

Abstract
cfDNA concentrations from patients with cancer are often elevated compared to that of healthy controls, but the sources of this extra cfDNA have never been determined. To address this issue, we assessed cfDNA methylation patterns in 178 patients with cancers of the colon, pancreas, lung, or ovary and 64 patients without cancer. Eighty-three of these individuals had cfDNA concentrations much greater than those generally observed in healthy subjects. The major contributor of cfDNA in all samples was leukocytes, accounting for ~76% of cfDNA, with neutrophils predominating. This was true regardless of whether the samples were derived from patients with cancer or the total plasma cfDNA concentration. High levels of cfDNA observed in patients with cancer did not come from either neoplastic cells or from surrounding normal epithelial cells from the tumor’s tissue of origin. These data suggest that cancers may have a systemic effect on cell turnover or DNA clearance.
Keywords: Cell-free DNA (cfDNA), circulating tumor DNA (ctDNA), DNA methylation, liquid biopsy, earlier cancer detection
INTRODUCTION
In the peripheral blood of healthy individuals, the vast majority of cell-free DNA (cfDNA) is derived from cells of hematopoietic lineage, consistent with lymphoid and myeloid cell death as the predominant source of cfDNA (1–4). The first measurements of cfDNA from patients with cancer were performed more than 50 years ago (5–8). Although these studies were performed before the development of technologies that could characterize the DNA fragments in detail, it was clear that the concentration of DNA was often elevated in patients with cancer compared to healthy controls (5,7,9,10). Numerous studies since then have confirmed that the cfDNA in patients with cancer is often elevated and that more advanced patients with cancer are more likely to have higher cfDNA concentrations (9,11). Sensitive studies of mutations and chromosome copy number changes have shown that part of the cfDNA derived from patients with cancer is derived from neoplastic cells within the tumor (9–16). However, many recent studies of cfDNA are based on characteristics that are not entirely specific to neoplastic cells. These include assays based on cfDNA fragment ends, lengths, enrichments in specific sequences such as promoters, and other epigenetic features that can be used to identify not only the presence of cancer but the cell type of origin. In general, previous studies have not been able to distinguish whether the great excess of cfDNA that is observed in some patients with cancer is derived from the neoplastic cells within the tumor rather than from normal epithelial cells surrounding the tumor that may have been damaged by the cancer (Supplementary Note 1 and (3,14–44). We sought to answer this question through a combination of genetic and epigenetic analysis of cfDNA, particularly in samples with very high concentrations of cfDNA (sample and analysis schema shown in Figure S1).
RESULTS
The amount of cfDNA in normal individuals and cancer patients
The normal concentration of cfDNA in healthy individuals generally ranges from 1 – 10 ng/ml of plasma, and the average concentration of cfDNA in the plasma of cancer patients is higher than in healthy individuals (7,9–11,33–36). As an example, the distribution of cfDNA concentrations as measured by quantitative real-time PCR among 812 healthy individuals and 1005 patients with cancer from a recently reported study (9) is shown in Figure 1A. The mean concentration of cfDNA in the plasma of the normal individuals was 4.3 ± 8.6 ng/mL of plasma (2.9 ± 1.6 ng/mL, median ± median absolute deviation (MAD), while the mean concentration of cfDNA in the plasma of patients with stages I-III cancer was 12.6 ± 18.1 ng/mL of plasma (6.30 ± 3.5 ng/mL, median ± MAD). The concentrations varied considerably with cancer type, with lung cancers having the lowest at 5.23 ± 6.4 ng/mL of plasma (3.3 ± 1.5 ng/mL, median ± MAD) and liver the highest at 46.0 ± 35.6 ng/mL of plasma (42.3 ± 29.4 ng/mL, median ± MAD) (Figure 1B). A particularly high concentration of cfDNA in liver cancers has been previously noted (3,37,38). There was a significant difference in cfDNA concentration between AJCC 7th edition stage I to stage III cancers as a whole (Figure S2A, p < 0.01), which varied within each cancer type (Figure S2B). Note that none of the 1005 cancer patients evaluated in Figures 1B, S2A, and S2B had distant metastatic disease at the time plasma was taken, though it is well known that patients with the most advanced disease have the highest cfDNA concentrations (7,10,11,38).
Figure 1. Plasma cfDNA concentrations in previously described patients.
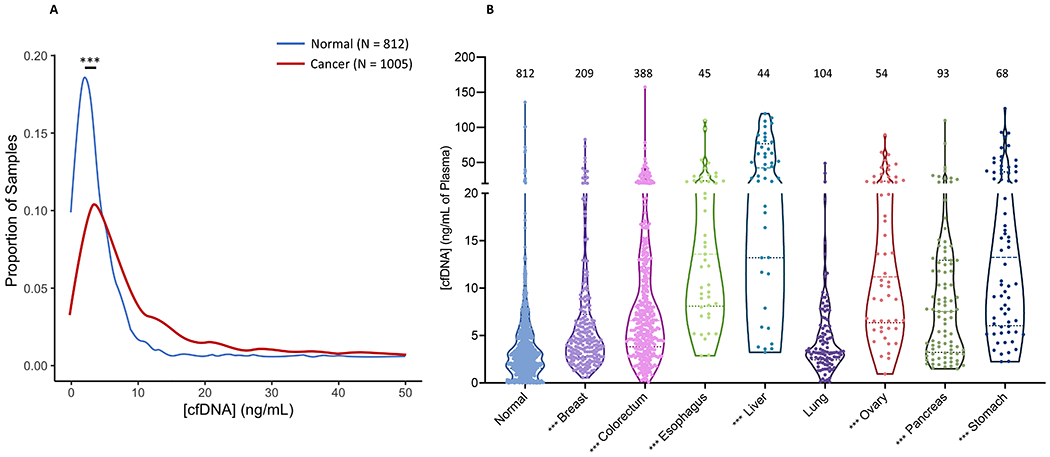
(A) Distribution of the average concentration of cfDNA in the plasma of patients with cancer as determined by qPCR shows that it is elevated compared to normal controls. Blue line = normal controls (N = 812). Red line = patients with cancer (N = 1005). (B) The concentration of cfDNA as determined by qPCR for normal controls (N = 812), and patients with breast (N = 209), colorectal (N = 388), esophageal (N = 45), liver (N = 44), lung (N = 104), ovarian (N = 54), pancreatic (N = 93), and stomach (N = 68) cancer. Data are derived from the previously published CancerSEEK study (9). *** p < 0.001
The concentration of cfDNA in the plasma of the new cohort of normal individuals included in the present study was very similar to the cohort described in ref. (9) (Supplementary Note 2). For 64 normal individuals, the mean concentration of cfDNA was 6.0 ± 10.5 ng/mL of plasma (3.4 ± 0.8 ng/mL, median ± MAD; p = 0.15 compared to data in ref (9). For 178 patients with stages I-IV cancer evaluated in the new cohort studied here, the mean concentration of cfDNA was greater at 21.8 ± 26.5 ng/mL of plasma (p < 0.001 compared to ref. (9); 11.8 ± 5.9, median ± MAD), reflecting the different cancer types and inclusion of patients with metastatic disease in the new cohort.
The tissue origin of cfDNA in normal individuals
In 64 normal individuals, whole genome bisulfite sequencing data showed that the vast majority of cfDNA arises from leukocytes, regardless of the total concentration of cfDNA in the plasma (Supplementary Note 3, Figure 2, blue dots, Figure S3, Table S1). Neutrophils accounted for roughly 2/3 of the leukocyte cfDNA, consistent with the ~2:1 ratio of neutrophils to lymphocytes in the circulation of healthy individuals. Interestingly, the fraction of cfDNA contributed by B cells was not consistent with the fraction of B cells in the circulation of healthy individuals. B cells are expected to account for only 10-15% of blood lymphocytes, while T cells account for most of the remainder (80%) (45). But B cell- and T cell-derived DNA accounted for 16.4 ± 12.0 % and 17.5 ± 4.3%, respectively, of lymphocyte cfDNA in the circulation. This difference between the ratio of B to T cells expected in the circulation and the ratio of B cell derived DNA to T cell derived DNA in the circulation was highly significant (p < 0.001) (Supplementary Note 4). Other minor tissue contributors to cfDNA were the liver, colon, heart, brain, and lung, accounting for 5.8 ± 6.6%, 4.0 ± 7.1%, 3.5 ± 3.4%, 3.1 ± 3.3%, and 2.4 ± 3.4% of the total, respectively (Figure 2, blue dots and Table S1). Deconvolution using NNLS (40–42) instead of QP (39) yielded nearly identical results (Figure S4).
Figure 2. Deconvolution of the plasma cfDNA methylation profile using the reference cell type matrix derived from Sun et al. (3).
