

ABSTRACT:
Methylation of DNA plays a key role in diverse biological processes spanning from bacteria to mammals. DNA methyltransferases (MTases) typically employ S-adenosyl-l-methionine (SAM) as a critical cosubstrate and the relevant methyl donor for modification of the C5 position of cytosine. Recently, work on the CpG-specific bacterial MTase, M.MpeI, has shown that a single N374K point mutation can confer the enzyme with the neomorphic ability to use the sparse, naturally occurring metabolite carboxy-S-adenosyl-l-methionine (CxSAM) in order to generate the unnatural DNA modification, 5-carboxymethylcytosine (5cxmC). Here, we aimed to investigate the mechanistic basis for this DNA carboxymethyltransferase (CxMTase) activity by employing a combination of computational modeling and in vitro characterization. Modeling of substrate interactions with the enzyme variant allowed us to identify a favorable salt bridge between CxSAM and N374K that helps to rationalize selectivity of the CxMTase. Unexpectedly, we also discovered a potential role for a key active site E45 residue that makes a bidentate interaction with the ribosyl sugar of CxSAM, located on the opposite face of the CxMTase active site. Prompted by these modeling results, we further explored the space-opening E45D mutation and found that the E45D/N374K double mutant in fact inverts selectivity, preferring CxSAM over SAM in biochemical assays. These findings provide new insight into CxMTase active site architecture and may offer broader utility given the numerous opportunities offered by using SAM analogs for selective molecular labeling in concert with nucleic acid or even protein-modifying MTases.
GRAPHICAL ABSTRACT:
INTRODUCTION
Chemical modifications to DNA encode an additional layer of information above the primary sequence.1 One of the most widely conserved modifications is DNA methylation, which plays a critical role in establishing cellular identity across species.2 Transfer of a methyl group onto DNA is catalyzed by DNA methyltransferases (MTases).3 In prokaryotes, DNA methylation is most associated with its role as a rudimentary immune system allowing bacteria to define “self” identity.1 In bacterial species, DNA MTases target either adenine (N6) or cytosine (N4 or C5) for methylation.4 In many cases, these MTases have coevolved with a partner restriction enzyme that shares the same sequence preference, allowing for targeted degradation of unmarked foreign DNA including bacteriophages.5 In higher organisms, DNA methylation has taken on new roles in establishing cellular identity. 5-Methylcytosine (5mC), in particular, serves as an epigenetic mark with regulatory roles in gene expression.6,7 5mC is critical to various processes, including embryonic development, genomic imprinting, X-chromosome inactivation, and gene silencing. Alterations in methylation status are also prominently associated with oncogenesis.8
DNA MTases can be identified, in part, by the presence of a Rossmann fold, one of the most omnipresent and functionally diverse protein folds.9–11 Rossmann folds bind a variety of cofactors, but specifically bind the substrate methyl donor, S-adenosyl-l-methionine (SAM), in DNA MTases.12,13 Given the power of selectively labeling target molecules, there has been widespread interest in the use of SAM analogs, rather than SAM itself, in concert with natural or engineered MTase variants.14,15 For example, sequence-specific methyltransferase-induced labeling of DNA (SMILing DNA) employs the DNA MTase, M.TaqI, to transfer an aziridine group from N-adenosylaziridine onto N6 of adenine within the 5′-TCGA-3′ recognition site of M.TaqI.16 With cytosine modification, bacterial enzymes that can modify sequences containing CpG dinucleotide motifs have been a particular area of focus, given the importance of CpGs to the mammalian epigenome. For example, an engineered CpG-specific MTase M.SssI (e.M.SssI) has an enlarged SAM binding site that permits tagging of unmodified CpGs at C5 of cytosine with a biotinylated analog.17 This biotin handle can then allow for DNA enrichment to permit profiling of the DNA “unmethylome”.17 With M.HhaI (a CCGG-specific MTase), S-adenosyl-l-homocysteine (SAH), in concert with small aldehydes, can be used to convert cytosine into 5-hydroxymethylcytosine or related bases.18
More recently, we evaluated the substrate specificity of the bacterial CpG MTase from Mycoplasma penetrans, M.MpeI.19 In exploring the SAM binding site, we performed saturation mutagenesis to a critical active site residue, N374, and introduced the expression plasmid into E. coli.20 We unexpectedly observed the formation of a novel unnatural base in DNA, 5-carboxymethylcytosine (5cxmC) by the N374K mutant (Figure 1). We further demonstrated 5cxmC results from the mutant taking on the neomorphic ability to use the sparse natural bacterial metabolite carboxy-S-adenosyl-l-methionine (CxSAM).21 This novel CpG DNA carboxymethyltransferase (CxMTase) has opened new biotechnological applications.22–24 Direct methylation sequencing (DM-Seq) uses a combination of the CxMTase with a DNA deaminase that can discriminate between cytosine modification states in the first all-enzymatic, base-resolution sequencing approach for localizing only 5mC without confounding by 5hmC.24
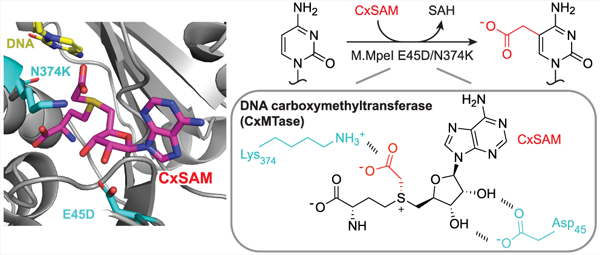
Figure 1.

Activities of M.MpeI Variants. (a) Left: M.MpeI canonically acts as an MTase, but an N374K mutation can confer CxMTase activity when CxSAM is present. Right: Previously established activities of M.MpeI WT and M.MpeI N374K. (b) Left: Structure of wild-type CpG-specific DNA methyltransferase M.MpeI (PDB 4DKJ) shown with 5-fluorocytosine containing DNA and S-adenosylhomocysteine (SAH) in the active site (zoomed image at left). The N374 and E45 residues are on different faces of the SAM binding site. Right: the chemical structures of SAM and CxSAM are shown.
