammation and cognitive decline at extended chronic time points after traumatic brain injury. Acta Neuropathol Commun, 2021. 9(1): p. 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alawieh A, et al. , Complement Drives Synaptic Degeneration and Progressive Cognitive Decline in the Chronic Phase after Traumatic Brain Injury. The Journal of Neuroscience, 2021. 41(8): p. 1830–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dejanovic B, et al. , Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nature Aging, 2022. 2(9): p. 837–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krukowski K, et al. , Traumatic Brain Injury in Aged Mice Induces Chronic Microglia Activation, Synapse Loss, and Complement-Dependent Memory Deficits. Int J Mol Sci, 2018. 19(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ritzel RM, et al. , Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiology of Aging, 2019. 77: p. 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Marschallinger J, et al. , Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nature Neuroscience, 2020. 23(2): p. 194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Singh Kushwaha S, Patro N, and Kumar Patro I, A Sequential Study of Age-Related Lipofuscin Accumulation in Hippocampus and Striate Cortex of Rats. Annals of Neurosciences, 2018. 25(4): p. 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ritzel RM, et al. , Brain injury accelerates the onset of a reversible age-related microglial phenotype associated with hyperphagocytosis and inflammatory neurodegeneration. bioRxiv, 2022: p. 2022.05.24.493292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen Y, et al. , Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab, 2003. 23(6): p. 748–55. [DOI] [PubMed] [Google Scholar]
- 77.Vogel DYS, et al. , Macrophages migrate in an activation-dependent manner to chemokines involved in neuroinflammation. Journal of Neuroinflammation, 2014. 11(1): p. 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alam A, et al. , Cellular infiltration in traumatic brain injury. Journal of Neuroinflammation, 2020. 17(1): p. 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morganti JM, et al. , Age exacerbates the CCR2/5-mediated neuroinflammatory response to traumatic brain injury. Journal of Neuroinflammation, 2016. 13(1): p. 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fenn AM, et al. , Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry, 2014. 76(7): p. 575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Muccigrosso MM, et al. , Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tapp ZM, et al. , Sleep fragmentation engages stress-responsive circuitry, enhances inflammation and compromises hippocampal function following traumatic brain injury. Exp Neurol, 2022. 353: p. 114058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Loane DJ, et al. , Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol, 2014. 73(1): p. 14–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dantzer R, Cytokine-induced sickness behavior: mechanisms and implications. Ann N Y Acad Sci, 2001. 933: p. 222–34. [DOI] [PubMed] [Google Scholar]
- 85.Henry CJ, et al. , Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both proinflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun, 2009. 23(3): p. 309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O’Neil SM, et al. , Astrocyte immunosenescence and deficits in interleukin 10 signaling in the aged brain disrupt the regulation of microglia following innate immune activation. Glia, 2022. [DOI] [PubMed] [Google Scholar]
- 87.Godbout JP, et al. , Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology, 2008. 33(10): p. 2341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Godbout JP, et al. , Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J, 2005. 19(10): p. 1329–31. [DOI] [PubMed] [Google Scholar]
- 89.Chen J, et al. , Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun, 2008. 22(3): p. 301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Meurs E, et al. , Molecular cloning and characterization of the human double-stranded RNAactivated protein kinase induced by interferon. Cell, 1990. 62(2): p. 379–390. [DOI] [PubMed] [Google Scholar]
- 91.Nayak D, et al. , Type I Interferon Programs Innate Myeloid Dynamics and Gene Expression in the Virally Infected Nervous System. PLOS Pathogens, 2013. 9(5): p. e1003395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Paul S, et al. , Type I interferon response in the central nervous system. Biochimie, 2007. 89(6): p. 770–778. [DOI] [PubMed] [Google Scholar]
- 93.Gessani S, et al. , Type I interferons as regulators of human antigen presenting cell functions. Toxins (Basel), 2014. 6(6): p. 1696–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang Y, et al. , An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci, 2014. 34(36): p. 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tran KM, et al. , A Trem2(R47H) mouse model without cryptic splicing drives age- and diseasedependent tissue damage and synaptic loss in response to plaques. Mol Neurodegener, 2023. 18(1): p. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Karve IP, et al. , Ablation of Type-1 IFN Signaling in Hematopoietic Cells Confers Protection Following Traumatic Brain Injury. eneuro, 2016. 3(1): p. ENEURO.0128–15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Decout A, et al. , The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nature Reviews Immunology, 2021. 21(9): p. 548–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fritsch LE, et al. , Type I Interferon Response Is Mediated by NLRX1-cGAS-STING Signaling in Brain Injury. Frontiers in Molecular Neuroscience, 2022. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yamamoto M, et al. , Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science, 2003. 301(5633): p. 640–3. [DOI] [PubMed] [Google Scholar]
- 100.Tabeta K, et al. , Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A, 2004. 101(10): p. 3516–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Abdullah A, et al. , STING-mediated type-I interferons contribute to the neuroinflammatory process and detrimental effects following traumatic brain injury. J Neuroinflammation, 2018. 15(1): p. 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Abe T and Barber GN, Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol, 2014. 88(10): p. 532841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yum S, et al. , TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci U S A, 2021. 118(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.de Weerd NA and Nguyen T, The interferons and their receptors--distribution and regulation. Immunol Cell Biol, 2012. 90(5): p. 483–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Todd BP, et al. , Traumatic brain injury results in unique microglial and astrocyte transcriptomes enriched for type I interferon response. Journal of Neuroinflammation, 2021. 18(1): p. 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bolte AC, et al. , The meningeal transcriptional response to traumatic brain injury and aging. Elife, 2023. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barrett JP, et al. , Interferon-beta plays a detrimental role in experimental traumatic brain injury by enhancing neuroinflammation that drives chronic neurodegeneration. J Neurosci, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barrett JP, et al. , Traumatic Brain Injury Induces cGAS Activation and Type I Interferon Signaling in Aged Mice. Frontiers in Immunology, 2021. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Barrett JP, et al. , Interferon-β Plays a Detrimental Role in Experimental Traumatic Brain Injury by Enhancing Neuroinflammation That Drives Chronic Neurodegeneration. The Journal of Neuroscience, 2020. 40(11): p. 2357–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]