

Abstract
Objective
Gastric cancer (GC) ranks fifth in incidence and fourth for mortality worldwide. The response to immune checkpoint blockade (ICB) therapy in GC is heterogeneous due to tumor-intrinsic and acquired immunotherapy resistance. We developed an immunophenotype-based subtyping of human GC based on immune cells infiltration to develop a novel treatment option.
Design
A algorithm was developed to reclassify GC into immune inflamed, excluded, and desert subtypes. Bioinformatics, human and mouse GC cell lines, syngeneic murine gastric tumor model, and CTLA4 blockade were utilized to investigate the immunotherapeutic effects by restricting receptor tyrosine kinase (RTK) signaling in immune desert (ICB-resistant) type GC.
Results
Our algorithm re-stratified subtypes of human GC in public databases and showed that immune desert- and excluded-type tumors are ICB-resistant compared with immune-inflamed GC. Moreover, epithelial-mesenchymal transition (EMT) signaling was highly enriched in immune desert-type GC, and syngeneic murine tumors exhibiting mesenchymal-like, compared with epithelial-like, properties are T cell-excluded and resistant to CTLA4 blockade. Our analysis further identified a panel of receptor tyrosine kinases (RTKs) as potential druggable targets in the immune desert-type GC. Dovitinib, an inhibitor of multiple RTKs, strikingly repressed EMT programming in mesenchymal-like immune desert syngeneic GC models. Dovitinib activated the tumor-intrinsic SNAI1/2-IFN-γ signaling axis and impeded the EMT program, converting immune desert-type tumors to immune inflamed-type tumors, sensitizing these mesenchymal-like ‘cold’ tumors to CTLA4 blockade.
Conclusion
Our findings identified potential druggable targets relevant to patient groups, especially for refractory immune desert-type/ ‘cold’ GC. Dovitinib, an RTK inhibitor, sensitized desert-type immune-cold GC to CTLA4 blockade by restricting EMT and recruiting T cells.
Keywords: immunophenotype, multiple RTKs, gastric cancer, EMT, tumor immune evasion
Introduction
Gastric cancer is the fifth most common cancer worldwide with an estimated 26,380 new cases and 11,090 deaths in the United States in 2022 [1, 2]. Furthermore, in countries without screening programs, most patients with gastric cancer present with distant metastasis showing poor response to standard chemotherapy with 5-year survival of 5% in these patients [3]. Therefore, there is a critical need to identify novel therapeutic strategies in the fight against gastric cancer.
Over the past decade, immunotherapy has revolutionized our treatment approaches in several cancer types[4, 5]. Immune checkpoint blockade (ICB) activates T-cells by blocking immune-inhibitory receptors and ligands, such as CTLA4[6, 7] and PD-1/PD-L1[8, 9]. ICB is changing the therapeutic strategies from broadly targeting tumors with chemotherapy to modulating immune responses against cancer cells. The clinical benefits of the successful immunotherapeutic intervention have been encouraging in multiple types of cancer, particularly in patients harboring hypermutated tumors [10, 11, 12]. However, the heterogeneous responses to ICB have limited its wide applicability [13, 14, 15]. A major unmet need in the field, therefore, is not only predicting which tumors are ICB resistant but also identifying and targeting novel underlying mechanisms of ICB resistance in tumors less sensitive to immunotherapy.
To better elaborate on the impacts of the number of infiltrated T cells and its spatial distribution on immunotherapeutic efficacy, Hegde et al. proposed the concept of tumor immunophenotype (IP) [16], including the inflamed-type, the excluded-type, and the desert-type. Immune-inflamed tumors, so-called ‘hot’ tumors, are characterized by high T cell infiltration and are more responsive to ICBs. Immune excluded-tumors and immune desert-tumors can be described as ‘cold’ tumors that are less or not responsive to ICBs. In immune excluded tumors, CD8+ T cells localize only at the periphery (invasive margin) of the tumors but do not penetrate deeply into the tumor core for destruction. In immune desert tumors, CD8+ T cells are absent from both the tumor core and periphery.
Beyond T-cell infiltration [17] [18], there are increasing reports providing evidence that epithelial-mesenchymal transition (EMT) plays a critical role in ICB resistance in various cancer types, such as breast cancer and lung cancer[19, 20]. The mesenchymal-like state of tumor cells has been correlated with immunosuppression, such as reduced CD4+/CD8+ T-cell infiltration, as well as enrichment of immunosuppressive M2-like macrophages and regulatory T-cells (Treg) in the tumor microenvironment (TME) [21]. EMT is a dynamic and reversible process in which quiescent epithelial cancer cells lose intercellular adhesion, transdifferentiate into aggressive mesenchymal-like cells, and trigger tumor metastasis[22, 23]. Typically, the activation of the EMT program sheds E-cadherin and overexpresses mesenchymal marker genes (Vimentin and Fibronectin) and core EMT-transcription factors (EMT-TFs), such as Snail, Slug, Twist, and Zeb1. EMT-TFs promote tumor immune evasion by upregulating multiple immune inhibitory chemokines to assemble an immunosuppressive TME[24], which may cause tolerance to ICB immunotherapy. However, the underlying mechanisms of EMT-assembled immunosuppressive TME remain largely unknown in GC.
In the present study, we develop a novel algorithm to accurately predict immunophenotypes from human gastric cancer specimens and GC patient-derived bulk transcriptomic data. Interrogation of putative signaling pathways underlying this immunophenotypic reclassification of GC reveals a panel of receptor tyrosine kinases (RTKs; i.e., FGFR1, PDGFRB, etc.) that are highly enriched in immune-desert tumors in GC and other cancers. A multivalent RTK inhibitor dovitinib restricts EMT programming, restores T-cell infiltration, and improves responses to anti-CTLA4 ICB in a syngeneic, immunocompetent murine model of mesenchymal-like GC. Our findings will contribute to treating ICB-refractory immune desert ‘cold’ tumors in a diverse spectrum of carcinomas.
Materials and Methods
The detailed materials and methods can be obtained in online supplementary methods.
Results
Immune desert-type human gastric tumors show EMT signaling enrichment and exhibit immunotherapeutic resistance and poor prognosis
Understanding the immunophenotypes of tumors is essential to predict patients’ responses to immunotherapies, such as anti-PD-L1 and anti-CTLA4 immune checkpoint blockades. In the past several years, increased studies have correlated the signature gene sets with different immunophenotypes[25, 26], making it possible to stratify the tumor immunophenotypes through ‘bulk’ transcriptome analysis. To determine molecular signatures for each immunophenotype, we performed RNA-sequencing analysis of 36 human gastric cancer (GC) tissue samples. These samples were stratified into immune-inflamed, immune-excluded, and immune-desert tumors based on the histological identification of spatial CD8+ T-cell distribution in the tumor microenvironment (TME) (Fig. 1A). Ultimately, we identified 12 immune-inflamed tumors, 16 immune-excluded tumors, and 8 immune-desert tumors (Fig. 1B). We then utilized tumor transcriptomes to perform gene set enrichment analysis (GSEA) and applied hallmark gene sets and GO biological processes such as immune cell signature, metabolism, and stem cell signature gene sets to interrogate heterogeneity among these three immunophenotypes. This revealed disproportionate enrichment of IFN-γ response, estrogen response, and EMT signatures in immune-inflamed, -excluded, and -desert tumors, respectively (Fig. 1C).
Figure 1. Construction of the new algorithm, the predicted immunophenotypes (predicted IP) via two signature gene sets.
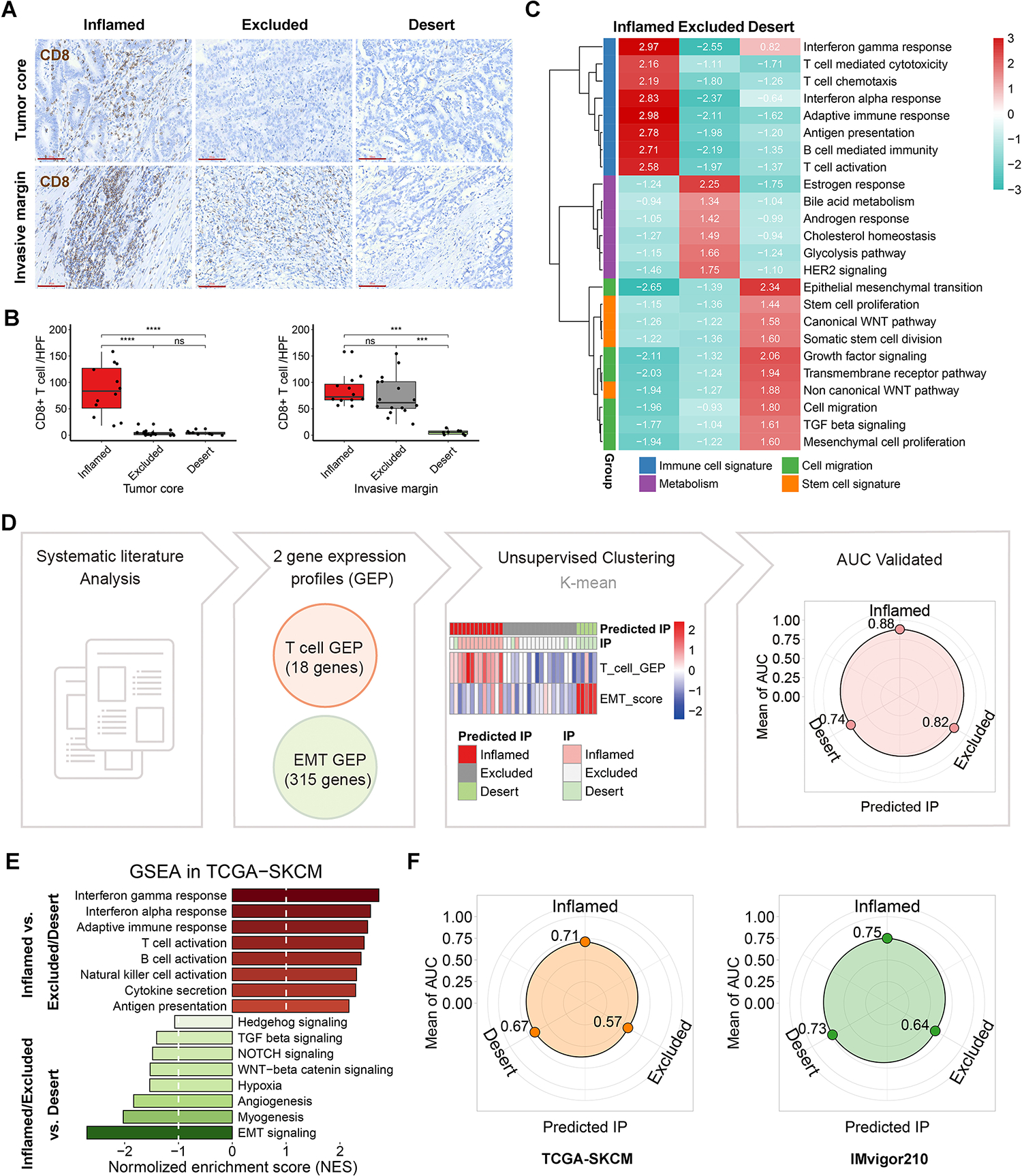
(A) Representative immunohistochemistry staining (scale bars, 80μm) of CD8 in tumor core and invasive margin of 36 human gastric tumors. (B) The quantification of CD8+ T cell per tumor (the black spot) was shown as the mean value of 3 independent high-power fields (HPF) in tumor. ns indicate no significance, **p < 0.01, ***p < 0.001, ****p < 0.0001. (C) Gene set enrichment analysis (GSEA) was employed in the local cohort, using a panel of functional gene sets from GO biological processes item and hallmarks item in MSigDB database of the Broad Institute. The values indicate normalized enrichment score (NES). (D) Unsupervised clustering of the 36 tumors was performed using the scores of T cell gene expression profile (GEP) and EMT signature. The predictive performance was verified by area under curve (AUC) of receiver operating curve analysis. (E) GSEA was performed in TCGA SKCM cohort. (F) The predictive performances of TCGA SKCM cohort and IMvigor210 cohort.