Given the broad utility of engineered MTases and SAM analogs, we recognized that a deeper understanding of how the N374K variant selects for the CxSAM cosubstrate could offer generalizable lessons or aid in the development of improved variants. Here, we employ computational modeling to elucidate the principles that govern CxSAM selectivity by M.MpeI N374K. While helping to rationalize the observed neomorphic activity, the modeling also revealed other residues in the conserved Rossmann fold that could contribute to discrimination between SAM and CxSAM. Building on these insights, we show that further engineering of the CxMTase enhances selectivity for CxSAM over SAM. Our results highlight both the complexity of SAM analogue recognition in the MTase active site and added opportunities for combined modeling and protein engineering approaches in the development of MTase variants in support of novel DNA or protein-labeling biotechnologies.
RESULTS AND DISCUSSION
Ligands Do Not Impact System Dynamic Motions.
To first explore the amenability of M.MpeI systems to modeling, we generated WT and N374K model systems with either SAM or CxSAM and evaluated if the overall structure or dynamic motion of the protein appeared stable. Multiple metrics of dynamic motion root-mean-squared fluctuation (RMSF), normal modes, motion cross-correlation, and principal component analysis were compared across all variant–ligand combinations to ensure protein stability. In all cases, the root-mean-squared deviation (RMSD) was constant over the span of the production time, indicating that the systems were stable with respect to the crystal structure and did not undergo any large conformational changes (Figure S1). RMSF values of all systems were consistent across multiple replicates of all variants and ligands (Figure S2). The normal modes, while different in magnitude between replicate trajectories, maintained similar overall motion profiles, indicating that the essential dynamics remained relatively unchanged across all systems (Figures S3 and S4). Correlated motion of residue pairs also showed similar patterns across systems (Figure S5). In our systems, the absence of significant changes in dynamic motion and overall structure suggested that the systems were well positioned for further analysis of specific intermolecular interactions.
N374K Mutation Stabilizes CxSAM.
Given the stability of the models, we next performed energy decomposition analysis (EDA) to determine how N374K or CxSAM impacts protein/cofactor interactions (Figure 2). EDA revealed that wild-type (WT) M.MpeI favors SAM over CxSAM by −133.9 kcal mol−1. The N374K variant also favors SAM over CxSAM, but by only −59.5 kcal mol−1. The N374K/SAM interaction differs from the WT/SAM interaction by +28.9 kcal mol−1 (less favorable), while the N374K/CxSAM interaction differs from the WT/CxSAM interaction by −45.6 kcal mol−1 (more favorable). The specific interactions contributing to these overall differences are highlighted in Figure 2, with residue 374 as the major driver, as predicted. At position N374 in the WT, the interaction favors SAM over CxSAM by 26.5 kcal mol−1. In the N374K variant, this selectivity changes to CxSAM by −61.2 kcal/mol, a total change in interaction energy of 87.7 kcal mol−1 when compared to that of the WT M.MpeI. EDA is based on the analysis of the nonbonded (electrostatic and Van der Waals) intermolecular interactions between different molecules. In the case of the N374K variant, we observe that the positive charge provides a significant stabilizing interaction with the negative charge of CxSAM and a direct hydrogen bond between the carboxylate moiety and the Lys side chain in the neomorphic enzyme. This model is also supported by our previously reported saturation mutagenesis study where we found that the substitution of N374 with the positively charged Arg similarly permits neomorphic carboxymethylation.20 The ionizable N374H mutant, however, was not found to generate 5cxmC by qualitative analysis but still maintained methylation activity. Similarly, when N374 was substituted with either negatively charged or neutral residues, such as N374D and N374A, both failed to produce 5cxmC while remaining proficient in 5mC generation.
Figure 2.
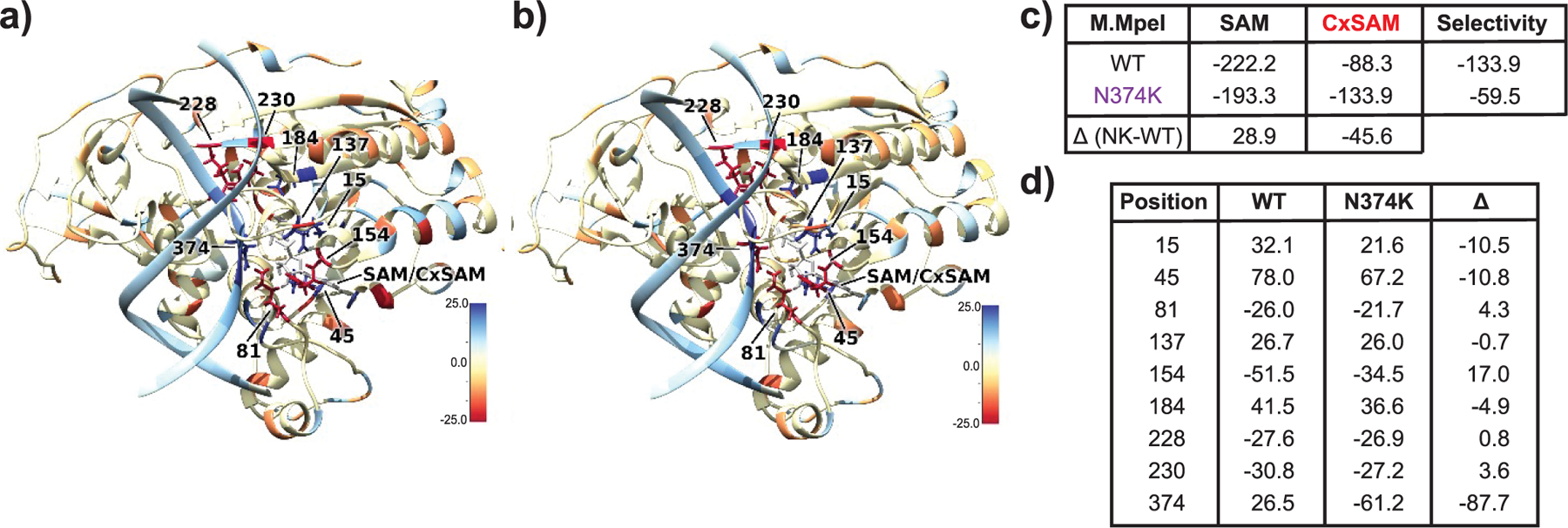
Comparison of WT versus N374K M.MpeI highlights stabilization of CXSAM by the mutant. (a) Difference in interaction energies between WT with CXSAM against SAM baseline. Residues highlighted in blue (red) interact more favorably with SAM (CXSAM). (b) Difference in interaction energies between N374K with CXSAM with respect to SAM. (c) Total interaction energies in kcal mol−1 between protein/DNA complex and SAM/CXSAM cosubstrates. (d) Interaction energy differences between highlighted residues and cosubstrates in kcal mol−1. Residues selected showed interaction energy ≥ 10 kcal/mol with SAM in either direction. Positive values indicate that the residue at that position interacts more favorably with SAM relative to CXSAM. Δ denotes difference for N374K with respect to WT.
