

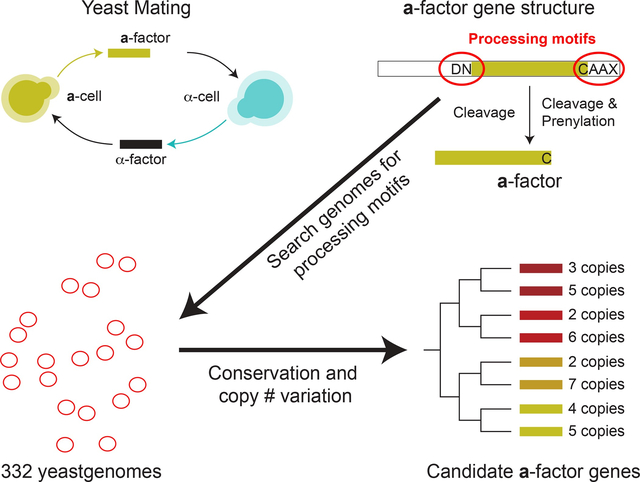
Summary
The mating of fungi depends on pheromones that mediate communication between two mating types. Most species use short peptides as pheromones, which are either unmodified (e.g., α-factor in Saccharomyces cerevisiae) or C-terminally farnesylated (e.g., a-factor in S. cerevisiae). Peptide pheromones have been found by genetics or biochemistry in a small number of fungi, but their short sequences and modest conservation make it impossible to detect homologous sequences in most species. To overcome this problem, we used a four-step computational pipeline to identify candidate a-factor genes in sequenced genomes of the Saccharomycotina, the fungal clade that contains most of the yeasts: we require that candidate genes have a C-terminal prenylation motif, are fewer than 100 amino acids long, contain a proteolytic processing motif upstream of the potential mature pheromone sequence, and that closely related species contain highly conserved homologs of the potential mature pheromone sequence. Additional manual curation exploits the observation that many species carry more than one a-factor gene, encoding identical or nearly identical pheromones. From 332 Saccharomycotina genomes, we identified strong candidate pheromone genes in 241 genomes, covering 13 clades that are each separated from each other by at least 100 million years, the time required for evolution to remove detectable sequence homology among small pheromone genes. For one small clade, the Yarrowia, we demonstrated that our algorithm found the a-factor genes: deleting all four related genes in the a-mating type of Yarrowia lipolytica prevents mating.
Keywords: Yeast mating, Pheromones, Gene annotation, Small peptides, Yarrowia
eTOC Blurb
Small, lipid-modified peptide pheromones are essential for yeast mating but remain unknown for most species. Srikant et al. identify pheromones in yeast genomes by using successive computational filters: functional motifs, conservation in sister species, and multiple gene copies. The predicted Yarrowia pheromone is essential for mating.
Graphical Abstract
Introduction
Fungi are an ancient lineage of eukaryotes whose extant members shared a common ancestor more than one billion years ago1. Within a species, mating depends on signaling by diffusible pheromones between haploid cells of two different mating types (Figure 1A)2. One important group is the Saccharomycotina, which emerged roughly 400 million years ago and contains most of the yeasts: unicellular fungi that lack fruiting bodies. Many yeast species play key roles as model systems, in the food industry, and as crop and human pathogens. Finding the pheromones of economically important yeast will help to understand and control their mating as well as shed light on the evolution of these molecules and the receptors that recognize them.
Figure 1. S. cerevisiae pheromones are produced by cleaving and modifying precursor peptides.
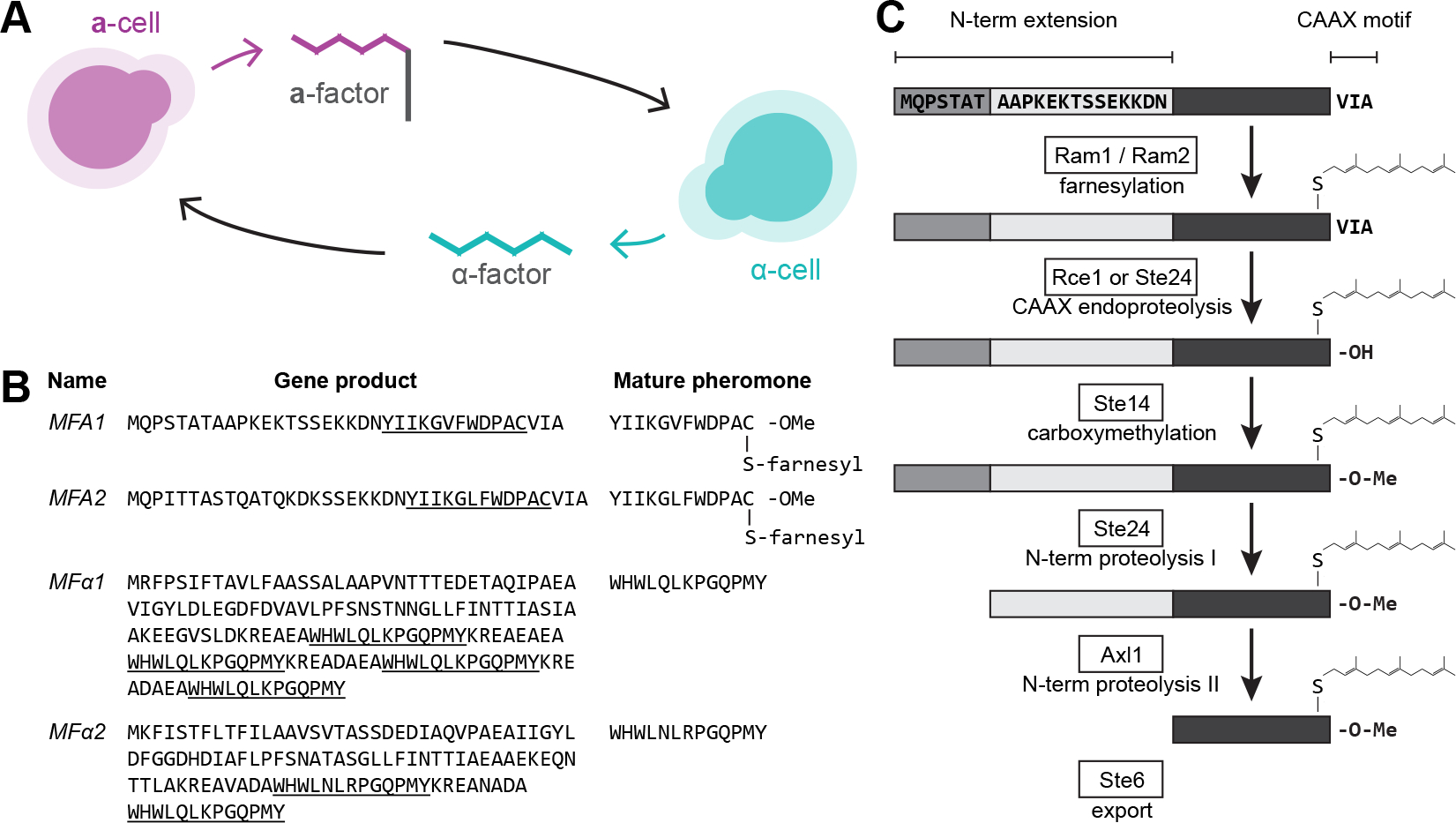
(A) Haploid S. cerevisiae have two mating types, a and α. Their mating with each other is initiated by the secretion of diffusible peptide pheromones that are recognized by G protein-coupled receptors (GPCRs): a-cells (magenta) secrete the lipidated peptide pheromone a-factor, which is recognized by the a-factor receptor expressed on α-cells, while α-cells (cyan) secrete the peptide pheromone α-factor, which is recognized by the α-factor receptor expressed by a-cells. (B) Mating pheromones (underlined) are encoded within precursor peptides by the MFA1 and MFA2 genes (for a-factor) and MFα1 and MFα2 genes (for α-factor). These peptides require several maturation steps before their secretion as biologically active molecules. (C) The modifications of initial products of the MFA1 and MFA2 genes that produce a-factor. Broadly, there are two stages of maturation, C-terminal modifications (S-thiol farnesylation, -AAX proteolysis (white bars), and carboxymethylation), followed by two steps of N-terminal proteolysis (two grey bars). The mature pheromone (black bar) is then exported from the cytosol through a dedicated ABC transporter, Ste6. See also Figure S1.
The best studied mating system is that of the baker’s yeast Saccharomyces cerevisiae. Genetic screens and biochemistry have identified pheromones, the proteins that produce and export them, pheromone receptors, and their associated signaling pathways. The two pheromones in S. cerevisiae are the 13-amino acid peptide α-factor produced by α-cells, and a-factor, the C-terminally farnesylated 12-amino acid peptide produced by a-cells (Figure 1B). Multiple copies of α-factor are present in each of two mating factor genes (MFα1 and MFα2) and are released by proteolysis in the endoplasmic reticulum allowing the pheromone to be exported in secretory vesicles3, 4. In contrast, a-factor is encoded in two loci (MFA1 and MFA2) as a precursor peptide that undergoes maturation in the cytosol to produce a single pheromone molecule (Figure 1C)5–7. a-factor maturation involves C-terminal farnesylation and carboxymethylation, followed by N-terminal proteolysis. These steps occur in the cytosol, after which a-factor is pumped across the plasma membrane by Ste6, a member of the ABC family of ATP-dependent transporters. In S. cerevisiae, maturation is essential for bioactivity; unmodified pheromone is trapped in the cytosol because it is not recognized and exported by Ste65, 8.
The Saccharomycotina fungi lie within the Ascomycota clade and the distinction between an unmodified α-factor and a farnesylated a-factor is maintained in the Ascomycota. In the Basidomycota, the sister clade to the Ascomycota, all mating pheromones are farnesylated, suggesting that the ancestral mating pheromones were farnesylated, poorly soluble molecules 9. The peptide sequences of both a-like and α-like pheromones diverge across fungal species with cognate receptors and transporters co-evolving to maintain activity10–12.
