

Abstract
PARP inhibitors (PARPi) kill cancer cells by stalling DNA replication and preventing DNA repair, resulting in a critical accumulation of DNA damage. Resistance to PARPi is a growing clinical problem in the treatment of high grade serous ovarian carcinoma (HGSOC). Acetylation of histone H3 lysine 14 (H3K14ac) and associated histone acetyltransferases (HATs) and epigenetic readers have known functions in DNA repair and replication. Our objectives are to examine their expression and activities in the context of PARPi-resistant HGSOC, and to determine if targeting H3K14ac or associated proteins has therapeutic potential. Using mass spectrometry profiling of histone modifications, we observed increased H3K14ac enrichment in PARPi-resistant HGSOC cells relative to isogenic PARPi-sensitive lines. By RT-qPCR and RNA-Seq, we also observed altered expression of numerous HATs in PARPi-resistant HGSOC cells and a PARPi-resistant PDX model. Knockdown of HATs only modestly altered PARPi response, although knockdown and inhibition of PCAF significantly increased resistance. Pharmacologic inhibition of HBO1 depleted H3K14ac but did not affect PARPi response. However, knockdown and inhibition of BRPF3, a bromodomain and PHD-finger containing protein that is known to interact in a complex with HBO1, did reduce PARPi resistance. This study demonstrates that depletion of H3K14ac does not affect PARPi response in HGSOC. Our data suggest that the bromodomain function of HAT proteins, such as PCAF, or accessory proteins, such as BRPF3, may play a more direct role compared to direct histone acetyltransferase function in PARPi response.
Keywords: HGSOC, ovarian cancer, PARP inhibitor, resistance, H3K14ac, BRPF3
Introduction
High grade serous ovarian carcinoma (HGSOC) is the deadliest gynecological malignancy. It is the fifth most common cause of cancer death in women and has one of the highest death-to-incidence ratios of all cancers (1). Most patients respond initially to standard practice cytoreductive surgery and platinum/taxol chemotherapy (2). The addition of PARP inhibitors (PARPi) as first-line maintenance therapy, supported by numerous clinical trials in homologous recombination-deficient and -proficient cohorts, has extended progression-free survival intervals for many patients (3–5). Despite these advances, a substantial proportion of patients do not respond to PARPi, and over 80% of cases recur and are expected to acquire resistance to all available therapies (2, 6). Since most HGSOC patients will be eligible to receive PARPi, it is essential to discover mechanisms of resistance and provide support for novel therapies to overcome resistance and improve outcomes.
Numerous studies and trials have focused on exploiting vulnerabilities to augment or restore PARPi sensitivity, including the use of topoisomerase inhibitors (7, 8), cyclin-dependent kinase knockdown or inhibition (9, 10), Wnt inhibitors (11), and histone methyltransferase inhibitors (12), among others [reviewed in (6)]. In the current study we examine histone modifications and expression of histone modifying enzymes in the context of PARPi-resistant HGSOC. Histone modifications play critical roles in PARPi resistance, as they control access to genomic DNA for vital processes including transcription, replication, and repair (13, 14). When dysregulated, these same modifications contribute to every aspect of cancer biology, from early oncogenesis to progression, metastasis, chemoresistance, and immune evasion (15–18). Acetylation of histone H3 lysine 14 (H3K14ac) neutralizes the positively charged lysine residue and reduces interactions between the histone octamer and encircling DNA, thus allowing greater access for transcriptional, replicative, and DNA repair machinery (19, 20). As such, H3K14ac is a modification associated with transcriptionally active chromatin.
While H3K14ac is only a single modification, its regulation is complex, as it may be catalyzed by numerous histone acetyltransferases (HATs) of different classes including (but not limited to) GCN5 and PCAF (encoded by KAT2A and KAT2B, respectively), CBP and P300 (encoded by KAT3A and KAT3B, respectively), as well as HBO1, also known as MYST2 (encoded by KAT7). H3K14ac is known to be a key factor in the process of single-stranded DNA break (SSB) and double-stranded DNA break (DSB) repair. For SSB repair, H3K14ac stabilizes chromatin interactions and promotes error-free Nucleotide Excision Repair (NER) (21). HATs GCN5 and PCAF are structurally similar and promote repair by acetylation of H3K14, and also by acetylation of non-histone targets including Replication Protein A1 (RPA1) (22). Acetylation of RPA1 by GCN5/PCAF activates NER and promotes accumulation of DNA repair proteins at sites of damage. For DSB, CBP and P300 are structurally similar and promote chromatin remodeling and recruitment of effectors involved in both homologous recombination (HR) and non-homologous end joining (NHEJ) (23, 24). Additionally, CBP/P300 promote transcription of HR factors BRCA1 and RAD51 recombinase (25). Finally, phosphorylated HBO1 has been shown to facilitate recruitment of the NER protein XPC to sites of cyclopyrimidine dimers (CPD) and is essential for NER at sites of ultraviolet (UV) irradiation damage (26).
Modifiers of histone acetylation, especially histone deacetylases (HDACs), have been investigated as therapeutic targets in HGSOC [reviewed in (27)]. HDAC inhibitors (HDACi) broadly increase histone acetylation and can improve the response of numerous cancers to DNA damaging agents (28). Conversely, HATs have not been frequently targeted, despite known functions in DNA repair, DNA replication, and other mechanisms that may contribute to PARPi resistance. The role of H3K14ac has also not been studied in the context of PARPi resistance. Our objective is to examine alterations to H3K14ac, including changes in expression or activity of associated epigenetic writers and readers, and to determine the therapeutic potential of perturbing H3K14ac in PARPi-resistant HGSOC.
Results
H3K14ac enrichment and expression of HATs are altered in PARPi-resistant HGSOC cell lines and PDX models
Two TP53-mutated HGSOC cell lines (PEO1 and OVSAHO) were subjected to stepwise dose escalation of the PARPi olaparib to select a population of resistant cells. PEO1 cells encode BRCA2 nonsense mutation 5193C>G and are BRCA2-deficient (29); OVSAHO have a BRCA2 homozygous deletion (30). Olaparib-resistant lines are noted as “-OR,” and resistance was confirmed in each line using dose response colony formation assays. Olaparib response in PEO1 (IC50 = 6.4 nM) and PEO1-OR (IC50 = 1223 nM), and mechanisms of resistance have been reported (11, 12). The olaparib IC50 increased from 322 nM in OVSAHO to 1300 nM in OVSAHO-OR (Supplemental Fig. S1). Histones from each parental and olaparib-resistant line were isolated and the percentage of histone H3 and H4 modifications were determined by mass spectrometry. We reported these data for PEO1/PEO1-OR (12), and we include these data here for comparison with the OVSAHO/OVSAHO-OR isogenic pair. Complete mass spectrometry data for PEO1/PEO1-OR are published (12), and data for all marks analyzed in the new lines are available in Supplemental File 1. All samples were analyzed in triplicate, yielding a “Total Average Percent” value. This number represents the percentage of the specific modification divided by the total detection of a histone tail residue. For example, in OVSAHO cells, H3K14ac was detected as 19.2% of all H3K14 (Fig. 1A and Supplemental File 1). Notably, H3K14ac was significantly increased in both resistant lines compared to the parental lines (Fig. 1A). We subsequently isolated histones from each cell line pair and immunoblotted for H3K14ac and total H3 and confirmed increased H3K14ac in both olaparib-resistant lines (Fig. 1B).
FIG 1. H3K14ac enrichment and histone acetyltransferase expression is altered in olaparib-resistant HGSOC cells and a PDX model.
