

Summary
Preclinical and clinical studies implicate endocannabinoids (eCBs) in fear extinction, but the underlying neural circuit basis of these actions is unclear. Here, we employed in vivo optogenetics, eCB biosensor imaging, ex vivo electrophysiology and CRISPR-Cas9 gene editing in mice to examine whether basolateral amygdala (BLA)-projecting medial prefrontal cortex (mPFC) neurons represent a neural substrate for the effects of eCBs on extinction. We found that photoexcitation of mPFC axons in BLA during extinction mobilizes BLA eCBs. eCB biosensor imaging showed that eCBs exhibit a dynamic stimulus-specific pattern of activity at mPFC→BLA neurons that tracks extinction learning. Furthermore, using CRISPR-Cas9-mediated gene editing we demonstrated that extinction memory formation involves eCB activity at cannabinoid CB1 receptors expressed at vmPFC→BLA synapses. Our findings reveal the temporal characteristics and a neural circuit basis of eCBs effects on fear extinction, and inform efforts to target the eCB system as a therapeutic approach in extinction-deficient neuropsychiatric disorders.
Keywords: Extinction, endocannabinoid, fear, amygdala, prefrontal cortex, optogenetics, GRABeCB2.0 biosensor, CRISPR-Cas9, anxiety, PTSD
Graphical Abstract
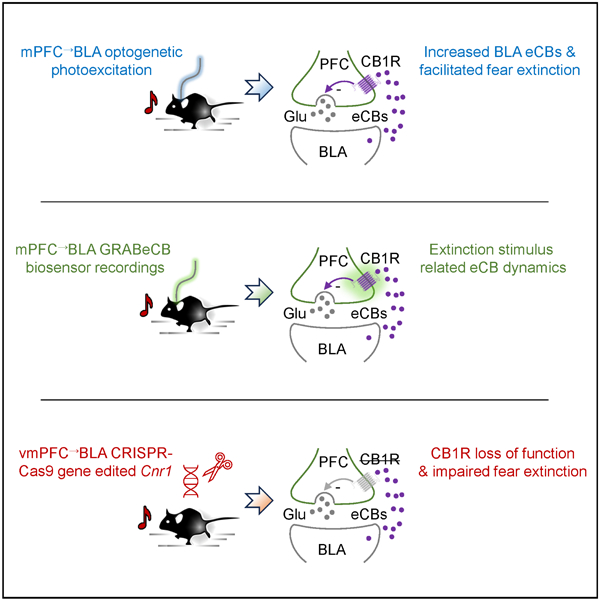
eTOC blurb
Gunduz-Cinar et al. find that fear extinction is associated with dynamic changes in endocannabinoids at mPFC input to BLA, and show that loss of endocannabinoid signaling at BLA-projecting mPFC neurons impairs fear extinction, thereby identifying a neural circuit substrate for endocannabinoid modulation of extinction.
Introduction
Forming associations between environmental cues and danger is an essential survival function enabling threat-related stimuli to generate defensive responses. When these stimuli are subsequently encountered in the absence of coincident threat, conditional defensive responding typically extinguishes, preventing excess fear 1–3. A growing literature demonstrates that endocannabinoids (eCBs) can promote extinction and thereby represent a potential therapeutic target for extinction-deficient conditions, including anxiety disorders and posttraumatic stress disorder (PTSD) 4–11. Yet, despite the enormous interest in the extinction-mediating effects of eCBs, the neural circuit substrates underlying these actions remain unclear.
Prior work has shown that eCB levels are elevated after extinction in rodent basolateral nucleus of the amygdala (BLA) 12,13, a brain region that is under neuromodulatory control and which is essential for fear extinction 14–16. Augmenting levels of the eCB, anandamide (AEA), in the rodent BLA by chronic antidepressant treatment or intra-BLA delivery of a fatty acid amide hydrolase (FAAH) inhibitor facilitates extinction and reverses the extinction-impairing effects of AEA depletion 17–19. Along similar lines, administration of cannabinoids or gene-related reductions in FAAH expression which improve extinction and reduce anxiety in mice and humans are associated with attenuated amygdala reactivity to stressors and stronger connectivity with medial prefrontal cortex (mPFC) 13,20–29.
In parallel with these findings, there is growing cross-species evidence implicating the mPFC→BLA pathway in fear extinction 30–34. Of particular note are recent studies showing that neurons in rodent mPFC (particularly the ventromedial PFC (vmPFC)/infralimbic cortex) instruct extinction memory formation 35–38 via inputs to BLA 30,39,40 and neighboring structures 41. The identification of the mPFC and its projections to BLA as a locus for extinction in turn relates to evidence that eCBs can alter mPFC activity to affect fear and extinction 27–29,42.
These convergent lines of research suggest that eCBs affect extinction by modulating the function of the mPFC→BLA pathway. Here, we address this possibility using a combination of in vivo optogenetics, pharmacology, eCB biosensor imaging, and CRISPR-Cas9-mediated gene editing. Our data provide novel evidence that eCBs dynamically signal at mPFC→BLA neurons during extinction and contribute to the formation of extinction memory. These findings advance our understanding of the neural substrates of extinction and inform ongoing efforts to therapeutically targeting the eCB system as a means to facilitate extinction in neuropsychiatric disorders characterized by deficient extinction.
Results
Prefrontal-amygdala excitation facilitates fear extinction and increases BLA eCBs
We began by asking whether the behavioral effects of bilateral optogenetic photoexcitation of channelrhodopsin (ChR2)-expressing mPFC axons in BLA were associated with coincident changes in BLA eCBs. Male C57BL6/J mice (male C57BL6/J-background mice were used throughout the study unless otherwise specified) first underwent fear conditioning, which involved repeated presentations of a tone (conditioned stimulus, CS), each co-terminating with footshock (unconditioned stimulus, US).
Conditioning was followed, one day later, by 10x CS-alone partial extinction training, then extinction retrieval testing one day (retrieval 1) and again two weeks (retrieval 2) later (Figure 1A,B). A partial extinction training procedure 30,39,43 was employed to enable detection of a hypothesized photoexcitation-induced facilitation of extinction. During each CS-presentation of extinction training, but not any other phase of testing, blue light was shone (20 Hz, in 5-millisecond pulses) at mPFC axons in BLA. Replicating prior findings using this procedure 30,39, we found that mPFC→BLA photoexcitation produced lower freezing, relative to non-excited YFP-expressing controls, on the light-free retrieval tests (Figure 1C,D and Figure S1A, see Table S1 for a summary of the statistical results of all experiments).
Figure 1: Photoexcitation of mPFC→BLA neurons facilitates fear extinction through BLA eCBs.
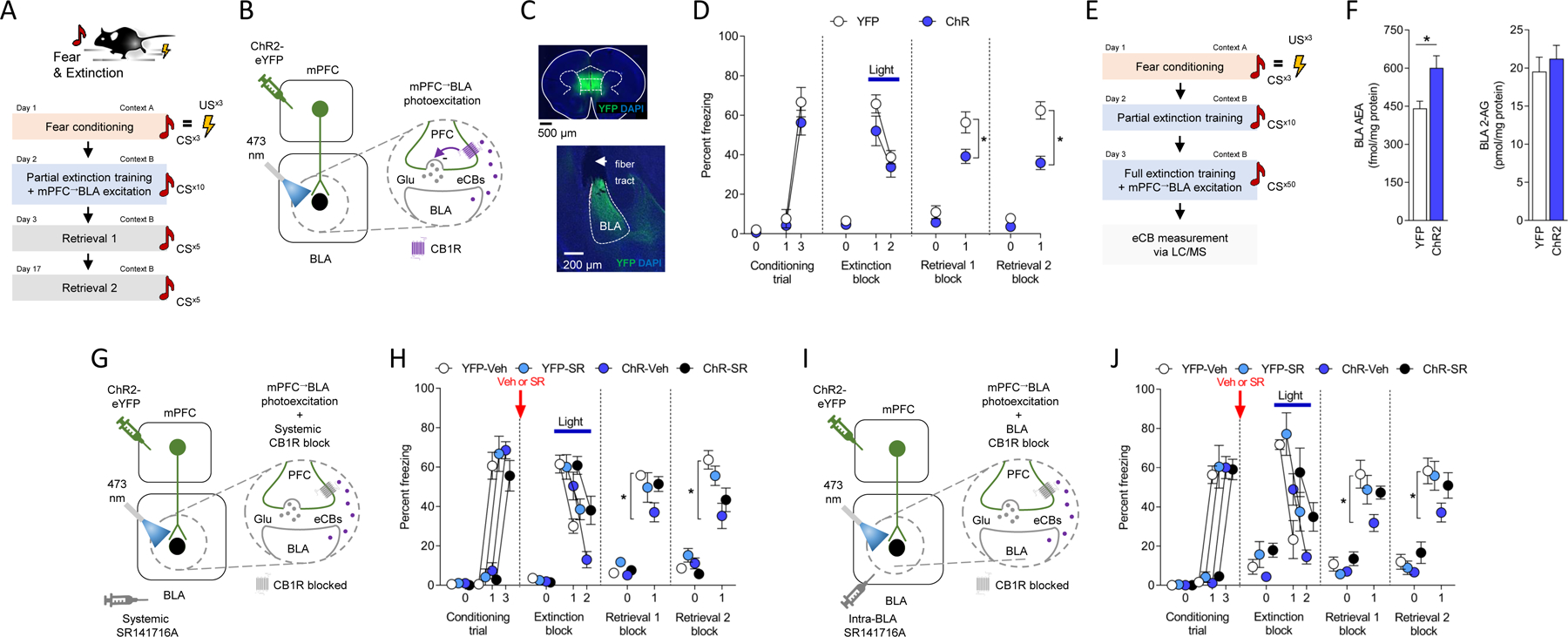
(A-C) Behavioral (A) and in vivo optogenetic (B) procedure for mPFC→BLA photoexcitation during partial (10-trial) extinction training (YFP n=11/ChR2 n=8). Virus expression in mPFC and BLA (C).
(D) Freezing increased over conditioning trials (RM-ANOVA: F(1,13)=63.960, P<0.0001, ηp2=0.73) and decreased over extinction trial-blocks (RM-ANOVA: F(1,17)=56.610, P<0.0001, ηp2=0.34). Lower freezing in opsin groups on early extinction (first 5-trial block) vs extinction retrieval 1 (RM-ANOVA: F(1,17)=5.683, P=0.0291, ηp2=0.17) and on extinction retrieval 2 (RM-ANOVA: F(1,17)=11.170, P=0.0039, ηp2=0.28).
(E,F) Measurement of photoexcitation-related BLA eCB levels (AEA-YFP n=15/AEA-ChR2 n=15, AEA-YFP n=15/AEA-ChR2 n=14) (E). Higher AEA, not 2-AG (P>0.05), levels in ChR2 versus YFP (unpaired t-test: t(28)=2.887, P=0.0074, ηp2=0.23 (F).
(G,H) Systemic SR141716A (SR, CB1R antagonist) prior to combined extinction training/mPFC→BLA photoexcitation (YFP-Vehicle (Veh) n=11/YFP-SR n=9/ChR2-Veh n=9/ChR2-SR n=12) (G). Freezing increased over conditioning trials (RM-ANOVA: F(1,36)=209.800, P<0.0001, ηp2=0.75). Freezing decreased over extinction trial-blocks (RM-ANOVA: F(1,37)=116.700, P<0.0001, ηp2=0.37). Lower freezing in ChR2 versus YFP on extinction retrieval 1 (ANOVA opsin x drug interaction: F(1,37)=4.742, P=0.0359, ηp2=0.10, Holm-Šídák's tests: YFP-Veh versus ChR2-Veh: P=0.0488, all others P>.05) and retrieval 2 (ANOVA opsin x drug interaction: F(1,37)=2.054, P=0.1602, ηp2=0.04, Holm-Šídák's tests: YFP-Veh versus ChR2-Veh: P=0.0023, all others P>.05) (H).
(I,J) Intra-BLA SR141716A prior to combined extinction training/mPFC→BLA photoexcitation (YFP-Veh n=10/YFP-SR n=8/ChR2=Veh n=15/ChR2-SR n=11) (I). Freezing increased over conditioning trials (RM-ANOVA: F(1,40)=306.400, P<0.0001, ηp2=0.77). Freezing decreased over extinction trial-blocks (RM-ANOVA: F(1,40)=76.000, P<0.0001, ηp2=0.28). Lower freezing in ChR2 versus YFP on extinction retrieval 1 (ANOVA opsin x drug interaction: F(1,40)=4.511, P=0.0399, ηp2=0.09, Holm-Šídák's tests: YFP-Veh versus ChR2-Veh: P=0.0029, all others P>.05) and retrieval 2 (ANOVA opsin x drug interaction: F(1,39)=2.323, P=0.1355, ηp2=0.05, Holm-Šídák's tests: YFP-Veh versus ChR2-Veh: P=0.0417, all others P>.05) (J).
Data are represented as mean±SEM. *P<0.05. Table S1 reports full statistical results for all figures.
We next tested whether the extinction-facilitating effects of mPFC→BLA photoexcitation were associated with the mobilization of eCBs in BLA. To do so, we conducted extinction training plus mPFC→BLA photoexcitation and then sacrificed animals to collect BLA tissue (and dorsal striatum for comparison) to measure eCB levels via liquid chromatography/tandem mass spectrometry 17. We found that animals receiving the combination of extinction and photoexcitation had higher levels of AEA, but not another major eCB, 2-arachidonoylglycerol (2-AG), in BLA (but not dorsal striatum), as compared to extinction-tested YFP-expressing controls (Figure 1E,F and Figure S1B).
These initial data show that optogenetically exciting mPFC axons in BLA facilitates extinction memory formation and causes a concomitant increase in BLA AEA levels.
Prefrontal-amygdala excitation facilitates extinction through eCB-CB1R signaling
We next tested whether increased BLA eCBs following the combination of extinction training and mPFC→BLA photoexcitation functionally contribute to the extinction-facilitating effects of photoexciting this neural pathway. To do so, we again performed mPFC→BLA photoexcitation during partial extinction training, but this time simultaneously blocked CB1 receptor (CB1R) mediated eCB-signaling by systemically administering animals with a selective CB1R antagonist (SR141716A, Rimonabant) prior to extinction training.
Using 2-factor analysis of variance (ANOVA), we found a significant interaction between drug treatment and opsin group interaction term for freezing during extinction retrieval (F(1,37)=4.742, P=0.0359, ηp2=0.10). Follow-up post hoc comparisons revealed that vehicle-treated control animals receiving mPFC→BLA excitation during extinction training had lower levels of freezing on light-free extinction retrieval tests, as compared to vehicle-treated YFP-expressing controls. By contrast, animals receiving the combination of photoexcitation and SR141716A treatment froze at levels that pairwise comparison showed were not statistically different from vehicle-treated YFP-expressing controls (Figure 1G,H and Figure S1C).
These data show that the facilitation of extinction following mPFC→BLA excitation depends upon eCB-CB1R signaling, but do not localize this effect to BLA. We therefore repeated the same optogenetic photoexcitation procedure but now microinfused SR141716A directly into BLA prior to extinction training. We found that BLA-targeted CB1R antagonism was sufficient to block the behavioral effects of mPFC→BLA excitation on light-free extinction retrieval. This conclusion was statistically supported by a significant 2-factor ANOVA drug treatment and opsin group interaction term (F(1,40)=4.511, P=0.0399, ηp2=0.09), and by similar freezing values in the excited SR141716A-treated and non-excited, vehicle-treated YFP groups - which pairwise comparison confirmed were not significantly different (Figure 1I,J and Figure S1D).
Together, these findings show that exciting mPFC axons in BLA during extinction training mobilizes BLA eCBs, which in turn promote extinction via CB1R-mediated signaling. It was nonetheless noteworthy that although freezing levels on extinction retrieval were lower in the ChR2-photoexcited group that was treated with vehicle than in the corresponding photoexcited group treated with SR141716A, pairwise comparison showed that this difference was not statistically significant. This was true for both the intra-BLA and systemic antagonist experiments and suggests that while blockade of eCB-CB1R signaling interferes with the ability of mPFC→BLA excitation to facilitate extinction, it is not sufficient to fully ablate this effect. The most parsimonious reason for this is that optogenetic photoexcitation of this pathway (particularly when using the ChR2 virus titer employed, see 30) engaged additional, eCB-independent mechanisms to promote extinction.
Dynamic prefrontal-amygdala eCB signaling during extinction
Our data thus far show that BLA eCBs are mobilized by mPFC→BLA excitation during extinction training and promote the formation of extinction memory. These findings led us to gain clearer insight into the temporal dynamics of eCB signaling by employing a genetically-encoded GPCR activation-based eCB biosensor (GRABeCB2.0) 44 to measure eCBs at mPFC→BLA neurons as animals underwent extinction. To do so, we virally transfected mPFC neurons with GRABeCB2.0 and used in vivo fiber photometry to record the GRABeCB2.0 signal emitted at mPFC inputs to BLA (Figure 2A–C). The GRABeCB2.0 signal was normalized to a simultaneously measured 405 nm fluorescence channel to evaluate motion-related artifacts.
Figure 2: Stimulus-related temporal patterning of eCBs at mPFC→BLA neurons during extinction.
