

Abstract
Introduction:
Impaired autophagy is a pathogenic mechanism in the synucleinopathies. Sirolimus, a potent mTOR inhibitor and autophagy activator, had no beneficial effects in a randomized placebo-controlled trial in patients with multiple system atrophy (MSA). Whether sirolimus effectively inhibited brain mTOR activity was unknown. We aimed to evaluate if patients with MSA treated with sirolimus had evidence of inhibited brain mTOR pathways by measuring neuron-derived serum extracellular vesicles (NEVs).
Methods:
Serum samples were collected from participants of the sirolimus-MSA trial, which randomized patients to sirolimus (2–6 mg/day) or placebo for 48-weeks. NEVs were immunoprecipitated with three antibodies-against neurons. Brain mTOR engagement was quantified as the change in the NEV phosphorylated mTOR (p-mTOR) to total-mTOR (tot-mTOR) ratio after 48-weeks of sirolimus.
Results:
Samples from 27 patients [mean (SD) age, 59.2 (7) years, 15 (55.5%) men] were analyzed (19 sirolimus, 8 placebo). Treated- and placebo-patients had similar p-mTOR:tot-mTOR ratio at 24- (placebo: 0.248 ± 0.03, sirolimus: 0.289 ± 0.02; P = 0.305) and 48-weeks (placebo: 0.299 ± 0.05, sirolimus: 0.261 ± 0.03; P = 0.544). The tot-mTOR, p-mTOR, or their ratio levels were not associated with Unified MSA Rating Scale (UMSARS) worsening.
Discussion:
These results are consistent with no brain mTOR engagement by sirolimus up to 6 mg/day. NEV-based biomarkers are a rational approach to investigating target engagement in clinical trials of brain-targeted therapeutics.
INTRODUCTION
Extracellular vesicles (EVs), including exosomes, are nanosized membranous particles secreted by cells, including neurons, that circulate in the blood and contain molecules representative of their origin. EVs cross the blood-brain barrier and EVs of neuronal origin (in short NEVs) can be isolated from peripheral blood to quantify their content [1]. We have used α-synuclein (αSyn) measurements in blood NEVs to distinguish Parkinson disease (PD) versus controls [2], and versus multiple system atrophy (MSA)[3]. We have also measured blood NEVs to evaluate levels of the brain insulin-signaling proteins and downstream effectors of the mechanistic target of rapamycin (mTOR) in patients with PD in the exenatide trial [4]. The utility of this technique in revealing the mechanisms of action of CNS drugs is increasingly recognized.
In synucleinopathies, impaired autophagy contributes to αSyn aggregation and spread [5]. mTOR activity inhibition with sirolimus (a.k.a. rapamycin) results in autophagy activation [6]. In synucleinopathy models, sirolimus reduced αSyn accumulation suggesting that treatment with sirolimus could prevent αSyn-induced neurodegeneration [7–9].
We recently used sirolimus for potential disease modification in a 48-week randomized, placebo-controlled clinical trial in patients with MSA. This study generated negative results [10]. Lack of target engagement was considered a likely cause for this failure, however, no measurements of the brain mTOR pathways were possible at the time because lumbar puncture and CSF collection were not included as assessments in the trial. Given our negative trial data, we hypothesized that sirolimus-treated MSA patients would show no differences in mTOR signaling proteins or downstream targets in NEVs compared to placebo-treated MSA patients, indicating no inhibition of brain mTOR pathways. To test this hypothesis, we measured changes in mTOR signaling biomarkers in NEVs from patients’ serum samples obtained at baseline, 24 weeks, and 48 weeks of treatment with sirolimus or placebo.
METHODS
Study design and participants
This is a post-hoc analysis of blood biomarkers from samples obtained in the sirolimus-MSA trial. The sirolimus trial was a randomized, participant- and investigator-blinded, placebo-controlled, single-center, clinical trial in patients with probable MSA according to the 2008 MSA Diagnostic Criteria, randomly assigned (3:1) to sirolimus (2–6 mg daily) for 48-weeks or placebo (ClinicalTrials.gov: NCT03589976). At each visit, patients donated blood samples for biomarker outcomes at baseline, week 24, and week 48 [10]. Patients consented to future analysis of all samples collected during this trial as part of the original trial consent process. The trial was conducted at the New York University Grossman School of Medicine and approved by its IRB (Study ID: S17-01392). All patients provided written informed consent and all data were deidentified. The trial was stopped after an interim analysis met their pre-defined futility criteria.