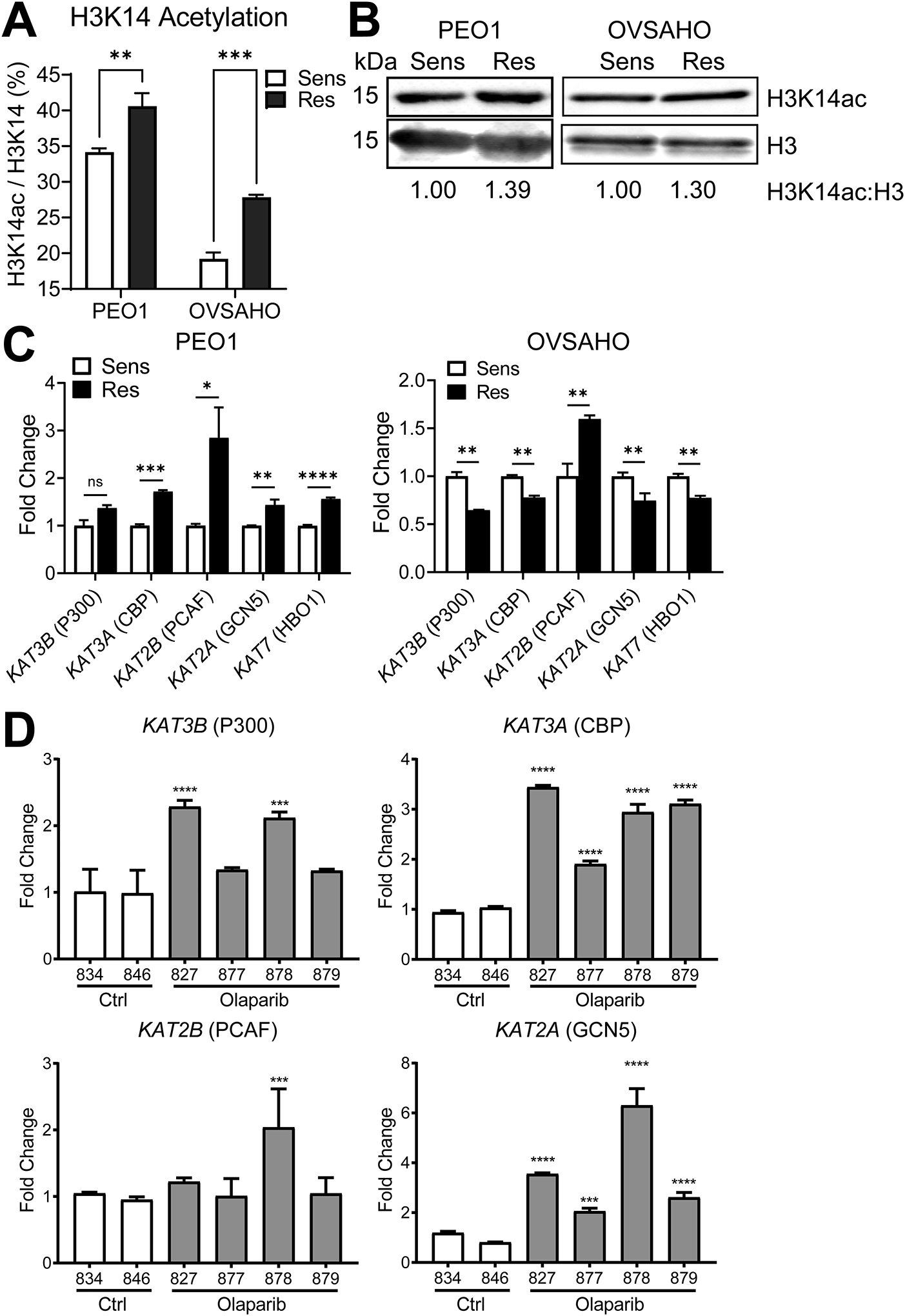
(A) Histone extracts from the indicated olaparib-sensitive and olaparib-resistant HGSOC cell lines were assayed in triplicate by mass spectrometry for histone modifications. Data are shown for each sensitive/resistant pair as the mean ± SD percentage of acetylated H3K14. N = 3; p-value by t-test. (B) Histone extracts from the indicated olaparib sensitive/resistant pairs were assayed by immunoblot for H3K14ac and total H3. The H3K14ac:H3 ratio was determined by band densitometry and normalized to the olaparib-sensitive line of each pair, which are set as 1. (C) mRNA from the indicated olaparib sensitive/resistant pairs was assayed for HAT expression by RT-qPCR. To demonstrate Fold Change, target gene expression was determined relative to GAPDH and then normalized to Control mice. Each bar represents the mean ± SD of three PCR reactions. p-values by t-test. (D) Human HGSOC PDX cells were injected into NSG mice. After seven days, mice received a 21-day treatment course of intraperitoneal vehicle control or 50 mg/kg olaparib. Following treatment, tumor cells were allowed to recur over an additional 21 days. Upon necropsy, ascites cells were collected. mRNA from ascites cells was analyzed by RT-qPCR for the indicated HATs. To demonstrate Fold Change, target gene expression was determined relative to GAPDH control and then normalized to Control mice. Each bar represents the mean ± SD of three PCR reactions. The numbers below bars are mouse ear tag IDs. p-values were determined by t-test comparing each olaparib-treated mouse (gray bars; #827, #877, #878, or #879) to the average of both control mice (white bars; #834 and #846). * p < 0.05, ** p < 0.01, *** p < 0.001; **** p < 0.0001. Sens = olaparib-sensitive, Res = olaparib-resistant.
We next examined the expression of HATs in each sensitive/resistant cell line pair. We examined expression of five HATs with known H3K14 acetylation activity by RT-qPCR, including KAT3B (P300), KAT3A (CBP), KAT2B (PCAF), KAT2A (GCN5), and KAT7 (HBO1) (31–36). We found significantly different expressions of multiple HATs in both olaparib-resistant lines relative to the parental lines (Fig. 1C). For example, PEO1-OR showed upregulation of all HATs except KAT3B (P300) relative to the olaparib-sensitive PEO1 cells, while OVSAHO-OR showed downregulation of multiple HATs relative to OVSAHO. Only KAT2B (PCAF) was upregulated in both PEO1-OR and OVSAHO-OR.
To establish an olaparib-resistant animal model, we inoculated NOD SCID gamma (NSG) mice by intraperitoneal injection with GTFB-PDX1009, a TP53-mutated, BRCA-wildtype HGSOC. We then treated tumor bearing mice daily with olaparib or vehicle control for 21 days, then monitored the mice for tumor recurrence and growth for two months [schematic published in (12)]. After recurrence and necropsy, tumor cells isolated from olaparib-treated mice were subsequently shown to be highly olaparib-resistant (11). By RT-qPCR, we found that mRNA expression of KAT3A and KAT2A were increased in all four olaparib-treated ascites, while KAT2B and KAT3B were upregulated in one and two mice, respectively (Fig. 1D). We further performed RNA-Seq to compare the transcriptomes of control mice (#834 and #836) with the two olaparib-treated mice that showed the greatest average HAT upregulation (#827 and #878). RNA-Seq data confirmed significant upregulation of KAT2A (logFC = 1.093; p = 5.11e-05) and KAT3A (logFC = 0.532; p = 0.041) in the olaparib-treated mice (Supplemental Table S1). Additional gene set analyses using the STRING database (37) revealed that the olaparib-treated tumors had activated KEGG pathways, including “Cell Cycle,” “DNA Replication,” and “Fanconi Anemia Pathway” (Supplemental Fig. S2 and Table S2). In summary, H3K14 acetylation and HAT expression are altered in PARPi-resistant models of HGSOC, including BRCA-deficient cell lines, as well as a BRCA-proficient PDX.
Genetic knockdown of specific HATs in olaparib-resistant HGSOC cells moderately alters olaparib response
H3K14ac is associated with multiple DNA repair mechanisms. Augmented DNA repair is an important component of PARPi resistance (6), and we have previously shown that knockdown or inhibition of epigenetic enzymes such as histone methyltransferases can significantly improve olaparib response (12). We therefore chose to knock down each HAT highlighted in Fig. 1C and examine olaparib response. Since PEO1-OR showed upregulation of four of the five HATs, we used lentiviral transduction to stably express short hairpin RNAs (shRNA) targeting each HAT, or a non-targeting scrambled shRNA as control (shCTRL), in these cells. We used two independent shRNA constructs per HAT; The RNAi Consortium Numbers and target sequences are given in Supplemental Table S3. We determined the degree of knockdown for each shRNA relative to shCTRL by RT-qPCR (Supplemental Fig. S3A–E). We also confirmed HAT knockdowns and assayed depletion of H3K14ac by immunoblot (Fig. 2A–E). Notably, the better knockdowns of P300 (shKAT3B#1), CBP (shKAT3A#1), and HBO1 (shKAT7#2) also partially depleted H3K14ac. Conversely, effective knockdown of PCAF (shKAT2B#1) did not deplete H3K14ac. GCN5 knockdown also did not affect H3K14ac levels. We then performed dose response colony formation assays. Cells were dosed with olaparib ranging from 4.8 nM to 30 μM or vehicle control and the olaparib IC50 was calculated for each shRNA. Full olaparib dose response curves are shown in Fig. 2 F–J and representative olaparib IC50 values are summarized in Table 1. Both shRNA against P300, one shRNA against CBP, and both shRNA against GCN5 showed a statistically significant (p < 0.05) reduction in olaparib IC50 relative to shCTRL. However, sensitization in such cases was modest. For example, the IC50 value for shKAT2A#1 was 1104 nM vs. the shCTRL value of 2620 nM. We performed a second set of dose response experiments to determine colony formation specifically at 120 nM, 600 nM, and 3 μM olaparib (Supplemental Fig. S3F–J). Colony formation for all shRNA was not significantly different from shCTRL at 120 nM or 600 nM. shKAT3A#1 (Fig. S3G) and shKAT2A#1 (Fig. S3I) showed small but significant reductions in PEO1-OR colony formation in 3 μM olaparib.
FIG 2. Knockdown of HATs elicits moderate alteration of olaparib response in PEO1-OR (olaparib-resistant) HGSOC cells.