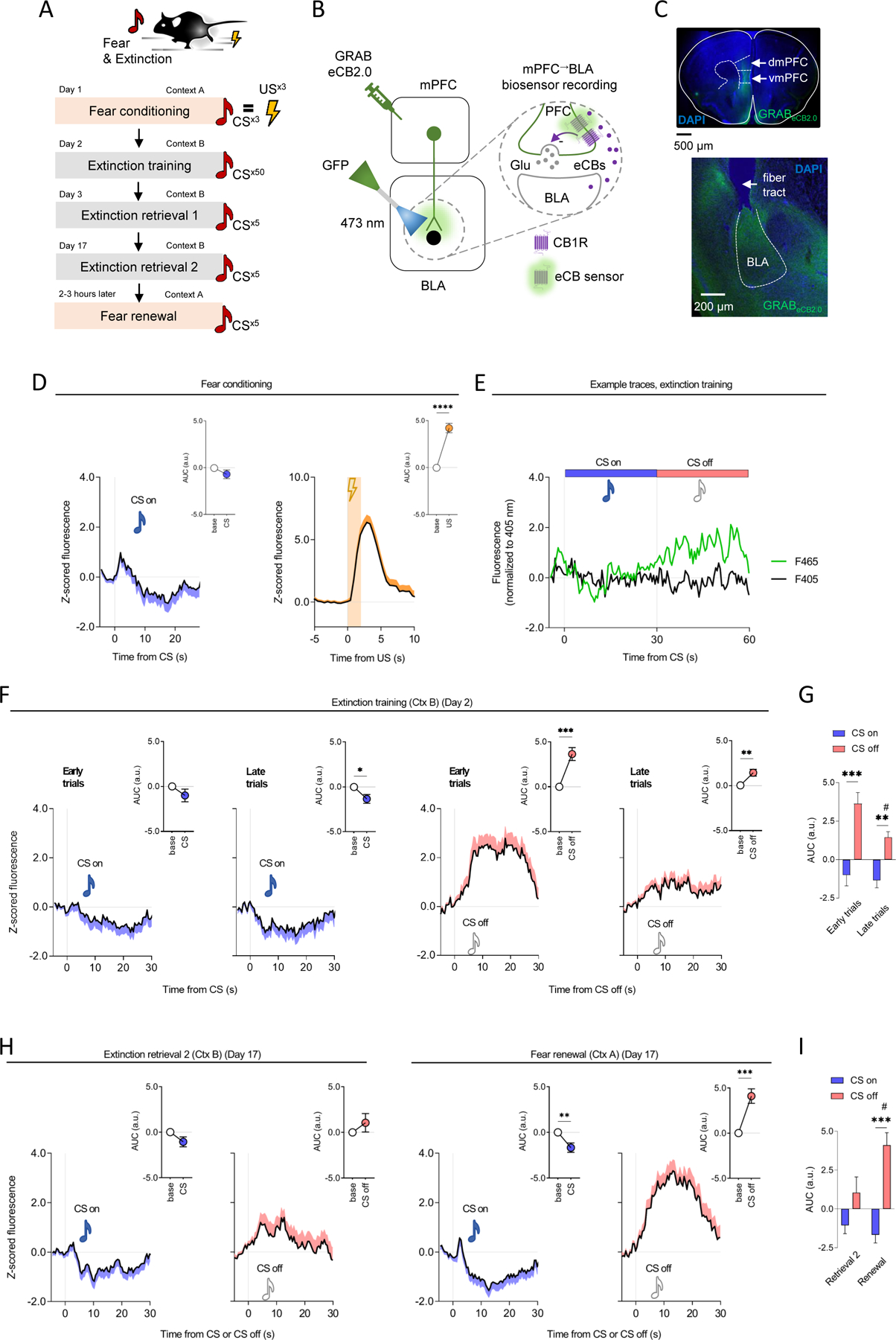
(A-C) Behavior (A) and in vivo fiber photometry (B) procedure for mPFC→BLA GRABeCB2.0 biosensor recordings (n=16 mice). Virus expression in mPFC and BLA (C).
(D) Increased GRABeCB2.0 signal during conditioning US (paired t-test for US versus baseline: t(15)=6.451, P<0.0001, ηp2=0.74) (not CS P>0.05) presentation.
(E) Example photometry traces showing increased GRABeCB2.0 signal during extinction training CS off periods.
(F,G) Increased GRABeCB2.0 signal at CS off (not CS on, P>0.05) during extinction training (paired t-test for first 5-trial block (early extinction) versus baseline: t(15)=5.100, P=0.0001, ηp2=0.63, paired t-test for last 5-trial block (late extinction) versus baseline: t(15)=3.754, P=0.0019, ηp2=0.48) (F). Reduced CS off-related GRABeCB2.0 signal on late versus early extinction trials (RM-ANOVA event-effect: F(1,30)=8.524, P=0.0066, ηp2=0.04, test-phase effect: F(1,30)=27.110, P<0.0001, ηp2=0.37, interaction: F(1,30)=1.700, P=0.2023, ηp2=0.02, Holm-Šídák's tests: early-CS off versus early-CS on: P=0.0001, late-CS off versus late-CS on: P=0.0098, late-CS off versus early-CS off: P=0.0212) (G).
(H,I) Increased GRABeCB2.0 signal at CS off (paired t-test for CS off versus baseline: t(12)=5.085, P=0.0003, ηp2=0.68), decreased at CS on (paired t-test for CS on versus baseline: t(12)=3.234, P=0.0072, ηp2=0.47) periods during fear renewal, but not during extinction retrieval (P>0.05) (H). Higher CS off-related GRABeCB2.0 signal on renewal versus retrieval 2 (RM-ANOVA event effect: F(1,26)=4.232, P=0.0498, ηp2=0.03, test-phase effect: F(1,26)=19.390, P=0.0002, ηp2=0.31, interaction: F(1,26)=4.204, P=0.0505, ηp2=0.07, Holm-Šídák's tests: renewal-CS off versus renewal-CS on: P=0.0003, renewal-CS off versus retrieval-CS off: P=0.0126) (I).
Data are represented as mean±SEM population average Z-scores normalized to 5-second pre-event baseline. Time-normalized AUC values correspond to 28 post-CS and 2 seconds post-US during conditioning (D), and 30 seconds post-CS on and CS off for extinction training, retrieval and renewal (F-I). ****P<0.0001, ***P<0.001, **P<0.01, #P<0.05.
We first sought to pharmacologically confirm the efficacy and specificity of the GRABeCB2.0 sensor as a tool to readout eCBs at mPFC input to BLA - as has been previously shown for cortical inputs to the striatum, and for neurons in the substantia nigra, hippocampus and BLA 44–47. After expressing GRABeCB2.0 in mPFC→BLA neurons, animals were systemically injected with compounds that either directly activate CB1R (WIN 55,212–22) or augment endogenous eCB levels via inhibition of eCB degradation (JZL195, URB597). We observed that all three compounds increased GRABeCB2.0 signal - albeit with differing kinetic profiles - and, importantly, found that in each case the effects were blocked by administration of a selective CB1R antagonist (SR141716A) (Figure S2A–D).
We next performed GRABeCB2.0 recordings as animals underwent fear conditioning, full (50-trial) extinction training and retrieval testing. During conditioning, GRABeCB2.0 signal was increased in response to the US, but not the CS. Interestingly, during extinction training there was a modest decrease to the CS (CS on period) that was particularly pronounced on late (last 5-trial block) as compared to early (first 5-trial block) extinction training trials (Figure 2D–E). Most strikingly, we observed a robust increase in GRABeCB2.0 signal in the periods following CS presentation (CS off). Notably, we also found that this CS off-related increase was attenuated on late, relative to early, extinction training trials (Figure 2F,G and Figure S3P). When we tested female mice, we found a similar pattern of eCB responses to the US during conditioning and, importantly, during the extinction training CS off periods (i.e., higher GRABeCB2.0 signal on early as compared to late trials) (Figure 3A–C). These data demonstrate that extinction-related changes in eCBs at mPFC→BLA neurons occur across sexes.
Figure 3: Extinction-related eCBs at mPFC→BLA neurons in females and non-conditioned males.
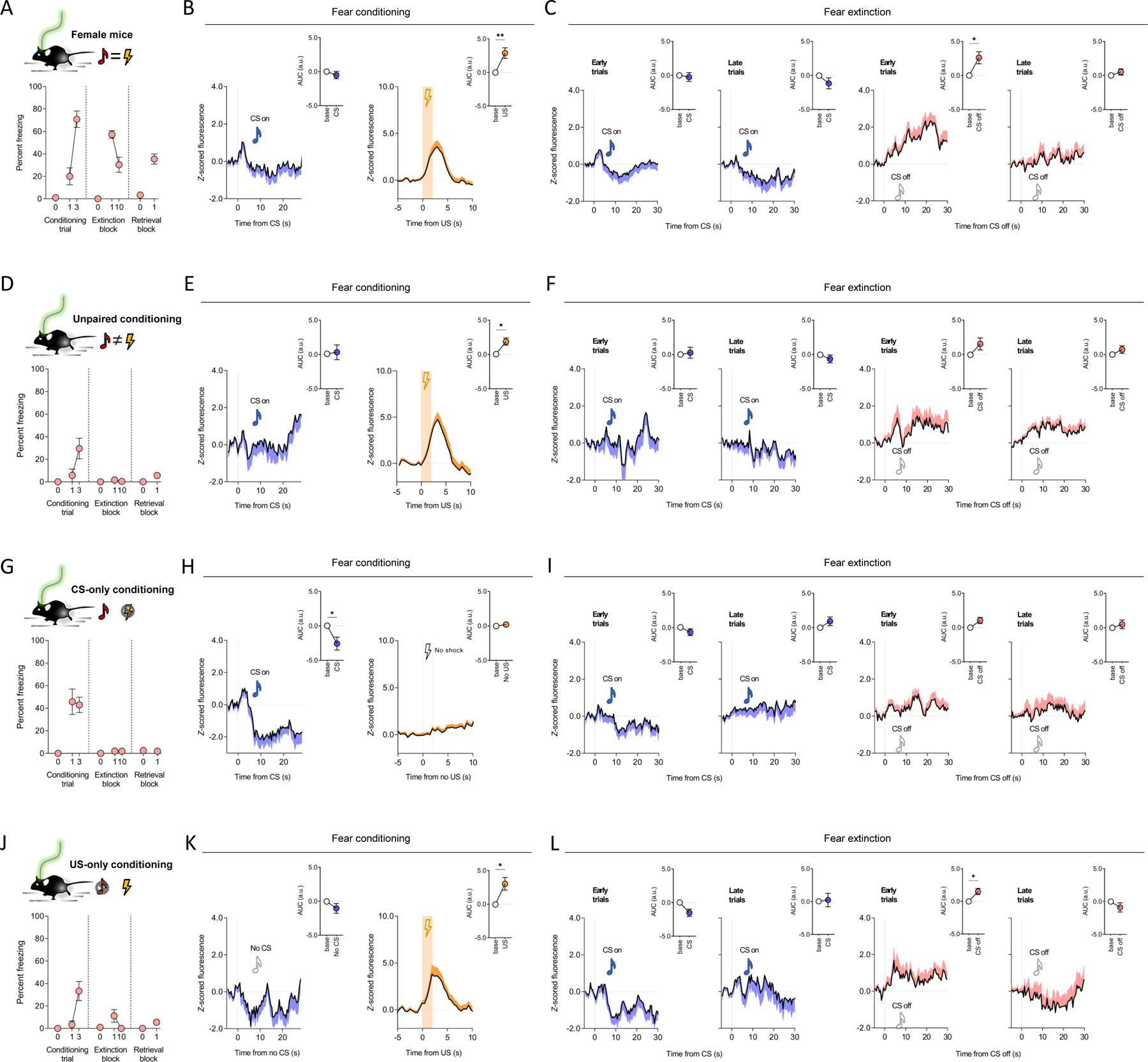
(A-C) In females (n=11 mice) freezing increased over conditioning trials (paired t-test for trial 1 versus trial 3: t(10)=5.369, P=0.0003, ηp2=0.74) and decreased from early (first trial-block) to late (last trial-block) extinction training (paired t-test: t(10)=4.006, P=0.0025, ηp2=0.62). Lower freezing on retrieval versus early extinction (paired t-test: t(10)=4.092, P=0.0022, ηp2=0.63) (A). Increased GRABeCB2.0 signal during conditioning US presentation (paired t-test for US versus baseline: t(10)=3.659, P=0.0044, η 2=0.57) (not CS P>0.05) presentation (B) and during CS off periods on early (paired t-test for CS off versus baseline: t(10)=2.927, P=0.0151, ηp2=0.46) (not late P>0.05) extinction training (C).
(D-F) In unpaired conditioned males (n=7 mice), freezing did not increase over conditioning trials (paired t-test for trial 1 versus trial 3: t(6)=2.443, P=0.0503, ηp2=0.50) and was low during extinction and retrieval
(D). Increased GRABeCB2.0 signal during conditioning US presentation (paired t-test for US versus baseline: t(6)=3.469, P=0.0133, ηp2=0.67) (not CS P>0.05) presentation (E) and no change on extinction trials (P<0.05) (F).
(G-H) In CS-only conditioned males (n=7 mice), freezing did not increase over conditioning trials (P<0.05) and was low during extinction training and retrieval (G). Decreased GRABeCB2.0 signal during conditioning CS presentation (paired t-test for CS versus baseline: t(6)=0.311, P=0.7663, ηp2=0.02) (not the US-equivalent period, P>0.05) (H) and showed no change during extinction training CS on (P>0.05) or CS off periods (P>0.05) (I).
(J-L) In US-only conditioned males (n=6 mice), freezing increased over conditioning trials (paired t-test for trial 1 versus trial 3: t(5)=3.503, P=0.0172, ηp2=0.71) and was low on extinction trials. (J). Decreased GRABeCB2.0 signal during conditioning US presentation (paired t-test for US versus baseline: t(5)=3.181, P=0.0245, ηp2=0.67) (not CS-equivalent period, P>0.05) (K) and CS off period during early (paired t-test for CS off versus baseline t(5)=3.183, P=0.0245, ηp2=0.67) (not late, P>0.05) extinction training (L).
Data are represented as mean±SEM population average Z-scores normalized to 5-second pre-event baseline. Time-normalized AUC values correspond to 28 post-CS and 2 seconds post-US during conditioning, and 30 seconds post-CS on and CS off for extinction training, retrieval and renewal. ***P<0.001, **P<0.01, *P<0.05 with multiple-comparison Bonferroni correction for extinction GRABeCB2.0 data.
In control experiments, male animals that received unpaired presentations of the CS and US during conditioning showed no significant change in GRABeCB2.0 signal during extinction training CS off periods (Figure 3D–F). Stimulus-related eCB correlates were also either absent or diminished in experiments in which either the CS or US was omitted during fear conditioning in male mice (Figure 3G–L). These data suggest that eCB responses observed during extinction CS off periods in conditioned animals were a primarily a function of the learned association of the CS with an aversive outcome. Lastly, we found that stimulus-related eCB responses were absent in fear conditioned mice systemically administered the CB1R antagonist, SR141716A, prior to extinction training (Figure S2E–I).
The finding that GRABeCB2.0 signal was increased during CS off periods suggest that eCBs might signal at mPFC→BLA to track the expectancy of shock-omission during extinction – which is high early in extinction and low late in extinction. If so, we reasoned that if eCB decreases over extinction training reflect a learned expectation that the US will be omitted, then this signal should be attenuated when fear levels are low during post-training testing. Furthermore, because learned expectancy of shock-omission is bound to the context in which extinction occurs 48, a change in extinction context that results in renewal of fear may result in shock-omission again being unexpected and, as a result, the shock-omission-related GRABeCB2.0 signal might increase in parallel. In line with these predictions, when we tested animals for extinction retrieval and fear renewal on the same day, we found that GRABeCB2.0 signal during shock-omission was low on retrieval and high on renewal, as were corresponding freezing levels (Figure 2H,I and Figure S3E).
In sum, these findings show for the first time that BLA eCBs dynamically signal at mPFC→BLA neurons during fear extinction in a manner that parallels the learned expectancy of shock-omission.
Prefrontal-amygdala eCB signaling related to movement and feeding
Extinction-related learning and the behavioral readout of this learning (i.e., freezing) change concurrently over the course of extinction. This raises the question of whether eCB levels could reflect movement, or its absence, during extinction, rather than extinction learning per se. In this context, although we simultaneously measured fluorescence at 405 nm as a reference to assess possible motion related changes, this wavelength might not be isosbestic for the GRABeCB2.0 sensor. However, arguing against the possibility that extinction-related changes in the GRABeCB2.0 signal reflected movement, we found only a modest, non-significant correlation across animals between GRABeCB2.0 signal and average levels of freezing during extinction training (Figure S3C). Weak, non-significant correlations were also evident when comparing the GRABeCB2.0 signal with freezing during the CS on and CS off periods on each of the individual 50 extinction training trials (Figure S3D,E). In fact, despite GRABeCB2.0 signal values being higher during the CS off period on the first than last extinction training trial-block, average freezing levels during these two periods were similar (both ~25%). Lastly, when animals were allowed to freely explore a neutral cage and we examined the relationship between the magnitude of GRABeCB2.0 signal and movement speed, we again found no clear association between the two measures (Figure S3F). Combined, these various analyses indicate that extinction-related GRABeCB2.0 signal changes report extinction-related learning rather than our behavioral measures of the manifestation of this learning or general movement per se.
That there is no clear relationship between the GRABeCB2.0 signal and measures of movement does not of course preclude the possibility that correlates of eCB signaling in the mPFC→BLA are only evident during extinction. Indeed, the possibility that eCB correlates in this pathway would only occur during fear extinction seems unlikely given the multiple functional roles of the mPFC and BLA. In this context, although the aim of the current study was not to broadly characterize the role of eCBs in this pathway, we did obtain preliminary insight into the potential eCB changes in the mPFC→BLA pathway by recording GRABeCB2.0 signal, via fiber photometry, as animals engaged in a non-aversive behavioral task - consumption of a palatable food. We found that food consumption was associated with an increase in GRABeCB2.0 signal. Notably, these eCB responses were only apparent when animals were in a fasted, but not sated state (Figure S3G–K), showing that the mPFC→BLA GRABeCB2.0 signal scales with the appetitiveness of food.
One interesting possibility raised by these findings is that eCBs acting in this pathway could serve to both suppress learned fear responses (via extinction) and enable foraging and food consumption. In this way, eCBs would adaptively modify the balance between two essential survival functions (predation and starvation). Of further potential relevance here is prior cross-species evidence that extinction and reward are encoded by overlapping neural mechanisms, including in BLA, wherein extinction-encoding BLA neurons respond to food in fasted mice 49. This overlap in function aligns with the idea that extinction itself generates a rewarding state 50–52. As such, while the current data remain preliminary, they may suggest mPFC→BLA eCB activity might be another example of an overlapping neural substrate for extinction and responding for reward.
eCB-CB1R signaling modulates prefrontal-amygdala synaptic neurotransmission
A key feature of eCBs is their ability to suppress presynaptic release of neurotransmitters 53–55 which, in the case of BLA-projecting mPFC principal neurons, is glutamate. However, although our GRABeCB2.0 data suggest that extinction engages eCB signaling at mPFC→BLA neurons, we have thus far not directly shown that CB1Rs modulate glutamatergic neurotransmission in this pathway. To address this question, we transfected mPFC neurons, specifically those located in vmPFC (because the vmPFC→BLA pathway is strongly implicated in extinction 30,39,40), with ChR2 and photoexcited their axons in BLA (via 2-millisecond light pulses, 0.6–1.5 mW LED intensity) to record optically-evoked excitatory postsynaptic currents (oEPSCs) in BLA neurons. We found that oEPSC paired-pulse ratio was increased in the presence of a CB1R agonist, a synthetic cannabinoid, (CP-55940) or selective inhibitors of either monoacylglycerol lipase (MAGL) (JZL184) or FAAH (PF-3845) (Figure 4A–D and Figure S4A–L). These are effects indicative of CB1R-mediated suppression of glutamate release probability at mPFC→BLA synapses. A secondary finding was that these effects were evident irrespective of whether BLA neurons were positive for Thy-1, a putative marker of extinction-mediating BLA neurons 56.