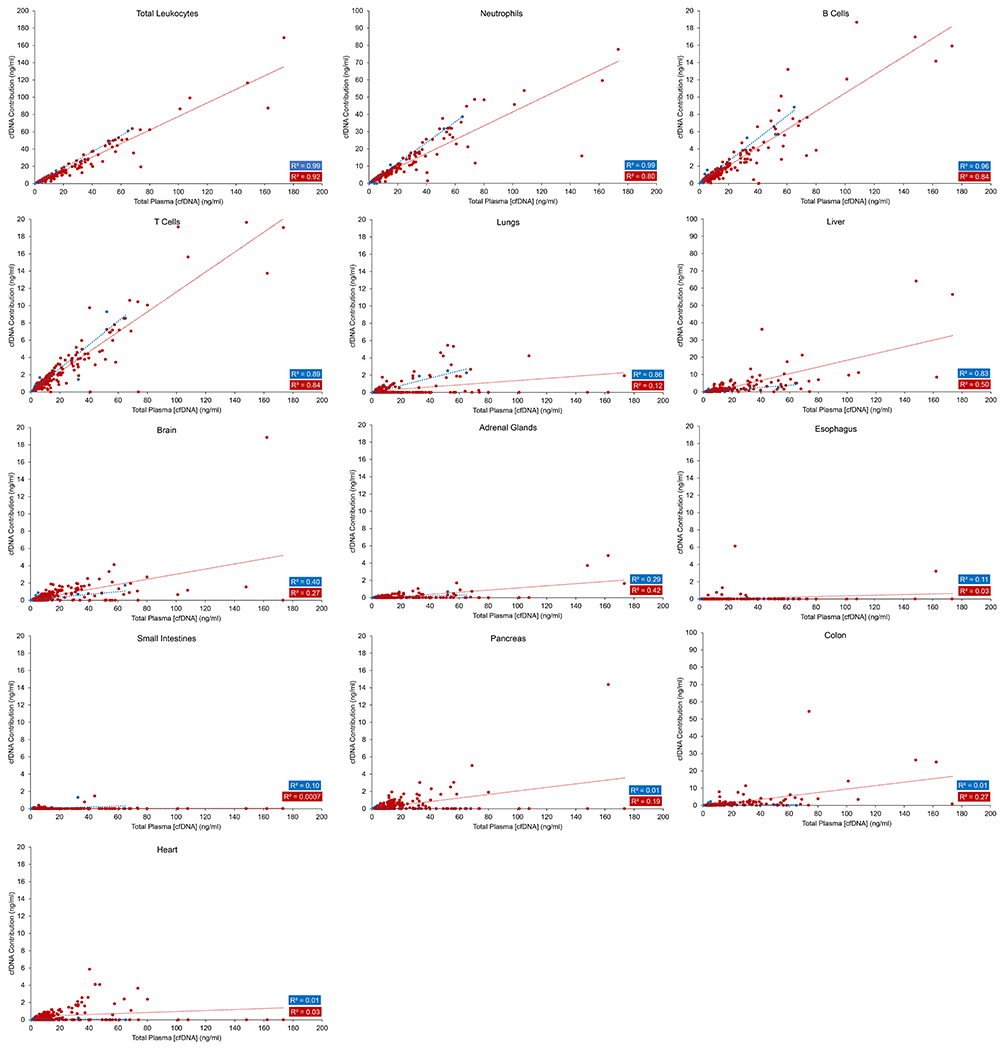
The total methylation profile of plasma cfDNA was deconvoluted into 12 different tissue types using quadratic programming. The total leukocyte concentration was taken to be the sum of the concentrations of neutrophils, B cells, and T cells. Note that the y-axes for the different tissue types shown are not the same.
Using a separate reference deconvolution matrix from Moss et al. (43) and QP (39) as the deconvolution algorithm, leukocytes were again the predominant contributor to plasma cfDNA at 61.2 ± 23.4% (Figure 3, blue dots and Table S1), with neutrophils contributing the most cfDNA, followed by monocytes, NK cells, myeloid progenitors, B cells, and T cells. Similar to data obtained with the Sun et al. (3) deconvolution reference matrix, other minor contributors included colon epithelial cells at 7.2 ± 8.5% and hepatocytes at 6.7 ± 9.8%, respectively. Again, deconvolution using NNLS (40–42) instead of QP (39) provided very similar results (Figure S5). Contributions of overlapping cell types as determined by the Moss et al. (43) and Sun et al. (3) reference matrices were similar for total leukocytes, neutrophils, B cells T cells, hepatocytes, colon epithelial cells, and brain (Tables S1 and S2).
Figure 3. Deconvolution of the plasma cfDNA methylation profile using the reference cell type matrix derived from Moss et al. (43).
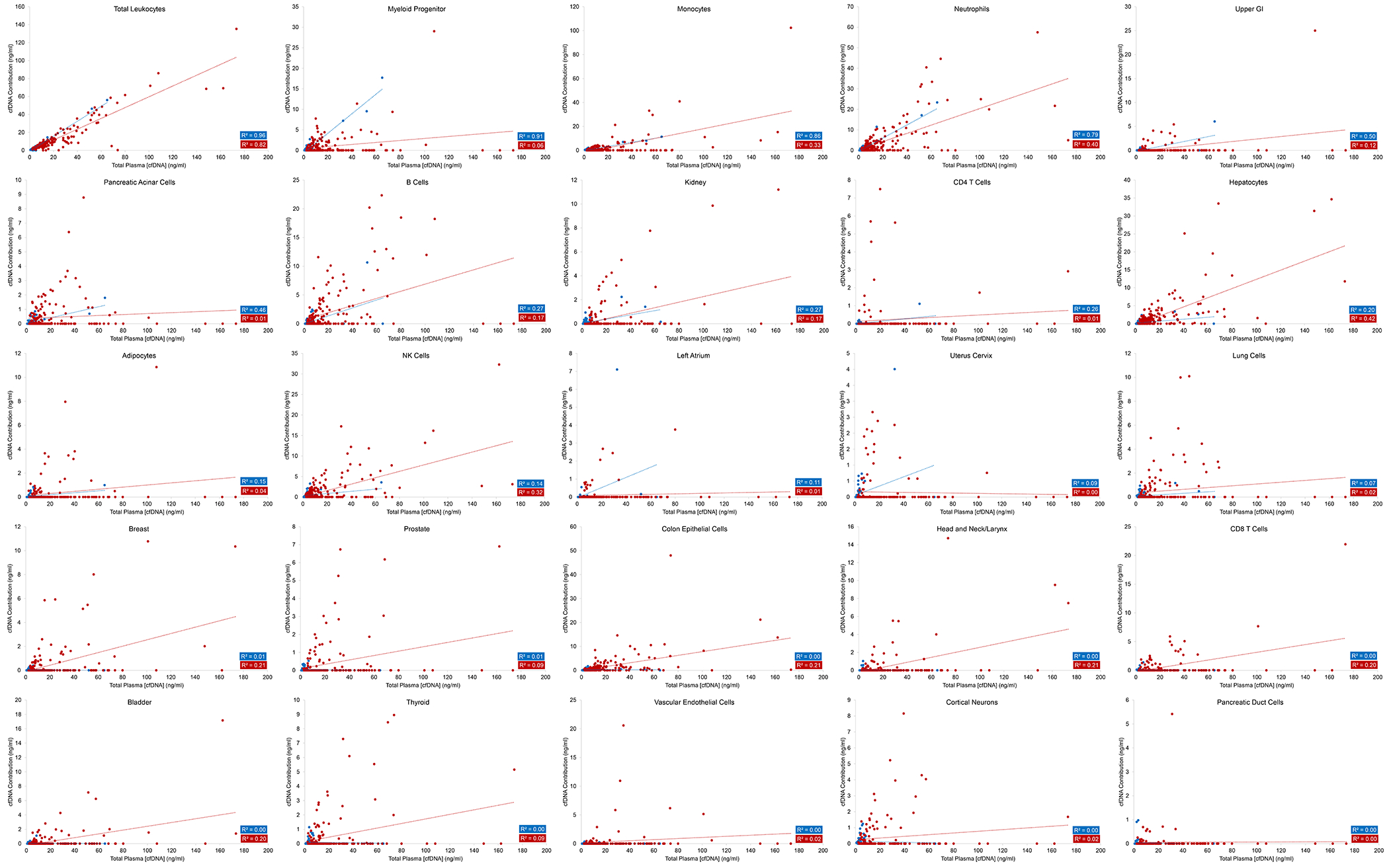
The total methylation profile of plasma cfDNA was deconvoluted into 25 different tissue types using quadratic programming The total leukocyte concentration was taken to be the sum of myeloid progenitors, monocytes, neutrophils, B cells, CD4 T cells, CD8 T cells, and NK cells. Note that the y-axes for the different tissue types shown are not the same.