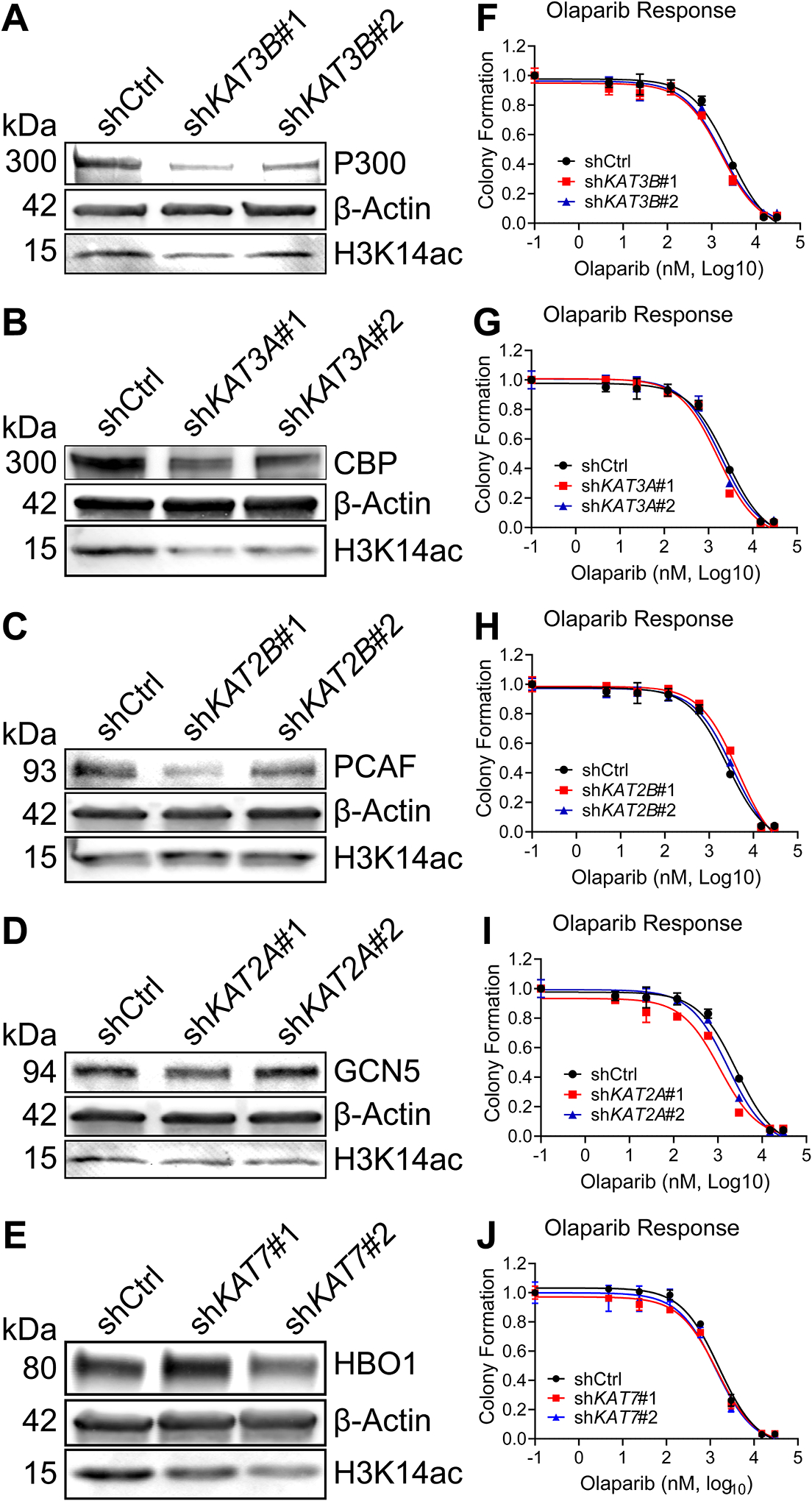
(A-E) PEO1-OR cells were transduced with the indicated targeted shRNA or a non-targeting scrambled shRNA (shCTRL) and then selected in puromycin. For each knockdown or control, immunoblot was performed for the specific HAT, β-Actin loading control, and H3K14ac. (F-J). Each shRNA line was seeded at 2500 cells per well in a 24-well plate and treated with vehicle control or increasing doses of olaparib for eight days, after which colonies were fixed and stained with crystal violet. Stain was dissolved and the absorbance from each well was read by spectrophotometer, then normalized to vehicle control. IC50 curves were plotted in GraphPad Prism.
Table 1. Olaparib IC50 values of control and HAT knockdown PEO1-OR HGSOC cells.
For each indicated shRNA, the olaparib IC50 values and p-values are shown. The column “2nd Run?” indicates if the shRNA significantly changed colony formation in a follow-up experiment.
| shRNA | Olaparib IC50 (nM) | p-value | 2nd Run? |
|---|---|---|---|
| shCTRL | 2620 | - | - |
| shKAT3B #1 | 1699 | 0.016 | No |
| shKAT3B #2 | 1716 | 0.021 | No |
| shKAT3A #1 | 1672 | 0.014 | Yes |
| shKAT3A #2 | 1955 | 0.099 | No |
| shKAT2B #1 | 4549 | 0.002 | Yes |
| shKAT2B #2 | 3409 | 0.105 | Yes |
| shKAT2A #1 | 1104 | < 0.0001 | Yes |
| shKAT2A #2 | 1704 | 0.009 | No |
| shRNA | Olaparib IC50 (nM) | p-value | |
| shCTRL | 1593 | - | - |
| shKAT7 #1 | 1431 | 0.444 | No |
| shKAT7 #2 | 1348 | 0.352 | No |
Despite differing levels of knockdown, both shRNA against KAT2B (PCAF) increased olaparib IC50 in PEO1-OR cells, with shKAT2B#1 showing a significant increase to 4549 nM vs. the shCTRL value of 2620 nM. (Table 1 and Fig. 2H). This result was confirmed for both KAT2B shRNA in PEO1-OR in the follow-up experiment, as both showed increased colony formation in 3 μM olaparib (Fig. S3H). Neither shRNA targeting KAT7 (HBO1) showed a significant effect in either experiment (Fig. 2J and S3J), despite depletion of H3K14ac by shKAT7#2 (Fig. 2E).
In summary, we determined that specific knockdown of CBP (using shKAT3A#1) and GCN5 (using shKAT2A#1) modestly improved olaparib response, while knockdown of PCAF increased olaparib resistance. All other knockdowns did not significantly alter olaparib response in PEO1-OR.
Pharmacologic depletion of H3K14ac does not sensitize olaparib-resistant HGSOC cells, but bromodomains may play roles in olaparib response
In the HAT knockdown experiments, depletion of H3K14ac was not predictive of decreased or increased resistance to olaparib. We next tested if pharmacologic depletion of H3K14ac would be effective in altering PARPi response. HBO1 inhibitor WM-3835 was demonstrated by MacPherson et al. (38) to rapidly deplete H3K14ac. We treated PEO1-OR cells with WM-3835 and then tested H3K14ac by immunoblotting. Densitometry analysis showed that only 27% of H3K14ac remained in PEO1-OR cells after a 6-hour incubation with 10 μM WM-3835 (Fig. 3A). 10 μM WM-3835 also depleted H3K14ac in OVSAHO-OR cells (Supplemental Fig. S4A). We then performed colony formation assays using olaparib alone or in combination with 10 μM WM-3835 to determine if depletion of H3K14ac was sufficient to sensitize PEO1-OR. Despite the efficacy of H3K14ac depletion, no change in olaparib response was found in PEO1-OR (Fig. 3B and Table 2) or in OVSAHO-OR (Fig. S4B).
FIG 3. Depletion of H3K14ac does not shift olaparib IC50, but specific BRD inhibitors moderately affect olaparib response in PEO1-OR HGSOC cells.