E45 Contributes to CxSAM Binding.
Although modeling highlighted the clear role for residue 374, we also observed differences in interaction energies between the WT and SAM/CxSAM cosubstrates and other regions spanning the active site. The largest of these differences occurred at position E45, corresponding to a change of 78.0 kcal mol−1 less favorable in its interactions with the CxSAM compared to SAM. In N374K, the interaction energy at this position is 67.2 kcal mol−1 less favorable in its interactions with CxSAM compared to SAM. Comparing WT and N374K mutants, the E45 residue has the second largest difference (10.8 kcal mol−1) in interaction energies (Figure 2). Prior investigations of Rossmann-fold enzymes suggest that E45 is the key acidic residue within the second β-sheet (termed β2-Asp/Glu motif) that makes a bidentate interaction between the ribosyl C2 and C3 hydroxyl groups of SAM.9 Intrigued by the unexpected contribution of E45 to differential interactions between SAM and CxSAM on the opposite face of the original N374K mutation (Figure 1), we next explored simulations with two variants: E45G to remove the negative charge and the bulk of the side chain and E45D to shorten the side chain and potentially expand the active site without removing the side chain critical for making the bidentate interaction.
After validating that both mutants with either SAM or CxSAM were consistent in dynamic motion and overall structure relative to the WT system (Figures S1–S5), we focused again on EDA. With the E45G system, we found an overall reduction in the ΔE between position 45 and the cosubstrates; however, the total difference in interaction energy between the protein/DNA complex and the cosubstrates is also less favorable for both when compared to other systems (Figure 3). These results suggest that, while E45G does alter selectivity between substrates, it would also be less likely to interact with either substrate when compared to other variants investigated. Conversely, the E45D variant shows an 81.8 kcal mol−1 more favorable selectivity for CxSAM over SAM, with comparatively minor changes at other residues relative to changes seen with the WT M.MpeI. Interestingly, the E45D variant also resulted in a significant improvement in discrimination at position N374 (Figure 3). The overall interaction energy between the protein/DNA complex and the SAM cosubstrate was similar to that of the N374K variant, while the interaction of the E45D variant with CxSAM was 17.4 kcal mol−1 more favorable than the N374K variant. Taken together, these data indicate that the charged residue at this position is important for binding; however, the smaller side chain of the aspartate variant appears to interact more favorably with the larger CxSAM substrate, potentially due to enlarging the active site.
Figure 3.

Analysis of mutants suggests a role for E45 in SAM and CXSAM discrimination. (a) Difference in interaction energies between E45G and E45D with CXSAM against SAM baseline. Residues highlighted in blue (red) interact more favorably with SAM (CXSAM). (b) Difference in interaction energies between E45D with CXSAM with respect to SAM. (c) Total interaction energies in kcal mol−1 between protein/DNA complex and SAM/CXSAM cosubstrates. (d) Interaction energy differences between highlighted residues and cosubstrates in kcal mol−1. Positive (negative) values indicate that the residue at that position interacts more favorably with SAM relative to CXSAM; Δ denotes difference with respect to WT (from Figure 2d).
E45D and N374K Both Impact Selectivity.
As the interaction energies for E45D and N374K were both shifted toward CxSAM selectivity in similar directions, we aimed to simulate the E45D/N374K double mutant (Figure 4). Interestingly, the interaction between D45 and the cosubstrate favors CxSAM over SAM to a greater extent with the double mutant relative to either single mutant. This observation suggests that the improvement of selectivity with these two positions is likely due to many body effects, as the double mutant includes both a positive and negative charge on opposite sides of the CxSAM cosubstrate that make productive interactions, leading to greater stabilization. The total interaction energies between the protein/DNA complex and the cosubstrates also select for CxSAM over SAM by 44.2 kcal mol−1, an interaction energy that is of similar magnitude to the SAM preference with single point mutants. Focusing on the E45D mutation, we looked at the occupancy of the bidentate interaction between the E45 or D45 residue in each structure with CxSAM bound. We observed that in the WT enzyme, a bidentate interaction (the presence of a double hydrogen bond) with the ribosyl group of CxSAM is observed in only 7% of the total simulation. In the E45D single mutant, a bidentate interaction is observed during 35% of the simulation time. In the E45D/N374K double mutation, we observed the bidentate interaction in 92% of the simulation time, highlighting the stabilization of the CxSAM.
Figure 4.

