

SUMMARY
Presenilin mutations that alter γ-secretase activity cause familial Alzheimer’s disease (AD) whereas ApoE4, an apolipoprotein for cholesterol transport, predisposes to sporadic AD. Both sporadic and familial AD feature synaptic dysfunction. Whether γ-secretase is involved in cholesterol metabolism and whether such involvement impacts synaptic function remains unknown. Here we show that, in human neurons, chronic pharmacological or genetic suppression of γ-secretase increases synapse numbers but decreases synaptic transmission by lowering the presynaptic release probability without altering dendritic or axonal arborizations. In search of a mechanism underlying these synaptic impairments, we discovered that chronic γ-secretase suppression robustly decreases cholesterol levels of neurons but not in glia, which in turn stimulates neuron-specific cholesterol synthesis gene expression. Suppression of cholesterol levels by HMG-CoA reductase inhibitors (statins) impaired synaptic function similar to γ-secretase inhibition. Thus, γ-secretase enables synaptic function by maintaining cholesterol levels, while chronic suppression of γ-secretase impairs synapses by lowering cholesterol levels.
eTOC Blurb:
Essayan-Perez and Südhof chronically suppressed γ-secretase in human induced neurons to record their synaptic activity and image their synapses. They show that γ-secretase regulates the probability of neurotransmitter release and synapse number. They identified that the mechanism underlying changes in synaptic transmission occurs via regulation of cholesterol levels by γ-secretase.
INTRODUCTION
γ-Secretase, an intramembrane protease, was named because it cleaves the C-terminal fragments (CTFs) of APP at its intramembranous γ-site after APP had been processed by α- or β-secretases. γ-Secretase cleavage of APP CTFs generates secreted Aβ and p3 peptides as well as the APP intracellular domain (AICD) that may act in transcriptional regulation1–5. The γ-cleavage of APP CTFs is of critical importance in Alzheimer’s disease (AD) pathogenesis, as Aβ forms soluble oligomers that are neurotoxic and accumulates into characteristic amyloid plaques observed in the brains of AD patients. Amyloid plaques are also observed in some cognitively normal individuals and their pathogenic role is disputed. Moreover, point mutations in the APP and the PSEN1 and PSEN2 genes (encoding the two presenilin isoforms that are the catalytic subunits of γ-secretase) alter Aβ peptide production and cause familial AD, providing further evidence for a close relationship of γ-secretase to AD pathogenesis6–12.
γ-Secretase is a multiprotein complex comprising presenilins and three additional proteins (nicastrin [NCSTN], PEN2 [PSENEN], and APH1) that are evolutionarily conserved and are universally co-expressed with presenilins in most cells2. Besides APP, γ-secretase cleaves multiple additional intramembrane protein substrates, such as Notch, suggesting that γ-secretase has broad additional functions in eukaryotic cells besides cleavage of APP13. Indeed, during development and in proliferating cells, the cleavage of Notch by γ-secretase is essential for ligand-induced Notch signaling that controls differentiation of cells, such as the differentiation of progenitor cells into neurons, and that regulates cancer cell division, and that is likely the evolutionarily older function of γ-secretase14–16. Although a vast number of papers examined the role of γ-secretase in development, AD pathogenesis, and cancer17–20, and although the atomic structures of γ-secretase and its mutants linked to familial AD have been elucidated21, few studies to date have explored the physiological functions of γ-secretase in non-proliferating cells such as neurons, and even fewer studies have probed its role in human neurons.
AD is a complex multifactorial disorder that features not only accumulations of extracellular Aβ, but also formation of intracellular tangles composed of tau and neuroinflammatory responses involving microglial and astrocytic activation. In patients, Aβ accumulation can be observed decades before AD becomes clinically manifest, consistent with a central role for Aβ accumulations in AD pathogenesis19,22,23. Pathophysiologically, AD leads to synaptic dysfunction and altered neural excitability, followed by neuronal cell death24–27, but the mechanisms involved are unclear. Recent progress in defining the genetic architecture of sporadic AD that accounts for the majority of AD cases identified variations in a large number of genes predisposing to AD25,28,29. However, the functions of many AD-linked genes are poorly understood, although their expression patterns suggest that a sizable number of AD-linked genes act in neuroinflammation pathways associated with AD pathogenesis30–32.
Prominent among the genetic risk factors for AD is ApoE, which encodes an apolipoprotein involved in intercellular cholesterol transport, with the ApoE4 variant being the most impactful genetic risk factor for AD10,12,33,34. ApoE synthesis is enhanced in astrocytes and microglia during neuroinflammation in AD, and ApoE may act by mediating cholesterol transport or by stimulating signaling pathways via a large number of receptors (>10)33–36. However, the precise role of ApoE in brain remains to be clarified, as does the possible relation of ApoE-mediated cholesterol transport and signaling to AD pathogenesis and to other AD-associated genes such as PSEN1 and PSEN2. Multiple AD risk genes are involved in cholesterol metabolism in addition to ApoE410–12,34. Moreover, lipid accumulations containing cholesterol are common in AD patient brains, as well as abnormal endosomes and lysosomes, suggesting a dysfunction of lipid metabolism in neurons in AD37,38. How cholesterol and other lipids contribute to the neuronal impairments and neuroinflammatory response in AD, and whether they might be related to γ-secretase, is poorly studied.
In previous studies, we and others described a role for γ-secretase in synaptic transmission, demonstrating that genetic suppression of γ-secretase activity in mice using presenilin or nicastrin mutations causes a profound impairment in neurotransmitter release and in short- and long-term synaptic plasticity39–44. Extensive evidence suggests that mutations in PSEN1 and PSEN2, the most common causes of familial early-onset AD, represent loss-of-function mutations for γ-secretase45,46, impairing processes such as APP cleavage47 and intracellular calcium homeostasis48,49. However, at least some of the PSEN1 and PSEN2 mutations are also associated with a gain-of-function production of pathogenic Aβ variants, especially Aβ−4210,12,34, as are rare mutations in APP that cause familial AD19,22,23. In a recent study in human neurons, Aβ production was enhanced by the APP Swedish mutation at the β-secretase cleavage site and was associated with increased synapse numbers and synaptic transmission, while APP deletion impaired synapses50. Therefore, APP cleavage by γ-secretase is likely involved in AD pathogenesis and in regulating synapses, but how γ-secretase and Aβ are related to the synaptic dysfunction of AD is incompletely understood.
Although a large literature examines the role of γ-secretase, Aβ, and ApoE in mouse models of AD, few mechanistic studies were performed on these molecules in mice and even fewer in human neurons whose dysfunction, after all, drives AD pathogenesis. In mouse embryonic fibroblasts (MEFs), γ-secretase inhibition altered lipid composition by lowering cellular cholesterol ester levels51 and reduced endocytosis of LDLR, thereby lowering cholesterol uptake52. Moreover, in MEFs, Aβ-mediated inhibition of HMG-CoA reductase seems to reduce cholesterol synthesis53. However, no studies were carried out in neurons, in particular human neurons, whose cholesterol metabolism is uniquely different from that of glia or peripheral cells54,55. Given the central role of γ-secretase in AD, the neuronal dysfunction underlying AD symptoms, and the lack of knowledge of γ-secretase function in human neurons, we investigated the function of γ-secretase in human neurons. Our data uncover a causal relationship between γ-secretase, cholesterol metabolism, and synapses that connects genetic traits in familial and sporadic AD to each other. Specifically, we show that chronic suppression of γ-secretase, either pharmacologically or genetically, causes a loss of synaptic function that is induced by a decrease in cholesterol, revealing a direct causal relationship between γ-secretase, cholesterol, and synapses.
RESULTS
Experimental strategy.
We trans-differentiated human embryonic stem (ES) cells into neurons (‘iN cells’) by forced Ngn2 expression, which produces a comparatively homogeneous population of excitatory forebrain neurons (Figure 1A)35,56,57. The induced human neurons were co-cultured with mouse glia on day 3 since glial factors are required for maturation and synapse formation of human neurons35. We chronically suppressed γ-secretase activity in the human neurons by two approaches. In our first approach, we incubated the human neurons for four weeks (from day 10 to 37–42) with DAPT or LY411575 (LY), two chemically distinct γ-secretase inhibitors58. DAPT (IC50 of 115 nM for total Aβ production) and LY411575 (IC50 of 0.082 nM for total Aβ production) are considered peptidomimetic inhibitors of presenilins58–61. Aβ levels in the cell media decreased with γ-secretase inhibition, as measured by ELISA (Figure 1B). As expected, APP and neurexin CTFs, conversely, increased manyfold when γ-secretase was chronically inhibited, as monitored by immunoblotting with multiple antibodies (Figure 1C–F; S1F–L). Dilution of cell lysates after chronic γ-secretase inhibition confirmed that the observed large increase in the levels of APP CTFs, although dependent on antibody affinities, was reliable and not distorted by massive non-linear effects. Alternative γ-secretase inhibitor treatments further demonstrated significant increases in APP CTFs (Figure S1F–L).
Figure 1: Chronic γ-secretase inhibition increases synapse numbers in human neurons (iN cells) without altering axonal or dendritic arborization.
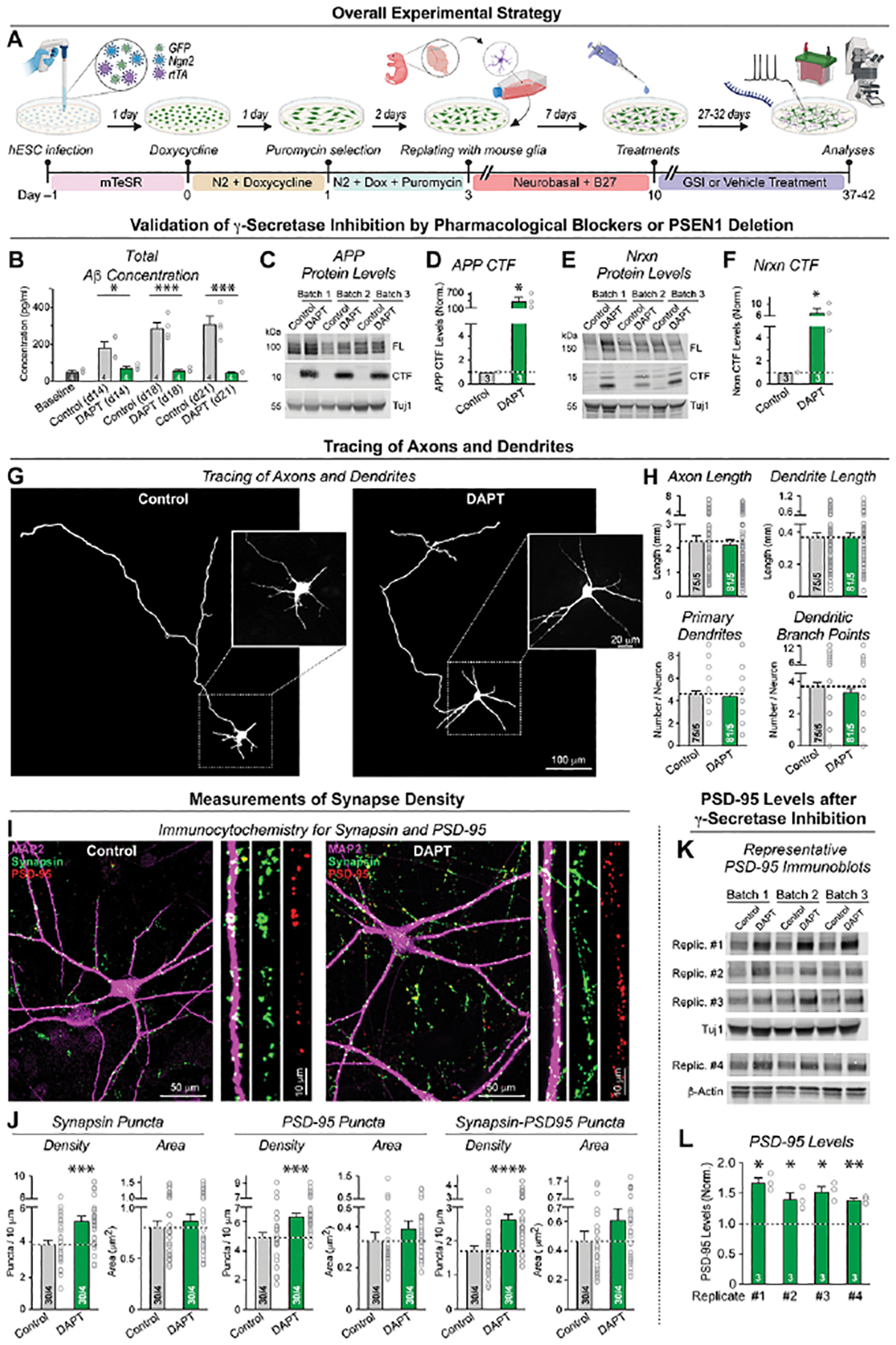
(A) Experimental strategy. Human neurons were trans-differentiated from naïve or PSEN1-mutant H1-ES cells by forced expression of Ngn2 (Zhang et al., 2013). Neurons were treated with vehicle or one of two chemically different γ-secretase inhibitors (GSIs) starting at 10 days and analyzed by imaging, electrophysiology, biochemistry, or RNAseq at day 37–42.
(B) Chronic γ-secretase inhibition with DAPT (40 μM) reduces Aβ production in cultured human neurons as monitored by ELISA in the medium at day 8 (2 days prior to treatment), day 14, 18 and 21.
(C-F) Chronic γ-secretase inhibition increases the levels of the C-terminal fragments (CTFs) of APP (C & D) and of neurexins (E & F) in human neurons (C, E: representative immunoblot; D, F: summary graphs of CTF levels analyzed at day 37).
(G & H) Chronic γ-secretase inhibition has no effect on axonal or dendritic outgrowth (G, representative images; H, summary graphs of the axon length (top left), dendrite length (top right), number of primary dendrites (bottom left), and dendritic branch points (bottom right)). Neurons treated with vehicle or DAPT starting at day 10 were transfected with tdTomato at day 19 and analyzed on DIV21 to identify early developmental changes in dendritic and axonal architecture.
(I & J) Chronic γ-secretase inhibition robustly enhances synapses numbers (I, representative images of human neurons treated with DAPT or vehicle from day 10 to 37 and stained for presynaptic synapsin, postsynaptic PSD-95, and microtubule-associated protein MAP2 [right, zoomed-in dendrite segments]; J, summary graphs of the density and size of synaptic puncta adjacent to MAP2-positive dendrites stained for synapsin (left) or PSD-95 (middle), or double-labeled for synapsin and PSD-95 (right)). For replication experiments, see Figure S2.
(K & L) Chronic γ-secretase inhibition elevates PSD-95 protein levels (K, representative immunoblots; L, summary graphs of PSD-95 levels as determined in 4 independently replicated experiments). Immunoblots were quantified with fluorescently labeled secondary antibodies; signals were normalized for Tuj1 (Rep1 to 3) or β-actin (Rep 4) as loading controls. For replication experiments, see Figure S2.
Data in (B), (D), (F), (H), (J), (L) are means +/− SEM (numbers in bars show cells/independent biological replicates (H, J), or biological replicates (B, D, F, L). Statistical analyses were performed with two-tailed unpaired t-tests, with * = p<0.05; ** = p<0.01, *** = p<0.001. For additional data, see Figures S1 and S2.
In our second approach, we genetically deleted presenilin-1 (PSEN1) in ES cells, and analyzed neurons derived from multiple PSEN1 knockout ES cell clones (Figure S1A–E). Our PSEN1 gene editing strategy targeted exon 4 with 3 single-guide RNAs. We isolated five cell clones homozygous for +1, −7 or −95 base pair indels causing frameshift mutations (Figure S1B, C). Neurons derived from PSEN1-mutant ES cells exhibited a significant increase in APP CTF levels as monitored by immunoblotting (Figure S1D). Presenilin-1 is subject to auto-cleavage producing a presenilin-1 CTFs that was not detectable in the neurons derived from the PSEN1 KO ES cells, indicating successful disruption of presenilin-1 function in γ-secretase by the genetic deletions (Figure S1E).
Thus, we established independent pharmacological and genetic approaches that chronically suppress γ-secretase function in human neurons. Note that with both approaches, γ-secretase activity is not completely abolished since pharmacological inhibition is intrinsically incomplete and the genetic PSEN1 deletion does not impair expression of the second presenilin gene, PSEN2. In the pharmacological approach, however, γ-secretase activity is suppressed both in neurons and in co-cultured glia, whereas in the genetic approach only neuronal but not glial γ-secretase activity is decreased.
γ-Secretase suppression does not alter neurite extension but increases synapse numbers.
γ-Secretase has multiple substrates with diverse functions13, suggesting that by impairing cleavage of these substrates, chronic γ-secretase inhibition could hinder neuronal development even when neurons are directly generated from ES cells. To address this possibility, we analyzed human neurons morphologically after chronic γ-secretase inhibition with DAPT and quantified their axons and dendrites. We observed no significant changes in axonal or dendritic arborization and only a slight decrease in the cell soma size (Figures 1G, H; S2A–D). Given the essential role of γ-secretase in Notch signaling14,15, it may seem surprising that chronic γ-secretase inhibition does not impair neuronal development, but we bypassed normal early neuronal with the Ngn2-induced trans-differentiation protocol. Also, our pharmacological inhibition begins 10 days after neuronal trans-differentiation was initiated, which may further explain a lack of a Notch phenotype.
