

Abstract
Viral infections are the most common cause of upper respiratory infections (URIs) and frequently infect adults 1–2 times and children 6–8 times annually. In most cases, these infections are self-limiting and resolve. However, many patients with chronic rhinosinusitis (CRS) relay that their initiating event began with a URI that progressed in both symptom severity and duration. Viruses bind to sinonasal epithelia through specific receptors, thereby entering cells and replicating within them. Viral infections stimulate interferon-mediated innate immune responses. Recent studies suggest that viral infections may also induce type 2 immune responses and stimulate the aberrant production of cytokines that can result in loss of barrier function, a hallmark in CRS. The main purpose of this review will be to highlight common viruses and their associated binding receptors and highlight pathophysiologic mechanisms associated with alterations in mucociliary clearance, epithelial barrier function and dysfunctional immune responses that might lead to a further understanding of the pathogenesis of CRS.
Keywords: Chronic rhinosinusitis, upper respiratory tract infection, airway epithelium, innate immunity, genetics, virus, barrier function
Introduction
Upper respiratory infections (URIs) are one of the most common medical diagnoses in the United States with costs exceeding $22 billion annually.1 The majority of URIs are due to human rhinovirus (RV), respiratory syncytial virus (RSV), influenza, and human coronaviruses (hCoV) infections.2, 3 These viruses bind to receptors in the nasal airway which mediate cell-entry, thereby allowing viral replication, which triggers an interferon antiviral host immune response. This inflammatory response can manifest with symptoms of fever, nasal congestion, hyposmia (decrease in smell), facial pain/pressure, and postnasal drainage. In most cases, this immune response is self-limiting and returns to a homeostatic baseline after the viral infection is cleared. However, an aberrant host immune response after viral infections characterized by chronic inflammation, decreased mucociliary clearance, and loss of the epithelial barrier function may be one hypothesis for the development of chronic rhinosinusitis (CRS).4, 5 This hypothesis is supported by a longitudinal study that identified that recurrent colds in childhood were a significant risk factor for the development of sinusitis in children and in adults.6
In this review, we highlight common respiratory viruses associated with sinusitis and their potential role in the development of CRS. We discuss the role of the host immune response to viruses, and how dysregulation might contribute to chronic inflammation and loss of the protective airway barrier. We also discuss the role of virus subtypes and genetic risk factors that might contribute to the pathogenesis of CRS.
Viral subtypes and clinical disease
Not all virus subtypes within each family have similar clinical effects. For example, RV infections are the most common cause of URIs7 and have also been linked to CRS and sinusitis exacerbations.4 HRVs are positive-strand RNA viruses in the picornaviridae family and divided into 3 species: HRV-A, -B, and -C. HRV-A and -B species were identified and sequenced in the 1980s, however HRV-C was only identified in 2006 as HRV-C did not grow in standard viral media.8 The advent of viral sequencing and subsequent distinctions of RV species have identified that HRV-A and HRV-C sinus infections result in more severe symptoms than HRV-B.9 This epidemiologic finding is supported by in vitro and in vivo findings that reveal a heightened immune response, as measured by gene expression and cytokine signals, after HRV-A and HRV-C infections compared to HRV-B infections.9, 10
RSV is the second most common virus identified in sinus infections and consists of two subtypes, RSV-A and RSV-B that differ by antigen sequences.11 In most epidemiologic studies, RSV-A tends to be the predominant virus identified in children, and RSV-A tends to have a more severe response and require ICU care in children secondary to bronchiolitis than RSV-B.12–14 More significant than virus subtype is patient age. Although RSV can infect people of all ages, infants / children and the elderly are at a significantly higher risk of lower airway morbidity and mortality due to impaired antiviral host responses.
Influenza viruses are negative-sense single-strand RNA viruses from the Orthomyxoviridae family. Although there are four types of influenza viruses: A, B, C, and D, only influenza A and B cause seasonal flu infections in humans.15, 16 Influenza subtypes are based on the combinations of the hemagglutin (HA) and neuraminidase (NA) proteins on the surface of the virus. Influenza A tends to produce more symptoms and is the only subtype known to cause epidemics. In 2009, the influenza A (H1N1) was declared a pandemic,17 with causes of severe acute respiratory distress and death reported in high risk populations, including the morbidly obese and pregnant patients.18
Human coronaviruses are enveloped positive-sense RNA viruses from the coronaviridae family. The common viral strains of human coronavirus: 1. 229E (alpha coronavirus), NL63 (alpha coronavirus), 3. OC43 (coronavirus), and 4. HKU1 (beta coronavirus) typically cause only mild symptoms. However, there are more severe strains including 5. MERS-CoV (beta coronavirus responsible for Middle East Respiratory Syndrome), 6. SARS-CoV (beta-coronavirus responsible for severe acute respiratory syndrome), and 7. SARS-CoV-2 (beta coronavirus responsible for COVID-19) that cause high rates of morbidity and mortality.19
Virus receptors, and binding:
Respiratory viruses are transmitted via mucosal surfaces in either the nasal or oral cavities. In the nasal airway, fusion proteins on the virus envelope bind to nasal epithelial cells through specific receptors present on the cell surface. This triggers translocation, in which the RNA virus can then enter the epithelial cell and replicate via the host-cell machinery. These viruses are then shed by the cell and can infect other host cells. Virus subtypes, their receptors, and their associated clinical symptoms are highlighted in Table 1.
Table 1.