Deconvolution with the Loyfer et al. (44) matrix and NNLS algorithm similarly showed that leukocytes were the predominant contributor to plasma cfDNA at 54.8 ± 20.3% (Table S2), with blood granulocytes contributing the most cfDNA, followed by megakaryocytes, blood monocytes/macrophages, keratinocytes, erythrocyte progenitors, endothelial cells, NK cells, and T cells. Analysis of our healthy cohort showed tissue contributions very similar to those reported in the healthy patients evaluated by Loyfer et al. (44).
For individuals with non-elevated concentrations of cfDNA, the results described above are consistent with prior studies showing that most cfDNA comes from cells of lymphoid and myeloid lineage (2–4,27). The novel aspect of the current study is the determination of these origins in individuals with elevated cfDNA. No prior studies had evaluated the tissue of origin of these elevated cfDNA concentrations in such individuals, and we hypothesized that such individuals might have had tissue-specific damage that accounted for their extremely high cfDNA levels. However, the results did not confirm our hypothesis; there was a linear correlation between the amount of DNA contributed by leukocytes and the total cfDNA concentration at all concentrations, regardless of whether the Sun et al. (3), Moss et al. (43), or Loyfer et al. (44) reference matrices were used for deconvolution, or whether the plasma cfDNA methylation signature was deconvoluted by QP or NNLS. (Figure 2, blue dots, R2 = 0.99, p < 0.001 and Figure 3, blue dots, R2 = 0.96, p < 0.001; Table S1). In other words, the great majority (average 79%, IQR 74% to 88%) of the cfDNA in healthy plasma, even when the total cfDNA concentration was more than 10x the normal level, arose from leukocytes. Similar fractions of DNA arising from neutrophils, B cells, and T cells were discovered, regardless of the total cfDNA concentrations (R2 = 0.99, 0.96, and 0.89, respectively, p < 0.001, Figure 2, blue dots). The amount of liver and lung DNA was proportionately increased with total cfDNA concentration, in the same way as observed in healthy individuals without elevated cfDNA (Figure 2, blue dots). Additionally, the same unexpectedly high contribution of B cells to total cfDNA was observed in normal individuals with high cfDNA concentrations as in those with low concentrations (Figure 2, blue dots). In addition to total leukocytes, analysis using the Moss et al. (43) reference matrix highlighted the contribution of myeloid progenitor cells (R2 = 0.91, p < 0.001), monocytes (R2 = 0.86, p < 0.001), and neutrophils (R2 = 0.79, p < 0.001) at all concentrations of total cfDNA (Figure 3, blue dots).
The tissue origins of cfDNA in patients with cancer
We analyzed plasma from 178 patients with colorectal (N = 18), lung (N = 31), ovarian (N = 36) or pancreatic cancer (N = 93) to determine the source of their cfDNA. As in the normal individuals described above, leukocyte lysis during sample collection or processing or other contribution from high molecular weight DNA was excluded in all cancer patients (Figure S3, Table S1).
In patients with cancer, the tissue source of cfDNA (Figure 2, red dots) was markedly similar to that in normal individuals (Figure 2, blue dots). Using the reference matrix from Sun et al. (3) and deconvolution via QP, 70.5 ± 13.7% (73.6 ± 5.4%, median ± MAD) of the cfDNA in these patients was contributed by leukocytes, with an average of 11.4 ± 11.4%, 5.9 ± 9.0%, 3.6 ± 2.8%, 3.1 ± 3.0%, 2.2 ± 3.7%, and 2.2 ± 2.7% contributed by liver, colon, brain, heart, lungs, and pancreas, respectively (Table S1). Of the leukocyte DNA, ~2/3 was derived from neutrophils in cancer patients, just as in normal individuals (Table S1). These results are consistent with previous studies on cancer patients without elevated cfDNA concentrations (3). Deconvolution strategies using the Moss et al. (43) reference matrix or using NNLS instead of QP yielded similar results (Figure 3, red dots compared to Figure 2, red dots; Figures S4 and S5; Table S1). Analysis using the Loyfer et al. (44) approach also produced similar results, with total leukocytes contributing 56.6 ± 13.3% to the total cfDNA pool, with predominant contributions by blood granulocytes, megakaryocytes, blood monocytes/macrophages, and hepatocytes, just as in healthy individuals (Table S2).
In patients with elevated concentrations of cfDNA, we expected that a major source of the large amounts of DNA in patients would be from the neoplastic cells and the surrounding non-neoplastic epithelial cells. This expectation was not confirmed by experiment. As with normal individuals, there was a linear correlation between the amount of DNA contributed by leukocytes and the total cfDNA concentration (Figure 2, red dots, R2 = 0.92, p < 0.001; Figure 3, red dots, R2 = 0.82, p < 0.001).
Interestingly, ten patients with cancer had approximately 5 ng/mL or more of their cfDNA derived from colonic epithelium (Table S1). These concentrations were significantly different from those derived from normal individuals, regardless of the total cfDNA concentration in the normal individuals (p < 0.001). Of the ten, 80% (8/10) of these patients had colorectal cancers. We sought to understand the origin of this epithelial DNA. In theory, it could have come from the neoplastic cells themselves or the surrounding non-neoplastic colorectal epithelial cells that had been destroyed by the cancer. It is well known that cancers destroy surrounding normal organ cells during the invasive process, and these dead or dying cells could in principle contribute to cfDNA (46–48). To distinguish between these two possibilities, tumor-specific mutations and copy number alterations (CNAs) in the cfDNA were used to determine the fraction of the cfDNA contributed by the neoplastic cells themselves.
We found a linear correlation between the fraction of cfDNA derived from colon epithelial cells and the fraction of cfDNA derived from neoplastic colon epithelial cells in patients with colorectal cancer (Figure S6, R2 = 0.95, p < 0.001; Supplementary Note 5). The former was assessed by whole genome bisulfite sequencing of plasma cfDNA while the latter was assessed by SafeSeqS analysis of mutations in the same plasma cfDNA samples, as described in Kinde et al. (49). The data in Figure S6 and Table S1 show that the amount of cfDNA derived from all colonic epithelial cells (assessed by methylation) was similar to that expected from the contribution of the neoplastic colonic epithelial cells alone (as assessed by mutation) (50). This conclusion was supported by copy number analysis of the cfDNA (Table S1).
The tissue origins of cfDNA following surgery
Other conditions besides cancer have been associated with elevated cfDNA concentrations (51–53). For example, it has been shown that large increases in cfDNA occur one day after surgery (10,54,55). To investigate the source of the extra cfDNA in such patients, we obtained plasma samples approximately 24 hours after surgery from nine of the patients with pancreatic cancer included in Table S1, all of whom had Whipple procedures for tumor resection. Prior to surgery, eight of the nine patients had total cfDNA concentrations in the normal range (Figure S7). Following surgery, there was a dramatic elevation of the total cfDNA, ranging from 2.3- to 18-fold (median of 8.7-fold) in these 8 patients (Figure S7, Table S1, p = 0.001). The only patient for whom the cfDNA concentration did not increase following surgery was the one (PANCA 1248) with elevated cfDNA (29.1 ng/mL of plasma) prior to surgery.
Bisulfite sequencing of plasma cfDNA in these eight patients revealed the following:
The amount of cfDNA from all evaluable tissue sources increased after surgery, though the additional contribution from the lungs, brain, esophagus, small intestines, pancreas, and heart appeared to be slightly elevated (p = 0.09, 0.13, 0.08, 0.09, 0.13, and 0.10, respectively) (Figure 4 and Table S1).
The majority (average 57%, range 42% to 70%) of the total cfDNA after surgery was from leukocytes, and the predominant contributors to leukocyte cfDNA were neutrophils (average 73%, range 67 to 82% of the leukocyte cfDNA; Figure S8, Table S1).
For most tissues, the proportional representation of each evaluable tissue either decreased or changed only slightly after surgery—except for the liver (Figure S8, Table S1). There was a striking increase (average 46-fold, range 6 to 144-fold) in the fraction of total cfDNA derived from hepatocytes following surgery, in marked contrast to the other tissues (Figure S8). The amount of the “neo-cfDNA” can be defined and calculated by subtracting the amount of cfDNA present pre-surgery from the amount of cfDNA present after surgery. This calculation showed that hepatocytes contributed an average of 38.2% (range 23.9% to 57.6%) and leukocytes contributed an average of 48.4% (range 23.3% to 67.1%) of the neo-cfDNA.
Figure 4. The amount of cfDNA from evaluable tissue sources before and ~24 hours after surgery.