We next asked whether chronic γ-secretase inhibition alters synapse formation since AD is thought to be a synaptic disorder24,26,27. We stained neurons for synapsin and PSD-95 as pre- and postsynaptic markers and quantified the density of synaptic puncta that are positive for either one or both of these markers (Figure 1I; S2F). Unexpectedly, chronic γ-secretase inhibition robustly increased, instead of decreasing, the synapse density (Figure 1J, S2G). Synaptic puncta that are stained by both presynaptic synapsin and postsynaptic PSD-95 exhibited a ~40% increase in density, suggesting that chronic γ-secretase inhibition enhances synapse formation (Figure 1J). Consistent with this conclusion, we detected a 40–60% increase in PSD-95 protein levels and a significant elevation in the levels of an additional subset of synaptic proteins (Figure 1K, L; S2G, H).
γ-Secretase is essential for normal neurotransmitter release.
To determine whether the increase in synapse numbers upon chronic γ-secretase inhibition is associated with increased synaptic transmission, we analyzed spontaneous mEPSCs in human neurons chronically treated with DAPT. In contrast to the increased synapse numbers, we detected no differences in the mEPSC frequency or amplitude (Figure 2A–C). We next monitored evoked EPSCs in chronically DAPT-treated or PSEN1 knockout neurons as a direct measure of synaptic function. Both chronic DAPT treatment and the PSEN1 deletion suppressed synaptic transmission as monitored via the evoked EPSC amplitude and charge transfer, with ~50% and ~30% decrease, respectively (Figure 2D–G). Thus, although chronic γ-secretase inhibition causes an increase in synapse number, it does not change spontaneous mEPSC frequencies and decreases evoked synaptic transmission. There were no changes in the kinetics of mEPSCs or evoked EPSCs (Figure S4E, F, H). The absence of a change in mEPSC frequency in these experiments could be explained as the sum of the increased mEPSC frequency expected from the enhanced synapse numbers, and the decreased mEPSC frequency expected from the weakened synapse function.
Figure 2: γ-Secretase activity is required for normal neurotransmitter release.
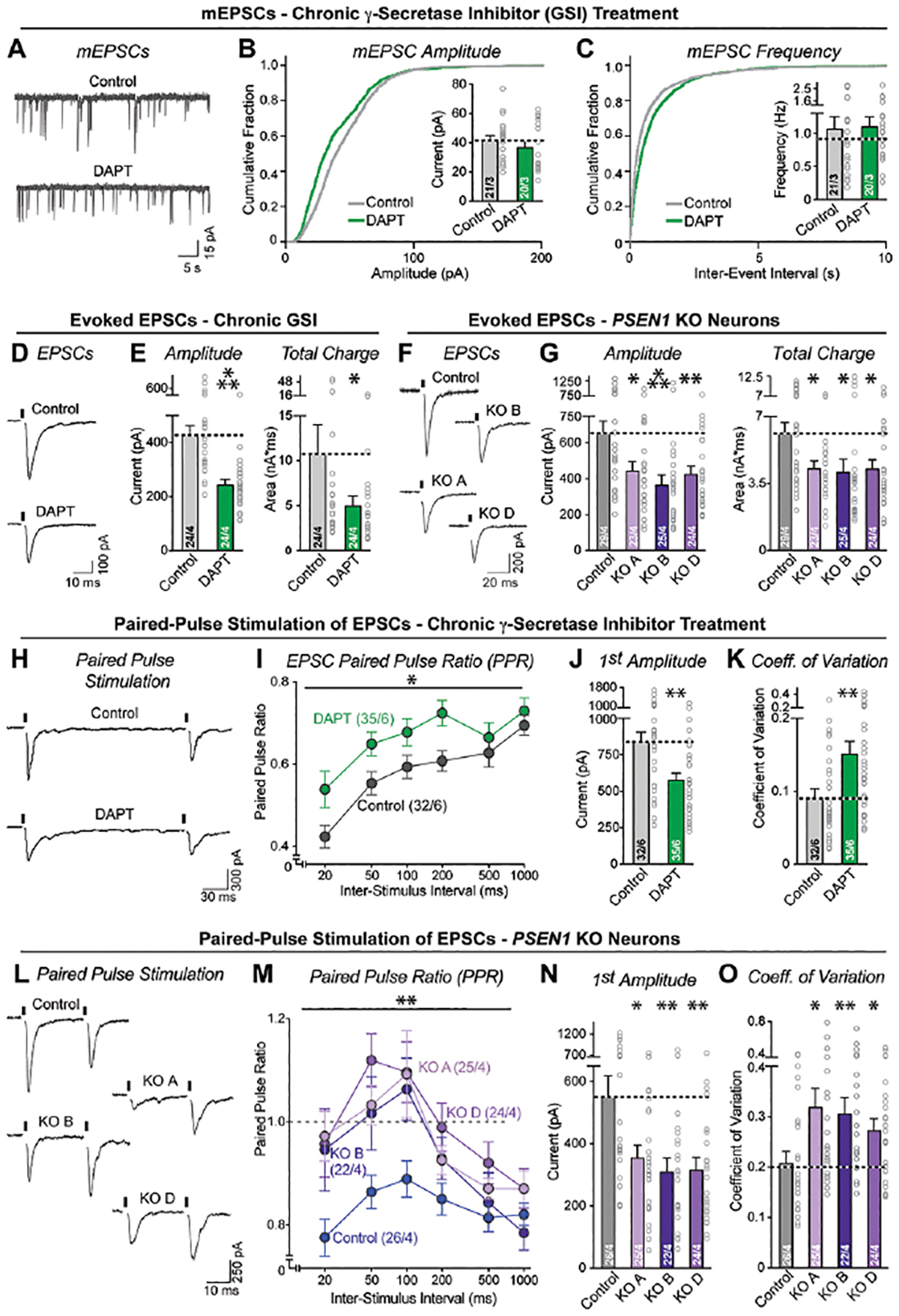
(A–C) Chronic suppression of γ-secretase activity produces no major impairments in mEPSCs (A, representative mESPC traces recorded on day 37–42 from human neurons treated chronically with vehicle (0.5%) or DAPT (40 μM); B & C, cumulative probability plots of the interevent intervals (B) and amplitudes (C) of mESPCs [insets: mean mEPSC frequency (B) and amplitude (C)]).
(D & E) Chronic suppression of γ-secretase activity impairs evoked synaptic transmission (D, representative traces; E, summary graphs of the EPSC amplitude (left) and charge transfer (right)).
(F & G) Same as D and E but measured in PSEN1 KO vs. control neurons.
(H & I) Chronic suppression of γ-secretase activity lowers paired-pulse depression, suggesting a decline in release probability (H, representative traces of EPSCs evoked by two closely spaced stimuli [stimulus intervals (20–1000 ms) are indicated above traces; for full traces, see Figure S3]; I, summary plot of the paired-pulse ratios as a function of the interstimulus interval).
(J) Confirmation that chronic suppression of γ-secretase activity lowers synaptic strength as monitored via the amplitude of first evoked EPSCs during paired-pulse stimulation (summary graph of EPSC amplitude).
(K) Chronic suppression of γ-secretase activity increases the coefficient of variation of evoked EPSC amplitudes, consistent with a reduced release probability.
(L–O) PSEN1 mutant vs. control neurons show increased coefficients of variation of evoked EPSC amplitudes, demonstrating that genetic suppression of γ-secretase activity also dramatically decreases the release probability of human neurons. Full traces are shown in Figure S3.
Numerical data are means ± SEM (numbers of cells/experiments are listed in bars). Statistical analyses were performed by two-tailed unpaired t-test (DAPT data) or two-way ANOVA with Bonferroni’s multiple comparison (PSEN1 KO data), with * = p<0.05; ** = p<0.01, *** = p<0.001. For intrinsic properties and controls, see Figure S3 and S4.
A potential cause of the decrease in evoked EPSCs could be a change in neuronal excitability and action potential generation, although this possibility would not explain the unchanged mEPSC frequency. To test the possibility that chronic γ-secretase inhibition alters the electrical properties of neurons, we conducted current clamp recordings. We observed no significant effects of chronic γ-secretase inhibition on action potential firing frequency, excitability, rheobase, peak amplitude, or kinetics (Figure S3A, B). Moreover, we measured the intrinsic electrical properties of the neurons and detected a modest decrease in capacitance and an increase in input resistance, consistent with the decreased soma size observed morphologically (Figure S3C).
An alternative explanation for the decrease in synaptic function upon chronic γ-secretase inhibition is a decline in the neurotransmitter release probability. To test possibility, we monitored EPSCs induced by two closely spaced stimuli (Figure S4B, G). Under our recording conditions in 3 mM extracellular Ca2+, human neurons exhibit a high release probability that results in paired-pulse depression (Figure 2H, L). Both chronic pharmacological and genetic γ-secretase inhibition reversed paired-pulse depression, consistent with a decrease in release probability (Figure 2I, M; S4B). As with the single stimuli, chronic γ-secretase inhibition lowered the amplitude of the first of the paired EPSCs by approximately 30% (Figure 2J, N) but significantly increased the coefficient of variation by 50%, which again argues for a decrease in release probability (Figure 2K, O). Membrane properties were also not grossly changed under these conditions, although a modest decrease in capacitance was again detected (Figure S4C–D). Thus, as assessed via two parameters, chronic γ-secretase inhibition impairs synaptic transmission by lowering the neurotransmitter release probability. Given that the mEPSC frequency and amplitude were unchanged upon chronic DAPT treatment whereas the evoked EPSC amplitude was decreased, these data indicate that chronic γ-secretase inhibition generates more synapses whose synaptic strength is decreased because of an impairment in release probability.
Is the decrease in synaptic release probability due to an acute or chronic effect of γ-secretase inhibition? To address this question, we measured the effects of shorter periods of γ-secretase inhibition on evoked EPSCs. We performed a 1-day γ-secretase inhibition in mature neurons on day 37–42 but observed no changes in synaptic transmission (Figure 3A; S5A). However, a 2-day γ-secretase inhibition in mature neurons produced a significant, albeit modest (~25%) decrease in EPSC amplitude (Figure 3B, S5B). The lack of an effect with only 1-day γ-secretase inhibition was not due to a sensitive period in development since γ-secretase inhibition for 1 day at day 21 also did not impair synaptic transmission (Figure 3C; S5C). Conversely, inhibition of γ-secretase from day 10 to 21 for 11 days did impair synaptic transmission, although the effect was smaller than that observed in mature neurons (Figure S5D, E). These results indicate that chronic, but not acute, γ-secretase inhibition impairs synaptic transmission, suggesting an indirect role for γ-secretase in neurotransmitter release.
Figure 3: Acute γ-secretase inhibition does not impair synaptic transmission.
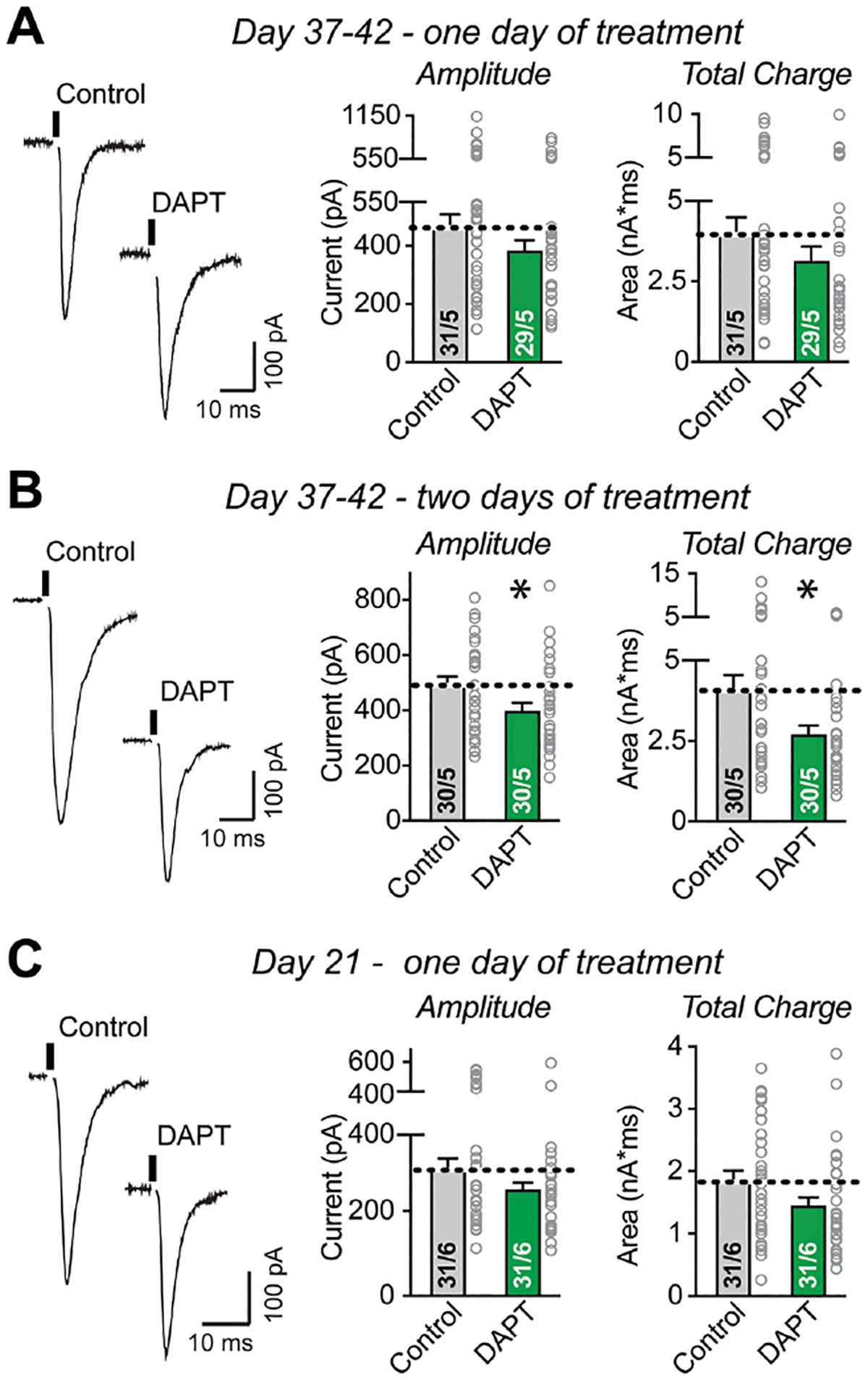
(A) Acute suppression of γ-secretase activity by one-day DAPT treatment at Day 37–42 has no detectable effect on evoked EPSCs (left, representative traces; right, summary graphs of evoked EPSC amplitudes and charge transfer).
(B) Semi-acute suppression of γ-secretase activity by two-day DAPT treatment at Day 37–42 significantly decreases synaptic strength as monitored via evoked EPSCs (left, representative traces; right, summary graphs of evoked EPSC amplitudes and charge transfer).
(C) Acute suppression of γ-secretase activity by a one-day DAPT treatment at Day 20, analyzed on Day 21, has no significant effect on evoked EPSCs (left, representative traces; right, summary graphs of evoked EPSC amplitudes and charge transfer).
Data are means +/− SEM; numbers of cells/replicates are indicated in the bars. Statistical analyses were performed using two-tailed unpaired t-test, with * = p<0.05; ** = p<0.01, *** = p<0.001. For kinetics and intrinsic properties, see Figure S5.
Chronic γ-secretase suppression selectively stimulates transcription of cholesterol synthesis genes.
To explore the mechanisms by which chronic γ-secretase inhibition acts on synapses, we performed RNAseq experiments on human neuron/mouse glia co-cultures after chronic inhibition of γ-secretase with either DAPT or LY411575 and analyzed the neuronal and glial transcriptomes separately based on their species of origin62,63. Six independent biological replicates were performed to ensure reproducibility. Strikingly, all top upregulated genes induced by chronic γ-secretase inhibition in human neurons, but none in mouse glia, are involved in cholesterol synthesis or transport (Figure 4A, C, E). Expression of nearly every major gene in cholesterol metabolism was stimulated by γ-secretase inhibition in the human neurons (Figure S6A; Luo et al., 2020). In contrast, in mouse glia, no cholesterol metabolism-related gene was upregulated by chronic γ-secretase inhibition (Figure 4B, D, F). Different from upregulated genes, the downregulated genes in human neurons were functionally heterogeneous (Figure 4A, C, G). Here, the largest subgroup in the top 20 downregulated genes included immediate early genes, which are constitutively expressed in cultured human neurons given their spontaneous network activity64. The lack of cholesterol metabolism gene changes in mouse glia suggests a cell type-specific transcriptional role of γ-secretase.
Figure 4: Chronic suppression of γ-secretase activity upregulates cholesterol synthesis gene expression in human neurons but not in mouse glia.