Serum sample collection
Blood samples were collected in accordance with preprocessing guidelines for EV-based biomarker analysis [11]. Samples from baseline, week 24, and week 48 were analyzed. Samples from 19 patients receiving sirolimus and 8 patients receiving placebo who completed the 24-week visit, and from 9 patients receiving sirolimus and 3 receiving placebo who completed the 48-week visit were analyzed for this secondary analysis of blood NEVs.
Quantification of EV mTOR signaling proteins in EVs enriched for neuronal origin
Total EV isolation, performed with the SmartSEC™ HT EV Isolation System, was followed by immunoaffinity-based EV capture for Neuroligin-1, GAP43, and L1CAM (5G3 clone). This two-step procedure allowed for the enrichment of EVs of neuronal origin from the patients’ plasma samples.
Enriched EVs in suspension were lysed with RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific). We quantified total-mTOR and phospho-mTOR using xMAP® bead-based 2-Plex Phospo/Total mTOR Luminex Assay (Millipore Sigma; 48-625MAG); moreover, to distinguish between the two mTOR complexes, mTORC1 and 2 were quantified using ELISAs (MyBioSource; MBS1600031 and MBS1600032). To characterize EV composition of the NEV isolate, we used the Exosome Characterization 6-Plex Human ProcartaPlex Luminex Assay (Thermo Fisher; EPX060-15845-901) to measure concentrations of the tetraspanins (CD9, CD63, CD81) as well as cytochrome c, syntenin-1, VLA-4. We used CD9 concentration to normalize NEV biomarkers for EV yield. This assay further demonstrated that NEV preparations had significantly higher levels of CD9 and CD63 compared to total EVs (p<0.001 and p<0.001 respectively) and EV-depleted plasma (p<0.001 and p<0.001 respectively), suggesting effective EV isolation and enrichment (Supplementary Table S1). All assays were conducted in duplicate, and the mean coefficients of variance were less than 30%.
Statistical Analysis
Biomarker levels were normalized to CD9 levels to control for EV abundance per sample, or reported as a ratio of phosphorylated mTOR (p-mTOR) to total-mTOR (tot-mTOR). The effect of sirolimus treatment on biomarker levels was evaluated by fitting a linear mixed-effects model with the treatment groups and visit as fixed effects and participant as a random effect, followed by a Wald test for each fixed factor. To assess whether longitudinal changes in biomarker levels were associated with longitudinal changes in clinical scores, we performed a Spearman’s correlation between the change in biomarker levels and the change in UMSARS-1 and UMSARS-2 scores from baseline to week 24 for the placebo- and sirolimus-treated groups separately. To test for differences in the slopes of linear regressions of each treatment group, an F-test of the slope of each regression was compared to a global model with a shared slope among both groups. P < 0.05 was considered statistically significant.
The Supplementary Methods provide additional details on the methodology to isolate and capture NEVs, as well as the quantification of mTOR signaling proteins in NEVs and the statistical analysis.
RESULTS
Patient characteristics
Samples from 27 patients [mean (±SD) age, 59.2±7 years, 15 (55.5%) men] were available for this study: 19 in the sirolimus group and 8 in the placebo group. Patient demographics and baseline characteristics were generally similar between the two groups, although sirolimus-treated participants were slightly older and had slightly more advanced UMSARS-1 scores than placebo-assigned participants (Table 1).
Table 1.