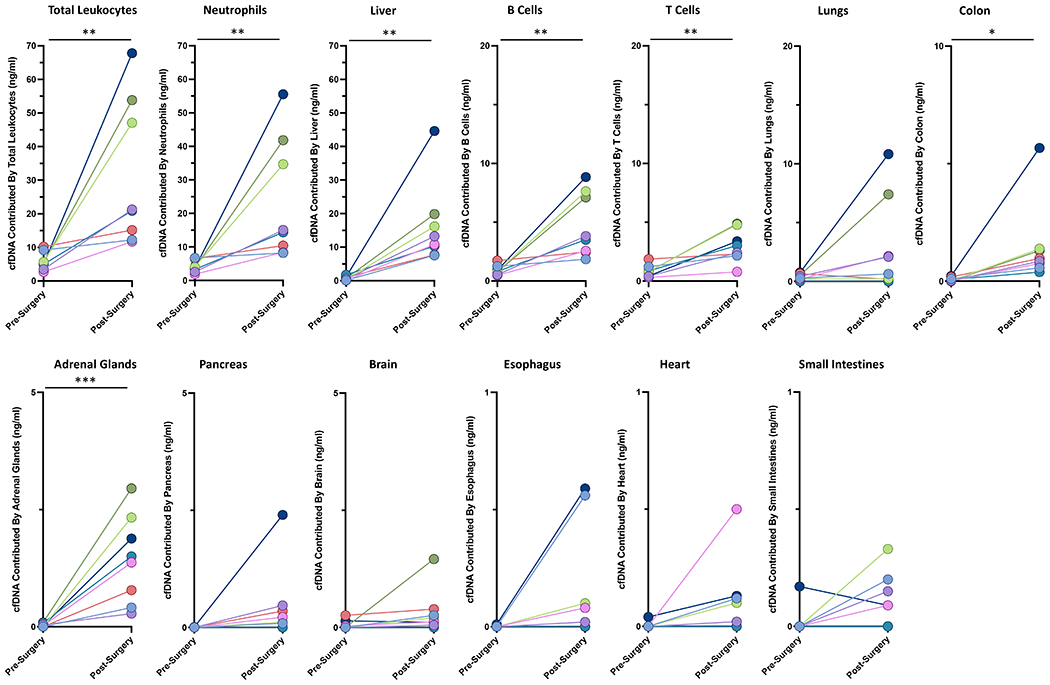
Each color represents a separate patient (see Supplemental Table 1). * p < 0.05, ** p < 0.01, *** p < 0.001.
Based on the clinical history of PANC 696 described above, as well as on previous data (3,52), we thought it likely that much of the extra cfDNA following surgery was due to liver damage. We were able to obtain standard measurements of liver function using alanine transaminase (ALT) and aspartate aminotransferase (AST) levels in five of the eight patients whose cfDNA concentrations increased following surgery. AST and ALT levels substantially increased in all five patients (p < 0.05; Figure S9). Notably, in the one patient (PANCA 1248) whose total cfDNA did not increase post-surgery, AST levels were already increased prior to surgery, unlike the other patients assessed (Table S1).
DISCUSSION
The results of this study lend support to previous observations about the origins of cfDNA in healthy individuals and patients with cancer that have normal to slightly elevated concentrations of cfDNA. Additionally, the results of this study lead to several important conclusions about the origin of excess cfDNA in patients with greatly elevated cfDNA concentrations:
In patients with colorectal, lung, ovarian, and pancreatic cancer with high concentrations of cfDNA in the present study, the increased cfDNA does not primarily come from either the neoplastic cells within the cancer or from adjacent non-neoplastic epithelial cells.
Instead, the increased cfDNA in these patients with cancer comes largely from leukocytes, primarily neutrophils.
The elevated cfDNA in patients with cancer studied is attributable to a systemic effect. It is not just neutrophils, but also B and T lymphocytes, and in some cases hepatocytes, colon epithelial cells, and lung epithelial cells, that release more DNA into the circulation when cfDNA concentrations are elevated.
Similarly, the elevated cfDNA that routinely occurs following surgery of patients with pancreatic cancer arises from a systemic effect, resulting in the release of cfDNA from leukocytes but in this case also from hepatocytes.
The cfDNA contributed by leukocytes is often associated with an overrepresentation of B cells compared to T cells, regardless of disease state and cfDNA concentration.
One of the major questions raised by our data is the nature of the systemic factor(s) that are responsible for increasing the contributions of cfDNA from all major tissue sources of cfDNA (56–73). One possibility is that the systemic factor(s) are one or more of the myriad of proteins and other molecules known to be secreted by neoplastic cells (74,75), or released upon the death of cancer cells in situ (46,75). Another possibility is that these factors come from endothelial cells within the cancers. There is convincing evidence indicating that the tumor vasculature is abnormal (76,77) and endothelial cells are in direct contact with the systemic circulation. Inflammatory cells within the tumor could also release cytotoxic products (78). A completely different, but enticing possibility, is that cell turnover is normal in these patients, but clearance of cfDNA is abnormal. We hope that the results of this study will stimulate research to identify the biochemical basis for the pronounced elevation of cfDNA observed in cancer patients and in other clinical scenarios.
MATERIALS AND METHODS
Sample Collection and DNA Isolation
All individuals participating in the study provided written informed consent after approval by the institutional review board at the patient’s participating institutions (including Johns Hopkins IRB00075499 and Melbourne Health HREC 2011.225), and the study complied with the Health Insurance Portability and Accountability Act and the Declaration of Helsinki. Peripheral blood was collected in K2-EDTA tubes after informed consent was obtained and prior to and/or 24 hours after patients underwent surgical resection. General demographics, surgical pathology, and American Joint Commission on Cancer (AJCC) stage (7th) were documented. The cohort is outline in Figure S1. The healthy cohort consisted of peripheral blood samples obtained from 64 individuals of median age 48.5 yrs (IQR interquartile range 28 to 58 yrs) with no history of cancer. The cancer and healthy control samples were processed in an identical manner. Plasma samples from 18 patients with colorectal cancer, 31 patients with lung cancer, 36 patients with ovarian cancer, and 93 patients with pancreatic cancer were included in the study (median age 67 yrs, IQR 56 to 74 yrs).
The 242 individuals included in this study were chosen from cfDNA samples collected for studies described in (48) and similar ongoing studies to evaluate the use of cfDNA for the earlier detection of cancer in patients prior to surgery or any other form of therapy. All individuals for whom sufficient plasma was available for construction of libraries for whole genome sequencing of bisulfite-treated DNA were considered. Any individual with a cfDNA concentration >15 ng/mL of plasma from this collection were chosen for analysis. Additionally, there were two different blood samples available from nine of the 21 patients with pancreatic cancer, one collected prior to surgery and the other collected approximately 24 hours after surgery, and these were chosen for analysis. Finally, normal individuals with cfDNA concentrations <15 ng/mL of plasma, as well as cancer patients with cfDNA concentrations <15 ng/mL, were chosen randomly. DNA from each of these 242 patients (251 total plasma samples) was purified with a BioChain cfDNA Extraction Kit (BioChain, cat #K5011610) using the manufacturer’s recommended protocol. DNA from peripheral WBCs was purified with the QIAsymphony DP DNA Midi Kit (Qiagen, cat #937255) as specified by the manufacturer. cfDNA was quantified using qPCR using Sso Advanced SYBR Green Supermix (BioRad Cat # 1725271) as directed by the manufacturer and employing the following primers: 5′-CACACAGGAAACAGCTATGACCATGGGTAACAGCTTTATCTATTGACATTATGC-3′ and 5′-CGACGTAAAACGACGGCCAGTNNNNNNNNNNNNNNAAACTTCATGCTTCATCTAGTCAGC-3′. National Institute of Standards and Technology (NIST) human DNA quantification standard NIST SRM 2372a, diluted to 1 ng/ml, served as the reference standard. 2.5 μL of cfDNA or NIST 2372a DNA was added to 97.5 μL of 1:1,000 SYBR Green I diluted in 1X PBS. Amplification and fluorescence detection conditions were as follows: one cycle of 98°C for 120 s, then 30 cycles of 98°C for 10 s, 57°C for 120 s, and 72°C for 120 s.
Bisulfite Treatment, Library Preparation, and Sequencing
For libraries prepared using the Accel-NGS Methyl-Seq DNA Library Kit (Swift BioSciences, cat #30024, Table S1), the EZ DNA Methylation Kit (Zymo Research, cat #D5001) was used to prepare DNA samples as follows. DNA was denatured in dilute M-Dilution buffer at 37°C for 15 minutes then bisulfite converted in the dark at 50°C for 16 hours before being placed on ice for 10 min (79). After a single wash with M-Wash buffer, the sample was desulphonated for 15 min at room temperature. The sample was washed twice in M-Wash Buffer then eluted in the Zymo Elution Buffer and stored at –20 °C (49). Sequencing libraries were then prepared using the Accel-NGS Methyl-Seq DNA Library Kit, with 9 PCR cycles used at the indexing stage. Samples assessed using MethylSaferSeqS (Table S1) were prepared as described in detail in Wang et al. (80), in which library preparation is performed prior to bisulfite-treatment (50). Methylation status as assessed by the two library preparation methods produced indistinguishable results (50). Each library was paired-end sequenced to 150 bp on a single lane of an Illumina HiSeq 4000 instrument. Reads passing Illumina CASAVA Chastity filters were used for subsequent analysis.
