

Abstract
Objectives:
DEPDC5 is a common causative gene in familial focal epilepsy with or without malformations of cortical development. Its pathogenic variants also confer a significantly higher risk for sudden unexpected death in epilepsy (SUDEP), providing opportunities to investigate the pathophysiology intersecting neurodevelopment, epilepsy, and cardiorespiratory function. There is an urgent need to gain a mechanistic understanding of DEPDC5-related epilepsy and SUDEP, identify biomarkers for patients at high risk, and develop preventive interventions.
Methods:
Depdc5 was specifically deleted in excitatory or inhibitory neurons in the mouse brain to determine neuronal subtypes that drive epileptogenesis and SUDEP. EEG, cardiac, and respiratory recordings were performed to determine cardiorespiratory phenotypes associated with SUDEP. Baseline respiratory function and the response to hypoxia challenge were also studied in these mice.
Results:
Depdc5 deletion in excitatory neurons in cortical layer 5 and dentate gyrus caused frequent generalized tonic-clonic seizures and SUDEP in young adult mice, but Depdc5 deletion in cortical interneurons did not. EEG suppression immediately following ictal offset was observed in fatal and non-fatal seizures, but low amplitude rhythmic theta frequency activity was lost only in fatal seizures. In addition, these animals developed baseline respiratory dysfunction prior to SUDEP, during which ictal apnea occurred long before terminal cardiac asystole.
Interpretation:
Depdc5 deletion in excitatory neurons is sufficient to cause DEPDC5-related epilepsy and SUDEP. Ictal apnea and respiratory dysregulation play critical roles in SUDEP. Our study also provides a novel mouse model to investigate the underlying mechanisms of DEPDC5-related epilepsy and SUDEP.
INTRODUCTION
Epilepsy affects more than 70 million people globally. Sudden unexpected death in epilepsy (SUDEP) has a global incidence of 0.22–1.2/1000 individuals/year(1), accounting for up to 17% of death in patients with epilepsy, second only to stroke in potential life-years lost due to neurological diseases (2). The etiology of SUDEP is heterogenous, including cardiac, respiratory, and autonomic dysregulation in different epilepsy syndromes(2). Previous SUDEP studies focused primarily on channelopathies. For example, SCN1A encodes the alpha-subunit of the voltage-gated sodium channel Nav1.1 in the mammalian brain and heart. Its pathogenic variants were identified in >70% of children with Dravet syndrome whose SUDEP risk was estimated to be 15-fold greater than other childhood-onset epilepsies(3). Other channelopathies also affect the brain and heart, suggesting that they could confer a higher risk for SUDEP via direct effects on cardiac functions. Therefore, a “brain-heart” hypothesis for SUDEP has been proposed(4). On the other hand, Kim et al. concluded that SUDEP in patients with Dravet syndrome resulted from primary central apnea(5). This hypothesis was supported in Scn8a mutant mice that developed apnea during the tonic phase of a seizure and failed to resume breathing(6).
DEPDC5 pathogenic variants were first identified in seven large families with familial focal epilepsy with variable foci (FFEVF) and in approximately 12% of smaller families with focal epilepsy(7). The Epi4K consortium and Epilepsy Phenome/Genome Project collaborations later demonstrated that among the top five most enriched genes for epilepsy risk, only DEPDC5 reached the study-wide significant enrichment for familial non-acquired focal epilepsy (NAFE)(8). Meanwhile, several studies suggested that pathogenic variants in DEPDC5 significantly increase the risk of SUDEP(9–12). For example, DEPDC5 variants were found in ~10% of cases in a retrospective SUDEP cohort (10). In contrast with channelopathies, DEPDC5-related epilepsy primarily manifests as a focal lesion in the brain(13, 14). While patients with tuberous sclerosis complex (TSC), the prototypical monogenic disorder of the mTOR pathway, develop widespread developmental lesions in the brain and other organ systems (e.g., skin, heart, and kidney)(15), patients with DEPDC5-related epilepsy have no extra-neural lesions(13, 14). For example, 16 patients with GATOR1-related epilepsy showed no evidence of cardiac arrhythmia or structural abnormality on Holter monitoring and transthoracic echocardiogram. More importantly, no clinical cardiac anomalies were detected in 3 of 3 patients carrying pathogenic DEPDC5 pathogenic variants who succumbed to SUDEP (9).
Understanding the pathophysiology of DEDPC5-related SUDEP has significant clinical implications. Such knowledge could identify preventable causes and biomarkers for patients at high risk of SUDEP. However, the lack of an animal model that reliably and faithfully recapitulates the human phenotype has made it challenging to parse out the mechanisms. For example, mice with Emx1-Cre mediated conditional knockout of Depdc5 in dorsal cortical progenitors that give rise to excitatory neurons and astrocytes died at P16–18. These mice showed repetitive tonic-clonic seizures, status epilepticus, wasting, and malnutrition, making it difficult to determine the cause of death (16, 17). On the other hand, mice with a pan-neuronal knockout of Depdc5 died after a single terminal seizure at 3 months of age (9, 18). Interestingly, the majority of these mice had no history of seizures, representing an extremely rare patient event. In this study, we showed that Depdc5 deletion in a subpopulation of mouse forebrain excitatory neurons caused frequent generalized tonic-clonic seizures in young adult mice. These mice eventually developed SUDEP. The respiratory recording showed interictal respiratory dysregulation and ictal apnea long before the terminal cardiac asystole. Our studies provide a refined framework to further investigate the pathophysiology of DEPDC5-related epilepsy and SUDEP at molecular, cellular, and circuit levels.
MATERIALS AND METHODS
Animals
Rbp4-Cre transgenic mice (B6.FVB(Cg)-Tg (Rbp4-Cre) KL100Gsat/Mmucd, RRID: MMRRC_037128-UCD, donated by Dr. Kenneth Kwon at the University of Michigan) were crossed with mice with floxed alleles of Depdc5F/F (Depdc5tm1c(EUCOMM)Hmgu, donated by Dr. Jun Hee Lee at the University of Michigan) to generate Depdc5 conditional knockout (CKO) mice. Ventral medial ganglionic eminence (MGE) progenitor-specific Nkx2.1-Cre transgenic mice (Jackson Laboratories #008661) were used to generate interneuron-specific Depdc5 CKO mice. Ai9 mice (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J, Jackson Laboratories #007909) were used to label cells with recombinase activity mediated by Rbp4-Cre. Male homozygous floxed Depdc5 mice (Depdc5F/F) were bred with female mice heterozygous for floxed Depdc5 (Depdc5F/W) and heterozygous for the Cre allele to generate litters of homozygous CKO (Rbp4-Cre; Depdc5F/F, Nkx2.1-Cre; Depdc5F/F), heterozygous CKO (Rbp4-Cre; Depdc5F/W, Nkx2.1-Cre; Depdc5F/W), and wild-type (Depdc5F/F or Depdc5F/W) mice. Because our pilot study showed that Rbp4-Cre; Dedpc5F/W displayed no pathological or epileptic phenotypes, in addition to previous studies showing that Emx1-Cre; Depdc5F/W and Syn1-Cre; Depdc5F/W had no phenotypes(16–18), Rbp4-Cre; Depdc5F/W mice were also used as littermate controls. DNA was extracted from mouse tail biopsies using the Extract-N-Amp Tissue PCR kit (Sigma, XNAT2–1KT) according to the manufacturer’s instructions. Genotyping of the Depdc5 gene was performed using primers (forward: 5′- CTGGAATTACAGGGGGTAAGCCAGTG-3′; reverse: 5′- CAGGACTACACAGAGAAACCCTGTCTC-3′) that flank LoxP sites surrounding exon 5, allowing concurrent detection of the WT and conditional alleles. Agarose gel electrophoresis was performed to detect a 177-base pair (bp) band for the WT allele and a 337-bp band for LoxP conditional allele. The presence of the Rbp4-Cre recombinase gene was determined using primers (forward: 5’- GGGCGGCCTCGGTCCTC −3′; reverse: 5’- CCCCAGAAATGCCAGATTACGTAT-3′) to detect a 600 bp band. The presence of the Nkx2.1-Cre recombinase gene was determined using primers (forward: 5’-CTCTGGTGGCTGCCTAAAAC-3′; reverse: 5’-CGGTTATTCAACTTGCACCA-3′) detecting a 410 bp band. All mice were housed in a 12-hr light-dark cycle, climate-controlled room, with access to food and water ad-lib. The protocol for the study has received approval by the Institutional Animal Care and Use Committee at the University of Michigan, and all studies were conducted in accordance with the United States Public Health Service’s Policy on Humane Care and Use of Laboratory Animals.