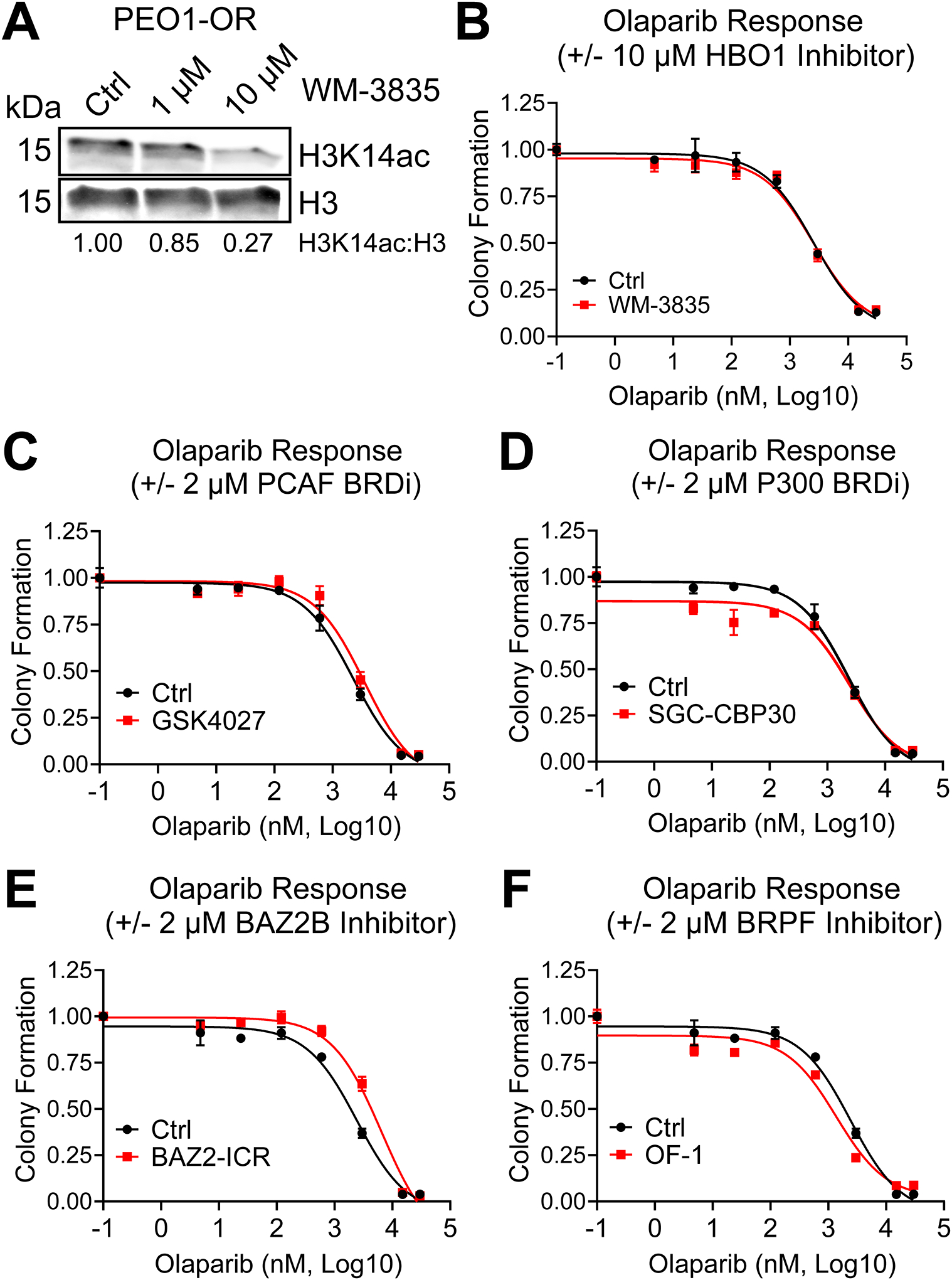
(A) PEO1-OR cells were treated with the indicated doses of HBO1 acetyltransferase inhibitor WM-3835 for 6 hours. Histone extracts were analyzed by immunoblot for H3K14ac and total H3. The H3K14ac:H3 ratio was calculated by densitometry to quantify H3K14ac depletion. (B) PEO1-OR were seeded at 2500 cells per well in a 24-well plate and treated with increasing doses of olaparib with or without 10 μM WM-3835. Media and drug were changed every 2–3 days for eight days, after which colonies were fixed and stained with crystal violet. Stain was dissolved and the absorbance from each well was read by spectrophotometer, then normalized to vehicle control. Error bars, SEM. Olaparib IC50 for each treatment condition was calculated in GraphPad Prism. (C-F) PEO1-OR cells in a 24-well plate were treated with increasing doses of olaparib with or without 2 μM of the indicated BRD inhibitors. Colony formation and olaparib IC50 were determined as described in Fig. 3B. Error bars, SEM.
Table 2. Olaparib IC50 values of PEO1-OR HGSOC cells treated with olaparib alone or in combination with HAT or BRD inhibitors.
For each treatment, the olaparib IC50 values and p-values are shown.
| Compound(s) | Olaparib IC50 (nM) | p-value |
|---|---|---|
| Olaparib Only | 2479 | - |
| Olap. + 10 μM WM-3835 | 2668 | 0.711 |
| Compound(s) | Olaparib IC50 (nM) | p-value |
| Olaparib Only | 2280 | - |
| Olap. + 2 μM GSK4027 | 3540 | 0.019 |
| Olap. + 2 μM SGC-CBP30 | 2488 | 0.719 |
| Compound(s) | Olaparib IC50 (nM) | p-value |
| Olaparib Only | 2436 | - |
| Olap. + 2 μM BAZ2-ICR | 6547 | < 0.0001 |
| Olap. + 2 μM OF-1 | 1379 | 0.016 |
Rather than continuing with direct inhibition of acetyltransferase activity, we elected to test if pharmacologic inhibition of the histone reader bromodomains (BRD) of HAT enzymes impacted olaparib response. Olaparib IC50 and p-values for all subsequent PEO1-OR inhibitor treatments are summarized in Table 2, and additional OVSAHO-OR inhibitor experiments are shown in Supplementary Fig. S4C–F. We performed colony formation assays in PEO1-OR using olaparib alone or in combination with 2 μM GCN5/PCAF BRD inhibitor GSK4027, or 2 μM P300/CBP BRD inhibitor SGC-CBP30. GSK4027 notably increased olaparib resistance in PEO1-OR cells (Fig. 3C and Table 2), but not in OVSAHO-OR (Fig. S4C). This agrees with our data showing that knockdown of PCAF also increases olaparib resistance in PEO1-OR. SGC-CBP30 had no effect on olaparib response in either cell line (Fig. 3D and Table 2 for PEO1-OR; Fig. S4D for OVSAHO-OR).
While the PCAF BRD may play a role in olaparib response, it must be noted that the GCN5/PCAF BRDs also read H3K36 acetylation and the P300/CBP BRDs read H3K56 acetylation (39). We therefore chose to specifically inhibit the BRDs of proteins that have direct reading function of H3K14ac. These include Bromodomain Adjacent to Zinc Finger Domain 2B (BAZ2B) and proteins in the Bromodomain and PHD-Finger Containing (BRPF)-family (39). We performed colony formation assays in PEO1-OR cells using olaparib alone or in combination with 2 μM BAZ2B inhibitor BAZ2-ICR, or 2 μM pan-BRPF inhibitor OF-1. Much like inhibition of PCAF, inhibition of BAZ2B with BAZ2-ICR significantly increased olaparib resistance in PEO1-OR (Fig. 3E and Table 2) and had no effect in OVSAHO-OR (Fig. S4E). However, inhibition of BRPF family proteins by OF-1 significantly decreased the olaparib IC50 in PEO1-OR (Fig. 3F and Table 2) and also sensitized OVSAHO-OR to high olaparib doses (3–30 μM) (Fig. S4F).
BRPF3 is elevated in HGSOC and associated with poor outcomes, and may play a role in olaparib response
Given that OF-1 improved olaparib response in both PEO1-OR and OVSAHO-OR, we searched for potential links between BRPF-family proteins, HAT complexes, and ovarian cancer. BRPF3 is notable among the BRPF family proteins for a known association with HBO1, in which it directs a multiprotein complex toward acetylation of H3K14, rather than lysines of histone H4 (40). We examined a publicly available microarray dataset (GSE10971) comparing mRNA expression in normal fallopian tube epithelium (FTE) versus HGSOC. KAT7 (HBO1) mRNA levels were elevated in HGSOC (Fig. 4A) but results were not significant (p = 0.0833). However, BRPF3 mRNA elevation was highly significant (p = 1.9e-06) in HGSOC compared to normal FTE (Fig. 4B). We also analyzed a microarray dataset of ovarian tumors from The Cancer Genome Atlas (TCGA) and analyzed overall survival. Using automatic cutoff selection and false-discovery correction in KMplot (41, 42) we split a cohort of patients into “low” or “high” expression of KAT7 or BRPF3. We observed that high KAT7 (N = 904) expressing patients trended toward longer survival than low KAT7 (N = 303) patients (high median survival = 46 months; low median survival = 43 months; p = 0.063) (Fig. 4C). High BRPF3 (N = 418) expressing patients, however, showed significantly shorter overall survival than low BRPF3 (N =237) patients (high median survival = 40 months; low median survival = 51 months; p = 0.00056) (Fig. 4D). We used the Cancer Science Institute of Singapore Ovarian Database (CSIOVDB) to perform a further meta-analysis of HGSOC microarray data (43–45). BRPF3 was significantly upregulated in higher stage and grade of HGSOC (Supplemental Fig. S5). A meta-analysis using canSAR Black (46) showed BRPF3 has the strongest association with ovarian cancer in combined molecular score, which includes mutation score, gene expression, and copy number variation score (Supplemental Fig. S6).
FIG 4. BRPF3 mRNA expression is elevated in HGSOC relative to normal fallopian tube, and elevated expression is associated with worse overall survival. BRPF3 knockdown moderately sensitizes PEO1-OR cells to olaparib.