DNA Sequencing Data Analysis
Illumina adapters and bases with quality scores below 25 were trimmed from the head and tail of each read using Trimmomatic (81). To improve mapping efficiency by reducing spurious mutations introduced by end repair, 15 bp were additionally cropped from the tail of Read 1 and the head of Read 2 using Trimmomatic per Swift’s recommendations. BSMAP was used to align each paired-end read to the bisulfite-converted hg19 genome, and the average methylation at each CpG computed using BSMAP’s methratio.py script (82).
Identification of Methylation Markers for Plasma cfDNA Tissue Deconvolution by Quadratic Programming
The average contribution of twelve tissue types (liver, lungs, colon, small intestines, pancreas, adrenal glands, esophagus, heart, brain, T cells, B cells, and neutrophils) to the total cfDNA pool was determined using 5,653 differentially methylated 500 bp regions described by Sun et al. (3) In brief, the approach was bioinformatically based on whole genome bisulfite sequencing of normal DNA from the liver, lungs, esophagus, heart, pancreas, colon, small intestines, adrenal glands, brain, and T cells, which was retrieved from the Human Epigenome Atlas from the Baylor College of Medicine (www.genboree.org/epigenomeatlas/index.rhtml). The bisulfite sequencing data for B cells and neutrophils were from Hodges et al. (83). All CpG islands (CGIs) and CpG shores on autosomes were assessed for potential inclusion into the methylation marker set. CGIs and CpG shores on sex chromosomes were not used, so as to minimize potential variations in methylation levels related to the sex-associated chromosome dosage difference in the source data. CGIs were downloaded from the University of California, Santa Cruz (UCSC) database (genome.ucsc.edu/, 27,048 CGIs for the human genome) (84), and CpG shores were defined as 2-kb flanking windows of the CGIs (85). Then, the CGIs and CpG shores were subdivided into nonoverlapping 500-bp units, and each unit was considered a potential methylation marker.
The methylation densities (i.e., the percentage of CpGs being methylated within a 500 bp unit) of all the potential marker loci were compared between the 12 tissue types. Using the methylation profiles of the 12 tissue types, two types of methylation markers were identified. Type I markers refer to any genomic loci with methylation densities that are 3 SDs below or above in one tissue compared with the mean level of the 12 tissue types. Type II markers are genomic loci that demonstrate highly variable methylation densities across the 12 tissue types. A locus is considered highly variable when (A) the methylation density of the most hypermethylated tissue is at least 20% higher than that of the most hypomethylated one; and (B) the SD of the methylation densities across the 12 tissue types when divided by the mean methylation density (i.e., the coefficient of variation) of the group is at least 0.25. To reduce the number of potentially redundant markers, only one marker would be selected in one contiguous block of two CpG shores flanking one CGI. The genomic locations of the Type I and Type II markers used in this study can be found in Supplementary Table 1 in Sun et al. (3).
Plasma cfDNA Tissue Deconvolution by Quadratic Programming (QP)
As described in Sun et al. (3), the mathematical relationship between the methylation densities of the different methylation markers in plasma and the corresponding methylation markers in different tissues can be expressed as
where represents the methylation density of the methylation biomarker i in the plasma; pk represents the proportional contribution of tissue k to the plasma; and MDik represents the methylation density of the methylation biomarker i in tissue k. The aim of the deconvolution process was to determine the proportional contribution of tissue k to the plasma, namely pk, for each member of the panel of tissues. Quadratic programming (39) was used to solve the simultaneous equations. A matrix was compiled including the panel of tissues and their corresponding methylation densities for each methylation marker on the combined list of type I and type II markers (a total of 5,653 markers). The program input a range of pk values for each tissue type and determined the expected plasma DNA methylation density for each marker. The tested range of pk values should fulfill the expectation that the total contribution of all candidate tissues, namely, the liver, neutrophils, and lymphocytes for this study, to plasma DNA would be 100% and the values of all pk would be nonnegative. The program then identified the set of pk values that resulted in expected methylation densities across the markers that most closely resembled the data obtained from the plasma DNA bisulfite sequencing.
The total contribution from T cells and B cells was regarded as the contribution from the lymphocytes, and the total contribution from leukocytes was regarded as the contribution from the lymphocytes and neutrophils. To obtain absolute levels of cfDNA (ng/ml) per cell type, the resulting contribution was multiplied by the total concentration of cfDNA present in the sample.
Identification of Methylation Markers for Plasma cfDNA Tissue Deconvolution by Non-Negative Least Squares Regression
The average contribution of 25 tissue types (neutrophils, monocytes, CD4 T cells, CD8 T cells, B cells, NK cells, myeloid progenitors, adipocytes, cortical neurons, hepatocytes, lung cells, pancreatic acinar cells, pancreatic duct cells, vascular endothelial cells, colon epithelial cells, left atrium, bladder, breast, head and neck/larynx, kidney, prostate, thyroid, upper GI, uterus/cervix) to the total cfDNA pool was determined using 7,890 differentially methylated CpG, as described in Moss et al. (43). In brief, all DNA methylation profiles were determined either on the Illumina Infinium Human Methylation 450K or EPIC BeadChip arrays. DNA methylation data for white blood cells (neutrophils, monocytes, B cells, CD4+ T cells, CD8+ T cells, NK cells, n = 6 each) were downloaded from GSE110555 (EPIC) (86). Data for myeloid progenitors (n = 5) were downloaded from GSE63409 (450K) (87), and data for left atrium (n = 4) were downloaded from GSE62727 (450K) (88). Data for bladder (n = 19), breast (n = 98), cervix (n = 3), colon (n = 38), esophagus (n = 16), oral cavity (n = 34), kidney (n = 160), prostate (n = 50), rectum (n = 7), stomach (n = 2), thyroid (n = 56), and uterus (n = 34) were downloaded from TCGA (89). DNA methylation data for adipocytes (n = 3, 450K), hepatocytes (n = 3, 450K and EPIC), alveolar lung cells (n = 3, EPIC), neurons (n = 3, 450K and EPIC), vascular endothelial cells (n = 2, EPIC) pancreatic acinar cells (n = 3, 450K and EPIC), duct cells (n = 3, 450K and EPIC), and colon epithelial cells (n = 3, EPIC) were generated by Moss et al. (43) and can be requested from the authors.
To analyze DNA methylation samples composed of admixed methylomes from various cell types, the authors approximated the plasma cfDNA methylation profile as a linear combination of the methylation profiles of cell types in the reference atlas. According to this model, the relative contributions of different cell types to plasma cfDNA can be determined using non-negative least squares linear regression (NNLS) as described in (40–42). To select candidate CpGs, the authors of (40–42) first excluded CpGs whose variance across the entire methylation atlas was below 0.1% or was missing. They then selected the K = 100 most specific hypermethylated CpGs for each cell type, denoting the methylation matrix X, composed of N rows (CpGs) by d columns (cell types). They then divided each row (the methylation pattern of one CpG over all cell types) by its sum:
For each cell type j, they identified the top K hypermethylated CpGs with the highest X’i,j values. To identify uniquely hypomethylated CpGs, they performed a similar process for the reversed methylation matrix (1−X). Finally, for each cell type they included both the top K hypermethylated and the top K unmethylated CpGs in the reference matrix. To this set of CpGs, they added neighboring CpGs, up to 50 bp. Pairwise-specific CpGs were iteratively selected as follows: given the current set S of CpGs, they projected the reference atlas on those coordinates and calculated the Euclidean distances between pairs of cell types. Once the closest pair of cell types was identified, they selected the CpG site where they differed the most and added it into the set S. This process was iteratively repeated, focusing on the most confusing pair of cell types in each iteration. Admixing experiments, similar to those performed in Sun et al. (3), were performed using buffy coat bisulfite sequencing data mixed with liver, lung, colon epithelial cell, or left atrium bisulfite sequencing data, showing excellent agreement between predicted fraction and actual fraction (as shown in Figures S10A–D).
Plasma cfDNA Tissue Deconvolution by Non-Negative Least Squares Regression (NNLS)
A custom python script adapted from the nnls package in MATLAB and described in Moss et al. (43) was used to perform non-negative least squares regression (40–42) to calculate the relative contribution of each cell type to a given sample. Given a matrix X of reference methylation values with N CpGs and d cell types, and a vector Y of methylation values of length N, non-negative coefficients β were identified by solving argminβ∥Xβ-Y∥2, subject to β ≥ 0. The resulting β was adjusted to have a sum of 1, where for each βj was defined as:
To obtain absolute levels of cfDNA (ng/ml) per cell type, the resulting βj′ was multiplied by the total concentration of cfDNA present in the sample, as measured by quantitative PCR.