An E45D/N374K double mutant interacts favorable with CxSAM. (a) Difference in interaction energies between E45D/N374K with CxSAM against SAM baseline. Residues highlighted in blue (red) interact more favorably with SAM (CxSAM). (b) Total interaction energies in kcal mol−1 between protein/DNA complex and SAM/CxSAM cosubstrates. (c) Interaction energy differences between highlighted residues and cosubstrates in kcal mol−1. Positive (negative) values indicate the residue at that position interacts more favorably with SAM (CxSAM)); Δ denotes difference with respect to WT (from Figure 2d).
Recognizing that differential binding might be accounted for, in part, by differences in the overall active site steric effects, we also calculated the active site cavity volume using the CASTp 3.0 server for each variant.25 With a probe radius of 1.4 Å, we find that the N374K variant has a slightly reduced volume (404.7 Å3) compared to that of the WT (409.9 Å3). For mutations at position 45, the volume for both E45G (429.9 Å3) and E45D (424.5 Å3) variants increases by 20 Å3 and 15 Å3, respectively, while the E45D/N374K double mutant (419.7 Å3) reduced in size compared to the E45D variant but is larger than the N374K variant alone. Taken collectively, these data suggest that the increased cavity volume in E45D/N374K over both WT and N374K could allow for less restricted motion of the larger CxSAM molecule while also maintaining the critical bidentate interaction that would be lost by other space-opening mutations such as E45G.
E45D/N374K has CxMTase Activity.
As our molecular dynamics analysis suggested that the active site E45D mutation could further improve the selectivity of M.MpeI for CxSAM, we cloned, expressed, and purified M.MpeI variants containing either the E45D or N374K mutations alone or the E45D/N374K double mutant (Figure S6). To initially assess enzymatic activities, we assayed each variant in vitro for the ability to use either SAM or CxSAM to modify an otherwise unmodified pUC19 plasmid DNA substrate (Figure 5A). In this assay, the MTase-reacted plasmid was linearized and digested with restriction enzymes that can report on modification. For analysis of MTase activity, we used the modification-sensitive restriction enzyme, HpaII (CCGG, 13 sites), which cleaves DNA when the central CpG is unmodified but not when it is 5mCpG. For analysis of CxMTase activity, we used MspI (CCGG, 13 sites), a restriction enzyme that cleaves DNA when the central CpG is unmodified but not when it is a 5cxmCpG. From this qualitative analysis, we observed that, as expected, the pUC19 DNA substrate is protected from HpaII-directed cleavage by reaction with M.MpeI WT + SAM. The WT enzyme, however, lacks substantive CxMTase activity, as the plasmid can be readily digested by MspI after reaction with M.MpeI WT and CxSAM. By contrast and consistent with our prior work, M.MpeI N374K appears proficient in both MTase and CxMTase activity, as the plasmid is protected from digestion with HpaII after reaction with SAM and protected from MspI cleavage after reaction with CxSAM.
Figure 5.

Qualitative evaluation of M.MpeI variants for methylation and carboxymethylation activity. (a) Experimental design. The pUC19 plasmid substrate was individually reacted with M.MpeI variants. The plasmid was then linearized with HindIII and digested with either HpaII or MspI to evaluate MTase or CxMTase activity, respectively. (b) Agarose gel of digestion products. M.MpeI WT and E45D display methylation activity, but not carboxymethylation activity. M.MpeI N374K and E45D/N374K display both methylation and carboxymethylation activity.
With the E45D mutant, we observed that MTase activity was preserved, suggesting the maintenance of SAM engagement. Contrary to our modeling predictions, we did not observe protection of pUC19 when the E45D single mutant was reacted with CxSAM. The absence of CxMTase activity could be due to multiple factors, including stabilization of substrate binding without appropriate positioning for transfer. In contrast to the E45D single mutant, however, and consistent with our modeling predictions, we observed that the E45D/N374K double mutant demonstrated the maintenance of MTase activity and proficient CxMTase activity. Thus, the N374K mutant, in the presence or absence of the E45D mutation, shows CxMTase activity. In line with these results, we separately found in an end point assay that the N374K single mutant and E45D/N374K double mutant are both able to completely label an oligo substrate with 5cxmC, while neither the WT or E45D single mutant demonstrate CxMTase activity (Figure S7).
E45D/N374K Enhances CxMTase Activity.
Across this series of substrates and variants, our experimental data suggested the primacy of N374K for being permissive for CxMTase activity. As the qualitative assay was unable to assess whether the E45D mutation altered selectivity, we next explored whether the quantitative preference for CxSAM versus SAM differs between the double and single mutants, as predicted computationally. We characterized the kinetic parameters for each mutant with either SAM or CxSAM as cosubstrates via a quantitative oligonucleotide-based assay (Figure S8, Figure 6A). In this assay, the fluorophore-labeled strand contains a single unmodified CpG site embedded in an HpaII (CCGG) cleavage site. The oligonucleotide is duplexed with a second strand containing a 5mCpG. After reaction with SAM or CxSAM, the unlabeled complement strand is exchanged for one containing an unmodified CpG. The product can then be analyzed by digestion with HpaII, where cleavage reports on the residual unmodified CpG fraction of the fluorophore-labeled strand, whereas the presence of either 5mCpG or 5cxmCpG blocks cleavage.
Figure 6.

Kinetic analysis of SAM and CxSAM use by M.MpeI variants. (a) Coupled assay experimental design. A hemimethylated, Cy3-labeled substrate containing a single unmodified CpG was incubated with an M.MpeI variant and either SAM or CxSAM. Following the reaction, the methylated bottom strand of the substrate was exchanged away to facilitate digestion with HpaII, with modified DNA resistant to cleavage, while unmodified DNS is readily cleaved. (b) Shown are the reaction velocities derived from each reaction performed with differing amounts of either SAM or CxSAM substrate. The observed rates were fit to a hyperbolic function to determine Vmax and Km. The selectivity constants (Vmax/Km) for each enzyme/substrate pair were separately calculated with an alternative form of the Michaelis Menten equation that allows for direct fitting and provides an associated error. Data shown are from three independent replicates with 95% confidence intervals provided.