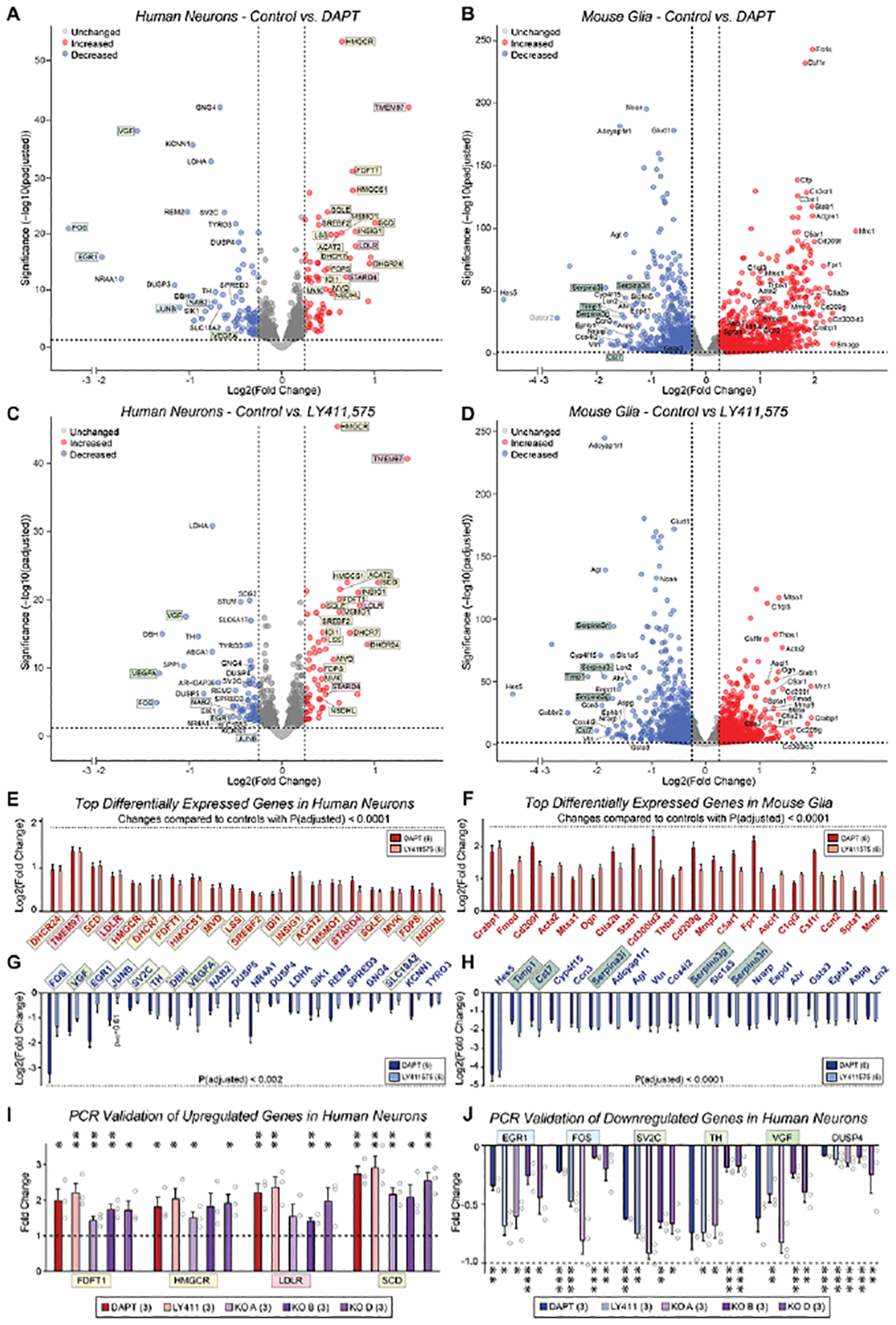
(A & B) Volcano plots of differentially expressed genes (DEGs) from co-cultured human neurons (A) and mouse glia (B) that were chronically treated with the γ-secretase inhibitor DAPT (40 μM) or vehicle (DMSO only) from day 10–40, and analyzed by bulk RNAseq. Data resulted from pairwise comparisons of each treatment group versus vehicle, normalized to raw counts, with a standard False Discovery Rate of 0.1; data underwent the Benjamini-Hochberg correction to obtain adjusted p-values (yellow, cholesterol synthesis genes; pink, cholesterol transport genes; blue, activity-dependent immediate early genes; light green, growth factors; dark green, protease inhibitors).
(C & D) Same as A and B, except that the cells were treated with the γ-secretase inhibitor LY411,575 (2.5 μM).
(E & F) Bar graphs for the top 20 upregulated genes identified by RNAseq analyses of DAPT-and LY411575-treated human neurons (E) and mouse glia (F) that exhibited adjusted p-values of <0.0001. The cutoff for log2(fold change) values (y-axis) was >0.3, corresponding to a fold change greater than 20% relative to controls.
(G & H) Same as E & F, but for the top 20 downregulated genes, using adjusted p-values of <0.002 (G) or <0.0001 (H).
(I) qRT-PCR measurement of 4 upregulated cholesterol synthesis pathway genes in three lines of PSEN1 KO human neurons, DAPT- and LY411,575-treated human neurons.
(J) qRT-PCR measurement of 4 upregulated cholesterol synthesis pathway genes in three lines of PSEN1 KO human neurons, DAPT- and LY411,575-treated human neurons.
(K) qRT-PCR measurement of 6 downregulated genes in three lines of PSEN1 KO human neurons in DAPT- and LY411,575-treated human neurons.
Data in (E-J) are means +/− SEM: (A-H) have 6 biological replicates; (I and J) have 3 biological replicates. Statistical analyses for were performed using the Wald test (A-H) and two-tailed unpaired t-test (I-J) and plotted as fold change relative to controls (*p<0.05; **p<0.01, ***p<0.001). For additional validation data, see Figure S6.
In mouse glia (whose γ-secretase activity is also pharmacologically inhibited in these experiments) both up- and downregulated genes were also functionally heterogeneous, except for a preponderance of secreted protease inhibitors among the glial downregulated genes (Figure 4B, D, F, H). In addition, chronic γ-secretase inhibition lowered expression in glia of Hes5, a Notch-target transcription factor65, and upregulated expression of colony stimulating factor 1 receptor (Csf1r), indicating possible regulation of microglia66. Prior studies suggested that γ-secretase dampens inflammation and Toll-like receptor 4-mediated immune response via the processing of LRP167, but this pathway was not a major component of the genes we identified. Despite the induction of microglial marker Csf1r, chronic γ-secretase inhibition did not appear to induce expression of stress or inflammatory responses in neurons or glia, suggesting that the inhibition did not cause an injury response.
Since the induction of cholesterol metabolism genes by chronic γ-secretase inhibition in human neurons was absent from mouse glia, it does not represent a general cellular pathway controlled by γ-secretase but is specific to neurons, which may explain why it has not been observed previously. To confirm the gene expression changes found by RNAseq and to ensure that these changes are not only caused by pharmacological inhibition of γ-secretase, we performed quantitative RT-PCR analyses of selected genes in human neurons with a chronic pharmacological or a genetic γ-secretase impairment (i.e., PSEN1 deletion). These analyses validated the RNAseq results both for the upregulation of cholesterol metabolism genes and the downregulation of immediate early genes (Figures 4I, J; S6B, C). Cholesterol gene mRNA levels showed an approximately two-fold increase while immediate-early gene mRNA levels exhibited an approximately two-fold decrease with pharmacological inhibition (Figures 4I, J; S6). PSEN1 KO neurons exhibited more modest but still significant changes, possibly because the continued expression of PSEN2 may produce a lesser suppression of γ-secretase activity (Figures 4I, J). Moreover, measurements of the mRNA levels at different times of pharmacological γ-secretase inhibition revealed that γ-secretase inhibition of 48 hours or more was required to induce changes in cholesterol synthesis gene expression (Figure S6D), consistent with the time course of synaptic impairments (Figure 3). Therefore, two transcriptomic approaches in multiple replicates confirmed that nearly all key genes in the cholesterol synthesis pathway were specifically upregulated in neurons upon chronic suppression of γ-secretase activity (Figure S6A).
γ-Secretase inhibition decreases cellular cholesterol levels.
We next asked whether the increased expression of cholesterol synthesis genes upon chronic γ-secretase inhibition might be due to a loss of cellular cholesterol, which would induce cholesterol synthesis gene expression, or conversely cause an increase in cellular cholesterol. To address this question, we used GFP-expressing human neurons that were chronically treated with γ-secretase inhibitors (DAPT or LY411575) or that contained the PSEN1 deletion. We stained the neurons with filipin, a fluorescent dye that inserts into the plasma membrane in a cholesterol-dependent manner68,69, and with antibodies to L1CAM, a surface marker of human neurons70. The co-localization of the filipin fluorescence, GFP, and L1CAM signals then allowed us to specifically quantify the filipin signal from neurons (Figure 5). Both chronic pharmacological γ-secretase inhibition and the decrease in γ-secretase activity induced by the PSEN1 mutation robustly decreased (~25%) the plasma membrane filipin signal in neurons (Figure 5). Given the tight regulation of plasma membrane cholesterol levels in cells, this is a large effect71.
Figure 5: Chronic suppression of γ-secretase activity reduces plasma membrane cholesterol in human neurons.
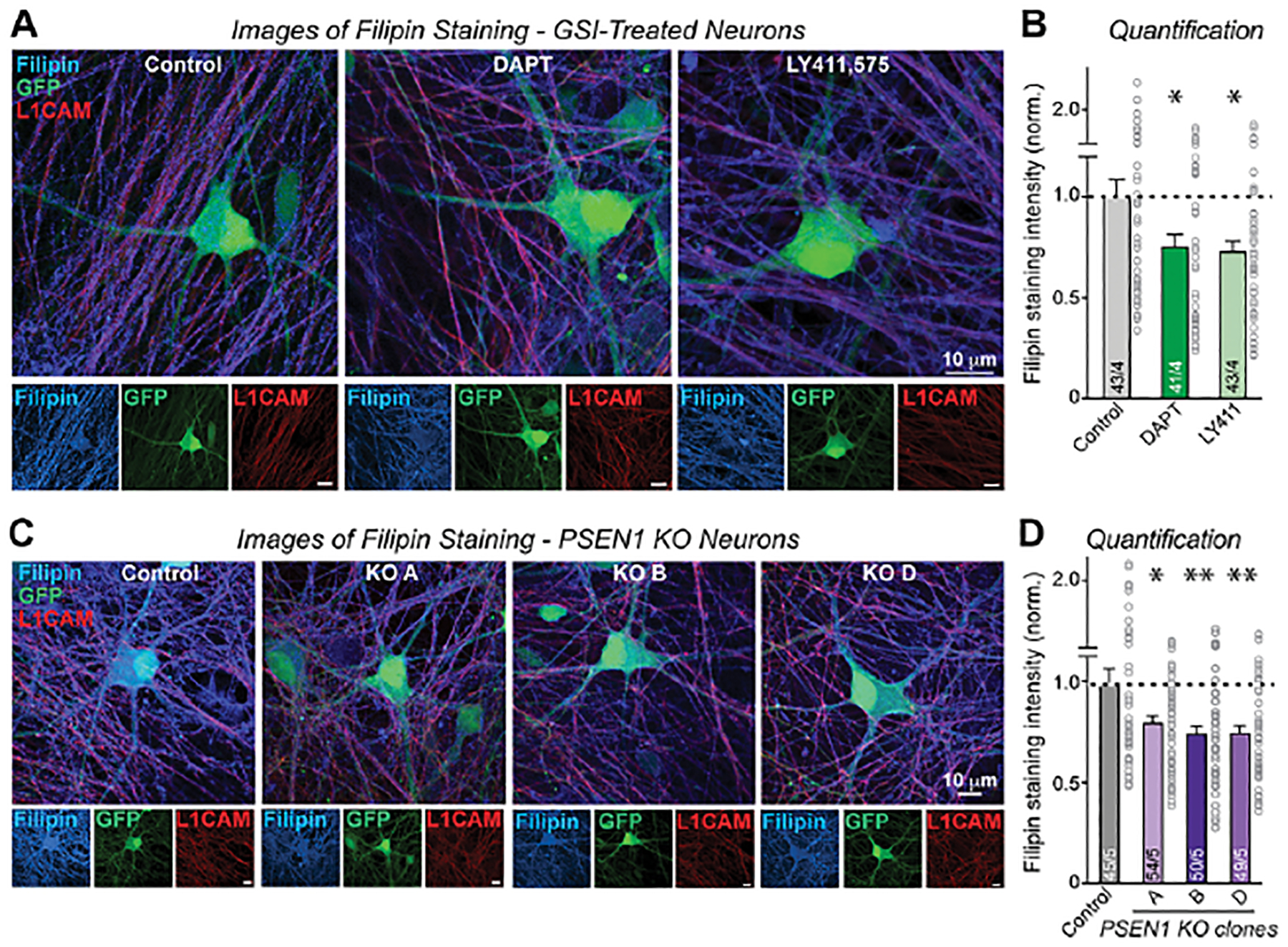
(A) Representative images illustrating reduced Filipin staining (blue) in human neurons upon chronic suppression of γ-secretase activity with DAPT or LY411,575. Neurons are labeled by GFP expression (green), and axons are labeled by L1CAM staining (red). Scale bars (white line) represent 10 μm.
(B) Chronic suppression of γ-secretase activity with DAPT or LY411,575 causes a robust decrease in the plasma membrane cholesterol content of human neurons as monitored by Filipin signal in GFP-positive human neurons.
(C & D) Same as A & B, but for PSEN1 KO neurons obtained from three independent KO lines.
Data in (B) and (D) are means ± SEM (numbers of cells/biological replicates) are shown in bars. Statistical analyses were conducted using two-tailed unpaired t-test, with * = p<0.05; ** = p<0.01, *** = p<0.001.
To independently validate the finding that chronic γ-secretase inhibition lowers cellular cholesterol levels in neurons, we isolated GFP-positive neurons and GFP-negative glia cells at 40 days post induction by fluorescence-activated cell sorting (FACS) (Figure 6A, S7). Cells were isolated after 30 days of pharmacological γ-secretase inhibition with DAPT or LY411575. We then measured the cellular content of unesterified and esterified cholesterol in both neurons and glia (Figure 6A). Again, we observed an approximately 20% decrease in total cholesterol in neurons, whereas cholesterol in glia was not significantly changed (Figures 6B, C). Given the lengthy time of the cell separation procedure, the consistency of the results between filipin staining and chemical cholesterol measurements is reassuring. Viewed together, these data indicate that chronic γ-secretase inhibition lowers neuronal but not glial cholesterol levels, which induces transcription of genes involved in cholesterol synthesis and transport in neurons in response to the decreased cellular content of cholesterol.
Figure 6: Chronic suppression in γ-secretase activity decreases neuronal but not glial levels of unesterified and esterified cholesterol.
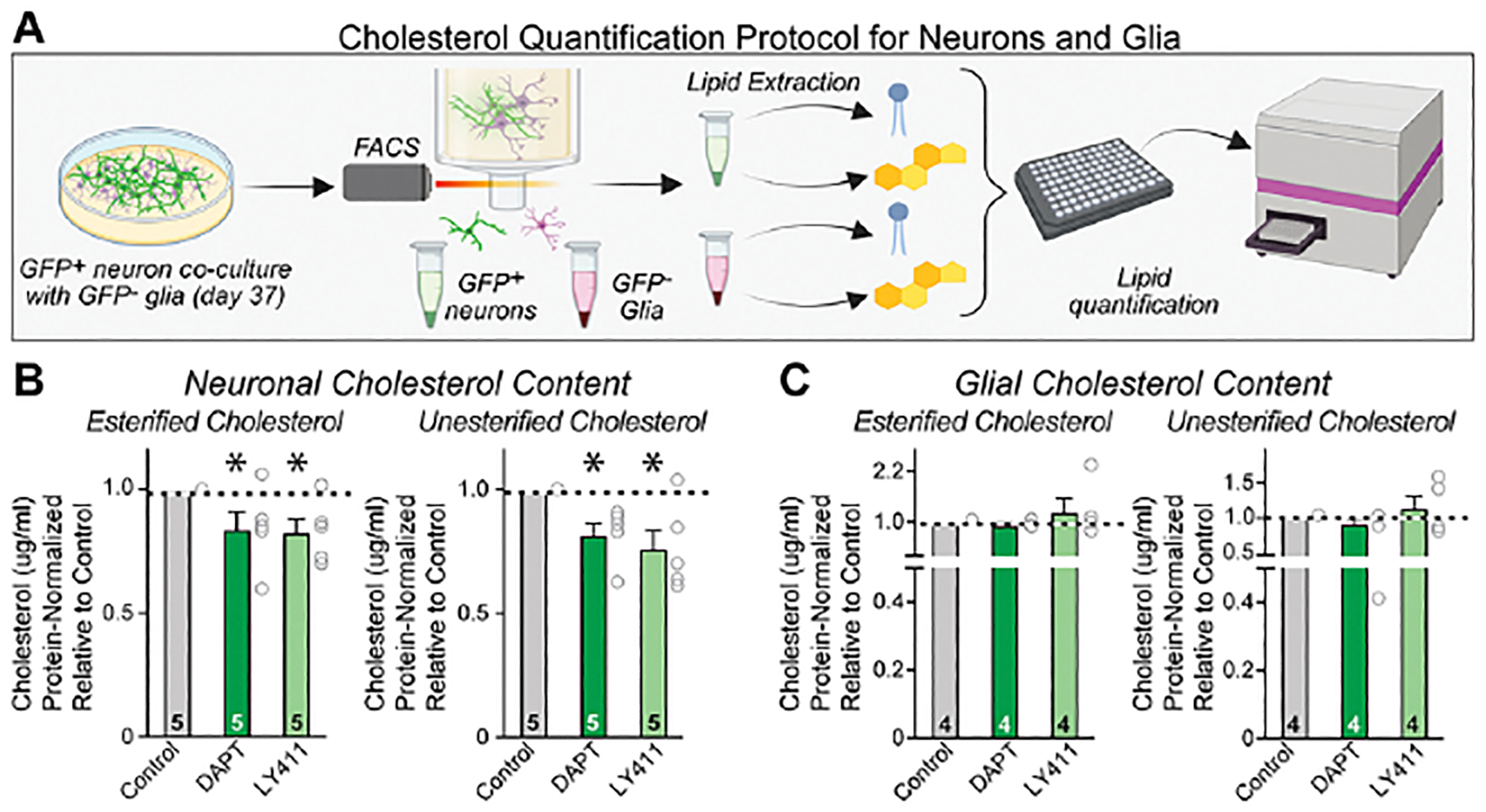
(A) Experimental approach: GFP-positive human neurons were separated from GFP-negative mouse glia using FACS and their free and esterified cholesterol contents were measured enzymatically.