Figure 4: eCB-CB1R signaling modulates mPFC→BLA neurotransmission.
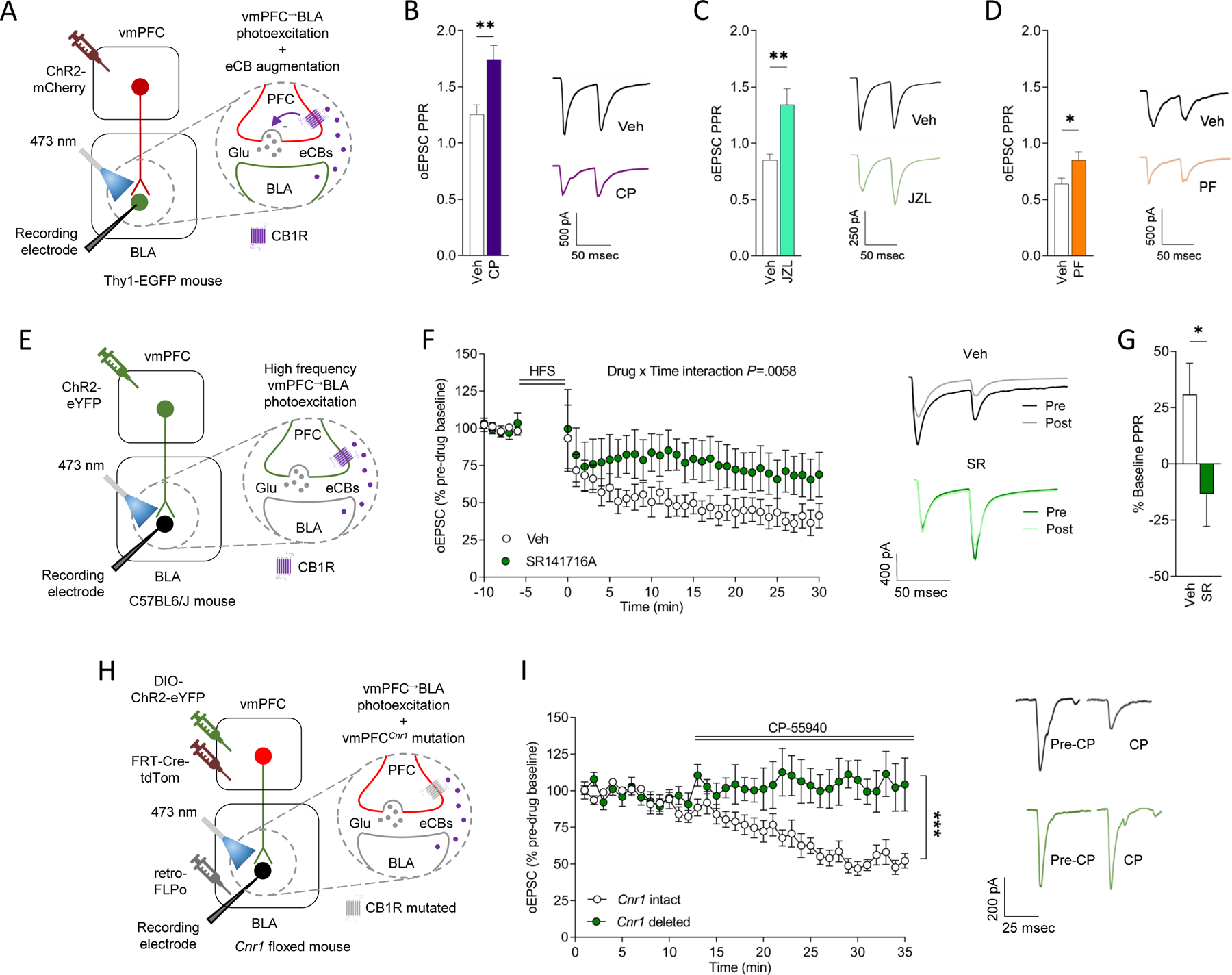
(A-D) In vitro recordings of CP55–940 (CP, CB1R agonist, Veh n=4 mice/22 cells, CP n=4 mice/15 cells), JZL184 (JZL, MAGL inhibitor, Veh n=3 mice/19 cells, JZL n=3 mice/14 cells) and PF-3845 (PF, FAAH inhibitor, Veh n=5 mice/22 cells, PF n=5 mice/18 cells) effects on ChR2-mediated optically-evoked EPSCs at vmPFC→BLA synapses (A). Higher oEPSC paired-pulse ratio (PPR) in slices incubated with CP (unpaired t-test for CP versus Veh: t(35)=3.344, P=0.0020, ηp2=0.24) (B), JZL (unpaired t-test for JZL versus veh: t(31)=3.587, P=0.0011, ηp2=0.29) (C) or PF (unpaired t-test versus veh: t(38)=2.389, P=0.0220, ηp2=0.13) (D), with example traces.
(E-G) In vitro recordings of SR141716A (SR, CB1R antagonist) effects on ChR2-mediated oEPSCs at mPFC→BLA synapses following vmPFC neuron high-frequency stimulation (HFS) (n=3 mice/6 cells Veh/n=3 mice/6 cells SR) (E). Versus Veh, SR attenuated an HFS-induced decrease in oEPSCs, with example traces (ANOVA drug x time interaction: F(35,385)=1.764, P=0.0058, ηp2=0.03) (F). Lower oEPSC PPR 25–30 minutes post-HFS in slices incubated with SR (unpaired t-test for SR versus Veh: t(10.97)=2.214, P=0.0489, ηp2=0.31) (G).
(H-I) In vitro recordings of effects of CP on ChR2-mediated oEPSCs at mPFC→BLA synapses following virus-mediated Cnr1 deletion from vmPFC→BLA neurons (n=3 mice/6 cells Veh/n=3 mice/6 cells SR) (H). CP decreased oEPSCs in Cnr1 intact not deleted mice (ANOVA drug x time interaction: F(1,17)=15.030, P=0.0012, ηp2=0.20), with example traces (I). Data are represented as mean±SEM .***P<0.001, **P<0.01, *P<0.05.
Next, we asked whether evoking activity at vmPFC→BLA synapses is sufficient to cause an increase in BLA eCBs and produce long-term depression (LTD) of glutamate release at mPFC→BLA synapses. To do so, we again expressed ChR2 in vmPFC neurons to enable photoexcitation-evoked glutamate release from vmPFC axons in BLA. After establishing a stable baseline (under 2-millisecond light pulses, 1–3 mW LED intensity), vmPFC axons were repeatedly photoexcited (via 5 trains of 30-second 50 Hz pulses, 30-second inter-train interval, the same intensity as baseline) while recording evoked responses in BLA neurons. We found that stimulation significantly decreased EPSCs relative to pre-stimulation baseline (i.e., produced LTD) in slices in which vehicle was applied. By contrast, there was no significant decrease in EPSCs when slices were incubated (for 30 minutes) and bathed in the CB1R antagonist, SR141716A, during recordings (Figure 4E–G). These findings show that mPFC activity is in and of itself sufficient to recruit eCBs and produce consequent CB1R-mediated suppression of glutamate release at vmPFC→BLA synapses.
To extend these findings, we employed a viral intersectional strategy to selectively delete Cnr1 from vmPFC→BLA neurons and then tested whether this caused loss of CB1R-mediated modulation of glutamate release. The approach entailed expressing a retrograde FLPo-expressing virus into BLA and a Flp-FRT-dependent Cre-expressing virus into vmPFC (along with ChR2) of Cnr1-floxed mice 57. In a viral control group in which Cnr1 was not deleted, application of the synthetic cannabinoid, CB1R agonist, CP-55940, reduced ChR2-mediated optically-evoked (achieved via 2-millisecond light pulses, 0.6–1.5 mW LED intensity) BLA neuronal EPSCs, indicating inhibition of glutamate transmission. By contrast, CP-55940 failed to prevent the effect of photoexcitation on BLA EPSCs in Cnr1-deleted animals, confirming that CB1R mediates the ability of CP-55940 to inhibit excitation-elicited synaptic glutamate release (Figure 4H–I).
Together, these data provide convergent evidence that eCBs, signaling via CB1R, modulate glutamatergic neurotransmission at vmPFC→BLA synapses.
CRISPR-Cas9-engineered prefrontal-amygdala Cnr1 mutation impairs extinction
Thus far, our findings show that eCBs dynamically signal and modulate neurotransmission in the mPFC→BLA pathway during extinction, but they do not address the question of whether behavior-related fluctuations in eCBs are causally relevant to extinction. To address this question we engineered a single-viral Staphylococcus aureus Cas9 (SaCas9)-guided CRISPR mRNA Cnr1-loss-of-function viral construct 58,59 and then used a viral intersectional strategy to selectively express the construct in mPFC→BLA neurons (Figure 5A,C,F,H). Combining BaseScope in situ hybridization with immunohistochemical staining indicated a qualitative absence of Cnr1 transcripts in Cre-expressing BLA-projecting vmPFC neurons of animals expressing the CRISPR-Cas9 Cnr1 virus, relative to Cnr1 intact virus controls (Figure 5B,C, Figure S5C,D).
Figure 5: CRISPR-Cas9 Cnr1 deletion from vmPFC→BLA not dmPFC→BLA neurons impairs extinction.
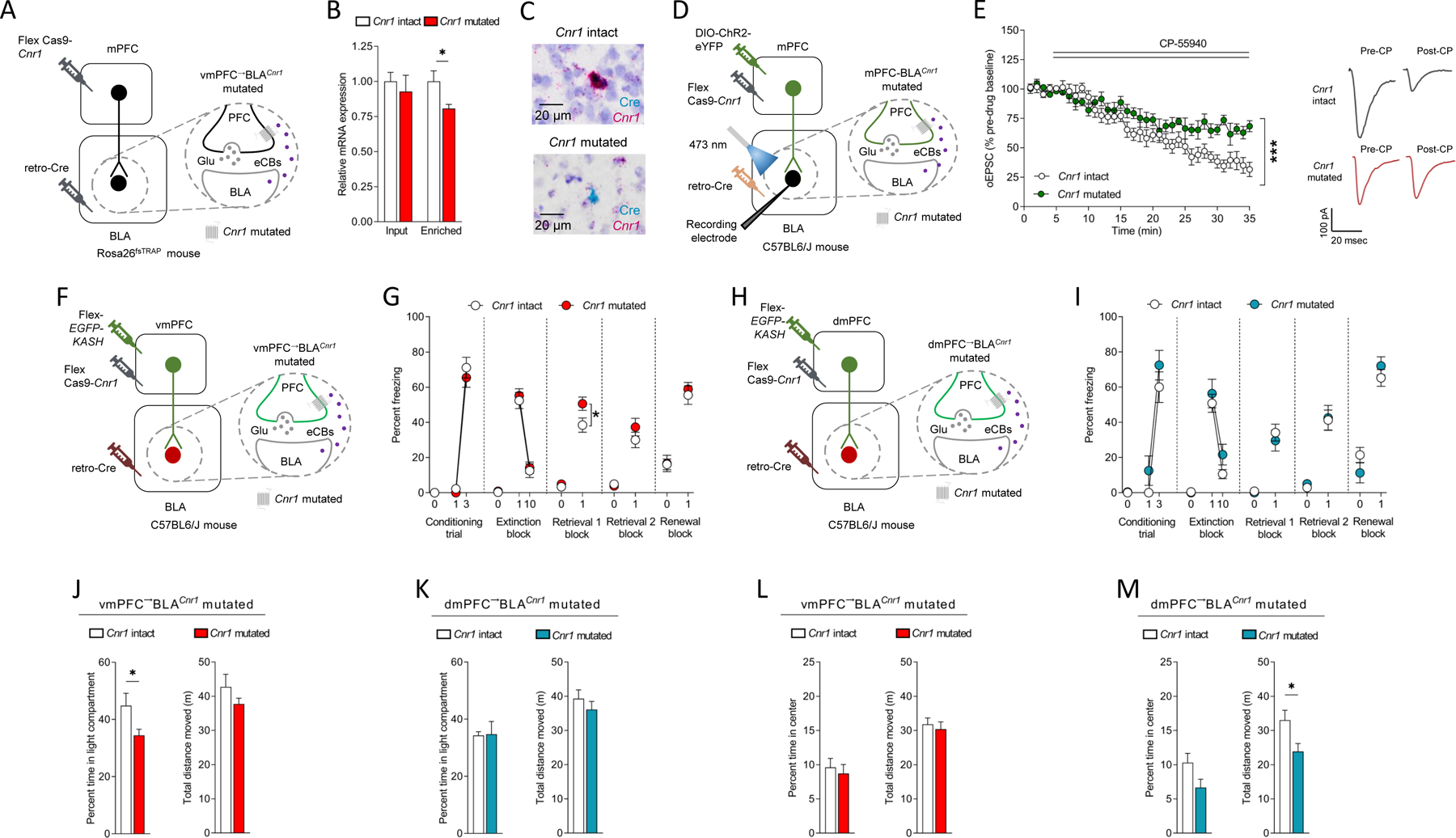
(A-C) Viral strategy for vmPFC→BLA CRISPR-Cas9 Cnr1 mutation in Rosa26fsTRAP mice (A). Less GFP-enriched purified RNA (not GFP-unbound input RNA) expression in mutated versus intact (unpaired t-test: t(11)=2.543, P=0.0273, ηp2=0.37, intact n=6/mutated n=7) (B). Example images from combined BaseScope in situ hybridization and immunohistochemistry showing Cnr1 mRNA loss in Cre-expressing BLA-projecting vmPFC neurons in mutated (not intact) (C).
(D,E) In vitro recordings of CP55–940 (CP, synthetic cannabinoid, CB1R agonist) on ChR2-mediated optically-evoked EPSCs at vmPFC→BLA synapses (D) showing lesser effect in mutated versus intact (ANOVA time x group: F(34,408=4.208, P<0.0001, ηp2=0.07, n=3 mice/6 cells intact/n=3 mice/6 cells mutated), with example traces (E).
(F,G) vmPFC→BLA Cnr1 mutation (F). Freezing increased over conditioning trials (RM-ANOVA: F(1,18)=279.100, P<0.0001, ηp2=0.88) and decreased over extinction trial-blocks (RM-ANOVA: F(1,18)=124.400, P<0.0001, ηp2=0.74). Less freezing on extinction retrieval 1 in mutated versus intact (RM-ANOVA F(1,18)=6.338, P=0.0215, ηp2=0.11) (intact n=9, mutated n=11) (G).
(H,I) dmPFC→BLA Cnr1 mutation (H). Freezing increased over conditioning trials (RM-ANOVA: F(1,13)=87.360, P<0.0001, ηp2=0.69), decreased over extinction trial-blocks (RM-ANOVA: F(1,13)=45.370, P<0.0001, ηp2=0.58) and was similar in mutated and intact across test-phases (intact n=7, mutated n=8) (I).
(J,K) Less light/dark test compartment time in vmPFC→BLA Cnr1 mutated (J) (unpaired t-test: t(18)=2.307, P=0.0331, ηp2=0.23, intact n=9/mutated n=11), not dmPFC→BLA Cnr1 mutated (intact n=8/mutated n=8) (K), versus intact.
(L,M) Less novel open field distance moved (unpaired t-test: t(14)=2.467, P=0.0272, ηp2=0.30, intact n=8/mutated n=8) in dmPFC→BLA Cnr1 mutated (M), not vmPFC→BLA Cnr1 mutated (L), versus intact.
Data are represented as mean±SEM. ***P<0.001, *P<0.05.
We used the same intersectional strategy to express the CRISPR-Cas9 virus in the BLA-projecting mPFC neurons of (male and female) Rosa26fsTRAP mice 60 and performed rt-PCR on mRNA isolated from these neurons via translating ribosome affinity purification (TRAP). This analysis indicated significantly decreased Cnr1 levels in the somata of GFP-enriched purified, but not input (GFP-unbound S20 post mitochondrial supernatant) RNA of BLA-projecting neurons in mPFC, relative to viral controls (Figure 5B, Figure S5C). Conversely, Cnr1 levels in the somata of GFP-enriched neurons within BLA were unaltered (Figure S5C).
While our assessment of Cnr1 mRNA levels provides a useful metric for demonstrating mutagenesis occurred, it is not a direct measure of total mutagenesis because CRISPR-Cas9 mutagenesis results in a reduction in mRNA through nonsense mediated decay and is not a direct measure of loss of protein function. Therefore, we again intersectionally expressed the CRISPR-Cas9 Cnr1 virus along with ChR2 in mPFC→BLA neurons and performed in vitro slice electrophysiological recordings of light-evoked (via 2-millisecond light pulses, 1–3 mW LED intensity) synaptic responses in BLA. We found that the ability of CP-55940 to suppress photoexcitation-generated EPSCs in BLA neurons was significantly reduced in animals expressing the Cnr1 virus (Cnr1 mutated) than in controls expression a control virus (Cnr1 intact). These data confirm that CRISPR-Cas9 mutagenesis resulted in functional loss of CB1R-mediated inhibition of glutamate release from mPFC→BLA synapses (Figure 5E).
On the basis of these results, we selectively expressed the CRISPR-Cas9 virus in vmPFC→BLA neurons and examined the consequences for behavior. We found that the vmPFC→BLACnr1 mutated animals exhibited similar freezing levels to Cnr1 intact viral controls during conditioning and extinction training. Critically, however, freezing levels on extinction retrieval testing were significantly higher in mutant animals than in Cnr1 intact controls - an effect consistent with impaired extinction (Figure 5G). Freezing levels were similar between groups on a second extinction retrieval test performed two weeks later, which either indicates that the extinction-impairing effects of the mutation were transient and/or that more remote extinction memories (which are meditated by different neural mechanisms than recent extinction memory 61–63) are relatively insensitive to loss of Cnr1 from vmPFC→BLA neurons.