Baseline clinical and demographic characteristics of randomized patients
| Sirolimus group (n=19) | Placebo group (n=8) | |
|---|---|---|
| Age, years | 60± 7.3 | 57± 6.2 |
| Sex | ||
| Women | 8 (42%) | 4 (50%) |
| Men | 11 (58%) | 4 (50%) |
| Ethnicity | ||
| Caucasian including Hispanic | 17 (90%) | 8 (100%) |
| Asian | 1 (5%) | 0 |
| Afro-American | 1 (5%) | 0 |
| Time since diagnosis, years | 1.2± 0.7 | 1.5± 0.8 |
| Predominant motor phenotype | ||
| Parkinsonian | 10 (52%) | 6 (75%) |
| Cerebellar | 9 (48%) | 2 (25%) |
| Diagnostic certainty | ||
| Probable | 19 (100%) | 8 (100%) |
| UMSARS | ||
| Activities of daily living (UMSARS-1) | 21± 4.6 | 18.9 ± 5.9 |
| Motor examination (UMSARS-2) | 22.8 ± 5.4 | 22.4 ± 5.8 |
| Global disability score (UMSARS-4) | 2.8± 0.9 | 3 ± 0.9 |
Data are mean (SD) or n (%). UMSARS=Unified Multiple System Atrophy Rating Scale.
Association of sirolimus with EV biomarker changes
As measures of target engagement, we assessed whether sirolimus treatment altered the p-mTOR:total-mTOR ratio levels in NEVs as an indicator of mTOR activity, along with the levels of total-mTOR, mTORC1, and mTORC2. Sirolimus-treated patients had similar EV-derived biomarker levels at baseline, week-24, and week-48 compared to placebo-treated participants (Figure 1A–C, Supplementary Table 2). The p-mTOR:tot-mTOR ratio levels were similar at week 24 and week 48 compared to baseline, for both sirolimus- and placebo-treated participants. However, p-mTOR:CD9 and tot-mTOR:CD9 levels were significantly higher at week 24 and week 48 compared to baseline, in both sirolimus- and placebo-treated participants (Figure 1A–C, Supplementary Table 2).
Figure 1. Effect of sirolimus on NEV mTOR biomarkers and associations with disease progression.
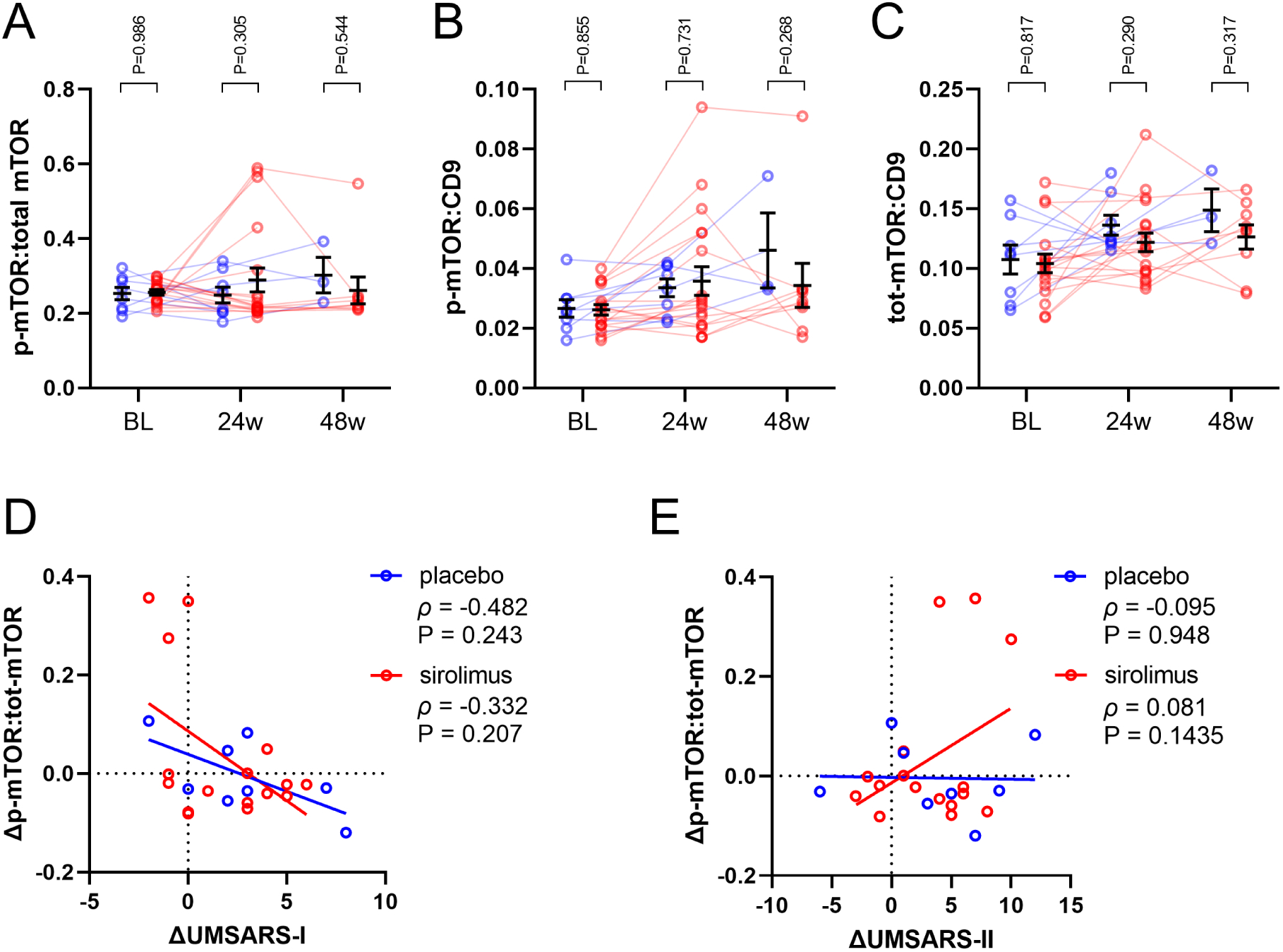
p-mTOR and tot-mTOR levels were measured from neuronally-enriched EVs from placebo- and sirolimus-treated subjects. Each measure was normalized to CD9 levels to account for EV abundance in the sample. Sirolimus treatment did not change (A) p-mTOR:tot-mTOR, (B) p-mTOR:CD9, or (C) tot-mTOR:CD9. Analyte values are plotted for each subject with a line connecting symbols from each individual. The horizontal line and error bars denote mean ± SEM, P-values for placebo vs. sirolimus post-test with Tukey’s correction for multiple comparisons. Spearman correlations were calculated for the change of p-mTOR:tot-mTOR levels vs. the change in (D) UMSARS-I or (E) UMSARS-II between the 24-week and baseline visits. Spearman ρ and P-values are reported.