We next examined whether these transcriptomic signatures are concordant with known immunophenotypic classifications. We identified two previously described transcriptomic signatures as the basis for our new algorithm: (1) pan-cancer T cell-inflamed gene expression profile (GEP) in 220 patients with 9 cancers[25]; (2) pan-cancer transcriptomic EMT signatures of ovarian, breast, bladder, lung, colorectal and gastric cancers[26]. Unsupervised clustering of the 36 samples was performed using the single sample GSEA scores of the two signatures/GEPs. Our predicted immunophenotypes (predicted IP) and the histology-identified immunophenotypes were highly identical. The prediction accuracy, as measured by the area under the curve (AUC), was 0.88 in the inflamed tumors, 0.82 in the excluded tumors, and 0.74 in the desert tumors (Fig. 1D). In line with the results from our local gastric cancer cohort, the predominant immune relative signatures in the inflamed-type and the strong EMT signature in the desert-type were confirmed in The Cancer Genome Atlas Program (TCGA) SKCM cohort (Fig. 1E). Receiver operating curve analysis further validated the high performance of the predicted IP, especially for the inflamed and desert types in TCGA SKCM cohort and urinary tract tumor cohort (IMvigor210) (Fig. 1F).
To validate the broad applicability of our IP methodology, we stratified tumors in multiple TCGA datasets, including stomach (STAD) (Fig. 2A), breast (BRCA), skin (SKCM), lung (LUAC), ovary (OV), colon (COAD), and esophagus (EAC) according to this immune classification with excellent concordance with T-cell inflamed and EMT-like signatures (Supplementary Fig. S1A–S1F). Additionally, we applied this algorithm to GCs from the Asian Cancer Research Group (ACRG) and European Nucleotide Archive (ENA) datasets (Supplementary Fig. S2A). In each of these cohorts (Fig. 2A–2D, Supplementary Fig. S1A–S1F, and Supplementary Fig. S2A–S2C), the inflamed IP was characterized by enrichment in computationally inferred CD8+ T-effector, Th1, activated dendritic cells (aDC), and M1 macrophage signatures, as well as immune checkpoints and antigen presentation machinery (APM) signatures. Conversely, the desert-type IP demonstrated significant enrichment in EMT-like and CD4+ Th2, MAIT, and M2 macrophage signatures. The excluded-type IP exhibited enrichment of CD8+ naïve T-cell, Th17, and neutrophil signatures. Taken together, we describe a novel immunophenotyping methodology derived from bulk transcriptome data to stratify gastric and other cancers into immune-inflamed or desert classifications, conflating with effector immune and EMT-like transcriptional identities, respectively.
Figure 2. Variation of clinical features, immunotherapeutic response and prognosis among the subtypes of predicted immunophenotype.
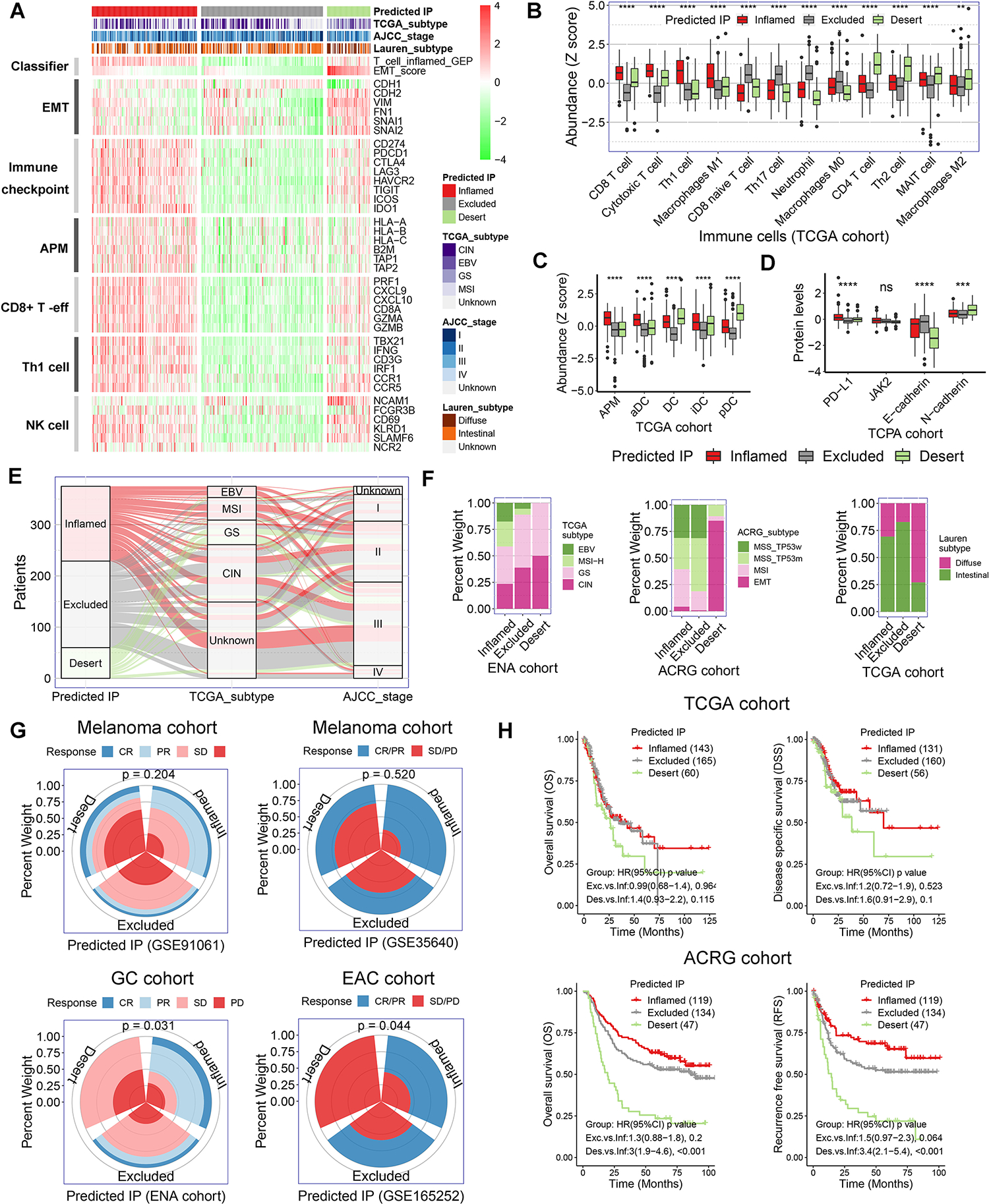
(A) Heatmap presented the expression of marker genes in the critical functional gene sets in TCGA gastric cancer cohort. The TCGA gastric cancer cohort were stratified as inflamed-type, excluded-type, and desert-type tumors by our predicted immunophenotype (predicted IP). AMP, antigen-presenting machinery. (B) The abundance of immune cells was evaluated by the immune cell abundance identifier (ImmuCellAI) and CIBERSORT tools in TCGA gastric cancer cohort. The values were normalized into Z score. (C) The abundance of various population of dendritic cells (DCs) in TCGA gastric cancer cohort. (D) Relative protein levels of PD-L1, JAK2, E-cadherin and N-cadherin in The Cancer Proteome Atlas (TCPA) gastric cancer cohort. (E and F) Correlation of predicted IP with clinicopathological characteristics in gastric cancer cohorts. (G) Immunotherapeutic response rate among the subtypes of predicted IP in ENA and three GEO cohorts (p = 0.204 for GSE91061, p = 0.520 for GSE35640, p = 0.031 for ENA cohort and p = 0.044 for GSE165252). (H) Kaplan-Meier survival analysis for patients with different predicted IP in TCGA and ACRG gastric cancer cohorts.
We next explored differences in clinical features, ICB responsivity, and prognosis across our IP classifier. Although the majority of Epstein–Barr virus (EBV)-associated and microsatellite instability (MSI) tumors in the TCGA-STAD dataset were classified as inflamed-type, and most of genomic stable (GS) and chromosomal instability (CIN) tumors were classified as desert-type and excluded-type, respectively, there was considerable subtype-mixing suggesting that TCGA consensus molecular subtyping may not adequately capture the nuances of our IP classification (Fig. 2E–2F). Not surprisingly, the desert-type IP correlated well with consensus EMT subtype based on the ACRG classification (Fig. 2F), while most Lauren diffuse tumors were also classified as desert-type (Fig. 2F, Supplementary Fig. S2D).