The first fungal pheromones were identified as lipidated peptides by biochemical isolation from the extracellular medium of two basidiomycetes Rhodosporidium toruloides13, 14 and Tremella mesenterica15, 16. Since then, pheromones have been biochemically isolated and characterized in laboratory yeasts, filamentous fungi, and crop pathogens by isolation from extracellular medium2, 9, 17, 18, but this approach depends on discovering the environmental conditions that stimulate mating and obtaining sufficient material to determine the pheromone’s peptide sequence.
An alternative route to pheromone identification is to look for homologs of known pheromones in sequenced fungal genomes. Because pheromones are small peptides and are only modestly conserved this approach only works over a limited phylogenetic distance. In principle, this problem could be solved by exploiting conservation in the order of genes along a chromosome (synteny): looking for orthologs of the longer genes on either side of the S. cerevisiae a-factor and then searching, more sensitively, for a pheromone ortholog that lies in the stretch of DNA between these two genes. Unfortunately, synteny is less conserved than pheromone sequence. A search for homologs of one of the S. cerevisiae genes that encode a-factor, MFA2, finds hits in two clades that diverged roughly 100 million years ago from the clade that enjoyed a whole genome duplication and includes S. cerevisiae: in the nearer clade (which contains Zygosaccharomyces rouxii and Torulospora delbrueckii) the a-factor homologs retain their syntenic location, but in the further one (which contains Kluyvermoyces lactis, Lachancea waltii and Eremothecium gossypii) synteny has been lost19.
We built a computational pipeline to identify farnesylated fungal pheromones in sequenced genomes. Our algorithm successively discards sequences that lack the C-terminal signal sequence for farnesylation, lack a sequence that controls proteolytic processing, and lack strongly conserved homologs in closely related species. Manual curation privileges sequences that occur more than once in the same genome and identifies all the known pheromones in the Saccharomycotina. Overall, we find known or candidate pheromone genes in 241 of 332 mined genomes. To test the ability to identify novel pheromones, we verified that deletion of all copies of the candidate farnesylated pheromone of Yarrowia lipolytica (whose last common ancestor with S. cerevisiae existed more than 300 million years ago) prevented mating of the a-cells of this species.
Results
Algorithmic sieve to find candidate pheromone open reading frames (ORFs)
Given the limits of homology searches for small unstructured peptides, we designed a new approach to identify farnesylated peptide pheromones from sequenced genomes: using successive identification of conserved features of known pheromone genes as a series of sieves to identify candidate pheromone sequences.
Biogenesis of a-factor in S. cerevisiae involves a number of essential post-translation modifications by dedicated enzymes5 (Figure 1C). We assume that these conserved enzymes play a role in all fungal pheromone maturation, and therefore our algorithm identifies candidate pheromone genes by searching for the sequence motifs that these enzymes recognize. The post-translational maturation can be broadly separated into two parts: C-terminal farnesylation and carboxymethylation, followed by N-terminal proteolytic cleavage (Figure 1C).
In eukaryotes, a conserved enzyme complex prenylates substrates by recognizing a conserved sequence motif9. The enzyme complex (Ram1/Ram2 (all protein names are those from S. cerevisiae)) adds a farnesyl group (a C15 isoprenoid) to the cysteine in the C-terminal sequence CAAX, with C representing the modified cysteine, A small aliphatic residues and X a small amino acid.
After prenylation, the C-terminus of the pheromone precursor must be further modified for bioactivity20, 21. The C-terminal AAX residues of S. cerevisiae a-factor are proteolytically removed (by Ste24 and Rce1), and carboxyl group of the terminal cysteine is methylated (by Ste14)5. To begin, we consolidated the CAAX motifs of known pheromones in the Ascomycota into a per-residue dictionary to identify farnesylation sites of potential pheromone candidate ORFs (Figure S1). To avoid biases based on the small number of known pheromones, we relaxed this CAAX detection dictionary based on experimental studies of the combinatorial signatures of farnesylation, proteolysis and carboxymethylation in S. cerevisiae22–24 (Figure S1).
We then selected the list of ORFs ending in CAAX for candidates that have an in-frame methionine within 6 to 100 residues upstream of the stop codon to find ORF candidates that are within the expected size range of pheromone genes in Ascomycota (Figure 2A). Our pipeline considers genome assemblies contig-by-contig and assumes that the pheromones can be positioned anywhere in the genome. Since our algorithm relies on tracing the ORF starting from the Stop codon, it is unlikely to find pheromone genes that contain introns, like those in S. pombe25. We believe this is a reasonable simplification of the algorithm given the lower frequency of introns in Hemiascomycota yeasts (2–14% of all genes), relative to other eukaryotes26.
Figure 2. Fungal pheromone candidates can be identified by a progressive filter for small open-reading frames that are farnesylated and cleaved by a protease associated with mating.
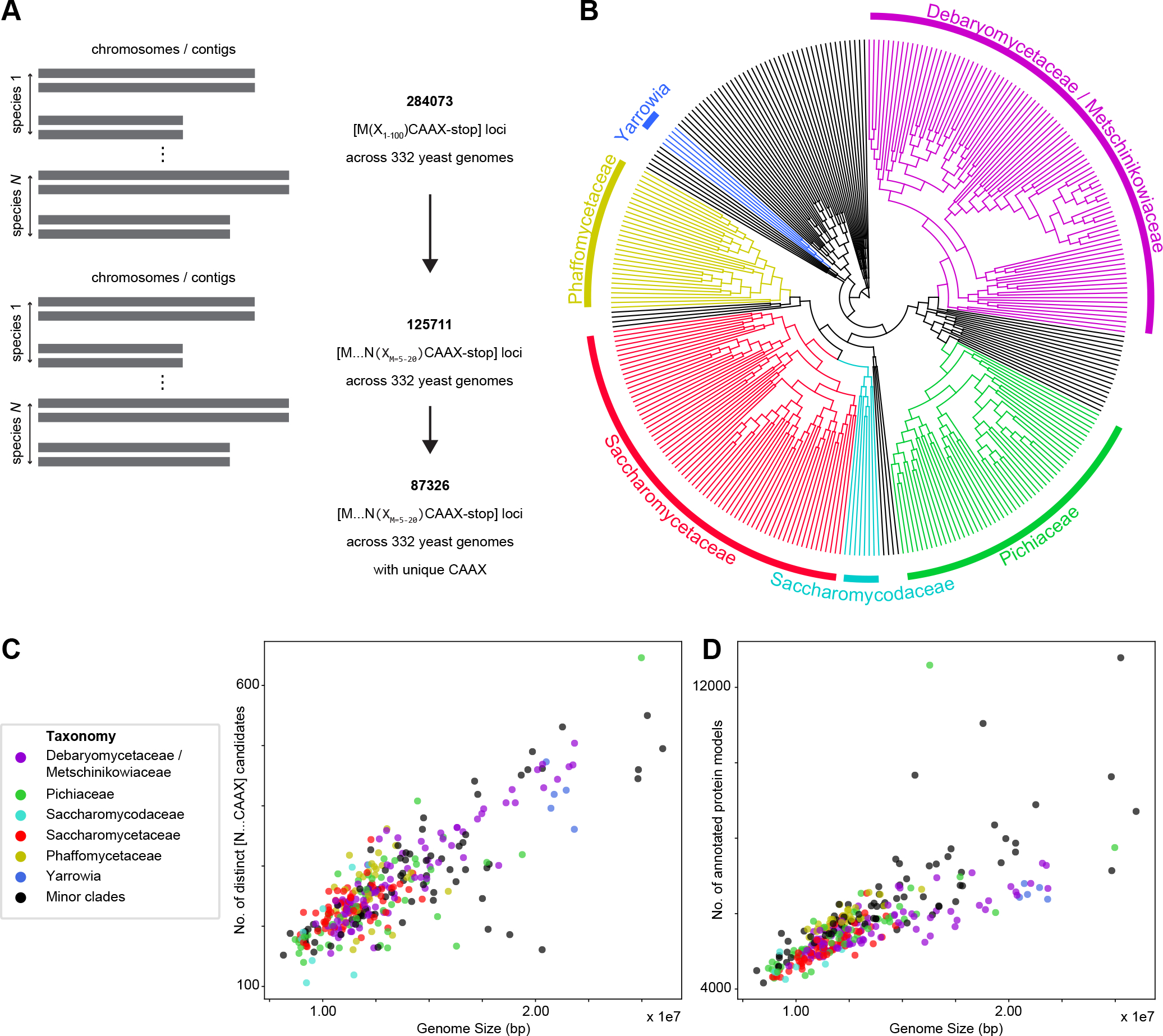
(A) Our algorithm begins by looking for all possible short open-reading frames with C-terminal farnesylation (CAAX-stop) in the 332 available fungal genomes, resulting in 284,073 candidates. We filter again for an in-frame proteolytic-motif asparagine (N) important for the final step of maturation to produce bio-active pheromone. This resulted in 125,711 candidates. Collapsing sets of candidate sequences that result from multiple Start codons upstream of a single CAAX to one sequence from the most upstream Start codon results in 87,326 unique farnesylated loci. (Figure S2E) (B) Phylogenetic tree of all 332 sequenced yeasts across Saccharomycotina, covering clades like Debaryomycetaceae/Metschinikowiaceae, Pichiaceae, Saccharomycodaceae, Saccharomycetaceae, Phaffomycetaceae and Yarrowia that contain species important for both basic biology and industrial production. The tree is based on those yeast genome sequences with estimated relative divergence times27. Selected clades are labeled. (C) Scatter plot showing that the number of unique pheromone candidate loci is linearly correlated with genome size, ranging from 100 to 650 candidates, as expected of a search for all possible pheromone candidates. (D) Scatter plot showing strong correlation between the number of annotated protein-coding genes and genome size, which ranges between 8 and 27 Mbp for the 332 yeast genomes. See also Figure S2.