Using this approach, we employed various concentrations of either SAM or CxSAM and analyzed the efficiency of CpG modification (Figure 6B). From these plots, we derived Km and apparent Vmax values for each enzyme–substrate pair and calculated enzyme specificity constants. With the N374K mutant, we observed a 3-fold higher Km for CxSAM over SAM, while Vmax for CxSAM was 0.6-fold that of SAM. With the E45D/N374K double mutant, we observed a similar Vmax relative to the N374K single mutant with both SAM and CxSAM; however, we also observed that the Km was now similar between the two substrates. The calculated specificity constants suggest that while the E45D mutation in concert with the N374K mutation showed no statistical difference in the ability of M.MpeI to utilize SAM as a substrate for 5mC generation, the mutation does result in significantly increased selectivity for CxSAM consistent with computational predictions.
E45D/N374K Prefers CxSAM over SAM.
To more rigorously evaluate the selectivity of M.MpeI variants for CxSAM versus SAM, we devised a novel assay to allow for direct competition between two candidate substrates. We modified the oligonucleotide-based assay so that the reactions were run with mixtures of different ratios of SAM and CxSAM, rather than each in isolation. After reaction of the oligonucleotide substrate with the M.MpeI variant and SAM/CxSAM mixture, we digested the oligonucleotide with HpaII, to account for generation of either 5mCpG or 5cxmCpG, or with MspI, to account for only 5cxmCpG. Comparing the reaction products allows for calculation of the fraction of 5mC versus 5cxmC products (Figure S9, Figure 7A). Carboxymethylation status can then be calculated as a percentage of the total modification status to normalize across conditions (Figure 7B).
Figure 7.

M.MpeI E45D/N374K shows greater selectivity for CxSAM over M.MpeI N374K when both SAM and CxSAM are present. (a) Competition-based experimental design. A hemimethylated, Cy3-labeled substrate containing a single unmodified CpG was incubated with an M.MpeI variant and varying ratios of SAM and CxSAM. Following the reaction, the methylated bottom strand of the substrate was exchanged away to facilitate digestion with HpaII (to monitor total 5mC and 5xcmC generation) and MspI (to monitor 5cxmC generation). (b) Results of a competition assay are plotted. Shown is the percent of the total product that is carboxymethylated (5cxmC) normalized to total modification activity (5mC + 5cxmC). Individual data points are shown from replicates, with mean value in the bar graph.
As expected, we detect that products are either fully carboxymethylated or fully methylated when the substrate pool contains only CxSAM or SAM, respectively. Mixtures of CxSAM and SAM revealed a pattern, however, that differed between the two enzyme variants. If the enzyme was equally likely to use CxSAM as the SAM, product formation should be observed in a ratio that matches that of the substrate mixture. For the N374K mutant, under conditions where 80% of the SAM mixture is CxSAM, 65% of the product contains 5cxmC; when CxSAM is 20% of the SAM mixture, only 12% of the product is 5cxmC. Thus, in direct competition, the N374K mutant favors SAM over CxSAM. By contrast, with the E45D/N374K double mutant, when CxSAM is 80% of the SAM mixture, 95% of the product is 5cxmC; when CxSAM is 20% of the mixture, 25% of the generated product is 5cxmC. Thus, the double mutant, in contrast to the N374K single mutant, shows a preference for CxSAM over SAM under these assay conditions.
CONCLUSIONS
Here, we describe how computational analysis of the CpG-specific DNA cytosine MTase, M.MpeI, reveals key determinants for the expansion of substrate selectivity. In our original report describing the generation of 5cxmC by M.MpeI N374K, we speculated that the positively charged lysine could form a salt bridge with the negatively charged carboxyl group.20 By first modeling CxSAM in the active site of M.MpeI variants and performing an EDA, we confirmed our hypothesis that the carboxylate of CxSAM participates in a highly energetically favorable interaction with the N374K. These models also unexpectedly suggested that alternative active site residues might be involved in differential interaction between SAM and CxSAM, with E45 being particularly intriguing. The computational results with E45G confirmed an anticipated role for the side chain carboxylate in making a critical bidentate interaction with the ribosyl moiety of SAM and CxSAM but also suggested that shortening the side chain with E45D could favor CxSAM over SAM.
Prompted by these computational findings, we performed a kinetic analysis of the N374K and E45D/N374K mutants, which together suggest that CxSAM can be used by the E45D/N374K mutant and that the preference for CxSAM is enhanced relative to N374K alone. Having two mutations on opposite sides of the CxSAM binding pocket (Figure 1b) was associated with a lowering of the Km, and we demonstrated an associated inversion in preferences using a direct competition assay. While N374K overall prefers its canonical SAM substrate over CxSAM, the E45D/N374K reverses this selectivity, preferring CxSAM over SAM. Notably, in recent work, the neomorphic CxMTase has been employed in novel epigenetic sequencing pipelines that permit specific detection of 5mCpG residues.22,23 As enzymatic sequencing approaches require high efficiency conversions, we anticipate that E45D/N374K could be exploited to further improve related sequencing approaches.
While the present study is rooted in a rational-design approach with molecular dynamics simulations guiding selection of mutagenesis candidates, our approach could be complemented by random mutagenesis studies or direct coupling analysis, which might further enhance the selectivity and/or activity profiles of CxMTases similar to those that have been used for other MTases.26–28 Directed evolution in particular has become one of the most powerful and widespread tools for improving desired protein function, especially when a selective pressure is applied.29 Indeed, the fact that the CxMTase activity can be observed in E. coli makes it viable that selection strategies could be employed to discover mutants that further alter either enzymatic efficiency or selectivity in favor of CXSAM over SAM.