We next classified molecularly annotated gastric and esophageal cancer, as well as non-gastric cancer, cohorts with available ICB response data[27, 28, 29, 30] with our IP stratification. In the gastric/esophageal, as well as melanoma, datasets, a higher incidence of complete response (CR) and partial response (PR) to ICB was observed in patients with inflamed-type IP tumors, while stable disease (SD) and progressive disease (PD) were more frequent in the desert-type IP (Fig. 2G, p > 0.05 in melanoma cohorts and p < 0.05 in gastric/esophageal cohorts). Finally, GC patients with tumors classified as desert-type IP demonstrated significantly worse overall and disease-free/specific survival compared with inflamed-type and/or excluded-type IP tumors (Fig. 2H). These data highlight the prognostic and predictive value of our IP classification system.
Multivalent RTK inhibitors restrict the EMT program in gastric cancer cells
Sensitizing ‘cold’ tumors to ICB is the central objective of cancer immunotherapy. We investigated molecular targeting approaches that can improve immune responses to ICBs in desert-type GC. First, we stratified the immunophenotypes of GC samples from TCGA (n=375) and ACRG (n=300) into our IP classifier. Second, we examined the differentially expressed genes (DEG) in desert-type compared to inflamed-type tumors, revealing 1854 overlapping DEGs highly expressed in immune desert-type tumors. Third, overlapping these DEGs with FDA-approved 182 cancer relative targets (obtained from Human Protein Atlas) revealed 29 potential druggable gene targets in immune desert-type tumors (Fig. 3A–3B). Among these targets, a panel of receptor tyrosine kinases (RTK), such as fibroblast growth factor receptor-1 (FGFR1) and platelet-derived growth factor receptor-B (PDGFRB), were strongly overexpressed in the desert-type GC, vs. inflamed-type or excluded-type GC (Fig. 3B–3C). Additionally, overexpression of FGFR1 and PDGFRB in desert-type tumors was confirmed in multiple TCGA cancer subtypes (COAD, EAC, BRCA, etc.; Supplementary Fig. S2E). Furthermore, we observed specifically enriched gene signatures and several druggable RTK targets, such as ERBB2 and FGFR3, in immune-exclude type GCs, suggesting potential therapeutic benefits to this group (Supplementary Fig. S3).
Figure 3. Dovitinib inhibits EMT program in mesenchymal-like gastric cancer cells.
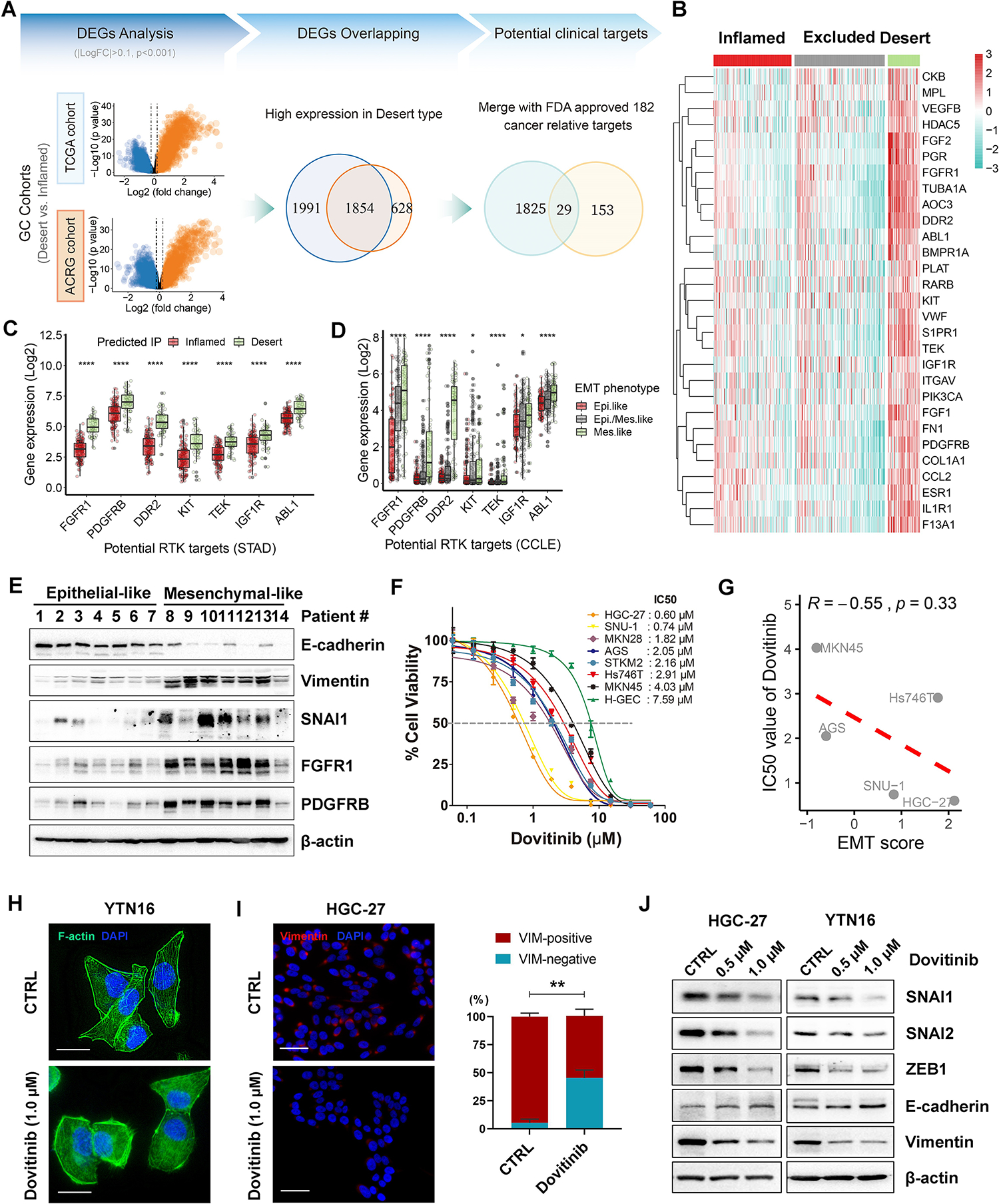
(A) Differentially expressed genes (DEGs) analysis were performed by comparing the predicted desert-type tumors with the predicted inflamed-type tumors in both TCGA and ACRG (GSE66229) cohorts. The 1854 common DEGs that are highly expressed in the desert tumors of TCGA and ACRG cohorts were selected for the overlapping with 182 FDA approved cancer-relative-targets (obtained from the Human Protein Atlas). (B) Gene expression heatmap of the final 29 DEGs that were identified as potential targets in the desert-type tumors in TCGA cohort. (C and D) The relative mRNA expression of a group of receptor tyrosine kinases (RTKs) in the desert-type tumors in TCGA STAD (gastric cancer) cohort, compared to the inflamed-type tumors (C), or in the mesenchymal-like cancer cell lines from CCLE database, compared to the epithelial-like cancer cell lines (D). (E) Western blots for E-cadherin, Vimentin, SNAI1, FGFR1 and PDGFRB in 14 cases of human gastric tumor samples. (F) ATP-Glo assays showed the IC50 curves of dovitinib in a panel of human gastric cancer cell lines and one human gastric epithelial cell line (H-GEC). (G) Pearson’s correlation between EMT score and IC50 value of dovitinib in the five accessible gastric cancer cell lines (R = −0.55, p = 0.33). (H) Phalloidin staining (scale bars, 25μm) for F-actin (green) and DAPI (blue) in YTN16 cells under vehicle control (CTRL) or dovitinib (1.0μM) treatment. (I) Immunofluorescence staining (scale bars, 50μm) for Vimentin (red) and DAPI (blue) in HGC-27 cells under vehicle control (CTRL) or dovitinib (1.0μM) treatment. The quantification of Vimentin-positive and -negative cells was shown as the mean ± SD of 3 independent fields, normalized to total cells (%); **p < 0.01. (J) Western blots for E-cadherin, Vimentin, SNAI1/2 and ZEB1 in HGC-27 and YTN16 cells under vehicle control and dovitinib treatment (0.5μM or 1.0μM).
Next, pan-cancer transcriptomic analysis from the Cancer Cell Line Encyclopedia (CCLE) compendium demonstrated overexpression of FGFR1 and PDGFRB RTKs in mesenchymal-like human cancer cell lines (Fig. 3D). We specifically investigated the EMT signaling enrichment of GC cell lines (n=37) from the Cancer Cell Line Encyclopedia (CCLE) database by EMT score[26], and stratified these as epithelial-like or mesenchymal-like (Supplementary Fig. S4A). Western blotting confirmed elevated expression of FGFR1 and PDGFRB in mesenchymal-like gastric tumors (designated by preferential expression of vimentin vs. E-cadherin; Fig. 3E), as well as in human and mouse mesenchymal-like GC cell lines (i.e., SNU-1, HGC-27, Hs746t, YTN16; Supplementary Fig. S4B).
Given that aberrantly overexpressed RTKs play pivotal roles in the EMT process in various cancers[31], we hypothesized that RTK inhibitors may restrict the EMT program in immunologically cold desert-type GC. Among several multivalent RTK inhibitors, dovitinib exhibited the most robust inhibitory effect and cancer cell killing in mesenchymal GC cells (Fig. 3F–3G, Supplementary Fig. S4C). In particular, dovitinib efficiently inhibited phosphorylation of FGFR1 (Tyr653/654) and PDGFRB (Tyr751), as well as their downstream targets in mesenchymal-type GC cells (human HGC-27 cells and mouse YTN16 cells), but not in epithelial-type GC cells (human MKN45 cells and mouse YTN2 cells) (Supplementary Fig. S4D–S4F). Notably, dovitinib treatment induced a cellular morphological shift from spindle-like to epithelial-like shape in mesenchymal-type YTN16 cells (Fig. 3H). These morphologic changes were associated with repression of EMT markers Vimentin, SNAI1, SNAI2, and ZEB1 and concomitant induction of E-cadherin (CDH1) transcription/expression with dovitinib treatment in these cells (Fig. 3I–3J, Supplementary Fig. S5A).