We ran our pipeline on a set of 332 published Saccharomycotina genomes27 (Figure 2B, S2A, B and Table 1, S1) and found 500–2000 candidate ORFs per genome (Figure S2C), which is far larger than can be experimentally validated even for a single species. After C-terminal farnesylation and carboxymethylation, Sca-factor precursors undergo N-terminal processing in two proteolytic steps5, 20. The second is the final step in maturation and is mediated by one of two Zn-dependent metalloendopeptidases (Axl1 and Ste23; Figure 1C)28. Random mutagenesis in S. cerevisiae a-factor revealed that the two residues immediately before and after the cleavage site are important for ScAxl1 cleavage20. Comparing known Ascomycota pheromones, we noticed that the cleavage site is typified by N-X with the peptidase cleaving C-terminal of the asparagine (N) and X representing the newly released N-terminal aromatic or uncharged residue of the pheromone (Figure 1C, S1A). Therefore, we required pheromone candidates to have an asparagine in frame with the conserved cysteine such that the C terminus of the mature pheromone would be between 5–20 residues downstream from the N-terminal residue X. Adding this sieve reduces the number of distinct pheromone candidates to 100–700 per genome (Figure 2C, S2D). As expected, the distribution of pheromone candidates per genome scales with genome size similar to the number of annotated protein models (Figure 2D). There are still too many candidates for experimental validation, but further knowledge of pheromone biology can be used to identify the best candidates.
Table 1:
Computational filters identify pheromone candidates in analyzed yeast genomes
| Major clade | Species count | Avg. genome size (Mbp)* | Avg. protein models* | Avg. CAAX candidates* | Avg. [N-CAAX] candidates* | Avg. distinct [N-CAAX] candidates* |
|---|---|---|---|---|---|---|
|
| ||||||
| Alloascoideaceae | 1 | 24.82 | 9631 | 1011 | 574 | 445 |
| CUG-Ala | 5 | 12.88 | 5944.4 | 691.2 | 359.8 | 248.8 |
| CUG-Ser1 | 94 | 13.71 | 5636.81 | 940.88 | 413.36 | 282.51 |
| CUG-Ser2 | 4 | 18.09 | 6718.25 | 855.75 | 369 | 265.5 |
| Dipodascaceae/Trichomonascaceae | 37 | 14.84 | 6284.49 | 1047.32 | 410.49 | 284.43 |
| Lipomycetaceae | 9 | 16.53 | 7218.33 | 1427.33 | 603.78 | 401.56 |
| Phaffomycetaceae | 34 | 12.26 | 5913.21 | 861.15 | 383.29 | 260.29 |
| Pichiaceae | 61 | 12.71 | 5722.87 | 782.11 | 359.23 | 251.72 |
| Saccharomycetaceae | 71 | 11.34 | 5107.11 | 687.3 | 320.55 | 230.65 |
| Saccharomycodaceae | 8 | 10.23 | 4679.88 | 589.38 | 284.5 | 195.75 |
| Sporopachydermia clade | 2 | 16.56 | 6086 | 962 | 442 | 313.5 |
| Trigonopsidaceae | 6 | 11.68 | 6127.17 | 619.67 | 253.33 | 184.5 |
Yeast pheromones are encoded in multiple gene copies and are conserved among closely related species
Yeast pheromones of closely related species are quite similar, although the number of copies in each genome varies (e.g., 1 each in Candida albicans and C. dubliensis29, 2 each in Komagataella phaffi and K. pastoris30, and 1 to 3 in different Saccharomycetaceae19). The amino acid sequence of the ORFs is most strongly conserved within the mature pheromone peptide sequence which must interact both with the a-factor exporter, Ste6, and the a-factor receptor, Ste2. The Saccharomycetaceae is the largest yeast lineage across which a pheromone candidate is known to be conserved and we used its time of divergence as an evolutionary horizon within which species are likely to show strong pheromone homology (Figure 3A). We identified all monophyletic clades on the yeast phylogram with ancestral nodes within this time horizon: 300 species give rise to 23 phylogroups, each containing at least 2 species, and 32 species are singleton orphans with no related species within this horizon (Figure 3A, B). Among these 23 phylogroups are large, recognized clades of yeasts, including Debaryomycetaceae/Metschinikowiaceae, Pichiaceae, Saccharomycodaceae, Saccharomycetaceae, Phaffomycetaceae and Yarrowia, which contain at least 5 species (Figure 3B). By pooling the candidates of species within these groups, we expect that the best candidate pheromones will share two properties: they are conserved within the phylogroup and conservation is greatest in the mature region, between the predicted N- and C-terminal processing signals.
Figure 3. Fungal pheromones are conserved within closely related species and are often encoded in multiple copies in a genome.

(A) Phylogenetic tree describing the evolutionary relationship of 332 yeasts with two horizons indicated. The solid grey line indicates the assumed maximum evolutionary distance at which mature pheromone sequences show detectable sequence homology, operationally defined by the divergence time of S. cerevisiae (Sc) and Kluveromyces lactis (Kl), whose pheromones show detectable sequence homology. The leaves corresponding to Sc, Kl and Vanderwaltozyma polyspora (Vp) are indicated on the tree. The leaves of the tree are also annotated in green for species with known pheromones prior to our work (first column) and species with pheromones identified in our work (second column). (B) Based on the pheromone homology time horizon (solid grey line in A), we separated 332 yeast genomes into 23 phylogroups of at least 2 species; there are also 32 singleton species. The most populated phylogroups correspond to the listed well-known clades where closely related species have been densely sequenced. These clades are represented in the tree by the corresponding colors. (C) Number of pheromone candidates per species plotted for each of the 23 conserved-pheromone phylogroups, where each circle corresponds to the number of candidates in a species, and each copy of a group of closely homologous sequences within a genome is counted separately. Selecting for candidates that have at least two homologous copies within the clade reduces the number of viable candidates per genome to 10–300 (center panel compared to left). Manual curation of candidates similar to known pheromones identifies the most likely pheromone(s) in each genome (right panel) for experimental validation. The curated candidates include both multiple copies of a single best candidate and multiple distinct candidates if a single best pheromone cannot be uniquely identified. There are 1–19 candidates encoded in each species for experimental testing. Some phylogroups contain too few species and thus no candidates rose above the rest through curation. See also Figure S3, S6, S7.
Farnesylated (a-factor-like) pheromones are often encoded in multiple copies in a yeast genome, possibly as a result of sexual selection during competitive mating31. The copy number of the pheromone genes seems to vary rapidly even among closely related species19, 32. To identify the most likely pheromones, we looked for candidates that are either present in at least two species within a phylogroup or have at least two homologous copies in the same genome of at least one species in the phylogroup. Since only the mature region of the pheromone is bioactive, we pre-filtered copies based on the conservation of the amino-acid sequence between the predicted protease site (asparagine) and the stop codon. Specifically, candidates that are greater than 70% identical to each other in the N- and C-terminal motifs and the mature region are considered copies encoding the same predicted pheromone in a phylogroup. We score a global alignment of the translated sequence from the cleavage-motif asparagine through the mature region and farnesylation motif (CAAX) with no gap penalties, score identical characters as 1 and 0 otherwise, and normalize to the length of the region. Our algorithm only considers ORFs that maintain the reading frame between the start and stop codons in a single exon and will not identify gene copies that contain introns or frameshifts.
Of the 78,206 candidate ORFs that have both farnesylation and proteolysis sites (N…CAAX candidates) across 300 species in the 23 phylogenetic clades, 17,125 candidates have at least one other homologous candidate in the phylogroup. There are between 10–300 candidate genes per species, including false positive ORFs that are conserved among closely related species present in a number of clades (Figure 3C, middle). To identify the best candidate pheromones among these ORFs we considered two criteria: (1) True pheromones are often encoded in multiple copies in a species’ genome and (2) pheromones are strongly conserved in closely-related species (Figure S3C). In three of the six major phylogroups with more than five species, a combination of these criteria unambiguously identifies a clear best candidate pheromone gene within genomes of closely-related yeasts. In the remaining three phylogroups there are smaller subclades that have one or more pheromone candidates that match one or both criterion and require experimental validation to determine the true pheromone genes. In seven of the 17 remaining smaller phylogroups (with fewer than 5 species), there are candidates that satisfy one or both criterion and can be considered the best pheromone candidate in the corresponding yeast genomes. This criterion-based curation identifies a total of 812 loci across 241 species, with an average of 3.4, and range of 1–19 potential pheromone-encoding ORFs per genome (Figure 3C, right). These 812 ORFs mostly account for a distinct “best” pheromone candidate often encoded in multiple copies per genome (Table 2).