Similar analysis using NNLS with an expanded matrix of 39 cell types is described in Loyfer et al. (44).
RealSeqS
RealSeqS was used to test the plasma samples for evidence of aneuploidy and contamination with high molecular weight DNA derived from leukocytes that were lysed during venipuncture or blood processing (12). RealSeqS uses a single primer pair to amplify ~750,000 loci scattered throughout the genome (12). PCR was performed in 25 μL reactions containing 7.25 μL of water, 0.125 μL of each primer, 12.5 μL of NEBNext Ultra II Q5 Master Mix (New England Biolabs, cat #M0544S), and 5 μL of DNA. The cycling conditions were: one cycle of 98°C for 120 s, then 15 cycles of 98°C for 10 s, 57°C for 120 s, and 72°C for 120 s. Each plasma DNA sample was assessed in eight independent reactions, and the amount of DNA per reaction varied from ~0.1 ng to 0.25 ng. A second round of PCR was then performed to add dual indexes (barcodes) to each PCR product prior to sequencing, as described in Douville et al. (12). The second round of PCR was performed in 25 μL reactions containing 7.25 μL of water, 0.125 μL of each primer, 12.5 μL of NEBNext Ultra II Q5 Master Mix (New England Biolabs, cat #M0544S), and 5 μL of DNA containing 5% of the PCR product from the first round. The cycling conditions were: one cycle of 98°C for 120 s, then 15 cycles of 98°C for 10 s, 65°C for 15 s, and 72°C for 120 s. Amplification products from the second round were purified with AMPure XP beads (Beckman, cat #a63880), as per the manufacturer’s instructions, prior to sequencing. As noted above, each sample was amplified in eight independent PCRs in the first round. Each of the eight independent PCRs was then re-amplified using index primers in the second PCR round. The sequencing reads from the 8 replicates were summed for the bioinformatic analysis but could also be assessed individually for quality control purposes. Massively parallel sequencing was performed on an Illumina HiSeq 4000. During the first round of PCR, degenerate bases at the 5’ end of one of the primers were used as molecular barcodes (unique identifiers, UIDs) to uniquely label each DNA template molecule (13). This ensured that each DNA template molecule was counted only once, as described in Kinde et al. (39). In all instances for RealSeqS in this paper, the term “reads” refers to uniquely identified reads (UIDs). If multiple reads had the same UID, at least 50% of the reads were required to map to the same genomic location. Reads with the same UID but with discordant genomic locations were discarded from analysis.
After massively parallel sequencing, gains or losses of each of the 39 chromosome arms covered by the assay were determined using a bespoke statistical learning method (13). A support vector machine (SVM) was used to discriminate between aneuploid and euploid samples. The SVM was trained using 2,651 aneuploid samples and 1,348 euploid plasma samples, to yield a “genome-wide aneuploidy score.” Samples were scored as positive when the genome-wide aneuploidy score was > 0.441.
Plasma samples were also analyzed for genomic DNA contamination using RealSeqS. RealSeqS enables the detection of genomic DNA by virtue of the differently-sized amplicons generated during PCR amplification (12). Because the average size of cell-free DNA is ~160-180 bp, almost all the ~750,000 amplicons are present in an average cfDNA sample. However, there were 1241 amplicons of size 200-500 bp, which represent contamination by genomic DNA. Coverage at these long amplicons is proportional to the background rate of gDNA contamination, as described in (13). In samples containing >15 ng of DNA per mL of plasma in which RealSeqS data were not available, an Agilent BioAnalyzer System was used to evaluate the fraction of DNA > 500 bp.
Somatic Mutations
For patients with colorectal cancer, a panel of 15 genes was designed to find mutations in DNA from primary tumors, as described in Tie et al. (90). This panel enabled detection of at least one mutation in 98% of CRC samples tested (90). The mutation with the highest mutant allele frequency in the primary tumor was then used to assess plasma DNA, as described by Tie et al. (90). The SafeSeqS approach, employing unique identifiers (UIDs, aka molecular barcodes), was then used to assess the plasma DNA for the mutation of interest (12). For patients with pancreatic cancer, plasma was directly assessed with SaferSeqS primers (9,91) for mutations at codons 12, 13, 59, 60, & 61, as >95% of pancreatic cancers harbor a mutation at one of these positions (92).
Copy Number Alterations
ichorCNA version 3.2 was downloaded on August 25, 2022 and applied to WGS data on bisulfite-treated cfDNA. Tumor fraction estimates were based on copy number analysis of 500kb intervals using default parameters. The lower limit of detection was considered to be 3% based on data from ref. (93).
Statistical Considerations
A η2 test was used to compare the number of individuals with elevated concentrations of cfDNA in 8 cancer types compared to healthy persons. A one-way ANOVA was used to compare the number of individuals with elevated concentrations of cfDNA in 8 cancer types by AJCC 7th edition stage. Pearson’s correlation coefficient was used to determine the relationship between total cfDNA concentration and relative contribution from individual tissues, and a t statistic was used to determine statistical significance. Student’s two tailed t-test was used to compare the total concentration of cfDNA pre- and post-surgery and aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels pre- and post-surgery. A p value ≤ 0.05 was considered statistically significant.
Data Availability Statement
Data on methylation (bisulfite sequencing) and copy number alterations in plasma DNA are deposited in the European Genome-Phenome Archive (EGAS00001005400). Similarly, data on mutations in plasma are available from the European Genome-phenome Archive (EGAS00001002764 and EGAS00001002444). Commercial use remains restricted due to Johns Hopkins Medicine legal requirements.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
The origin of excess cfDNA in patients with cancer is unknown. Using cfDNA methylation patterns, we determined that neither the tumor nor the surrounding normal tissue contribute this excess cfDNA - it comes from leukocytes. This finding suggests cancers have a systemic impact on cell turnover or DNA clearance.
FUNDING
Lustgarten Foundation for Pancreatic Cancer Research, The Virginia and D.K. Ludwig Fund for Cancer Research, The Sol Goldman Center for Pancreatic Cancer Research, The Marcus Foundation, John Templeton Foundation, National Institutes of Health Grants (GM136577, CA06973, GM008752). Hong Kong Research Grants Council Theme-Based Research Grant (T12-401/16-W and T12-403/15N), Endowed chair from the Li Ka Shing Foundation and funding from the Innovation and Technology Commission of the Hong Kong SAR Government.
Footnotes
CONFLICT OF INTEREST STATEMENT
BV, KWK, & NP are founders of Thrive Earlier Detection, an Exact Sciences Company. BV, KWK, NP, and CD hold equity in Exact Sciences. BV, KWK, and NP are founders of Personal Genome Diagnostics. KWK, BV, & NP hold equity in are consultants to CAGE Pharma. KWK, and NP own equity in Neophore and KWK and NP are consultants to Neophore. BV is a consultant to and holds equity in Catalio Capital Management. CB is a consultant to Depuy-Synthes and Bionaut Labs. The companies named above, as well as other companies, have licensed previously described technologies related to the work described in this paper from Johns Hopkins University. BV, KWK, NP, CB, RH, CT, CD, AKM, and JDC are inventors on some of these technologies. Licenses to these technologies are or will be associated with equity or royalty payments to the inventors as well as to Johns Hopkins University. Patent applications on the work described in this paper may be filed by Johns Hopkins University. The terms of all these arrangements are being managed by Johns Hopkins University in accordance with its conflict-of-interest policies. YMDL is a scientific cofounder, past member of scientific advisory board and past consultant of Grail. YMDL and KCAC hold equities and are board members of Take2 and DRA Limited. YMDL, KCAC and PJ receive patent licensing incomes from Illumina, Sequenom, Xcelom, Grail, Take2 and DRA Limited. KCAC is a past consultant to Grail. PJ holds equities in Grail. PJ is a consultant of Take2 Technologies Limited. PJ is a Director of KingMed Future.