An added strength of our approach is that, in addition to rationalizing CXMTase activity, our study offers an important precedent for combined computational and experimental approaches to further enhance the tolerance of SAM analogs. Intriguing prior work from the Tawfik group identified the bidentate ribose-carboXylate interaction, with its strict associated geometry, as a marker of the common ancestry for diverse proteins including MTases or enzymes that use other nucleotide cofactors including NAD or FAD.9,12 Using our modeling approaches and considering the SAM analog, CXSAM, we were independently drawn toward the same motif in a model Rossmann enzyme: the cytosine DNA MTase, M.MpeI. Our data support the conclusion that the M.MpeI E45D mutation maintains the critical bidentate ribosyl interaction while opening additional active site space to accommodate CXSAM, in a way that is unnecessary for the natural parent cosubstrate SAM. Given the broad interest in employing SAM analogs as substrates for other classes of methyltransferases,14,15 it will be interesting to investigate whether engineering of other enzymes with this phylogenetically conserved β2-Asp/Glu motif in the Rossmann-fold would yield improved or expanded biochemical properties. As this motif is highly conserved, such insights could extend beyond SAM-dependent enzymes to those using other nucleoside-associated cofactors including FAD and NAD9,12 and could be especially important for applications of cofactor analogs where competition with ubiquitous metabolites needs to be overcome to achieve selectivity.
MATERIALS AND METHODS
Computational Methods.
Five protein variants were prepared for modeling from the original M.MpeI crystal structure (PDB: 4DKJ21): wildtype (WT), N374K, E45G, E45D, and E45D/N374K. For each variant, systems were prepared with SAM or CXSAM cosubstrates, totaling 10 unique systems. Protonation states of amino acid side chains were determined using the H++ server.30–32 Custom force-field parameters were generated for the SAM and CXSAM ligands using the PyRED server.33–35 These force-fields were used along with FF14SB for the protein, TIP3P for the water, counterions, and metal ions and OL15 for the DNA. Each system was neutralized to zero net charge using K+ counterions and solvated in TIP3P water with a minimum distance of 12 Å between the protein surface and the edge of the periodic boundary, resulting in a periodic unit cell measuring 71 × 64 × 51Å.
Systems were each minimized over 50 steps of steepest descent, followed by 450 steps of a conjugate gradient at 10 K with the protein, ligand, and DNA frozen to allow the density of the solvent box to equilibrate. The system was heated from 10 to 300 K over 20 stages, with each stage taking 12.5 ps. After heating, restraints on all nonsolvent molecules were gradually removed from 100 kcal mol−1 Å−2 in 10 stages. Equilibration and production were run at temperatures of 300 K and 1.0 atm using a Berendsen thermostat in an NVT ensemble. The nonbonded cutoff was set to 8.0 Å, and a smooth particle-mesh Ewald method was used for long-range Coulomb interactions.36 Each system was equilibrated for 50 ns before production to ensure stability. Production was run with a 1 fs time step for 250 ns using the pmemd.cuda module in AMBER18.37 All bonds involving hydrogen atoms were constrained by using the SHAKE algorithm. All systems were simulated in triplicate for a total of 750 ns of MD sampling per combination.
Correlated motion, hydrogen bonding interactions, root-mean-square deviation (RMSD) and fluctuation (RMSF), normal modes, and distances were calculated by using cpptraj. Energy decomposition analysis (EDA) was performed using AMBER-EDA.38 Active site cavity volume was calculated using the CASTp server.25
Enzyme Cloning and Expression.
Cloning and expression of M.MpeI variants were performed as previously described.20 All mutations were made via Q5 Site Directed Mutagenesis (New England Biolabs, NEB) and confirmed by Sanger sequencing. Briefly, variants were purified via affinity chromatography using a C-terminal His tag and ultimately dialyzed into 20 mM Tris HCl at pH 7.5 and 25 °C, 0.2 mM EDTA, 2 mM DTT, 150 mM NaCl, and 10% glycerol (v/v) before the addition of an equal volume of cold 40% (v/v) glycerol and flash freezing with liquid nitrogen for long-term storage at − 80 °C. All proteins were visualized for purity via SDS-PAGE and quantified using a Qubit fluorometer (Figure S6).
CxSAM Synthesis.
Reactions were performed as previously described.39 Briefly, 2-iodoacetic acid (667 mg) was added to a solution of S-adenosyl-l-homocysteine (20 mg) in 3.3 mL of 150 mM aqueous ammonium bicarbonate. The reaction was incubated at 37 °C for 24 h. 80 mL of methanol was then added and incubated at 4 °C overnight. Precipitates were collected by centrifugation at 4 °C (2000g, 30 min) and washed twice with cold methanol to yield CxSAM. CxSAM was dissolved in nuclease-free water and stored at −20 °C. CxSAM was quantified using absorbance measured at 260 nm (15 400 L mol−1 cm−1). High resolution mass spectrometry (HRMS) was performed with an observed mass of 443.1360 (mDa = −0.2, PPM = −0.5, theoretical mass: 443.1343).
pUC19 Plasmid Assay for MTase and CxMTase Activity.
M.MpeI variants (1 μM) were incubated with 160 μM SAM or CxSAM substrate and pUC19 plasmid DNA (100 ng) for 4 h at 37 °C in M.MpeI reaction buffer (10 mM Tris Cl, 50 mM NaCl, 1 mM DTT, 1 mM EDTA, pH 7.9 at 25 °C) in a 5 μL volume. 2.5 μL of the reaction was then incubated with the appropriate restriction enzyme, and the plasmid DNA was simultaneously linearized with the methylation-insensitive HindIII-HF (NEB) in a 25 μL volume. HpaII (NEB) cleaves CCGG (13 sites) if unmodified but does not if modifications are present. MspI (NEB) recognizes the same CCGG sites but cleaves if the DNA is either unmodified or methylated, but not if it is carboxymethylated. Samples were treated with 1 μL of Proteinase K at 37 °C for 10 min, separated on an agarose gel, and visualized with SYBR Safe DNA Gel Stain (Thermo Fisher).