Virus subtypes, surface receptors, and clinical disease
| Virus | Subtypes | Surface Receptors | Clinical Disease |
|---|---|---|---|
| Rhinovirus | RV-A | ICAM-1, LDLR | Associated with more severe sinus symptoms |
| RV-B | ICAM-1, LDLR | Associated with milder sinus symptoms | |
| RV-C | CDHR3 | Associated with more severe sinus symptoms and childhood asthma exacerbations | |
| Respiratory syncytial virus | RSV-A and RSV-B | RSV G protein binds CX3CR1 and HSPG, RSV F protein can interact with NCL, EGFR, IGF1R, AND ICAM-1. | Frequent cause of wheezing and severe bronchiolitis in infants and young children. Associated with URIs and LRTIs. Increased morbidity in infants and young children RSV-A more common in children and associated with a higher severity of illness compared to RSV-B. |
| Influenza | Influenza A and B | Sialic acid-containing receptors on airway cells, α(2,6)-linked sialic acid receptors in humans | Common presenting signs include fever, cough, rhinorrhea, and vomiting. Influenza A associated with more severe symptoms and its rapid spread can be associated with epidemic/pandemics. Severe acute respiratory distress and death reported in high-risk populations. |
| Coronavirus | HCoV-229E | Human aminopeptidase N | Mild URI symptoms, within the range of typical common colds. |
| SARS-CoV | S glycoprotein attachment to ACE2 | Virus responsible for SARS. Highly contagious and may present initially as fever, myalgia, headache, and progress to cough, dyspnea, and respiratory distress. | |
| SARS-CoV-2 | S glycoprotein attachment to ACE2 | Virus responsible for COVID-19. Highly contagious with symptoms ranging from mild URI symptoms, hyposmia/anosmia and can progress to severe lower respiratory symptoms with high rates of morbidity and mortality. |
There are three different receptors for RV-infections. ICAM-1 is a cell surface glycoprotein that mediates leukocyte adhesion in endothelial cells and cell migration, barrier function, and proliferation in epithelial cells.20 For RV-A and RV-B, the major group of RV serotypes, (90%) use ICAM-1 as their receptor 21, 22 while the minor group of RV serotypes (10%) use LDLR as their receptor.23 In 2014, Bonnelyke et al. identified that a missense variant in the rs6967330 single nucleotide polymorphism (SNP) resulting in a cysteine to tyrosine substation was highly associated with severe childhood asthma exacerbations.24 Further analysis of this SNP revealed that it was associated with differential expression of the cadherin related family member 3 gene (CDHR3) in 2015. CDHR3 is a transmembrane protein that is highly expressed at the cell surface during mucociliary differentiation. Bochov et al. in an elegant study using differentiated sinonasal airway epithelial cells identified that CDHR3 was a receptor for HRV-C.25 Subsequent studies by Basnet et al. suggest that cells expressing the rs6967330 SNP have increased binding and replication of HRV-C.26 This genetic risk factor may explain why persons with the rs6967330 SNP have a 2-fold increased odds to have adult CRS in a multi-center study.27
RSV enters cells by binding the G protein attachment to either heparin sulfate proteoglycans (HSPGs) or CX3CR1 receptors present on airway cells.28, 29 This binding then allows the RSV F protein attachment to bind to nucleolin (NCL), insulin-like growth factor-1 receptor (IGF1R), epidermal growth factor (EGFR), and ICAM-1 and enter the cell.30 RSV disease susceptibility has been associated with SNPs in genes regulating innate immune host responses including the vitamin D receptor, JUN, a gene involved in pro-inflammatory cytokine production, and IFNA5, an interferon gene.31
Influenza uses the hemagglutinin (HA) protein on its cell surface to bind to sialic acid-containing receptors on airway cells and other cell types.32 Influenza virus typically enters the human body through the upper respiratory tract, causing sinusitis symptoms. However, the virus can spread to the lower respiratory tract, which can lead to life threatening illnesses secondary to uncontrolled cytokine production. Chatzopoulou et al. assessed complement-related genetic risk factors in over 200 individuals during the 2009 H1N1 influenza pandemic grouped according to disease severity. They identified that persons with SNPs in the CD55 gene (rs2564978) and in the C1QBP gene (rs3786054) were associated with increased mortality but not disease severity.33
Different strains of human coronavirus bind to epithelial cells via different receptors. For example, common coronavirus HCoV-229E is a pathogen that is frequently responsible for URI disease and uses human aminopeptidase N (APN) as its entry receptor to invade cells.34 Alternatively, both SARS-CoV and SARS-CoV-2 use angiotensin-converting enzyme 2 (ACE2) to enter cells. ACE2 is expressed on the apical surface of epithelial cells in the lung and nasal airways.35 Attachment of the virus to the host cell membrane is mediated by the S glycoprotein on the viral surface.36 The S protein on SARS-CoV strains is split into two subunits: the S1 subunit binds ACE2, while the S2 subunit anchors the S protein to the membrane.37 Ziegler et al recently identified that ACE2 was an interferon-stimulated gene,38 and our lab reported that RV infections in asthmatics increased ACE2 expression.39 This finding suggests that common viral infections, including RV, RSV, and influenza, might increase ACE2 expression and COVID-19 disease severity.
Host immune response (interferons):
Viruses can activate the immune system via different mechanisms. The immediate immune response is via the innate immune system, designed to block/inhibit viral infection, protect host cells, and kill virus-infected cells. The innate immune system is initiated by activation of pattern recognition receptors in the host cell. Viral RNA can activate Toll-like receptors (TLR) on the cell surface,40 and/or cytoplasmic receptors can be activated within the cell including retinoic acid-inducible gene I (RIG-I), or melanoma differentiation-associated gene 5 (MDA-5). These pattern recognition receptors can then activate signal integrators such as myeloid differentiation factor 88 (MyD88), TIR-domain-containing adapter-inducing interferon-β (TRIF), nuclear factor-κB (NFK-κB), and interferon regulator factor (IRF) transcription factors.41 Activation of these signal integrators promote expression of antiviral interferons, interferon-stimulated genes, and inflammatory cytokines.42
Interferons are a family of cytokines that interfere with virus replication and can be divided into three groups. Type 1 interferons (IFN-I) are the most studied anti-viral cytokines and include IFN-α and IFN-β interferons. Type 1 interferons can directly inhibit virus replication and stimulate adaptive immune responses of B cells to produce antibodies and T-cells to recognize and destroy virus-infected cells. Type 2 interferons (IFN-γ) can orchestrate macrophage, neutrophil, dendritic cell, and NK cell responses. Type 3 interferons (IFNλ and IL-10) can mediate antiviral responses.43 Although interferon responses are critical for antiviral innate immunity, there is evidence that prolonged and sustained interferon production may also contribute to chronic inflammation.44
There have been several in vivo and in vitro studies of host innate immune response to RV in sinusitis with conflicting results. Tan et al. was one of the first groups to use air-liquid-interface (ALI) cultures that replicated nasal epithelia to assess the immunological response to RV-infection. They found that RV infection induced CXCL-11, IP-10, CXCL-9, and RANTES expression and activated Toll-like receptor 7 (TLR7) and retinoic acid-inducible gene 1 (RIG-1) pattern recognition signaling cascades to induce type 1 and 3 interferon signaling pathways.45 Kim et al. assessed epithelial immune responses to RV in ALI cultures from control and CRS patients. They found that although antiviral cytokine production in CRS cultures were not different than healthy controls, there was a slight reduction in IFN-β and MDA5 mRNA expression which might result in decreased clearance of virus.46 Lee et al. in a similar in vitro study identified that RV-induced production of anti-viral interferon responses was not different between control and CRS with nasal polyp tissues.47 Hwang et al. then compared type 1 and type 3 interferon responses to respiratory viruses in the nasal tissues of healthy controls and patients with CRS. They found that type 1 and type 3 interferon responses were significantly decreased in patients with CRS compared to controls.48 One possible explanation for these findings might be in timing. In studies of viral responses in asthma, asthmatics were found to have IFN impairment at baseline resulting in enhanced RV replication. This resulted in higher viral loads which then upregulated antiviral signals thereby resulting in an exaggerated IFN response later during asthma exacerbation.49 This hypothesis is supported by in vivo host response changes after CRS exacerbations in which there were significantly increased of IL-6, MBP, and MPO in nasal lavage fluids compared to baseline and healthy controls.50, 51
The host immune response to RSV infection differs based on the age of the patient. In infants, a very low IFN-λ response in nasal fluids is related to a higher degree of symptoms and disease severity including nasal congestion, sinusitis and productive cough.52 However, in adults, RSV infection induces increased levels of IFN-β, IFN-λ1, and IFN-γ in nasal fluid.53 This age-related finding suggest that an immature interferon response in infants might be related to an increase in RSV symptoms.