Immunohistochemistry
Brains were removed and fixed in 4% paraformaldehyde in PBS after transcardial perfusion, sectioned at 50μm on a Leica VT1000S vibratome, and processed for immunocytochemistry as free-floating sections. Primary antibodies included rabbit anti-Parvalbumin (1:1000, Abcam, Cat# ab181086), rabbit anti-GFAP (1:1000, Abcam, Cat# ab7260), and rabbit anti-phospho-S6 ribosomal protein Ser 235/236 (1:2000, Cell Signaling). Fluorescently conjugated secondary antibodies (Alexa Fluor 488, 594, or 647, 1:500) were obtained from Molecular Probes, and nuclei were labeled with bisbenzimide (Molecular Probes, Cat# H1398).
EEG implantation and analysis
To determine if Rbp4-Cre; Depdc5F/F and Nkx2.1-Cre; Depdc5F/F CKO mice exhibit electroclinical seizures and interictal epileptiform discharges (IEDs), we monitored animals with continuous video-EEG (Natus, Middleton, WI). Mice were implanted with three epidural screw electrodes (E363/96; P1 Technologies, Roanoke, VA) at P30. Procedures for affixing electrodes were performed as previously described(16). Three electrodes were positioned and fastened through burr holes, with two electrodes implanted over the left and right parietal lobes and a reference electrode implanted over the cerebellum. The sockets were fitted into a 6-pin electrode pedestal, and the entire apparatus was secured with dental cement (Stoelting). 5 days after surgery, animals were monitored continuously for up to 60 days or until death. Recordings were sampled at 4096 Hz and analyzed offline with concurrent video. Seizures and epileptiform activity were assessed manually in their entirety by fellowship-trained and board-certified epileptologists blinded to the genotype. Interictal epileptiform discharges (IED) were defined as transients distinguishable from background activity with characteristic morphology typically, but neither exclusively nor invariably, found in interictal EEGs of people with epilepsy (19) and had to meet at least 3 of following criteria: 1) Di- or tri-phasic waves with a sharp wave or spike morphology (<200 msec duration); 2) Different wave duration than the ongoing background activity; 3) Asymmetry of the waveform: a sharply ascending phase and a slower descending phase, or vice versa; 4) The transient is followed by an associated after-going slow wave; 5) The background activity is disrupted by the presence of the epileptiform discharges. Seizures are defined as EEG phenomena consisting of repetitive epileptiform discharges at >2 cycles/second and/or a characteristic pattern with quasi-rhythmic spatiotemporal evolution (i.e., gradual change in frequency, amplitude, morphology, and location) lasting at least several seconds (usually >10 s). Two other short-duration (<10 s) EEG seizure patterns are defined as electro-decrement or low voltage fast activity seen during clinically apparent epileptic seizures. EEG seizure patterns unaccompanied by behavioral epileptic manifestations are referred to as electrographic or subclinical seizures (19). Mice with dislodged EEG head caps were excluded from the study.
Recordings in European Data Format (EDF) were transformed using the “edfread” function in MATLAB. Spectrograms were constructed using the MATLAB “fft” function to analyze data from 10 minutes before to 10 minutes after seizure offset in rectangular 1-second windows without overlap. Signal power at 1– 70 Hz was represented in the spectrograms on a log scale. Differences in the time-frequency spectrograms between fatal and non-fatal seizures were evaluated with a shuffle test. For the 24 seizures reported (16 non-fatal and 8 fatal), the difference between the mean non-fatal and mean fatal spectrograms were calculated for 600 seconds preceding and following seizure offset. To generate a surrogate distribution of differences in the mean spectrograms, the labels “fatal” and “non-fatal” were randomly assigned to each seizure, and the difference in mean spectrograms was recalculated. This was repeated 10,000 times. The p-value testing the null hypothesis of no difference between fatal and non-fatal seizures at each time-frequency point was determined as the fraction of surrogate differences greater than the true difference. P-values were smoothed using a 2D Gaussian filter with a standard deviation of 0.5.
Simultaneous recording of EEG, EKG, and breathing during SUDEP
Mice around two months old were implanted with EEG and EKG electrodes. For EEG electrodes, two holes were drilled into the skull (one on the parietal bone and the other above the cerebellum), and electrodes were positioned and fastened using mounting screws (E363/96). For the EKG electrode, the wires were bilaterally connected to the tissue under the skin on the side of the body and extended subcutaneously to the head. Both EEG and EKG electrodes were connected to the head mount, which was stabilized by dental cement. After one week of recovery, mice were placed in the whole-body plethysmograph chamber (Buxco, ~500 mL), and the head mount was connected to the amplifier. EEG, EKG, and breathing data were collected by PowerLab (AD instruments). The sampling rate was 1000 Hz. The EEG signal was digitally filtered using a band-pass filter (25–300 Hz) and integrated (time constant of 0.1 s) by LabChart (AD instruments). The EKG signal was digitally filtered using a high-pass filter (25 Hz). The mice were recorded daily until they developed SUDEP.
Breathing recording in hypoxia challenge
For breathing recording in the basal and hypoxia condition, mice were placed in the whole-body plethysmography (WBP) chamber at room temperature. Mice were allowed to acclimate to the chamber in normoxia (21% O2 balanced with nitrogen) at 1L/min flow rate before recording breathing. For the hypoxia challenge, mice were exposed to normoxia for 20 min and then switched to hypoxia (10% O2 balanced by nitrogen) for 20 min before switching back to normoxia. During the switches, the chamber was flushed with the new gas mixture for 1 minute before data collection. Breathing parameters were recorded and analyzed by Emka IOX2 software (EMKA Technologies) that calculates the tidal volume based on the Drorbaugh and Fenn formula (20). Body temperature was tested by a rectal probe connected to a temperature controller (Physitemp Instruments, TCAT-2LV) in the hypoxia challenge.
Image acquisition, soma size, cortical thickness measurement, and statistical analyses
Multi-channel imaging was performed using a Leica SP5 confocal microscope. All images were further processed in Adobe Photoshop software. The soma size of Ai9+ or PV+ neurons from P21~30 brains were compared between control and Depdc5 CKO. Cortical thickness was measured from the pial surface to the white matter from the somatosensory cortex. Automated measurement was performed in ImageJ. Statistical analysis was performed using GraphPad. The number of animals used in each experiment was listed in the results and figure legends. p values were determined by the student t-test. Analysis of variance was conducted with Bonferroni post hoc correction for multiple comparisons. All data were shown as mean ± s.e.m. A p-value less than 0.05 was considered to be statistically significant. In figure panels throughout the manuscript, the following labeling conventions for statistical testing are used: no significance (ns): p>0.05; *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
RESULTS
Depdc5 deletion in a subpopulation of forebrain excitatory neurons causes epilepsy.
We and others have shown that Emx1-Cre mediated Depdc5 deletion in dorsal cortical progenitors causes severe epilepsy, megalencephaly, and premature death at postnatal (P) weeks 2–3 (16, 17). Surprisingly, Syn1-Cre mediated Depdc5 deletion in all neuronal lineages (excitatory and inhibitory neurons) and neural structures often caused just one terminal seizure followed by death at 3 months old (9, 18). These striking phenotypic differences suggest that epileptogenesis and SUDEP in DEPDC5-related epilepsy are cell-type dependent.
To test whether Depdc5 deletion in excitatory neurons alone causes epilepsy, we crossed Rbp4-Cre transgenic mice (21) to mice with floxed Depdc5 alleles (16) to delete Depdc5 in a subset of forebrain excitatory neurons as early as at embryonic day (E) 16.5 (21) (hereafter referred to as Rbp4-Cre; Depdc5F/F CKO mice). To examine the recombination pattern, we first crossed Rbp4-Cre to the tdTomato Cre reporter line Ai9 (22). tdTomato was observed in the somata, dendrites, and axons of layer 5 pyramidal and dentate gyrus granule excitatory neurons (Fig. 1A), consistent with the previous report (21). Furthermore, these tdTomato labeled cells were not immunopositive for markers of interneurons (Fig. 1B), astrocytes (Fig. 1C), or oligodendrocytes (data not shown), suggesting that Rbp4-Cre recombinase activity is both spatially and cell-type specific. We then crossed Rbp4-Cre to both Ai9 and Depdc5F/F transgenic lines to generate Depdc5 conditional knockout (Rbp4-Cre; Ai9; Depdc5F/F) and littermate controls (Rbp4-Cre; Ai9; Depdc5F/W or Rbp4-Cre; Ai9; Depdc5W/W). Because of the lack of a specific antibody against DEPDC5, immunohistochemistry and Western blot could not be performed to directly detect the Cre- mediated Depdc5 deletion. Instead, we examined the expression level of ribosomal protein S6 (pS6), the downstream target of the mTOR signaling pathway. Compared to layer 4 or 6 neurons (tdTomato negative), layer 5 Depdc5 KO neurons (tdTomato positive) in the same Rbp4-Cre; Depdc5F/F CKO mice brain showed a robust increase in pS6 immunoreactivity (Fig. 1D–D”), indicating elevated mTOR signaling. Compared to layer 5 WT neurons, layer 5 Depdc5 KO neurons also showed significantly increased pS6 staining (data not shown) with a much larger soma surface area (Fig. 1E–G), a pathological hallmark of mTORopathies. Cortical thickness, however, was not significantly increased in Rbp4-Cre; Dedpc5F/F CKO mice (Fig. 1H).