Additional behavioral testing showed that vmPFC→BLACnr1 mutated animals had heightened levels of anxiety-like behavior in the light/dark exploration test (but not open field, elevated plus-maze or marble burying tests) (Figure 5J,L and Figure S5F–H), and ate less food (Figure S5E) under the same fasted conditions in which we had observed elevated GRABeCB2.0 signal at mPFC→BLA neurons. These data demonstrate that selective loss of Cnr1 from vmPFC→BLA neurons is sufficient to impair extinction memory formation and cause other behavioral alterations including increased anxiety-like behavior and reduced seeking of an appetitive stimulus, depending on its motivational salience. Taken together with the results of our GRABeCB2.0 biosensor experiments, these findings further suggest eCB signaling at mPFC→BLA neurons play a role in modulating a range of behavioral states that extends beyond fear extinction.
In rats, projections of vmPFC and dmPFC neurons to the BLA are anatomically distinguishable 64 and BLA neurons activated by extinction are preferentially innervated by vmPFC axons 65. Moreover, while earlier studies ascribe an extinction-facilitating role to the vmPFC→BLA pathway 20–22,34,39,40, the dmPFC (prelimbic/ventral anterior cingulate cortex) and its projections to BLA is thought to be involved in the promotion and discrimination of fear 66,67. Given this literature, it was notable that when we tested the effects of the same manipulation in dmPFC→BLA neurons, we found it had no consequences for fear extinction or the other behaviors we tested (with the exception of a decrease in open field exploration (Figure 5H, I, K,M and Figure S5I–L). These data indicate that the extinction-related consequences of mutating Cnr1 in BLA-projecting neurons are specific to the vmPFC→BLA pathway.
These pathway-related differences were not related to lesser Cnr1 mRNA expression in dmPFC→BLA (~95%), as compared to vmPFC→BLA (~97%), neurons (Figure S5D); although Cnr1 expression does not necessarily equate to the functional ability of CB1R to modulate transmitter release 68. Hence, it could be that other factors, such as preferential innervation of BLA extinction-activated neurons by vmPFC axons 65, explain the selective extinction-mediating role of eCBs in the vmPFC→BLA pathway. Resolving this question would be an interesting area for future study.
Discussion
The eCB system plays an important role in a range of neurobiological and behavioral processes. Among these, there is growing evidence that eCBs modulate fear extinction and that targeting this system may be therapeutically tractable. However, the precise neural circuit basis of these actions has remained unclear. In the current study, we use a range of complementary approaches to identify the neural basis of eCB effects on fear extinction. We demonstrate that optogenetic excitation of mPFC→BLA neurons leads to the mobilization of eCBs and the facilitation of fear extinction. Using a novel eCB biosensor we reveal that eCBs at mPFC→BLA neurons change dynamically during extinction as animals learn that the CS no longer predicts shock. Furthermore, we show that eCBs, via CB1R, suppress glutamate release at vmPFC→BLA synapses and find that loss of CB1R specifically from vmPFC→BLA neurons is sufficient to impair extinction. Taken together, these findings provide a novel and important advance in our understanding of the neural substrates through which eCBs signal to modulate fear extinction.
We found that optogenetic excitation of mPFC→BLA terminals increases BLA eCB (AEA) levels and produces an associated enhancement in extinction memory formation. This behavioral effect could reflect a stimulation-induced augmentation of BLA AEA production that enhances the extinction-instructive actions of eCBs signaling at mPFC→BLA neurons. Relatedly, we found that optogenetic vmPFC excitation increases glutamatergic transmission at vmPFC→BLA synapses and that this effect is suppressed by CB1R-mediated signaling at these synapses. While prior studies suggest that fear extinction involves glutamatergic neurotransmission and associated synaptic plasticity in cortico-amygdala circuits 14,33,69, the current data demonstrate that eCBs modulate the vmPFC→BLA pathway and show this effect is critical for extinction – as evidenced by CRISPR-Cas9-mediated Cnr1 deletion from vmPFC→BLA neurons.
Another key novel finding came from our GRABeCB2.0 biosensor recordings of eCBs. These recordings showed that eCBs at mPFC→BLA neurons were increased, most prominently during early, extinction CS off periods. Hence, eCBs signal at mPFC→BLA neurons in relation to the two most salient events encountered by the animal during extinction - presentation of the shock-associated CS and the omission of expected shock delivery when the CS is terminated - and do so in a manner that changes over the course of extinction training. This finding generally echoes prior work suggesting that BLA-eCBs potentiate the emotional salience of fear stimuli and relay this information via connections with mPFC 42,70,71. However, our data advance this notion by showing that the stimulus-related temporal patterning of eCB activity at mPFC→BLA neurons may be critical to extinction, potentially by dynamically sculpting glutamatergic transmission at mPFC→BLA synapses. Hence, high eCB activity could signal unexpected shock-omission on early extinction CS off periods through suppression of synaptic glutamate release and, additionally, may disinhibit synaptic glutamate release when the CS is repeatedly presented over extinction training in the absence of shock.
Such a scheme would align with prior evidence that optogenetically exciting mPFC→BLA neurons specifically during CS presentation facilitates extinction30,39. More direct interrogation of the model will require novel tools that allow for precise temporal control over eCB signaling at mPFC→BLA synapses during CS on and off periods. Notwithstanding, our data generally fit with observations in other experimental settings showing that dynamic changes in eCB levels are associated with learning in other brain circuits; for example, GRABeCB2.0-measured eCB activity at substantia nigra dopamine (DA) neurons correlates with motor-learning 47.
Recent studies using in vivo single-unit and calcium photometry recordings have posited that extinction-related increases in medial ventral tegmental DA neuronal activity 52,72 report an extinction-mediating prediction error (see also 73,74–76). Although we suggest the eCB dynamics observed during extinction CS off periods resemble those characteristics of a neural substrate of a prediction error, shock-omission responses have been observed in some populations of mPFC 77 and amygdala neurons 43, it is currently unclear whether the eCB signal we observed in mPFC→BLA neurons represents a prediction error rather than another phenomenon, such as relief at removal of the threat cue 69. It should be borne in mind that the kinetics of the GRABeCB2.0 changes are also noticeably slower than the DA neuron calcium transients reported in the aforementioned studies, and more similar to those measured at optogenetically-stimulated cortical input to the dorsal striatum 46. These slow responses likely reflect the time taken for eCB production, release and retrograde transport to mPFC terminals and do not discount the possibility that eCBs and DA could functionally interact during extinction as has been reported for other processes 10,78,79.
Earlier work has shown that pharmacologically blocking CB1R in the BLA disrupts extinction 12. Here, using CRISPR-Cas9 gene editing we show that selective loss of Cnr1 from vmPFC→BLA neurons impaired extinction memory formation as evidenced on a retrieval test one day after extinction training whereas, by contrast, Cnr1 deletion from dmPFC→BLA neurons was without effect. However, vmPFC→BLA Cnr1 deletion did not impair extinction memory on a second retrieval test conducted two weeks later, which could indicate that the extinction-impairing effects of the mutation were transient and/or that more remote extinction memories are insensitive to loss of Cnr1 from vmPFC→BLA neurons. Given the neural mechanisms maintaining extinction memories are known to change as a function of the time since extinction learning 61–63, neuromodulators other than eCBs and/or neural pathways other than vmPFC→BLA may be greater functional importance at later extinction retrieval timepoints.
In this context, in addition to the effects of eCBs at mPFC→BLA synapses we report in the current study, eCB-CB1R signaling at cell types within BLA could also play a role in extinction, including astrocytes 80,81 and GABAergic interneurons 82. In line with this idea, eCBs suppress GABA release in the BLA 4,17 (see Figure S4) and the loss of this modulatory action is associated with decreased conditioned freezing and anxiety-like behavior 83–87. Additionally, CB1Rs are expressed on BLA cholecystokinin-expressing interneurons which, when optogenetically excited, promote extinction 88. Hence, CB1R-mediated signaling at GABAergic interneurons might contribute to the extinction-facilitating effects resulting from mobilization of BLA eCBs triggered by mPFC input.
Further adding to the potential breadth of BLA-eCB effects on extinction is that multiple eCBs with divergent effects could be involved. Whereas augmenting levels of AEA via either systemic or intra-BLA administration of a FAAH inhibitor improves extinction/decreases fear 18,85,89–91, systemic or BLA-specific increases in 2-AG produced by a MAGL inhibitor increases fear 85,92. We found that mPFC→BLA excitation during partial extinction training increased BLA levels of AEA but not 2-AG, and that GRABeCB2.0 signal at mPFC input to BLA were increased in response to pharmacological AEA augmentation. However, extinction per se associates with BLA increases in both these eCBs in some, not all, studies 12,18 and the GRABeCB2.0 biosensor measures both AEA and 2-AG transients 44–46. Further studies utilizing novel approaches that are able dissociate the in vivo dynamics of AEA and 2-AG will be required to parse the specific roles of these two eCBs to fear extinction.
In conclusion, taken together our findings suggest a novel model in which engagement of mPFC→BLA neurons during fear extinction mobilizes eCBs in BLA which, in turn, dynamically report changes in learned expectancy of the CS-shock association back to these neurons to support instantiation of extinction memory. These findings provide an important advance in our understanding of the neural circuit mechanisms underlying the extinction-facilitating effects of eCBs, with possible implications for the development and application of eCB-targeting drugs for extinction-deficient neuropsychiatric disorders.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Holmes (Andrew.Holmes@nih.gov).
Materials availability
No new materials were generated in this manuscript.
Data and code availability
The data reported in this paper will be shared by the Lead Contact upon reasonable request.
This paper does not report original code. Existing code from the David Root lab (https://www.root-lab.org/code) was modified for current analyses. The modified version of this code is deposited and publicly available at Zenodo. The associated DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Subjects
Male and female C57BL/6J (JAX stock #000664), Tg(Thy1-EGFP)MJrs/J (Thy1-EGFP, JAX stock #007788), and male and female B6.129S4-Gt(ROSA)26Sortm1(CAG-EGFP/Rpl10a,-birA)Wtp/J (Rosa26fsTRAP, JAX stock #022367) mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Breeding pairs of C57BL/6J-background Cnr1-floxed were generously provided by Dr. Josephine Egan (National Institute of Aging, Bethesda, MD, USA) 93 and bred by intercrossing heterozygous mice. Homozygous female Rosa26fsTRAP mice were crossed with male C57BL/6J mice and heterozygous offspring to produce mice from experiments. Mice (8–12 weeks old) were housed two/cage in a temperature- and humidity-controlled vivarium under a 12h light/dark cycle (lights on 0600 h, experiments conducted in the light phase), except following chronic fiber implantations – when they were single-housed to prevent cage-mates from damaging the implants by cage-mate allogrooming. Testing was conducted at least 4 weeks after surgery to allow for recovery from surgical procedures and, where applicable, to allow for virus expression. Experimental procedures were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the National Institute on Alcohol Abuse and Alcoholism Animal Care and Use Committees.
METHOD DETAILS
Stereotaxic surgery
Stereotaxic surgery for intracranial virus infusions and implantation of fluid cannulae and ferrules was performed using the laboratory’s standard procedures for targeting mPFC and BLA, as described previously 13,39. For intracranial virus infusions and implantation of fluid cannulae and ferrules, mice were placed in a stereotaxic frame after isoflurane anesthesia (David Kopf Instruments, Tujunga, CA, USA). Viral constructs were bilaterally infused at a constant rate over 10 minutes using a 0.5 µL syringe (Neuros model #7001 KH, Hamilton Robotics, Reno, NV, USA) connected to a SYS-Micro4 Controller (World Precision Instruments, Sarasota, FL, USA), which was left in place for 5 minutes after the virus had been completely infused to ensure diffusion into the tissue.
The coordinates for vmPFC injections were +1.80 mm anteroposterior (AP), ±0.35 mm mediolateral (ML) and −2.73 mm dorsoventral (DV) relative to bregma or +1.87 mm AP, ±1.40 mm ML and −2.84 mm DV at a 20° angle relative to bregma. The coordinates for dmPFC injections were +1.95 mm AP, ±1.00 mm ML and −1.83 mm DV at a 20° angle relative to bregma. The coordinates for BLA injections were −1.40 mm AP, −3.25 mm ML and −4.95 mm DV relative to bregma. Where required, during the same surgery, ferrules (optogenetic experiments) (200 μm diameter, numerical aperture, 0.37, product #CFMLC12U, ThorLabs, Newton, NJ, USA), dual opto-fluid cannulae (combined optogenetic/pharmacology experiments) (product #DiOFC-LG_P=6.5_320/430_5.0, Doric Lenses, Quebec, Canada) or wide ferrules (biosensor experiments) (400 μm, 0.57 numerical aperture, product #MFC_400/430-0.57-6mm_SM3-FLT, B280-4706-6, or MFC-400/430-0.66-6mm_SM3-FLT, B280–4653.6, Doric Lenses) were chronically implanted in BLA; either bilaterally (optogenetic and combined optogenetics/pharmacology experiments) or unilaterally, ipsilateral to the virus injection (biosensor experiments), and affixed to the skull with dental cement.
Behavioral testing
Pavlovian cued fear conditioning and extinction
Mice underwent cued fear conditioning and extinction using previously described methods 13,94,95. Fear conditioning was conducted on day 1 in a 30 × 25 × 25 cm operant chamber with metal walls and a metal rod floor (Context A). Chambers were cleaned between mice with fragrance-free detergent and soap. The chamber was then scented with a solution of 79.5% water:19.5% ethanol:1% vanilla extract.
Following a 180-second baseline, mice received 3 pairings of a 30-second, 75-dB white noise cue (conditioned stimulus, CS) that co-terminated with a 2-second 0.6-mA scrambled footshock (unconditioned stimulus, US). Each CS/US pairing was separated by a 60–90 second inter-trial interval (ITI). There was a 120-second stimulus-free period after the final pairing. The Med Associates Video Freeze Monitor System controlled presentation of the CS and US (Med Associates, Inc., Fairfax, VT USA).
Extinction training was conducted on day 2 in a 27 × 27 × 14 cm chamber with transparent walls and a floor covered with wood chips (Context B) and scented between mice with a solution of 99% water:1% acetic acid. After a 180-second baseline, there were either 10 (partial extinction procedure, used to reveal photoexcitation effects in optogenetics experiments) or 50 (full extinction procedure, used in all other experiments) CS presentations. Extinction retrieval was tested on day 3 and again on day 17 in Context B via 5 CS presentations. Fear renewal was tested on day 17 in Context A, 2h after extinction retrieval, via 5 CS presentations. On extinction training, retrieval and renewal, each CS was separated by a 5-second ITI, except for the in vivo fiber photometry experiments, in which there was a 30-second ITI to better dissociate eCB activity at CS-offset and onset. Throughout testing, freezing (defined as the absence of any visible movement other than respiration) was scored live or from video every 5-seconds by an experienced observer blind to experimental group.
Unpaired, no-US and no-CS conditioning
Unpaired conditioning (and subsequent extinction) was conducted as described above with the exception that the CS and US were temporally separated (33–50 variable interval between CS offset and US during conditioning. no-US and no-CS conditioning (and subsequent extinction) was conducted as described above with the exception that either the US or CS, respectively, was absent during conditioning.
Light/dark exploration test
One week after day 17 extinction retrieval/renewal testing, CRISPR-Cas9 Cnr1 mutated and intact mice were tested on the light-dark exploration test for anxiety-related behavior, based on previously described methods 96. The apparatus comprised an opaque black-Plexiglas compartment (39 × 13 × 16 cm) with a 13 × 8 cm aperture at floor level that allowed access to a larger (39 × 39 × 35 cm) white-walled square Plexiglas arena illuminated to ∼95 lux. Individual mice began a 15-minute test session by being gently placed at the entrance facing the opaque shelter. Time spent in the light and dark compartments and the total distance traveled and average movement velocity within the apparatus was measured by the Ethovision video tracking system (Noldus Information Technology Inc., Leesburg, VA, USA).
Elevated plus-maze test
Approximately 1 week after the light/dark exploration test, CRISPR-Cas9 Cnr1 mutated and intact mice were tested in the elevated plus-maze, based on previously described methods 97. The apparatus (San Diego Instruments, San Diego, CA, USA), elevated to a height of 38 cm above the floor and illuminated to ~95 lux, comprised 2 open arms (30 x 5 cm, with a 0.5 cm raised lip around the edge) and 2 closed arms (30 x 5, with a 15 cm wall around the edge), each extending from a common central platform (5 x 5 cm). Individual mice began a 5-minute test session by being gently placed in the center square facing an open arm. Time spent in the open and closed arms, and the total distance traveled and average movement velocity within the apparatus was measured by Ethovision (Noldus Information Technology Inc).
Novel open field test
Approximately 1 week after the elevated plus-maze test, CRISPR-Cas9 Cnr1 mutated and intact mice were tested in the novel open field test, using a previously described apparatus 98. The apparatus was a white opaque Plexiglas square arena (40 x 40 x 45 cm) illuminated to ~95 lux. Individual mice began a 10-minute test session by being gently placed in the center. Time spent in the 18 x 18 cm center square of the open field, and the total distance traveled and average movement velocity within the apparatus was measured by Ethovision (Noldus Information Technology Inc).
Marble burying test
Approximately 1 week after the novel open field test, CRISPR-Cas9 Cnr1 mutated and intact mice were tested in the marble burying test. A clean cage was lined with an even floor of 5 cm fresh sawdust bedding. Twelve glass marbles (clear, 13 mm diameter, 3.7 g in weight) were placed on top of the sawdust bedding and evenly distributed in a 4 X 3 array. Individual mice began a 30-minute test by being gently placed in the right corner of the cage. The number of buried marbles (submerged in the bedding to least 2/3 of their depth) was counted at the end of the test by an experimenter blind to experimental group. Marbles were cleaned with 70% ethanol and dried between mice.