The role of interferons as protective against influenza infection was reported by Klinkhammer et al. using murine models.54 In their study, both IFN-α and IFN-γ protected against the spread of influenza virus infection from the upper airway to the lungs and inhibited viral transmission from infected to healthy individuals.54 However, other studies have reported that IFN-γ is involved in the pathogenesis of influenza virus.55, 56 They showed that deficiency of IFN-γ signaling protected mice with upper airway infection from developing disease severity and improved survival.55, 56 The success of the influenza virus in reducing the morbidity and mortality of flu-related disease, suggest that priming the immune system to develop antibodies to circulating influenza subtypes can reduce the severity of disease.
Several studies have suggested that early antiviral immune responses in the upper respiratory tract may serve as an early indicator of disease severity in SARS-CoV-2 infected individuals.57 In particular, IFNs have been shown to have an inverse relationship with the severity of COVID-19. Weakened IFN responses in the upper respiratory airway and impaired type I and type III IFN levels in the blood have been correlated with severe symptoms of COVID-19.57–59 Zhang et al. hypothesized that decreased antiviral immunity seen in non-eosinophilic CRS cultures might result in the increased SARS-CoV2 binding and replication compared to healthy controls.60 Lei et al. reported IFN-β treatment effectively inhibits SARS-CoV-2 replication and that SARS-CoV-2 evades type 1 IFN production and signaling, indicating that IFNs are targeted by SARS-CoV-2 to disrupt host immune response for their replication.61, 62 These findings suggest that type I and type III IFNs may be potential therapies for COVID-19, and in fact, many clinical trials utilizing IFNs as therapeutic agents are underway,63 including the phase 3 trial recently reported that COVID-19 related hospitalization or death reduced by 47% in the single dose of pegylated IFN-λ group compared to the placebo group.64
T-cell mediated responses:
T-cell mediated adaptive immune responses are critical in recognizing and destroying virus-infected cells. Type 1 immune responses, characterized by IFN-γ and produced by Th1 cells help to protect against intracellular pathogens, such as viruses. Type 2 immune responses, characterized by the production of IL-4, IL-5, and IL-13, are characteristic of allergic responses. Type 3 immune responses, characterized by Th17 cells and the production of IL-17 and IL-22, are characteristic of antimicrobial responses. Classically type 1 immune responses are the best studied in antiviral responses, however there is increasing evidence that type 2 and type 3 immune responses are instrumental.
Jackson et al. sought to answer how RV infections, which classically triggered Th1 IFN-γ antiviral responses, might induce Th2 immune responses. They found that RV infections in both in vitro and in vivo models stimulated the production of IL-33, which in turn induced the secretion of type 2 cytokines IL-4, IL-5, and IL-13.65 Bosco et al sampled the nose of children presenting to the ER with RV-induced asthma/wheeze.66 He found that children with a high Th1 interferon response had mild symptoms, while those with a low IFN-γ response but upregulation of EGF, IL-4, IL-6, IL-10, and TGF-β had more severe symptoms. Using gene network reconstruction, they identified IRF7 as the hub connecting interferon-mediated antiviral response.67 When IRF7 was knocked down in airway epithelial cells, RV-induced antiviral responses were reduced and IL-33 Th2 responses were increased suggesting a role of interferons and IRF7 in regulating Th1 and Th2 immune responses.68 This Th2 shift in more severe RV-related airway disease might explain why the use of omalizumab, an IgE blocker, improved IFN-α responses to RV and reduced asthma exacerbations in inner-city youth.69 It will be interesting to see if the use of biologics targeting type 2 pathways reduces viral-mediated CRS exacerbations in the future.
Zhang et al. reported that IL-33, an alarmin cytokine that triggers type 2 immunity, was elevated in nasal aspirates from infants with severe RSV infection. Furthermore, blocking IL-33 has been shown to effectively inhibit RSV-induced immunopathogenesis in several respiratory animal studies,70–72 suggesting IL-33 as a potential therapeutic target to treat airway RSV infection. Recently, a phase 3 trial of an RSV vaccine showed significant reductions in RSV-related respiratory infections and severity in older adults. Further studies assessing the host response in those who receive the RSV vaccine might shed light on if the type 2 immune response is decreased.73
Type 2 inflammation and interferon production has also been shown to increase ACE2 expression in the airway epithelia, suggesting an increased risk for COVID-19 viral infections.74 75, 76 77, 78 Although there is very limited data for COVID-19 infection and CRS risk, Lee et al. did identify that persons with CRS had a higher risk for SARS-CoV-2 infection and severe COVID-19 in a Korean cohort.79 Interestingly, this finding was unique to patients with CRS without nasal polyps, classically a Th1-mediated disorder.
Mucociliary function:
Mucociliary clearance is a primary innate defense in the upper and lower airways. Pathogens, including viruses, are trapped in the mucus layer and then removed from the airways by motile cilia. Viruses can target this mucociliary defense through various mechanisms. CDHR3, the receptor for RV-C, is expressed exclusively on ciliated airway cells.80 Similarly, the ACE2 receptor, which binds SARS-CoV and SARS-CoV-2, is highly expressed in ciliated airway cells which might explain why these viruses preferentially bind to nasal airway epithelia.81, 82 For RSV infections, viral entry into the cell is mediated by two surface glycoproteins that bind to the CX3CR1 receptor expressed on motile cilia.83 Viral infections can also increase the expression of mucin proteins, including MUC5AC and MUC5B, which can result in increased mucus production thereby slowing mucociliary clearance.84–87 Finally, SARS-CoV and SARS-CoV-2 have been shown to destroy motile cilia, possibly due to downregulation of FOXJ1, a transcription factor necessary for ciliogenesis. The end result of decreased mucociliary clearance after viral infections might promote viral spread and increase disease severity.82
Epithelial barrier function:
An intact barrier of the airway epithelium provides a physical barrier against inhaled viruses in the nose. This epithelial barrier is comprised of tight junction (TJ) including Zona-occludens 1 (ZO-1), occludins, and claudins that mediate paracellular transport and adherens junctions (AJ) including the cadherin and catenin families that provide cell-cell adhesion and cellular integrity by associating with the actin cytoskeleton. Several studies have suggested that the epithelial barrier might be compromised in patients with CRS.88–90 The Gern lab suggested an interesting hypothesis that barrier function loss might predispose to viral infections. In their model, reduced airway barrier function in asthmatics might expose viral binding receptors, thereby leading to increased viral binding and replication.91 Although they speculate that allergic disease might trigger reduced airway barrier function leading to viral infections, it also possible that repeated viral infections might also result in decreased barrier function. This hypothesis is also supported by work in our lab investigating the early pathogenesis of sinusitis. In our study, we followed over 700 individuals from birth to adulthood in a longitudinal cohort. We found that viral infections/colds, allergies, and asthma were significant risk factors for an early onset chronic sinusitis phenotype.6
RV, RSV, and influenza have all been shown to decrease epithelial barrier function via in vivo and in vitro models. After RV infection, Yeo et al. found that TJ (ZO-1, occludin, and claudin-1) mRNA and protein levels were significantly reduced with a corresponding decrease in transepithelial resistance (TER).92 Looi et al. found similar decreases in TJ proteins after RV infection, and network analyses suggested this downregulation was due to the expression of antiviral IL-15 responses.93 RSV infections can also lead to tight junction disassembly and alterations to epithelial permeability. A study by Rezaee et al. showed tight junction and barrier dysfunction post-RSV infection through immunofluorescence labeling and a reduction in TER in nasal airway cells.94, 95 Similar findings have been shown in vivo using RSV murine models.96 Similarly, influenza virus has also been shown to decrease tight junction proteins and reduce TER.97, 98 This decrease in barrier function is believed to be secondary to antiviral cytokines induced after viral infection.