To further clarify the role of RTKs in promoting EMT in GC, we utilized agonists of dovitinib targets, such as FGF2 (i.e., FGFR1 agonist) and PDGF-BB (i.e., PDGFRB agonist), to treat epithelial-type GC cells (human AGS cells and mouse YTN2 cells). The results indicated these agonists enhanced EMT characteristics by increasing SNAI1, SNAI2, and Vimentin expression and decreasing E-cadherin expression (Supplementary Fig. S5B), confirming multivalent RTK signaling can induce EMT programming in GC. Given that desert-type tumors exhibit higher metastatic characteristic (Supplementary Fig. S5C), we next examined whether dovitinib could inhibit this property in vitro. As expected, dovitinib significantly repressed migration and invasion of mesenchymal-like gastric cancer cells (Supplementary Fig. S5D–S5E). Therefore, we investigated the possibility of clinical application of dovitinib on metastatic GC. Remarkably, after removing batch effect between the different datasets, the metastatic gastric tumors in GSE198136 dataset (n=21), showed an active EMT signaling and repressed immune response signature compared to inflamed tumors in TCGA (n=146) (Supplementary Fig. S5F). Given that YTN16 is a higher metastatic mouse GC cell line than YTN2[32], the hyperactive EMT signature and positive response to dovitinib of YTN16 suggest a potential clinical approach towards metastatic GC by dovitinib. Collectively, these results indicate that dovitinib significantly restricts EMT programming in mesenchymal-like immune desert-type GCs.
Dovitinib reprograms TME by recruiting CD8+ T cells into immune desert-type syngeneic tumors
Our bioinformatics analysis suggested that EMT-related genes correlate with desert-type gastric tumors. To further investigate the role of EMT in GC immunosuppression, we compared tumor formation and T lymphocyte infiltration of syngeneic tumors derived from epithelial-like GC cells (YTN2) or mesenchymal-like GC cells (YTN16). Interestingly, the number of formed tumors and tumor volume of YTN2-derived xenografts was significantly less than YTN16-derived tumors (frequency: 30% vs. 90%, Fig. 4A; mean of tumor volume: 185.2 vs. 552.9 mm3, p<0.05, Supplementary Fig. S6A). The epithelial-like or mesenchymal-like state of YTN2- and YTN16-derived tumors was confirmed by immunofluorescent staining and Western blots of E-cadherin and Vimentin (Fig. 4B, Supplementary Fig. S4B). Importantly, CD3/CD8 staining revealed abundant CD8+ T cells infiltration in the core and invasive margin of YTN2-derived tumors but not in YTN16-derived tumors (Fig. 4C). In line with the previous studies[33], our results suggested an intimate relationship between EMT and the desert-type of TME in GC.
Figure 4. Dovitinib reprograms TME by promoting CD8+ T cell recruitment.
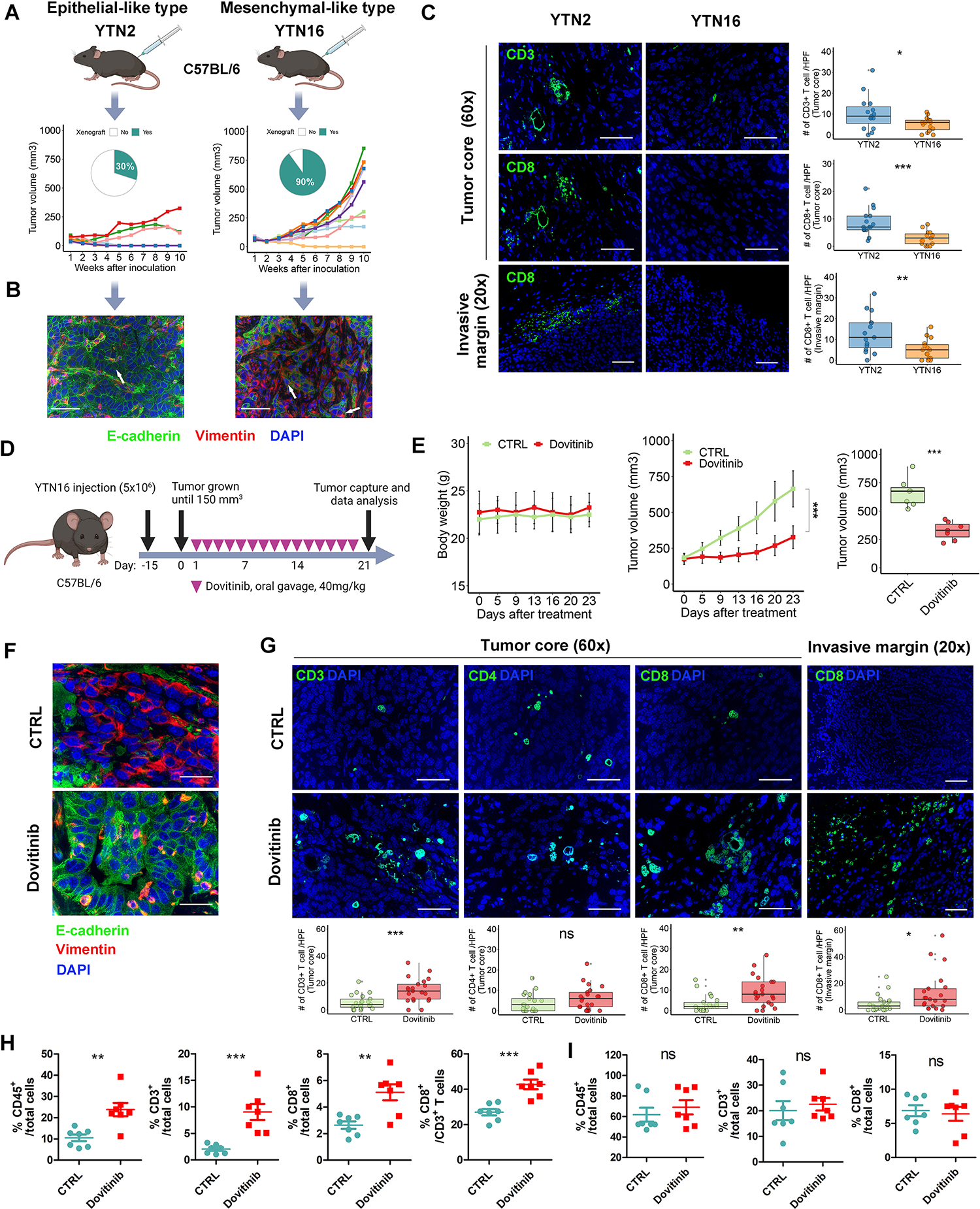
(A) Tumor initiation rate and tumor volume of syngeneic tumors derived from YTN2 and YTN16 cells. Each group included 10 tumors. (B) Immunofluorescence staining (scale bars, 50μm) for E-cadherin (green), Vimentin (red) and DAPI (blue) in the relevant tumors. White arrows indicate representative staining of tumor cells. (C) Immunofluorescence staining (scale bars, 50μm) for CD3/CD8 (green) and DAPI (blue) in tumor core or invasive margin in the syngeneic tumors from (A). The quantification of CD3+/CD8+ T cells were shown as the cell numbers of 3 independent high-power fields for each tumor. Each group included 5 tumors. (D) A schematic view of the treatment plan for the syngeneic tumors derived from YTN16 cells. (E) Plots of mice body weight (Left) and tumor volumes (Middle), which were measured twice per week. The box whisker plots of the tumor volumes on day23 were shown (Right). Each group included 7 tumors. (F and G) Immunofluorescence staining (scale bars, 50μm) for E-cadherin/Vimentin/DAPI in tumor core (F), or CD3/CD4/CD8/DAPI in the core or invasive margin (G) of the syngeneic tumors from (E). The quantification of T cells shown as the cell numbers of 3 independent high-power fields for each tumor (bottom). (H and I) Flow cytometry analysis for CD45+/CD3+/CD8+ T cells in syngeneic tumors (H) and relevant mouse spleens (I). A one-way ANOVA test was performed for the between-group difference. ns indicate no significance, **p < 0.01, ***p < 0.001.
Next, we investigated whether targeting multiple RTKs will reprogram the TME in YTN16-derived tumors. Once tumor volume reached 150mm3, dovitinib (40mg/kg) or the vehicle control (CTRL) was administrated daily by oral gavage for 3 weeks (Fig. 4D). Our results showed that dovitinib dramatically delayed tumor growth in YTN16-derived tumors without compromising mouse body weight (Fig. 4E). Western blotting confirmed that dovitinib treatment inactivated downstream targets (Supplementary Fig. S6B), consistent with our in vitro results. We also detected increased E-cadherin and decreased Vimentin by dovitinib treatment, suggesting that blockade of multiple RTKs reprograms the EMT program in vivo (Fig. 4F, Supplementary Fig. S6B). Most importantly, immunofluorescence staining, and flow cytometry analysis showed that dovitinib significantly increased CD3+CD8+ T cells within tumors (Fig. 4G–4H, Supplementary Fig. S6C–S6D). By contrast, splenic CD3+CD8+ T cells seemed unaffected in the relevant tumor-bearing dovitinib-treated vs. vehicle-treated mice (Fig. 4I, Supplementary Fig. S6E). Additionally, as mesenchymal-type carcinoma cells were reported to recruit a large number of M2-like tumor-associated macrophages (TAM) into the tumor core [19, 20], we investigated whether dovitinib could repress M2 macrophages by restricting EMT. Our results indicated that dovitinib significantly decreased F4/80+CD206+ M2-like macrophages within YTN16-derived tumors (Supplementary Fig. S7). In summary, we demonstrate that targeting multiple RTKs via dovitinib represses EMT-related gene expression and reprograms TME of mouse syngeneic mesenchymal-like immune desert gastric tumors in vivo.
Dovitinib promotes CD8+ T cell infiltration via activation of IFN-γ signaling in tumor cells
To identify potential mechanisms for dovitinib-induced T cells infiltration in GC, we performed a gene set enrichment analysis (GSEA) using the TCGA GC cohort. This analysis revealed enrichment of several critical immune pathways, such as IFN-α/γ response and T cells recruitment chemotaxis, in the inflamed-type compared to the desert-type GC (Fig. 5A). These results suggested that lack of IFN-α/γ response and T cells recruitment chemotaxis promotes immune evasion in the desert-type GC. At first, we excluded CD103 + dendritic cells (DCs)-dependent effector T cells recruitment[34], since dovitinib didn’t increase CD103+ DCs in YTN16-derived mouse tumors (Supplementary Fig. S8A). Next, we found dovitinib treatment dramatically upregulated IFN-γ expression (Fig. 5B–5E, Supplementary Fig. S8B) and activated downstream STAT1/IRF1 signaling axis in both humans and mice mesenchymal-type GC cells (Fig. 5E, Supplementary Fig. S8B). Moreover, the chemotaxis assays confirmed that neutralization of IFN-γ attenuated the dovitinib-induced recruitment of CD3+ T cells toward gastric tumor cells (Fig. 5F, all p<0.05).