Food consumption assay
One week after day 17 extinction retrieval/renewal testing (eCB biosensor experiment) or one week after the marble buying test (CRISPR-Cas9 Cnr1 mutation experiment), mice were tested on a food consumption assay, based on previously described methods 99. The apparatus was a neutral cage (37 x 15.5 x 12.5 cm) containing a bottom part of a petri dish (35 X 10 mm). During an initial habituation session, non-fasted mice were individually placed in the cage for 10 minutes and free over a further 10 minutes to eat small pieces of a highly palatable food, ‘bacon softies’ (Bio-Serv, Flemington, NJ, USA) placed in the petri dish. The following day, the same procedure was repeated, and total food consumed was measured by weighing the petri dish before and after the session: representing the sated state test. Immediately after the test, mice were food (not water) deprived for 24 hours until the procedure was repeated: representing the fasted state test. Food consumption behavior was videorecorded and analyzed using Behavioral Observation Research Interactive Software (BORIS) software 100.
In vivo optogenetics
To selectively optogenetically photoexcite mPFC axons in BLA, a viral construct containing the excitatory opsin, channelrhodopsin (ChR2) (rAAV5/CaMKII-hChR2(H134R)-eYFP, University of North Carolina (UNC) Vector Core, Chapel Hill, NC, USA, custom preparation, titer 1.1×1013 gc/mL), or the corresponding YFP-control construct (rAAV5/CaMKII-eYFP, UNC Vector Core, titer: 6×1012 gc/mL) was bilaterally injected (0.20–0.22 µl/hemisphere) into mPFC of C57BL6/J mice. Ferrules for optic fibers were chronically implanted, bilaterally, in BLA as previously described 30,39.
Prior to behavioral testing, mice were handled for 2 minutes/day for 5 days and then connected to the optic fiber cables in the home cage for 40 minutes/day for 3 days. During each extinction training CS presentation, blue light (λ = 473 nm) was shone (20 Hz, in 5-millisecond pulses) through the optic fibers using a laser (Opto Engine, Midvale, UT, USA), as previously described 94. Laser power was calibrated before each test by measuring the power at the tip of the patch cord with a PM100D optical power meter and S120C sensor (Thorlabs, Newton, NJ, USA) and multiplying the power value by the transmittance of the ferrule connection on each optic fiber 101,102. Light was not shone during the extinction retrieval and renewal tests, but the optic fiber cables (a possible contextual feature of extinction training) were attached.
eCB measurements via liquid chromatography/tandem mass spectrometry (LC/MS)
Following fear conditioning and fear retrieval/partial extinction training (to confirm fear memory formation), mice underwent optogenetic mPFC→BLA photoexcitation (as above) during a full extinction training session. Within 5 minutes of completing testing, mice were sacrificed by cervical dislocation and decapitation. Brains were removed on ice and snap frozen. Dorsal striatum (DS) and BLA dissected on dry ice using a 2-mm and 1-mm diameter micropunch, respectively and stored at −80℃ until LC/MS. Tissues were homogenized in 80–100 µL Tris (pH:8.0) buffer and protein concentrations determined using the Bradford assay, with BSA as a standard. Lipids were extracted and AEA and 2-AG levels quantified by LC/MS using multiple reactions monitoring, as previously described 95.
Briefly, the mass spectrometer was set for electrospray ionization operated in positive ion mode. The molecular ion and fragments for each compound measured were as follows: m/z 352.3→66.1 and 352.3→91 for [2H4] AEA (CID-energy: 12V and 56V, respectively), m/z 348.3→62.1 and 348.3→91 for AEA (CID-energy: 12V and 48V, respectively), and m/z 379.3→91 and 379.3→67.1 for 2-AG (CID-energy: 64V and 56V, respectively). Analytes were quantified using MassHunter Workstation LC/QQQ Acquisition and MassHunter Workstation Quantitative Analysis (Agilent Technologies, Santa Clara, CA, USA). AEA and 2-AG levels were determined against standard curves and expressed as fmol/mg and pmol/mg of protein, respectively.
Combined in vivo optogenetics and pharmacology
In one experiment, optogenetic mPFC→BLA photoexcitation (procedure as described above) was combined with intraperitoneal injection of 1.0 mg/kg (10 mL/kg injection volume) of the CB1R antagonist, SR141716A (Tocris Bioscience, Minneapolis, MN, USA, catalogue #0923), or an equivalent volume of DMSO:Tween 80:saline (1:1:8) vehicle, 50 minutes prior to extinction training. In a second experiment, optogenetic mPFC→BLA photoexcitation (as above) was combined with bilateral intra-BLA microinfusion of 2 µg/µL SR141716A (0.5 µl/min infusion rate, over 2 minutes, then 3 minutes to allow diffusion), or an equivalent volume of DMSO:Tween 80:saline (1:1:8) vehicle, 30 minutes prior to extinction training.
Verification of virus, optic fiber and cannula localization for optogenetics and pharmacology experiments
After completion of behavioral testing, mice were terminally overdosed with pentobarbital and transcardially perfused with ice cold PBS followed by ice cold 4% paraformaldehyde in phosphate buffer. Brains were removed from the skull and were post-fixed overnight at 4°C. Coronal sections (50-μm thick) were cut using a vibratome (Leica Biosystems, Buffalo Grove, IL, USA, model #VT1000S) in 0.1 M phosphate buffer and/or stored in 2% sodium azide in PBS for later usage. Sections were mounted and coverslipped with Vectashield HardSet mounting medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA).
Sections were imaged using either a Zeiss confocal microscope (Carl Zeiss Microscopy, White Plains, NY, USA, model #LSM 700) under Plan-Apochromat 20x/0.8 M27 objectives, an Olympus fluorescent microscope (Olympus, Center Valley, PA, USA, model #BX41) under UPlanFL N 4x/0.13 or PlanApo N 2x/0.08 objective, or an Olympus slide scanning microscope (Olympus, Virtual Slide Microscope system, model #VS120) under UPlanSApo 10x/0.40 or 40x/0.95 objectives. Mice adjudged to have inadequate or misplaced virus expression and/or ferrule/cannula placement were excluded from analysis.
In vivo fiber photometry measurement of GRABeCB2.0 biosensor activity
A viral construct containing the GRABeCB2.0 biosensor (AAV9-hSyn-eCB2.0, Vigene, Rockville, MD, catalogue #YL10070, titer 3.77 x 1013 gc/mL) was unilaterally injected (0.2 µL) into mPFC and optic fibers were chronically implanted, unilaterally (ipsilateral to the mPFC injection), in BLA of male and female C57BL6/J mice. Prior to behavioral testing, mice were handled for 2 minutes/day for 5 days and then connected to the optic fiber cables in the home cage for 1 hour/day for 3 days. To record fluorescence from the GRABeCB2.0 biosensor, a photometry system (Doric Lenses) used 2 continuous sinusoidally-modulated LEDs (Thorlabs) at 465 nm (1017 Hz) and 405 nm (1017 Hz) as a light source to excite the sensor and an isosbestic autofluorescence signal, respectively, as previously described 16. Light intensity at the tip of the patch cable (i.e., the interface of patch cable and fiber implant) was in the 50–100 µW range for each channel.
The LEDs were connected to a mini cube (Doric Lenses) and each bandpass was filtered before being coupled to a single large core (400 μm), high NA (0.48) optical fiber patch cord (Doric Lenses). Emitted light was unilaterally projected through the same mini cube, passed through a GFP emission bandpass filter (500–525 nm) and then focused onto a Newport Visible Femtowatt Photoreceiver (Doric Lenses). A RZ5P Processor acquisition system (Tucker-Davis Technologies, Alachua, FL, USA), equipped with a real-time signal processor controlled the LEDs and independently demodulated the fluorescence brightness due to 465 nm and 405 nm excitation. Fluorescence data were analyzed by applying a least-squares linear fit to the 405 nm signal to align it to the 465 nm signal. The resulting fitted 405 nm signal was then used to normalize the 465 nm signal as follows: ΔF= (465 nm signal − fitted 405 nm signal). Signals were downsampled to ~2 Hz for analysis.
Recordings during behavior.
US, CS onset and CS offset related changes in GRABeCB2.0 activity were measured during fear conditioning and extinction testing. Z-scores were calculated to compare ΔF values following each of these 3 events with the ΔF values during a 5 second period immediately preceding each event (z= [ΔF-mean(ΔF(t= −5 to 0))]/std, where std is the standard deviation of ΔF values during the pre-event period. Values for area under the curve (AUC) were calculated using MATLAB’s built-in ‘trapz’ function, which uses trapezoidal numerical integration to calculate the AUC (Z-score) between inputted x-values (time) on a graph of z-score versus time. Time-normalized AUC values for the US (2 seconds post-US) during conditioning, CS onset (28 seconds – i.e., excluding the final 2 seconds when the US was presented – post-CS onset) during conditioning, and CS onset and CS offset (30 seconds post-CS onset or CS-offset) during extinction training, retrieval and renewal were compared to each event’s respective (5-second) pre-event baseline.
Recordings during behavior + CB1R blockade.
The same optogenetic procedure as described above was employed, with the exception that mice received injection (intraperitoneally, in a volume of 10 mL/kg body weight) 5.0 mg/kg of the CB1R inverse agonist/antagonist, SR141716A (Tocris Bioscience catalogue #0923), or an equivalent volume of DMSO:Tween 80:saline (1:1:8) vehicle, 50 minutes prior to extinction training.
Recordings during drug challenge.
To assess drug-related changes in GRABeCB2.0 biosensor activity, recordings were performed in the home cage. After being connected to the cables, mice were given a 30-minute habituation period before the first drug was injected (intraperitoneally, in a volume of 10 mL/kg body weight). Separate groups of mice were used for each of the 7 challenge experiments. The drug, dose and duration of the recordings were as follows: 1) vehicle DMSO:Tween 80:saline (1:1:8) recorded for 60 minutes post-injection, 2) 5.0 mg/kg of the CB1R inverse agonist/antagonist SR141716A recorded for 60 minutes post-injection, 3) 5.0 mg/kg of the CB1R agonist WIN 55,212–22 (Tocris Bioscience catalogue #1038) recorded for 60 minutes post-injection, 3) a combination of 5.0 mg/kg SR141716A recorded for 30 minutes post-injection, then 5.0 mg/kg WIN 55,212–22 recorded for a further 60 minutes, 4) 20.0 mg/kg of the dual FAAH/MAGL inhibitor JZL195 (Tocris Bioscience catalogue #4715) recorded for 120 minutes post-injection, 5) a combination of 5.0 mg/kg SR141716A recorded for 30 minutes post-injection, then 20 mg/kg JZL195 recorded for a further 120 minutes, 6) 3.0 mg/kg of the FAAH inhibitor URB597 (Tocris Bioscience catalogue #4612) recorded for 120 minutes post-injection, 7) a combination of 5.0 mg/kg SR141716A recorded for 30 minutes post-injection, then 3.0 mg/kg URB597 recorded for a further 100 minutes. ΔF values were Z-scored to (5 minute) pre-drug baseline and expressed in a timeseries and as AUC averages (averaged over the entire post-drug period).
GRABeCB2.0 virus expression and optic fiber localization.
At the completion of testing, mice were terminally overdosed with pentobarbital and transcardially perfused with ice cold PBS followed by ice cold 4% paraformaldehyde in phosphate buffer. Coronal sections (50-μm thick) were cut using a vibratome (Leica Biosystems) in 0.1 M phosphate buffer and/or stored in 2% sodium azide in PBS. Sections were blocked for 2 hours in a blocking buffer containing PBS buffer solution, 0.3% Triton-X, 10% normal goat serum, and 1% bovine serum albumin, then rinsed and incubated overnight at 4°C with a chicken anti-green fluorescent protein (GFP) antibody (Abcam, Cambridge, MA, USA,1:1K, catalogue #ab13970) in PBS containing 10% blocking buffer and 0.3% Triton X-100. The next day, sections were rinse and incubated for 2 hours at room temperature in an Alexa 488 (goat anti-chicken, 1:1K, catalogue #A11039, Life Technologies, Carlsbad, CA, USA) secondary antibody in the PBS containing 10% blocking buffer and 0.3% Triton X-100 solution. The sections were then re-rinsed, mounted and coverslipped with Vectashield HardSet mounting media (Vector Laboratories) or mowiol-based mounting media (Polysciences Inc, Warrington, PA, USA, catalogue #17951). At the completion of testing, coronal brain sections were prepared and imaged as described above (under Verification of virus, optic fiber and cannula localization for optogenetics and pharmacology experiments).
eCB activity/freezing and movement correlations.
GRABeCB2.0 activity (AUC values) during extinction training were correlated (using Pearson’s correlation coefficient) with corresponding freezing levels during 1) the entire-session (averaged, excluding the 180-second baseline period), 2) CS on (averaged over 50 trials) and 3) CS off (averaged over 50 trials). Additionally, GRABeCB2.0 activity (AUC values) during 10-minute exploration of a neutral (37 x 15.5 x 12.5 cm) bare cage were correlated (using Pearson’s correlation coefficient) with Z-scored (to the entire session value) movement velocity (measured using the Ethovision video tracking system, Noldus Information Technology Inc).
In vitro slice electrophysiology recordings of eCB-mediated responses
eCB drug effects.
Thy1 mutant mice had a viral construct expressing ChR2 (AAV5-CaMKIIa-hChR2(H134R)-mCherry, UNC catalogue #AV7954, Addgene catalogue #26975, titer 1.6 x 1013 gc/ml) bilaterally injected into vmPFC (0.2 µl/hemisphere). At least 5 weeks later, mice were briefly anesthetized with isoflurane and transcardially perfused with ice-cold oxygenated (95% v/v O2, 5% v/v CO2) N-methyl-D-glucamine (NMDG) based ACSF 103 comprised (in mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na-ascorbate, 3 Na-pyruvate, 5 N-acetylcyctine, 0.5 CaCl2·4H2O and 10 MgSO4·7H2O. The brain was quickly removed and 250 µm coronal slices containing BLA were cut using a vibratome (Leica Biosystems, model #VT1000S) in the NMDG solution. Slices were incubated for 8–15 minutes at 32°C in oxygenated NMDG-ACSF then stored at 24°C until recordings were performed in HEPES-based ACSF containing (in mM): 92 NaCl, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 ascorbate, 3 Na-pyruvate, 5 N-acetylcyctine, 2 CaCl2·4H2O and 2 MgSO4·7H2O.
Recordings were performed in a submerged recording chamber during continuous perfusion of oxygenated ACSF containing (in mM): 113 NaCl, 2.5 KCl, 1.2 MgSO4∙7H2O, 2.5 CaCl2∙2H2O, 1 NaH2PO4, 26 NaHCO3, 1 ascorbate, 3 Na-pyruvate and 20 glucose; at a flow rate of 2.5 – 3 ml/minute. Slices were visualized using a Nikon microscope (Eclipse FN1, Nikon Instruments Inc., Melville, NY) equipped with differential interference contrast microscopy. Whole-cell current clamp recordings were obtained under visual control using a 40x objective. 2 – 6 MΩ borosilicate glass pipettes were filled with a Cs+ based internal solution (in mM): 120 CsOH, 120 D-gluconic acid, 2.8 NaCl, 20 HEPES, 5 TEA-Cl, 2.5 Mg-ATP, 0.25 Na-GTP. Optically-evoked responses were achieved by using a Mightex 455 nm LED system (BLS-Series) (Mightex, Toronto, Ontario, Canada) to pulse light (2 milliseconds, 0.6–1.5 mW LED intensity) through the 40x objective at 0.03–0.05 Hz.
Optically-evoked excitatory post-synaptic currents (oEPSCs) and feed-forward (FF) inhibitory post-synaptic currents (IPSCs) were measured at −70 mV and +10 mV, respectively; paired-pulse ratio (PPR) was obtained in voltage-clamp with an inter-stimulus interval of 50 milliseconds. Drug wash-on experiments were carried out after assessing at least six minutes of stable baseline recordings. Vehicle, CP-55940 (10 µM in this experiment; 5 µM for CRISPR-Cas9 Cnr1 mutation experiment, Tocris Bioscience catalogue #0949), PF-3845 (5 µM; Tocris Bioscience cataloguer #4175) or JZL184 (1 µM; Tocris Bioscience catalogue #3836) was then washed on. Stock solutions of these drugs were prepared in DMSO, and final recording solutions contained 0.05% w/v Bovine Serum Albumin (BSA, Sigma-Aldrich, St. Louis, MO, USA).
Thy1+ and Thy1-were identified by the presence or absence of fluorophore expression. Input/output curves were generated by increasing the intensity of the applied light pulses and the maximal oEPSC and oIPSC amplitude was used to compare the excitatory/inhibitory ratio. Optical stimulation as well as data collection was coordinated using pClamp 10 (Molecular Devices, San Jose, CA, USA). Cell electrical properties were monitored using a Molecular Devices 700B MultiClamp amplifier and Digidata 1440A low-noise data acquisition digitizer. Responses were filtered at 2 kHz and digitized at 10 kHz.
High-frequency stimulation-induced long-term depression.
C57BL/6J mice had a viral construct expressing ChR2 (AAV5-CaMKIIa-hChR2(H134R)-mCherry, Addgene catalogue #26969, titer 1×10¹³ gc/ml) bilaterally injected into vmPFC (0.2 µl/hemisphere). At least 5 weeks later, oEPSCs were recorded after establishing a stable baseline (under 2-millisecond light pulses, 1–3 mW LED intensity) prior to the high frequency stimulation of BLA principal neurons as described above. The LTD induction protocol consisted of 5 trains of 50 Hz 2-millisecond light pulses, for 30 seconds, with a 30-second inter-train interval. Prior to recordings, approximately half the slice were incubated and bathed in 10 µM of the CB1R antagonist, SR141716A (Cayman Chemical, Ann Arbor, MI, USA, catalogue #9000484) for at least 30 minutes. PPR was obtained in voltage-clamp with an inter-stimulus interval of 50 milliseconds. The stimulation-related change in PPR (averaged over minutes 25–30 post-stimulation) was calculated as a percent of, and compared to, a (5-minute) pre-stimulation baseline.
Intersectional virus mediated Cnr1 deletion
An intersectional virus approach was used to selectively delete Cnr1 from vmPFC→BLA neurons in Cnr1-floxed mice, based on previously described methods 57,104. Homozygous Cnr1-floxed mice and wild-type littermate controls had a retrogradely-traveling viral construct expressing Flipase (AAVretro-EF1a-Flpo, Addgene catalogue #55637, titer 1.63 x 1013 gc/mL) bilaterally injected into BLA (0.30 µl/hemisphere) and 2 constructs respectively expressing ChR2 (AAV5-EF1-DIO-hChR(H134R)-eYFP, UNC catalogue #AV43134, Addgene catalogue #55640, titer 5.5 x 1012) and Cre recombinase in the presence of Flp (AAV5-CMV-p2A-Frt-Cre-mcherry, Vector Builder, Chicago, IL catalogue #VB160808–1035zgm, titer 1.23 x 1012 gc/ml) bilaterally injected into vmPFC (0.2 µl/hemisphere). At least 5 weeks later, oEPSCs were recorded from BLA principal neurons ipsilateral to the hemisphere where injections were performed using the same recording parameters and CP-55940 (or vehicle) bath application described in In vitro slice electrophysiology recordings of eCB-mediated responses (eCB drug effects).