Figure 1. Rbp4-Cre mediates Depdc5 deletion in cortical layer V and dentate gyrus excitatory neurons.
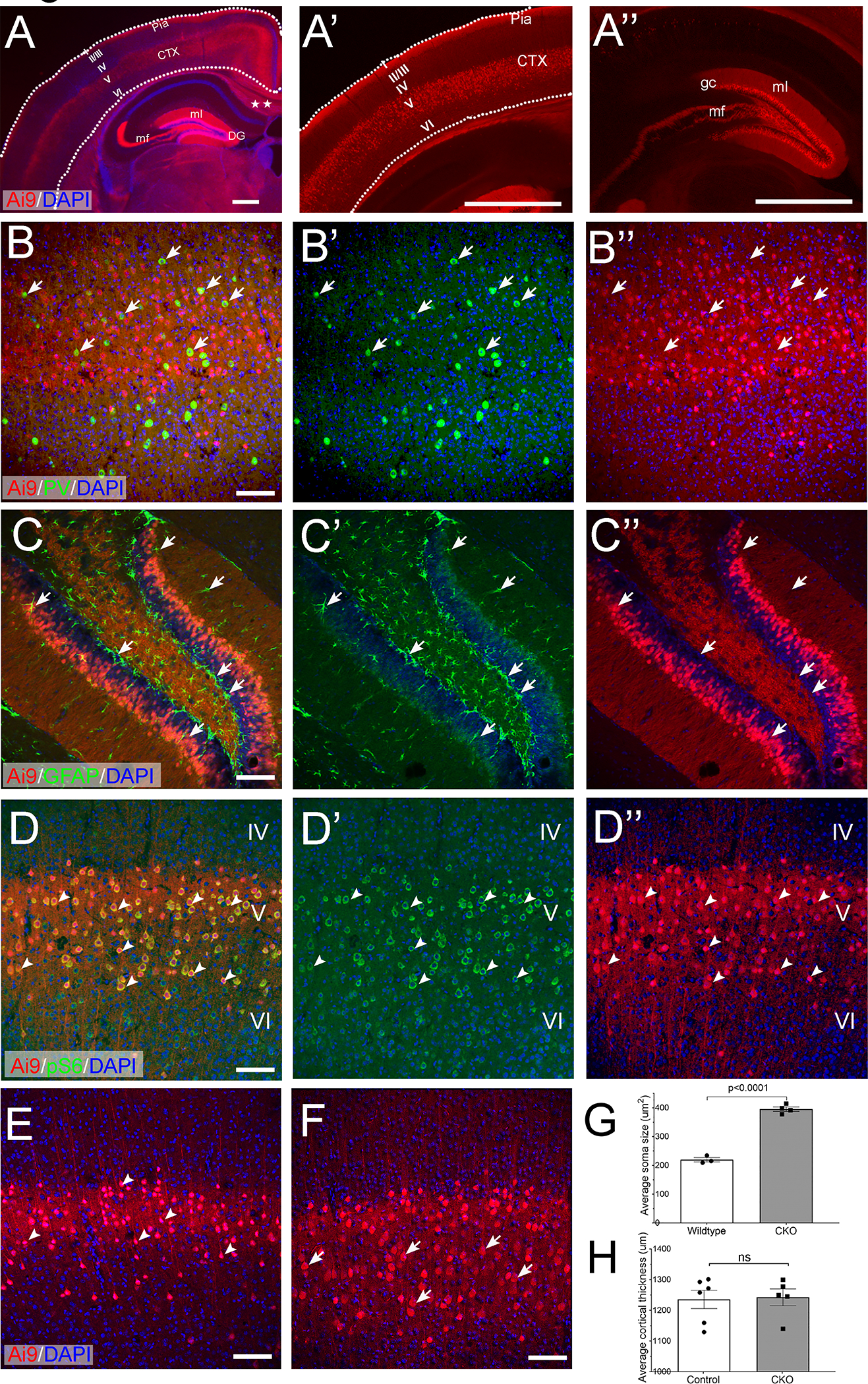
A) Representative coronal section of an Rbp4-Cre; Ai9 mouse brain. In the cortex, tdTomato-positive neurons are in cortical layer V and send out interhemispheric cortical-to-cortical axonal projection (stars). In the hippocampus, granular cells (gc) in the dentate gyrus (DG), mossy fibers (mf), and dendrites in the molecular layer (ml) are intensely labeled. A’ and A”) are higher magnification images of the cortex and hippocampus, respectively. Dotted lines are drawn to outline the cortex. Roman numerals indicate different cortical layers. CTX: cortex. B-B”) tdTomato-positive cells are not positive for the interneuron marker, Parvalbumin (green, arrows), or C-C”) the astrocyte marker, GFAP (green, arrows). D-D”) Representative images obtained from an Rbp4-Cre; Depdc5F/F; Ai9+/− mouse brain. In the cortex, dtTomato-positive Depdc5 KO neurons in layer V have significantly increased immunoreactivity of pS6 (green), as compared to dtTomato-negative neurons in layers IV and VI. E-G) Compared to tdTomato-positive layer V neurons in control animals (E, arrowheads), tdTomato-positive Depdc5 KO neurons (F, arrowheads) are cytomegalic and have a significantly increased surface area (G, control: 219.7± 7.46um2, n=3, vs. CKO 395.5 ± 7.64um2, n=4. p<0.0001). H) Cortical thickness is not increased in Rbp4-Cre; Depdc5 CKO mice (control: 1235 ± 49.97um, n=6 vs. CKO 1262 ± 34.9um, n=5, p=0.6). Scale bar: 250 μm in A; 1 mm in A’ and A”; 100 um in B-F.
To determine whether Depdc5 deletion mediated by Rbp4-Cre is sufficient to cause spontaneous seizures, continuous video-electroencephalogram (vEEG) monitoring was performed (Fig. 2A, n = 19). All Rbp4-Cre; Depdc5F/F CKO mice developed interictal epileptiform discharges (IED), including spike and wave discharges that at times were periodic or rhythmic and lasted up to several seconds (Fig. 2B, B’). Mice developed their first seizures by 2 months old (postnatal day 57.25 ± 10.92 days old). The seizure frequency was 1.49 ± 0.83 per day (Fig. 2D, F). Most seizures were characterized by generalized tonic-clonic (GTC) activity followed by wild running (Racine score 5, n = 122 out of 126) (23), and a few were subclinical seizures (n = 4 out of 126). All seizures showed bi-hemispheric onset with rhythmic spike-and-wave discharges followed by evolution in frequency and amplitude (Fig. 2C, C’). Seizures were brief (Fig. 2E), and no animals developed status epilepticus (defined as lasting longer than 3 min) or seizure clusters (defined as >2 seizures within one hour). In contrast, Rbp4-Cre; Depdc5F/W mice (n = 11) did not show IED or spontaneous seizures.
Figure 2. Rbp4-Cre; Depdc5F/FCKO mice develop spontaneous generalized tonic-clonic seizures.