Figure 5. Dovitinib promotes CD8+ T cell infiltration via activation of intrinsic IFN-γ/IFNGR/STAT1 signaling in tumor cells.
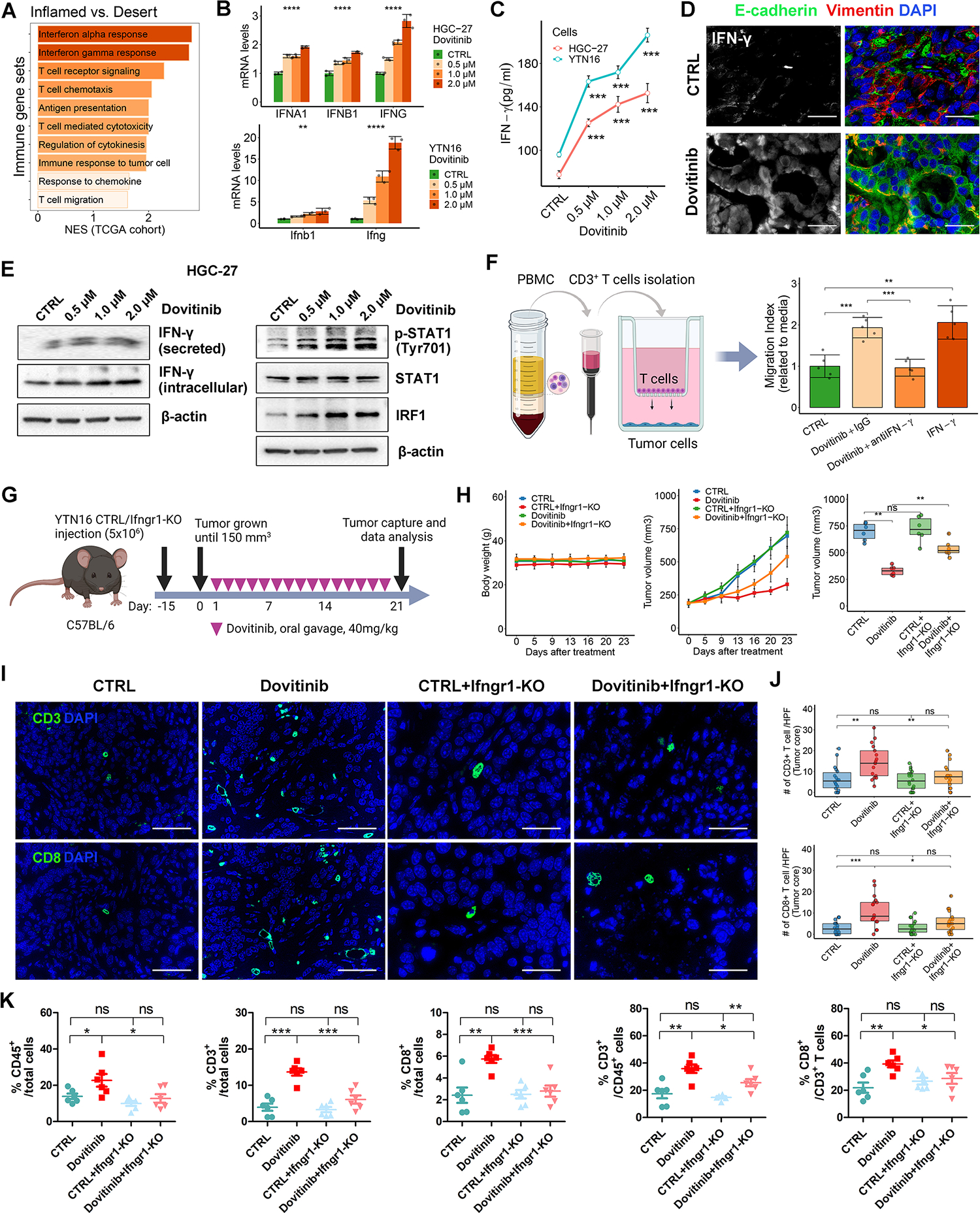
(A) GSEA were performed in TCGA cohort, using a panel of immune relative gene sets from MSigDB database. NES, normalized enrichment score. (B and C) HGC-27 or YTN16 cells were treated with indicated doses of dovitinib. CTRL: vehicle control. (B) qRT-PCR analysis of IFNA1, IFNB1/Ifnb1, or IFNG/Ifng. (C) ELISA of IFN-γ. (D) Immunofluorescence staining (scale bars, 50μm) for IFN-γ (white), E-cadherin (green), Vimentin (red), and DAPI (blue) in YTN16-derived tumors. (E) Western blots for p-STAT1 (Tyr701), STAT1, IRF1, IFN-γ, and β-actin in HGC-27 cells treated with indicated doses of dovitinib. IFN-γ (secreted), secreted IFN-γ in the conditional medium. (F) Chemotaxis assay of CD3+ T cells, isolated from peripheral blood mononuclear cells (PBMC), toward tumor cells. HGC-27 cells were pre-treated with Dovitinib or incubated with IgG, IFN-γ neutralization antibody or recombinant IFN-γ. The T cell migration ratio toward tumor cells were quantified by T cells number in the media of bottom wells, normalized to the media from the well of tumor cells alone (CTRL). (G) A schematic view of the treatment plan in C57BL/6 mice implanted with YTN16 cells with or without Ifngr1 knockout. (H) Plots of mice body weight (Left) and tumor volumes (Middle), which were measured twice per week. The box whisker plots of the tumor volumes on day23 were shown (Right). Each group included 6 tumors. (I) Immunofluorescence staining (scale bars, 50μm) for CD3/CD8/DAPI in the core of syngeneic tumors from (h). (J) The quantification of T cell/HPF of (I) were shown as the cell numbers of 3 independent fields for each tumor. (K) Flow cytometry analysis for CD45+/CD3+/CD8+ T cells in syngeneic tumors from (H). A one-way ANOVA test was performed to examine the between-group difference. In all panels, ns indicate no significance, *p < 0.05, **p < 0.01, ***p < 0.001.
Additionally, the transcriptome analysis in TCGA and ACRG cohorts uncovered a group of chemokines as potential downstream mediators of IFN-γ/STAT1/IRF1 in GC, such as CXCL9 and CCL5, known to play an important role in T cell recruitment in GC. These were specifically activated in the inflamed-type tumors (Supplementary Fig. S8C) and correlated with IFNG expression and cytotoxic T cell infiltration (Supplementary Fig. S8D). The qRT-PCR results demonstrated that dovitinib induced CXCL9 and CCL5 mRNA expressions in HGC-27 and YTN16 cells (Supplementary Fig. S8E, all p<0.01). ELISA and Western blots further confirmed that dovitinib increased intracellular and secreted levels of CXCL9 (Supplementary Fig. S8F–S8G). Furthermore, antibody neutralization assays using IFN-γ antibody abrogated dovitinib-induced mRNA and/or protein expression of CXCL9 and CCL5 (Supplementary Fig. S8H, all p<0.05). Our findings suggest that dovitinib promotes T cells infiltration into desert-type syngeneic tumors via activation of intrinsic IFN-γ/STAT1/IRF1 signaling axis and downstream chemokines in GC cells.
Ifngr-knockout in tumor cells abrogates dovitinib-induced T cells infiltration in immune desert-type syngeneic mouse gastric tumor
To test whether IFN-γ signaling blockage attenuated the effect of dovitinib therapy, we introduced Ifngr1 knockout (KO) into our mouse GC cell line. Following the instruction of the previous study[35], we established a stable Ifngr1 knockout (KO) clone of YTN16 cells (Supplementary Fig. S9A). We used dovitinib to treat the syngeneic mouse tumors derived from YTN16 cells or Ifngr1-KO YTN16 cells as per the indicated schedule (Fig. 5G). Compared to the untreated control group (CTRL), Ifngr1-KO did not affect mouse weight and tumor volume. However, Ifngr1 knockout significantly impaired the tumor suppression of dovitinib therapy (Fig. 5H, p<0.01). The qRT-PCR results indicated that Ifngr1 knockout significantly abrogated dovitinib-upregulated Cxcl9 and Ccl5 (all p<0.05, Supplementary Fig. S9B). At the end of the treatment, tumor samples were harvested for subsequent analysis. The knockout of Ifngr1 was verified in vivo (Supplementary Fig. S9C). Immunofluorescence staining and flow cytometry analysis indicated that Ifngr1-deficiency limited dovitinib-induced CD8+ T cells infiltration in the desert-type syngeneic tumors (Fig. 5I–5K). Collectively, our results suggest that dovitinib treatment promotes CD8+ T cell recruitment depending on intrinsic IFN-γ mediated IFNGR/STAT1 signaling in tumor cells.
Dovitinib transcriptionally upregulates IFNG expression by repressing SNAI1/2
Our previous bioinformatics analysis and in vivo syngeneic tumor model verified activation of IFN-γ signaling in epithelial-type GC. Consistently, Pearson’s correlation analysis in the TCGA cohort exhibited an inverse correlation between IFNG mRNA expression and EMT score or EMT transcription factors (EMT-TFs) expression in the inflamed-type and desert-type GC (Fig. 6A). These findings led us to hypothesize that activation of the EMT program could regulate IFNG expression in GC. To address it, we used TGF-β, FGF2, and PDGF-BB to transiently induce EMT characteristics in epithelial-like gastric cancer cells (Fig. 6B, Supplementary Fig. S5B). qRT-PCR results indicated that IFNG mRNA expressions were significantly decreased with EMT program activation (Fig. 6B–6C). To identify the potential upstream transcription factors, sequence homology analysis identified a 619 bp sequence of IFNG promoter highly conserved between human and mouse. Next, the JASPAR tool (http://jaspar.genereg.net/) predicted the high scores of binding possibilities of SNAI1, SNAI2, TWIST1, and ZEB1 within the conserved sequence of IFNG promoter (Fig. 6D). There were two putative SNAI1/2 binding regions named P1 and P2 (Fig. 6E). Transient overexpression of SNAI1 or SNAI2 inhibited IFNG expression in AGS cells (Fig. 6F). Conversely, SNAI1 or SNAI2 knockdown significantly promoted IFNG mRNA expression in mesenchymal-like HGC-27 cells (Fig. 6G). In addition, luciferase reporter assay indicated that both dovitinib treatment and SNAI1/2 silencing increased IFNG promoter activity, whereas overexpression of SNAI1/2 or TGF-β treatment reversed these results (Fig. 6H, all p < 0.05). Quantitative chromatin immunoprecipitation (ChIP) assay confirmed direct binding of SNAI1/2 on the two IFNG promoter regions with or without overexpression of SNAI1/2 compared to the IgG controls (Fig. 6I, all p < 0.05). These data suggest that dovitinib treatment upregulates IFNG mRNA in gastric cancer cells by repressing SNAI1/2 expression.