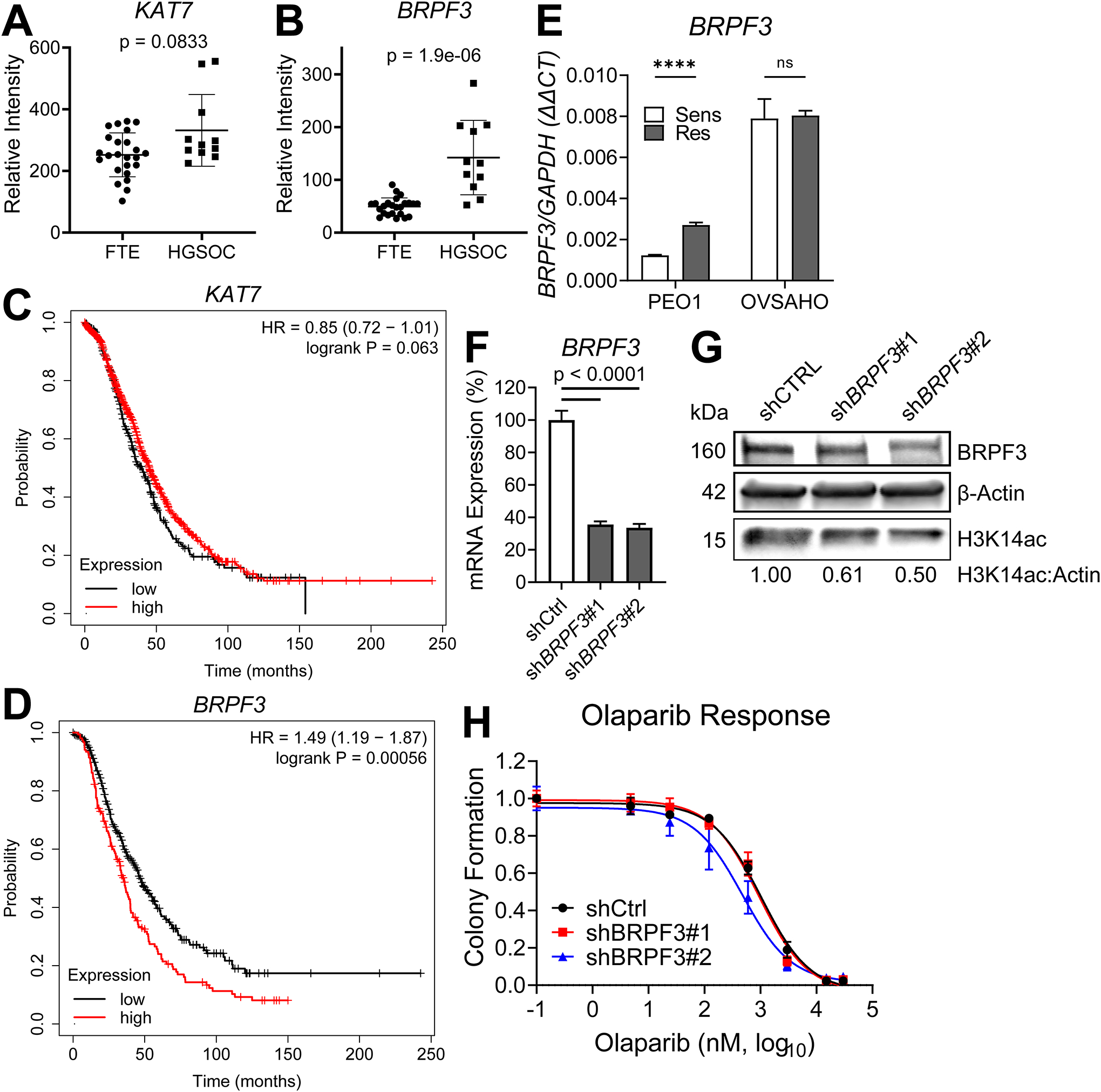
(A-B) mRNA expression of KAT7 and BRPF3 in microarray dataset GSE10971 was compared between HGSOC and normal FTE. p-value by t-test. (C-D) TCGA microarray data for KAT7 and BRPF3 were analyzed using KMplot and Kaplan-Meier curves were generated for overall survival of high and low expressing patients for each gene. (E) mRNA from the indicated olaparib sensitive/resistant pairs was assayed for BRPF3 expression by RT-qPCR. Target gene expression was determined relative to GAPDH control by ΔΔCt method. Each bar represents the mean ± SD of three PCR reactions. p-values by t-test. **** p < 0.0001. PEO1-OR cells were then transduced with shRNA targeting BRPF3 or a non-targeting scrambled shRNA (shCTRL) and then selected in puromycin. Knockdown of BRPF3 was determined by RT-qPCR (F). Knockdown of BRPF3, as well as protein levels of H3K14ac and β-Actin loading control were assayed by immunoblot. (G). mRNA expression in (F) was determined relative to GAPDH by ΔΔCt method, then normalized to shCTRL for each knockdown. p-value by t-test. (H) Cells from F-G were treated with increasing doses of olaparib, and colony formation was assayed as described in Fig. 3B.
We examined BRPF3 mRNA expressions in our two olaparib sensitive/resistant cell line pairs. It was increased in PEO1-OR vs sensitive PEO1, but unchanged in OVSAHO-OR vs OVSAHO (Fig. 4E). BRPF3 was also upregulated in RNA-Seq analysis of olaparib-treated ascites of GTFB-PDX1009 (logFC = 0.642; p = 0.019) (Supplemental Table S1). We used lentiviral transduction to stably express shCTRL or two independent shRNA targeting BRPF3 in PEO1-OR. We determined the degree of knockdown for each shRNA relative to shCTRL by RT-qPCR (Fig. 4F) and immunoblot (Fig. 4G). While mRNA expression appeared similar between the two BRPF3 shRNA lines, the amount of protein in shBRPF3#2 was notably lower, which corresponded with depleted H3K14ac (Fig. 4G). We next compared olaparib response in BRPF3 knockdown cells compared to control. Olaparib IC50 for shCTRL in this experiment was 1036 nM. shBRPF3#1 (IC50 = 938 nM, p = 0.517) did not significantly alter olaparib IC50 in PEO1-OR. However, in agreement with better protein knockdown, shBRPF3#2 (IC50 = 479 nM, p = 0.0002) did significantly reduce the olaparib IC50 (Fig. 4H). In a duplicate experiment, the olaparib IC50 of shCTRL cells was 1593 nM, while the IC50 was significantly reduced to 835 nM in shBRPF3#2 cells (Supplemental Fig. S6), confirming that knockdown of BRPF3 partially sensitized PEO1-OR to olaparib.
Discussion
Regardless of BRCA status, numerous clinical trials showed significant progression-free survival benefit from PARPi treatment to both newly diagnosed and recurrent HGSOC cases (3, 4, 47). Going forward, nearly all HGSOC patients will be eligible to receive PARPi, and it is essential to identify and specifically target novel vulnerabilities to improve response rates and increase the number of patients who can benefit from PARPi treatment. PARPi cause ssDNA breaks and can cause replication fork collapse due to altered fork speed and stability (48, 49). Epigenetic modifications and enzymes are likely to be important mechanisms of PARPi resistance, as they control access to DNA for transcription, replication, and repair. In this report, we profiled the histone landscape of two BRCA-deficient PARPi-resistant HGSOC cell lines. We observed an increase in H3K14ac in both PARPi-resistant lines compared to isogenic PARPi-sensitive cells (Fig. 1A–B). It is crucial to note that even modest changes in the percentage of a specific histone modification may represent altered enrichment or depletion at hundreds or even thousands of genomic loci in each cell and may have considerable consequences for access to DNA. Gene expression of HATs was also altered in PARPi-resistant lines, as well as in a PDX model of BRCA-proficient PARPi-treated HGSOC (Fig. 1C–D). The olaparib-treated PDX notably showed activation of cell cycle and DNA replication pathways, as well as the Fanconi anemia pathway, indicating augmented DNA repair. While HATs are not included in gene sets in analysis of expression, they are highly relevant to pathway function. As noted previously, all five of the HATs analyzed in this study have roles in DNA repair, including acetylation of non-histone proteins or recruitment of repair factors. HBO1-mediated histone acetylation contributes to MCM2–7 loading, indicating roles in cell cycle and DNA replication (50).
Using both genetic and pharmacologic approaches, we examined if depletion or inhibition of specific HATs or accessory BRD (i.e., H3K14ac reader) proteins altered the response to the PARPi olaparib. Notably, rapid and robust pharmacologic depletion of H3K14ac (using HBO1 inhibitor WM-3835) did not affect PARPi response. However, knockdown or inhibition of PCAF, which did not alter H3K14ac enrichment, increased PARPi resistance. Knockdown and inhibition of BRPF3, a BRD protein associated with HBO1, modestly sensitized HGSOC cells to olaparib.