Table 2:
Distribution of predicted pheromone genes across phylogenetic groups
| Phylogenetic group | Genus* | Family* | Clade name | Major Clade | Species count | Distinct pheromone candidates** (species avg) | Pheromone copy number*** (species avg) |
|---|---|---|---|---|---|---|---|
|
| |||||||
| phylogroup 1 | Debaryomycetaceae/Metschnikowiaceae | Debaryomycetaceae/Metschinikowiaceae | CUG-Ser1 | 70 | 1.4 | 5.3 | |
| phylogroup 6 | Komagataella | Saccharomycetales incertae sedis | Pichiaceae | 3 | 1.0 | 2.0 | |
| phylogroup 7 | Citeromyces | Saccharomycetales incertae sedis | Pichiaceae | 3 | 1.0 | 12.7 | |
| phylogroup 10 | Pichiaceae/Saccharomycetales incertae sedis | Pichiaceae | Pichiaceae | 41 | 1.7 | 2.3 | |
| phylogroup 12 | Ascoidea | Ascoideaceae | CUG-Ser2 | 1 | 1.0 | 3.0 | |
| phylogroup 13 | Hanseniaspora | Saccharomycodaceae | Saccharomycodaceae | Saccharomycodaceae | 8 | 1.0 | 2.0 |
| phylogroup 14 | Saccharomycetaceae | Saccharomycetaceae | Saccharomycetaceae | 71 | 1.1 | 1.7 | |
| phylogroup 16 | Starmera | Phaffomycetaceae | Phaffomycetaceae | 3 | 1.0 | 1.3 | |
| phylogroup 17 | Phaffomycetaceae | Phaffomycetaceae | Phaffomycetaceae | 30 | 1.6 | 3.4 | |
| phylogroup 18 | Sporopachydermia | Saccharomycetales incertae sedis | Sporopachydermia clade | 2 | 1.0 | 2.5 | |
| phylogroup 22 | Yarrowia | Dipodascaceae | Yarrowia | Dipodascaceae/Trichomonascaceae | 5 | 1.0 | 8.6 |
| phylogroup 24 | Dipodascus | Dipodascaceae | Dipodascaceae/Trichomonascaceae | 2 | 1.0 | 1.0 | |
| phylogroup 30 | Starmerella | Dipodascaceae | Dipodascaceae/Trichomonascaceae | 2 | 1.0 | 2.5 | |
Genus is listed if there is only 1 genus represented in the phylogenetic group. Most phylogroups contain species of a single family, and often of a single genus. Left blank when species across multiple genus are present in the phylogroup.
Distinct pheromones are determined in every species and averaged across species in the phylogenetic group.
Copynumber of each distinct pheromone is averaged within a species (for species that have more than one distinct candidate), and then averaged across species in a phylogenetic group.
See also Table S2.
The well-characterized farnesylated pheromones of Saccharomyces species6, 19, C. albicans and C. dubliensis29, and K. pastoris and K. phaffii30 are among the identified candidates (Figure 1 and Table 2, S2). The Saccharomyces a-factor genes are a large group of homologous sequences: every one of the 71 sequenced Saccharomycetaceae genomes has at least one copy and 33 of these species contain at least two homologous pheromone genes. Similarly, the Komagatella pheromones are identified by the presence of the same pheromone in the sequenced sister species K. pseudopastoris and K. populi. Our curation criteria result in pheromone candidates that are often encoded in a range of gene copies in a genome (Figure 4A). In species of Metschinikowiaceae, Citeromyces and Yarrowia, pheromone candidates are encoded in 5 to 19 copies in genomes, with dramatic copy number variation among closely related species (Figure 4A, Table 2). More generally however, most pheromone candidates across the yeast lineage are encoded in 1 to 3 copies per genome (Figure S4A). The pheromones of C. albicans and C. dubliensis are homologous to each other and encoded in one copy in each genome29. In the related species of C. tropicalis and C. corydalis, there are pheromone candidates that have <70% sequence identity but are likely evolutionarily related (Table S2). In three clades, Debaryomycetaceae/Metschinikowiaceae, Pichiaceae and Phaffomycetaceae the criteria do not unambiguously identify a single “best” pheromone across the entire clade. There are candidates that fit both criteria in subclades (e.g., in Metschinikowiaceae), or multiple candidates in a genome that fit one or both criteria (e.g., in Pichiaceae). In 72 of 141 species in these clades we have identified 2 or 3 distinct candidates that need further experimental testing to determine the true pheromone (Figure S4B, C). Despite this ambiguity, in each of these clades we have identified what we believe is the most likely set of pheromone genes, which is shown as the left most consensus sequence in Figure S4C. In the Phaffomycetaceae, there is one candidate that is present in 29 of the 30 species where a pheromone candidate was detected and is present in multiple copies in 26 of these species, whereas the next most widespread candidate was found in only 10 species, always as a single copy. In the Pichiaceae, the most widespread sequence was found in 30 out of 40 species and although it was always present as a single copy, the most widespread sequence with multiple copies was found in only 5 species, all of which contained a copy of the other pheromone candidate. The most complex clade was the Debaryomycetaceae/Metschinikowiaceae. In this clade, 64 of the 71 species that revealed a pheromone candidate had a match to a sequence with a conserved N-terminal motif (KDN) and a conserved glycine located six residues N-terminal to the C-terminal cysteine of the mature pheromone. In 53 out of the 64 species, this candidate sequence was present in multiple copies. This group contains the verified pheromone sequence of C. albicans whose sequence is strongly conserved between closely related species. In several cases similar sequences are found in widely separated species (for example two widely separated trios of species (C. auris, C. heveicola, and C. intermedia; T. gatunensis, T. kruzii, and T. cretensis) all contain the motif GxVPxxC seven to nine residues C-terminal to the N-terminal KDN motif.
Figure 4. Mature pheromones are peptides between 6–20 amino acids and encoded in multiple copies in a genome.
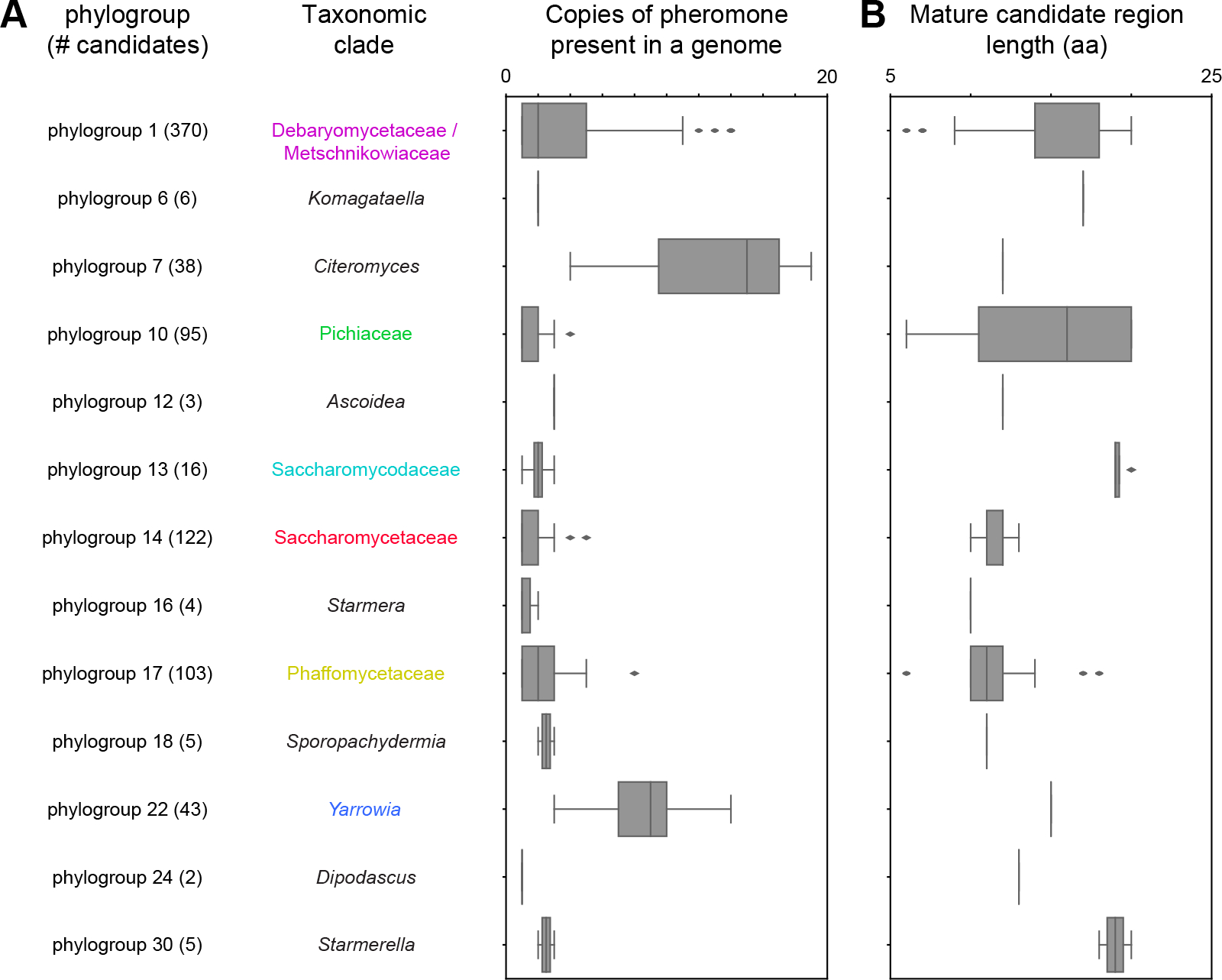
(A) Box-and-whisker plot of number of copies of pheromones encoded per genome categorized by the conserved-pheromones within phylogroups. The box represents the inner quartiles, the whiskers the outer quartiles and outliers are highlighted as diamonds. Thirteen phylogroups with at least two species contain a total of 812 candidates, with the number of candidates in each phylogroup listed in parentheses (also see Table 2). The taxonomic clade corresponding to phylogroups are provided for reference, with the most populated clades containing majority of the candidates (colored similar to Figures 2 and 3). For species with multiple distinct candidate pheromones, they are treated separately and the count of each in the genome is included in the distribution. (B) Box-and-whisker plot of the length of the mature region of pheromone candidates categorized by the conserved-pheromone phylogroups. The box represents the inner quartiles, the whiskers the outer quartiles and outliers are highlighted as diamonds. Mature sequences of all 812 sequences are defined between the upstream proteolysis site (N) and the C-terminal farnesylated cysteine. See also Figure S4, S6.