Summary
The interaction between viruses and airway epithelia and the resulting host immune response is an important field of research for providing insight into sinonasal inflammation. In this review, we discuss risk factors including genetics, virus subtypes, and aberrant immune responses that might shed light on the pathogenesis of chronic rhinosinusitis. We hypothesize that in acute self-limiting viral infections, an elevated IFN-I response is triggered which transiently increases epithelial barrier permeability and orchestrates macrophage, neutrophil, dendritic cell, and NK cells to kill virus-infected cells. Importantly, this response is attenuated shortly after the viral infection and the host immune response returns to a healthy homeostatic state (Figure 1). Alternatively, we hypothesize that aberrant immune responses might result in chronic inflammation of the sinonasal mucosa thereby contributing to a hallmark of CRS. In this model, a persistently elevated IFN-I response induces type 2 immune responses, characterized by IL-4, IL-5, IL-13 secretion and the recruitment of eosinophils, mast cells, and IgE. Another possibility might be due to increased virulent strains, such as RV-C or SARS-CoV2 infections or increased viral load in subjects with genetic risk factors, such as the rs6967330 SNP in the RV-C receptor CDHR3. In this case, this persistent inflammatory state could increase epithelial barrier permeability and result in tissue damage and airway remodeling seen in CRS (Figure 2).
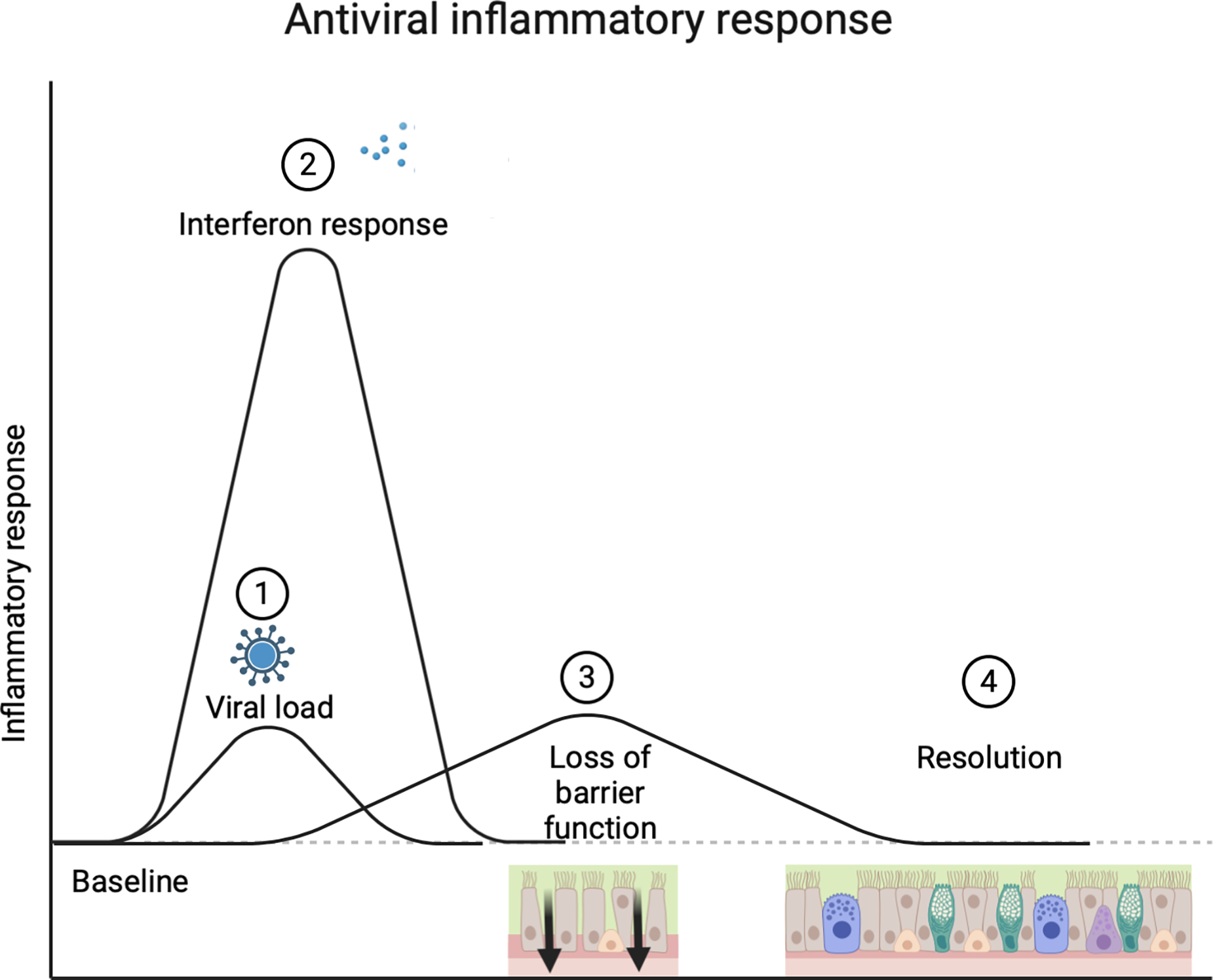
Figure 1. Antiviral immune response.
Virus binds to sinonasal airway epithelial cells (1), and replicates within the cell. This triggers antiviral interferon responses (2) and cytokine induction that rapidly increases and can transiently induce loss of epithelial barrier function to allow for cell-mediated viral killing (3). These host immune responses than resolve to a homeostatic baseline (4).
