

The risk of progression for Barrett’s esophagus (BE) is estimated to range from 0.12% to 0.5% per year.1 Identification of clinical risk factors such as age, sex, obesity, smoking, presence of hiatal hernia, and length of BE, are insufficient to wholly account for the few individuals who progress from BE to adenocarcinoma.2 To explain some of the unaccounted risk, we hypothesized that a significant fraction of individuals with BE who progress to adenocarcinoma harbor pathogenic germline mutations in cancer predisposing genes.
We examined the prevalence of monoallelic, pathogenic germline mutations associated with moderate to high risk of cancer in 640 study participants with esophageal adenocarcinoma (EAC) enrolled in publicly available genomic cohorts that performed either whole genome sequencing (ICGC-ARGO) or whole-exome sequencing (TCGA Pan-Cancer Cohort, Broad Institute Esophageal Adenocarcinoma Cohort, and Memorial Sloan Kettering Prospective Clinical Cohort, Figure 1A).3–6 Pathogenic germline mutations were discovered in 59 out of 640 individuals (9.2%, Figure 1B, Supplementary Table 1). ATM was the most frequently mutated gene, occurring in 10 individuals (1.6%) followed by CHEK2 (1.25%). Five individuals (0.8%) harbored germline mutations in TP53. Two individuals (0.3%) harbored distinct, splice-donor mutations in CDH1 at intron 10. Despite this prevalence, somatic coding mutations that represent likely loss-of-heterozygosity events, were only present in 3/60 tumors (5.0%, 1 BRCA2- and 2 TP53 mutation carriers).
Figure 1: Germline Mutational Landscape Across Esophageal Adenocarcinoma.
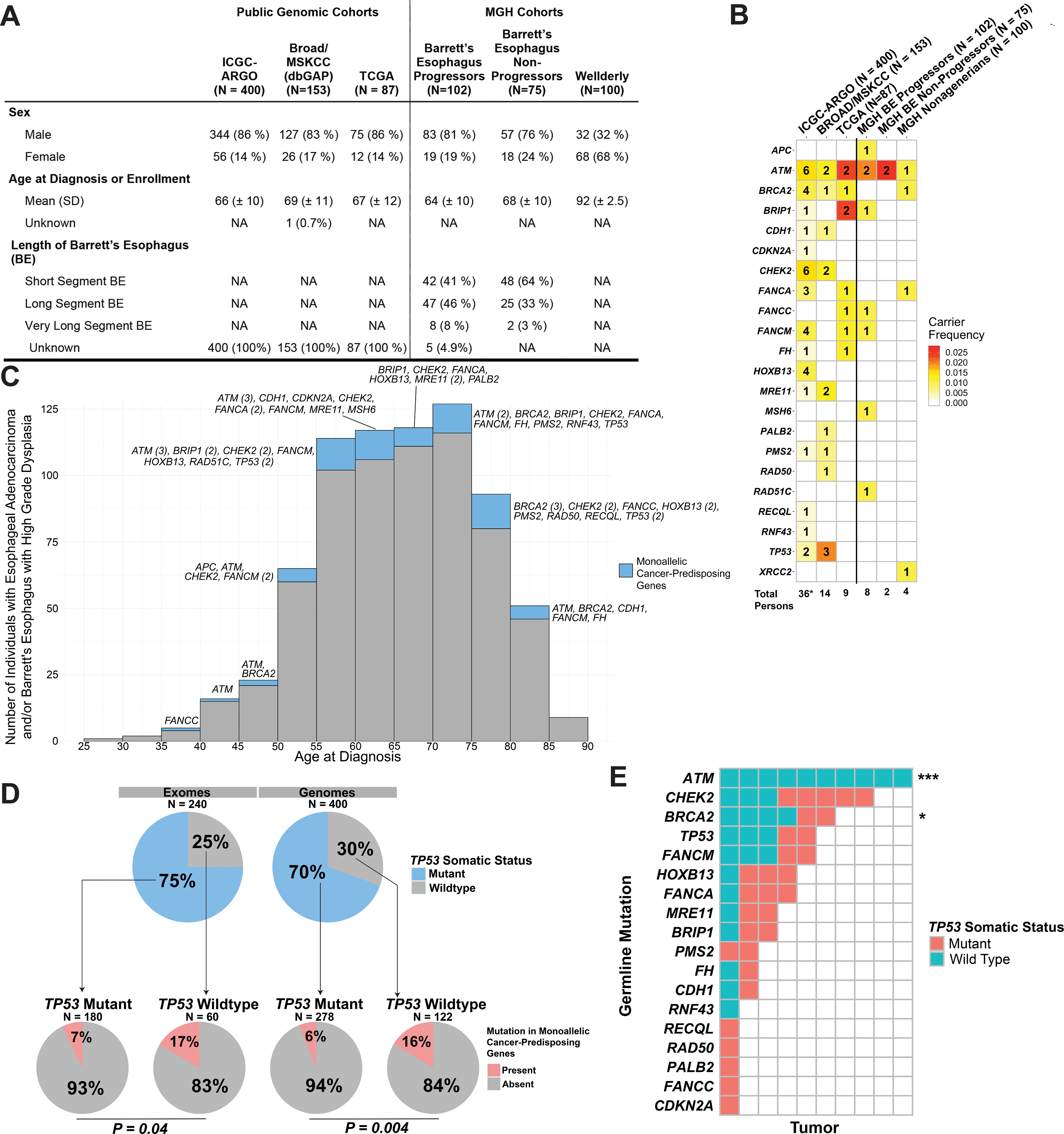
(A) Clinical characteristics of study participants from public genomic and MGH cohorts. ICGC-ARGO refers to International Cancer Genome Consortium Project Accelerating Research in Genomic Oncology; Broad/MSKCC Cohort refers to the pooled public exomes of esophageal adenocarcinoma available on dbGAP; TCGA refers to The Cancer Genome Atlas; Wellderly refers