Placing these results in the context of prior studies, we reported an epigenetic mechanism of PARPi resistance in HGSOC. Specifically, we previously showed that H3K9me2 was enriched in PARPi-resistant HGSOC cell line and PDX models, concomitant with upregulation of euchromatic histone-lysine N-methyltransferase-1 and −2 (EHMT1 and EHMT2), the enzymes responsible for writing the H3K9me2 mark (12). Genetic knockdown or pharmacologic inhibition of EHMT1/2 sensitized PARPi-resistant HGSOC cells and ablated both HR and NHEJ DNA repair. H3K9 methylation is known to be linked to H3K14ac by the triple Tudor domain of SETDB1, which may play a role in silencing of active genomic regions (51). We observed significant upregulation of both H3K9me2 and H3K14ac in PEO1-OR cells relative to parental, olaparib-sensitive PEO1. Profiling studies such as chromatin immunoprecipitation (ChIP) will be required to determine if regions of these two marks overlap. H3K9me2 was not as strongly upregulated in OVSAHO-OR, suggesting that a strong linkage of H3K9 methylation with H3K14ac is likely context-dependent. Further studies will be necessary to evaluate the efficacy of combined inhibition of EHMT1/2 with HAT or BRD inhibitors.
The knockdown of specific HATs had only modest effects at sensitizing PEO1-OR to olaparib (Table 1). Despite PCAF being upregulated in PEO1-OR cells relative to olaparib-sensitive PEO1, knockdown of PCAF surprisingly further increased olaparib resistance (Table 1 and Supplemental Fig. S3). In a prior study, Zhao et al. showed that acetylation of RPA1 by PCAF/GCN5 promoted NER (22), so we would expect knockdown of PCAF and/or GCN5 to lessen DNA repair and promote PARPi sensitivity. However, a separate study by Liu et al. showed that PCAF and CBP/P300 acetylate p53 in response to DNA damage (52). We may speculate that the presence of TP53 mutations in nearly all HGSOC tumors could alter this response. Additionally, Kim et al. published that PCAF acetylates H4K8 in BRCA-deficient cells at stalled replication forks and promotes fork degradation (53). Thus, knockdown of PCAF in PEO1-OR may stabilize forks and prevent cell death.
The depletion of H3K14ac by HBO1 inhibitor WM-3835 indicates that the majority of H3K14 acetylation in PEO1-OR cells is mediated by HBO1 (Fig. 3A). A prior study by Xiao et al. demonstrated that HBO1 was enriched on the transcriptional start sites of actively transcribed genes, and that HBO1 contributed to bulk histone acetylation at these sites (54). In that study, HBO1 enrichment correlated with the level of promoter H3K14 acetylation. Given that the addition of 10 μM WM-3835 did not affect the olaparib IC50 in PEO1-OR (Fig. 3B), we conclude that the acetyltransferase activity of HBO1 and bulk H3K14 acetylation do not have a major effect on PARPi response. The Xiao et al. study also showed that HBO1 enrichment, despite being correlated with H3K14ac, did not correlate with transcription levels of specific genes. Thus, even though WM-3835 depletes H3K14ac, RNA-seq data would be needed to determine if WM-3835 had significant effects on transcription. Further, despite other HATs having a lesser role in bulk genomic H3K14ac, it is possible that other HATs acetylate histones at other genomic locations and genes, some of which may alter PARPi response. We must also consider that acetylation of non-histone targets by other HATs may have effects on PARPi response. ChIP studies of specific loci and specific targeting with other HAT inhibitors will be required to tease out precise effects.
Unlike HBO1 acetyltransferase inhibition, the use of specific BRD inhibitors did affect olaparib response. PCAF BRD inhibitor GSK2027 increased olaparib resistance in PEO1-OR (Fig. 3C and Table 2), which agrees with increased olaparib IC50 in PCAF knockdown cells (Table 1) and indicates that the BRD of PCAF, rather than the HAT domain, may play a significant role. Also, despite being readers of the same H3K14ac mark, inhibition of BAZ2B or BRPF-family proteins had opposite effects on olaparib response. Inhibition of BAZ2B with BAZ2-ICR increased resistance in PEO1-OR (Fig. 3E and Table 2), while inhibition of the BRPF family with OF-1 decreased resistance in both PEO1-OR (Fig. 3F and Table 2) and OVSAHO-OR (Supplemental Fig. 4F), demonstrating the highly complex regulatory nature of histone modifications. Overall, we found that the blunt method of HBO1 inhibition was ineffective at reversing PARPi resistance. Changes in IC50 due to BRD inhibition suggest that histone readers and other accessory factors in HAT protein complexes may play a more direct role in PARPi response than the HATs themselves. The exact subunit composition of such complexes in HGSOC, and how these may be altered in acquired PARPi resistance, remain unknown.
The potential importance of accessory factors is further emphasized by the increased expression of BRPF3 in HGSOC compared to normal FTE (Fig. 4B), as well as the association of elevated BRPF3 expression with higher ovarian cancer stage and grade (Supplemental Fig. S5 and S6), and with worse overall survival (Fig. 4D). While OF-1 is a pan-BRPF inhibitor, specific knockdown of BRPF3 effectively reduced olaparib IC50 in PEO1-OR (Fig. 4H and S7), indicating its importance in PARPi response. A study by Feng et al. showed that the protein complex containing BRPF3 and HBO1 regulates H3K14 acetylation, while complexes of HBO1 with other cofactors preferentially acetylated lysines of histone H4. BRPF3 was found to be crucial for H3K14 acetylation, as well as loading of CDC45 at DNA origins of replication and activation of those origins (55). Since PARPi-induced DSBs are generated during DNA replication at collapsed replication forks, this role of BRPF3 in DNA replication suggests a possible mechanism for loss or inhibition of BRPF3 to sensitize olaparib-resistant cells.
In terms of therapeutic potential, the observed effects are modest. Other classes of drugs (e.g., HDACi) have shown greater efficacy, at least in combination with carboplatin or other DNA damaging agents. However, development of further BRD inhibitors may improve the response. As an alternative, BRPF3-HBO1 complexes may be enriched in tumor cells, and therefore targeting protein-protein interaction domains may be a way to selectively improve PARPi response in tumor cells while avoiding bystander effects in non-cancerous cells.
Conclusions
Our reported findings highlight the altered epigenetic landscape of PARPi-resistant HGSOC, including histone modifications and changes in expression of associated epigenetic-modifying and reading enzymes. While non-histone targets for acetylation have yet to be examined, our data suggest that the reader function of specific BRDs may play a more direct role than acetyltransferase domains. This may be especially important in the context of HAT proteins such as PCAF or HBO1, which can acetylate lysine residues of H3 or H4 depending on their protein complex partners. In conclusion, specific BRD proteins such as BRPF3, in association with HATs, may play a role in PARPi response even if total H3K14ac levels are not predictive or associated with resistance. Further examination of HAT complex composition in HGSOC and the activities of each subunit may reveal novel vulnerabilities.
Methods
Cell culture, shRNA, and lentivirus
Cell lines were obtained from the Gynecologic Tumor and Fluid Bank (GTFB) at the University of Colorado and were authenticated at the University of Arizona Genomics Core using short tandem repeat DNA profiling. Cells were confirmed to be free of mycoplasma during all experiments, including revisions, using MycoLookOut PCR (Sigma). HGSOC lines were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. 293FT lentiviral packaging cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. All cells were grown at 37 °C supplied with 5% CO2. shRNA in pLKO.1 lentiviral vector plasmid were purchased from the University of Colorado Functional Genomics Facility. Sequences and The RNAi Consortium numbers are listed in Supplemental Table S3. A scrambled non-targeting shRNA was used as control (Sigma-Aldrich #SHC016). Lentivirus was packaged as previously described (56) in 293FT using 3rd generation packaging plasmids (Virapower, Invitrogen) with polyethyleneimene (PEI) transfection in a 1:3 DNA:PEI ratio. Culture supernatant was harvested at 48–72 hours post-transfection and processed through 0.45 μM filters. Viruses encode a puromycin resistance gene. Transduced HGSOC cells were selected in 1 μg/mL puromycin and used for experiments as a heterogeneous population.
Histone modification profiling
Profiling of histone modifications in olaparib-sensitive and -resistant cells was performed by the Northwestern University Proteomics Core. Briefly, we provided frozen pellets of 5 × 106 PEO1, PEO1-OR, OVSAHO, and OVSAHO-OR cells. Histone extracts were trypsin digested and histone residues were assayed in triplicate as previously reported (57, 58) by liquid chromatography coupled to mass spectrometry using a TSQ Quantiva Ultra Triple Quadrupole Mass Spectrometer.
PDX mouse model of PARPi-resistant HGSOC
All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the University of Colorado Institutional Animal Care and Use Committee. Primary ovarian cancer sample GTFB1009 (BRCA1/2-wildtype) was provided by the University of Colorado Gynecologic Tumor and Fluid Bank. Intraperitoneal injections of 5 million GTFB1009 ascites cells were given to 6- to 8-week-old NOD SCID gamma (NSG) mice (Jackson Labs). Following a 7-day incubation period, mice were given once daily intraperitoneal injections of 50 mg/kg olaparib or vehicle control (10% 2-hydroxypropyl-β-cyclodextrin, Sigma-Aldrich #C0926) for 21 days. After treatment, tumors were allowed to recur for two months. Mice were then euthanized, and ascites cells were collected for analysis. This model is further characterized in (11).