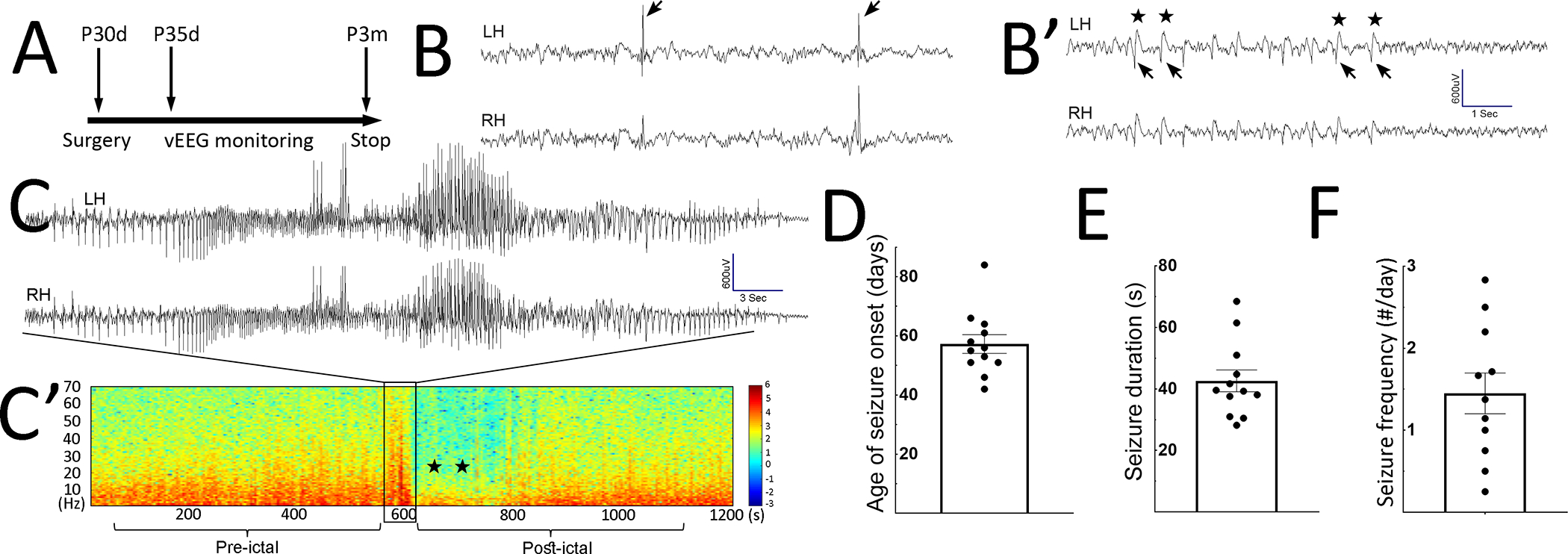
A) Timeline of continuous video-EEG monitoring experiments. EEG implantation was performed between P30–31 days (d). vEEG started 5 days after implantation and continues until P3 months (m). B, B’) Representative examples of interictal epileptiform discharges (IED), including singly occurred spikes in B) and periodic spike-and-wave discharges in B’). In B’) arrows indicate spikes, and stars indicate the after-going slow waves. LH: left hemisphere; RH: right hemisphere. C) Representative EEG trace during generalized tonic-clonic seizures. C’) Representative spectrogram during the pre-ictal, ictal, and post-ictal stages. The boxed area displays a surge of increased power across all frequency bands, representing the seizure in C). Postictally, decreased power in all frequency bands (stars) is evident and represents EEG background suppression, lasting up to 200 seconds (s). The background then gradually recovers. D-F) Age of seizure onset: 57.25 ± 3.16 postnatal days (n=12), seizure duration (42.65 ± 3.544 sec, n=12), and seizure frequency (1.45 ± 0.25 per day, n=11).
To test if Depdc5 deletion in interneurons (IN) causes epilepsy, we deleted Depdc5 in ventral cortical progenitors within the medial ganglionic eminence (MGE) using Nkx2.1-Cre transgenic mice (24). MGE is the primary source of parvalbumin (PV) and somatostatin (SST) interneurons(25) that respectively provide the major contribution to perisomatic and dendritic inhibition(26). Cre expression in Nkx2.1-Cre mice was detected in the MGE as early as E10.5 and postnatally in more than 80% of PV- and SST- positive interneurons(24). Although PV+ IN were cytomegalic with significantly increased pS6 immunostaining (Fig. 3A–C), Nkx2.1-Cre; Depdc5F/F CKO mice showed no change in cortical thickness (Fig. 3D) and no seizures during 2–6 weeks of continuous vEEG recording (data not shown). In summary, our data suggest that Depdc5 deletion in a subpopulation of excitatory neurons in the brain is sufficient to drive epileptogenesis and cause seizures.
Figure 3. Depdc5 deletion in interneurons does not cause seizures.
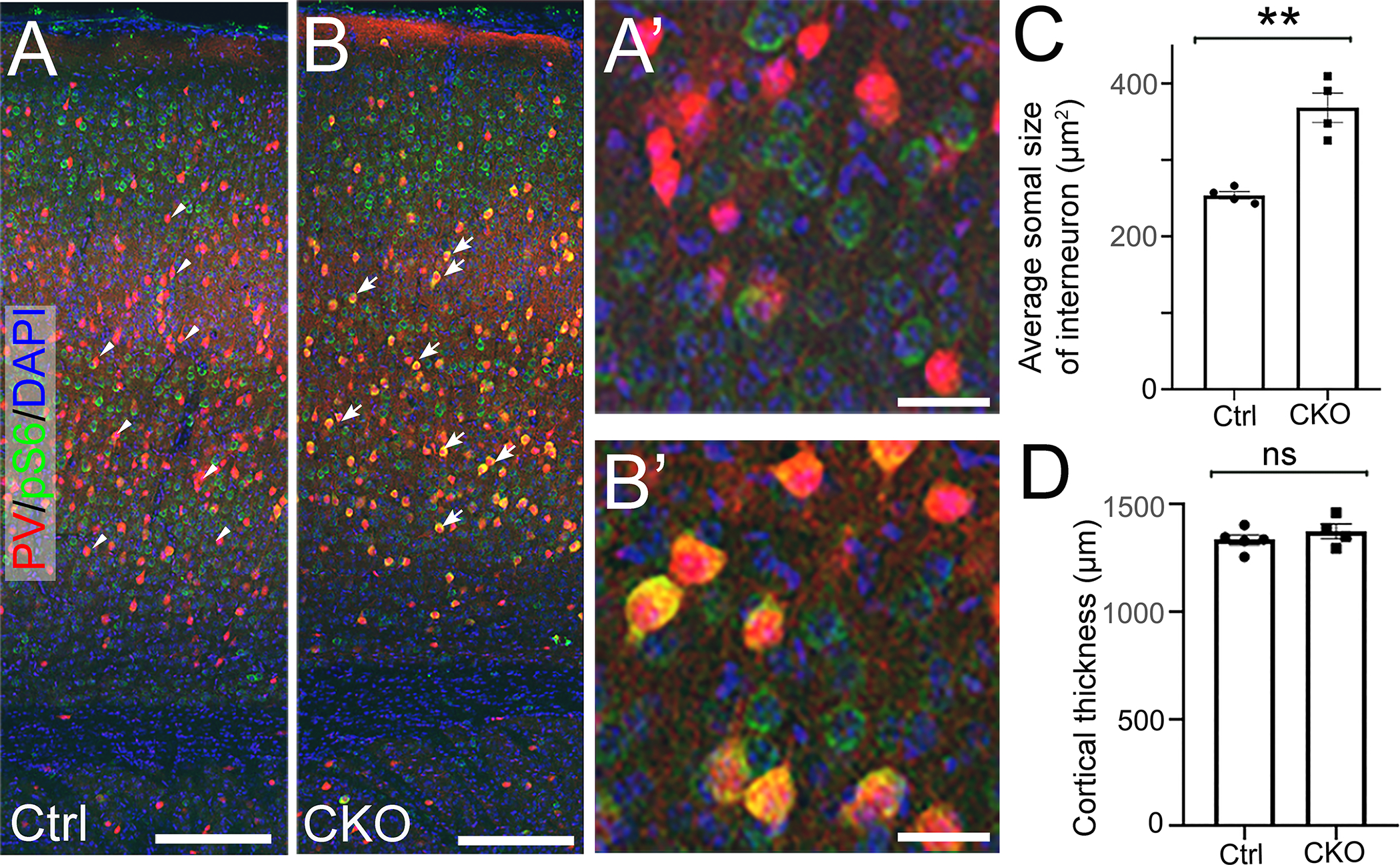
A, B) Representative images of anti-pS6 (green) and anti-PV (red) co-immunohistochemistry staining on P21 brain coronal sections from control A) and Nkx2.1-Cre; Depdc5 CKO mice. Compared to control PV+ interneurons (arrowheads in A), Depdc5 KO PV+ interneurons (arrows in B) show significantly increased immunoreactivity of pS6 (yellow). A’) and B’) are higher magnification images. C) Quantification shows that cortical PV+ interneurons in Nkx2.1-Cre; Depdc5 CKO mice have an enlarged soma size (n=4 in control and CKO groups, student t-test, p<0.001). D) Nkx2.1-Cre; Depdc5 CKO mice do not display increased cortical thickness. Scale bars: 500 um in A, B; 25um in A’, B’.
Rbp4-Cre; Depdc5F/F CKO mice develop SUDEP.
Rbp4-Cre; Depdc5F/F CKO mice grew normally into young adulthood but had a shortened life span (Fig.4A). Nearly all Rbp4-Cre; Depdc5F/F CKO mice died suddenly without physical or behavioral concerns. The carcasses were often in a tonic posture with extended hindlimbs, raising concern for SUDEP. Littermate wildtype and Rbp4-Cre; Depdc5F/W mice all survived beyond 6 months. Continuous video-EEG monitoring was performed to identify SUDEP. 8 of 12 Rbp4-Cre; Depdc5F/F CKO mice died immediately following a generalized tonic-clonic seizure (Racine score of 5). The fatal and the last non-fatal seizures from the same animal were analyzed to investigate if EEG features could predict SUDEP. The EEG signals started recovering within 10–30 seconds from the onset of post-ictal suppression in non-fatal seizures, while the EEG signals never recovered from the suppression in fatal seizures (Fig. 4B–C). Visual inspection of the post-ictal EEG revealed that all non-fatal seizures preserved low amplitude rhythmic theta frequency activity while fatal seizures did not (Fig. 4B, C inserts), which was confirmed by a statistically significant difference in theta power immediately after seizures (Fig. 4D and D’). The absence of this specific theta frequency (3–12 Hz) band could suggest that “death” already occurred before the seizure offset or could represent a unique electrographic marker indicating imminent death or an irreversible pathological state. Because the seizure was brief and cardiac asystole did not occur ictally (see next section), the immediate EEG suppression likely represented an abrupt and widespread circuit perturbation that is not reversible and results in death. Semiologically, there was no difference in severity between the fatal and last non-fatal seizure since most seizures in each group were GTC (Fig. 4E). The seizure duration did not differ between the fatal and last non-fatal seizure (Fig. 4F). The frequency of non-fatal seizures did not increase before the terminal seizure (Fig. 4G). Fatal seizures occurred during both the nocturnal and diurnal phases (4 in the light phase and 4 in the dark phase). The age at SUDEP ranged from P46–116 days (Fig. 4H). Our data show that the Rbp4-Cre; Depdc5F/F CKO mouse model recapitulates many features of epilepsy and SUDEP in humans.