We aligned the protein sequences of the likely mature pheromones, identified by our criteria, within each conserved-pheromone phylogroup to understand sequence divergence within closely-related pheromones. The sequences between farnesylation motifs at the C-terminal end and the presumed proteolytic site define the mature pheromone candidates across each phylogroup (Figure 4B, S4C). The mature pheromone region of the best candidates are peptides 6–20 amino acids in length (Figure 4A). There is dramatic protein sequence variation across phylogroups, but the length and sequence of the candidate pheromones are conserved within a phylogroup (Figure 4B, S4C).
The exceptions to this conservation pattern are the pheromone candidates in Debaryomycetaceae/Metschinikowiaceae, Pichiaceae, and Phaffomycetaceae, which are more variable in length (Figure 4B) and sequence (Figure S4C) even within the phylogroup. In these three phylogroups, we identified multiple candidates that could be the true pheromone, with groups of species within a phylogroup containing candidates that are >70% sequence identical. This is either because pheromone divergence has accelerated in some subclades leading to a shorter time horizon of pheromone conservation (like Metschnikowia species having related candidates but not shared in the wider Debaryomycetaceae/Metschinikowiaceae clade), or the true pheromone is one of these candidates and must be validated experimentally.
Even with the lower threshold for similarity to detect pheromone homologs in closely related species, some species in the large clades of Debaryomycetaceae/Metschinikowiaceae (21 of 91 species) and Pichiaceae (11 of 52 species) are missing candidates that are homologous to the best candidates in the most closely related species (Figure S3B and Table S2). Possible explanations for this observation include lineage-specific selection for faster pheromone evolution, incomplete or poorly assembled genomes, or the presence of introns in pheromone genes.
Identifying the Y. lipolytica a-factor
The Yarrowia clade of yeasts is used for the heterologous expression of proteins using hydrocarbons as a metabolic carbon source33, 34, and the production of secondary metabolites35. In basic research, Y. lipolytica is used to study fatty acid metabolism in peroxisomes36 and mitochondrial complex I in an obligate aerobe37, 38. Other than the five Yarrowia genomes included in the 332 genomes, we also analyzed a sixth genome available from NCBI for pheromone candidates (Y. sp. 30695; Figure 5A). The best candidate pheromone is present in multiple copies in each of the genomes (Figure 5A and Table S2, S3). Comparing the flanking regions of copies in all the genomes confirms that all identified loci are unique within a genome and are not being overcounted due to poor genome assembly (Figure S5C). The presumed mature peptide sequence of the pheromone is completely conserved within the clade except for a single conservative mutation (F to Y; Figure 5A). Although there are several DNA polymorphisms in the region encoding the mature peptide sequence, they are synonymous mutations. In contrast, there are both non-synonymous variation and indels in the N-terminal region of the pheromone precursor in the Yarrowia lineage (Figure 5A). This variation is consistent with previous experiments in S. cerevisiae which showed that the sequence identity of the proteolyzed N-terminal sequence does not affect the production of mature a-factor20.
Figure 5. All species in Yarrowia clade of yeasts have a homologous farnesylated pheromone that is encoded in multiple copies per genome.
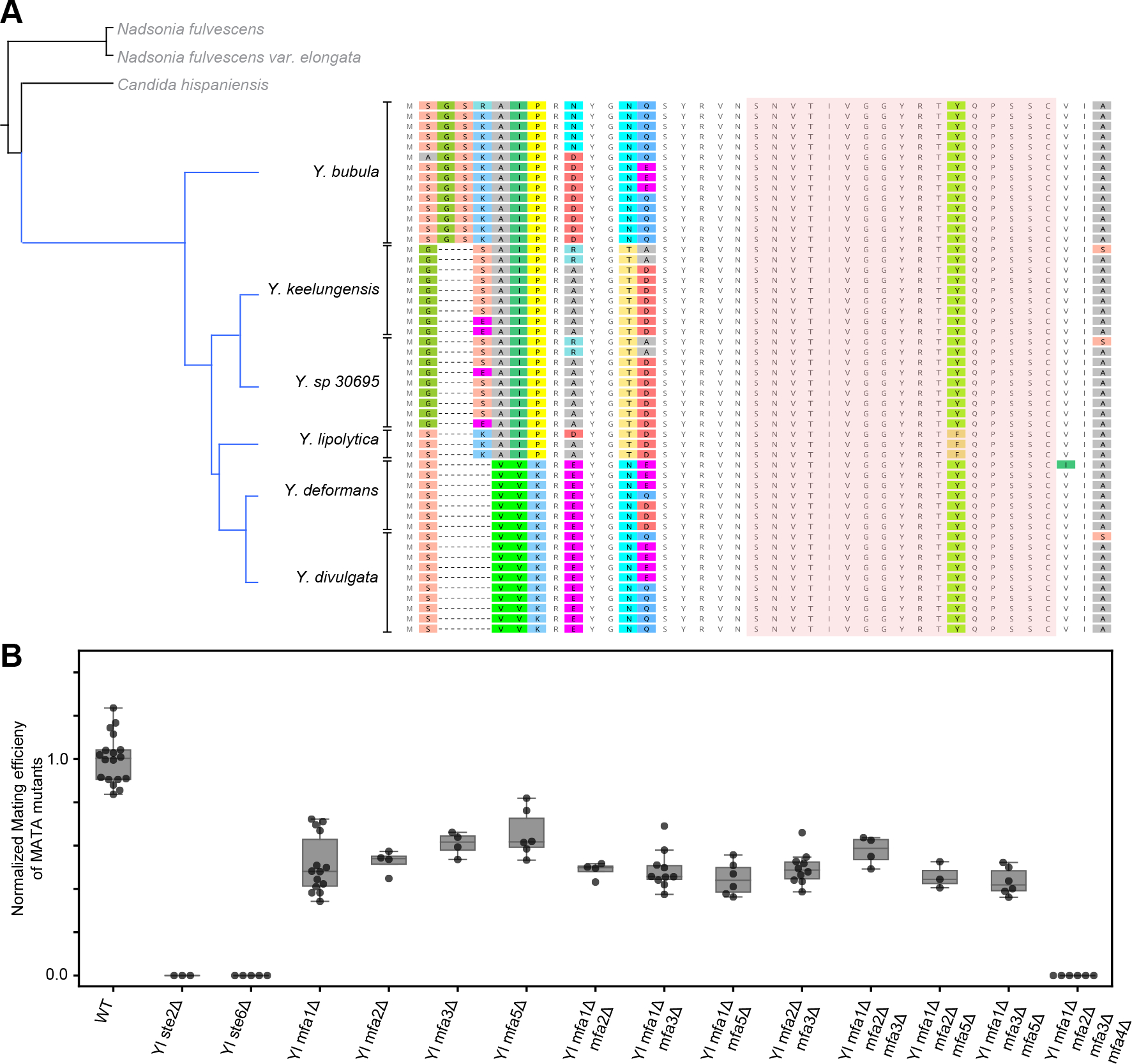
(A) Phylogenetic tree of Yarrowia species (and outgroup in grey) from the set 332 of sequenced yeast of genomes27, along with Y. sp 30695 which is a sister species of Y. keelungensis. Analysis of the genome of Y. sp. 30695 also produced copies of an identical pheromone candidate. The translated ORFs of curated candidates from each species are aligned by the nucleotide sequence of the coding region and ordered according to the phylogenetic relationship between the species. The red shaded region represents the candidate mature pheromone sequence, showing no non-synonymous variation across Yarrowia, except for a conservative change of phenylalanine (F) to tyrosine (Y) in Y. lipolytica. (B) The mating efficiency of MATA haploid derivatives of Y. lipolytica with combinations of the four pheromone loci deleted was evaluated using a semi-quantitative mating protocol. Measurements for each genotype are represented as a group of at least 2 biological replicates each with 2 technical replicate measurements. Single- double- and triple- mutants of YlMFA genes show reduced mating, but only the quadruple deletion of all pheromone loci is deficient in mating to a comparable degree as the receptor (Ylste2Δ) and pheromone exporter (Ylste6Δ) deleted strains. See also Figure S5 and Table S3, S4.
In the Yarrowia lineage we also observe that syntenic conservation falls away much more rapidly than conservation of the mature pheromone sequence (Figure 5A, S5C). Trivially, this is true when gene duplication leads to new pheromone locus in a sister genome, as is likely to have happened to generate the 3 extra copies in Y. divulgata compared to Y. deformans (Figure S5C).
We began our experimental analysis of the Yarrowia pheromone candidates by showing that the Y. lipolytica MATA mating-type is functionally homologous to MATa of S. cerevisiae: Ylste2Δ (YALI0F03905g, ortholog of the α-factor receptor) and Ylste6Δ (YALI0E05973g, ortholog of Ste6, the a-factor exporter) deletions are mating-deficient in MATA, and a Ylste3Δ (YALI0F11913g, ortholog of the a-factor receptor) deletion is mating-deficient in MATB (Figure S5A). We deleted the best pheromone gene candidates identified by the algorithm (YlMFA1, YlMFA2, YlMFA3) in MATA strains as single-, double-, and triple-deletions (Figure 5B). We saw a decrease in the mating efficiency in all the deletion strains, suggesting that the candidates play a role in mating. However, we were surprised to find that the triple mutant did not eliminate the mating of MATA cells. Comparing the copy number of the pheromone candidate in different Yarrowia species revealed that Y. lipolytica is an outlier with fewer pheromone copies (3) than any of the other Yarrowia species (7, 9, 9, 10 and 14) (Figure 5A and Table S3, S4). Because the triple mutant still retained mating activity, we hypothesized that there is at least one additional, intron-containing copy of the pheromone in the Y. lipolytica genome.