CRISPR-Cas9 mediated Cnr1 mutation
A single-viral Staphylococcus aureus Cas9 (SaCas9)-guided CRISPR mRNA construct was engineered and packaged into an AAV, based on previously described methods 58. Briefly, the 12 splice variants of the Cnr1 gene (Mouse Genome Informatics http://www.informatics.jax.org/) were aligned to the mouse genomic sequence (UCSC Genome Browser Gateway https://genome.ucsc.edu/cgi-bin/hgGateway) and the sequence corresponding to the common start site (exon 8) was queried using CRISPOR (http://crispor.tefor.net/). A guide 430 base pairs downstream of the start site was cloned into pAAV-AAV1-CMV-Flex-SaCas9-U6-sgCNR1 (Addgene catalogue #124844) using the forward primer CACCGAAGGCCTGCATCGGAGACTGC and reverse primer AAACGCAGTCTCCGATGCAGGCCTTC).
For behavioral experiments, we elected to use the CRISPR-Cas9 approach in C57BL6/J mice for this experiment, instead of an intersectional Cre-Lox viral knockout strategy in Cnr1-floxed mutant mice, to ensure the same background strain was used for behavioral testing throughout the study 105. To selectively mutate Cnr1 in vmPFC→BLA neurons or, in a separate experiment, dmPFC→BLA neurons, mice received bilateral injection (0.3 μl/hemisphere) of a retrogradely-traveling viral construct expressing Cre recombinase (AAVretro-mCherry-IRES-Cre, Addgene catalogue #55632, titer 8.0 10^12 gc/ml) into BLA, and either a Cre-dependent CRISPR-Cas9 construct (AAV1-CMV-Flex-SaCas9-U6-sgCNR1, L. Zweifel, University of Washington, titer ~1x10^12 gc/ml) or Cre-dependent control construct (AAV1-CMV-Flex-SaCas9-U6-sgRosa26, as described above, titer ~1x10^12 gc/ml, titer ~1x10^12 gc/ml), together (8:1 ratio) into vmPFC or dmPFC (0.2 µl/hemisphere) of C57BL6/J mice. To aid neuron visualization, with a Cre-dependent neuron labeling construct (AAV1-Flex-EGFP-KASH, L. Zweifel, titer ~5x10^12 gc/ml) was also injected into vmPFC or dmPFC (0.2 µl/hemisphere). For validation of CRISPR-Cas9 mediated Cnr1 disruption, the intersectional virus strategy was repeated in heterozygous female Rosa26fsTRAP mice, with the PFC injection volume increased (0.4 µl/hemisphere) to transfect mPFC and increase the yield of transfected neurons for analysis.
Immunocytochemistry.
To visualize of Cre recombinase and GFP, immunocytochemistry was performed at the completion of behavioral testing, as described above under In vivo fiber photometry measurement of GRABeCB2.0 biosensor activity, with the exception that the primary antibodies used were chicken anti-GFP and rat anti-tdTomato (1:1000, catalogue #EST203, Kerafast, Boston, MA, USA) and the secondary antibodies were Alexa 488 (goat anti-chicken, 1:1K, catalogue #A11039, Life Technologies, Carlsbad, CA, USA) and Alexa 555 (goat anti-rat, 1:1K, catalogue #A21434, Life Technologies, Carlsbad, CA, USA).
TRAP and rt-PCR.
Brain tissue from Rosa26fsTRAP mice was analyzed via cell type-specific mRNA purification by translating ribosome affinity purification (TRAP), followed by rt-PCR. Mice were sacrificed via cervical dislocation and decapitation, brains removed on ice, and washed in ice cold dissection buffer (1× HBSS, 2.5 mM HEPES-KOH (pH 7.3), 35 mM glucose, 4 mM NaHCO3 and 100 μg/ml cycloheximide (added freshly) in RNase-free water). mPFC and BLA were dissected on ice using 1-mm and 2 mm-diameter micropunches respectively, then washed in ice cold dissection buffer and stored at −80℃. Tissue was homogenized in ice cold homogenization buffer (20 mM HEPES KOH (pH 7.4), 150 mM KCl and 10 mM MgCl2 in RNase-free water with the following freshly added: 100 μg/ml cycloheximide (Sigma-Aldrich, St Louis, MO, catalogue #C7698), EDTA-free protease inhibitors (1tbl/10ml, Sigma-Aldrich, catalogue #11836170001), 0.5 mM DL-Dithiothreitol (DTT) (Sigma-Aldrich, catalogue #D9779), 2 μl/ml RNasin® ribonuclease inhibitor (80U/ml, ProMega, Madison, WI, USA, catalogue #N2615) and SUPERase•In™ RNase inhibitor 40U/ml, Thermo Fisher Scientific, Waltham MA, USA, catalogue #AM2694) with 12 strokes of a motor driven Teflon-glass homogenizer (Glas-Col Homogenizer) at ~900 rpm on ice.
Homogenized samples were centrifuged at 4°C for 10 min at 2,000g. The post-nuclear supernatant (S2) was mixed with 10% NP-40 and 300 mM, 2-diheptanoyl-sn-glycero-3-phospho-choline (DHPC, 200 mg, Avanti Polar Lipids, Alabaster, AL, USA, catalogue #850306P) incubated for 5 minutes on ice and centrifuged at 4°C for 15 minutes at 17,000g to obtain post-mitochondrial supernatant (S20). An aliquot of S20 material was used as input RNA to compare with the sample collected after enrichment of purified material with GFP. For immunopurification, samples were incubated with the affinity matrix at 4°C gentle end-over-end mixing in a tube rotator overnight. The affinity matrix was prepared in advance following the protocol outlined previously 106. Briefly, Streptavidin MyOne T1 Dynabeads (300 μl per IP, Thermo Fisher Scientific, catalogue #65601) were washed twice on a magnetic rack with PBS and incubated with biotinylated Protein L (120 μg, Thermo Fisher Scientific, catalogue #29997) for 35 minutes at room temperature. The Protein L loaded beads were blocked by 5X washes in PBS with 3% IgG-free and protease-free BSA (Jackson ImmunoResearch, West Grove, PA, USA, catalogue #001000162).
Next, beads were resuspended in low salt buffer (20 mM HEPES KOH (pH 7.3), 150 mM KCl, 10 mM MgCl2, 1% (vol/vol) NP40) in RNAase-free water with the following added freshly: 100 μg/ml cycloheximide and 0.5 mM DTT) and incubated with 50 μg of each GFP antibodies 19C8 and 19F7 (clones Htz-GFP-19F7 and Htz-GFP-19C8, bioreactor supernatant purity, Memorial Sloan-Kettering Monoclonal Antibody Facility, New York, NU, USA) at room temperature for 1 hour by gentle end-over-end mixing in a tube rotator. The following day, beads were collected on the magnetic rack, whole fraction was saved, and beads were washed 4X with high salt buffer (20 mM HEPES KOH (pH 7.3), 350 mM KCl, 10 mM MgCl2 and 1% (vol/vol) NP-40 in RNase-free water with the following freshly added: 100 μg/ml cycloheximide and 0.5 mM DTT). After the final wash, RNA was eluted by adding 100 μl Lysis Buffer plus 2-mercaptoethanol (Sigma-Aldrich, catalogue #63689) and purified using the micro-RNA isolation Kit (QIAGEN Sciences Inc, Germantown, MD, USA, catalogue #74004) and instructions from the company. Reverse transcription was performed with 3 ng of RNA using the Iscript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA, catalogue #1708891) and a C1000 thermal cycler (Bio-Rad).
Gene expression was quantified for the Cnr1 gene using the Mm_Cnr1_2_SG QuantiTect Primer Assay (QIAGEN Sciences Inc, catalogue #QT01748831) and a custom Cnr1 primer for the CRISPR-Cas9 construct PAGE primers from Thermo Fisher Scientific (Forward TTCACTCCCGCAGTCTCCGAT TM 63.29 % GC 57.14 and Reverse GGGACTATCTTTGCGGTGGAA TM 63.29 % GC 52.38) and power SYBR Green PCR master mix (Applied Biosystems, Grand Island, NY, USA, catalogue #4385612) using a StepOnePlus Real-Time PCR instrument (Applied Biosystems). Cycle threshold (CT) values normalized to the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (Gapdh) (QIAGEN Sciences Inc, catalogue #QT01658692) using the QuantiTect Primer Assay (QIAGEN Sciences Inc).
BaseScope in situ hybridization.
Brain tissue from C57BL6/J mice was examined via BaseScope in situ hybridization using the BaseScope RED v2 Assay (Advanced Cell Diagnostics, Santa Ana, CA, USA), combined with the Immunohistochemistry Integrated Co-Detection Workflow (Advanced Cell Diagnostics). Mice were sacrificed via cervical dislocation and decapitation, brains removed and frozen in 2-methyl butane on dry ice and stored at −80°C. Coronal sections (16-um thick) were cut using a cryostat (Leica Biosystems, model #CM3050S) and mounted onto Super Frost Plus slides (Thermo Fisher Scientific, catalogue #12-550-15), then stored at −80°C. Sections were then incubated in 4°C 10% neutral buffered formalin (Fisher Scientific, catalogue #245–684) for 20 minutes, then dehydrated in an ethanol dilution series (50%, 70%, 100%) twice. Slides were air dried, and a hydrophobic barrier was drawn around each section using an ImmEdge barrier pen (Vector Laboratories, Burlingame, CA, USA). Guinea pig polyclonal anti-Cre recombinase primary antibody (Synaptic Systems, catalogue #257 004, Gottingen, Germany) diluted with Co-Detection Antibody Diluent (Advanced Cell Diagnostics, catalogue #323160) at a 1:500 dilution was applied to each section and incubated at 4°C overnight.
The next day, sections were incubated in 10% NBF at room temperature for 30 minutes for post-primary fixation. After washing with PBS-T (1XPBS with 0.1% Tween-20), RNAScope Protease IV solution (Advanced Cell Diagnostics, catalogue #322336) was applied for 30 minutes at room temperature. Sections were then washed in distilled water and incubated with the custom Cnr1 probe (Advanced Cell Diagnostics, BA-Mm-Cnr1-1zz-st-C1) (1014–1053 base pairs on NCBI reference sequence NM_007726.5) based on the sgRNA for the CRISPR-Cas9 mutation or control probes (Advanced Cell Diagnostics, positive: BA-Mm-Ppib-1zz, negative: BA-DapB-1zz) for 2 hours at 40°C in a HybEZ II hybridization oven (Advanced Cell Diagnostics). Sections were washed twice in 1x wash buffer (Advanced Cell Diagnostics, catalogue #310091) after the probe incubation and each amplification step. Eight amplifiers (Advanced Cell Diagnostics, catalogue #323910) were applied to each section: AMP1 at 40°C for 30 min, AMP2 at 40°C for 30 minutes, AMP3 at 40°C for 15 minutes, AMP4 at 40°C for 30 minutes, AMP5 at 40°C for 30 minutes, AMP6 at 40°C for 15 minutes, AMP7 at room temperature for 30 minutes, and AMP8 at room temperature for 15 minutes. BaseScope Fast RED A+B solution (Advanced Cell Diagnostics, catalogue #323910) was prepared at a 60:1 ratio and applied at room temperature for 10 minutes. Sections were rinsed with fresh tap water.
To block the tissues for immunohistochemistry, tissues were first washed with 1x wash buffer twice, then Co-Detection Blocker (Advanced Cell Diagnostics, catalogue #323170) was applied for 15 minutes at 40°C. Sections were subsequently washed twice with 1x wash buffer and once with 1x PBST. Goat Anti-guinea pig IgG secondary antibody (Vector laboratories, catalogue #BA-7000-1.5) was diluted with the Co-Detection Antibody Diluent at a 1:200 ratio, and sections were incubated in the secondary antibody solution for 45 minutes at room temperature. Sections were washed 2x with PBS-T, and VECTASTAIN ABC Reagent (Vector laboratories, catalogue #PK-4007) was applied for 30 minutes at room temperature. The 2.5 LS Green Accessory Pack (Advanced Cell Diagnostics, catalogue #322550) was used to detect Cre immunostaining. Green solution was mixed at a 1:50 ratio of Green-B to Green-A, and tissues were incubated with the solution for 15 minutes at room temperature. Sections were then counterstained for 30 seconds at room temperature with 50% Hematoxylin I (Sigma-Aldrich, catalogue #GHS132), and blue reaction was induced by 0.02% Ammonia water (Sigma-Aldrich, catalogue #221228). Sections were dried at 60°C and coverslipped by dipping the slides in fresh xylene with EcoMount (Biocare Medical, Pacheco, CA, USA, catalogue #EM897L). Slides were imaged using an Olympus slide scanning microscope (model #VS120).
In vitro slice electrophysiology recordings.
Selective deletion/mutation of Cnr1 from vmPFC projections to BLA in C57BL6/J mice was achieved using an intersectional viral approach consisting of 3 sequential injections. First, retrograde viral construct coding for Cre recombinase (AAVretro-hSyn-Cre-P2A-dTomato, Addgene catalogue #107738, titer 7.0 10^12 gc/ml) was unilaterally injected (0.3 μl) into BLA (AP:−1.45, ML +3.4, DV −4.8). Second, either the Cre-dependent CRISPR-Cas9 or control construct as described earlier was injected (0.2 μl) into the ipsilateral vmPFC (24° Angle AP +1.8, ML +0.4, DV − 2.6) along with Cre-dependent ChR2 (0.2 µl) (AAV5-EF1a-DIO-hChR2(H134R)-EYFP-WPRE-HGHpA, Addgene catalogue #20298, titer 1 x 10^13). At least 5 weeks later, oEPSCs were recorded from BLA principal neurons ipsilateral to the hemisphere where injections were performed using the same recording parameters and CP-55940 (or vehicle) bath application described in In vitro slice electrophysiology recordings of eCB-mediated responses (eCB drug effects).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed using Prism (GraphPad, San Diego, CA, USA) and MATLAB (MathWorks, Natick, MA, USA) software. Group effects were analyzed using Student’s or Welch’s t-test or analysis of variance (ANOVA), depending on the number of independent variables, followed by Holm-Šídák's post hoc tests or paired Student’s t-test, with Bonferroni correction applied in cases involving multiple comparisons. Effect size was estimated using partial eta squared (ηp2). The threshold for statistical significance was set at P<0.05 and subject to Bonferroni correction where indicated. Power analysis were performed to determine how much the groups are different from each other. The measure of effect size ηp2 for ANOVA is calculated manually from the ratio of the sum of squares for effect of variables and the total sum of squares. The statistical details of experiments can be found in Table S1.