Figure 4. Rbp4-Cre; Depdc5F/F CKO mice develop SUDEP.
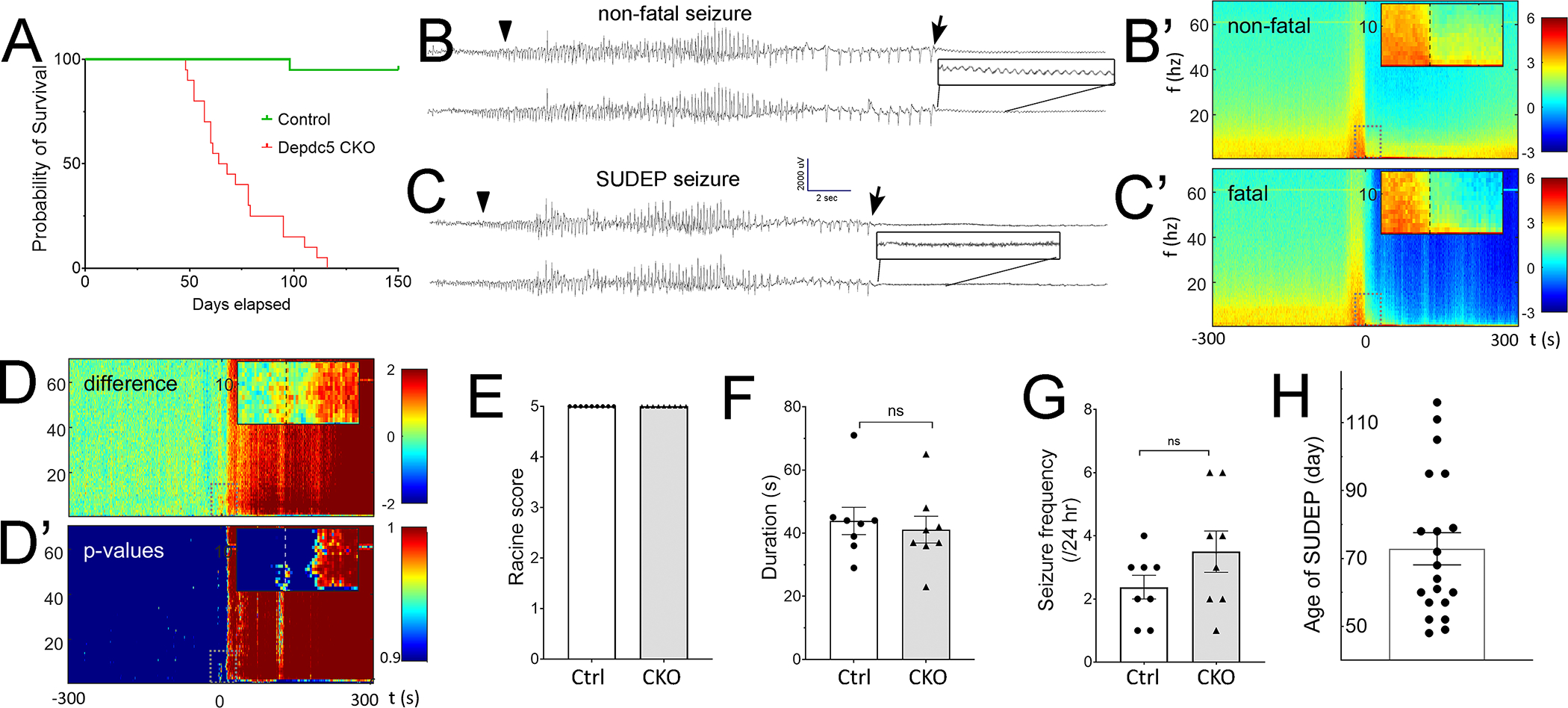
A) Rbp4-Cre; Depdc5F/F CKO mice (red line, n=20) have a shortened lifespan and die by 4 months of age. In contrast, control (green line, n=19) and Nkx2.1-Cre; Dedpc5 CKO (data not shown) mice survive beyond 6 months. B, C) Representative EEG examples of the last non-fatal B) and fatal seizure C) recorded from the same animal. The arrowhead and arrow indicate the ictal onset and ictal offset, respectively. Immediately following the ictal offset, generalized EEG suppression is seen in both non-fatal and fatal seizures. However, Close-up visual examination (black boxes) reveals that low amplitude monotonous theta frequency activity is preserved only in non-fatal seizures. B’, C’) Mean spectrograms showing log10 (power) for non-fatal (B’, n=16) and fatal seizures (C’, n=8) in the 0–70 Hz frequency bands. Insets show −20 s to +30 s from seizure offset (time 0, dashed line) and frequencies from 0 to 15 Hz. Immediately after the ictal offset, spectrograms display decreased power in the 0–10Hz band in fatal seizures, confirming the electrographic findings. D) Difference between mean log10(power) spectrograms (non-fatal – fatal). D’) p-values that the difference between fatal and non-fatal seizures arose due to chance alone as a function of time and frequency. A significant difference (p<0.05, inset) is observed immediately after the ictal offset in the theta frequency band. E) The last non-fatal seizures and fatal seizures are generalized tonic-clonic seizures. F) The last non-fatal and fatal seizures have similar seizure duration. G) The seizure frequency does not increase significantly on the day of non-fatal seizures. H) Quantification of the age of SUDEP.
Rbp4-Cre; Depdc5F/F CKO mice develop ictal apnea and interictal respiratory dysregulation.
To better understand the cause of post-ictal death in Rbp4-Cre; Depdc5F/F CKO mice, a custom mouse epilepsy monitoring unit (EMU) was used to record EEG, electrocardiogram (EKG), behavior, and plethysmography. Mice were singly housed in a plethysmography recording chamber and underwent daily 12-hour recording sessions until they developed SUDEP. While the majority of SUDEP events occurred when the recording was temporarily halted or after the recording apparatus was dislodged, 3 SUDEP events were recorded and analyzed. Pre-ictally, Rbp4-Cre; Depdc5F/F CKO mice exhibited rhythmic breathing and heartbeats (Fig. 5 A, B, B1 enlarged from B). Interestingly, slightly before ictal onset detected by EEG (Fig. 5C1, arrow), Rbp4-Cre; Depdc5F/F CKO mice already increased their breathing rate (Fig. 5C1, arrowhead) without clear body movements detected on video or by movement artifacts on EEG. During fatal seizures, tachypnea continued into the ictal period and then abruptly developed into complete apnea (Fig. 5D1, red arrowhead), which persisted into the post-ictal period and never recovered (Fig. 5E). Modest bradycardia was seen during the post-ictal phase, but the heartbeat continued for several minutes with slowly worsening bradycardia, eventually leading to terminal asystole (Fig. 5E1 enlarged from E). It is noteworthy that the clinical and electrographic seizure was still ongoing after the onset of apnea. In contrast, tachypnea was observed through the ictal and post-ictal periods during non-fatal seizures. These results suggest the critical role of ictal apnea in the death of Rbp4-Cre; Depdc5F/F CKO mice.
Figure 5. Rbp4-Cre; Depdc5F/F CKO mice develop ictal apnea and interictal respiratory dysregulation.