to healthy nonagenarians without history of gastrointestinal neoplasia. (B) Number of pathogenic mutations itemized by cancer-predisposing genes across multiple cohorts. Color-coding of entries demonstrates carrier-frequency in their respective cohorts. (C) Histogram showing the age at diagnosis of Barrett’s esophagus with high-grade dysplasia or esophageal adenocarcinoma. Mutation carriers and non-carriers are color-coded by blue and gray, respectively. (D) Correlation of germline pathogenic mutations with somatic TP53 status in tumors, segregated by exomes and genomes. (E) Correlations between individual genes mutated in the germline and somatic TP53 status. *** designates P < 0.001 and * designates P = 0.06.
As validation, we performed germline WES on prospective cohorts at Massachusetts General Hospital that encompass BE progressors who developed high-grade dysplasia or intramucosal carcinoma (102 individuals), BE without progression to dysplasia over 10+ years (75 individuals), and healthy nonagenarians without any prior known history of gastrointestinal neoplasia (100 individuals). Again, germline ATM mutations were the most frequent pathogenic alteration, occurring in 2% and 2.7% of progressors and non-progressors (short-segment BE), respectively. Despite the lack of enrichment of ATM carriers among progressors, immunohistochemistry demonstrated loss of ATM staining among progressors and retained expression among non-progressors, implying epigenetic mechanisms for LOH (Supplementary Figure 1A).
Across all HGD/EAC cohorts, the prevalence of germline mutations in genes associated with monoallelic cancer predisposition within the Fanconi Anemia pathway (BRCA2, PALB2, BRIP1, RAD51C, FANCA, FANCC, FANCM) demonstrated enrichment over the carrier rate for all Fanconi Anemia genes in the general population (overall 2.3% vs. 0.6%). The age at diagnosis of those with high-grade dysplasia or adenocarcinoma did not differ between those with or without any germline mutations (Figure 1C).