REFERENCES
- 1.Lui YY, Chik KW, Chiu RW, Ho CY, Lam CW, Lo YM. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin Chem 2002;48(3):421–7. [PubMed] [Google Scholar]
- 2.Zheng YW, Chan KC, Sun H, Jiang P, Su X, Chen EZ, et al. Nonhematopoietically derived DNA is shorter than hematopoietically derived DNA in plasma: a transplantation model. Clin Chem 2012;58(3):549–58 doi 10.1373/clinchem.2011.169318. [DOI] [PubMed] [Google Scholar]
- 3.Sun K, Jiang P, Chan KC, Wong J, Cheng YK, Liang RH, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci U S A 2015;112(40):E5503–12 doi 10.1073/pnas.1508736112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016;164(1–2):57–68 doi 10.1016/j.cell.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mandel P, Metais P. Nuclear Acids In Human Blood Plasma. C R Seances Soc Biol Fil 1948;142(3-4):241–3. [PubMed] [Google Scholar]
- 6.Steinman CR. Free DNA in serum and plasma from normal adults. J Clin Invest 1975;56(2):512–5 doi 10.1172/JCI108118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 1977;37(3):646–50. [PubMed] [Google Scholar]
- 8.Stroun M, Anker P, Lyautey J, Lederrey C, Maurice PA. Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol 1987;23(6):707–12 doi 10.1016/0277-5379(87)90266-5. [DOI] [PubMed] [Google Scholar]
- 9.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018;359(6378):926–30 doi 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14(9):985–90 doi 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6(224):224ra24 doi 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Douville C, Cohen JD, Ptak J, Popoli M, Schaefer J, Silliman N, et al. Assessing aneuploidy with repetitive element sequencing. Proc Natl Acad Sci U S A 2020;117(9):4858–63 doi 10.1073/pnas.1910041117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Douville C, Springer S, Kinde I, Cohen JD, Hruban RH, Lennon AM, et al. Detection of aneuploidy in patients with cancer through amplification of long interspersed nucleotide elements (LINEs). Proc Natl Acad Sci U S A 2018;115(8):1871–6 doi 10.1073/pnas.1717846115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan KC, Jiang P, Chan CW, Sun K, Wong J, Hui EP, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc Natl Acad Sci U S A 2013;110(47):18761–8 doi 10.1073/pnas.1313995110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan KC, Jiang P, Zheng YW, Liao GJ, Sun H, Wong J, et al. Cancer genome scanning in plasma: detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin Chem 2013;59(1):211–24 doi 10.1373/clinchem.2012.196014. [DOI] [PubMed] [Google Scholar]
- 16.Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013;497(7447):108–12 doi 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 17.Mouliere F, El Messaoudi S, Gongora C, Guedj AS, Robert B, Del Rio M, et al. Circulating Cell-Free DNA from Colorectal Cancer Patients May Reveal High KRAS or BRAF Mutation Load. Transl Oncol 2013;6(3):319–28 doi 10.1593/tlo.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larson MH, Pan W, Kim HJ, Mauntz RE, Stuart SM, Pimentel M, et al. A comprehensive characterization of the cell-free transcriptome reveals tissue- and subtype-specific biomarkers for cancer detection. Nat Commun 2021;12(1):2357 doi 10.1038/s41467-021-22444-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diehl F, Schmidt K, Durkee KH, Moore KJ, Goodman SN, Shuber AP, et al. Analysis of mutations in DNA isolated from plasma and stool of colorectal cancer patients. Gastroenterology 2008;135(2):489–98 doi 10.1053/j.gastro.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A 2005;102(45):16368–73 doi 10.1073/pnas.0507904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev 2016;35(3):347–76 doi 10.1007/s10555-016-9629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chabon JJ, Hamilton EG, Kurtz DM, Esfahani MS, Moding EJ, Stehr H, et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020;580(7802):245–51 doi 10.1038/s41586-020-2140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med 2019;25(12):1928–37 doi 10.1038/s41591-019-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mouliere F, Thierry AR. The importance of examining the proportion of circulating DNA originating from tumor, microenvironment and normal cells in colorectal cancer patients. Expert Opin Biol Ther 2012;12 Suppl 1:S209–15 doi 10.1517/14712598.2012.688023. [DOI] [PubMed] [Google Scholar]
- 25.Bronkhorst AJ, Ungerer V, Diehl F, Anker P, Dor Y, Fleischhacker M, et al. Towards systematic nomenclature for cell-free DNA. Hum Genet 2021;140(4):565–78 doi 10.1007/s00439-020-02227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anker P, Stroun M, Maurice PA. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res 1975;35(9):2375–82. [PubMed] [Google Scholar]
- 27.Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Consortium C. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol 2020;31(6):745–59 doi 10.1016/j.annonc.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nuzzo PV, Berchuck JE, Korthauer K, Spisak S, Nassar AH, Abou Alaiwi S, et al. Detection of renal cell carcinoma using plasma and urine cell-free DNA methylomes. Nat Med 2020;26(7):1041–3 doi 10.1038/s41591-020-0933-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo S, Diep D, Plongthongkum N, Fung HL, Zhang K, Zhang K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet 2017;49(4):635–42 doi 10.1038/ng.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guler GD, Ning Y, Ku CJ, Phillips T, McCarthy E, Ellison CK, et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat Commun 2020;11(1):5270 doi 10.1038/s41467-020-18965-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lo YMD, Han DSC, Jiang P, Chiu RWK. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science 2021;372(6538) doi 10.1126/science.aaw3616. [DOI] [PubMed] [Google Scholar]
- 32.Nelson WG, De Marzo AM, Yegnasubramanian S. Epigenetic alterations in human prostate cancers. Endocrinology 2009;150(9):3991–4002 doi 10.1210/en.2009-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mouliere F, El Messaoudi S, Pang D, Dritschilo A, Thierry AR. Multi-marker analysis of circulating cell-free DNA toward personalized medicine for colorectal cancer. Mol Oncol 2014;8(5):927–41 doi 10.1016/j.molonc.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, et al. High fragmentation characterizes tumour-derived circulating DNA. PLoS One 2011;6(9):e23418 doi 10.1371/journal.pone.0023418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meddeb R, Dache ZAA, Thezenas S, Otandault A, Tanos R, Pastor B, et al. Quantifying circulating cell-free DNA in humans. Sci Rep 2019;9(1):5220 doi 10.1038/s41598-019-41593-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014;20(5):548–54 doi 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan L, Chen Y, Zhou J, Zhao H, Zhang H, Wang G. Diagnostic value of circulating cell-free DNA levels for hepatocellular carcinoma. Int J Infect Dis 2018;67:92–7 doi 10.1016/j.ijid.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 38.Banini BA, Sanyal AJ. The use of cell free DNA in the diagnosis of HCC. Hepatoma Res 2019;5 doi 10.20517/2394-5079.2019.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van den Meersche K, Soetaert K, Van Oevelen D. xsample(): An R Function for Sampling Linear Inverse Problems. Journal of Statistical Software, Code Snippets 2009;30(1):1–15 doi 10.18637/jss.v030.c01. [DOI] [Google Scholar]
- 40.Accomando WP, Wiencke JK, Houseman EA, Nelson HH, Kelsey KT. Quantitative reconstruction of leukocyte subsets using DNA methylation. Genome Biol 2014;15(3):R50 doi 10.1186/gb-2014-15-3-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012;13:86 doi 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Houseman EA, Christensen BC, Yeh RF, Marsit CJ, Karagas MR, Wrensch M, et al. Model-based clustering of DNA methylation array data: a recursive-partitioning algorithm for high-dimensional data arising as a mixture of beta distributions. BMC Bioinformatics 2008;9:365 doi 10.1186/1471-2105-9-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun 2018;9(1):5068 doi 10.1038/s41467-018-07466-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loyfer N, Magenheim J, Peretz A, Cann G, Bredno J, Klochendler A, et al. A DNA methylation atlas of normal human cell types. Nature 2023;613(7943):355–64 doi 10.1038/s41586-022-05580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoffman R Hematology : basic principles and practice. New York: Churchill Livingstone; 2000. xxxi, 2584 p. p. [Google Scholar]
- 46.Heitzer E, Auinger L, Speicher MR. Cell-Free DNA and Apoptosis: How Dead Cells Inform About the Living. Trends Mol Med 2020;26(5):519–28 doi 10.1016/j.molmed.2020.01.012. [DOI] [PubMed] [Google Scholar]
- 47.Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 2001;61(4):1659–65. [PubMed] [Google Scholar]