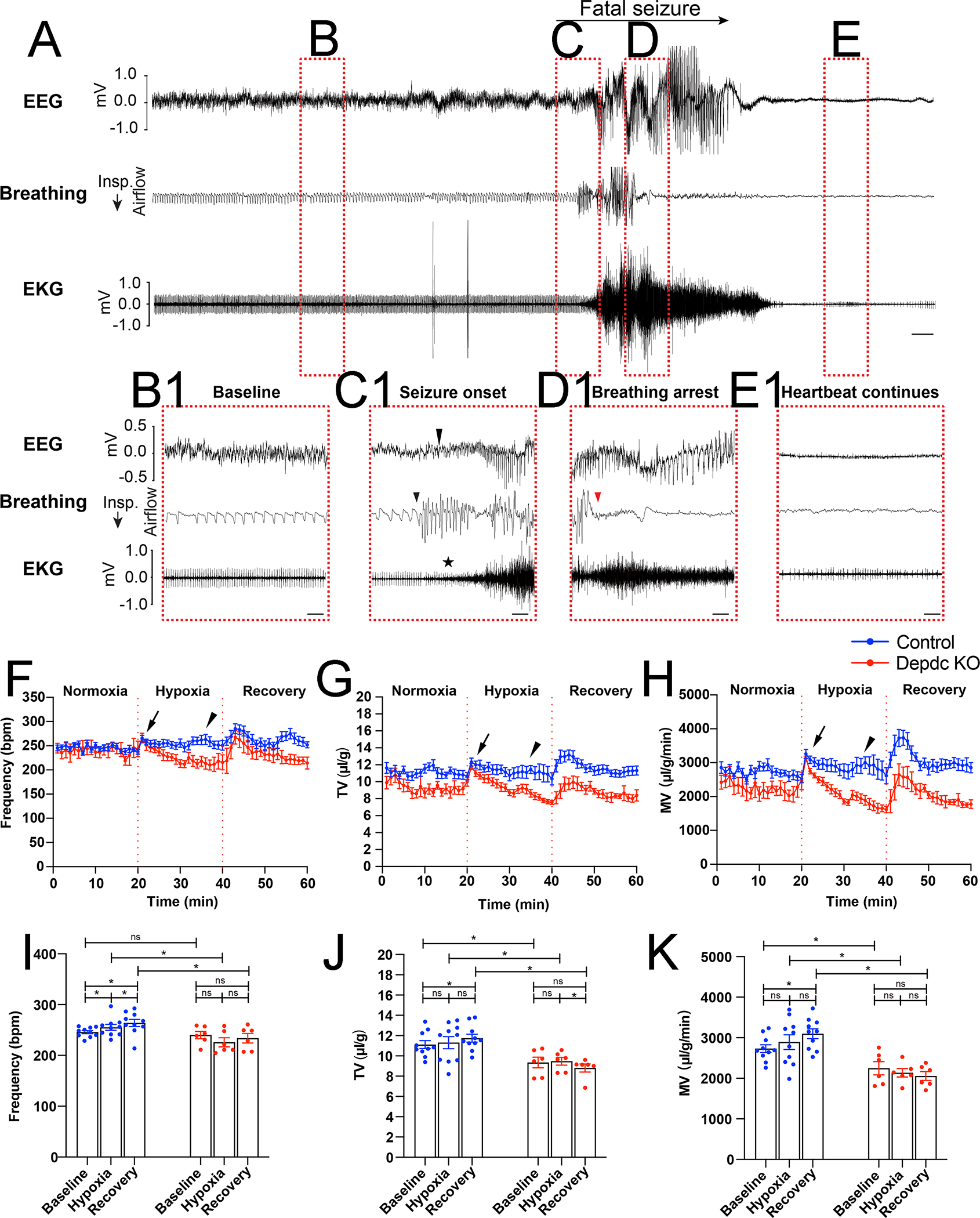
A) Representative EEG, breathing, and EKG traces during a spontaneous fatal seizure (SUDEP). Red dotted rectangles indicate B) the baseline/pre-ictal stage, C) the ictal onset, D) the seizure progression, and E) the post-ictal stage. B1-E1) Expanded traces at the stages labeled B-E). B1) Pre-ictally, heartbeat and breathing are rhythmic. C1) Ictal onset is marked by the arrow on the EEG trace. The mouse displays tachypnea with increased respiratory rate slightly before the definite electrographic onset (arrowhead on the breathing flow trace), suggesting a possible hypersynchrony involving respiratory circuits. Although the EKG trace is soon masked by muscle artifacts (star), there is no clear cardiac arrhythmia or irregular heartbeat at the seizure onset. D1) Tachypnea persists in the ictal period and suddenly transitions to complete apnea (red arrowhead) in the middle of the seizure (~15 seconds after the onset). E1) During the post-ictal stage, the background suppression on EEG and the complete apnea on plethysmography continue and never recover during SUDEP. In contrast, mild bradycardia is evident on EKG and continues to worsen for several minutes leading to terminal asystole. Scale bars: 5 s in A; 1 s in B1-E1. F-H) Respiratory rate (bpm) F), tidal volume (TV) G), and minute ventilation (MV) H) of the Rbp4-Cre; Depdc5F/FCKO mice (n = 6, red) and their littermate controls (n = 10, blue) before, during, and after hypoxia challenges (each period 20 min). Upon exposure to a hypoxia condition, CKO mice and their littermate controls initially develope a similar hypoxia ventilatory response, including increased respiratory rate, TV, and MV (arrows). However, during the last 5 min of the challenge, CKO mice exhibit significantly diminished respiratory function in all 3 parameters (arrowheads). I-K) Quantification of respiratory rate I), tidal volume J), and minute ventilation K) show that Depdc5 CKO mice (n = 6) and their littermate controls (n = 10) before, during, and after the hypoxia challenge.
Because these animals failed to recover from ictal apnea, we next asked if they have interictal respiratory phenotypes and performed hypoxia challenge test. At baseline (normoxia), Rbp4-Cre; Depdc5F/F CKO mice and their littermate controls had similar respiratory rates (Fig. 5F and I), but Rbp4-Cre; Depdc5F/F CKO mice showed decreased tidal volume (TV) (Fig. 5G, H) and minute ventilation (MV) (Fig. 5J, K). Upon exposure to 10% O2 (hypoxia), Rbp4-Cre; Depdc5F/F CKO mice and their littermate controls showed similar body temperature (36.0 ± 0.4 ℃ in control vs. 36.2 ± 0.2 ℃ in CKO mice, n = 3) and developed a similar hypoxia ventilatory response, including increased respiratory rate, TV, and MV (Fig. 5F–H, arrows). During the 20-minute challenge, control mice gradually exhibited hypoxic ventilatory depression, in which their respiratory parameters returned to baseline (27). In contrast, Rbp4-Cre; Depdc5F/F CKO mice showed a sharp and continuous decline of respiratory function (Fig. 5F–K). By the end of hypoxia challenge, their respiratory rate, TV, and MV were not only significantly lower than control mice but also lower than their own baseline. These results suggest that Rbp4-Cre; Depdc5F/F CKO mice have intrinsic circuit disturbances resulting in maladaptation to hypoxia.
DISCUSSION
Depdc5 deletion in cortical layer 5 and hippocampal dentate gyrus excitatory neurons is sufficient to cause epilepsy.
DEPDC5 encodes the DEP domain-containing protein 5 (DEPDC5) that, together with nitrogen permease regulator 2-like (NPRL2) and nitrogen permease regulator 3-like (NPRL3), forms GTPase-activating protein (GAP) activity toward Rags-1 (GATOR1) complex, the main negative regulator of the amino acids sensing machinery in mTORC1 signaling pathway(28). The mTOR pathway plays critical functions in various physiological and behavioral processes, and its aberrant hyperactivation has been linked to different pathological conditions including epilepsy. Cell type-specific loss of mTOR inhibition in cerebellar, dopaminergic, serotonergic, or striatal neurons induces different cognitive and behavioral deficits in the absence of epilepsy (29–34), suggesting that distinct cell types and circuits are responsible for specific neuropsychiatric manifestations. Here, we showed that Depdc5 deletion in a subset of forebrain excitatory neurons was sufficient to cause epilepsy and SUDEP. In contrast, Nkx2.1-Cre mediated interneuron-specific Depdc5 deletion did not cause spontaneous seizures, consistent with previous studies. For example, mice with Tsc1 deletion in PV+ or SST+ interneurons did not develop spontaneous seizures and grew normally into adulthood(35). Mice conditionally expressing the constitutively active Pik3ca p.H1047R pathogenic variant in MGE-derived interneurons (Nkx2.1-Cre)(36), or more specifically in PV+ interneurons (Pvalb-Cre) (37), did not develop spontaneous seizures. These results suggest that mTORopathies-related epilepsy is not caused by the deletion of mTOR pathway genes in interneurons. Interestingly, a recent study used a human cerebral organoid model of TSC and identified the over-proliferation of caudal late interneuron progenitor (CLIP) cells as a vulnerability for brain tumors and cortical malformation(38). Although human-specific neurodevelopmental programs could be unique, it remains unclear whether these findings from cultured organoids representing very early embryogenesis can be translated into functional changes in more mature brains.
Although patients with familial DEPDC5-related epilepsy harbor a germline mutation, they primarily present with focal pathology in the brain (13, 14). Genetic evidence has emerged that the focal pathology is the consequence of a pathogenic germline mutation and a somatic mutation leading to mosaic biallelic DEPDC5 loss-of-function (LOF)(39–41), concordant with Knudson’s “two-hit” hypothesis(42). Although our studies did not precisely recapitulate this “two-hit” genetic mechanism, Rbp4-Cre; Depdc5F/F CKO mice modeled the mosaic biallelic LOF mutation in the brain.
Rbp4-Cre; Depdc5F/F CKO mice recapitulate DEPDC5-related epilepsy and SUDEP.