(B & C) Summary graphs showing that free and esterified cholesterol levels are diminished in human neurons (B) but unchanged in mouse glia (C). Data in are means +/− SEM (numbers in bars indicate biological replicates). Statistical analyses were conducted using one-sample t-tests with theoretical mean of 1.0, with * = p<0.05; ** = p<0.01, *** = p<0.001. For representative FACS plots, see Figure S7.
Direct suppression of cholesterol synthesis impairs neurotransmitter release.
Does γ-secretase regulate the presynaptic release probability directly, or does it act indirectly by controlling the neuronal cholesterol content? To address this question, we suppressed cholesterol synthesis in neurons using chronic treatment with two unrelated HMG-CoA reductase inhibitors (rosuvastatin and simvastatin; referred to as statins)72,73, either alone or in combination with DAPT as a γ-secretase inhibitor (Figure S8A). As expected, both statins induced transcription of cholesterol synthesis genes by four-fold since they lower cellular cholesterol levels (Figure 7A). The two statins also decreased immediate-early gene expression in a similar manner to DAPT and LY411575 (Figure 7B). For both transcriptional phenotypes, the effects of chronic cholesterol synthesis inhibition by statins and chronic γ-secretase inhibition by DAPT were not additive, suggesting that the changes operate via the same pathway.
Figure 7: HMG-CoA reductase inhibitors (statins) upregulate cholesterol synthesis gene and downregulate activity-dependent immediate-early gene expression but do not increase synapse numbers.
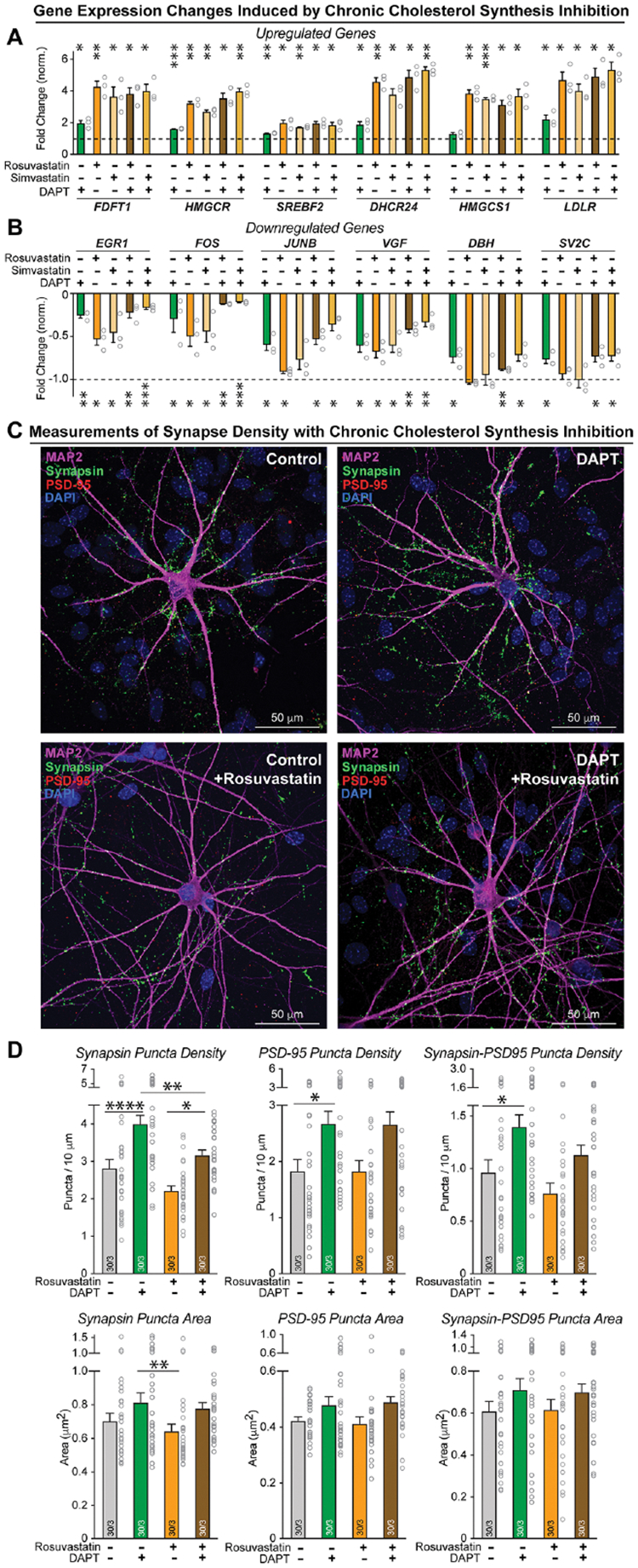
(A & B) qRT-PCR measurements reveal that chronic inhibition of HMG-CoA reductase, a rate-limiting enzyme in cholesterol biosynthesis, induces upregulation of the same cholesterol-synthesis genes (A) and downregulation of the same set of genes including immediate-early genes (B) as chronic inhibition of γ-secretase. Neuron / glia co-cultures were treated with vehicle (0.5% DMSO), rosuvastatin (1 μM), or simvastatin (1 μM) without or with DAPT (40 μM) from day 10 to 37.
(C) Representative immunocytochemistry images of human neurons treated with vehicle, DAPT, vehicle + Rosuvastatin, or DAPT + Rosuvastatin from day 10 to 37. Neurons were stained for presynaptic synapsin, postsynaptic PSD-95, and microtubule-associated protein MAP2.
(D) Chronic treatment of human neurons with statins, different from chronic treatment of human neurons with γ-secretase inhibitors that robustly enhance synapse numbers, do not induce an increase in synapse density. Panels show summary graphs of the density (top graphs) and size (bottom graphs) of synaptic puncta labeled for synapsin (left) or PSD-95 (middle), or double-labeled for co-localized synapsin and PSD-95 (right). Only puncta adjacent to MAP2-positive dendrites were quantified.
Data in (A), (B), (D) +/− SEM (numbers in bars show cells/independent biological replicates). Statistical analyses were performed with unpaired t-tests for (A) and (B), and with two-way ANOVA with Bonferroni’s multiple comparison for (D), with * = p<0.05; ** = p<0.01, *** = p<0.001.
Next, we tested whether statins had an effect on synapse numbers by measuring the density of synapsin- and PSD95-positive puncta in cells treated with vehicle, DAPT, vehicle and rosuvastatin, or DAPT and rosuvastatin (Figure 7C). Chronic γ-secretase inhibition increased synapse density consistently by 30 to 50%, replicating the results described in Figure 3, whereas statins did not (Figure 7D). Instead, chronic lowering of cholesterol produced a non-significant trend of lowering synapse numbers without preventing the increase in synapse numbers caused by chronic γ-secretase inhibition (Figure 7D). These results suggest that chronic γ-secretase inhibition changes synapse number via cholesterol-independent mechanisms.
Finally, we measured evoked EPSCs to analyze synapse function. Chronic cholesterol synthesis inhibition by statins suppressed synaptic strength, as monitored via the EPSC amplitude and EPSC charge transfer, by 30% and 50% respectively (Figure 8A–C). No effect by statins on membrane properties or EPSC kinetics were detected (Figure S8B, C). The phenotype induced by chronic cholesterol synthesis inhibition was as strong as that of chronic γ-secretase inhibition. Moreover, the effects of the γ-secretase and cholesterol synthesis inhibitions were also not additive, corroborating the notion that they operate in the same pathway (Figures 8A–C). To further compare the effects of chronic γ-secretase and cholesterol synthesis inhibition on synapses, we measured paired-pulse responses (Figure 8D). Chronic γ-secretase and cholesterol synthesis inhibition induced similar effects on the paired-pulse ratio indicative of a decrease in release probability, with a 50% decrease in the amplitude of the first EPSCs compared to controls (Figure 8E–G). Again, their combined application was not additive, indicating that chronic cholesterol synthesis inhibition decreased the release probability (Figures 8E, F). Finally, chronic γ-secretase and cholesterol synthesis inhibition similarly enhanced the coefficient of variation of EPSCs, and here their combined effects were also not additive (Figure 8G). Together, these data support the hypothesis that chronic γ-secretase inhibition suppresses synaptic strength in human neurons by decreased neuronal cholesterol synthesis and reduced neuronal cholesterol content.
Figure 8: HMG-CoA reductase inhibitors (statins) suppress the neurotransmitter release probability in human neurons.
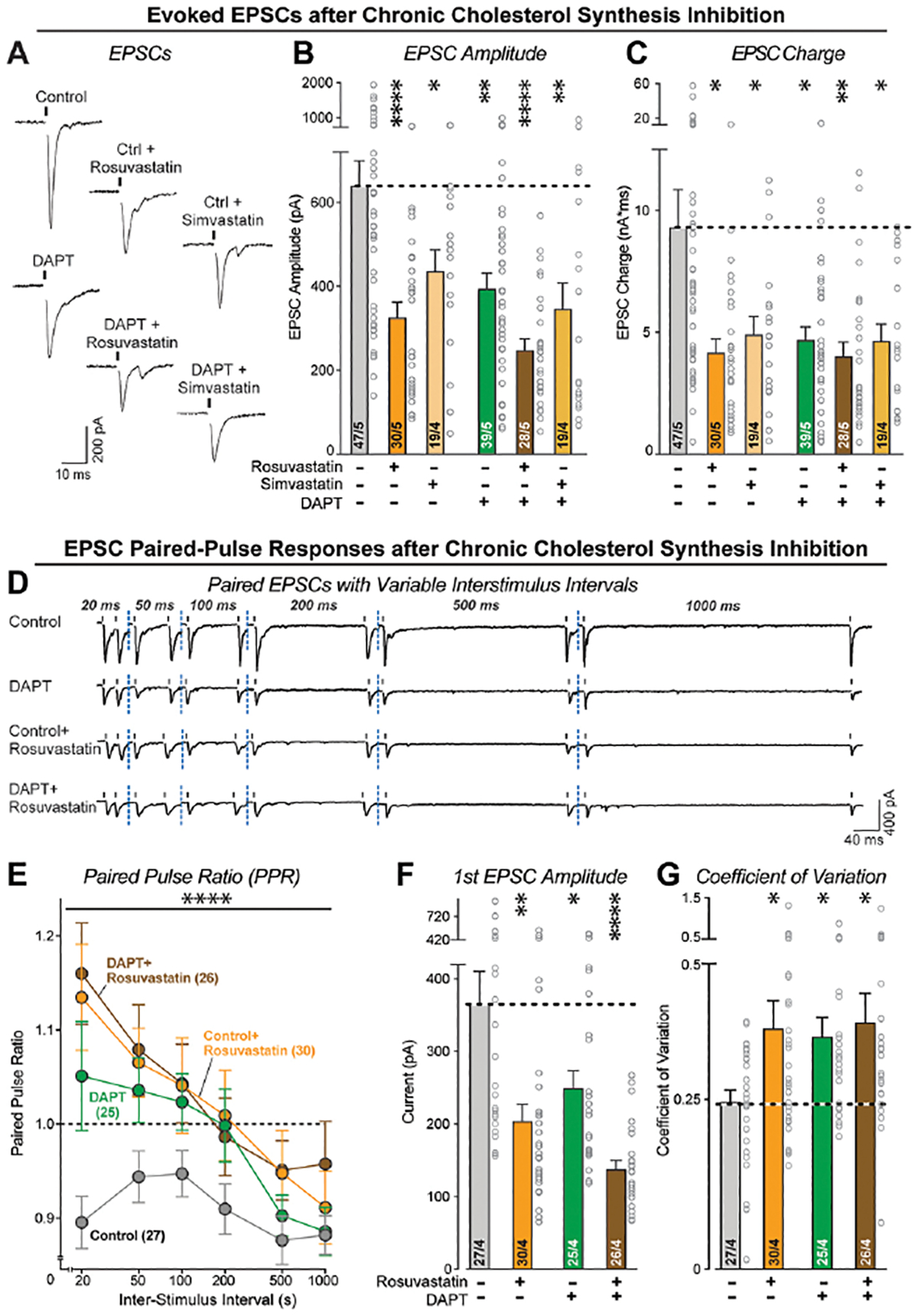
(A) Representative EPSC traces from neurons treated with DAPT and HMG-CoA reductase inhibitors as described for A & B.
(B & C) Chronic inhibition of HMG-CoA reductase causes the same suppression in synaptic strength, measured via the amplitude (B) or charge transfer (C) of evoked EPSCs, as chronic inhibition of γ-secretase. Note that the effects of chronic inhibition of HMG-CoA reductase of γ-secretase are not additive.
(D) Representative traces of EPSCs evoked by two closely spaced stimuli (paired-pulse stimulation), monitored in human neurons as a function of chronic treatments with γ-secretase and/or HMG-CoA reductase inhibitors.
(E) Chronic suppression of the HMG-CoA reductase and/or γ-secretase activity reverses paired-pulse depression, suggesting a decline in release probability. The summary plot depicts the paired-pulse ratios (second/first EPSC amplitude) as a function of the interstimulus interval.
(F) As shown for the isolated EPSCs, chronic suppression of the HMG-CoA reductase and/or γ-secretase activity decreases the amplitude of evoked EPSCs monitored via the first response during paired-pulse stimulation.
(G) Chronic suppression of the HMG-CoA reductase and/or γ-secretase activity also increases the coefficient of variation of evoked EPSC amplitudes, consistent with a reduced release probability.
Data are means +/− SEM (the number of cells/biological replicates are shown in the bars). For (B), (C), (F), (G), statistical analysis was measured using two-tailed unpaired t-test. For (E), statistical analysis was assessed by two-way ANOVA with Bonferroni’s multiple comparison test. Statistical significance: *p<0.05; **p<0.01, ***p<0.001. For additional data, see Figure S8.
DISCUSSION
Here we show that chronic γ-secretase inhibition induced by pharmacological treatments or by deletion of PSEN1 in human neurons causes a decrease in cellular cholesterol levels that results in synaptic dysfunction by suppressing the presynaptic neurotransmitter release probability. The decrease in cholesterol by chronic γ-secretase inhibition is only observed in neurons but not in glia, and strongly induces neuronal cholesterol synthesis gene expression. The fact that cholesterol synthesis genes are the major mRNAs that are upregulated in human neurons by chronic γ-secretase inhibition suggests that cellular cholesterol metabolism is a prominent target of γ-secretase function in neuronal physiology. Our results thus identify a neuron-specific role for γ-secretase in regulating cellular cholesterol levels, thereby defining a functional link between PSEN1, the predominant gene involved in familial AD, and cholesterol metabolism, a major pathway regulated by ApoE that is implicated in sporadic AD. In addition, we show that chronic γ-secretase inhibition causes an increase in synapse numbers that is not induced by the decrease in cholesterol levels with statins. Lowering neuronal cholesterol levels with statins results in the same decrease in neurotransmitter release probability as with chronic γ-secretase inhibition, but does not increase synapse numbers, which is consistent with a multifaceted function of γ-secretase in neurons.
A relation of cholesterol metabolism to the presynaptic release probability was previously described in rodent neurons. Specifically, lowering cholesterol using methyl-beta-cyclodextrin, chronic statin (HMG-CoA reductase inhibitor) treatments, or knockdown of the cytoplasmic cholesterol transport protein ORP2 decreased the release probability of hippocampal neurons74–78. These findings agree well with our results in human neurons using two chemically distinct statins as cholesterol-synthesis inhibitors, therefore providing a cholesterol-dependent mechanism by which γ-secretase regulates synaptic function. Both chronic γ-secretase and cholesterol synthesis inhibition not only increased cholesterol synthesis gene expression, but also lowered immediate-early gene transcription, consistent with the impairment in synaptic transmission. Somewhat surprisingly, chronic γ-secretase inhibition did not impair development of neuronal axons and dendrites but did produce a small decrease in membrane capacitance and soma size. Since the capacitance decrease was also observed with chronic statin treatments (soma sizes were not measured), it is likely due to a decline in available cholesterol in neurons. Such decline may limit soma expansion during neuronal maturation, suggesting that neurons with a lowered cholesterol content prioritize extension of axonal and dendritic arbors over soma growth.
Further, our data suggest that γ-secretase controls synapse numbers by a cholesterolin-dependent process since chronic γ-secretase inhibition increased the synapse density, whereas statins did not. In fact, statins produced a trend towards a decrease in synapse number. Moreover, lowering cholesterol with statins also did not prevent the increase in synapse density produced by chronic γ-secretase inhibition. The increase in synapse numbers induced by chronic γ-secretase inhibition is unlikely to represent a compensatory process in response to decreased synaptic transmission, because impairments in neurotransmitter release induced by other mechanisms, including statins, do not increase synapse numbers. Given that γ-secretase has multiple substrates not only in developing but also in mature neurons, the presence of multiple pathways regulated by γ-secretase is plausible. Important regulators of neuronal functions and synaptic organizers, such as neurexins79,80, neuroligins81, cadherins82, protocadherins83 and Notch pathway ligands84 are known to be γ-secretase substrates and may be involved. In this regard, we show that chronic γ-secretase inhibition does not cause a multitude of impairments in developing neurons but appears to have a major positive effect on synapse numbers and a large negative effect on the presynaptic release probability. The restricted range of major γ-secretase functions in committed but developing human neurons emerging from our experiments agrees well with pioneering studies from the Shen lab that also identified a decrease in presynaptic release probability as the major consequence of presenilin and nicastrin deletions in rodent hippocampus40–42. It is therefore possible that the phenotype of the presenilin deletions in rodent hippocampus was also caused by a lowering of neuronal cholesterol levels.