We searched translated genome fragments (using tBLASTN) for loci that might code for another copy of the best candidate protein sequence and found a hit on Chromosome F. This locus encodes a copy of the pheromone synonymous to another locus (YlMFA3) but with an intron that disrupts the ORF, masking it from our algorithm (Figure S5B). The intron matches the precise expectations of a Y. lipolytica intron in length, 5’ splice site (5’ ss), branch point (BP), and 3’ splice site (3’ ss) implying this is a true extra copy of the pheromone26.
A single gene copy of the pheromone can weakly support mating in other yeasts that mate under nutrient starvation like S. pombe25. We therefore made the quadruple-deletion that eliminated all copies of the candidate pheromone genes (YlMFA1, YlMFA2, YlMFA3, YlMFA4). This manipulation abrogates mating as effectively as deleting the pheromone transporter or receptor (Figure 5B). Thus, our experiments in the model yeast Y. lipolytica confirm that our pheromone detection algorithm can identify the true farnesylated pheromone of a yeast clade based on the conservation of maturation motifs and multiple homologous copies in closely related genomes. This algorithm will enable annotation of genomes to include the short pheromone genes across the full yeast lineage.
Discussion
We designed an algorithm that takes raw genome sequences as its input and outputs candidate farnesylated pheromones in fungi. These candidates are of particular importance for pathogenic fungi that are of medical (C. albicans and C. neoformans)39 and agricultural (U. maydis (corn smut)40, P. striiformis (wheat stripe rust)41 and M. oryzae (rice blast)42) relevance, because pheromone signaling and mating have been correlated with the development of alternate cell morphologies that play a role in virulence. We experimentally validated the candidate pheromone of the Yarrowia clade by genetic deletions and mating assays in Y. lipolytica. Our method is currently limited by the inability to identify candidates that contain introns, but can be expanded to include models of introns in fungi26. We did not consider the possibility of introns in this work since introns are rare in Saccharomycotina clade (compared to Eukaryotes) with 2–14%26 of genes having an intron, with no introns in known pheromones of Saccharomycotina (till the Y. lipolytica pheromone described in this work). Though broad features of introns are conserved across the lineage, there seem to be significant variation amongst clades, which would need to be accounted for to identify candidate pheromone genes.
In addition to the sieves described above, we considered the possibility that conserved regulatory DNA sequences could help identify candidate pheromone genes43, 44. We searched for and found candidate regulatory sequences within individual phylogroups upstream of the homologs of genes involved in fungal mating (Figure S6). We examined the 1000 DNA base pairs upstream of 15 mating genes, either as a whole set or in two subsets (8 genes whose expression is induced by either a-factor or α-factor in S. cerevisiae, and 7 genes that are uniquely expressed in a-cells). In each phylogroup we identified sequence motifs that are significantly (p < 0.01) associated with these genes. This search produced a total of 133 motifs, 52 of which were associated with the full set of mating genes, 44 associated with pheromone-induced genes, and 37 associated with a-specific genes. Within a phylogroup, we counted the number times each motif is associated with all the copies of a candidate sequence found in the phylogroup, comparing the motif counts for candidates that passed the first three criteria (prenylation and proteolysis sequences, and mature pheromone length) and the subset of these candidates that we identified as the best pheromone candidates. The best pheromone gene candidates had a significantly higher frequency of these putative regulatory motifs, but the difference was small, and neither the overall motif count, nor the presence of individual motifs reliably discriminated between all candidate sequences and those that are either known pheromone sequences or the candidates that we argue are the likely pheromone sequences in other species. The weak correlation and lack of discrimination is consistent with the work that reveals that transcriptional regulation is more evolutionary malleable than protein function45.
In yeasts that have been used as laboratory models (S. cerevisiae and S. pombe) or for the industrial production of proteins and secondary metabolites (K. pastoris and Y. lipolytica), pheromones can be used as tools to control their cell biology and transcriptional program. In Ascomycota (specifically in yeast-like fungi), α-factor-like pheromones remain the more frequently studied pheromone signaling cascade because the genes that encode them are easily identified by the presence of multiple repeats encoded in a single prepro-peptide and the pheromone molecules are easy to produce and biochemically well-behaved. Although farnesylated pheromones are the ancestral form of pheromones, with the Basidiomycota, such as U. maydis and T. mesenterica, only possessing farnesylated pheromones2, 18, 46, they have been harder to identify. Our method relies not on standard sequence homology search, instead, it is an algorithmic filter that uses a series of conserved features to narrow down the list of all possible ORFs to a smaller list of candidates that can be experimentally tested. The current version identifies most copies of a predicted pheromone gene but may not identify all copies (due to the presence of introns, for example). This tool provides a platform to add other filters that might be relevant to specific clades of fungi, like tandem copies within a single pheromone gene47, or the genetic linkage of basidiomycete pheromones to their receptors in a mating locus48, 49. In the 332 Saccharomycotina genome analyzed, candidate pheromones were identified in 241 genomes. Of the remaining species, 32 are orphans within the evolutionary horizon of pheromone conservation we considered, making them inaccessible to our methods, while the remaining 59 had no strong candidate identified. For the second group, our failure could be due to either technical (incomplete genomes, inappropriate phylogenetic grouping, lack of sufficient sister-species), or biological (rapidly evolving sub-lineages, difference in genetic architecture, the loss of pheromones in asexual lineages) factors. Across the Saccharomycotina, mating has been elaborated and altered such that various sub-clades show dramatic differences in their cell identity, gene regulation and propensity to mate48, 49. Many species of yeast that had only been observed to reproduce asexually (anamorphs) were classified as Candida and are now being appropriately renamed and classified based on genome sequencing and the identification of their sexually reproductive forms (teleomorphs)48. Analysis of the genomes of the 332 species considered here also identified the genetic organization of the mating loci, highlighting the flexibility of the mating system across the lineage49. Mating loci are found in all but 2 species, Lodderomyces elongisporus and Candida sojae, for which we also cannot find strong candidate pheromones, though closely-related sister species have predicted pheromones. Our method identifies candidate pheromones in several of the 332 yeasts, including Starmerella (2 species), Yamadazyma (4 species) and Barnettozyma (3 species) where mating is not well characterized, suggesting that the appropriate conditions for inducing and observing mating have not been found. On the other hand, genera like Priceomyces (4 species), Meyerozyma (2 species) form ascospores during sexual reproduction and have mating loci and homologs of mating genes but lack a genus-wide pheromone candidate, suggesting a technical failure of detection. More generally, among the 91 genomes for which pheromones were not found, the most common pattern is the lack of a large clade of closely-related species that have been sequenced. If these species also have only a single pheromone gene, one of our criteria, the presence of multiple homologous ORFs within a single genome, can no longer be used.
The divergence of pheromones across the yeast lineage gives us a window into the evolution of short, unstructured peptides that perform a conserved function. The peptide sequence of pheromones are presumably constrained by needing to co-evolve with their pheromone transporters11 and receptors10, 12. In addition to sequence divergence over long evolutionary timescales, pheromones also show copy number variation between the genomes of sister species indicating selection for gene duplications that increase the level of pheromone secretion and thus promote efficient mating50. Modern methods indicate that short peptide-coding genes are common in genomes across the tree of life, but their identification and functional characterization remain difficult51, 52. The pheromones of the fungal lineage offer a class of short coding genes with clear physiological roles to study sequence and copy number evolution across a lineage of a billion years.
STAR Methods
Resource Availability
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Murray (awm@mcb.harvard.edu).
Materials Availability
All unique/stable reagents (strains) generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
The data that support the findings of this study are available within the paper, its supplementary information files, and in a dataset containing predicted pheromone candidates, which has been uploaded to the GitHub repository.
All code and analysis are publicly available in the GitHub repository (https://github.com/sriramsrikant/pheromoneFinder). The GitHub repository README file describes the location and use of various scripts and Jupyter notebooks (.ipynb) needed to replicate the analysis presented in this paper.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Y. lipolytica
The alkane-using yeast Yarrowia lipolytica strains used in this study are derived from the standard CLIB122 strain (listed in Table S5). Strains were generally grown in YPD or CSM media at 30°C or as specified in the Methods Details. Established protocols to work with this species were adapted as described in the Method Details section below.
Method Details
Strains and plasmids
The Y. lipolytica pair of mating strains (ML16507 and ML16510) were a gracious gift from Joshua Truehart (DSM Ltd.) and are derivatives of the sequenced CLIB122 strain53. Genomic transformation was done using standard protocols54, 55. Gene deletions were done by replacing the ORF with an auxotrophic marker for YlLEU2 or YlURA3. To re-use the URA3 marker, deletions were made with a YlURA3 fragment flanked by repeat regions (from plasmid PMB5082, a gift from Joshua Truehart) that can excise the URA3 cassette upon selection on 5-fluoro-orotic acid (5FOA). Geneious Prime 2021.2.2 was used to align and compare candidate sequences.
Y. lipolytica semi-quantitative mating assay
All strains listed in Table S5 were constructed from our wildtype MATA strain (ML16507) using a chemical transformation protocol55. We modified a quantitative mating protocol used in56 to test the mating efficiency of MATA strains with deletions of pheromone candidates against our MATB partner (ML16510). Briefly, exponential cultures in yeast extract, peptone, dextrose medium (YPD) of the genetically manipulated MATA strain and partner MATB strain were harvested, and 2.5 × 106 cells of each partner were mixed in 150 μL sterile water + 0.02% (w/v) bovine serum albumin. Mating mixtures were transferred onto filters using a filter assembly (with the cells spreading to about 5mm radius), and the filters (with cells) were moved onto YM mating media plates (3 g/L yeast extract, 5 g/L Bacto-peptone, 5 g/L malt extract and 20 g/L Bacto-agar)56. These plates were incubated at 28°C in the dark for three days (70–74 h). After 3 days filters with the mating mixtures were moved into 3 mL YP + 2% (v/v) glycerol + 0.05% (w/v) dextrose and incubated on a roller drum at 30°C for 3 h to recover cells from filters. The resuspended cells were transferred to microfuge tubes and sonicated to disrupt clumps, cells were counted using a Coulter counter, and pelleted by centrifugation. The pelleted cells were resuspended in water containing 0.02% (w/v) bovine serum albumin to a density of 108 cells/mL and plated on diploid selective media (CSM-Lys-Ade). The mating efficiency was calculated as the number of diploid cells for a sample normalized to the number of diploid cells from the control mating (ML16507 + ML16510) performed on the same day (code on Github at https://github.com/sriramsrikant/pheromoneFinder). The experiment was repeated with biological replicates and replica plating from each biological replicate to account for the intrinsic noise of mating mixtures.