Rbp4-Cre; Depdc5F/F CKO mice developed fatal seizures between ~P50–120 days, equivalent to young adulthood in humans. It is noteworthy that SUDEP occurs at 20~40 years old on average, with a higher incidence at the younger end(43). We also did literature search and identified 8 well-documented DEPDC5-related SUDEP cases with the age of death ranging from 18 to 58 years old(9, 11, 12). Rbp4-Cre; Depdc5F/F CKO mice developed frequent spontaneous tonic-clonic seizures, the greatest risk factor for SUDEP, and their death did not occur in the context of status epilepticus or repetitive seizures, phenocopying humans with SUDEP. These features are remarkably different from previous animal models of DEPDC5-related epilepsy. We previously generated dorsal progenitor-specific Emx1-Cre; Depdc5F/F CKO mice that developed severe seizures and died during the 3rd postnatal week(16, 17). These mice developed seizure clusters, immobility, malnutrition, and wasting preceding death, making it challenging to use them for SUDEP studies. On the other hand, most Syn1-Cre; Depdc5F/F CKO mice died of a single seizure at ~3 months without any prior spontaneous (clinical or subclinical) seizures, which represents an extremely rare event in patients with SUDEP(9, 18). This is also in sharp contrast to the clinical hallmark of mTORopathies: frequent seizures. Although the strain background could slightly differ in these transgenic experiments (it is unclear if previous studies used a congenic C57BL/6 Syn1-Cre strain)(9, 18), the drastic difference in seizure severity was likely caused by Depdc5 deletion in different cell populations.
Electrographically, all fatal seizures showed immediate flattening of EEG activities upon ictal offset, which could represent post-ictal generalized EEG suppression (PGES) (5, 43). This immediate and abrupt switch from brief high amplitude ictal discharges to absolute inactivity suggests an intrinsic cerebral shutdown or an irreversible failure resulting in imminent death. PGES was proposed as a strong predictor of SUDEP, with the odds of SUDEP increasing by a factor of 1.7% (p<0.005) with each 1-second increase in PGES duration(44, 45). However, several follow-up studies did not reproduce these findings and showed that non-fatal generalized tonic-clonic seizures could also cause PGES. These discrepancies could be explained by heterogeneous patient populations and inherent difficulties defining PGES. PGES was defined as diffuse EEG background attenuation (e.g., <10 μV) based on the standard human 10–20 system scalp-EEG recording(45, 46) that has relatively low sensitivity and often cannot detect low-voltage cortical activity. Therefore, non-fatal seizures could result in PGES on scalp EEG but still show clear cortical activity on intracranial EEG. The mouse EEG in our studies was obtained from screw electrodes fixed in the skull similar to epidural EcoG, which could explain the high specificity of PGES in our study. Interestingly, in non-fatal seizures, the post-ictal low voltage rhythmic EEG activity and the preserved breathing rate were both in the theta frequency range. It is well-known that nasal respiration could drive neuronal oscillations(47). Prominent respiration-entrained oscillations have been observed in the barrel cortex(48), medial prefrontal cortex (mPFC) (49), and parietal cortex(50), during immobility, sleep, and active exploration. The post-ictal theta-frequency on EEG could represent activity driven by respiration, not the classically defined theta rhythm.
Rbp4-Cre; Depdc5F/F CKO mice develop ictal apnea and interictal respiratory dysfunction.
In the landmark MORTEMUS study, patients who died of SUDEP in epilepsy monitoring units showed terminal apnea preceding terminal asystole(51). Several animal models also recapitulated a seizure-induced respiratory arrest. For example, death could be prevented in a Dravet mouse model by mechanical ventilation and intra-cerebroventricular infusion of atropine, suggesting a central apnea mechanism of SUDEP (5, 52, 53). Importantly, the tonic phase-related apnea has been observed in both Dravet mice and wild-type mice with chemically induced seizures, suggesting a possible common SUDEP mechanism independent of epilepsy etiology(6). In agreement with these studies, Rbp4-Cre; Depdc5F/F CKO mice developed ictal apnea before cardiac asystole during fatal seizures and, more interestingly, had baseline respiratory dysregulation.
It remains unclear how seizure leads to respiratory arrest in SUDEP, but there are several possibilities. First, direct and indirect projections from the cortex to brainstem respiratory centers could be the anatomical substrate for seizure-induced respiratory dysfunction. Generalized seizures drive dual inhibition on the central respiratory and cardiac pathways resulting in ictal apnea and bradycardia (54). In addition, it is possible that seizures alter cellular or neural network function, leading to progressive respiratory disruption and increased SUDEP propensity. Radiographic and pathological studies indeed showed structural and cytoarchitectural abnormalities in the ventral lateral and raphe regions of the medulla, which both play important roles in central respiratory regulation(55). Recurrent seizures could trigger extensive gliosis in those areas resulting in respiratory abnormalities and premature mortality (56). Concordantly, the inadequate response to hypoxia in Rbp4-Cre; Depdc5F/FCKO mice suggests that the breathing circuit develops increased vulnerability to severe seizures over time that results in respiratory decompensation and, ultimately, failure. A second possibility is that defective molecular programs in respiratory control regions make the breathing circuit more vulnerable to seizures. For example, neurons in breathing control regions express ion channels important for respiratory functions. In a Dravet syndrome mouse model, expression of an SCN1A pathogenic variant in inhibitory neurons in the retrotrapezoid nucleus, a key brainstem region in the breathing circuit, resulted in defective chemosensation and SUDEP (57). A third possibility is that developmentally accumulated molecular and cellular defects induced by recurrent seizures, together with intrinsically perturbed homeostasis due to gene mutations, progressively increase the susceptibility of critical respiratory centers to failure during generalized tonic-clonic seizures.
In summary, our study provides a novel animal model of DEPDC5-related epilepsy and SUDEP with good construct and face validity, paving the way to parse out the pathophysiology at molecular, cellular, and circuit levels.
ACKNOWLEDGEMENTS
We thank Dr. Kenneth Kwan for donating the Rbp4-Cre transgenic mice. We thank Dr. Jun Hee Lee for donating the Dedpc5 conditional knockout mice.
This work was supported by NIH R01 NS113824 (YW), U54 NS117170 (YW), NIH R01 HL156989 (PL), and NIH R01 AT011652 (PL).
Footnotes
POTENTIAL CONFLICTS OF INTEREST
All authors declared no potential conflicts of interest.
DATA AVAILABILITY
Raw data, materials, and reagents of this study are available from the corresponding authors (Y.W. and P.L.) on request.