Since chronic γ-secretase inhibition downregulated cholesterol levels and upregulated cholesterol synthesis and transport gene expression only in neurons but not in glia, the function of γ-secretase in cholesterol metabolism does not represent a general cellular pathway controlled by γ-secretase, but at least in brain appears to be neuron-specific. Chronic γ-secretase inhibition lowered expression in glia of Hes5, a Notch-target transcription factor65, and upregulated expression of colony stimulating factor 1 receptor (Csf1r), indicating possible regulation of microglia66. However, no upregulation of genes associated with ‘reactive microglia’, such as those observed for the disease-associated microglial gene expression signature85,86, was detected. Prior studies suggest that γ-secretase dampens inflammation and Toll-like receptor 4-mediated immune response via the processing of LRP167, but this pathway was also not a major component of the genes we identified. Chronic γ-secretase inhibition did not appear to induce expression of stress or inflammatory responses in neurons or glia, such as reactive oxygen species-related genes and cytokine-response genes87,88. Given the differences in surface proteins of neurons and glia, the repertoire of γ-secretase targets is likely distinct between neurons and various types of glia. It is therefore plausible that γ-secretase activity results in cell type-specific CTFs and ICDs with separate effects on cellular functions, including gene transcription, and that chronic γ-secretase inhibition induces distinct phenotypes in neurons and glia as a result.
The impairment in synaptic function by chronic γ-secretase inhibition that we observed in human neurons agrees well with previous results obtained in mouse models41,89 and similar impairments induced by mutations of the AD risk gene BIN190, although others have reported a decrease in synapse numbers upon γ-secretase inhibition91. Early studies reported that the PSEN1 deletion in the hippocampus caused no significant change in paired-pulse facilitation, suggesting no change in release probability92, but later studies of the hippocampus with a PSEN1/2 double deletion uncovered a large decrease in release probability41. The differences in results between these studies and our data, which agree with Zhang et al. [2009], may be due to the extent of the suppression of γ-secretase activity or the time period of suppression. The studies by Zhang et al. [2009] and Yu et al. [2001] on mouse hippocampus analyzed different gene dosages at slightly different time points. However, impaired neurotransmitter release in the mouse hippocampus following suppression of γ-secretase by deletion of nicastrin93 supports the general notion that a decrease in γ-secretase activity causes a decrease in presynaptic release probability.
The finding that statins (HMG-CoA reductase inhibitors), which are taken by millions of people worldwide, cause a large effect on neurotransmitter release in human neurons replicates earlier findings in mouse neurons74,76. Generally, statins are not known to impair cognition in people, but there appears to be limited consensus on this subject despite the widespread use of statins. A small number of randomized clinical studies with lovastatin and simvastatin, lipophilic statins that can cross the blood-brain barrier, reported cases of short-term cognitive impairment and mild depressive symptoms that correlated with initiation of statins and that subsided once statins were discontinued94–96. Importantly, a meta-analysis of 25 randomized controlled trials including over 29,000 patients showed no significant adverse cognitive effects due to statin therapy97. Our findings raise the question of why the impairment in neurotransmitter release that we observed upon treatment of neurons with these drugs at therapeutic doses does not affect the cognitive performance of human subjects. Likely, for hydrophilic statins, cognitive side effects are not observed in human subjects due to the blood brain barrier that limits the extent to which these drugs can exert their effects on the brain. More generally for all statins, compensatory neural circuit adjustments by other neuronal subtypes may operate in the human brain. Our data also suggest that chronic statin use upregulates cholesterol synthesis genes in neurons, which may offset any changes in the brain following the initiation of these therapies.
The field of AD has long debated the potential use of statins as a treatment strategy, though there is also no consensus to date on this subject98. Prior comparative studies across 22,000 medical records revealed a significantly decreased likelihood of an AD diagnosis among individuals taking lovastatin and pravastatin for cardiac reasons, as compared to individuals taking non-statin-based cardiac therapies99,100. However, a large randomized controlled trial in the general elderly population detected no effect on cognitive decline when statins were used for 42 months101, although this study was not specifically investigating AD. Therefore, the potential of targeting cholesterol for AD therapeutics remains unclear, despite the substantial evidence for cholesterol as a risk factor for AD with ApoE4 as a prominent risk factor. While our study does not focus on AD modeling, our results provide insights into an emerging link between γ-secretase, typically associated with early-onset AD, and cholesterol, relevant for sporadic AD.
Limitations of our study.
Our findings uncover a mechanism whereby γ-secretase regulates synapse function, namely via control of the neuronal cholesterol content, but our data have not identified which γ-secretase substrates among the hundreds described13 are responsible for controlling cholesterol metabolism. The same limitation applies to the control of synapse numbers by γ-secretase that is independent of cholesterol.
Moreover, our findings do not provide evidence directly related to AD pathogenesis and we do not know how the control of neuronal cholesterol levels by γ-secretase is related to AD, although the prominent involvement of cholesterol metabolism in sporadic AD suggests a strong connection. Prior studies describe that modifying neuronal cholesterol levels can regulate APP cleavage by γ-secretase, tau phosphorylation, and tau aggregation73,102–104. These data suggest possible pathways that connect cholesterol metabolism to AD pathogenesis, but other mechanisms are also plausible. Independent of whether the neuron-specific functions of γ-secretase in cholesterol metabolism are relevant to AD pathogenesis, the control of cholesterol by γ-secretase sheds new light on the role of this fascinating enzyme in neurons.
A different limitation of our study is that it does not address a puzzling clinical observation on a rare genetic skin disease, hidradenitis suppurativa. A subset of patients with this disorder harbors loss-of-function mutations in various γ-secretase subunits but these patients do not appear to suffer from an increased risk for AD105,106. This observation suggests that a decrease in γ-secretase activity does not necessarily lead to AD pathogenesis, although the change in γ-secretase activity caused by the hidradenitis suppurativa mutations may be more modest in neurons than in skin cells and may be qualitatively different. The γ-secretase mutations in hidradenitis suppurativa primarily target NCSTN and PESENEN whereas in familial AD the mutations are observed in PSEN1 or PSEN2, suggesting that various γ-secretase mutations may differentially affect γ-secretase activity in distinct tissues and for diverse substrates.
Furthermore, we validated the pharmacological inhibition of γ-secretase with a genetic PSEN1 deletion, but inhibited cholesterol biosynthesis only pharmacologically and not genetically. Although we used two well-characterized chemically distinct HMG-CoA reductase inhibitors (statins) in our experiments, additional experiments using a CRISPR-mediated deletion of cholesterol synthesis enzymes to confirm the selective action of statins would provide further confidence.
Finally, an additional limitation of our study is that we analyzed a relatively homogeneous population of glutamatergic human neurons produced by trans-differentiation from ES cells with Ngn2 overexpression. It is possible that these neurons are not representative of human glutamatergic neurons in general and may be particularly sensitive to chronic γ-secretase inhibition and statins.
Outlook.
In summary, our data describe a mechanism by which γ-secretase regulates synaptic transmission, namely via its control of neuronal cholesterol levels that in turn determine the presynaptic release probability. We reveal a previously unknown primary function of γ-secretase in maintaining the cholesterol levels of neurons but not glia. Our work demonstrates that decreases in cholesterol levels, produced by suppression of γ-secretase or of cholesterol synthesis, severely impair synaptic transmission. This is a general cell-biological process that may be related to AD pathogenesis, given that cholesterol metabolism is clearly affected by AD-linked genes and by the pathogenesis of AD. The link of γ-secretase to cholesterol metabolism that we identified in the present study connects γ-secretase dysfunction to cholesterol changes and to synaptic dysfunction in AD, opening a new avenue to the identification of AD drug targets.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Thomas C. Südhof (tcs1@stanford.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All data and renewable materials reported in this paper will be shared by the lead contact upon request (lead contact, Thomas C. Südhof (tcs1@stanford.edu)). This paper does not report original code. Bulk RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti Syt1 | Synaptic Systems | Cat# 105-011C2: RRID:AB_2619760 |
| Chicken polyclonal anti MAP2 | Encor | CPCA-MAP2, RRID:AB_2138173 |
| Rabbit polyclonal anti Syn1 | Sudhof Lab | Clone YZ6078, RRID: AB_2861225 |
| Mouse monoclonal anti PSD95 | Thermo | Cat# MA146: RRID:AB_2092361 |
| Rabbit polyclonal anti APP | Ho et al., 2008 | Clone U955 |
| Mouse monoclonal anti APP | Sigma | Cat# MABN380: RRID: AB_2714163 |
| Rabbit polyclonal anti CASK | Neuromab | Cat# O14936: RRID: AB_2877188 |
| Rabbit polyclonal anti Fe65 | Cao and Südhof, 2001 | Clone 4194 |
| Rabbit polyclonal anti Mint1 | Ho et al., 2008 | Clone P730 |
| Rabbit polyclonal anti Nlgn1 | Jahn Monoclonals | Clone 4C12 |
| Rabbit polyclonal anti pan-Nrxn | Sclip and Südhof, 2020 | Clone G394, RRID: AB_2800397 |
| Rabbit polyclonal anti RIM1/2 | Acuna et al., 2015 | Clone U952 |
| Rabbit polyclonal anti Stx1 a/b | Zhou et al., 2012 | Clone 438B |
| Rabbit polyclonal anti PSD95 | Patzke et al., 2019 | Clone L667: RRID:AB_2800396 |
| Mouse monoclonal anti L1CAM | Sigma | Cat# L4543: RRID:AB_609903 |
| Rabbit monoclonal anti APP | Abcam | Cat# Ab32136: RRID: AB_2289606 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Accutase | Innovative Cell | Cat# AT104 |
| Gem21 Neuroplex supplement | Gemini Bio | Cat# 400–160 |
| BDNF | PeproTech | Cat# 450–02 |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| DMEM media | Thermo Fisher | Cat# 11965092 |
| DMEM/F-12 media | Thermo Fisher | Cat# 11320082 |
| HyClone fetal bovine serum (FBS) | GE Healthcare | Cat# SH30071.03 |
| MEM media | Thermo Fisher | Cat# 51200038 |
| Matrigel | BD Biosciences | Cat# 356230 |
| mTeSR1 media | Stem Cell Technologies | Cat# 85850 |
| N-2 supplement | Thermo Fisher | Cat# 17502048 |
| Neurobasal-A Medium | Thermo Fisher | Cat# 10888022 |
| NT-3 | PeproTech | Cat# 450–03 |
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| Thiazovivin | Bio Vision | Cat# 1681 |
| Laminin-1 | Invitrogen | Cat# 23017–015 |
| Trypsin / EDTA | Thermo Fisher | Cat# 25300120 |
| Papain, Suspension | Worthington Biochemical | Cat# LS003127 |
| Cytosine arabinofuranoside (AraC) | Sigma-Aldrich | Cat# C6645 |
| Glutamax | Thermo Fisher | Cat# 35050079 |
| TTX | Fisher Scientific | Cat# 50-753-2807 |
| EDTA | Sigma-Aldrich | Cat# E6758-100G |
| Non-Essential Amino Acids | Thermo Fisher | Cat# 11140076 |
| Human Holotransferrin | Gemini Bio | Cat# 800-131P |
| Total Human Abeta ELISA | Clonetech | Cat# 27729A |
| DMSO, Cell Culture Grade | Sigma | Cat# D2650 |
| DAPT | Tocris | Cat# 2634 |
| LY411575 | Selleckchem | Cat# S2714 |
| Rosuvastatin | Selleckchem | Cat# S2169 |
| Simvastatin | Selleckchem | Cat# S1792 |
| TRIzol | Invitrogen | Cat# 15596026 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74104 |
| Filipin | Sigma | Cat# SAE0088 |
| Amplex Red Cholesterol Kit | Invitrogen | Cat# A12216 |
| Experimental Models: Cell Lines | ||
| Human H1-ESCs | WiCell Research Institute | Cat# WA01 |
| HEK293T cells | ATCC | Cat# CRL-11268 |
| Experimental Models: Organisms/Strains | ||
| CD-1 IGS mouse | Charles River | Cat# Crl:CD1(ICR) |
| Quantitative qRT-PCR | ||
| Human ACAT2 | IDT | Hs.PT.56a.26153626 |
| Human ACTB | IDT | Hs.PT.39a.22214847 |
| Human DBH | IDT | Hs.PT.58.39251730 |
| Human DHCR24 | IDT | Hs.PT.58.4561516 |
| Human DHCR7 | IDT | Hs.PT.58.38763048 |
| Human DUSP4 | IDT | Hs.PT.58.1640850 |
| Human DUSP5 | IDT | Hs.PT.58.1558918 |
| Human EGR1 | IDT | Hs.PT.58.40805543.g |
| Human FDFT1 | IDT | Hs.PT.58.25526506 |
| Human FOS | IDT | Hs.PT.58.15540029 |
| Human HMGCR | IDT | Hs.PT.58.41105492 |
| Human HMGCS1 | IDT | Hs.PT.58.4084870 |
| Human IDI1 | IDT | Hs.PT.58.19303620 |
| Human JUNB | IDT | Hs.PT.58.27174746.g |
| Human LDHA | IDT | Hs.PT.58.22929122 |
| Human LDLR | IDT | Hs.PT.58.2004261 |
| Human LSS | IDT | Hs.PT.58.24976485 |
| Human MAP2 | IDT | Hs.PT.58.20680759 |
| Human MVD | IDT | Hs.PT.58.39035173 |
| Human NAB2 | IDT | Hs.PT.58.24622150 |
| Human NR4A1 | IDT | Hs.PT.58.20779713 |
| Human SCD | IDT | Hs.PT.58.45714389 |
| Human SREBF2 | IDT | Hs.PT.58.45335433 |
| Human SV2C | IDT | Hs.PT.58.4640250 |
| Human TH | IDT | Hs.PT.58.369742 |
| Human TMEM97 | IDT | Hs.PT.58.40917738 |
| Human VEGFA | IDT | Hs.PT.58.1149801 |
| Human VGF | IDT | Hs.PT.58.26072166 |
| Recombinant DNA | ||
| FUW-TetO-Ng2-T2A-puromycin | Zhang et al., 2013 | 52047 Addgene (lentiviral vector) |
| FUW-rtTA | Zhang et al., 2013 | 20342 Addgene (lentiviral vector) |
| FUW-TetO-EGFP | Vierbuchen et al., 2010 | 30130 Addgene (lentiviral vector) |
| pREV | Dull et al., 1998 | 12253 Addgene (lentiviral helper vector) |
| pRRE | Dull et al., 1998 | 12251 Addgene (lentiviral helper vector) |
| pVSVg | Patzke et al., 2019 | 12259 Addgene (lentiviral helper vector) |
| Deposited Datasets | ||
| Bulk RNA-seq data | GEO | GSE206102 |
| Software and Algorithms | ||
| ImageJ | NIH | RRID: SCR_003070 |
| ImageStudio software | Li-Cor | RRID:SCR_013715 |
| pClamp | Molecular Devices | RRID:SCR_011323 |
| Nikon NIS-Elements | Nikon | RRID:SCR_014329 |
| GraphPad Prism | GraphPad Prism | RRID:SCR_002798 |
| RStudio | RStudio Team | RRID:SCR_000432 |
| DESeq2 | Bioconductor (Love et al., 2014) | RRID:SCR_015687 |
EXPERIMENTAL MODEL
ESC Culture.
Male human embryonic stem cells (ESC), line H1, were obtained from WiCell (line WA01). The stem cells were maintained feeder-free in mTeSR1 medium (Stem Cell Technologies), from frozen cell stocks of passage 50. With each passage, cells were detached with Accutase at 37°C, centrifuged and resuspended in mTeSR1 with 2 μM thiazovivin (BioVision), and then replated on Matrigel-coated 6-well plates. The Stem Cell Research Oversight (SCRO) at Stanford University approved the protocols used in this work (SCRO 518).
Mouse Lines.
CD1 wild type female and male pups were used between postnatal days 0 to 2 for primary astrocyte cultures. All experiments were approved by Stanford University’s Administrative Panel on Laboratory Animal Care (APLAC). All mice were housed in the Stanford SIM1 animal facility under supervision of the Stanford animal care unit; all mice were healthy and were not involved in previous experiments.
HEK293T Cell Lines.
HEK293T cells were purchased from ATCC and low-passage cells were expanded and stored. HEK293T cells below passage 20 were cultured in DMEM + 10% FBS and reached confluency every 3 days, at which point they were passaged (1:20 split ratio).
METHOD DETAILS
Gene Targeting for Generating PSEN1 KO ESC Lines.
Three single-guide RNAs (sgRNAs) were designed for PSEN1 (purchased from Synthego). The sgRNA sequences were (5’ to 3’): CACAACGACAGACGGAGCCT, CCAGGGTAACTCCCGGCAGG, CCCTGTGACTCTCTGCATGG. The three sgRNAs were combined with 20 μM recombinant Cas9 (IDT), and Lonza nucleofector solution (P3 4D Nucleofector X kit S, Lonza). The Ribonucleoprotein (RNP) complexes were incubated for 10 minutes at room temperature. H1 ESCs were detached with Accutase, centrifuged, and resuspended in mTeSR1 with thiazovivin; 4.5e5 H1 cells were used for nucleofection with the RNP complexes, per nucleofector cuvette well. Following nucleofection, cells were transferred very gently to 6-well plates with pre-warmed media of mTeSR1 and thiazovivin (2 μM). Once confluent, part of the well was kept for genomic DNA extraction (QuickExtract DNA solution, Lucigen) and Sanger Sequencing to determine the population knockout score, while the remaining cells were plated at 1,000 cells per 6-well plate. When these wells had small and well-defined rounded colonies, the colonies were picked and further expanded prior to freezing. With subsequent genotyping and validation, three PSEN1 KO ESC cell lines (KO A, B, D) with frameshift mutations and 100% knockout scores (ICE Analysis V2, Synthego) were used for experiments.