RNA-Seq of GTFB-PDX1009
RNA was isolated from control (n = 2) and olaparib-treated (n = 2) GTFB-PDX1009 ascites using the RNeasy Plus Mini kit (Qiagen). RNA quality was confirmed using an Agilent TapeStation and all RNA used for library preparation had a RIN>9. Libraries were generated and sequencing was performed by Novogene. HISAT (59) was used for alignment against GRCh37 version of the human genome. Samples were normalized using TPM (Transcripts per Kilobase Million) measurement and gene expression using the GRCh37 gene annotation was calculated using home-made scripts. The Division of Translational Bioinformatics and Cancer Systems Biology at the University of Colorado School of Medicine performed analysis. Data are available at accession GSE131231. Additional gene set analysis, including KEGG pathways, was performed using the STRING database (37).
Compounds
Olaparib (SelleckChem #S1060), P300/CBP inhibitor SGC-CBP30 (SelleckChem #S7256), PCAF inhibitor GSK4027 (Cayman #23421), BAZ2B inhibitor BAZ2-ICR (Tocris #5266), and BRPF inhibitor OF-1 (SelleckChem #S7681) were purchased from the indicated suppliers. HBO1 inhibitor WM-3835 was kindly provided by Professor Jonathan Baell of the Monash Institute of Pharmaceutical Sciences (Victoria, Australia) and Professor Mark Dawson of the Peter MacCallum Cancer Centre, University of Melbourne (Melbourne, Australia).
Colony formation assay
PEO1-OR: On 24-well plates, 2500 cells were seeded per well and treated with increasing doses of olaparib alone or in combination with the indicated compounds. Media and drugs were changed every three days until control wells were confluent. Colonies were washed twice with PBS, then incubated in fixative (10% methanol and 10% acetic acid in PBS). Fixed colonies were stained with 0.4% crystal violet in 20% ethanol/PBS. After imaging, crystal violet was dissolved in fixative and absorbance was measured at 570 nm using a Molecular Devices SpectraMax M2e plate reader. OVSAHO-OR: On 96-well plates, 7500 cells were seeded per well and treated as noted for six days, then fixed and stained.
Reverse-transcriptase quantitative PCR (RT-qPCR)
RNA was isolated from cells using the RNeasy Plus Mini Kit (Qiagen). mRNA expression was determined using SYBR green Luna One Step RT-qPCR Kit (New England BioLabs) on a C1000 Touch (Bio-Rad) or QuantStudio 6 (Applied Biosystems) thermocycler. Expression was quantified by the ΔΔCt method using target-specific primers and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) control primers. mRNA-specific primers were designed to span exon-exon junctions to avoid detection of genomic DNA. Primer sequences are shown in Supplemental Table S4.
Immunoblotting and Densitometry
For histone blots, extracts were made using the Histone Extraction Kit (Abcam #ab113476). For total protein, cells were lysed and briefly sonicated in RIPA buffer (150 mM NaCl, 1% TritonX-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 8.0) supplemented with cOmplete EDTA-free protease inhibitors (Roche #11873580001) and phosphatase inhibitors NaF and Na3VO4. Protein was separated using 4–20% Mini-PROTEAN TGX SDS-PAGE gels (BioRad) and transferred to PVDF membrane using the TransBlot Turbo (BioRad). Membranes were blocked in LI-COR Odyssey buffer (#927–50000) for 1 hour at room temperature. Primary antibody incubation was performed overnight in blocking buffer at 4 °C. Membranes were washed 3 times for 5 minutes each in TBST (50 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween-20), then secondary antibodies were applied in blocking buffer for one hour at room temperature. Membranes were washed again 3 times for 5 minutes each in TBST. Bands were visualized using the LI-COR Odyssey Imaging System. Primary antibodies are listed in Supplemental Table S5. Secondary antibodies were LI-COR fluorophore-labeled secondary goat anti-rabbit (IRDye 680RD or IRDye 800CW, LI-COR Biosciences Cat# 925–68071, RRID:AB_2721181 or LI-COR Biosciences Cat# 926–32211, RRID:AB_621843) or goat anti-mouse (IRDye 680RD or IRDye 800CW, LI-COR Biosciences Cat# 926–68070, RRID:AB_10956588 or LI-COR Biosciences Cat# 925–32210, RRID:AB_2687825) at 1:20,000 dilution. Band fluorescence intensity was analyzed using LI-COR ImageStudio 4. Immunoblots of H3K14ac were normalized to band intensity of total H3.
Ovarian cancer dataset analysis
Microarray data from publicly available ovarian cancer database GSE10971 (60, 61) was analyzed to compare gene expression between normal FTE and HGSOC. KMplot (41, 42) was used to interrogate TCGA ovarian cancer gene expression data and compare overall survival in high and low expressing patients. Patient cohorts were split using default/automatic parameters and p-values were corrected for false discovery. CSIOVDB (43–45) and canSAR Black (46) were used to further interrogate associations between BRPF3 and ovarian cancer microarray datasets.
Software and statistical analysis
Statistical analysis and calculation of P value was performed using GraphPad Prism v9. Quantitative data are expressed as mean ± SD unless otherwise stated. A two-tailed t-test was used for single comparisons. Analysis of variance (ANOVA) with Fisher’s Least Significant Difference (LSD) was used in multiple comparisons. Olaparib IC50 were calculated and compared using the Nonlinear regression (curve fit) analysis, log (inhibitor) vs. response (three parameters) model, with extra sum-of-squares F-test. For all statistical analyses, the level of significance was set at 0.05.
Supplementary Material
Acknowledgements
We thank the University of Colorado OB-GYN Scientific Editor Dr. Heather Aldrich for her assistance in manuscript preparation and revision. We also thank Professor Jonathan Baell of the Monash Institute of Pharmaceutical Sciences (Victoria, Australia) and Professor Mark Dawson of the Peter MacCallum Cancer Centre, University of Melbourne (Melbourne, Australia) and their labs for their generous gift of HBO1 inhibitor WM-3835.
Funding:
We acknowledge philanthropic contributions from the Kay L. Dunton Endowed Memorial Professorship in Ovarian Cancer Research, the McClintock-Addlesperger Family, Karen M. Jennison, Don and Arlene Mohler Johnson Family, Michael Intagliata, Duane and Denise Suess, Mary Normandin, and Donald Engelstad. This work was primarily supported by the Cancer League of Colorado (BGB, 183478-BB; ZLW, 193527-ZW), with additional support from The Department of Defense Ovarian Cancer Research Program (BGB, OC170228, OC200302, OC200225; ZLW, OC210257), The American Cancer Society (BGB, RSG-19-129-01-DDC), the NIH/National Cancer Institute (BGB, R37CA261987; ZLW, R03CA249571), and the Colorado Clinical & Translational Sciences Institute (ZLW, CO-J-20-006). Use of the University of Colorado Functional Genomics Facility was supported by NCI grant P30CA046934 and by NIH/NCATS Colorado CTSA Grant Number UL1TR002535. Histone profiling was performed by the Northwestern University Proteomics Core, supported by NCI CCSG P30CA060553 and P41GM108569.
Abbreviations
- BRD
bromodomain
- BRDi
BRD inhibitor
- CPD
cyclopyrimidine dimers
- CSIOVDB
Cancer Science Institute of Singapore Ovarian Database
- DSB
double-stranded break
- H3K14ac
acetylated histone H3 lysine 14
- HAT
histone acetyltransferase
- HATi
HAT inhibitor
- HDAC
histone deacetylase
- HDACi
HDAC inhibitor
- HGSOC
high grade serous ovarian carcinoma
- HR
homologous recombination
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- NER
nucleotide excision repair
- NHEJ
non-homologous end joining
- PARP
poly (ADP-ribose) polymerase
- PARPi
PARP inhibitor
- PDX
patient derived xenograft
- PEI
polyethyleneimene
- shRNA
short hairpin RNA
- SSB
single-stranded break
- TCGA
The Cancer Genome Atlas
- UV
ultraviolet
Footnotes
Competing interests: The authors declare that they have no competing interests.