REFERENCES
- 1.Harden C, Tomson T, Gloss D, Buchhalter J, Cross JH, Donner E, et al. Practice guideline summary: Sudden unexpected death in epilepsy incidence rates and risk factors: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2017;88(17):1674–80. [DOI] [PubMed] [Google Scholar]
- 2.Li R, and Buchanan GF. Scurrying to Understand Sudden Expected Death in Epilepsy: Insights From Animal Models. Epilepsy Curr. 2019;19(6):390–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kearney J Sudden unexpected death in dravet syndrome. Epilepsy Curr. 2013;13(6):264–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Costagliola G, Orsini A, Coll M, Brugada R, Parisi P, and Striano P. The brain-heart interaction in epilepsy: implications for diagnosis, therapy, and SUDEP prevention. Ann Clin Transl Neurol. 2021;8(7):1557–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim Y, Bravo E, Thirnbeck CK, Smith-Mellecker LA, Kim SH, Gehlbach BK, et al. Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest. 2018;128(3):1141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wenker IC, Teran FA, Wengert ER, Wagley PK, Panchal PS, Blizzard EA, et al. Postictal Death Is Associated with Tonic Phase Apnea in a Mouse Model of Sudden Unexpected Death in Epilepsy. Ann Neurol. 2021;89(5):1023–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dibbens LM, de Vries B, Donatello S, Heron SE, Hodgson BL, Chintawar S, et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. 2013;45(5):546–51. [DOI] [PubMed] [Google Scholar]
- 8.Kc Epi, and Epilepsy Phenome/Genome P. Ultra-rare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol. 2017;16(2):135–43. [DOI] [PubMed] [Google Scholar]
- 9.Bacq A, Roussel D, Bonduelle T, Zagaglia S, Maletic M, Ribierre T, et al. Cardiac Investigations in Sudden Unexpected Death in DEPDC5-Related Epilepsy. Ann Neurol. 2022;91(1):101–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bagnall RD, Crompton DE, Petrovski S, Lam L, Cutmore C, Garry SI, et al. Exome-based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol. 2016;79(4):522–34. [DOI] [PubMed] [Google Scholar]
- 11.Weckhuysen S, Marsan E, Lambrecq V, Marchal C, Morin-Brureau M, An-Gourfinkel I, et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia. 2016;57(6):994–1003. [DOI] [PubMed] [Google Scholar]
- 12.Nascimento FA, Borlot F, Cossette P, Minassian BA, and Andrade DM. Two definite cases of sudden unexpected death in epilepsy in a family with a DEPDC5 mutation. Neurol Genet. 2015;1(4):e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baldassari S, Picard F, Verbeek NE, van Kempen M, Brilstra EH, Lesca G, et al. The landscape of epilepsy-related GATOR1 variants. Genet Med. 2019;21(2):398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Figlia G, Muller S, Hagenston AM, Kleber S, Roiuk M, Quast JP, et al. Brain-enriched RagB isoforms regulate the dynamics of mTORC1 activity through GATOR1 inhibition. Nat Cell Biol. 2022;24(9):1407–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holmes GL, Stafstrom CE, and Tuberous Sclerosis Study G. Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia. 2007;48(4):617–30. [DOI] [PubMed] [Google Scholar]
- 16.Yang T, Hu S, Chang WC, Kao HY, and Wang Y. Perineuronal Nets Degradation and Parvalbumin Interneuron loss in a Mouse Model of DEPDC5-related Epilepsy. Dev Neurosci. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klofas LK, Short BP, Zhou C, and Carson RP. Prevention of premature death and seizures in a Depdc5 mouse epilepsy model through inhibition of mTORC1. Hum Mol Genet. 2020;29(8):1365–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuskaitis CJ, Jones BM, Wolfson RL, Super CE, Dhamne SC, Rotenberg A, et al. A mouse model of DEPDC5-related epilepsy: Neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol Dis. 2018;111:91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu S, Knowlton RC, Watson BO, Glanowska KM, Murphy GG, Parent JM, et al. Somatic Depdc5 deletion recapitulates electroclinical features of human focal cortical dysplasia type IIA. Ann Neurol. 2018;84(1):140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Drorbaugh JE, and Fenn WO. A barometric method for measuring ventilation in newborn infants. Pediatrics. 1955;16(1):81–7. [PubMed] [Google Scholar]
- 21.Leone DP, Heavner WE, Ferenczi EA, Dobreva G, Huguenard JR, Grosschedl R, et al. Satb2 Regulates the Differentiation of Both Callosal and Subcerebral Projection Neurons in the Developing Cerebral Cortex. Cereb Cortex. 2015;25(10):3406–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32(3):281–94. [DOI] [PubMed] [Google Scholar]
- 24.Xu Q, Tam M, and Anderson SA. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J Comp Neurol. 2008;506(1):16–29. [DOI] [PubMed] [Google Scholar]
- 25.Wonders CP, Taylor L, Welagen J, Mbata IC, Xiang JZ, and Anderson SA. A spatial bias for the origins of interneuron subgroups within the medial ganglionic eminence. Dev Biol. 2008;314(1):127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tremblay R, Lee S, and Rudy B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron. 2016;91(2):260–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yao Y, Chen J, Li X, Chen ZF, and Li P. A carotid body-brainstem neural circuit mediates sighing in hypoxia. Curr Biol. 2023;33(5):827–37 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, et al. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013;340(6136):1100–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clipperton-Allen AE, and Page DT. Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism-relevant behavioral tests. Hum Mol Genet. 2014;23(13):3490–505. [DOI] [PubMed] [Google Scholar]
- 30.Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488(7413):647–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kosillo P, Doig NM, Ahmed KM, Agopyan-Miu A, Wong CD, Conyers L, et al. Tsc1-mTORC1 signaling controls striatal dopamine release and cognitive flexibility. Nat Commun. 2019;10(1):5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMahon JJ, Yu W, Yang J, Feng H, Helm M, McMahon E, et al. Seizure-dependent mTOR activation in 5-HT neurons promotes autism-like behaviors in mice. Neurobiol Dis. 2015;73:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benthall KN, Cording KR, Agopyan-Miu A, Wong CD, Chen EY, and Bateup HS. Loss of Tsc1 from striatal direct pathway neurons impairs endocannabinoid-LTD and enhances motor routine learning. Cell Rep. 2022;38(12):110563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Normand EA, Crandall SR, Thorn CA, Murphy EM, Voelcker B, Browning C, et al. Temporal and mosaic Tsc1 deletion in the developing thalamus disrupts thalamocortical circuitry, neural function, and behavior. Neuron. 2013;78(5):895–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao JP, and Yoshii A. Hyperexcitability of the local cortical circuit in mouse models of tuberous sclerosis complex. Mol Brain. 2019;12(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Gama AM, Woodworth MB, Hossain AA, Bizzotto S, Hatem NE, LaCoursiere CM, et al. Somatic Mutations Activating the mTOR Pathway in Dorsal Telencephalic Progenitors Cause a Continuum of Cortical Dysplasias. Cell Rep. 2017;21(13):3754–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma V, Sood R, Lou D, Hung TY, Levesque M, Han Y, et al. 4E-BP2-dependent translation in parvalbumin neurons controls epileptic seizure threshold. Proc Natl Acad Sci U S A. 2021;118(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eichmuller OL, Corsini NS, Vertesy A, Morassut I, Scholl T, Gruber VE, et al. Amplification of human interneuron progenitors promotes brain tumors and neurological defects. Science. 2022;375(6579):eabf5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baldassari S, Ribierre T, Marsan E, Adle-Biassette H, Ferrand-Sorbets S, Bulteau C, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol. 2019;138(6):885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee WS, Stephenson SEM, Howell KB, Pope K, Gillies G, Wray A, et al. Second-hit DEPDC5 mutation is limited to dysmorphic neurons in cortical dysplasia type IIA. Ann Clin Transl Neurol. 2019;6(7):1338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ribierre T, Deleuze C, Bacq A, Baldassari S, Marsan E, Chipaux M, et al. Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J Clin Invest. 2018;128(6):2452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1(2):157–62. [DOI] [PubMed] [Google Scholar]
- 43.Tomson T, Walczak T, Sillanpaa M, and Sander JW. Sudden unexpected death in epilepsy: a review of incidence and risk factors. Epilepsia. 2005;46 Suppl 11:54–61. [DOI] [PubMed] [Google Scholar]
- 44.Carlson C Generalized Postictal EEG Background Suppression: A Marker of SUDEP Risk. Epilepsy Curr. 2011;11(3):86–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lhatoo SD, Faulkner HJ, Dembny K, Trippick K, Johnson C, and Bird JM. An electroclinical case-control study of sudden unexpected death in epilepsy. Ann Neurol. 2010;68(6):787–96. [DOI] [PubMed] [Google Scholar]
- 46.Tao JX, Yung I, Lee A, Rose S, Jacobsen J, and Ebersole JS. Tonic phase of a generalized convulsive seizure is an independent predictor of postictal generalized EEG suppression. Epilepsia. 2013;54(5):858–65. [DOI] [PubMed] [Google Scholar]
- 47.Tort ABL, Brankack J, and Draguhn A. Respiration-Entrained Brain Rhythms Are Global but Often Overlooked. Trends Neurosci. 2018;41(4):186–97. [DOI] [PubMed] [Google Scholar]
- 48.Ito J, Roy S, Liu Y, Cao Y, Fletcher M, Lu L, et al. Whisker barrel cortex delta oscillations and gamma power in the awake mouse are linked to respiration. Nat Commun. 2014;5:3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Biskamp J, Bartos M, and Sauer JF. Organization of prefrontal network activity by respiration-related oscillations. Sci Rep. 2017;7:45508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tort ABL, Ponsel S, Jessberger J, Yanovsky Y, Brankack J, and Draguhn A. Parallel detection of theta and respiration-coupled oscillations throughout the mouse brain. Sci Rep. 2018;8(1):6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ryvlin P, Nashef L, and Tomson T. Prevention of sudden unexpected death in epilepsy: a realistic goal? Epilepsia. 2013;54 Suppl 2:23–8. [DOI] [PubMed] [Google Scholar]
- 52.Massey CA, Sowers LP, Dlouhy BJ, and Richerson GB. Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol. 2014;10(5):271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goldman AM. When Apnea Turns Terminal: When, How, Why? Epilepsy Curr. 2021;21(6):449–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schilling WP, McGrath MK, Yang T, Glazebrook PA, Faingold CL, and Kunze DL. Simultaneous cardiac and respiratory inhibition during seizure precedes death in the DBA/1 audiogenic mouse model of SUDEP. PLoS One. 2019;14(10):e0223468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patodia S, Tachrount M, Somani A, Scheffer I, Yousry T, Golay X, et al. MRI and pathology correlations in the medulla in sudden unexpected death in epilepsy (SUDEP): a postmortem study. Neuropathol Appl Neurobiol. 2021;47(1):157–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dhaibar HA, Hamilton KA, and Glasscock E. Kv1.1 subunits localize to cardiorespiratory brain networks in mice where their absence induces astrogliosis and microgliosis. Mol Cell Neurosci. 2021;113:103615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuo FS, Cleary CM, LoTurco JJ, Chen X, and Mulkey DK. Disordered breathing in a mouse model of Dravet syndrome. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data, materials, and reagents of this study are available from the corresponding authors (Y.W. and P.L.) on request.