Glial Cell Culture.
Mouse glia were prepared from cortices of newborn CD1 pups, dissected in HBS on postnatal days 0 to 2 (P0–2). The cortex was digested with 80 μl of papain in 5 ml for 20 minutes at 37°C. Tissues were washed 3 times with DMEM + 10% fetal bovine serum (FBS), then harshly triturated and plated in T75 cell culture flasks with 12 ml of DMEM + 10% FBS media. Once 90% confluent, approximately 7 days after dissection, glial cultures were detached with 0.05% trypsin and replated in 3 new T75 flasks (1:3 split ratio) in DMEM + 10% FBS. The replating step and media prevent mouse neurons from surviving. No antibiotics were used in these cultures.
Plasmids.
Lentiviral packaging plasmids Rev, RRE, and VSV were previously described in Zhang et al. (2013). The Ngn2 overexpression plasmids were FUW-TetO-Ngn2-T2A-puromycin or FUW-TetO-Ngn2-P2A-GFP-T2A-puromycin56. The FUGW-GFP plasmid was a gift from David Baltimore (Addgene plasmid #14883). The tdTomato overexpression used the pCAG-tdTomato107.
Virus Generation.
Transfection for lentivirus production was performed in HEK293T cells at 70% confluence on the day of transfection, using calcium phosphate and HBS (Takara). In total, 12 μg of lentiviral packaging DNA were transfected per T75 flask of HEK293T cells: Rev (4 μg), RRE (8 μg), and VSV (6 μg). 48 hours after transfection, cell media was harvested and centrifuged at 19,000g for 2 hours. Pellets were resuspended overnight at 4°C in 100 μl of DMEM, aliquoted and frozen at −80°C. For each 6-well plate, 3 μl of each lentivirus were used.
Generation of iN Cells and Pharmacological Treatments.
H1 ESCs were directly reprogrammed into iN as previously described56. In summary, H1 ESCs were plated in mTeSR1 at a ratio of 1:18 and infected with two lentiviruses, TetO-Puro-Ngn2 and rtTA (prepared as described above) in mTeSR1 media with thiazovivin. For a subset of experiments requiring green fluorescent labeling of neurons, a third lentivirus carrying GFP was added at the time of infection. On day 0, Ngn2 expression was induced with doxycycline in the induction medium containing: DMEM-F12, doxycycline (2 μg/ml, Sigma), Non-Essential Amino Acids, BDNF (10 ng/ml, PeproTech), human NT3 (10 ng/ml, PeproTech), mouse Laminin-1 (0.2 μg/ml, PeproTech). On day 1 and 2, puromycin (1 μg/ml) was added to the induction medium. On day 3, iN were dissociated from the 6-well plates in which they were induced using Accutase, and were replated on Matrigel-coated coverslips in 24-well plates. The media contained Neurobasal with B27 (Gemini21), doxycycline, BDNF, NT3, Laminin-1, and Glutamax. Glia were simultaneously detached (as previously described), resuspended in the same Neurobasal-based media with the neurons, and the human neuron-mouse glia cell mixture was replated in each well with a Matrigel-coated coverslip. Half-media changes were performed with the Neurobasal-based media every 2 days until day 10 in vitro.
On day 10, cells were transitioned to Growth Medium: 900 ml MEM, 100 mg transferrin, 50 ml FBS, 25 ml of 20% D-Glucose, 2.5 ml Glutamax, 2.5 ml of 8% NaHCO3, 20 ml of B27 (Gemini21), doxycycline (2 μg/ml) and 4AraC (2 μM). Between day 10 and the experimental day, half-media changes were performed once a week. If iN were made from PSEN1 knockout ESCs (previously described), no additional components were added to the media starting on day 10. For iN made from wild-type ESCs for experiments with γ-secretase inhibition, the co-cultures were treated with vehicle (Cell Culture-Grade DMSO, Sigma, 0.5%) or with the GSIs DAPT (40 μM, Tocris) or LY411575 (2.5 μM, SelleckChem) starting day 10. The treatments were subsequently provided in the weekly media changes. For co-culture experiments treated with both GSIs and statins, rosuvastatin (1 μM, SelleckChem) or simvastatin (1 μM, SelleckChem) was added starting day 10, and treated weekly until the experimental day. Unless noted otherwise, experiments in this work were performed at days 37 to 42 in vitro.
Immunofluorescence.
Human iN were washed 3 times with PBS and fixed with 4% PFA and 4% sucrose in PBS for 15 minutes at room temperature. Cells were then washed three times with PBS, followed by permeabiliziation with 0.1% Triton-X-100 for 15 minutes at room temperature. Primary antibodies used for immunocytochemistry were: rabbit polyclonal anti-Synapsin (Südhof Lab, YZ6078 RRID: AB_2920537, 1:1000 dilution), mouse monoclonal anti-PSD95 (Thermo, 7E3–1B8, 1:250 dilution), chicken polyclonal anti-MAP2 (Encor, CPCA-MAP2, 1:1000 dilution), mouse monoclonal anti-L1CAM (Sigma, clone UJ 127.11, 1:300 dilution). For synapsin-only staining, blocking was performed in 5% goat serum (Sigma); for synapsin and PSD95 co-labeling, blocking was performed in 1% goat serum with 4% BSA for one hour at room temperature. Incubation with primary antibodies was performed overnight at 4°C in the corresponding blocking buffers. Cells were washed three times with PBS and incubated for one hour at room temperature with Alexa conjugated secondary antibodies: goat anti-chicken Alexa 647, goat anti-rabbit Alexa 488, and goat anti-mouse Alexa 546, all 1:500. Coverslips were washed once with PBS and once with water to remove salts before mounting, then were mounted using Fluoromount (with DAPI) on glass slides. All images were taking with the Nikon A1RSi confocal microscope at 60x for analysis of synaptic puncta using Nikon Analysis Software. For L1CAM staining, live cells were incubated with L1CAM antibody (mouse, Millipore-Sigma L4543, 1:500) at 37°C for 15 minutes, then fixed and incubated with goat anti-mouse Alexa 546 (1:500). Permeabilization (as described above) was only performed on these L1CAM-stained coverslips if co-labeling with another antibody was needed.
Cell Morphology and Tracing.
Human iN co-cultured with mouse glia were sparsely transfected with 0.5 μg of TdTomato per well of a 24-well plate. Transfections were performed using 2M calcium phosphate and HBS on day in vitro 19. Two days later (day 21), cells were fixed using 4% paraformaldehyde and 4% sucrose in PBS, for 15 minutes at room temperature, and mounted on glass slides using Fluoromount with DAPI. Images were taken with the Nikon A1RSi confocal microscope on 20x magnification, with stitching to follow isolated axons and dendrites. Axons and dendrites were traced using Fiji software.
Immunoblotting and Protein Quantification.
Neurons were lysed in ice-cold RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton-X-100, plus protease inhibitors (Roche). Lysates were then centrifuged at 14,000 g for 30 minutes. Protein levels were measured by BCA (Pierce) per the manufacturer’s instructions, or loaded on stain-free 10% gels (Bio-Rad), along with standards of known concentrations. The supernatant was diluted in 4X Laemmli Buffer (Bio-Rad) with beta-mercapto-ethanol. Lysates were analyzed by SDS-PAGE on 4–20% polyacrylamide Criterion gels (Bio-Rad), followed by semi-dry transfer for mixed molecular weights. Blocking was performed using 5% BSA in TBST at room temperature. Primary antibodies used were: rabbit polyclonal anti-APP (Südhof Lab, U955, 1:1000 dilution); mouse monoclonal anti-APP (Millipore, MABN380, 1:1000 dilution); rabbit monoclonal anti-APP (Abcam, Y188, 1:1000 dilution); rabbit polyclonal anti-CASK (Neuromab, clone K56A/50, 1:1000 dilution); rabbit polyclonal anti-Fe65 (Südhof Lab, 1:1000 dilution); rabbit polyclonal anti-Mint1 (Südhof Lab, P730, 1:1000 dilution); rabbit polyclonal anti-Nlgn1 (Südhof Lab, K138, 1:500), rabbit polyclonal anti-pan-Neurexin (Südhof Lab, G394, 1:1000 dilution); rabbit polyclonal anti-PSD-95 (Südhof Lab, L667, 1:1000 dilution); rabbit polyclonal anti-RIM1/2 (Südhof Lab, 1:1000 dilution); rabbit polyclonal anti-synapsin (Südhof Lab, YZ6078, 1:1000 dilution); mouse monoclonal anti-Synaptotagmin-1 (Synaptic Systems, clone 41.1, 1:1000 dilution); rabbit polyclonal anti-Syntaxin-1a/b (Südhof Lab, 438B, 1:1000 dilution). Primary antibodies were diluted in 5% BSA in TBST and incubated overnight at 4°C. Following 3 washes with TBST, membranes were incubated with fluorescently labeled secondary antibodies (800CW and 680LT) for one hour at room temperature in 5% BSA in TBST. Signals were normalized for Tuj1 (mouse, Biolegend, 1:2,500). Blots were imaged using the Odyssey Infrared Imager CLX and analyzed using Image Study 5.2.5. (LI-COR Biosciences).
ELISA.
Conditioned culture media was harvested on days in vitro 8 (baseline, prior to γ-secretase inhibitor treatment), 14, 18, and 21 to measure the secreted total Ab content. Media was stored at −20°C until usage, to allow for all timepoints to be measured on the same ELISA plate. Once thawed, media was loaded in the ELISA plates and the wash steps were performed per the manufacturer’s instructions for total human Aβ (Clonetech, #27729A). Absorbance was measured using the Apollo LB915 (Berthold Technologies), with background subtraction prior to analysis.
Electrophysiology.
Whole-cell patch-clamp electrophysiology was performed on coverslips of human iN grown on mouse glia on days 37 to 42 (unless specified for day 21). For all experiments, coverslips were mounted in a recording chamber with an inverted phase-contrast microscope (Nikon) with DIC. Extracellular bath solution contained: 145 mM NaCl, 5 mM KCl, 2 mM MgCl2, 10 mM HEPES, 3 mM CaCl2, 10 mM glucose monohydrate, pH 7.4 and osmolarity 305 mOsm. Borosilicate glass pipettes were prepared from the Narishige PC-10 puller (Japan), with resistances of 3–3.5 MΩ. For voltage-clamp recordings, the recording pipette was filled with internal solution containing: 135 mM CsCl, 5 mM NaCl, 4 mM ATP-Magnesium, 10 mM HEPES, 0.3 mM GTP-Sodium, 5 mM EGTA, 5 mM QX314-Bromide, pH 7.2 and osmolarity 300 mOsm. For current-clamp recordings, the recording pipette contained internal solution with: 123 mM K-gluconate, 10 mM KCl, 1 mM MgCl2, 10 mM HEPES, 1 mM EGTA, 1.5 ATP-Magnesium, 0.3 mM GTP-Sodium, 5 mM glucose, pH 7.2 and osmolarity 300 mOsm. Electrical signals were recorded with a two-channel Axoclamp 700B amplifier (Axon Instruments), digitalized with a Digidata 1440 digitizer (Molecular Devices), controlled under Clampex 10.1 software. Whole-cell capacitance, membrane resistance, and series resistance were recorded and compensated more than 80%. All recordings were performed at room temperature.
Neurons were maintained at −70 mV holding potentials for whole-cell voltage-clamp recordings. To record miniature excitatory postsynaptic currents (mEPSCs), TTX (0.5 μM) was applied to the bath solution. To record evoked excitatory postsynaptic currents (eEPSC) or paired-pulse recordings, extracellular stimulation was achieved with a bipolar electrode (WPI) placed 200 μm away from patched cells. Square pulses (100 μs) of voltage were applied via a stimulus isolation unit (Model 2100 Isolated Pulse Stimulator, A-M Systems). Single evoked currents were recorded at −70 mV holding potentials in voltage clamp, and with 10 sweeps per recording. Coefficients of variation were calculated from the standard deviation of the amplitudes, divided by the mean EPSC amplitudes of each cell. For paired-pulse recordings, two identical stimuli were applied in succession, with interstimulus intervals (ISIs) of 20 ms, 50 ms, 100 ms, 200 ms, 500 ms, and 1000 ms between stimuli. The first response amplitude and coefficients of variation were measured for recordings with ISIs of 20 ms. At each ISI, 3 sweeps were recorded. The ratio between peak amplitude of the second and the first responses were computed to obtain the Paired Pulse Ratio (PPR). To trigger action potentials, cells were held in current clamp near −70 mV resting potential. The recording Current steps were injected through the recording pipette over 1 second in 10 pA increments from −20 pA to 240 pA.
RNA Sequencing.
Co-cultures of human neurons and mouse glia (day 40) in 6-well plates were lysed using TRIzol (Invitrogen). Samples were homogenized in TRIzol, then mixed with chloroform to achieve phase-separation. The upper layer containing RNA was loaded onto columns of the RNeasy Plus Mini Kit (Qiagen) and purified per manufacturer’s instructions. RNA was eluted in 30 ul of RNAse-free water (Invitrogen) and the concentration was measured with the NanoDrop 1000 Spectrophotometer (ThermoFisher). For RNAseq experiments, RNA concentration and integrity was verified independently at the Stanford PAN Facility; all RNA integrity numbers were greater than 9. RNA was stored at −80°C and only thawed once for either RNAseq or cDNA synthesis.
Bulk RNAseq was performed for a total of 6 biological replicates. Illumina TruSeq Stranded mRNA was used for library preparation, and sequencing was performed with the Illumina NovaSeq with 100 bp paired-end reads. Sequencing depth was 40 million total reads (20 million paired). Raw data was processed using Kallisto for pseudo-alignment of the reads to a chimeric mouse and human transcriptome, as described in Bray et al. (2016) and used in Marro et al. (2019). The R package DESeq2 was used to analyze differential gene expression108. The DESeq data set included the matrix of raw, un-normalized counts. The DESeq2 model internally corrected for library size. The mean expected counts of each gene within each group were adjusted by normalization factors. Batch effects were controlled for in the DESeq2 data set by using “batch” and “condition” variables. The “results” function filtered the mean normalized counts, followed by the Log2FoldChange (LFC) shrinkage function to ensure that the largest fold changes were not due to genes with unreasonably low counts. P-values were also corrected for low counts using the Benjamini-Hochberg correction. The False Discovery Rate (FDR) threshold for adjusted p-values was set to alpha < 0.1, and the adjusted p-value threshold used to determine significance was 0.05. The log2(Fold Change) threshold was greater than 0.25 for upregulated DEGs and was less than −0.25 for downregulated DEGs; this corresponded to an approximately 20% increase or decrease in gene expression, respectively. The top 20 genes across both DAPT and LY411575 groups, relative to vehicle, were determined first by significance of the adjusted p-value, followed by the log2(Fold Change) cutoff. Volcano plots were generated using the VolcaNoseR package and the “EnhancedVolcano” package in R Studio.
Quantitative RT-PCR.
Total RNA was extracted as described above, at day in vitro 40 following acute or chronic GSI treatment. qRT-PCR measurements were performed on cDNA synthesized from RNA (RNA to cDNA kit, ThermoFisher). The IDT 5X Master Mix was used along with PrimeTime qPCR probe assays from IDT, which consist of two primers and one FAM-labeled ZEN/IBFQ-quenched 5’ nuclease probe. The PrimeTime qPCR primer-probes were human-specific probes for: ACAT2 (Hs.PT.56a.26153626); ACTB (Hs.PT.39a.22214847); DBH (Hs.PT.58.39251730); DHCR24 (Hs.PT.58.4561516); DHCR7 (Hs.PT.58.38763048); DUSP4 (Hs.PT.58.1640850); DUSP5 (Hs.PT.58.1558918); EGR1 (Hs.PT.58.40805543.g); FDFT1 (Hs.PT.58.25526506); FOS (Hs.PT.58.15540029); HMGCR (Hs.PT.58.41105492); HMGCS1 (Hs.PT.58.4084870); IDI1 (Hs.PT.58.19303620); JUNB (Hs.PT.58.27174746.g); LDHA (Hs.PT.58.22929122); LDLR (Hs.PT.58.2004261); LSS (Hs.PT.58.24976485); MAP2 (Hs.PT.58.20680759); MVD (Hs.PT.58.39035173); NAB2 (Hs.PT.58.24622150); NR4A1 (Hs.PT.58.20779713); SCD (Hs.PT.58.45714389); SREBF2 (Hs.PT.58.45335433); SV2C (Hs.PT.58.4640250); TH (Hs.PT.58.369742); TMEM97 (Hs.PT.58.40917738); VEGFA (Hs.PT.58.1149801); VGF (Hs.PT.58.26072166). qRT-PCR was performed with the Applied Biosystems 7900HT machine. Both ACTB and MAP2 were used as endogenous controls (reference genes) in every qPCR experiment. Data were normalized to the mean of ACTB and MAP2 using the 2−ΔΔCt method. Ct values greater than 35 were omitted.
Filipin Staining.
Coverslips with GFP-positive human neurons grown on GFP-negative mouse glia were incubated with L1CAM primary antibody for 15 minutes at 37°C, as described above, to live-stain the axons of human neurons. Filipin stock solution (Sigma, 1 mg/ml) was diluted to a final concentration of 0.05 mg/ml in PBS with 5% goat serum. Coverslips were then rinsed three times with PBS at room temperature, and then were fixed with 4% paraformaldehyde and 4% sucrose in PBS. Coverslips were not permeabilized to avoid loss of free cholesterol with detergent use. Coverslips were incubated with glycine (1.5 mg/ml) in PBS for 10 minutes at room temperature to quench paraformaldehyde. Diluted filipin was added to coverslips for 2 hours at room temperature, in the dark. Cells were rinsed once with PBS and once briefly with water to remove salts before mounting with Fluoromount (no DAPI). Images were taken with the Nikon A1RSi confocal microscope on 60x magnification, with NyQuist zoom. Nikon Analysis software was used to measure Filipin fluorescence intensity within the GFP-positive (neuronal) area.