Figure 6. Dovitinib transcriptionally upregulates IFNG expression by repressing SNAI1/2.
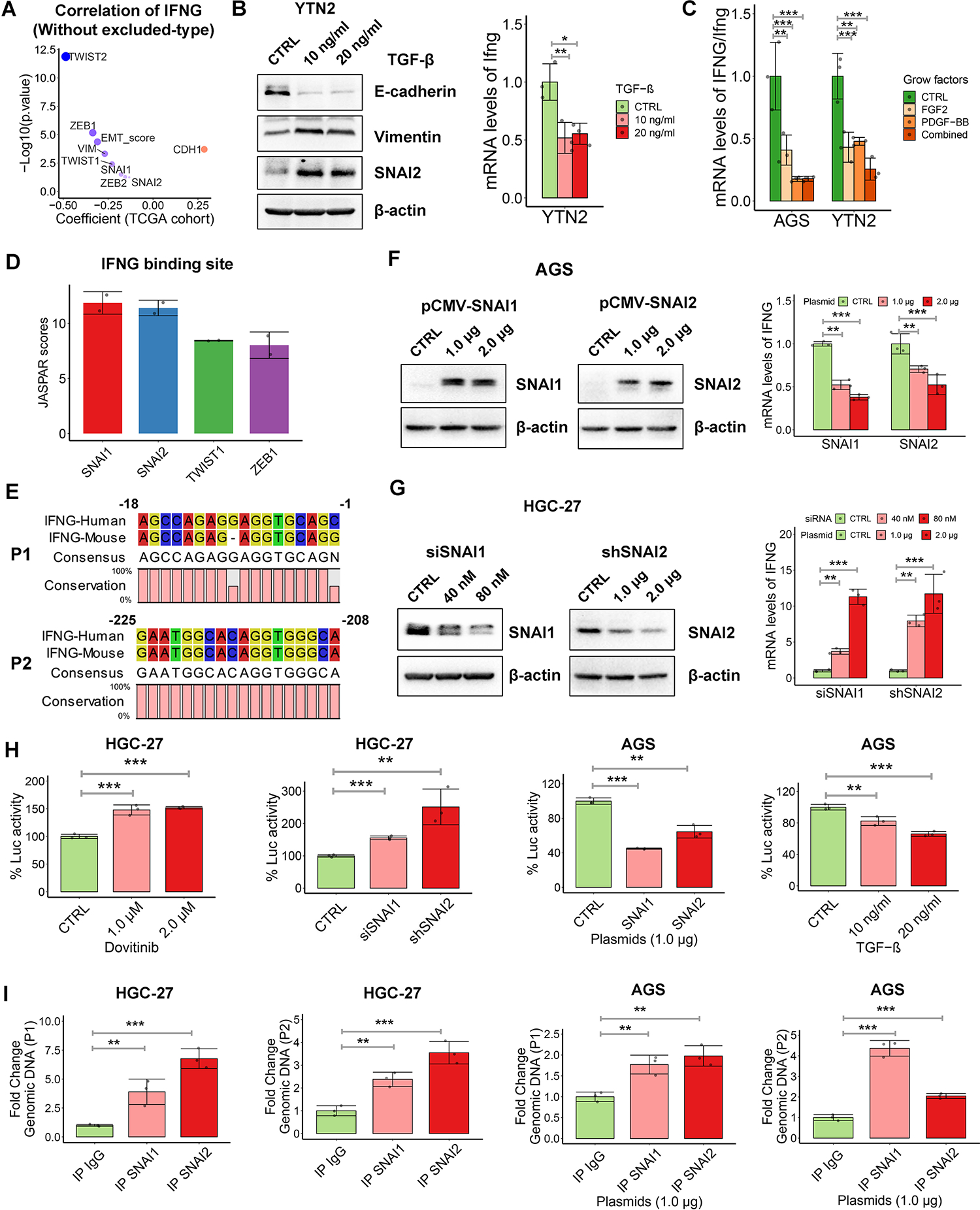
(A) Pearson’s correlation between IFNG expression and EMT score or EMT marker genes expression in the inflamed-type and desert-type of TCGA cohort. (B) Western blots of E-cadherin, Vimentin and SNAI2 in YTN2 cell with the treatment of indicated doses of TGF-β for 3 days (left). qRT-PCR analysis of Ifng in YTN2 cell with treatment of TGF-β (right), (C) qRT-PCR analysis of IFNG/Ifng in gastric cancer cells with treatment of FGF2, PDGF-BB or the combination. (D and E) JASPAR scores (https://jaspar.genereg.net/) of EMT-TFs binding sites (D) and two putative SNAI1/2 binding regions named P1 and P2 (E) in the conserved sequence of IFNG promoters. (F and G) Western blots for SNAI1/2 and qRT-PCR analysis of IFNG in AGS cells with overexpression of SNAI1 or SNAI2 (F), or in HGC-27 cells with knockdown of SNAI1 or SNAI2 (G). (H) IFNG promoter luciferase reporter assays were performed in HGC-27 cells with dovitinib treatment or transient knockdown of SNAI1/2, and in AGS cells with transfection of SNAI1/2 plasmids or TGF-βtreatment. (I) Chromatin immunoprecipitation (ChIP) assays were performed by using SNAI1 or SNAI2 antibodies, followed by qPCR applying primers covering P1 or P2 region. IgG works as non-specific pull-down control. AGS cells were transfected with SNAI1/2 plasmids. In all panels, *p < 0.05, **p < 0.01, ***p < 0.001.
Targeting multiple RTKs sensitizes mesenchymal-like desert-type syngeneic tumors to CTLA4 blockade
Although we found that dovitinib treatment significantly promoted CD8+ T cells infiltration (Fig. 4G) in the desert-type murine tumors, relatively few of the CD8+ T cells are cytotoxic with Granzyme B (GZMB) expression (Supplementary Fig. S10A). Thus, we hypothesized that, beyond recruitment, activation of CD8+ T cells is also suppressed in the TME of YTN16-derived tumors. The transcriptome analysis of TCGA and ACRG gastric cancer cohorts revealed that the essential immune checkpoint genes, programmed cell death ligand 1 (PD-L1) and CD80, are highly expressed in the predicted inflamed tumors compared to the predicted desert/excluded tumors (Supplementary Fig. S10C). Western blots confirmed overexpression of CD80 in epithelial-like GC cells but not in mesenchymal-like ones (Supplementary Fig. S10D). Interestingly, dovitinib upregulated the expression of CD80 (not PD-L1) in mesenchymal-like GC cells (Supplementary Fig. S10E–S10F) and mesenchymal-like murine tumors (Supplementary Fig. S10G). Consistently, we observed increased exhausted T cells, such as CTLA4+/CD8+ cells (Fig. 7A, p < 0.001) and PD1+/CD8+ cells (Supplementary Fig. S10B, p < 0.01), in dovitinib-treated mesenchymal mouse tumors. Furthermore, T cell-mediated tumor cell-killing assays demonstrated that dovitinib renders the mesenchymal GC cells more resistant to T cells; however, CTLA4 blockade improved the sensitivity of dovitinib-treated GC cells to lymphocytes attack (Fig. 7B). These results suggest that the combination of dovitinib and ICB may achieve a better outcome.
Figure 7. Targeting multiple RTKs sensitizes the desert-type syngeneic tumors to CTLA4 blockade.