Algorithmic filter to identify pheromone candidates
As described in the main text, we identified all stop codons (TAA, TAG, TGA) in the six frames of translation of a genome that are immediately preceded by the farnesylation motif, CAAX (Figure S1B). We further only considered loci that have a start codon (ATG) within 100 codons upstream of the stop codon in the same frame of translation. The last filter to eliminate pheromone candidates was to only consider loci that have an asparagine (N) 5–20 aa upstream of the CAAX motif, which is important for the proteolytic processing of fungal pheromones. The presence of multiple, in-frame start (Met) codons in the 300 bases upstream of a CAAX-stop motif gave rise to multiple pheromone candidates (Figure S2E) that share a common CAAX-stop terminus and at least one internal Asn/N cleavage site (Figure 2A; 125,711 ORFs encoded in 87,326 unique CAAX-stop loci). In analyzing pheromone candidates from unique CAAX-stop loci, the start codon for the largest protein was considered for all our analysis. We used the chromosomes or contigs as a natural split given that their length in fungi allows for reasonable single-core performance of the sieve for stop codons in all frames of translation. To ensure our pipeline finds all possible candidates the algorithm considers ORFs in all contigs since some fungi have not been assembled to chromosomes. Due to the incomplete assembly of chromosomes in many yeasts, pheromone candidates that bridge contigs or lie in gaps between them cannot be identified. We distributed the calculation by contig across processors on the Cannon cluster (Research computing, Harvard FAS).
All scripts to identify candidate loci and subsequent search for homologous pheromone candidates and mating-regulation motifs are custom-written in Python. Pheromone candidate homologs are determined by measuring the pairwise sequence identity of the candidate ORFs from the potential protease site (N) to the stop codon (N(XM)CAAX) across species within the phylogroup where pheromones are conserved (Figure 3A). We score a pairwise global alignment of the candidate sequences with no gap penalties, score identical characters as 1 and all mismatches as 0, and normalize to the length of the larger region. About a quarter of candidates (17,125 of 78,206 [N…CAAX] candidates in conserved-pheromone phylogroups that have at least 2 species – 300 of 332 species) have at least one other candidate that is >70% sequence identical and are considered to have a homologous copy. We considered true pheromones as having two features: (1) They are often encoded in multiple copies in the same genome producing the same mature pheromone, and (2) closely-related species are likely to share very similar pheromones. We thus considered two criteria for curating candidates: (1) candidates that have >70% sequence identity with other candidates within the same genome, and (2) candidates that have >70% sequence identity to a candidate in the genome of a different species genome in the same phylogroup. The best candidates are those that satisfy both criteria but if such a candidate is lacking, we also considered those that satisfy one or the other criterion. A total of 812 candidates from 241 species pass our criteria-based curation and are listed in Table 2.
Motifs involved in mating regulation were identified by using MEME57 to look for motifs upstream of known mating genes (FIG1, PRM1, KAR4, KAR5, SST2, FUS3, STE12, FUS1 as pheromone-activated genes and STE2, STE6, ASG7, AXL1, RAM2, BAR1, AGA2 as a-specific genes in S. cerevisiae)45 in all 332 yeast genomes. We identified at most 3 significant mating-related motifs upstream of all mating genes, pheromone-activated genes, or a-specific genes by grouping species into phylogroups where mating regulation is expected to be conserved (Figure S7)45. FIMO58 was then used to identify significant hits of motifs from each phylogroup upstream of the candidates of species within that phylogroup.
Analysis and plotting of statistics of filters are written in Python using Jupyter notebooks. All scripts and Jupyter notebooks are available on GitHub (https://github.com/sriramsrikant/pheromoneFinder).
Quantification and Statistical Analysis
All quantitative analysis and statistical tests were performed in Python. Sequence analysis was done in Python using the BioPython package (https://pypi.org/project/biopython/). Manuscript figures were generated by the Jupyter notebook to analyze the pheromone candidates identified.
Supplementary Material
Table S1: Results of candidate pheromone gene pipeline on 332 Saccharomycotina genomes, Related to Table 1.
Table S2: Candidate pheromone genes in 332 Saccharomycotina genomes. Related to Table 2.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Yeast genome sequences and annotations | Figshare Dataset associated with Shen et al27 | Figshare Dataset available at https://doi.org/10.6084/m9.figshare.5854692.v1 |
| Experimental Models: Organisms/Strains | ||
| Yeast strains | See Table S5 | N/A |
| Oligonucleotides | ||
| Primers | See Table S6 | Custom oligo synthesis |
| Software and Algorithms | ||
| Codebase | https://github.com/sriramsrikant/pheromoneFinder | Python scripts |
| Geneious Prime 2021.2.2 | Dotmatics | Geneious (RRID:SCR_010519) |
Highlights.
Their small size makes yeast pheromone genes are hard to identify.
Filtering ORFs for pheromone-like features finds candidates in 241 yeast species.
Pheromone sequence is conserved but gene copy number varies widely.
Acknowledgements
We thank the members of the Murray lab and Gaudet lab for helpful discussions and Michael Laub and Sean Eddy for comments on the manuscript. S.S. was a Howard Hughes Medical Institute International Student Research fellow. This work was funded in part by NIGMS grant R01GM120996 to R.G. and NIH grant R01GM43987 and the NSF-Simons Center for Mathematical and Statistical Analysis of Biology at Harvard (#1764269 [NSF] and #594596 [Simons]) to A.W.M.
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lücking R, Huhndorf S, Pfister DH, Plata ER, and Lumbsch HT (2017). Fungi evolved right on track. Mycologia 101, 810–822. [DOI] [PubMed] [Google Scholar]
- 2.Jones SK Jr., and Bennett RJ (2011). Fungal mating pheromones: choreographing the dating game. Fungal Genet Biol 48, 668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kurjan J, and Herskowitz I (1982). Structure of a yeast pheromone gene (MF alpha): a putative alpha-factor precursor contains four tandem copies of mature alpha-factor. Cell 30, 933–943. [DOI] [PubMed] [Google Scholar]
- 4.Singh A, Chen EY, Lugovoy JM, Chang CN, Hitzeman RA, and Seeburg PH (1983). Saccharomyces cerevisiae contains two discrete genes coding for the alpha-factor pheromone. Nucleic Acids Res 11, 4049–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen P, Sapperstein SK, Choi JD, and Michaelis S (1997). Biogenesis of the Saccharomyces cerevisiae Mating Pheromone a-Factor. The Journal of Cell Biology 136, 251–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michaelis S, and Herskowitz I (1988). The a-factor pheromone of Saccharomyces cerevisiae is essential for mating. Molecular and Cellular Biology 8, 1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Michaelis S, and Barrowman J (2012). Biogenesis of the Saccharomyces cerevisiae pheromone a-factor, from yeast mating to human disease. Microbiol Mol Biol Rev 76, 626–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michaelis S (1993). STE6, the yeast a-factor transporter. Seminars in Cell Biology 4, 17–27. [DOI] [PubMed] [Google Scholar]
- 9.Caldwell GA, Naider F, and Becker JM (1995). Fungal lipopeptide mating pheromones: a model system for the study of protein prenylation. Microbiol Rev 59, 406–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seike T, Nakamura T, and Shimoda C (2015). Molecular coevolution of a sex pheromone and its receptor triggers reproductive isolation in Schizosaccharomyces pombe. Proceedings of the National Academy of Sciences of the United States of America 112, 4405–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srikant S, Gaudet R, and Murray AW (2020). Selecting for Altered Substrate Specificity Reveals the Evolutionary Flexibility of ATP-Binding Cassette Transporters. Curr Biol 30, 1689–1702 e1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin SH, Wingfield BD, Wingfield MJ, and Steenkamp ET (2011). Causes and consequences of variability in peptide mating pheromones of ascomycete fungi. Mol Biol Evol 28, 1987–2003. [DOI] [PubMed] [Google Scholar]
- 13.Kamiya Y, Sakurai A, Tamura S, Takahashi N, Abe K, Tsuchiya E, and Fukui S (1978). Isolation of RhodotorucineA, a Peptidyl Factor Inducing the Mating Tube Formation inRhodosporidium toruloides. Agricultural and Biological Chemistry 42, 1239–1243. [Google Scholar]
- 14.Kamiya Y, Sakurai A, Tamura S, Takahashi N, Abe K, Tsuchiya E, Fukui S, Kitada C, and Fujino M (1978). Structure of rhodotorucine A, a novel lipopeptide, inducing mating tube formation in Rhodosporidiumtoruloides. Biochemical and biophysical research communications 83, 1077–1083. [DOI] [PubMed] [Google Scholar]
- 15.Sakagami Y, Isogai A, Suzuki A, Tamura S, Kitada C, and Fujino M (1979). Structure of Tremerogen A–10, a Peptidal Hormone Inducing Conjugation Tube Formation inTremella mesenterica. Agricultural and Biological Chemistry 43, 2643–2645. [Google Scholar]
- 16.Sakagami Y, Isogai A, Suzuki A, Tamura S, Tsuchiya E, and Fukui S (1978). Isolation of a Novel Sex Hormone, Tremerogen A-10, Controlling Conjugation Tube Formation inTremella mesentericaFries. Agricultural and Biological Chemistry 42, 1093–1094. [Google Scholar]