Supplementary Material
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Chicken anti-green fluorescent protein (GFP) | Abcam | Cat#ab13970; RRID: AB_300798 |
| Rat anti-tdTomato | Kerafast | Cat#EST203; RRID: AB_2732803 |
| Guinea pig polyclonal anti-Cre recombinase | Synaptic Systems | Cat#257 004; RRID: AB_2782969 |
| Goat anti-chicken Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A11039; RRID: AB_2534096 |
| Goat anti-rat Alex Fluor 555 | Thermo Fisher Scientific | Cat#A21434, RRID: AB_2535855 |
| Goat anti-guinea pig IgG secondary antibody | Vector laboratories | Cat#BA-7000-1.5, RRID: AB_2336132 |
| anti-GFP 19C8 TRAP purification antibody | Memorial Sloan-Kettering Monoclonal Antibody Facility | Cat#HtzGFP-19C8_as_BRS; RRID: AB_2716737 |
| anti-GFP 19F7 TRAP purification antibody | Memorial Sloan-Kettering Monoclonal Antibody Facility | Cat#HtzGFP-19F7_as_BRS; RRID: AB_2716736 |
| Bacterial and virus strains | ||
| rAAV5/CaMKII-hChR2(H134R)-eYFP | University of North Carolina Vector Core | N/A Custom preparation |
| AAV5-CaMKIIa-hChR2(H134R)-mCherry | pAAV-CaMKIIa-hChR2(H134R)-EYFP was a gift from Karl Deisseroth (Addgene viral prep # 26969-AAV5; http://n2t.net/addgene:26969 ; RRID:Addgene_26969); PubMed 20473285 | Addgene Cat# 26969-AAV5 |
| rAAV5/CaMKII-eYFP | University of North Carolina Vector Core | N/A Custom preparation |
| AAV9-hSyn-eCB2.0 | Vigene | Cat#YL10070 |
| AAV5-CaMKIIa-hChR2(H134R)-mCherry | pAAV-CaMKIIa-hChR2(H134R)-mCherry was a gift from Karl Deisseroth University of North Carolina Vector Core, Addgene pAAV-CaMKIIa-hChR2(H134R)-mCherry was a gift from Karl Deisseroth (Addgene viral prep # 26975-AAV5; http://n2t.net/addgene:26975 ; RRID:Addgene_26975) | UNC Cat#AV7954, Addgene viral prep # 26975-AAV5 |
| AAVretro-EF1a-Flpo | pAAV-EF1a-Flpo was a gift from Karl Deisseroth (Addgene viral prep # 55637-AAVrg; http://n2t.net/addgene:55637; RRID:Addgene_55637); PubMed 24908100 | Addgene Cat#55637-AAVrg |
| AAV5-EF1-DIO-hChR(H134R)-eYFP | pAAV-Ef1a-dDIO hChR2(H134R)-EYFP was a gift from Karl Deisseroth (Addgene plasmid # 55640 ; http://n2t.net/addgene:55640 ; RRID:Addgene_55640); PubMed 24908100; Addgene; University of North Carolina Vector Core, PubMed 24908100 | UNC Cat# AV43134 Addgene Cat# 55640 |
| AAV5-CMV-p2A-Frt-Cre-mcherry | Vector Builder | Cat#VB160808-1035zgm |
| pAAV- AAV1-CMV-Flex-SaCas9-U6-sgCNR1 | pAAV-FLEX-SaCas9-U6-sgRNA was a gift from Larry Zweifel (Addgene plasmid # 124844 ; http://n2t.net/addgene:124844 ; RRID:Addgene_124844) PubMed 32209486 | Addgene Cat#124844 |
| AAVretro-mCherry-IRES-Cre | pAAV-Ef1a-mCherry-IRES-Cre was a gift from Karl Deisseroth (Addgene viral prep # 55632-AAVrg; http://n2t.net/addgene:55632 ; RRID:Addgene_55632); PubMed 24908100 | Addgene Cat#55632 |
| AAVretro-hSyn-Cre-P2A-dTomato | pAAV-hSyn-Cre-P2A-dTomato was a gift from Rylan Larsen (Addgene viral prep # 107738-AAVrg; http://n2t.net/addgene:107738 ; RRID:Addgene_107738) | Addgene Cat# 107738-AAVrg |
| AAV5-EF1a-DIO-hChR2(H134R)-EYFP-WPRE-HGHpA | pAAV-EF1a-double floxed-hChR2(H134R)-EYFP-WPRE-HGHpA was a gift from Karl Deisseroth (Addgene viral prep # 20298-AAV5; http://n2t.net/addgene:20298 ; RRID:Addgene_20298) | Addgene Cat# 20298-AAV5 |
| AAV1-CMV-Flex-SaCas9-U6-sgCNR1 | Dr. Larry Zweifel, (University of Washington) | N/A |
| AAV1-CMV-Flex-SaCas9-U6-sgRosa26 | Dr. Larry Zweifel, (University of Washington) | N/A |
| AAV1-Flex-EGFP-KASH | Dr. Larry Zweifel, (University of Washington) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| SR141716A (Rimonabant) - CB1R antagonist | Tocris | Cat#0923; CAS: 158681-13-1 |
| WIN 55,212-22 - CB1R agonist | Tocris | Cat#1038; CAS: 131543-23-2 |
| JZL195 - FAAH/MAGL inhibitor | Tocris | Cat#4715; CAS: 1210004-12-8 |
| URB597 - FAAH inhibitor | Tocris | Cat#4612; CAS: 546141-08-6 |
| CP-55,940 - CB1R agonist | Tocris | Cat#0949; CAS: 83002-04-4 |
| PF-3845 - FAAH inhibitor | Tocris | Cat#4175; CAS: 1196109-52-0 |
| JZL184 - MAGL inhibitor | Tocris | Cat#3836; CAS: 1101854-58-3 |
| VECTASTAIN ABC Reagent | Vector Laboratories | Cat#PK-4007; RRID: AB_2336816 |
| Critical commercial assays | ||
| Micro-RNA isolation Kit | QIAGEN Sciences Inc | Cat#74004 |
| Iscript cDNA synthesis kit | Bio-rad | Cat#1708891 |
| Mm_Cnr1_2_SG QuantiTect Primer Assay | QIAGEN Sciences Inc | Cat#QT01748831 |
| Power SYBR Green PCR master mix | Applied Biosystems | Cat#4385612 |
| QuantiTect Primer Assay | QIAGEN Sciences | Cat#249900 |
| BaseScope RED v2 Assay | Advanced Cell Diagnostics | Cat#323900 |
| 2.5 LS Green Accessory Pack | Advanced Cell Diagnostics | Cat#322550 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratories | JAX stock #000664 |
| Mouse: B6.Cnr1-floxed | Dr. Josephine Egan (National Institute of Aging, Bethesda, MD, USA) | N/A |
| Mouse: B6.Tg(Thy1-EGFP)MJrs/J (Thy1-EGFP) | Jackson Laboratories | JAX stock #007788 |
| Mouse: B6.129S4-Gt(ROSA)26Sortm1(CAG-EGFP/Rpl10a,-birA)Wtp/J (Rosa26fsTRAP) | Jackson Laboratories | JAX stock #022367 |
| Oligonucleotides | ||
| Primer for constructing CNR1 CRISPR-Cas9 virus Forward: CACCGAAGGCCTGCATCGGAGACTGC Reverse: AAACGCAGTCTCCGATGCAGGCCTTC |
This paper | N/A |
|
Cnr1 Primer for CRISPR-Cas9 construct PAGE Primer Forward: TTCACTCCCGCAGTCTCCGAT Reverse: GGGACTATCTTTGCGGTGGAA |
ThermoFisher Scientific | N/A |
| Custom Cnr1 probe BA-Mm-Cnr1-1zz-st-C1 for BaseScope | Advanced Cell Diagnostics | N/A |
| Positive control probe BA-Mm-Ppib-1zz for BaseScope | Advanced Cell Diagnostics | N/A |
| Negative control probe BA-DapB-1zz for BaseScope | Advanced Cell Diagnostics | N/A |
| Software and algorithms | ||
| Video Freeze Monitor System | MedAssociates | https://www.med-associates.com/product/video-fear-conditioning/ |
| Ethovision XT | Noldus | https://www.noldus.com/ethovision-xt; RRID: SCR_000441 |
| MassHunter Workstation LC/QQQ Acquisition | Agilent Technologies | https://www.agilent.com/en-us/support/software-informatics/b0600servicepack1 |
| MassHunter Workstation Quantitative Analysis | Agilent Technologies | https://www.agilent.com/en-us/products/software-informatics/masshunter-suite/masshunter-quantitative-analysis; RRID: SCR_015040 |
| Mouse Genome Informatics | Jackson Laboratory | http://www.informatics.jax.org; RRID: SCR_006460 |
| UCSC Genome Browser Gateway | UCSC | https://genome.ucsc.edu/cgi-bin/hgGateway |
| CRISPOR | Concordet and Haeussler 2018 | http://crispor.tefor.net/ |
| Behavioral Observation Research Interactive Software (BORIS) | Friard and Gamba 2016 | http://www.boris.unito.it/ |
| MATLAB | MathWorks | https://www.mathworks.com; RRID:SCR_001622 |
| Prism 9 | GraphPad | http://www.graphpad.com; RRID:SCR_002798 |
| QuPath | Bankhead et al. 2017 | https://qupath.github.io/ |
| Other | ||
| Bacon softies | Bio-Serv | Cat#F3580 |
| Code Availability | ||
| MATLAB Code | This paper | DOI: 10.5281/zenodo.8047753 |
Highlights.
Optogenetic excitation of mPFC axons in BLA during extinction mobilizes BLA eCBs
eCB-biosensor activity at mPFC→BLA neurons dynamically tracks extinction
eCB-CB1R regulates glutamate release and plasticity at vmPFC→BLA synapses
CRISPR-Cas9-mediated vmPFC→BLA-CB1R mutation impairs extinction memory formation
Acknowledgements
We are very grateful to Dr. Josephine M. Egan for providing breeding pairs of Cnr1-floxed mutant mice, to Dr. Eliot Smith for technical advice on the ACD BaseScope procedure and to Dr. Guohong Cui, Dr. Jie Liu and Julia Schaffer for valuable discussions. Research supported by the NIAAA Intramural Research Program (A.H.), NIDA grant P30 DA048736 (L.S.Z.) and NIMH grants MH107435 and MH119817 (S.P.).
Abbreviations
- ANOVA
analysis of variance
- RM
repeated-measures
- base
baseline
- Veh
vehicle
- YFP
yellow fluorescent protein
- ChR2
channelrhodopsin2
- SR
SR141716A
- AEA
anandamide
- 2-AG
2-Arachidonoyl glycerol
- PPR
paired-pulse ratio
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interests
S.P. is/has been also a scientific consultant for H. Lundbeck A/S, Psy therapeutics and Sophran Therapeutics, within the past three years, unrelated to the current work. The other authors declare no competing interests.
For comparing two groups Student’s t-test (paired or unpaired) was used except where indicated. Effect size is given as ηp2.
References
- 1.Craske MG, Stein MB, Eley TC, Milad MR, Holmes A, Rapee RM, and Wittchen HU (2017). Anxiety disorders. Nature reviews. Disease primers 3, 17024. 10.1038/nrdp.2017.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pavlov IP (1927). Conditioned reflexes (Oxford University Press).
- 3.Hariri AR, and Holmes A (2015). Finding translation in stress research. Nat Neurosci 18, 1347–1352. 10.1038/nn.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gunduz-Cinar O, Hill MN, McEwen BS, and Holmes A (2013). Amygdala FAAH and anandamide: mediating protection and recovery from stress. Trends Pharmacol Sci 34, 637–644. 10.1016/j.tips.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayo LM, Rabinak CA, Hill MN, and Heilig M (2021). Targeting the Endocannabinoid System in the Treatment of Posttraumatic Stress Disorder: A Promising Case of Preclinical-Clinical Translation? Biol Psychiatry 10.1016/j.biopsych.2021.07.019. [DOI] [PMC free article] [PubMed]
- 6.Araque A, Castillo PE, Manzoni OJ, and Tonini R (2017). Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 124, 13–24. 10.1016/j.neuropharm.2017.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lutz B, Marsicano G, Maldonado R, and Hillard CJ (2015). The endocannabinoid system in guarding against fear, anxiety and stress. Nat Rev Neurosci 16, 705–718. 10.1038/nrn4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gunduz-Cinar O (2021). The endocannabinoid system in the amygdala and modulation of fear. Prog Neuropsychopharmacol Biol Psychiatry 105, 110116. 10.1016/j.pnpbp.2020.110116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bisby MA, Richardson R, and Baker KD (2020). Developmental differences in the effects of CB1/2R agonist WIN55212–2 on extinction of learned fear. Progress in neuro-psychopharmacology & biological psychiatry 99, 109834. 10.1016/j.pnpbp.2019.109834. [DOI] [PubMed] [Google Scholar]
- 10.Tan H, Ahmad T, Loureiro M, Zunder J, and Laviolette SR (2014). The role of cannabinoid transmission in emotional memory formation: implications for addiction and schizophrenia. Frontiers in psychiatry 5, 73. 10.3389/fpsyt.2014.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singewald N, Sartori SB, Reif A, and Holmes A (2023). Alleviating anxiety and taming trauma: Novel pharmacotherapeutics for anxiety disorders and posttraumatic stress disorder. Neuropharmacology 226, 109418. 10.1016/j.neuropharm.2023.109418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, et al. (2002). The endogenous cannabinoid system controls extinction of aversive memories. Nature 418, 530–534. 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 13.Gunduz-Cinar O, Brockway E, Lederle L, Wilcox T, Halladay LR, Ding Y, Oh H, Busch EF, Kaugars K, Flynn S, et al. (2019). Identification of a novel gene regulating amygdala-mediated fear extinction. Molecular psychiatry 24, 601–612. 10.1038/s41380-017-0003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tovote P, Fadok JP, and Luthi A (2015). Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16, 317–331. 10.1038/nrn3945. [DOI] [PubMed] [Google Scholar]
- 15.Likhtik E, and Johansen JP (2019). Neuromodulation in circuits of aversive emotional learning. Nat Neurosci 22, 1586–1597. 10.1038/s41593-019-0503-3. [DOI] [PubMed] [Google Scholar]
- 16.Sengupta A, and Holmes A (2019). A Discrete Dorsal Raphe to Basal Amygdala 5-HT Circuit Calibrates Aversive Memory. Neuron 103, 489–505 e487. 10.1016/j.neuron.2019.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunduz Cinar O, Flynn S, Kaugars K, Ramikie TS, Cinar R, Kunos G, Patel S, and Holmes A (2016). Fluoxetine facilitates fear extinction through amygdala endocannabinoids. Neuropsychopharmacology 41, 1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunduz-Cinar O, MacPherson KP, Cinar R, Gamble-George J, Sugden K, Williams B, Godlewski G, Ramikie TS, Gorka AX, Alapafuja SO, et al. (2013). Convergent translational evidence of a role for anandamide in amygdala-mediated fear extinction, threat processing and stress-reactivity. Molecular psychiatry 18, 813–823. 10.1038/mp.2012.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mock ED, Mustafa M, Gunduz-Cinar O, Cinar R, Petrie GN, Kantae V, Di X, Ogasawara D, Varga ZV, Paloczi J, et al. (2020). Discovery of a NAPE-PLD inhibitor that modulates emotional behavior in mice. Nat Chem Biol 16, 667–675. 10.1038/s41589-020-0528-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hariri AR, Gorka A, Hyde LW, Kimak M, Halder I, Ducci F, Ferrell RE, Goldman D, and Manuck SB (2009). Divergent effects of genetic variation in endocannabinoid signaling on human threat- and reward-related brain function. Biol Psychiatry 66, 9–16. 10.1016/j.biopsych.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dincheva I, Drysdale AT, Hartley CA, Johnson DC, Jing D, King EC, Ra S, Gray JM, Yang R, DeGruccio AM, et al. (2015). FAAH genetic variation enhances fronto-amygdala function in mouse and human. Nature communications 6, 6395. 10.1038/ncomms7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayo LM, Asratian A, Linde J, Holm L, Natt D, Augier G, Stensson N, Vecchiarelli HA, Balsevich G, Aukema RJ, et al. (2018). Protective effects of elevated anandamide on stress and fear-related behaviors: translational evidence from humans and mice. Molecular psychiatry 10.1038/s41380-018-0215-1. [DOI] [PubMed]
- 23.Zabik NL, Iadipaolo AS, Marusak HA, Peters C, Burghardt K, and Rabinak CA (2022). A common genetic variant in fatty acid amide hydrolase is linked to alterations in fear extinction neural circuitry in a racially diverse, nonclinical sample of adults. Journal of neuroscience research 100, 744–761. 10.1002/jnr.24860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rabinak CA, Angstadt M, Sripada CS, Abelson JL, Liberzon I, Milad MR, and Phan KL (2013). Cannabinoid facilitation of fear extinction memory recall in humans. Neuropharmacology 64, 396–402. 10.1016/j.neuropharm.2012.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rabinak CA, Angstadt M, Lyons M, Mori S, Milad MR, Liberzon I, and Phan KL (2014). Cannabinoid modulation of prefrontal-limbic activation during fear extinction learning and recall in humans. Neurobiology of learning and memory 113, 125–134. 10.1016/j.nlm.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammoud MZ, Peters C, Hatfield JRB, Gorka SM, Phan KL, Milad MR, and Rabinak CA (2019). Influence of Delta9-tetrahydrocannabinol on long-term neural correlates of threat extinction memory retention in humans. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 44, 1769–1777. 10.1038/s41386-019-0416-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gidyk DC, Diwan M, Gouveia FV, Giacobbe P, Lipsman N, and Hamani C (2021). Investigating the role of CB1 endocannabinoid transmission in the anti-fear and anxiolytic-like effects of ventromedial prefrontal cortex deep brain stimulation. J Psychiatr Res 135, 264–269. 10.1016/j.jpsychires.2021.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Kuhnert S, Meyer C, and Koch M (2013). Involvement of cannabinoid receptors in the amygdala and prefrontal cortex of rats in fear learning, consolidation, retrieval and extinction. Behavioural brain research 250, 274–284. 10.1016/j.bbr.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Rossignoli MT, Lopes-Aguiar C, Ruggiero RN, Do Val da Silva, R.A., Bueno-Junior, L.S., Kandratavicius, L., Peixoto-Santos, J.E., Crippa, J.A., Cecilio Hallak, J.E., Zuardi, A.W., et al. (2017). Selective post-training time window for memory consolidation interference of cannabidiol into the prefrontal cortex: Reduced dopaminergic modulation and immediate gene expression in limbic circuits. Neuroscience 350, 85–93. 10.1016/j.neuroscience.2017.03.019. [DOI] [PubMed] [Google Scholar]
- 30.Bukalo O, Nonaka M, Weinholtz CA, Mendez A, Taylor WW, and Holmes A (2021). Effects of optogenetic photoexcitation of infralimbic cortex inputs to the basolateral amygdala on conditioned fear and extinction. Behav Brain Res 396, 112913. 10.1016/j.bbr.2020.112913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arruda-Carvalho M, and Clem RL (2015). Prefrontal-amygdala fear networks come into focus. Frontiers in systems neuroscience 9, 145. 10.3389/fnsys.2015.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marek R, Sun Y, and Sah P (2019). Neural circuits for a top-down control of fear and extinction. Psychopharmacology (Berl) 236, 313–320. 10.1007/s00213-018-5033-2. [DOI] [PubMed] [Google Scholar]