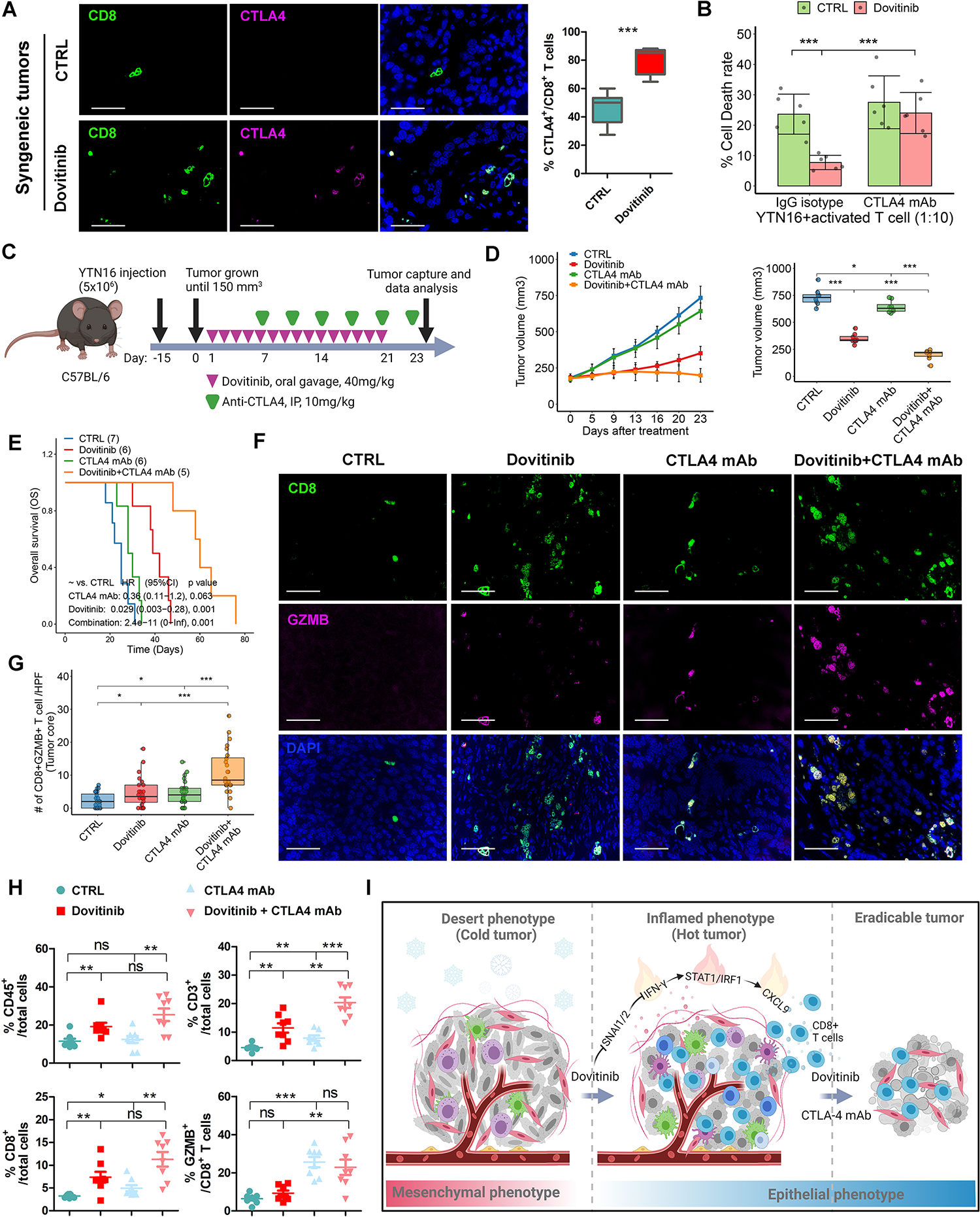
(A) Immunofluorescence staining (scale bars, 50μm) for CD8/CTLA4/DAPI in YTN16-derived syngeneic tumors treated with dovitinib or vehicle control. 3 independent fields for each tumor and 7 tumors for each group were utilized for quantification of CTLA4+/CD8+ T cells. (B) T cell-mediated tumor cell-killing assays showed the relative dead YTN16 cells treated with dovitinib and/or CTLA4 neutralization antibody. (C) A schematic view of the experiment plan in C57BL/6 mice implanted with YTN16 cells treated with dovitinib, CTLA-4 monoclonal antibody (mAb) or the combination. (D) Plots of tumor volumes (Left), which were measured twice per week. The box whisker plots of the tumor volumes on day23 were shown (Right). Each group included 8 tumors. (E) Kaplan-Meier survival analysis for the tumor host mice. (F) Immunofluorescence staining (scale bars, 50μm) for CD8/GZMB/DAPI in the core of the syngeneic tumors from (D). (G) The quantification of T cell/HPF of (F) were shown as the cell numbers of 3 independent fields for each tumor. (H) Flow cytometry analysis for CD45+/CD3+/CD8+/GZMB+ T cells in the syngeneic tumors from (d). (I) Representative summary of findings. A one-way ANOVA test was performed to examine the between-group difference. In all panels, ns indicate no significance, *p<0.05, **p<0.01, ***p<0.001.
The following treatments on YTN16-derived syngeneic mouse tumors included dovitinib, CTLA-4 monoclonal antibody (mAb), or the combination (Fig. 7C). Compared to the untreated control group (CTRL), none of treatments significantly affected mouse body weight (Supplementary Fig. S10H). Although dovitinib alone significantly repressed the tumor growth in the early two weeks, this suppression was gradually attenuated after two weeks of treatment. As we expected, CTLA4 mAb alone had a weak anti-tumor effect and slightly prolonged survival time compared to the control group (CTRL) in our mesenchymal-like desert-type tumor model. However, a combination of dovitinib and CTLA4 mAb further decreased tumor growth and extended animal survival time compared to control (CTRL), dovitinib, or CTLA-4 mAb alone (Fig. 7D–7E). The survival analysis also indicated that combined therapy of dovitinib and CTLA4 mAb had the best effects on decreasing the hazard risk (HR) of mouse death compared to the other groups (Fig. 7E). Accordingly, immune profiling by immunofluorescence staining and flow cytometry showed dovitinib treatment activated CD3+ and CD8+ T cell infiltration (Fig. 7F–7H, Supplementary Fig. S10I). More importantly, the combined treatment group exhibited a striking increase of the cytotoxic T cell (GZMB+/CD8+) infiltration in the tumor core compared to the other groups (Fig. 7F–7H). Our findings identified that dovitinib improves the responses of desert-type gastric cancer to CTLA4 blockade by increasing the infiltration of cytotoxic CD8+ T cells.
Discussion
Gastric cancer is the fifth most common cancer with a poor 5-year overall survival rate worldwide[2, 36]. In absence of screening programs, most patients with gastric cancer present with distant metastasis showing poor response to standard chemotherapy with 5-year survival of 5% in these patients [3]. Although some ICB clinical trials showed encouraging response in MSI-high and EBV-positive GC [27, 37, 38], most clinical trials of pembrolizumab/nivolumab or ipilimumab failed to improve the overall survival (OS) or progression-free survival (PFS), compared to standard of care [39, 40, 41]. The therapeutic effects of ICBs are minimal in immune-desert type GC. Here, we demonstrate a new approach to stratify immunophenotypes in gastric cancer. We analyzed patient-derived transcriptomic data to develop immunophenotype classification and identified a panel of multiple RTKs enriched in immune desert-like tumors. Targeting RTKs using dovitinib restricted the EMT program and sensitizes the mesenchymal-like syngeneic tumors to CTLA4 blockade.
We classified GCs, based on patient-derived bulk transcriptomic data and histological identification of spatial CD8+ T-cell distribution in the relevant tumors. This approach allowed us to predict tumors’ immunophenotypes (IP) in public databases that usually lack histological IP stratification. The predicted IP was validated in our gastric cancer cohort and the TCGA SKCM cohort with similar clinical immunophenotype classification. More specifically, the predicted IP satisfies the large sample size for the further discovery of predominant signaling pathways and potential therapeutic vulnerabilities in different immune subtypes of tumors. We also report EMT signature as a critical phenotype in shaping the tumor-immune microenvironment (TIME), supporting earlier studies of EMT signature [21, 24]. Applying the predicted IP to multiple molecularly annotated human GC datasets, we discovered a group of receptor tyrosine kinases (RTKs), specifically FGFR1 and PDGFRB, as potential druggable targets in the predicted desert tumors. RTKs like HER3, FGFR, EGFR, MET, VEGFR and AXL act as major mediators of EMT[42]. Recently, dovitinib, an inhibitor targeting multiple RTKs, has been submitted to FDA as a third-line therapy for renal cell carcinoma patients after completing a phase 3 clinical trial[43]. In the present study, dovitinib effectively inhibited phosphorylation of FGFR1 and PDGFRB and restricted the EMT program. Dovitinib also reshaped the immunosuppressive TME by improving CD8+ T lymphocytes infiltration in the desert-type syngeneic tumors derived from mesenchymal-like mouse GC cells.
Several attempts have combined EMT inhibitors with ICB therapy, such as anti-PD-L1 combined with targeting TGFβR[44, 45], or MEK inhibitor[46, 47]. EMT inhibitors may activate immune response to anti-PD-L1 by reconstituting T cell infiltration into the TME of ‘cold’ tumors. Although Dongre et al. demonstrated that abrogation of quasi-mesenchymal (qM) breast carcinoma cell-derived factors sensitizes refractory qM tumors to anti-CTLA4 ICB[19], there is lack of data on combination therapy of EMT inhibitors and CTLA4 blockade. We found that dovitinib significantly sensitized the ICB-resistant desert tumors to anti-CTLA4 treatment by recruiting T cells into TME. Dovitinib also restricted EMT program and restored CD80 expression in the mesenchymal tumors, suggesting CD80-CTLA4 inhibitory interaction plays a core role in the immune evasion of the GC inflamed tumors.
Although clinical trials reported frequent toxicities of long-term dovitinib administration[48], we utilized a low dose of dovitinib in our study. This dose was safe to mice and dramatically improved the effectiveness of ICB therapy. Compared to dovitinib, other multitarget RTK inhibitors, such as nintedanib, lenvatinib, and others, may work similarly, providing alternative options for the combined treatments. Based on these findings, we suggest that anti-CTLA4 in combination with dovitinib as a therapeutic strategy in the GC mesenchymal/desert-type tumors. Our GSEA and ‘predicted immunophenotype’ analysis revealed enriched EMT and repressed immune response signatures in metastatic gastric tumors, compared to inflamed tumors. These findings rationalize the use of a combination of dovitinib and anti-CTLA4 as a second line therapy in metastatic immune desert-type tumors.
Interferon-γ (IFN-γ) has a prominent role in tumor immunity with paradoxical anti-tumor and pro-tumor functions in cancer. Studies of IFN-γ-neutralization and knockout of IFN-γ receptor (IFNGR) or STAT1 showed that endogenous IFN-γ prevents tumor development[49, 50, 51]. On the other hand, IFN-γ promotes expression of the inhibitory molecules, such as PD-L1, PD-L2, IDO1, iNOS, FAS, and FASL, to restrict anti-tumor immunity[52]. In our study, we demonstrated high expression of IFN-γ was associated with improved T-cell infiltration in epithelial-like GC compared to mesenchymal-type tumor cells, although it is difficult to decipher from bulk transcriptomic data whether the source of high IFN-γ is tumor cell-intrinsic or derived from lymphocytes such as NK cells in epithelial-type GC human tumors. The results may also suggest that the presence of autocrine activation of STAT1 signaling via tumor cell-derived IFN-γ. Dovitinib treatment not only restricted the EMT program but also restored the IFN-γ expression levels in desert-type tumors. The immune-activating functions of IFN-γ on tumor cells are largely attributed to inducing the expression of MHC class I, CXCL9, CXCL10, and CXCL11 in tumor cells[53]. In dovitinib-treated desert-type tumors, the IFN-γ-IFNGR1-STAT1 signaling axis promoted the expression and secretion of trafficking chemokines, such as Cxcl9, Cxcl10, and Ccl5, sequentially improving T cell recruitment and infiltration. Mechanically, dovitinib upregulated IFNG gene expression in GC cells by decreasing the expression of SNAI1 and SNAI2, which we identified as transcriptional repressors of IFNG. Therefore, tumor cell-intrinsic IFN-γ may play critical anti-tumor functions by recruiting T-cells via a chemoattractant program to sensitize GC responses to ICB. Future avenues in the laboratory are pursuing whether dovitinib promotes antigen presentation via upregulating IFN-γ-MHC class I or affects non-tumor cells in the TME, which remain outstanding questions.