Association of biomarkers with clinical rating scales
To determine whether clinical measures of disease progression correlated with changes in the levels of these proteins, we calculated the change-change correlations between each protein and UMSARS-1 and UMSARS-2 scores between the 24-week and baseline visits. There was no correlation between changes in these proteins and clinical scores (Figure 1D–E, Supplementary Table 3). Additionally, the slopes of the linear regression between the sirolimus and control groups did not differ for either measure (Supplementary Table 4).
DISCUSSION
Our results indicate that sirolimus at dosages 2–6 mg/day was unable to inhibit the brain mTOR pathway in patients with MSA, thus confirming our hypothesis of lack of target engagement and providing a putative explanation for the failure of the sirolimus trial.
In preclinical models, mTOR inhibition with sirolimus reduced αSyn accumulation suggesting that treatment with sirolimus could prevent αSyn-induced neurodegeneration [7–9]. Despite this promising preclinical data, our clinical trial, the first testing an mTOR inhibitor in any neurodegenerative disorder, showed no evidence of clinical or biomarker benefit [10]. Sirolimus is lipophilic, and early human evidence hinted at the possibility that it might cross the brain-blood barrier and inhibit mTOR activity in the CNS, specifically in patients with refractory epilepsy and glioblastoma multiforme, disorders in which the blood-brain barrier might be compromised [12, 13]. Our study, however, supports the notion that oral sirolimus at dosages 2–6 mg/day is unable to inhibit brain mTOR in patients with MSA. Whether a higher dose could have resulted in brain target engagement is unknown. However, chronic sirolimus dosages > 6 mg/day are challenging in the clinic, as they are associated with a higher risk of intolerable side effects. It remains to be seen if improved mTOR inhibitors (e.g., with higher brain permeability and selectivity) could result in disease-modifying effects in patients with neurodegeneration.