- 33.Bukalo O, Pinard CR, and Holmes A (2014). Mechanisms to medicines: elucidating neural and molecular substrates of fear extinction to identify novel treatments for anxiety disorders. Br J Pharmacol 171, 4690–4718. 10.1111/bph.12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, and Rauch SL (2009). Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biological psychiatry 66, 1075–1082. 10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Do-Monte FH, Manzano-Nieves G, Quinones-Laracuente K, Ramos-Medina L, and Quirk GJ (2015). Revisiting the role of infralimbic cortex in fear extinction with optogenetics. The Journal of neuroscience : the official journal of the Society for Neuroscience 35, 3607–3615. 10.1523/JNEUROSCI.3137-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maroun M, Kavushansky A, Holmes A, Wellman C, and Motanis H (2012). Enhanced extinction of aversive memories by high-frequency stimulation of the rat infralimbic cortex. PloS one 7, e35853. 10.1371/journal.pone.0035853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fitzgerald PJ, Whittle N, Flynn SM, Graybeal C, Pinard CR, Gunduz-Cinar O, Kravitz AV, Singewald N, and Holmes A (2014). Prefrontal single-unit firing associated with deficient extinction in mice. Neurobiology of learning and memory 113, 69–81. 10.1016/j.nlm.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim HS, Cho HY, Augustine GJ, and Han JH (2016). Selective Control of Fear Expression by Optogenetic Manipulation of Infralimbic Cortex after Extinction. Neuropsychopharmacology 41, 1261–1273. 10.1038/npp.2015.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bukalo O, Pinard CR, Silverstein S, Brehm C, Hartley ND, Whittle N, Colacicco G, Busch E, Patel S, Singewald N, and Holmes A (2015). Prefrontal inputs to the amygdala instruct fear extinction memory formation. Sci Adv 1, e1500251. 10.1126/sciadv.1500251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bloodgood DW, Sugam JA, Holmes A, and Kash TL (2018). Fear extinction requires infralimbic cortex projections to the basolateral amygdala. Transl Psychiatry 8, 60. 10.1038/s41398-018-0106-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adhikari A, Lerner TN, Finkelstein J, Pak S, Jennings JH, Davidson TJ, Ferenczi E, Gunaydin LA, Mirzabekov JJ, Ye L, et al. (2015). Basomedial amygdala mediates top-down control of anxiety and fear. Nature 527, 179–185. 10.1038/nature15698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laviolette SR, and Grace AA (2006). Cannabinoids Potentiate Emotional Learning Plasticity in Neurons of the Medial Prefrontal Cortex through Basolateral Amygdala Inputs. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 6458–6468. 10.1523/JNEUROSCI.0707-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hagihara KM, Bukalo O, Zeller M, Aksoy-Aksel A, Karalis N, Limoges A, Rigg T, Campbell T, Mendez A, Weinholtz C, et al. (2021). Intercalated amygdala clusters orchestrate a switch in fear state. Nature 594, 403–407. 10.1038/s41586-021-03593-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong A, He K, Dudok B, Farrell JS, Guan W, Liput DJ, Puhl HL, Cai R, Wang H, Duan J, et al. (2021). A fluorescent sensor for spatiotemporally resolved imaging of endocannabinoid dynamics in vivo. Nat Biotechnol 10.1038/s41587-021-01074-4. [DOI] [PMC free article] [PubMed]
- 45.Farrell JS, Colangeli R, Dong A, George AG, Addo-Osafo K, Kingsley PJ, Morena M, Wolff MD, Dudok B, He K, et al. (2021). In vivo endocannabinoid dynamics at the timescale of physiological and pathological neural activity. Neuron 109, 2398–2403 e2394. 10.1016/j.neuron.2021.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liput DJ, Puhl HL, Dong A, He K, Li Y, and Lovinger DM (2022). 2-Arachidonoylglycerol mobilization following brief synaptic stimulation in the dorsal lateral striatum requires glutamatergic and cholinergic neurotransmission. Neuropharmacology 205, 108916. 10.1016/j.neuropharm.2021.108916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Z, Yang N, Dong J, Tian W, Chang L, Ma J, Guo J, Tan J, Dong A, He K, et al. (2022). Deficiency in endocannabinoid synthase DAGLB contributes to early onset Parkinsonism and murine nigral dopaminergic neuron dysfunction. Nature communications 13, 3490. 10.1038/s41467-022-31168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bouton ME (2002). Context, ambiguity, and unlearning: sources of relapse after behavioral extinction. Biol Psychiatry 52, 976–986. 10.1016/s0006-3223(02)01546-9. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Kim J, and Tonegawa S (2020). Amygdala Reward Neurons Form and Store Fear Extinction Memory. Neuron 10.1016/j.neuron.2019.12.025. [DOI] [PubMed]
- 50.Solomon RL, and Corbit JD (1974). An opponent-process theory of motivation. I. Temporal dynamics of affect. Psychol Rev 81, 119–145. 10.1037/h0036128. [DOI] [PubMed] [Google Scholar]
- 51.Nasser HM, and McNally GP (2012). Appetitive-aversive interactions in Pavlovian fear conditioning. Behav Neurosci 126, 404–422. 10.1037/a0028341. [DOI] [PubMed] [Google Scholar]
- 52.Salinas-Hernandez XI, Vogel P, Betz S, Kalisch R, Sigurdsson T, and Duvarci S (2018). Dopamine neurons drive fear extinction learning by signaling the omission of expected aversive outcomes. Elife 7. 10.7554/eLife.38818. [DOI] [PMC free article] [PubMed]
- 53.Katona I, and Freund TF (2012). Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci 35, 529–558. 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lovinger DM, Mateo Y, Johnson KA, Engi SA, Antonazzo M, and Cheer JF (2022). Local modulation by presynaptic receptors controls neuronal communication and behaviour. Nat Rev Neurosci 23, 191–203. 10.1038/s41583-022-00561-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Domenici MR, Azad SC, Marsicano G, Schierloh A, Wotjak CT, Dodt HU, Zieglgansberger W, Lutz B, and Rammes G (2006). Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J Neurosci 26, 5794–5799. 10.1523/JNEUROSCI.0372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jasnow AM, Ehrlich DE, Choi DC, Dabrowska J, Bowers ME, McCullough KM, Rainnie DG, and Ressler KJ (2013). Thy1-expressing neurons in the basolateral amygdala may mediate fear inhibition. J Neurosci 33, 10396–10404. 10.1523/JNEUROSCI.5539-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marcus DJ, Bedse G, Gaulden AD, Ryan JD, Kondev V, Winters ND, Rosas-Vidal LE, Altemus M, Mackie K, Lee FS, et al. (2020). Endocannabinoid Signaling Collapse Mediates Stress-Induced Amygdalo-Cortical Strengthening. Neuron 10.1016/j.neuron.2019.12.024. [DOI] [PMC free article] [PubMed]
- 58.Hunker AC, Soden ME, Krayushkina D, Heymann G, Awatramani R, and Zweifel LS (2020). Conditional Single Vector CRISPR/SaCas9 Viruses for Efficient Mutagenesis in the Adult Mouse Nervous System. Cell Rep 30, 4303–4316 e4306. 10.1016/j.celrep.2020.02.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Castro DC, Oswell CS, Zhang ET, Pedersen CE, Piantadosi SC, Rossi MA, Hunker AC, Guglin A, Moron JA, Zweifel LS, et al. (2021). An endogenous opioid circuit determines state-dependent reward consumption. Nature 598, 646–651. 10.1038/s41586-021-04013-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou P, Zhang Y, Ma Q, Gu F, Day DS, He A, Zhou B, Li J, Stevens SM, Romo D, and Pu WT (2013). Interrogating translational efficiency and lineage-specific transcriptomes using ribosome affinity purification. Proc Natl Acad Sci U S A 110, 15395–15400. 10.1073/pnas.1304124110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Albo Z, and Graff J (2018). The mysteries of remote memory. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 373. 10.1098/rstb.2017.0029. [DOI] [PMC free article] [PubMed]
- 62.Fitzgerald PJ, Pinard CR, Camp MC, Feyder M, Sah A, Bergstrom HC, Graybeal C, Liu Y, Schluter OM, Grant SG, et al. (2015). Durable fear memories require PSD-95. Molecular psychiatry 20, 913. 10.1038/mp.2015.44. [DOI] [PubMed] [Google Scholar]
- 63.Do Monte FH, Quirk GJ, Li B, and Penzo MA (2016). Retrieving fear memories, as time goes by. Molecular psychiatry 21, 1027–1036. 10.1038/mp.2016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McDonald AJ (1998). Cortical pathways to the mammalian amygdala. Prog Neurobiol 55, 257–332. 10.1016/s0301-0082(98)00003-3. [DOI] [PubMed] [Google Scholar]
- 65.Knapska E, Macias M, Mikosz M, Nowak A, Owczarek D, Wawrzyniak M, Pieprzyk M, Cymerman IA, Werka T, Sheng M, et al. (2012). Functional anatomy of neural circuits regulating fear and extinction. Proceedings of the National Academy of Sciences of the United States of America 109, 17093–17098. 10.1073/pnas.1202087109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Likhtik E, and Paz R (2015). Amygdala-prefrontal interactions in (mal)adaptive learning. Trends Neurosci 38, 158–166. 10.1016/j.tins.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sotres-Bayon F, and Quirk GJ (2010). Prefrontal control of fear: more than just extinction. Curr Opin Neurobiol 20, 231–235. 10.1016/j.conb.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dudok B, Barna L, Ledri M, Szabo SI, Szabadits E, Pinter B, Woodhams SG, Henstridge CM, Balla GY, Nyilas R, et al. (2015). Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nature neuroscience 18, 75–86. 10.1038/nn.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bouton ME, Maren S, and McNally GP (2021). Behavioral and Neurobiological Mechanisms of Pavlovian and Instrumental Extinction Learning. Physiol Rev 101, 611–681. 10.1152/physrev.00016.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tan H, Lauzon NM, Bishop SF, Bechard MA, and Laviolette SR (2010). Integrated cannabinoid CB1 receptor transmission within the amygdala-prefrontal cortical pathway modulates neuronal plasticity and emotional memory encoding. Cerebral cortex 20, 1486–1496. 10.1093/cercor/bhp210. [DOI] [PubMed] [Google Scholar]
- 71.Tan H, Lauzon NM, Bishop SF, Chi N, Bechard M, and Laviolette SR (2011). Cannabinoid transmission in the basolateral amygdala modulates fear memory formation via functional inputs to the prelimbic cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 5300–5312. 10.1523/JNEUROSCI.4718-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cai LX, Pizano K, Gundersen GW, Hayes CL, Fleming WT, Holt S, Cox JM, and Witten IB (2020). Distinct signals in medial and lateral VTA dopamine neurons modulate fear extinction at different times. Elife 9. 10.7554/eLife.54936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jo YS, Heymann G, and Zweifel LS (2018). Dopamine Neurons Reflect the Uncertainty in Fear Generalization. Neuron 100, 916–925 e913. 10.1016/j.neuron.2018.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Luo R, Uematsu A, Weitemier A, Aquili L, Koivumaa J, McHugh TJ, and Johansen JP (2018). A dopaminergic switch for fear to safety transitions. Nature communications 9, 2483. 10.1038/s41467-018-04784-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Badrinarayan A, Wescott SA, Vander Weele CM, Saunders BT, Couturier BE, Maren S, and Aragona BJ (2012). Aversive stimuli differentially modulate real-time dopamine transmission dynamics within the nucleus accumbens core and shell. J Neurosci 32, 15779–15790. 10.1523/JNEUROSCI.3557-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yau JO, and McNally GP (2022). The activity of ventral tegmental area dopamine neurons during shock omission predicts safety learning. Behav Neurosci 136, 276–284. 10.1037/bne0000506. [DOI] [PubMed] [Google Scholar]
- 77.Yau JO, and McNally GP (2015). Pharmacogenetic excitation of dorsomedial prefrontal cortex restores fear prediction error. J Neurosci 35, 74–83. 10.1523/JNEUROSCI.3777-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Covey DP, Mateo Y, Sulzer D, Cheer JF, and Lovinger DM (2017). Endocannabinoid modulation of dopamine neurotransmission. Neuropharmacology 124, 52–61. 10.1016/j.neuropharm.2017.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wenzel JM, and Cheer JF (2018). Endocannabinoid Regulation of Reward and Reinforcement through Interaction with Dopamine and Endogenous Opioid Signaling. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 43, 103–115. 10.1038/npp.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Covelo A, Eraso-Pichot A, Fernandez-Moncada I, Serrat R, and Marsicano G (2021). CB1R-dependent regulation of astrocyte physiology and astrocyte-neuron interactions. Neuropharmacology 195, 108678. 10.1016/j.neuropharm.2021.108678. [DOI] [PubMed] [Google Scholar]
- 81.Navarrete M, and Araque A (2008). Endocannabinoids mediate neuron-astrocyte communication. Neuron 57, 883–893. 10.1016/j.neuron.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 82.Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, and Freund TF (2001). Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci 21, 9506–9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morena M, Aukema RJ, Leitl KD, Rashid AJ, Vecchiarelli HA, Josselyn SA, and Hill MN (2019). Upregulation of Anandamide Hydrolysis in the Basolateral Complex of Amygdala Reduces Fear Memory Expression and Indices of Stress and Anxiety. J Neurosci 39, 1275–1292. 10.1523/JNEUROSCI.2251-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kamprath K, Plendl W, Marsicano G, Deussing JM, Wurst W, Lutz B, and Wotjak CT (2009). Endocannabinoids mediate acute fear adaptation via glutamatergic neurons independently of corticotropin-releasing hormone signaling. Genes Brain Behav 8, 203–211. 10.1111/j.1601-183X.2008.00463.x. [DOI] [PubMed] [Google Scholar]
- 85.Llorente-Berzal A, Terzian AL, di Marzo V, Micale V, Viveros MP, and Wotjak CT (2015). 2-AG promotes the expression of conditioned fear via cannabinoid receptor type 1 on GABAergic neurons. Psychopharmacology (Berl) 232, 2811–2825. 10.1007/s00213-015-3917-y. [DOI] [PubMed] [Google Scholar]
- 86.Rey AA, Purrio M, Viveros MP, and Lutz B (2012). Biphasic effects of cannabinoids in anxiety responses: CB1 and GABA(B) receptors in the balance of GABAergic and glutamatergic neurotransmission. Neuropsychopharmacology 37, 2624–2634. 10.1038/npp.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Metna-Laurent M, Soria-Gomez E, Verrier D, Conforzi M, Jego P, Lafenetre P, and Marsicano G (2012). Bimodal control of fear-coping strategies by CB(1) cannabinoid receptors. J Neurosci 32, 7109–7118. 10.1523/JNEUROSCI.1054-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rovira-Esteban L, Gunduz-Cinar O, Bukalo O, Limoges A, Brockway E, Muller K, Fenno L, Kim YS, Ramakrishnan C, Andrasi T, et al. (2019). Excitation of Diverse Classes of Cholecystokinin Interneurons in the Basal Amygdala Facilitates Fear Extinction. eNeuro 6. 10.1523/ENEURO.0220-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chhatwal JP, Davis M, Maguschak KA, and Ressler KJ (2005). Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology 30, 516–524. 10.1038/sj.npp.1300655. [DOI] [PubMed] [Google Scholar]
- 90.Bitencourt RM, Pamplona FA, and Takahashi RN (2008). Facilitation of contextual fear memory extinction and anti-anxiogenic effects of AM404 and cannabidiol in conditioned rats. Eur Neuropsychopharmacol 18, 849–859. 10.1016/j.euroneuro.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 91.Pamplona FA, Bitencourt RM, and Takahashi RN (2008). Short- and long-term effects of cannabinoids on the extinction of contextual fear memory in rats. Neurobiol Learn Mem 90, 290–293. 10.1016/j.nlm.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 92.Hartley ND, Gunduz-Cinar O, Halladay L, Bukalo O, Holmes A, and Patel S (2016). 2-arachidonoylglycerol signaling impairs short-term fear extinction. Transl Psychiatry 6, e749. 10.1038/tp.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gonzalez-Mariscal I, Montoro RA, Doyle ME, Liu QR, Rouse M, O'Connell JF, Santa-Cruz Calvo S, Krzysik-Walker SM, Ghosh S, Carlson OD, et al. (2018). Absence of cannabinoid 1 receptor in beta cells protects against high-fat/high-sugar diet-induced beta cell dysfunction and inflammation in murine islets. Diabetologia 61, 1470–1483. 10.1007/s00125-018-4576-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bukalo O, Pinard CR, Silverstein S, Brehm C, Hartley ND, Whittle N, Colacicco G, Busch E, Patel S, Singewald N, and Holmes A (2015). Prefrontal inputs to the amygdala instruct fear extinction memory formation. Sci Adv 1. 10.1126/sciadv.1500251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gunduz-Cinar O, Flynn S, Brockway E, Kaugars K, Baldi R, Ramikie TS, Cinar R, Kunos G, Patel S, and Holmes A (2016). Fluoxetine Facilitates Fear Extinction Through Amygdala Endocannabinoids. Neuropsychopharmacology 41, 1598–1609. 10.1038/npp.2015.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ferrazzo S, Gunduz-Cinar O, Stefanova N, Pollack GA, Holmes A, Schmuckermair C, and Ferraguti F (2019). Increased anxiety-like behavior following circuit-specific catecholamine denervation in mice. Neurobiol Dis 125, 55–66. 10.1016/j.nbd.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Holmes A, and Rodgers RJ (2003). Prior exposure to the elevated plus-maze sensitizes mice to the acute behavioral effects of fluoxetine and phenelzine. Eur J Pharmacol 459, 221–230. 10.1016/s0014-2999(02)02874-1. [DOI] [PubMed] [Google Scholar]
- 98.Boyce-Rustay JM, and Holmes A (2006). Ethanol-related behaviors in mice lacking the NMDA receptor NR2A subunit. Psychopharmacology (Berl) 187, 455–466. 10.1007/s00213-006-0448-6. [DOI] [PubMed] [Google Scholar]
- 99.Hardaway JA, Halladay LR, Mazzone CM, Pati D, Bloodgood DW, Kim M, Jensen J, DiBerto JF, Boyt KM, Shiddapur A, et al. (2019). Central Amygdala Prepronociceptin-Expressing Neurons Mediate Palatable Food Consumption and Reward. Neuron 102, 1088. 10.1016/j.neuron.2019.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Friard O, and Gamba M (2016). BORIS: a free, versatile open-source event-logging software for video/audio coding and live observations. Methods in Ecology and Evolution 7, 1325–1330. [Google Scholar]
- 101.Halladay LR, Kocharian A, Piantadosi PT, Authement ME, Lieberman AG, Spitz NA, Coden K, Glover LR, Costa VD, Alvarez VA, and Holmes A (2020). Prefrontal Regulation of Punished Ethanol Self-administration. Biol Psychiatry 87, 967–978. 10.1016/j.biopsych.2019.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bergstrom HC, Lipkin AM, Lieberman AG, Pinard CR, Gunduz-Cinar O, Brockway ET, Taylor WW, Nonaka M, Bukalo O, Wills TA, et al. (2018). Dorsolateral Striatum Engagement Interferes with Early Discrimination Learning. Cell Rep 23, 2264–2272. 10.1016/j.celrep.2018.04.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ting JT, Daigle TL, Chen Q, and Feng G (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol 1183, 221–242. 10.1007/978-1-4939-1096-0_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gremel CM, Chancey JH, Atwood BK, Luo G, Neve R, Ramakrishnan C, Deisseroth K, Lovinger DM, and Costa RM (2016). Endocannabinoid Modulation of Orbitostriatal Circuits Gates Habit Formation. Neuron 90, 1312–1324. 10.1016/j.neuron.2016.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hefner K, Whittle N, Juhasz J, Norcross M, Karlsson RM, Saksida LM, Bussey TJ, Singewald N, and Holmes A (2008). Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. J Neurosci 28, 8074–8085. 10.1523/JNEUROSCI.4904-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heiman M, Kulicke R, Fenster RJ, Greengard P, and Heintz N (2014). Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nat Protoc 9, 1282–1291. 10.1038/nprot.2014.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data reported in this paper will be shared by the Lead Contact upon reasonable request.
This paper does not report original code. Existing code from the David Root lab (https://www.root-lab.org/code) was modified for current analyses. The modified version of this code is deposited and publicly available at Zenodo. The associated DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.