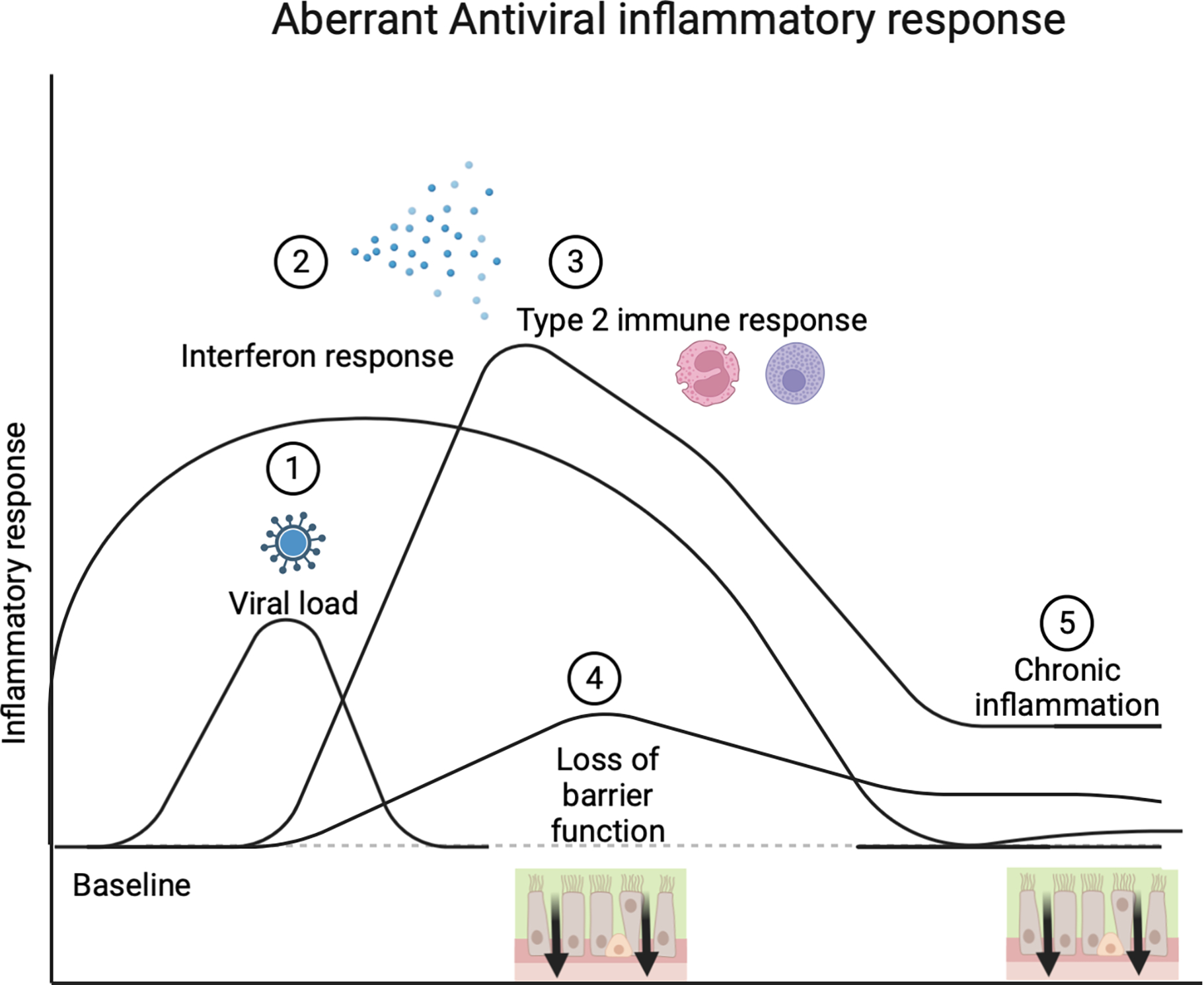
Figure 2. Aberrant antiviral inflammatory response.
Viruses bind to sinonasal airway epithelial cells and can have an increased viral load either due to increased virulence of the virus subtype or enhanced viral binding (1). A delayed interferon response (2) results in increased viral replication and a prolonged interferon response can induce type 2 immune responses characterized by eosinophil and basophil activation (3). The release of interferon and type 2 mediated cytokines can increase epithelial barrier permeability and damage, resulting in a prolonged loss of barrier function (4) and chronic inflammation (5).
The increasing knowledge of the molecular pathophysiology of virus mediated CRS also brings the potential of novel therapeutics. Although honey tea and chicken soup might continue to be the main treatment for respiratory viral infections, it is increasingly clear that more targeted approaches might be beneficial in those at risk for more severe viral disease. The SARS-CoV2 epidemic accelerated approaches for mRNA-based vaccines that would prime the immune system to create antibodies to virus binding and these have been very successful in reducing the morbidity and mortality associated with COVID-19. Similar approaches have been used in creating the first RSV vaccine, a breakthrough that was recently FDA approved for clinical use. Biologic therapies targeting immune pathways are also available, and it will be interesting to see if biologics used to treat CRS with nasal polyps, a type 2 immune mediated disease, also reduces viral exacerbations. Further understanding of the virus-mediated immune responses in CRS might bring us closer to the goal of slowing the progression of this common chronic condition.
Acknowledgements:
Figures made in biorender.com
Funding declarations:
This work was supported by the NIH (R01 AI 146131).
Abbreviations
- (URIs)
Upper respiratory infections
- (CRS)
chronic rhinosinusitis
- (RV)
rhinovirus
- (RSV)
respiratory syncytial virus
- (hCoV)
human coronavirus
- (HA)
hemagglutin
- (NA)
neuraminidase
- (H1N1)
influenza A
- (MERS-CoV)
beta coronavirus responsible for Middle East Respiratory Syndrome
- (SARS-CoV)
beta-coronavirus responsible for severe acute respiratory syndrome
- (SARS-CoV2)
beta coronavirus responsible for COVID-19
- (ICAM-1)
intracellular adhesion molecule 1
- (LDLR)
low density lipoprotein receptor
- (SNP)
single nucleotide polymorphism
- (CDHR3)
cadherin related family member 3 gene
- (HSPGs)
heparin sulfate proteoglycans
- (NCL)
nucleolin
- (IGF1R)
insulin-like growth factor-1 receptor
- (EGFR)
epidermal growth factor
- (APN)
human aminopeptidase N
- (ACE2)
angiotensin-converting enzyme 2
- (TLR)
toll-like receptors
- (RIG-I)
retinoic acid-inducible gene I
- (MDA-5)
melanoma differentiation-associated gene 5
- (TRIF) TIR
domain-containing adapter-inducing interferon-β
- (NFK-κB)
nuclear factor-κB
- (IRF)
interferon regulator factor
- (IFN-I)
Type 1 interferons
- (IFN-γ)
Type 2 interferons
- (IFNλ and IL-10)
Type 3 interferons
- (ALI)
air-liquid-interface
- (TJ)
tight junction
- (ZO-1)
zona-occludens 1
- (AJ)
adherens junctions
- (TER)
transepithelial resistance
Footnotes
Conflict of interest:
Chang EH: Grant funding from NIH and Sanofi-Regeneron, advisor for Sanofi-Regeneron. The rest of the authors declare that they have no relevant conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Fendrick AM, Monto AS, Nightengale B, Sarnes M. The Economic Burden of Non–Influenza-Related Viral Respiratory Tract Infection in the United States. Archives of Internal Medicine 2003; 163:487–94. [DOI] [PubMed] [Google Scholar]
- 2.Mäkelä MJ, Puhakka T, Ruuskanen O, Leinonen M, Saikku P, Kimpimäki M, et al. Viruses and bacteria in the etiology of the common cold. J Clin Microbiol 1998; 36:539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nokso-Koivisto J, Pitkäranta A, Blomqvist S, Jokinen J, Kleemola M, Takala A, et al. Viral Etiology of Frequently Recurring Respiratory Tract Infections in Children. Clinical Infectious Diseases 2002; 35:540–6. [DOI] [PubMed] [Google Scholar]
- 4.Basharat U, Aiche MM, Kim MM, Sohal M, Chang EH. Are rhinoviruses implicated in the pathogenesis of sinusitis and chronic rhinosinusitis exacerbations? A comprehensive review. Int Forum Allergy Rhinol 2019; 9:1159–88. [DOI] [PubMed] [Google Scholar]
- 5.Rosenfeld RM, Piccirillo JF, Chandrasekhar SS, Brook I, Ashok Kumar K, Kramper M, et al. Clinical practice guideline (update): adult sinusitis. Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery 2015; 152:S1–S39. [DOI] [PubMed] [Google Scholar]
- 6.Chang EH, Stern DA, Willis AL, Guerra S, Wright AL, Martinez FD. Early life risk factors for chronic sinusitis: A longitudinal birth cohort study. National burden of antibiotic use for adult rhinosinusitis. 2018; 141:1291–7.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winther B Rhinovirus infections in the upper airway. Proceedings of the American Thoracic Society 2011; 8:79–89. [DOI] [PubMed] [Google Scholar]
- 8.Gern JE. The ABCs of rhinoviruses, wheezing, and asthma. J Virol 2010; 84:7418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willis AL, Calton JB, Calton J, Kim AS, Lee R, Torabzadeh E, et al. RV-C infections result in greater clinical symptoms and epithelial responses compared to RV-A infections in patients with CRS. Allergy 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakagome K, Bochkov YA, Ashraf S, Brockman-Schneider RA, Evans MD, Pasic TR, et al. Effects of rhinovirus species on viral replication and cytokine production. National burden of antibiotic use for adult rhinosinusitis. 2014; 134:332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramadan HH, Farr RW, Wetmore SJ. Adenovirus and respiratory syncytial virus in chronic sinusitis using polymerase chain reaction. Laryngoscope 1997; 107:923–5. [DOI] [PubMed] [Google Scholar]