Given this enrichment of pathogenic germline mutations in progressors, we examined if such germline alterations could influence the somatic mutanome. We examined the association of germline mutations with the development of pathogenic somatic TP53 alterations, since such alterations have been associated with BE progression and genome doubling events (Figure 1D).7 Pathogenic, somatic TP53 mutations were detected among 75% of tumor exomes and 70% of tumor genomes. When stratified by somatic TP53 mutant status, pathogenic germline mutations were present in 16.7% of cancer exomes with wild-type TP53 versus 7.2% withTP53 mutations (OR 2.6, 95% C.I. 0.9–6.8, P = 0.04, Fisher’s exact test). Among cancer genomes, germline mutations were present in 15.6% of cases with wild-type TP53 versus 6.1% of TP53 mutants (OR 2.8, 95% C.I. 1.3–6.2, P = 0.004 Fisher’s exact test).
To examine if the overall enrichment of germline mutations among TP53 wildtype tumors is driven by select genes, we stratified somatic TP53 mutant status by each cancer-predisposing gene (Figure 1E). ATM germline mutations demonstrated 100% mutual exclusivity with pathogenic somatic TP53 mutations (OR 0, 95% CI 0–0.2, P = 2.9 × 10−6, Fisher’s exact test). We validated this mutual exclusivity with an independent cohort of 475 publicly available and non-redundant gastroesophageal adenocarcinomas previously sequenced on the MSK-IMPACT platform, with 7/7 ATM carriers harboring wild-type TP53 (Supplementary Figure 1B). Exclusion of ATM carriers still demonstrated a persistent enrichment of germline mutations among TP53 wildtype tumors, occurring in 10.1% and 12.1% of exomes and genomes, respectively.
Pathogenic germline BRCA2 mutations also demonstrated a trend toward mutual exclusivity with TP53 mutation (OR 0.2, 95% C.I. 0.2–1.4, P = 0.06, Fisher’s exact test). Given the strong association of homologous recombination deficiency with somatic TP53 mutations, we examined HRD status from tumor genomes utilizing the HRDetect algorithm. We observed HRD present in only 14/400 (3.5%) of tumor whole genomes, with only 1/4 BRCA2 carriers demonstrating HRD (Supplementary Figure 1C). Among tumor exomes with either BRCA2 or PALB2 germline alterations, no samples demonstrated dominance by the single base substitution signature associated with HRD (Sig3, Supplementary Figure 1D).
Among 742 individuals with BE with HGD or EAC, we identified pathogenic germline mutations in monoallelic, cancer-predisposing genes among 9.0% of participants, compared to 2.7% of non-progressors. This overall enrichment suggests that these mutations facilitate the progression of Barrett’s esophagus to adenocarcinoma. The ages of onset for those with germline mutations did not cluster among earlier-onset cases but occurred throughout the age spectrum, implying that these inherited mutations may require the development of BE and additional environmental factors as prerequisites to promote esophageal carcinogenesis.
Somatic TP53 alterations have been identified as a key driver in the progression of nondysplastic Barrett’s esophagus to dysplasia, functioning as a checkpoint for genome doubling events and chromosomal instability.7 Validating its role as a key driver of progression, we did observe an overrepresentation of germline TP53 mutations (0.7% among progressors). However, 25–30% of esophageal adenocarcinomas lacked somatic alterations in TP53. We discovered that such TP53 wild-type tumors were significantly enriched for pathogenic germline mutations compared to TP53-mutant cancers (overall 15.9% vs. 6.6%, OR 2.7, 95% CI 1.5–4.8, P = 4.2 × 10−4, Fisher’s exact test). This enrichment implies an early and causative role for even heterozygous germline mutations in BE progression since they can obviate the selection pressures for the acquisition of somatic TP53 coding mutations. Multiple studies have demonstrated that heterozygosity of cancer predisposing genes can promote genomic instability.8,9 Genome-wide association studies have quantified moderate effects associated with rare, heterozygous germline mutations.
Genetic testing has been recommended for all individuals diagnosed with pancreatic adenocarcinoma, where the prevalence of germline mutations is 7–10% and second hit mutations are uncommon.10 Given the similar prevalence in EAC, universal genetic testing should be considered.