- 48.Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 2011;11(6):426–37 doi 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 49.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A 2011;108(23):9530–5 doi 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ili C, Buchegger K, Demond H, Castillo-Fernandez J, Kelsey G, Zanella L, et al. Landscape of Genome-Wide DNA Methylation of Colorectal Cancer Metastasis. Cancers (Basel) 2020;12(9) doi 10.3390/cancers12092710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao L, Quan XB, Zeng WJ, Yang XO, Wang MJ. Mechanism of Hepatocyte Apoptosis. J Cell Death 2016;9:19–29 doi 10.4137/JCD.S39824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang P, Chan KCA, Lo YMD. Liver-derived cell-free nucleic acids in plasma: Biology and applications in liquid biopsies. J Hepatol 2019;71(2):409–21 doi 10.1016/j.jhep.2019.04.003. [DOI] [PubMed] [Google Scholar]
- 53.Aucamp J, Bronkhorst AJ, Badenhorst CPS, Pretorius PJ. The diverse origins of circulating cell-free DNA in the human body: a critical re-evaluation of the literature. Biol Rev Camb Philos Soc 2018;93(3):1649–83 doi 10.1111/brv.12413. [DOI] [PubMed] [Google Scholar]
- 54.Gogenur M, Burcharth J, Gogenur I. The role of total cell-free DNA in predicting outcomes among trauma patients in the intensive care unit: a systematic review. Crit Care 2017;21(1):14 doi 10.1186/s13054-016-1578-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lo YM, Rainer TH, Chan LY, Hjelm NM, Cocks RA. Plasma DNA as a prognostic marker in trauma patients. Clin Chem 2000;46(3):319–23. [PubMed] [Google Scholar]
- 56.Hu Z, Chen H, Long Y, Li P, Gu Y. The main sources of circulating cell-free DNA: Apoptosis, necrosis and active secretion. Crit Rev Oncol Hematol 2021;157:103166 doi 10.1016/j.critrevonc.2020.103166. [DOI] [PubMed] [Google Scholar]
- 57.Grabuschnig S, Bronkhorst AJ, Holdenrieder S, Rosales Rodriguez I, Schliep KP, Schwendenwein D, et al. Putative Origins of Cell-Free DNA in Humans: A Review of Active and Passive Nucleic Acid Release Mechanisms. Int J Mol Sci 2020;21(21) doi 10.3390/ijms21218062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ungerer V, Bronkhorst AJ, Van den Ackerveken P, Herzog M, Holdenrieder S. Serial profiling of cell-free DNA and nucleosome histone modifications in cell cultures. Sci Rep 2021;11(1):9460 doi 10.1038/s41598-021-88866-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pastor B, Abraham JD, Pisareva E, Sanchez C, Kudriavstev A, Tanos R, et al. Association of neutrophil extracellular traps with the production of circulating DNA in patients with colorectal cancer. iScience 2022;25(2):103826 doi 10.1016/j.isci.2022.103826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang P, Sun K, Peng W, Cheng SH, Ni M, Yeung PC, et al. Plasma DNA End-Motif Profiling as a Fragmentomic Marker in Cancer, Pregnancy, and Transplantation. Cancer Discov 2020;10(5):664–73 doi 10.1158/2159-8290.CD-19-0622. [DOI] [PubMed] [Google Scholar]
- 61.Lo YM, Chan KC, Sun H, Chen EZ, Jiang P, Lun FM, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010;2(61):61ra91 doi 10.1126/scitranslmed.3001720. [DOI] [PubMed] [Google Scholar]
- 62.Sin STK, Jiang P, Deng J, Ji L, Cheng SH, Dutta A, et al. Identification and characterization of extrachromosomal circular DNA in maternal plasma. Proc Natl Acad Sci U S A 2020;117(3):1658–65 doi 10.1073/pnas.1914949117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma ML, Zhang H, Jiang P, Sin STK, Lam WKJ, Cheng SH, et al. Topologic Analysis of Plasma Mitochondrial DNA Reveals the Coexistence of Both Linear and Circular Molecules. Clin Chem 2019;65(9):1161–70 doi 10.1373/clinchem.2019.308122. [DOI] [PubMed] [Google Scholar]
- 64.Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res 2014;24(6):766–9 doi 10.1038/cr.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tan M, Xu FF, Peng JS, Li DM, Chen LH, Lv BJ, et al. Changes in the level of serum liver enzymes after laparoscopic surgery. World J Gastroenterol 2003;9(2):364–7 doi 10.3748/wjg.v9.i2.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cho YJ, Park YJ, Min SH, Ryu HG. The Effect of General Anesthesia on Aminotransferase Levels in Patients with Elevated Aminotransferase Levels: A Single-Center 5-Year Retrospective Study. Anesth Analg 2015;121(6):1529–33 doi 10.1213/ANE.0000000000001030. [DOI] [PubMed] [Google Scholar]
- 67.Sender R, Milo R. The distribution of cellular turnover in the human body. Nat Med 2021;27(1):45–8 doi 10.1038/s41591-020-01182-9. [DOI] [PubMed] [Google Scholar]
- 68.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015;347(6217):78–81 doi 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bauernhofer T, Kuss I, Henderson B, Baum AS, Whiteside TL. Preferential apoptosis of CD56dim natural killer cell subset in patients with cancer. Eur J Immunol 2003;33(1):119–24 doi 10.1002/immu.200390014. [DOI] [PubMed] [Google Scholar]
- 70.Czystowska M, Gooding W, Szczepanski MJ, Lopez-Abaitero A, Ferris RL, Johnson JT, et al. The immune signature of CD8(+)CCR7(+) T cells in the peripheral circulation associates with disease recurrence in patients with HNSCC. Clin Cancer Res 2013;19(4):889–99 doi 10.1158/1078-0432.CCR-12-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saito T, Kuss I, Dworacki G, Gooding W, Johnson JT, Whiteside TL. Spontaneous ex vivo apoptosis of peripheral blood mononuclear cells in patients with head and neck cancer. Clin Cancer Res 1999;5(6):1263–73. [PubMed] [Google Scholar]
- 72.Hoffmann TK, Dworacki G, Tsukihiro T, Meidenbauer N, Gooding W, Johnson JT, et al. Spontaneous apoptosis of circulating T lymphocytes in patients with head and neck cancer and its clinical importance. Clin Cancer Res 2002;8(8):2553–62. [PubMed] [Google Scholar]
- 73.McCracken JM, Allen LA. Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death 2014;7:15–23 doi 10.4137/JCD.S11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gelman MS, Ye XK, Stull R, Suhy D, Jin L, Ng D, et al. Identification of cell surface and secreted proteins essential for tumor cell survival using a genetic suppressor element screen. Oncogene 2004;23(49):8158–70 doi 10.1038/sj.onc.1208054. [DOI] [PubMed] [Google Scholar]
- 75.Welsh JB, Sapinoso LM, Kern SG, Brown DA, Liu T, Bauskin AR, et al. Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc Natl Acad Sci U S A 2003;100(6):3410–5 doi 10.1073/pnas.0530278100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005;307(5706):58–62 doi 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 77.Kerbel RS. Tumor angiogenesis. N Engl J Med 2008;358(19):2039–49 doi 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140(6):883–99 doi 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mattox AK, Wang Y, Springer S, Cohen JD, Yegnasubramanian S, Nelson WG, et al. Bisulfite-converted duplexes for the strand-specific detection and quantification of rare mutations. Proc Natl Acad Sci U S A 2017;114(18):4733–8 doi 10.1073/pnas.1701382114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang Y, Douville C, Cohen JD, Mattox A, Curtis S, Silliman N, et al. Detection of rare mutations, copy number alterations, and methylation in the same template DNA molecules. Proc Natl Acad Sci U S A 2023;120(15):e2220704120 doi 10.1073/pnas.2220704120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30(15):2114–20 doi 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 2009;10:232 doi 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell 2011;44(1):17–28 doi 10.1016/j.molcel.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res 2002;12(6):996–1006 doi 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009;41(2):178–86 doi 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 2012;7(7):e41361 doi 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jung N, Dai B, Gentles AJ, Majeti R, Feinberg AP. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat Commun 2015;6:8489 doi 10.1038/ncomms9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhou J, Gao J, Liu Y, Gu S, Zhang X, An X, et al. Human atrium transcript analysis of permanent atrial fibrillation. Int Heart J 2014;55(1):71–7 doi 10.1536/ihj.13-196. [DOI] [PubMed] [Google Scholar]
- 89.Weisenberger DJ. Characterizing DNA methylation alterations from The Cancer Genome Atlas. J Clin Invest 2014;124(1):17–23 doi 10.1172/JCI69740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tie J, Cohen JD, Wang Y, Christie M, Simons K, Lee M, et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol 2019;5(12):1710–7 doi 10.1001/jamaoncol.2019.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cohen JD, Douville C, Dudley JC, Mog BJ, Popoli M, Ptak J, et al. Detection of low-frequency DNA variants by targeted sequencing of the Watson and Crick strands. Nat Biotechnol 2021;39(10):1220–7 doi 10.1038/s41587-021-00900-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321(5897):1801–6 doi 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Adalsteinsson VA, Ha G, Freeman SS, Choudhury AD, Stover DG, Parsons HA, et al. Scalable whole-exome sequencing of cell-free DNA reveals high concordance with metastatic tumors. Nat Commun 2017;8(1):1324 doi 10.1038/s41467-017-00965-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data on methylation (bisulfite sequencing) and copy number alterations in plasma DNA are deposited in the European Genome-Phenome Archive (EGAS00001005400). Similarly, data on mutations in plasma are available from the European Genome-phenome Archive (EGAS00001002764 and EGAS00001002444). Commercial use remains restricted due to Johns Hopkins Medicine legal requirements.