- 12.Esposito S, Piralla A, Zampiero A, Bianchini S, Di Pietro G, Scala A, et al. Characteristics and Their Clinical Relevance of Respiratory Syncytial Virus Types and Genotypes Circulating in Northern Italy in Five Consecutive Winter Seasons. PLoS One 2015; 10:e0129369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laham FR, Mansbach JM, Piedra PA, Hasegawa K, Sullivan AF, Espinola JA, et al. Clinical Profiles of Respiratory Syncytial Virus Subtypes A AND B Among Children Hospitalized with Bronchiolitis. Pediatr Infect Dis J 2017; 36:808–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walsh EE, McConnochie KM, Long CE, Hall CB. Severity of respiratory syncytial virus infection is related to virus strain. J Infect Dis 1997; 175:814–20. [DOI] [PubMed] [Google Scholar]
- 15.Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. The genomic and epidemiological dynamics of human influenza A virus. Nature 2008; 453:615–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watts G A/H1N1 influenza virus: the basics. Bmj 2009; 339:b3046. [DOI] [PubMed] [Google Scholar]
- 17.Emergence of a Novel Swine-Origin Influenza A (H1N1) Virus in Humans. New England Journal of Medicine 2009; 360:2605–15. [DOI] [PubMed] [Google Scholar]
- 18.Domínguez-Cherit G, Ñamendys-Silva SA, de la Torre A, Macias AE, Cordova-Villalobos JA. H1N1 Influenza Pandemic of 2009 Compared With Other Influenza Pandemics: Epidemiology, Diagnosis, Management, Pulmonary Complications, and Outcomes. Current Infectious Disease Reports 2010; 12:204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu DX, Liang JQ, Fung TS. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encyclopedia of Virology 2021:428–40. [Google Scholar]
- 20.Choi H, Fleming NW, Serikov VB. Contact Activation via ICAM-1 Induces Changes in Airway Epithelial Permeability in vitro. Immunological Investigations 2007; 36:59–72. [DOI] [PubMed] [Google Scholar]
- 21.Staunton DE, Merluzzi VJ, Rothlein R, Barton R, Marlin SD, Springer TA. A cell adhesion molecule, ICAM-1, is the major surface receptor for rhinoviruses. Cell 1989; 56:849–53. [DOI] [PubMed] [Google Scholar]
- 22.Tomassini JE, Graham D, DeWitt CM, Lineberger DW, Rodkey JA, Colonno RJ. cDNA cloning reveals that the major group rhinovirus receptor on HeLa cells is intercellular adhesion molecule 1. Proc Natl Acad Sci U S A 1989; 86:4907–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofer F, Gruenberger M, Kowalski H, Machat H, Huettinger M, Kuechler E, et al. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc Natl Acad Sci U S A 1994; 91:1839–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonnelykke K, Sleiman P, Nielsen K, Kreiner-Moller E, Mercader JM, Belgrave D, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet 2014; 46:51–5. [DOI] [PubMed] [Google Scholar]
- 25.Bochkov YA, Watters K, Ashraf S, Griggs TF, Devries MK, Jackson DJ, et al. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc Natl Acad Sci U S A 2015; 112:5485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basnet S, Bochkov YA, Brockman-Schneider RA, Kuipers I, Aesif SW, Jackson DJ, et al. CDHR3 Asthma-Risk Genotype Affects Susceptibility of Airway Epithelium to Rhinovirus C Infections. American journal of respiratory cell and molecular biology 2019:rcmb.2018–0220OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang EH, Willis AL, McCrary HC, Noutsios GT, Le CH, Chiu AG, et al. Association between the CDHR3 rs6967330 risk allele and chronic rhinosinusitis. J Allergy Clin Immunol 2017; 139:1990–2.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chirkova T, Lin S, Oomens AGP, Gaston KA, Boyoglu-Barnum S, Meng J, et al. CX3CR1 is an important surface molecule for respiratory syncytial virus infection in human airway epithelial cells. J Gen Virol 2015; 96:2543–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tripp RA, Jones LP, Haynes LM, Zheng H, Murphy PM, Anderson LJ. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nature Immunology 2001; 2:732–8. [DOI] [PubMed] [Google Scholar]
- 30.Feng Z, Xu L, Xie Z. Receptors for Respiratory Syncytial Virus Infection and Host Factors Regulating the Life Cycle of Respiratory Syncytial Virus. Front Cell Infect Microbiol 2022; 12:858629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janssen R, Bont L, Siezen CL, Hodemaekers HM, Ermers MJ, Doornbos G, et al. Genetic susceptibility to respiratory syncytial virus bronchiolitis is predominantly associated with innate immune genes. J Infect Dis 2007; 196:826–34. [DOI] [PubMed] [Google Scholar]
- 32.Connor RJ, Kawaoka Y, Webster RG, Paulson JC. Receptor specificity in human, avian, and equine H2 and H3 influenza virus isolates. Virology 1994; 205:17–23. [DOI] [PubMed] [Google Scholar]
- 33.Chatzopoulou F, Gioula G, Kioumis I, Chatzidimitriou D, Exindari M. Identification of complement-related host genetic risk factors associated with influenza A(H1N1)pdm09 outcome: challenges ahead. Med Microbiol Immunol 2019; 208:631–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT, et al. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992; 357:420–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med 2020; 26:681–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol 2022; 23:3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A 2020; 117:11727–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020; 181:1016–35.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang EH, Willis AL, Romanoski CE, Cusanovich DA, Pouladi N, Li J, et al. Rhinovirus Infections in Individuals with Asthma Increase ACE2 Expression and Cytokine Pathways Implicated in COVID-19. Am J Respir Crit Care Med 2020; 202:753–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawai T, Akira S. TLR signaling. Cell Death Differ 2006; 13:816–25. [DOI] [PubMed] [Google Scholar]
- 41.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol 2006; 7:131–7. [DOI] [PubMed] [Google Scholar]
- 42.García-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in détente. Science 2006; 312:879–82. [DOI] [PubMed] [Google Scholar]
- 43.Katze MG, He Y, Gale M. Viruses and interferon: a fight for supremacy. Nature Reviews Immunology 2002; 2:675–87. [DOI] [PubMed] [Google Scholar]
- 44.Makris S, Paulsen M, Johansson C. Type I Interferons as Regulators of Lung Inflammation. Front Immunol 2017; 8:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan KS, Ong HH, Yan Y, Liu J, Li C, Ong YK, et al. In Vitro Model of Fully Differentiated Human Nasal Epithelial Cells Infected With Rhinovirus Reveals Epithelium-Initiated Immune Responses. J Infect Dis 2018; 217:906–15. [DOI] [PubMed] [Google Scholar]