Assessment of Labeling Efficiency and Kinetic Parameters by Oligonucleotide Assay.
The assay format was modified from prior work on MTases.40 A Cy3-labeled top strand oligonucleotide with a single unmethylated CCGG and unlabeled complementary bottom strand oligonucleotides with either an unmethylated or methylated CCGG were obtained from IDT. For determination of labeling efficiency, 150 nM of the duplexed, hemimethylated oligonucleotide was reacted with 1 uM M.MpeI and 160 μM SAM or CxSAM substrate at 37 °C in M.MpeI reaction buffer in a 5 μL reaction volume for 4 h before heat inactivation at 95 °C for 5 min. A 25× excess of the unmethylated bottom strand was added and reannealed before restriction digestion with either HpaII or MspI in a 10 μL volume. Undigested and digested strands were resolved on 20% TBE acrylamide denaturing PAGE (Figure S7). For determination of kinetic parameters, the same assay was used except with 200 nM M.MpeI, 2-fold serial dilutions of 160 μM SAM or CxSAM, and a 1 h, 37 °C incubation before heat inactivation at 95 °C for 5 min (Figure S8). To calculate Vmax/Km values with associated error, the data were fit using nonlinear regression to an alternative form of the Michaelis Menten equation that allows for direct fitting to Vmax/Km.
Oligonucleotide Competition Assay.
The oligonucleotide assay from above was modified to allow for competition between the SAM and CxSAM. 150 nM duplexed, hemimethylated oligonucleotide was reacted with 200 nM M.MpeI and mixtures of SAM and CxSAM (at a final concentration of 25 μM) that varied in ratio. The reaction was performed at 37 °C in M.MpeI reaction buffer in a 5 μL reaction volume for 1 h before heat inactivation at 95 °C for 5 min. A 25× excess of the unmethylated bottom strand was reannealed before restriction digestion with either HpaII (to monitor for either 5mC or 5cxmC) or MspI (to monitor for 5cxmC) in a final volume of 10 μL. Undigested and digested strands were resolved on a 20% TBE Acrylamide Denaturing PAGE (Figure S9).
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the National Institutes of Health through R01-HG10646 (to R.M.K.) and R01-GM108583 (to G.A.C.). C.E.L. was supported by F31-HG012892. Computing time from XSEDE through allocation TG-CHE160044 and UNT CASCaM (partially funded by NSF Grant Nos. CHE1531468 and OAC-2117247) is gratefully acknowledged.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.3c00184.
MD analysis for replicate trajectories of all variants and ligands, along with added details for biochemical evaluation of variants (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.3c00184
The authors declare the following competing financial interest(s): T.W. and R.M.K through the University of Pennsylvania have filed a patent application on MTase enzymes engineered to have CxMTase activity.
Contributor Information
Christian E. Loo, Graduate Group in Biochemistry and Biophysics, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
Mark A. Hix, Department of Chemistry, Wayne State University, Detroit, Michigan 48202, United States.
Tong Wang, Graduate Group in Biochemistry and Biophysics, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
G. Andrés Cisneros, Department of Physics, University of Texas at Dallas, Richardson, Texas 75801, United States; Department of Chemistry and Biochemistry, University of Texas at Dallas, Richardson, Texas 75801, United States.
Rahul M. Kohli, Department of Medicine and Department of Biochemistry and Biophysics, University of Pennsylvania, Philadelphia, Pennsylvania 19104, United States.
REFERENCES
- (1).Nabel CS; Manning SA; Kohli RM The curious chemical biology of cytosine: Deamination, methylation, and oxidation as modulators of genomic potential. ACS Chem. Biol 2012, 7, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Schubeler D Function and information content of DNA methylation. Nature 2015, 517, 321–326. [DOI] [PubMed] [Google Scholar]
- (3).Iyer LM; Abhiman S; Aravind L Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci 2011, 101, 25–104. [DOI] [PubMed] [Google Scholar]
- (4).Beaulaurier J; Schadt EE; Fang G Deciphering bacterial epigenomes using modern sequencing technologies. Nat. Rev. Genet 2019, 20, 157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wilson GG; Murray NE Restriction and modification systems. Annual Review of Genetics 1991, 25, 585–627. [DOI] [PubMed] [Google Scholar]
- (6).Jurkowska RZ; Jurkowski TP; Jeltsch A Structure and function of mammalian DNA methyltransferases. Chembiochem 2011, 12, 206–222. [DOI] [PubMed] [Google Scholar]
- (7).Jurkowska RZ; Jeltsch A Enzymology of mammalian DNA methyltransferases. Adv. Exp. Med. Biol 2022, 1389, 69–110. [DOI] [PubMed] [Google Scholar]
- (8).Saghafinia S; Mina M; Riggi N; Hanahan D; Ciriello G Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell. Rep 2018, 25, 1066–1080.e8. [DOI] [PubMed] [Google Scholar]
- (9).Laurino P; Tóth-Petróczy A; Meana-Pañeda R; Lin W; Truhlar DG; Tawfik DS An ancient fingerprint indicates the common ancestry of rossmann-fold enzymes utilizing different ribose-based cofactors. PLoS Biol 2016, 14, e1002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schubert HL; Blumenthal RM; Cheng X Many paths to methyltransfer: A chronicle of convergence. Trends Biochem. Sci 2003, 28, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Rao ST; Rossmann MG Comparison of super-secondary structures in proteins. J. Mol. Biol 1973, 76, 241–256. [DOI] [PubMed] [Google Scholar]
- (12).Chouhan BPS; Maimaiti S; Gade M; Laurino P Rossmann-fold methyltransferases: Taking a ″β-turn″ around their cofactor, S-adenosylmethionine. Biochemistry 2019, 58, 166–170. [DOI] [PubMed] [Google Scholar]