In summary, our studies provide a novel approach to reclassify GC and other solid tumors into immunophenotypes using transcriptomic data and discover a multivalent RTK-driven EMT-like transcriptional program in immune desert-like GCs. Dovitinib, a multi-RTK inhibitor, restricts the EMT program and successfully induces conversion from immune desert-type tumors to immune inflamed-type tumors, sensitizing these mesenchymal-like ‘cold’ tumors to CTLA4 blockade. These findings support the development of a clinical trial combining dovitinib and ICB in immune desert GCs to improve therapeutic responses and clinical outcomes following immunomodulatory therapies in GC patients.
Supplementary Material
Significance of this study.
What is already known about this subject?
The response to immune checkpoint blockade (ICB) therapy in gastric cancer (GC) is heterogeneous due to tumor-intrinsic and acquired immunotherapy resistance.
Epithelial-Mesenchymal Transition (EMT) signaling contributes to tumor progression, metastasis, and chemoresistance in GC.
What are the new findings?
We established an algorithm to reclassify the immuno-subtypes of human GC by analyzing public databases.
EMT signaling is highly enriched in immune desert-type GC, compared with immune inflamed-type GC.
Receptor tyrosine kinases (RTKs) are potential druggable targets in the immune desert-type GC. Inhibition of multiple RTKs restricts EMT programming in mesenchymal-like immune desert syngeneic GC models.
Inhibition of RTKs converts immune desert-type GC to immune inflamed-type via tumor-intrinsic SNAI1/2-IFN-γ signaling axis, promoting recruitment T cells and enhancing immunotherapeutic efficacy of CTLA4 blockade.
How might it impact on clinical practice in the foreseeable future?
Gastric cancer ranks fifth for incidence and fourth for mortality worldwide, where immunotherapies have limited efficacy. Our findings provide a new perspective on converting immune-cold tumors to immune-hot tumors by repressing EMT programming.
Multivalent tyrosine kinase inhibitors can restrict EMT, promote T cell infiltration, and improve the therapeutic efficacy of CTLA4 blockade, suggesting that it can be a promising strategy for combined therapy with ICB in GC patients.
Acknowledgments
The authors would like to thank Changyin Feng of the Department of Pathology for assistance with assessing human gastric tumor pathology.
Funding
This study was supported by grants from the U.S. National Institutes of Health (R01CA249949 and 1R01CA266528) and the U.S. Department of Veterans Affairs (1IK6BX003787 and I01BX001179). The Sylvester Comprehensive Cancer Center supported the use of shared resources (P30CA240139). This work’s content is solely the responsibility of the authors. It does not necessarily represent the official views of the Department of Veterans Affairs, National Institutes of Health, or the University of Miami.
Footnotes
Competing interests
The authors declare that they do not have conflict of interest.
Ethics approval
The ethical review community approved the study of Fujian medical university union hospital. All study participants provided written informed consent.
Provenance and peer review
Not commissioned; externally peer-reviewed
Data availability statement
All data are available from the corresponding author upon reasonable request.
References:
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33. [DOI] [PubMed] [Google Scholar]
- 2.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 3.Jim MA, Pinheiro PS, Carreira H, Espey DK, Wiggins CL, Weir HK. Stomach cancer survival in the United States by race and stage (2001–2009): Findings from the CONCORD-2 study. Cancer 2017;123 Suppl 24:4994–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pires da Silva I, Ahmed T, Reijers ILM, Weppler AM, Betof Warner A, Patrinely JR, et al. Ipilimumab alone or ipilimumab plus anti-PD-1 therapy in patients with metastatic melanoma resistant to anti-PD-(L)1 monotherapy: a multicentre, retrospective, cohort study. Lancet Oncol 2021;22:836–47. [DOI] [PubMed] [Google Scholar]
- 8.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017;545:60–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015;372:2018–28. [DOI] [PubMed] [Google Scholar]
- 12.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 2015;372:311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 2019;25:462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 2016;375:1823–33. [DOI] [PubMed] [Google Scholar]
- 15.Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N Engl J Med 2018;378:2078–92. [DOI] [PubMed] [Google Scholar]
- 16.Hegde PS, Karanikas V, Evers S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin Cancer Res 2016;22:1865–74. [DOI] [PubMed] [Google Scholar]
- 17.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 18.Rashidian M, Ingram JR, Dougan M, Dongre A, Whang KA, LeGall C, et al. Predicting the response to CTLA-4 blockade by longitudinal noninvasive monitoring of CD8 T cells. J Exp Med 2017;214:2243–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dongre A, Rashidian M, Eaton EN, Reinhardt F, Thiru P, Zagorulya M, et al. Direct and Indirect Regulators of Epithelial-Mesenchymal Transition-Mediated Immunosuppression in Breast Carcinomas. Cancer Discov 2021;11:1286–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH, et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin Cancer Res 2016;22:3630–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, et al. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res 2017;77:3982–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell 2016;166:21–45. [DOI] [PubMed] [Google Scholar]
- 23.Chou MY, Yang MH. Interplay of Immunometabolism and Epithelial-Mesenchymal Transition in the Tumor Microenvironment. Int J Mol Sci 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taki M, Abiko K, Ukita M, Murakami R, Yamanoi K, Yamaguchi K, et al. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition. Clin Cancer Res 2021;27:4669–79. [DOI] [PubMed] [Google Scholar]
- 25.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med 2014;6:1279–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 2018;24:1449–58. [DOI] [PubMed] [Google Scholar]
- 28.van den Ende T, de Clercq NC, van Berge Henegouwen MI, Gisbertz SS, Geijsen ED, Verhoeven RHA, et al. Neoadjuvant Chemoradiotherapy Combined with Atezolizumab for Resectable Esophageal Adenocarcinoma: A Single-arm Phase II Feasibility Trial (PERFECT). Clin Cancer Res 2021;27:3351–9. [DOI] [PubMed] [Google Scholar]
- 29.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017;171:934–49 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ulloa-Montoya F, Louahed J, Dizier B, Gruselle O, Spiessens B, Lehmann FF, et al. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol 2013;31:2388–95. [DOI] [PubMed] [Google Scholar]
- 31.Saraon P, Pathmanathan S, Snider J, Lyakisheva A, Wong V, Stagljar I. Receptor tyrosine kinases and cancer: oncogenic mechanisms and therapeutic approaches. Oncogene 2021;40:4079–93. [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto M, Nomura S, Hosoi A, Nagaoka K, Iino T, Yasuda T, et al. Established gastric cancer cell lines transplantable into C57BL/6 mice show fibroblast growth factor receptor 4 promotion of tumor growth. Cancer Sci 2018;109:1480–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagaoka K, Shirai M, Taniguchi K, Hosoi A, Sun C, Kobayashi Y, et al. Deep immunophenotyping at the single-cell level identifies a combination of anti-IL-17 and checkpoint blockade as an effective treatment in a preclinical model of data-guided personalized immunotherapy. J Immunother Cancer 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017;31:711–23 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu M, Peng Z, Qin M, Liu Y, Wang J, Zhang C, et al. Interferon-gamma induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol Cell 2021;81:1216–30 e9. [DOI] [PubMed] [Google Scholar]
- 36.Jin H, Pinheiro PS, Callahan KE, Altekruse SF. Examining the gastric cancer survival gap between Asians and whites in the United States. Gastric Cancer 2017;20:573–82. [DOI] [PubMed] [Google Scholar]
- 37.Chao J, Fuchs CS, Shitara K, Tabernero J, Muro K, Van Cutsem E, et al. Assessment of Pembrolizumab Therapy for the Treatment of Microsatellite Instability-High Gastric or Gastroesophageal Junction Cancer Among Patients in the KEYNOTE-059, KEYNOTE-061, and KEYNOTE-062 Clinical Trials. JAMA Oncol 2021;7:895–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol 2020;38:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shitara K, Ozguroglu M, Bang YJ, Di Bartolomeo M, Mandala M, Ryu MH, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet 2018;392:123–33. [DOI] [PubMed] [Google Scholar]
- 40.Bang YJ, Ruiz EY, Van Cutsem E, Lee KW, Wyrwicz L, Schenker M, et al. Phase III, randomised trial of avelumab versus physician’s choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: primary analysis of JAVELIN Gastric 300. Ann Oncol 2018;29:2052–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bang YJ, Cho JY, Kim YH, Kim JW, Di Bartolomeo M, Ajani JA, et al. Efficacy of Sequential Ipilimumab Monotherapy versus Best Supportive Care for Unresectable Locally Advanced/Metastatic Gastric or Gastroesophageal Junction Cancer. Clin Cancer Res 2017;23:5671–8. [DOI] [PubMed] [Google Scholar]
- 42.From the American Association of Neurological Surgeons ASoNC, Interventional Radiology Society of Europe CIRACoNSESoMINTESoNESOSfCA, Interventions SoIRSoNS, World Stroke O, Sacks D, Baxter B, et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int J Stroke 2018;13:612–32. [DOI] [PubMed] [Google Scholar]
- 43.Motzer RJ, Porta C, Vogelzang NJ, Sternberg CN, Szczylik C, Zolnierek J, et al. Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: an open-label, randomised phase 3 trial. Lancet Oncol 2014;15:286–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov 2012;11:790–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holmgaard RB, Schaer DA, Li Y, Castaneda SP, Murphy MY, Xu X, et al. Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J Immunother Cancer 2018;6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Limagne E, Nuttin L, Thibaudin M, Jacquin E, Aucagne R, Bon M, et al. MEK inhibition overcomes chemoimmunotherapy resistance by inducing CXCL10 in cancer cells. Cancer Cell 2022;40:136–52 e12. [DOI] [PubMed] [Google Scholar]
- 47.Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH Jr, Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med 2019;25:936–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hahn NM, Bivalacqua TJ, Ross AE, Netto GJ, Baras A, Park JC, et al. A Phase II Trial of Dovitinib in BCG-Unresponsive Urothelial Carcinoma with FGFR3 Mutations or Overexpression: Hoosier Cancer Research Network Trial HCRN 12–157. Clin Cancer Res 2017;23:3003–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity 1994;1:447–56. [DOI] [PubMed] [Google Scholar]
- 50.Alspach E, Lussier DM, Schreiber RD. Interferon gamma and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb Perspect Biol 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A 1998;95:7556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gocher AM, Workman CJ, Vignali DAA. Interferon-gamma: teammate or opponent in the tumour microenvironment? Nat Rev Immunol 2022;22:158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Metzemaekers M, Vanheule V, Janssens R, Struyf S, Proost P. Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front Immunol 2017;8:1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the corresponding author upon reasonable request.