Availability of data and materials:
Data generated and/or analyzed during this study are available from the corresponding author(s) on reasonable request. RNA-Seq dataset(s) are deposited as described in materials and methods and are publicly available.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: a cancer journal for clinicians. 2018;68(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Lisio MA, Fu L, Goyeneche A, Gao ZH, Telleria C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. International journal of molecular sciences. 2019;20(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. The New England journal of medicine. 2018. [DOI] [PubMed] [Google Scholar]
- 4.Domchek SM, Aghajanian C, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecologic oncology. 2016;140(2):199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matulonis UA, Penson RT, Domchek SM, Kaufman B, Shapira-Frommer R, Audeh MW, et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Ann Oncol. 2016;27(6):1013–9. [DOI] [PubMed] [Google Scholar]
- 6.Bitler BG, Watson ZL, Wheeler LJ, Behbakht K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecologic oncology. 2017;147(3):695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel AG, Flatten KS, Schneider PA, Dai NT, McDonald JS, Poirier GG, et al. Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. The Journal of biological chemistry. 2012;287(6):4198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Znojek P, Willmore E, Curtin NJ. Preferential potentiation of topoisomerase I poison cytotoxicity by PARP inhibition in S phase. British journal of cancer. 2014;111(7):1319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer research. 2014;74(1):287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell reports. 2016;17(9):2367–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamamoto TM, McMellen A, Watson ZL, Aguilera J, Ferguson R, Nurmemmedov E, et al. Activation of Wnt signaling promotes olaparib resistant ovarian cancer. Molecular carcinogenesis. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watson ZL, Yamamoto TM, McMellen A, Kim H, Hughes CJ, Wheeler LJ, et al. Histone methyltransferases EHMT1 and EHMT2 (GLP/G9A) maintain PARP inhibitor resistance in high-grade serous ovarian carcinoma. Clin Epigenetics. 2019;11(1):165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okonogi TM, Alley SC, Harwood EA, Hopkins PB, Robinson BH. Phosphate backbone neutralization increases duplex DNA flexibility: a model for protein binding. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(7):4156–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Audia JE, Campbell RM. Histone Modifications and Cancer. Cold Spring Harbor perspectives in biology. 2016;8(4):a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esteller M Cancer epigenomics: DNA methylomes and histone-modification maps. Nature reviews Genetics. 2007;8(4):286–98. [DOI] [PubMed] [Google Scholar]
- 17.Wang R, Xin M, Li Y, Zhang P, Zhang M. The Functions of Histone Modification Enzymes in Cancer. Curr Protein Pept Sci. 2016;17(5):438–45. [DOI] [PubMed] [Google Scholar]
- 18.Lu J, He X, Zhang L, Zhang R, Li W. Acetylation in Tumor Immune Evasion Regulation. Front Pharmacol. 2021;12:771588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Ramirez M, Rocchini C, Ausio J. Modulation of chromatin folding by histone acetylation. The Journal of biological chemistry. 1995;270(30):17923–8. [DOI] [PubMed] [Google Scholar]
- 20.Tse C, Sera T, Wolffe AP, Hansen JC. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Molecular and cellular biology. 1998;18(8):4629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duan MR, Smerdon MJ. Histone H3 lysine 14 (H3K14) acetylation facilitates DNA repair in a positioned nucleosome by stabilizing the binding of the chromatin Remodeler RSC (Remodels Structure of Chromatin). The Journal of biological chemistry. 2014;289(12):8353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao M, Geng R, Guo X, Yuan R, Zhou X, Zhong Y, et al. PCAF/GCN5-Mediated Acetylation of RPA1 Promotes Nucleotide Excision Repair. Cell reports. 2017;20(9):1997–2009. [DOI] [PubMed] [Google Scholar]
- 23.Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, et al. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011;30(18):2135–46. [DOI] [PubMed] [Google Scholar]
- 24.Tamburini BA, Tyler JK. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Molecular and cellular biology. 2005;25(12):4903–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogiwara H, Kohno T. CBP and p300 histone acetyltransferases contribute to homologous recombination by transcriptionally activating the BRCA1 and RAD51 genes. PloS one. 2012;7(12):e52810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niida H, Matsunuma R, Horiguchi R, Uchida C, Nakazawa Y, Motegi A, et al. Phosphorylated HBO1 at UV irradiated sites is essential for nucleotide excision repair. Nat Commun. 2017;8:16102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matthews BG, Bowden NA, Wong-Brown MW. Epigenetic Mechanisms and Therapeutic Targets in Chemoresistant High-Grade Serous Ovarian Cancer. Cancers. 2021;13(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li G, Tian Y, Zhu WG. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front Cell Dev Biol. 2020;8:576946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer research. 2009;69(16):6381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuo MH, Brownell JE, Sobel RE, Ranalli TA, Cook RG, Edmondson DG, et al. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature. 1996;383(6597):269–72. [DOI] [PubMed] [Google Scholar]
- 32.Grant PA, Eberharter A, John S, Cook RG, Turner BM, Workman JL. Expanded lysine acetylation specificity of Gcn5 in native complexes. The Journal of biological chemistry. 1999;274(9):5895–900. [DOI] [PubMed] [Google Scholar]
- 33.Schiltz RL, Mizzen CA, Vassilev A, Cook RG, Allis CD, Nakatani Y. Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. The Journal of biological chemistry. 1999;274(3):1189–92. [DOI] [PubMed] [Google Scholar]
- 34.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nature reviews Molecular cell biology. 2007;8(4):284–95. [DOI] [PubMed] [Google Scholar]
- 35.Nagy Z, Tora L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene. 2007;26(37):5341–57. [DOI] [PubMed] [Google Scholar]
- 36.Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. The EMBO journal. 2011;30(2):249–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic acids research. 2023;51(D1):D638–d46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacPherson L, Anokye J, Yeung MM, Lam EYN, Chan YC, Weng CF, et al. HBO1 is required for the maintenance of leukaemia stem cells. Nature. 2020;577(7789):266–70. [DOI] [PubMed] [Google Scholar]
- 39.Wu Q, Heidenreich D, Zhou S, Ackloo S, Kramer A, Nakka K, et al. A chemical toolbox for the study of bromodomains and epigenetic signaling. Nat Commun. 2019;10(1):1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng Y, Vlassis A, Roques C, Lalonde ME, González-Aguilera C, Lambert JP, et al. BRPF3-HBO1 regulates replication origin activation and histone H3K14 acetylation. The EMBO journal. 2016;35(2):176–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocrine-related cancer. 2012;19(2):197–208. [DOI] [PubMed] [Google Scholar]
- 42.Lánczky A, Győrffy B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J Med Internet Res. 2021;23(7):e27633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan TZ, Miow QH, Huang RY, Wong MK, Ye J, Lau JA, et al. Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol Med. 2013;5(7):1051–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med. 2014;6(10):1279–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tan TZ, Yang H, Ye J, Low J, Choolani M, Tan DS, et al. CSIOVDB: a microarray gene expression database of epithelial ovarian cancer subtype. Oncotarget. 2015;6(41):43843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitsopoulos C, Di Micco P, Fernandez EV, Dolciami D, Holt E, Mica IL, et al. canSAR: update to the cancer translational research and drug discovery knowledgebase. Nucleic acids research. 2021;49(D1):D1074–d82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matulonis UA, Penson RT, Domchek SM, Kaufman B, Shapira-Frommer R, Audeh MW, et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Ann Oncol. 2016;27(6):1013–9. [DOI] [PubMed] [Google Scholar]
- 48.Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. High speed of fork progression induces DNA replication stress and genomic instability. Nature. 2018;559(7713):279–84. [DOI] [PubMed] [Google Scholar]
- 49.D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst). 2018;71:172–6. [DOI] [PubMed] [Google Scholar]
- 50.Miotto B, Struhl K. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Molecular cell. 2010;37(1):57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jurkowska RZ, Qin S, Kungulovski G, Tempel W, Liu Y, Bashtrykov P, et al. H3K14ac is linked to methylation of H3K9 by the triple Tudor domain of SETDB1. Nat Commun. 2017;8(1):2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Molecular and cellular biology. 1999;19(2):1202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim JJ, Lee SY, Choi JH, Woo HG, Xhemalce B, Miller KM. PCAF-Mediated Histone Acetylation Promotes Replication Fork Degradation by MRE11 and EXO1 in BRCA-Deficient Cells. Molecular cell. 2020;80(2):327–44.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao Y, Li W, Yang H, Pan L, Zhang L, Lu L, et al. HBO1 is a versatile histone acyltransferase critical for promoter histone acylations. Nucleic acids research. 2021;49(14):8037–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feng Y, Vlassis A, Roques C, Lalonde ME, Gonzalez-Aguilera C, Lambert JP, et al. BRPF3-HBO1 regulates replication origin activation and histone H3K14 acetylation. The EMBO journal. 2016;35(2):176–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nature medicine. 2015;21(3):231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng Y, Thomas PM, Kelleher NL. Measurement of acetylation turnover at distinct lysines in human histones identifies long-lived acetylation sites. Nat Commun. 2013;4:2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.LaFave LM, Beguelin W, Koche R, Teater M, Spitzer B, Chramiec A, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nature medicine. 2015;21(11):1344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nature methods. 2015;12(4):357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tone AA, Virtanen C, Shaw PA, Brown TJ. Decreased progesterone receptor isoform expression in luteal phase fallopian tube epithelium and high-grade serous carcinoma. Endocrine-related cancer. 2011;18(2):221–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tone AA, Begley H, Sharma M, Murphy J, Rosen B, Brown TJ, et al. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14(13):4067–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated and/or analyzed during this study are available from the corresponding author(s) on reasonable request. RNA-Seq dataset(s) are deposited as described in materials and methods and are publicly available.