- 17.Bennett RJ, and Turgeon BG (2016). Fungal Sex: The Ascomycota. Microbiol Spectr 4. [DOI] [PubMed] [Google Scholar]
- 18.Coelho MA, Bakkeren G, Sun S, Hood ME, and Giraud T (2017). Fungal Sex: The Basidiomycota. Microbiol Spectr 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.OhEigeartaigh SS, Armisen D, Byrne KP, and Wolfe KH (2011). Systematic discovery of unannotated genes in 11 yeast species using a database of orthologous genomic segments. Bmc Genomics 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huyer G, Kistler A, Nouvet FJ, George CM, Boyle ML, and Michaelis S (2006). Saccharomyces cerevisiae a-factor mutants reveal residues critical for processing, activity, and export. Eukaryot Cell 5, 1560–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marr RS, Blair LC, and Thorner J (1990). Saccharomyces-Cerevisiae-Ste14 Gene Is Required for Cooh-Terminal Methylation of a-Factor Mating Pheromone. Journal of Biological Chemistry 265, 20057–20060. [PubMed] [Google Scholar]
- 22.Berger BM, Kim JH, Hildebrandt ER, Davis IC, Morgan MC, Hougland JL, and Schmidt WK (2018). Protein Isoprenylation in Yeast Targets COOH-Terminal Sequences Not Adhering to the CaaX Consensus. Genetics 210, 1301–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein V, Kubala MH, Steen J, Grimmond SM, and Alexandrov K (2015). Towards the systematic mapping and engineering of the protein prenylation machinery in Saccharomyces cerevisiae. PLoS One 10, e0120716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trueblood CE, Boyartchuk VL, Picologlou EA, Rozema D, Poulter CD, and Rine J (2000). The CaaX Proteases, Afc1p and Rce1p, Have Overlapping but Distinct Substrate Specificities. Molecular and Cellular Biology 20, 4381–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kjaerulff S, Davey J, and Nielsen O (1994). Analysis of the structural genes encoding M-factor in the fission yeast Schizosaccharomyces pombe: identification of a third gene, mfm3. Mol Cell Biol 14, 3895–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neuveglise C, Marck C, and Gaillardin C (2011). The intronome of budding yeasts. C R Biol 334, 662–670. [DOI] [PubMed] [Google Scholar]
- 27.Shen XX, Opulente DA, Kominek J, Zhou X, Steenwyk JL, Buh KV, Haase MAB, Wisecaver JH, Wang M, Doering DT, et al. (2018). Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 175, 1533–1545 e1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adames N, Blundell K, Ashby MN, and Boone C (1995). Role of Yeast Insulin-Degrading Enzyme Homologs in Propheromone Processing and Bud Site Selection. Science (New York, N.Y.) 270, 464–467. [DOI] [PubMed] [Google Scholar]
- 29.Dignard D, El-Naggar AL, Logue ME, Butler G, and Whiteway M (2007). Identification and characterization of MFA1, the gene encoding Candida albicans a-factor pheromone. Eukaryot Cell 6, 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heistinger L, Moser J, Tatto NE, Valli M, Gasser B, and Mattanovich D (2018). Identification and characterization of the Komagataella phaffii mating pheromone genes. FEMS Yeast Res 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith C, and Greig D (2010). The cost of sexual signaling in yeast. Evolution 64, 3114–3122. [DOI] [PubMed] [Google Scholar]
- 32.Seike T, Shimoda C, and Niki H (2019). Asymmetric diversification of mating pheromones in fission yeast. PLoS Biol 17, e3000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicaud JM (2012). Yarrowia lipolytica. Yeast 29, 409–418. [DOI] [PubMed] [Google Scholar]
- 34.Liu HH, Ji XJ, and Huang H (2015). Biotechnological applications of Yarrowia lipolytica: Past, present and future. Biotechnol Adv 33, 1522–1546. [DOI] [PubMed] [Google Scholar]
- 35.Markham KA, and Alper HS (2018). Synthetic Biology Expands the Industrial Potential of Yarrowia lipolytica. Trends Biotechnol 36, 1085–1095. [DOI] [PubMed] [Google Scholar]
- 36.Dulermo R, Gamboa-Melendez H, Ledesma-Amaro R, Thevenieau F, and Nicaud JM (2015). Unraveling fatty acid transport and activation mechanisms in Yarrowia lipolytica. Biochimica et biophysica acta 1851, 1202–1217. [DOI] [PubMed] [Google Scholar]
- 37.Kerscher S, Dröse S, Zwicker K, Zickermann V, and Brandt U (2002). Yarrowia lipolytica, a yeast genetic system to study mitochondrial complex I. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1555, 83–91. [DOI] [PubMed] [Google Scholar]
- 38.Parey K, Brandt U, Xie H, Mills DJ, Siegmund K, Vonck J, Kuhlbrandt W, and Zickermann V (2018). Cryo-EM structure of respiratory complex I at work. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boral H, Metin B, Dogen A, Seyedmousavi S, and Ilkit M (2018). Overview of selected virulence attributes in Aspergillus fumigatus, Candida albicans, Cryptococcus neoformans, Trichophyton rubrum, and Exophiala dermatitidis. Fungal Genet Biol 111, 92–107. [DOI] [PubMed] [Google Scholar]
- 40.Chacko N, and Gold S (2012). Deletion of the Ustilago maydis ortholog of the Aspergillus sporulation regulator medA affects mating and virulence through pheromone response. Fungal Genet Biol 49, 426–432. [DOI] [PubMed] [Google Scholar]
- 41.Zhu X, Liu W, Chu X, Sun Q, Tan C, Yang Q, Jiao M, Guo J, and Kang Z (2018). The transcription factor PstSTE12 is required for virulence of Puccinia striiformis f. sp. tritici. Mol Plant Pathol 19, 961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y, Que Y, Liu Y, Yue X, Meng X, Zhang Z, and Wang Z (2015). The putative Ggamma subunit gene MGG1 is required for conidiation, appressorium formation, mating and pathogenicity in Magnaporthe oryzae. Curr Genet 61, 641–651. [DOI] [PubMed] [Google Scholar]
- 43.Sengupta P, and Cochran BH (1990). The PRE and PQ box are functionally distinct yeast pheromone response elements. Mol Cell Biol 10, 6809–6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wong Sak Hoi J, and Dumas B (2010). Ste12 and Ste12-like proteins, fungal transcription factors regulating development and pathogenicity. Eukaryot Cell 9, 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sorrells TR, Booth LN, Tuch BB, and Johnson AD (2015). Intersecting transcription networks constrain gene regulatory evolution. Nature 523, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raudaskoski M, and Kothe E (2010). Basidiomycete mating type genes and pheromone signaling. Eukaryot Cell 9, 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmoll M, Seibel C, Tisch D, Dorrer M, and Kubicek CP (2010). A novel class of peptide pheromone precursors in ascomycetous fungi. Mol Microbiol 77, 1483–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wolfe KH, and Butler G (2017). Evolution of Mating in the Saccharomycotina. Annu Rev Microbiol 71, 197–214. [DOI] [PubMed] [Google Scholar]
- 49.Krassowski T, Kominek J, Shen XX, Opulente DA, Zhou X, Rokas A, Hittinger CT, and Wolfe KH (2019). Multiple Reinventions of Mating-type Switching during Budding Yeast Evolution. Curr Biol 29, 2555–2562 e2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moore TI, Chou CS, Nie Q, Jeon NL, and Yi TM (2008). Robust spatial sensing of mating pheromone gradients by yeast cells. PLoS One 3, e3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Couso JP, and Patraquim P (2017). Classification and function of small open reading frames. Nat Rev Mol Cell Biol 18, 575–589. [DOI] [PubMed] [Google Scholar]
- 52.Andrews SJ, and Rothnagel JA (2014). Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet 15, 193–204. [DOI] [PubMed] [Google Scholar]
- 53.Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, De Montigny J, Marck C, Neuveglise C, Talla E, et al. (2004). Genome evolution in yeasts. Nature 430, 35–44. [DOI] [PubMed] [Google Scholar]
- 54.Burke D, Dawson D, and Stearns T (2000). Methods in Yeast genetics: A Cold Spring Harbor Laboratory course manual, (Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- 55.Verbeke J, Beopoulos A, and Nicaud JM (2013). Efficient homologous recombination with short length flanking fragments in Ku70 deficient Yarrowia lipolytica strains. Biotechnol Lett 35, 571–576. [DOI] [PubMed] [Google Scholar]
- 56.Rosas-Quijano R, Gaillardin C, and Ruiz-Herrera J (2008). Functional analysis of the MATB mating-type idiomorph of the dimorphic fungus Yarrowia lipolytica. Curr Microbiol 57, 115–120. [DOI] [PubMed] [Google Scholar]
- 57.Bailey TL, and Elkan C (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2, 28–36. [PubMed] [Google Scholar]
- 58.Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tamura K, Battistuzzi FU, Billing-Ross P, Murillo O, Filipski A, and Kumar S (2012). Estimating divergence times in large molecular phylogenies. Proceedings of the National Academy of Sciences of the United States of America 109, 19333–19338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Results of candidate pheromone gene pipeline on 332 Saccharomycotina genomes, Related to Table 1.
Table S2: Candidate pheromone genes in 332 Saccharomycotina genomes. Related to Table 2.
Data Availability Statement
The data that support the findings of this study are available within the paper, its supplementary information files, and in a dataset containing predicted pheromone candidates, which has been uploaded to the GitHub repository.
All code and analysis are publicly available in the GitHub repository (https://github.com/sriramsrikant/pheromoneFinder). The GitHub repository README file describes the location and use of various scripts and Jupyter notebooks (.ipynb) needed to replicate the analysis presented in this paper.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.