MSA is characterized by αSyn accumulation predominantly in glia, in contrast to predominant neuronal αSyn accumulation in PD. Because our goal was to understand if sirolimus engaged the mTOR pathway in the CNS, irrespective of the predominant cell form affected in MSA, we measured NEVs only and not glial-derived EVs. In any case, a differential target engagement in neurons vs. glia would be unexpected and inconsistent with the lack of benefit observed in the sirolimus MSA trial.
Our NEV isolation approach has limitations. No currently available technique is flawless for NEV isolation; however, combining two techniques (i.e., particle isolation with Size Exclusion Chromatography, and immune capture) as we did, is a promising strategy to enrich for NEVs [11]. To isolate a population of NEVs, we used antibodies against three neuronal proteins expressed in different cellular compartments, deviating from the common approach of only relying on L1CAM. Paradoxically, the negative findings of this study are consistent with effective neuronal enrichment, given that there is abundant evidence documenting mTOR inhibition by sirolimus and similar rapalogs in non-CNS tissues [14]. We found no evidence of such inhibition with our NEV isolation technique, indicating that we effectively isolated a population of EVs originating from cells not reached by sirolimus. Another potential limitation is the small sample size.
Intriguingly, we found that the levels of NEV p-mTOR and tot-mTOR normalized to CD9 increased at week-24 and week-48, compared to baseline, in all participants regardless of treatment allocation. The interpretation of this finding remains elusive. While this might suggest that these NEV biomarkers could track MSA disease progression, none of these correlated with longitudinal UMSARS changes, making such an interpretation unlikely (Supplementary Table 3). Notably, we previously described correlations between other biomarkers and longitudinal UMSARS changes [10].
In summary, we present human biomarker evidence that oral sirolimus at dosages up to 6 mg/day does not engage or inhibit brain mTOR pathways in patients with MSA, suggesting that oral sirolimus at such doses does not cross the brain-blood barrier at concentrations sufficient to inhibit mTOR activity. Our results have implications for the use of mTOR inhibitors for CNS disorders. The use of NEVs obtained from blood provides a potential method to determine CNS target engagement that should be further investigated in future trials of disease-modifying interventions.
Supplementary Material
Highlights.
Sirolimus is an autophagy activator with neuroprotective effects in synucleinopathy models.
A placebo-controlled sirolimus trial in multiple system atrophy (MSA) showed no benefit
Using blood samples from the trial participants, we isolated neuron-derived extracellular vesicles to measure mTOR biomarkers
There were no differences in mTOR biomarkers in sirolimus-treated vs placebo patients.
Our results suggest that oral sirolimus did not engage the brain mTOR pathway.
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institute on Aging, NIH; and in part by NINDS R01NS107596 and U01NS122419
Financial disclosures:
KAP: research support from the Intramural Research Program from the NIH. TM: No disclosures. WY: research support from the Intramural Research Program from the NIH. HK: research funding from the NIH, Michael J. Fox Foundation, MSA Coalition, Familial Dysautonomia Foundation, FDA, and Biogen; is an advisory Board Member for Lundbeck, Biogen, Biohaven, Theravance, PTC Therapeutics, ONO, Takeda, Vaxxinity, and Lilly; is Editor-in-Chief of Clinical Autonomic Research. UJK: research funding from the NIH. Advisory board member of Amprion. DK: research support from the Intramural Research Program from the NIH. JAP: Research funding from the NIH. Salary from Novartis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- [1].Mustapic M, Eitan E, Werner JK Jr., Berkowitz ST, Lazaropoulos MP, Tran J, Goetzl