- 46.Kim JH, Kim YS, Cho GS, Kim NH, Gong CH, Lee BJ, et al. Human Rhinovirus-induced Proinflammatory Cytokine and Interferon-β Responses in Nasal Epithelial Cells From Chronic Rhinosinusitis Patients. Allergy Asthma Immunol Res 2015; 7:489–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee SH, Han MS, Lee TH, Lee DB, Park JH, Lee SH, et al. Rhinovirus-induced anti-viral interferon secretion is not deficient and not delayed in sinonasal epithelial cells of patients with chronic rhinosinusitis with nasal polyp. Front Immunol 2022; 13:1025796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hwang JW, Lee KJ, Choi IH, Han HM, Kim TH, Lee SH. Decreased expression of type I (IFN-β) and type III (IFN-λ) interferons and interferon-stimulated genes in patients with chronic rhinosinusitis with and without nasal polyps. J Allergy Clin Immunol 2019; 144:1551–65.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altman MC, Gill MA, Whalen E, Babineau DC, Shao B, Liu AH, et al. Transcriptome networks identify mechanisms of viral and nonviral asthma exacerbations in children. Nat Immunol 2019; 20:637–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Divekar RD, Samant S, Rank MA, Hagan J, Lal D, O'Brien EK et al. Immunological profiling in chronic rhinosinusitis with nasal polyps reveals distinct VEGF and GM-CSF signatures during symptomatic exacerbations. Clin Exp Allergy 2015; 45:767–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rank MA, Hagan JB, Samant SA, Kita H. A proposed model to study immunologic changes during chronic rhinosinusitis exacerbations: data from a pilot study. American journal of rhinology & allergy 2013; 27:98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aberle JH, Aberle SW, Rebhandl W, Pracher E, Kundi M, Popow-Kraupp T. Decreased interferon-gamma response in respiratory syncytial virus compared to other respiratory viral infections in infants. Clin Exp Immunol 2004; 137:146–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hijano DR, Vu LD, Kauvar LM, Tripp RA, Polack FP, Cormier SA. Role of Type I Interferon (IFN) in the Respiratory Syncytial Virus (RSV) Immune Response and Disease Severity. Front Immunol 2019; 10:566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klinkhammer J, Schnepf D, Ye L, Schwaderlapp M, Gad HH, Hartmann R, et al. IFN-lambda prevents influenza virus spread from the upper airways to the lungs and limits virus transmission. Elife 2018; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicol MQ, Campbell GM, Shaw DJ, Dransfield I, Ligertwood Y, Beard PM, et al. Lack of IFNgamma signaling attenuates spread of influenza A virus in vivo and leads to reduced pathogenesis. Virology 2019; 526:155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Califano D, Furuya Y, Roberts S, Avram D, McKenzie ANJ, Metzger DW. IFN-gamma increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol 2018; 11:209–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baker JR, Mahdi M, Nicolau DV, Jr., Ramakrishnan S, Barnes PJ, Simpson JL, et al. Early Th2 inflammation in the upper respiratory mucosa as a predictor of severe COVID-19 and modulation by early treatment with inhaled corticosteroids: a mechanistic analysis. Lancet Respir Med 2022; 10:545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020; 369:718–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020; 181:1036–45 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Z, Peng H, Lai J, Jiang L, Wang L, Jin S, et al. Differential susceptibility to SARS-CoV-2 in the normal nasal mucosa and in chronic sinusitis. Eur J Immunol 2022; 52:1308–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun 2020; 11:3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, et al. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep 2020; 33:108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Calabrese LH, Lenfant T, Calabrese C. Interferon therapy for COVID-19 and emerging infections: Prospects and concerns. Cleve Clin J Med 2020. [DOI] [PubMed] [Google Scholar]
- 64.Reis G, Moreira Silva EAS, Medeiros Silva DC, Thabane L, Campos VHS, Ferreira TS, et al. Early Treatment with Pegylated Interferon Lambda for Covid-19. N Engl J Med 2023; 388:518–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jackson DJ, Makrinioti H, Rana BMJ, Shamji BWH, Trujillo-Torralbo M-B, Footitt J, et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am J Respir Crit Care Med 2014; 190:1373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khoo SK, Read J, Franks K, Zhang G, Bizzintino J, Coleman L, et al. Upper Airway Cell Transcriptomics Identify a Major New Immunological Phenotype with Strong Clinical Correlates in Young Children with Acute Wheezing. J Immunol 2019; 202:1845–58. [DOI] [PubMed] [Google Scholar]
- 67.Bosco A ES, Panyala S, Martinez FD. Interferon regulatory factor 7 is a major hub connecting interferon-mediated responses in virus-induced asthma exacerbations in vivo. J Allergy Clin Immunol 2012; 129(1):88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bosco A, Wiehler S, Proud D. Interferon regulatory factor 7 regulates airway epithelial cell responses to human rhinovirus infection. BMC Genomics 2016; 17:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Teach SJ, Gill MA, Togias A, Sorkness CA, Arbes SJ Jr., Calatroni A, et al. Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. J Allergy Clin Immunol 2015; 136:1476–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang L, Wan Y, Ma L, Xu K, Cheng B. Inhibition of NF-kappaB/IL-33/ST2 Axis Ameliorates Acute Bronchiolitis Induced by Respiratory Syncytial Virus. J Immunol Res 2021; 2021:6625551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Warren KJ, Poole JA, Sweeter JM, DeVasure JM, Dickinson JD, Peebles RS Jr., et al. Neutralization of IL-33 modifies the type 2 and type 3 inflammatory signature of viral induced asthma exacerbation. Respir Res 2021; 22:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nikonova A, Shilovskiy I, Galitskaya M, Sokolova A, Sundukova M, Dmitrieva-Posocco O, et al. Respiratory syncytial virus upregulates IL-33 expression in mouse model of virus-induced inflammation exacerbation in OVA-sensitized mice and in asthmatic subjects. Cytokine 2021; 138:155349. [DOI] [PubMed] [Google Scholar]