- (13).Struck A; Thompson ML; Wong LS; Micklefield J S-adenosyl-methionine-dependent methyltransferases: Highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. Chembiochem 2012, 13, 2642–2655. [DOI] [PubMed] [Google Scholar]
- (14).Zhang J; Zheng YG SAM/SAH analogs as versatile tools for SAM-dependent methyltransferases. ACS Chemical Biology 2016, 11, 583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Rudenko AY; Mariasina SS; Sergiev PV; Polshakov VI Analogs of S-adenosyl-l-methionine in studies of methyltransferases. Mol. Biol 2022, 56, 229–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Pljevaljcic G; Schmidt F; Weinhold E Sequence-specific methyltransferase-induced labeling of DNA (SMILing DNA). Chembiochem 2004, 5, 265–269. [DOI] [PubMed] [Google Scholar]
- (17).Kriukienė E; Labrie V; Khare T; Urbanavičiūtė G; Lapinaite A; Koncevicius K; Li D; Wang T; Pai S; Ptak C; Gordevicius J; Wang S; Petronis A; Klimasauskas S DNA unmethylome profiling by covalent capture of CpG sites. Nat. Commun 2013, 4, 2190. [DOI] [PubMed] [Google Scholar]
- (18).Liutkeviciute Z; Lukinavicius G; Masevicius V; Daujotyte D; Klimasauskas S Cytosine-5-methyltransferases add aldehydes to DNA. Nat. Chem. Biol 2009, 5, 400–402. [DOI] [PubMed] [Google Scholar]
- (19).Wojciechowski M; Czapinska H; Bochtler M CpG underrepresentation and the bacterial CpG-specific DNA methyltransferase M.MpeI. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang T; Kohli RM Discovery of an unnatural DNA modification derived from a natural secondary metabolite. Cell. Chem. Biol 2021, 28, 97–104.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kim J; Xiao H; Bonanno JB; Kalyanaraman C; Brown S; Tang X; Al-Obaidi NF; Patskovsky Y; Babbitt PC; Jacobson MP; Lee Y; Almo SC Structure-guided discovery of the metabolite carboxy-SAM that modulates tRNA function. Nature 2013, 498, 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xiong J; Chen K; Xie N; Ji T; Yu S; Tang F; Xie C; Feng Y; Yuan B Bisulfite-free and single-base resolution detection of epigenetic DNA modification of 5-Methylcytosine by methyltransferase-directed labeling with APOBEC3A deamination sequencing. Anal. Chem 2022, 94, 15489–15498. [DOI] [PubMed] [Google Scholar]
- (23).Wang T; Loo CE; Kohli RM Enzymatic approaches for profiling cytosine methylation and hydroxymethylation. Mol. Metab 2022, 57, 101314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang T; Fowler JM; Liu L; Loo CE; Luo M; Schutsky EK; Berríos KN; DeNizio JE; Dvorak A; Downey N; Montermoso S; Pingul BY; Nasrallah M; Gosal WS; Wu H; Kohli RM Direct enzymatic sequencing of 5-methylcytosine at single-base resolution. Nat. Chem. Biol 2023, DOI: 10.1038/s41589-023-01318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tian W; Chen C; Lei X; Zhao J; Liang J CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res 2018, 46, W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gerasimaite R; Vilkaitis G; Klimasauskas S A directed evolution design of a GCG-specific DNA hemimethylase. Nucleic Acids Res 2009, 37, 7332–7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Laurino P; Rockah-Shmuel L; Tawfik DS Engineering and directed evolution of DNA methyltransferases. Adv. Exp. Med. Biol 2016, 945, 491–509. [DOI] [PubMed] [Google Scholar]
- (28).Ravishankar K; Jiang X; Leddin EM; Morcos F; Cisneros GA Computational compensatory mutation discovery approach: Predicting a PARP1 variant rescue mutation. Biophys. J 2022, 121, 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Arnold FH Directed evolution: Bringing new chemistry to life. Angew. Chem., Int. Ed. Engl 2018, 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Anandakrishnan R; Aguilar B; Onufriev AV H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res 2012, 40, W537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Myers J; Grothaus G; Narayanan S; Onufriev A A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules. Proteins 2006, 63, 928–938. [DOI] [PubMed] [Google Scholar]
- (32).Gordon JC; Myers JB; Folta T; Shoja V; Heath LS; Onufriev A H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res 2005, 33, W368–W371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Vanquelef E; Simon S; Marquant G; Garcia E; Klimerak G; Delepine JC; Cieplak P; Dupradeau F R.E.D. server: A web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res 2011, 39, W511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Dupradeau F; Pigache A; Zaffran T; Savineau C; Lelong R; Grivel N; Lelong D; Rosanski W; Cieplak P The R.E.D. tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys 2010, 12, 7821–7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Case DA; Ben-Shalom IY; Brozell SR; Cerutti DS; Cheatham TE III; Cruzeiro V; Darden TA; Duke RE; Ghoreishi D; Gilson MK AMBER; University of California: San Francisco, 2018. [Google Scholar]
- (36).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A smooth particle mesh ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- (37).Roe DR; Cheatham TE, 3 PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- (38).Leddin EM; Cisneros GA CisnerosResearch/AMBER-EDA, first release (v0.1); Zenodo, 2021. [Google Scholar]
- (39).Kim J; Xiao H; Koh J; Wang Y; Bonanno JB; Thomas K; Babbitt PC; Brown S; Lee Y; Almo SC Determinants of the CmoB carboxymethyl transferase utilized for selective tRNA wobble modification. Nucleic acids research 2015, 43, 4602–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Nabel CS; DeNizio JE; Carroll M; Kohli RM DNA methyltransferases demonstrate reduced activity against arabinosylcytosine: Implications for epigenetic instability in AML. Biochemistry 2017, 56, 2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.