EJ, Kapogiannis D, Plasma Extracellular Vesicles Enriched for Neuronal Origin: A Potential Window into Brain Pathologic Processes, Front Neurosci 11 (2017) 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Blommer J, Pitcher T, Mustapic M, Eren E, Yao PJ, Vreones MP, Pucha KA, Dalrymple-Alford J, Shoorangiz R, Meissner WG, Anderson T, Kapogiannis D, Extracellular vesicle biomarkers for cognitive impairment in Parkinson’s disease, Brain 146(1) (2023) 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dutta S, Hornung S, Kruayatidee A, Maina KN, Del Rosario I, Paul KC, Wong DY, Duarte Folle A, Markovic D, Palma JA, Serrano GE, Adler CH, Perlman SL, Poon WW, Kang UJ, Alcalay RN, Sklerov M, Gylys KH, Kaufmann H, Fogel BL, Bronstein JM, Ritz B, Bitan G, alpha-Synuclein in blood exosomes immunoprecipitated using neuronal and oligodendroglial markers distinguishes Parkinson’s disease from multiple system atrophy, Acta Neuropathol 142(3) (2021) 495–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Athauda D, Gulyani S, Karnati HK, Li Y, Tweedie D, Mustapic M, Chawla S, Chowdhury K, Skene SS, Greig NH, Kapogiannis D, Foltynie T, Utility of Neuronal-Derived Exosomes to Examine Molecular Mechanisms That Affect Motor Function in Patients With Parkinson Disease: A Secondary Analysis of the Exenatide-PD Trial, JAMA Neurol 76(4) (2019) 420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Scrivo A, Bourdenx M, Pampliega O, Cuervo AM, Selective autophagy as a potential therapeutic target for neurodegenerative disorders, Lancet Neurol 17(9) (2018) 802–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kim YC, Guan KL, mTOR: a pharmacologic target for autophagy regulation, J Clin Invest 125(1) (2015) 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bove J, Martinez-Vicente M, Vila M, Fighting neurodegeneration with rapamycin: mechanistic insights, Nature reviews. Neuroscience 12(8) (2011) 437–52. [DOI] [PubMed] [Google Scholar]
- [8].Lopez-Cuina M, Guerin P, Bezard E, Meissner W, Fernagut PO, Rapamycin for treating MSA: A preclinical proof of concept study, Movement Disord 33 (2018) S441–S441. [Google Scholar]
- [9].Gao J, Perera G, Bhadbhade M, Halliday GM, Dzamko N, Autophagy activation promotes clearance of alpha-synuclein inclusions in fibril-seeded human neural cells, J Biol Chem 294(39) (2019) 14241–14256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Palma JA, Martinez J, Millar Vernetti P, Ma T, Perez MA, Zhong J, Qian Y, Dutta S, Maina KN, Siddique I, Bitan G, Ades-Aron B, Shepherd TM, Kang UJ, Kaufmann H, mTOR Inhibition with Sirolimus in Multiple System Atrophy: A Randomized, Double-Blind, Placebo-Controlled Futility Trial and 1-Year Biomarker Longitudinal Analysis, Mov Disord 37(4) (2022) 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Coumans FAW, Brisson AR, Buzas EI, Dignat-George F, Drees EEE, El-Andaloussi S, Emanueli C, Gasecka A, Hendrix A, Hill AF, Lacroix R, Lee Y, van Leeuwen TG, Mackman N, Mager I, Nolan JP, van der Pol E, Pegtel DM, Sahoo S, Siljander PRM, Sturk G, de Wever O, Nieuwland R, Methodological Guidelines to Study Extracellular Vesicles, Circ Res 120(10) (2017) 1632–1648. [DOI] [PubMed] [Google Scholar]
- [12].Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, Hsueh T, Chen Y, Wang W, Youngkin D, Liau L, Martin N, Becker D, Bergsneider M, Lai A, Green R, Oglesby T, Koleto M, Trent J, Horvath S, Mischel PS, Mellinghoff IK, Sawyers CL, Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma, PLoS Med 5(1) (2008) e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Parker WE, Orlova KA, Parker WH, Birnbaum JF, Krymskaya VP, Goncharov DA, Baybis M, Helfferich J, Okochi K, Strauss KA, Crino PB, Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder, Sci Transl Med 5(182) (2013) 182ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lamming DW, Inhibition of the Mechanistic Target of Rapamycin (mTOR)-Rapamycin and Beyond, Cold Spring Harb Perspect Med 6(5) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.