Supplementary Material
Acknowledgements:
Members of the MGH-MIT Gastrointestinal Cohorts Working Group are:
Alvin Jeon, M.D.1, Eugeniu Stratulat, M.D.1, Rachid Zagani, Ph.D.1, Martin S. Taylor, M.D., Ph.D.2, Steffen Rickelt, Ph.D.3, Dmitriy Kedrin, M.D.,Ph.D.1, Anupam Batra, M.D.4,5,6, Ardeshir Hashmi, M.D.4,7, Norman S. Nishioka, M.D.1, Chin Hur, M.D., M.P.H.1,8,9, Lipika Goyal, M.D.10, Fateh Bazerbachi, M.D.1,11, Lawrence Zukerberg, M.D.2#
1. Division of Gastroenterology, Massachusetts General Hospital and Harvard Medical School, Boston, MA
2. Department of Pathology, Massachusetts General Hospital and Harvard Medical School, Boston, MA
3. David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA
4. Massachusetts General Hospital Division of Senior Health and Harvard Medical School, Boston, MA
5. Department of Oncology, Cambridge Memorial Hospital, Cambridge, Ontario, Canada
6. Department of Oncology, Grand River Regional Cancer Center, Kitchener, Ontario, Canada
7. Center for Geriatric Medicine, Cleveland Clinic, Cleveland, OH, USA
8. Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center, New York, NY
9. Division of Digestive and Liver Diseases, Columbia University Vagelos School of Medicine, New York, New York
10. Division of Oncology, Massachusetts General Hospital Cancer Center and Harvard Medical School, Boston, MA
11. Interventional Endoscopy Program, CentraCare and St. Cloud Hospital, St. Cloud, MN
12. Department of Pathology, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA
13. Department of Biology, Massachusetts Institute of Technology, Cambridge, MA
14. Clinical and Translational Epidemiology Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA
Grant Support:
This work was supported by grants from the Elsa U. Pardee Foundation (Manish Gala), American College of Gastroenterology Junior Faculty Career Development Award (Manish Gala), and the NIDDK (K23DK103119)
Abbreviations:
- BE
Barrett’s esophagus
- HGD
high-grade dysplasia
- HRD
homolgous recombination deficient
- HRR
homologous recombination repair
- MGH
Massachusetts General Hospital
Footnotes
Conflict of Interest Statement: The authors have no relevant conflicts to disclose.
Data Transparency Statement: Genomic data from OCCAMS-UK is available from ICGC-ARGO (https://www.icgc-argo.org) and dbGAP (phs000178.v10, phs000598.v2 and phs001783.v1). Sequencing results from MGH Cohorts will be provided with reasonable requests and data protections.
References.
- 1.Hvid-Jensen F, et al. N Engl J Med 2011;365:1375–83. [DOI] [PubMed] [Google Scholar]
- 2.Parasa S, et al. Gastroenterology 2018;154:1282–1289 e2. [DOI] [PubMed] [Google Scholar]
- 3.Fitzgerald R, et al. OCCAMS-UK. ICGC-ARGO. 2022. https://platform.icgc-argo.org/
- 4.The Cancer Genome Atlas Network. Esophageal Carcinoma. dbGAP. 2022. phs000178.v10. [Google Scholar]
- 5.Bass A, et al. Exome Sequencing of Esophageal Adenocarcinoma. dbGAP. 2021. phs000598.v2. [Google Scholar]
- 6.Solit D, et al. Exome recapture and sequencing of prospectively characterized clinical specimens from cancer patients. dbGAP. 2022. phs001783.v1 [Google Scholar]
- 7.Stachler MD, Taylor-Weiner A, et al. Nat Genet 2015;47:1047–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karaayvaz-Yildirim M, et al. Sci Adv 2020;6:eaay2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oliveira C, et al. Gastroenterology 2009;136:2137–48. [DOI] [PubMed] [Google Scholar]
- 10.Yurgelun MB, Chittenden AB, Morales-Oyarvide V, et al. Genet Med 2019;21:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.