FACS.
Co-cultures of GFP-positive human iN and GFP-negative mouse glia were dissociated with Accutase for 20 minutes at 37°C on day in vitro 40, in order to ensure single-cell suspensions. The Accutase was quenched with Neurobasal cell media. The dissociated cells were passed through 100 μm filters, and were then sorted using a BD Bioscience FACS Aria II SORP flow cytometer. GFP-negative human iN were used as a negative control in order to set the gating scheme for isolation of GFP-negative and GFP-positive cell populations. Gating was conducted using BD Biosciences FACS Diva Software and was further analyzed using FlowJo flow cytometry analysis software. Cells were sorted into individual tubes containing Neurobasal media with supplements (as described above). Sorted cells were then centrifuged at 1.4 rpm for 5 minutes to obtain cell pellets of human iN or of mouse glia, which were then resuspended in 1X reaction buffer of the Amplex Red Cholesterol Kit (Invitrogen, A12216).
Amplex Red Cholesterol Quantifications.
Sorted human neurons and sorted mouse glia from co-cultures at day 40 were lysed for cholesterol analysis by sonication in 1X reaction buffer of the Amplex Red Cholesterol Assay Kit (Invitrogen, A12216). The homogenized mixture underwent snap-freezing and was subjected to enzymatic reactions per the Amplex kit protocol, as previously described (2). Cholesterol esterase was omitted for the free cholesterol measurements with the same kit. Measurements were normalized using cholesterol standards of known concentrations, as well as Resorufin standards, supplied in the Amplex kit. Fluorescence was measured using the Tecan Infinite M1000 plate reader at the Stanford High-Throughput Bioscience Center.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results are reported as means with SEM, with the number of cells and number of biological replicates reported on each graph or in the figure legend. Statistical analyses were performed with GraphPad Prism 9. Statistical tests were performed using unpaired two-tailed Student’s t-tests for two groups, one-way ANOVA with Dunnett’s multiple comparison tests for multiple groups, or two-way ANOVA with Bonferroni’s multiple comparison tests for multiple groups with two variables (* p < 0.05, ** p < 0.01, *** p < 0.001). RNA-seq statistical analysis were performed using DESeq2108, using the Wald test and are described in the “RNA Sequencing” section of the Method Details.
Supplementary Material
Highlights:
Pharmacologic and genetic suppression of γ-secretase decrease synaptic transmission
Chronic γ-secretase inhibition decreases cholesterol levels in neurons but not glia
Regulation of synaptic release probability by γ-secretase is cholesterol-dependent
γ-Secretase suppression increases synapse numbers independently of cholesterol
ACKNOWLEDGEMENTS
We thank Drs. Justin Trotter, Christopher Patzke, Jinye Dai, Jinzhao Wang, Francisco Galdos and ChuChu Wang for advice. This study was supported by grants from the National Institute of Aging (AG070919 and AG048131 to TCS) and a predoctoral fellowship from the National Institutes of Health (5F30AG064819 to SEP). SEP was also supported by Stanford University Medical Scientist Training Program Grants T32-GM007365 and T32-GM145402.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare that they have no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Cao X, and Südhof TC (2001). A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120. 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 2.Johnson DS, Li Y-M, Pettersson M, and St George-Hyslop PH (2017). Structural and Chemical Biology of Presenilin Complexes. Cold Spring Harb. Perspect. Med 7, a024067. 10.1101/cshperspect.a024067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steiner H, Fluhrer R, and Haass C (2008). Intramembrane proteolysis by gamma-secretase. J. Biol. Chem 283, 29627–29631. 10.1074/jbc.R800010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jurisch-Yaksi N, Sannerud R, and Annaert W (2013). A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta 1828, 2815–2827. 10.1016/j.bbamem.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Wolfe MS (2020). Unraveling the complexity of γ-secretase. Semin. Cell Dev. Biol 105, 3–11. 10.1016/j.semcdb.2020.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Annaert W, Cupers P, Saftig P, and De Strooper B (2000). Presenilin function in APP processing. Ann. N. Y. Acad. Sci 920, 158–164. 10.1111/j.1749-6632.2000.tb06917.x. [DOI] [PubMed] [Google Scholar]
- 7.St George-Hyslop PH (2000). Molecular genetics of Alzheimer’s disease. Biol. Psychiatry 47, 183–199. 10.1016/s0006-3223(99)00301-7. [DOI] [PubMed] [Google Scholar]
- 8.Tanzi RE, and Bertram L (2001). New frontiers in Alzheimer’s disease genetics. Neuron 32, 181–184. 10.1016/s0896-6273(01)00476-7. [DOI] [PubMed] [Google Scholar]
- 9.Tcw J, and Goate AM (2017). Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med 7, a024539. 10.1101/cshperspect.a024539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardy J, and Escott-Price V (2019). Genes, pathways and risk prediction in Alzheimer’s disease. Hum. Mol. Genet 28, R235–R240. 10.1093/hmg/ddz163. [DOI] [PubMed] [Google Scholar]
- 11.Neuner SM, Tcw J, and Goate AM (2020). Genetic architecture of Alzheimer’s disease. Neurobiol. Dis 143, 104976. 10.1016/j.nbd.2020.104976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sierksma A, Escott-Price V, and De Strooper B (2020). Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science 370, 61–66. 10.1126/science.abb8575. [DOI] [PubMed] [Google Scholar]
- 13.Güner G, and Lichtenthaler SF (2020). The substrate repertoire of γ-secretase/presenilin. Semin. Cell Dev. Biol 105, 27–42. 10.1016/j.semcdb.2020.05.019. [DOI] [PubMed] [Google Scholar]
- 14.Ehebauer M, Hayward P, and Martinez-Arias A (2006). Notch signaling pathway. Sci. STKE Signal Transduct. Knowl. Environ 2006, cm7. 10.1126/stke.3642006cm7. [DOI] [PubMed] [Google Scholar]
- 15.Bray SJ, and Gomez-Lamarca M (2018). Notch after cleavage. Curr. Opin. Cell Biol 51, 103–109. 10.1016/j.ceb.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Hunter GL, and Giniger E (2020). Phosphorylation and Proteolytic Cleavage of Notch in Canonical and Noncanonical Notch Signaling. Adv. Exp. Med. Biol 1227, 51–68. 10.1007/978-3-030-36422-9_4. [DOI] [PubMed] [Google Scholar]
- 17.Bai G, and Pfaff SL (2011). Protease regulation: the Yin and Yang of neural development and disease. Neuron 72, 9–21. 10.1016/j.neuron.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Tetering G, and Vooijs M (2011). Proteolytic cleavage of Notch: “HIT and RUN.” Curr. Mol. Med 11, 255–269. 10.2174/156652411795677972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Escamilla-Ayala A, Wouters R, Sannerud R, and Annaert W (2020). Contribution of the Presenilins in the cell biology, structure and function of γ-secretase. Semin. Cell Dev. Biol 105, 12–26. 10.1016/j.semcdb.2020.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Nie P, Vartak A, and Li Y-M (2020). γ-Secretase inhibitors and modulators: Mechanistic insights into the function and regulation of γ-Secretase. Semin. Cell Dev. Biol 105, 43–53. 10.1016/j.semcdb.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou R, Yang G, and Shi Y (2020). Macromolecular complex in recognition and proteolysis of amyloid precursor protein in Alzheimer’s disease. Curr. Opin. Struct. Biol 61, 1–8. 10.1016/j.sbi.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Trambauer J, Fukumori A, and Steiner H (2020). Pathogenic Aβ generation in familial Alzheimer’s disease: novel mechanistic insights and therapeutic implications. Curr. Opin. Neurobiol 61, 73–81. 10.1016/j.conb.2020.01.011. [DOI] [PubMed] [Google Scholar]
- 23.Hampel H, Hardy J, Blennow K, Chen C, Perry G, Kim SH, Villemagne VL, Aisen P, Vendruscolo M, Iwatsubo T, et al. (2021). The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 26, 5481–5503. 10.1038/s41380-021-01249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palop JJ, and Mucke L (2009). Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol 66, 435–440. 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, Cummings J, and van der Flier WM (2021). Alzheimer’s disease. Lancet Lond. Engl 397, 1577–1590. 10.1016/S0140-6736(20)32205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zott B, and Konnerth A (2022). Impairments of glutamatergic synaptic transmission in Alzheimer’s disease. Semin. Cell Dev. Biol, S1084–9521(22)00080–5. 10.1016/j.semcdb.2022.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Fu W-Y, and Ip NY (2023). The role of genetic risk factors of Alzheimer’s disease in synaptic dysfunction. Semin. Cell Dev. Biol 139, 3–12. 10.1016/j.semcdb.2022.07.011. [DOI] [PubMed] [Google Scholar]
- 28.Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, et al. (2022). New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet 54, 412–436. 10.1038/s41588-022-01024-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagisetty Y, Bourquard T, Al-Ramahi I, Mangleburg CG, Mota S, Soleimani S, Shulman JM, Botas J, Lee K, and Lichtarge O (2022). Identification of risk genes for Alzheimer’s disease by gene embedding. Cell Genomics 2, 100162. 10.1016/j.xgen.2022.100162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen DV, Hanson JE, and Sheng M (2018). Microglia in Alzheimer’s disease. J. Cell Biol 217, 459–472. 10.1083/jcb.201709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Y, Ulland TK, and Colonna M (2018). TREM2-Dependent Effects on Microglia in Alzheimer’s Disease. Front. Aging Neurosci 10, 202. 10.3389/fnagi.2018.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griciuc A, and Tanzi RE (2021). The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol 34, 228–236. 10.1097/WCO.0000000000000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou X, Fu AK, and Ip NY (2021). APOE signaling in neurodegenerative diseases: an integrative approach targeting APOE coding and noncoding variants for disease intervention. Curr. Opin. Neurobiol 69, 58–67. 10.1016/j.conb.2021.02.001. [DOI] [PubMed] [Google Scholar]
- 34.Martens YA, Zhao N, Liu C-C, Kanekiyo T, Yang AJ, Goate AM, Holtzman DM, and Bu G (2022). ApoE Cascade Hypothesis in the pathogenesis of Alzheimer’s disease and related dementias. Neuron 110, 1304–1317. 10.1016/j.neuron.2022.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y-WA, Zhou B, Wernig M, and Südhof TC (2017). ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell 168, 427–441.e21. 10.1016/j.cell.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lane-Donovan C, and Herz J (2017). ApoE, ApoE Receptors, and the Synapse in Alzheimer’s Disease. Trends Endocrinol. Metab. TEM 28, 273–284. 10.1016/j.tem.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paasila PJ, Aramideh JA, Sutherland GT, and Graeber MB (2022). Synapses, Microglia, and Lipids in Alzheimer’s Disease. Front. Neurosci 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szabo MP, Mishra S, Knupp A, and Young JE (2022). The role of Alzheimer’s disease risk genes in endolysosomal pathways. Neurobiol. Dis 162, 105576. 10.1016/j.nbd.2021.105576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Priller C, Dewachter I, Vassallo N, Paluch S, Pace C, Kretzschmar HA, Van Leuven F, and Herms J (2007). Mutant presenilin 1 alters synaptic transmission in cultured hippocampal neurons. J. Biol. Chem 282, 1119–1127. 10.1074/jbc.M605066200. [DOI] [PubMed] [Google Scholar]
- 40.Tabuchi K, Chen G, Südhof TC, and Shen J (2009). Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci 29, 7290–7301. 10.1523/JNEUROSCI.1320-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Südhof TC, and Shen J (2009). Presenilins are essential for regulating neurotransmitter release. Nature 460, 632–636. 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang D, Zhang C, Ho A, Kirkwood A, Südhof TC, and Shen J (2010). Inactivation of presenilins causes pre-synaptic impairment prior to post-synaptic dysfunction. J. Neurochem 115, 1215–1221. 10.1111/j.1471-4159.2010.07011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xuan Z, Barthet G, Shioi J, Xu J, Georgakopoulos A, Bruban J, and Robakis NK (2013). Presenilin-1/γ-secretase controls glutamate release, tyrosine phosphorylation, and surface expression of N-methyl-D-aspartate receptor (NMDAR) subunit GluN2B. J. Biol. Chem 288, 30495–30501. 10.1074/jbc.M113.499004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fazzari P, Snellinx A, Sabanov V, Ahmed T, Serneels L, Gartner A, Shariati SAM, Balschun D, and De Strooper B (2014). Cell autonomous regulation of hippocampal circuitry via Aph1b-γ-secretase/neuregulin 1 signalling. eLife 3, e02196. 10.7554/eLife.02196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelleher RJ, and Shen J (2017). Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 114, 629–631. 10.1073/pnas.1619574114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou R, Yang G, and Shi Y (2017). Dominant negative effect of the loss-of-function γ-secretase mutants on the wild-type enzyme through heterooligomerization. Proc. Natl. Acad. Sci. U. S. A 114, 12731–12736. 10.1073/pnas.1713605114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, and Kelleher RJ (2015). Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 85, 967–981. 10.1016/j.neuron.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee S-F, Hao Y-H, Serneels L, De Strooper B, Yu G, and Bezprozvanny I (2006). Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126, 981–993. 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nelson O, Supnet C, Liu H, and Bezprozvanny I (2010). Familial Alzheimer’s disease mutations in presenilins: effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes. J. Alzheimers Dis. JAD 21, 781–793. 10.3233/JAD-2010-100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou B, Lu JG, Siddu A, Wernig M, and Südhof TC (2022). Synaptogenic effect of APP-Swedish mutation in familial Alzheimer’s disease. Sci. Transl. Med 14, eabn9380. 10.1126/scitranslmed.abn9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gutierrez E, Lütjohann D, Kerksiek A, Fabiano M, Oikawa N, Kuerschner L, Thiele C, and Walter J (2020). Importance of γ-secretase in the regulation of liver X receptor and cellular lipid metabolism. Life Sci. Alliance 3, e201900521. 10.26508/lsa.201900521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tamboli IY, Prager K, Thal DR, Thelen KM, Dewachter I, Pietrzik CU, St George-Hyslop P, Sisodia SS, De Strooper B, Heneka MT, et al. (2008). Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J. Neurosci. Off. J. Soc. Neurosci 28, 12097–12106. 10.1523/JNEUROSCI.2635-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grimm MOW, Grimm HS, Pätzold AJ, Zinser EG, Halonen R, Duering M, Tschäpe JA, De Strooper B, Müller U, Shen J, et al. (2005). Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat. Cell Biol 7, 1118–1123. 10.1038/ncb1313. [DOI] [PubMed] [Google Scholar]
- 54.Lütjohann D (2006). Cholesterol metabolism in the brain: importance of 24S-hydroxylation. Acta Neurol. Scand. Suppl 185, 33–42. 10.1111/j.1600-0404.2006.00683.x. [DOI] [PubMed] [Google Scholar]
- 55.Moutinho M, Nunes MJ, and Rodrigues E (2016). Cholesterol 24-hydroxylase: Brain cholesterol metabolism and beyond. Biochim. Biophys. Acta 1861, 1911–1920. 10.1016/j.bbalip.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pak C, Danko T, Zhang Y, Aoto J, Anderson G, Maxeiner S, Yi F, Wernig M, and Südhof TC (2015). Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell 17, 316–328. 10.1016/j.stem.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang G, Zhou R, Guo X, Yan C, Lei J, and Shi Y (2021). Structural basis of γ-secretase inhibition and modulation by small molecule drugs. Cell 184, 521–533.e14. 10.1016/j.cell.2020.11.049. [DOI] [PubMed] [Google Scholar]
- 59.Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, et al. (2001). Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J. Neurochem 76, 173–181. 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- 60.Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, Engstrom L, Pinzon-Ortiz M, Fine JS, Lee H-JJ, et al. (2004). Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem 279, 12876–12882. 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- 61.Morohashi Y, Kan T, Tominari Y, Fuwa H, Okamura Y, Watanabe N, Sato C, Natsugari H, Fukuyama T, Iwatsubo T, et al. (2006). C-terminal fragment of presenilin is the molecular target of a dipeptidic gamma-secretase-specific inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester). J. Biol. Chem 281, 14670–14676. 10.1074/jbc.M513012200. [DOI] [PubMed] [Google Scholar]