- 73.Papi A, Ison MG, Langley JM, Lee DG, Leroux-Roels I, Martinon-Torres F, et al. Respiratory Syncytial Virus Prefusion F Protein Vaccine in Older Adults. N Engl J Med 2023; 388:595–608. [DOI] [PubMed] [Google Scholar]
- 74.Sajuthi SP, DeFord P, Li Y, Jackson ND, Montgomery MT, Everman JL, et al. Type 2 and interferon inflammation regulate SARS-CoV-2 entry factor expression in the airway epithelium. Nat Commun 2020; 11:5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kimura H, Francisco D, Conway M, Martinez FD, Vercelli D, Polverino F, et al. Type 2 inflammation modulates ACE2 and TMPRSS2 in airway epithelial cells. J Allergy Clin Immunol 2020; 146:80–8 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jackson DJ, Bacharier KB, Kattan M, O’Connor GT, Wood RA, et al. Association of respiratory allergy, asthma, and expression of the SARS-CoV-2 receptor ACE2. J Allergy Clin Immunol 2020; 146:203–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ziegler CGK, Nyquisst SK, Mbano IM, Miao VN, C.N. Tzouanas, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and enriched in specific cell subsets across tissues. Cell 2020; 181:1016–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang M, Fang G, Luan G, Huang Y, Akdis CA, et al. Distinct expression of SARS-CoV-2 receptor ACE2 correlates with endotypes of chronic rhinosinusitis with nasal polyps. Allergy 2021; 76:789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee SW, Kim SY, Moon SY, Yang JM, Ha EK, Jee HM, et al. Estimating COVID-19 Infection and Severity Risks in Patients with Chronic Rhinosinusitis: A Korean Nationwide Cohort Study. J Allergy Clin Immunol Pract 2021; 9:2262–71.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Griggs TF, Bochkov YA, Basnet S, Pasic TR, Brockman-Schneider RA, Palmenberg AC, et al. Rhinovirus C targets ciliated airway epithelial cells. Respir Res 2017; 18:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jia HP, Look DC, Shi L, Hickey M, Pewe L, Netland J, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol 2005; 79:14614–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robinot R, Hubert M, de Melo GD, Lazarini F, Bruel T, Smith N, et al. SARS-CoV-2 infection induces the dedifferentiation of multiciliated cells and impairs mucociliary clearance. Nature Communications 2021; 12:4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeong KI, Piepenhagen PA, Kishko M, DiNapoli JM, Groppo RP, Zhang L, et al. CX3CR1 Is Expressed in Differentiated Human Ciliated Airway Cells and Co-Localizes with Respiratory Syncytial Virus on Cilia in a G Protein-Dependent Manner. PLoS One 2015; 10:e0130517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barbier D, Garcia-Verdugo I, Pothlichet J, Khazen R, Descamps D, Rousseau K, et al. Influenza A induces the major secreted airway mucin MUC5AC in a protease-EGFR-extracellular regulated kinase-Sp1-dependent pathway. Am J Respir Cell Mol Biol 2012; 47:149–57. [DOI] [PubMed] [Google Scholar]
- 85.Du X, Yang Y, Xiao G, Yang M, Yuan L, Qin L, et al. Respiratory syncytial virus infection-induced mucus secretion by down-regulation of miR-34b/c-5p expression in airway epithelial cells. J Cell Mol Med 2020; 24:12694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hewson CA, Haas JJ, Bartlett NW, Message SD, Laza-Stanca V, Kebadze T, et al. Rhinovirus induces MUC5AC in a human infection model and in vitro via NF-κB and EGFR pathways. European Respiratory Journal 2010; 36:1425–35. [DOI] [PubMed] [Google Scholar]
- 87.Lee S, Na HG, Choi YS, Bae CH, Song SY, Kim YD. SARS-CoV-2 Induces Expression of Cytokine and MUC5AC/5B in Human Nasal Epithelial Cell through ACE 2 Receptor. Biomed Res Int 2022; 2022:2743046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rogers GA, Den Beste K, Parkos CA, Nusrat A, Delgaudio JM, Wise SK. Epithelial tight junction alterations in nasal polyposis. Int Forum Allergy Rhinol 2011; 1:50–4. [DOI] [PubMed] [Google Scholar]
- 89.Soyka MB, Wawrzyniak P, Eiwegger T, Holzmann D, Treis A, Wanke K, et al. Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN-γ and IL-4. J Allergy Clin Immunol 2012; 130:1087–96.e10. [DOI] [PubMed] [Google Scholar]
- 90.Pothoven KL, Norton JE, Hulse KE, Suh LA, Carter RG, Rocci E, et al. Oncostatin M promotes mucosal epithelial barrier dysfunction, and its expression is increased in patients with eosinophilic mucosal disease. Journal of Allergy and Clinical Immunology 2015; 136:737–46.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bochkov YA, Gern JE. Rhinoviruses and Their Receptors: Implications for Allergic Disease. Curr Allergy Asthma Rep 2016; 16:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yeo NK, Jang YJ. Rhinovirus infection-induced alteration of tight junction and adherens junction components in human nasal epithelial cells. Laryngoscope 2010; 120:346–52. [DOI] [PubMed] [Google Scholar]
- 93.Looi K, Troy NM, Garratt LW, Iosifidis T, Bosco A, Buckley AG, et al. Effect of human rhinovirus infection on airway epithelium tight junction protein disassembly and transepithelial permeability. Exp Lung Res 2016; 42:380–95. [DOI] [PubMed] [Google Scholar]
- 94.Rezaee F, DeSando SA, Ivanov AI, Chapman TJ, Knowlden SA, Beck LA, et al. Sustained protein kinase D activation mediates respiratory syncytial virus-induced airway barrier disruption. J Virol 2013; 87:11088–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rezaee F, Harford TJ, Linfield DT, Altawallbeh G, Midura RJ, Ivanov AI, et al. cAMP-dependent activation of protein kinase A attenuates respiratory syncytial virus-induced human airway epithelial barrier disruption. PLoS One 2017; 12:e0181876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Smallcombe CC, Linfield DT, Harford TJ, Bokun V, Ivanov AI, Piedimonte G, et al. Disruption of the airway epithelial barrier in a murine model of respiratory syncytial virus infection. Am J Physiol Lung Cell Mol Physiol 2019; 316:L358–l68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pharo EA, Williams SM, Boyd V, Sundaramoorthy V, Durr PA, Baker ML. Host-Pathogen Responses to Pandemic Influenza H1N1pdm09 in a Human Respiratory Airway Model. Viruses 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tian T, Zi X, Peng Y, Wang Z, Hong H, Yan Y, et al. H3N2 influenza virus infection enhances oncostatin M expression in human nasal epithelium. Exp Cell Res 2018; 371:322–9. [DOI] [PubMed] [Google Scholar]