- 62.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 63.Marro SG, Chanda S, Yang N, Janas JA, Valperga G, Trotter J, Zhou B, Merrill S, Yousif I, Shelby H, et al. (2019). Neuroligin-4 Regulates Excitatory Synaptic Transmission in Human Neurons. Neuron 103, 617–626.e6. 10.1016/j.neuron.2019.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun Z, and Südhof TC (2021). A simple Ca2+-imaging approach to neural network analyses in cultured neurons. J. Neurosci. Methods 349, 109041. 10.1016/j.jneumeth.2020.109041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, and Kageyama R (1999). Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation. EMBO J 18, 2196–2207. 10.1093/emboj/18.8.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Luo J, Elwood F, Britschgi M, Villeda S, Zhang H, Ding Z, Zhu L, Alabsi H, Getachew R, Narasimhan R, et al. (2013). Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J. Exp. Med 210, 157–172. 10.1084/jem.20120412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zurhove K, Nakajima C, Herz J, Bock HH, and May P (2008). Gamma-secretase limits the inflammatory response through the processing of LRP1. Sci. Signal 1, ra15. 10.1126/scisignal.1164263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nicholson AM, and Ferreira A (2009). Increased membrane cholesterol might render mature hippocampal neurons more susceptible to beta-amyloid-induced calpain activation and tau toxicity. J. Neurosci. Off. J. Soc. Neurosci 29, 4640–4651. 10.1523/JNEUROSCI.0862-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cho YY, Kwon O-H, Park MK, Kim T-W, and Chung S (2019). Elevated cellular cholesterol in Familial Alzheimer’s presenilin 1 mutation is associated with lipid raft localization of β-amyloid precursor protein. PloS One 14, e0210535. 10.1371/journal.pone.0210535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patzke C, Brockmann MM, Dai J, Gan KJ, Grauel MK, Fenske P, Liu Y, Acuna C, Rosenmund C, and Südhof TC (2019). Neuromodulator Signaling Bidirectionally Controls Vesicle Numbers in Human Synapses. Cell 179, 498–513.e22. 10.1016/j.cell.2019.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lange Y, and Steck TL (2020). Active cholesterol 20 years on. Traffic Cph. Den 21, 662–674. 10.1111/tra.12762. [DOI] [PubMed] [Google Scholar]
- 72.Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, et al. (2001). Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A 98, 5856–5861. 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, Steenvoorden E, Rynearson KD, Brouwers JF, Helms JB, et al. (2019). Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 24, 363–375.e9. 10.1016/j.stem.2018.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wasser CR, Ertunc M, Liu X, and Kavalali ET (2007). Cholesterol-dependent balance between evoked and spontaneous synaptic vesicle recycling. J. Physiol 579, 413–429. 10.1113/jphysiol.2006.123133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frank C, Rufini S, Tancredi V, Forcina R, Grossi D, and D’Arcangelo G (2008). Cholesterol depletion inhibits synaptic transmission and synaptic plasticity in rat hippocampus. Exp. Neurol 212, 407–414. 10.1016/j.expneurol.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 76.Mailman T, Hariharan M, and Karten B (2011). Inhibition of neuronal cholesterol biosynthesis with lovastatin leads to impaired synaptic vesicle release even in the presence of lipoproteins or geranylgeraniol. J. Neurochem 119, 1002–1015. 10.1111/j.1471-4159.2011.07474.x. [DOI] [PubMed] [Google Scholar]
- 77.Korinek M, Gonzalez-Gonzalez IM, Smejkalova T, Hajdukovic D, Skrenkova K, Krusek J, Horak M, and Vyklicky L (2020). Cholesterol modulates presynaptic and postsynaptic properties of excitatory synaptic transmission. Sci. Rep 10, 12651. 10.1038/s41598-020-69454-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Weber-Boyvat M, Kroll J, Trimbuch T, Olkkonen VM, and Rosenmund C (2022). The lipid transporter ORP2 regulates synaptic neurotransmitter release via two distinct mechanisms. Cell Rep 41, 111882. 10.1016/j.celrep.2022.111882. [DOI] [PubMed] [Google Scholar]
- 79.Bot N, Schweizer C, Ben Halima S, and Fraering PC (2011). Processing of the synaptic cell adhesion molecule neurexin-3beta by Alzheimer disease alpha- and gamma-secretases. J. Biol. Chem 286, 2762–2773. 10.1074/jbc.M110.142521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Saura CA, Servián-Morilla E, and Scholl FG (2011). Presenilin/γ-secretase regulates neurexin processing at synapses. PloS One 6, e19430. 10.1371/journal.pone.0019430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Suzuki K, Hayashi Y, Nakahara S, Kumazaki H, Prox J, Horiuchi K, Zeng M, Tanimura S, Nishiyama Y, Osawa S, et al. (2012). Activity-dependent proteolytic cleavage of neuroligin-1. Neuron 76, 410–422. 10.1016/j.neuron.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 82.Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, et al. (2002). A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21, 1948–1956. 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Haas IG, Frank M, Véron N, and Kemler R (2005). Presenilin-dependent processing and nuclear function of gamma-protocadherins. J. Biol. Chem 280, 9313–9319. 10.1074/jbc.M412909200. [DOI] [PubMed] [Google Scholar]
- 84.Haapasalo A, and Kovacs DM (2011). The many substrates of presenilin/γ-secretase. J. Alzheimers Dis. JAD 25, 3–28. 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y, and Colonna M (2021). Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J. Exp. Med 218, e20202717. 10.1084/jem.20202717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Romero-Molina C, Garretti F, Andrews SJ, Marcora E, and Goate AM (2022). Microglial efferocytosis: Diving into the Alzheimer’s disease gene pool. Neuron 110, 3513–3533. 10.1016/j.neuron.2022.10.015. [DOI] [PubMed] [Google Scholar]
- 87.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 88.Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, Duong DM, Pennington MW, Lah JJ, Seyfried NT, et al. (2018). Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener 13, 24. 10.1186/s13024-018-0254-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee SH, Bolshakov VY, and Shen J (2021). Inactivation of Presenilin in inhibitory neurons results in decreased GABAergic responses and enhanced synaptic plasticity. Mol. Brain 14, 85. 10.1186/s13041-021-00796-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.De Rossi P, Nomura T, Andrew RJ, Masse NY, Sampathkumar V, Musial TF, Sudwarts A, Recupero AJ, Le Metayer T, Hansen MT, et al. (2020). Neuronal BIN1 Regulates Presynaptic Neurotransmitter Release and Memory Consolidation. Cell Rep 30, 3520–3535.e7. 10.1016/j.celrep.2020.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bittner T, Fuhrmann M, Burgold S, Jung CKE, Volbracht C, Steiner H, Mitteregger G, Kretzschmar HA, Haass C, and Herms J (2009). Gamma-secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J. Neurosci. Off. J. Soc. Neurosci 29, 10405–10409. 10.1523/JNEUROSCI.2288-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu H, Saura CA, Choi SY, Sun LD, Yang X, Handler M, Kawarabayashi T, Younkin L, Fedeles B, Wilson MA, et al. (2001). APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron 31, 713–726. 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]
- 93.Lee SH, Sharma M, Südhof TC, and Shen J (2014). Synaptic function of nicastrin in hippocampal neurons. Proc. Natl. Acad. Sci. U. S. A 111, 8973–8978. 10.1073/pnas.1408554111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Muldoon MF, Barger SD, Ryan CM, Flory JD, Lehoczky JP, Matthews KA, and Manuck SB (2000). Effects of lovastatin on cognitive function and psychological well-being. Am. J. Med 108, 538–546. 10.1016/s0002-9343(00)00353-3. [DOI] [PubMed] [Google Scholar]
- 95.Muldoon MF, Ryan CM, Sereika SM, Flory JD, and Manuck SB (2004). Randomized trial of the effects of simvastatin on cognitive functioning in hypercholesterolemic adults. Am. J. Med 117, 823–829. 10.1016/j.amjmed.2004.07.041. [DOI] [PubMed] [Google Scholar]
- 96.King T, Bland Y, Webb S, Barton S, and Brown NA (2002). Expression of Peg1 (Mest) in the developing mouse heart: involvement in trabeculation. Dev. Dyn. Off. Publ. Am. Assoc. Anat 225, 212–215. 10.1002/dvdy.10142. [DOI] [PubMed] [Google Scholar]
- 97.Ott BR, Daiello LA, Dahabreh IJ, Springate BA, Bixby K, Murali M, and Trikalinos TA (2015). Do statins impair cognition? A systematic review and meta-analysis of randomized controlled trials. J. Gen. Intern. Med 30, 348–358. 10.1007/s11606-014-3115-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shepardson NE, Shankar GM, and Selkoe DJ (2011). Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch. Neurol 68, 1239–1244. 10.1001/archneurol.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jick H, Zornberg GL, Jick SS, Seshadri S, and Drachman DA (2000). Statins and the risk of dementia. Lancet Lond. Engl 356, 1627–1631. 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 100.Wolozin B, Kellman W, Ruosseau P, Celesia GG, and Siegel G (2000). Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol 57, 1439–1443. 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 101.Trompet S, van Vliet P, de Craen AJM, Jolles J, Buckley BM, Murphy MB, Ford I, Macfarlane PW, Sattar N, Packard CJ, et al. (2010). Pravastatin and cognitive function in the elderly. Results of the PROSPER study. J. Neurol 257, 85–90. 10.1007/s00415-009-5271-7. [DOI] [PubMed] [Google Scholar]
- 102.Runz H, Rietdorf J, Tomic I, de Bernard M, Beyreuther K, Pepperkok R, and Hartmann T (2002). Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J. Neurosci. Off. J. Soc. Neurosci 22, 1679–1689. 10.1523/JNEUROSCI.22-05-01679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Song Y, Hustedt EJ, Brandon S, and Sanders CR (2013). Competition between homodimerization and cholesterol binding to the C99 domain of the amyloid precursor protein. Biochemistry 52, 5051–5064. 10.1021/bi400735x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Langness VF, van der Kant R, Das U, Wang L, Chaves RDS, and Goldstein LSB (2021). Cholesterol-lowering drugs reduce APP processing to Aβ by inducing APP dimerization. Mol. Biol. Cell 32, 247–259. 10.1091/mbc.E20-05-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vellaichamy G, Dimitrion P, Zhou L, Ozog D, Lim HW, Liao W, Hamzavi IH, and Mi Q-S (2021). Insights from γ-Secretase: Functional Genetics of Hidradenitis Suppurativa. J. Invest. Dermatol 141, 1888–1896. 10.1016/j.jid.2021.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang B, Yang W, Wen W, Sun J, Su B, Liu B, Ma D, Lv D, Wen Y, Qu T, et al. (2010). Gamma-secretase gene mutations in familial acne inversa. Science 330, 1065. 10.1126/science.1196284. [DOI] [PubMed] [Google Scholar]
- 107.Wang J, Miao Y, Wicklein R, Sun Z, Wang J, Jude KM, Fernandes RA, Merrill SA, Wernig M, Garcia KC, et al. (2021). RTN4/NoGo-receptor binding to BAI adhesion-GPCRs regulates neuronal development. Cell 184, 5869–5885.e25. 10.1016/j.cell.2021.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and renewable materials reported in this paper will be shared by the lead contact upon request (lead contact, Thomas C. Südhof (tcs1@stanford.edu)). This paper does not report original code. Bulk RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti Syt1 | Synaptic Systems | Cat# 105-011C2: RRID:AB_2619760 |
| Chicken polyclonal anti MAP2 | Encor | CPCA-MAP2, RRID:AB_2138173 |
| Rabbit polyclonal anti Syn1 | Sudhof Lab | Clone YZ6078, RRID: AB_2861225 |
| Mouse monoclonal anti PSD95 | Thermo | Cat# MA146: RRID:AB_2092361 |
| Rabbit polyclonal anti APP | Ho et al., 2008 | Clone U955 |
| Mouse monoclonal anti APP | Sigma | Cat# MABN380: RRID: AB_2714163 |
| Rabbit polyclonal anti CASK | Neuromab | Cat# O14936: RRID: AB_2877188 |
| Rabbit polyclonal anti Fe65 | Cao and Südhof, 2001 | Clone 4194 |
| Rabbit polyclonal anti Mint1 | Ho et al., 2008 | Clone P730 |
| Rabbit polyclonal anti Nlgn1 | Jahn Monoclonals | Clone 4C12 |
| Rabbit polyclonal anti pan-Nrxn | Sclip and Südhof, 2020 | Clone G394, RRID: AB_2800397 |
| Rabbit polyclonal anti RIM1/2 | Acuna et al., 2015 | Clone U952 |
| Rabbit polyclonal anti Stx1 a/b | Zhou et al., 2012 | Clone 438B |
| Rabbit polyclonal anti PSD95 | Patzke et al., 2019 | Clone L667: RRID:AB_2800396 |
| Mouse monoclonal anti L1CAM | Sigma | Cat# L4543: RRID:AB_609903 |
| Rabbit monoclonal anti APP | Abcam | Cat# Ab32136: RRID: AB_2289606 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Accutase | Innovative Cell | Cat# AT104 |
| Gem21 Neuroplex supplement | Gemini Bio | Cat# 400–160 |
| BDNF | PeproTech | Cat# 450–02 |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| DMEM media | Thermo Fisher | Cat# 11965092 |
| DMEM/F-12 media | Thermo Fisher | Cat# 11320082 |
| HyClone fetal bovine serum (FBS) | GE Healthcare | Cat# SH30071.03 |
| MEM media | Thermo Fisher | Cat# 51200038 |
| Matrigel | BD Biosciences | Cat# 356230 |
| mTeSR1 media | Stem Cell Technologies | Cat# 85850 |
| N-2 supplement | Thermo Fisher | Cat# 17502048 |
| Neurobasal-A Medium | Thermo Fisher | Cat# 10888022 |
| NT-3 | PeproTech | Cat# 450–03 |
| Puromycin | Sigma-Aldrich | Cat# P8833 |
| Thiazovivin | Bio Vision | Cat# 1681 |
| Laminin-1 | Invitrogen | Cat# 23017–015 |
| Trypsin / EDTA | Thermo Fisher | Cat# 25300120 |
| Papain, Suspension | Worthington Biochemical | Cat# LS003127 |
| Cytosine arabinofuranoside (AraC) | Sigma-Aldrich | Cat# C6645 |
| Glutamax | Thermo Fisher | Cat# 35050079 |
| TTX | Fisher Scientific | Cat# 50-753-2807 |
| EDTA | Sigma-Aldrich | Cat# E6758-100G |
| Non-Essential Amino Acids | Thermo Fisher | Cat# 11140076 |
| Human Holotransferrin | Gemini Bio | Cat# 800-131P |
| Total Human Abeta ELISA | Clonetech | Cat# 27729A |
| DMSO, Cell Culture Grade | Sigma | Cat# D2650 |
| DAPT | Tocris | Cat# 2634 |
| LY411575 | Selleckchem | Cat# S2714 |
| Rosuvastatin | Selleckchem | Cat# S2169 |
| Simvastatin | Selleckchem | Cat# S1792 |
| TRIzol | Invitrogen | Cat# 15596026 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74104 |
| Filipin | Sigma | Cat# SAE0088 |
| Amplex Red Cholesterol Kit | Invitrogen | Cat# A12216 |
| Experimental Models: Cell Lines | ||
| Human H1-ESCs | WiCell Research Institute | Cat# WA01 |
| HEK293T cells | ATCC | Cat# CRL-11268 |
| Experimental Models: Organisms/Strains | ||
| CD-1 IGS mouse | Charles River | Cat# Crl:CD1(ICR) |
| Quantitative qRT-PCR | ||
| Human ACAT2 | IDT | Hs.PT.56a.26153626 |
| Human ACTB | IDT | Hs.PT.39a.22214847 |
| Human DBH | IDT | Hs.PT.58.39251730 |
| Human DHCR24 | IDT | Hs.PT.58.4561516 |
| Human DHCR7 | IDT | Hs.PT.58.38763048 |
| Human DUSP4 | IDT | Hs.PT.58.1640850 |
| Human DUSP5 | IDT | Hs.PT.58.1558918 |
| Human EGR1 | IDT | Hs.PT.58.40805543.g |
| Human FDFT1 | IDT | Hs.PT.58.25526506 |
| Human FOS | IDT | Hs.PT.58.15540029 |
| Human HMGCR | IDT | Hs.PT.58.41105492 |
| Human HMGCS1 | IDT | Hs.PT.58.4084870 |
| Human IDI1 | IDT | Hs.PT.58.19303620 |
| Human JUNB | IDT | Hs.PT.58.27174746.g |
| Human LDHA | IDT | Hs.PT.58.22929122 |
| Human LDLR | IDT | Hs.PT.58.2004261 |
| Human LSS | IDT | Hs.PT.58.24976485 |
| Human MAP2 | IDT | Hs.PT.58.20680759 |
| Human MVD | IDT | Hs.PT.58.39035173 |
| Human NAB2 | IDT | Hs.PT.58.24622150 |
| Human NR4A1 | IDT | Hs.PT.58.20779713 |
| Human SCD | IDT | Hs.PT.58.45714389 |
| Human SREBF2 | IDT | Hs.PT.58.45335433 |
| Human SV2C | IDT | Hs.PT.58.4640250 |
| Human TH | IDT | Hs.PT.58.369742 |
| Human TMEM97 | IDT | Hs.PT.58.40917738 |
| Human VEGFA | IDT | Hs.PT.58.1149801 |
| Human VGF | IDT | Hs.PT.58.26072166 |
| Recombinant DNA | ||
| FUW-TetO-Ng2-T2A-puromycin | Zhang et al., 2013 | 52047 Addgene (lentiviral vector) |
| FUW-rtTA | Zhang et al., 2013 | 20342 Addgene (lentiviral vector) |
| FUW-TetO-EGFP | Vierbuchen et al., 2010 | 30130 Addgene (lentiviral vector) |
| pREV | Dull et al., 1998 | 12253 Addgene (lentiviral helper vector) |
| pRRE | Dull et al., 1998 | 12251 Addgene (lentiviral helper vector) |
| pVSVg | Patzke et al., 2019 | 12259 Addgene (lentiviral helper vector) |
| Deposited Datasets | ||
| Bulk RNA-seq data | GEO | GSE206102 |
| Software and Algorithms | ||
| ImageJ | NIH | RRID: SCR_003070 |
| ImageStudio software | Li-Cor | RRID:SCR_013715 |
| pClamp | Molecular Devices | RRID:SCR_011323 |
| Nikon NIS-Elements | Nikon | RRID:SCR_014329 |
| GraphPad Prism | GraphPad Prism | RRID:SCR_002798 |
| RStudio | RStudio Team | RRID:SCR_000432 |
| DESeq2 | Bioconductor (Love et al., 2014) | RRID:SCR_015687 |