

Abstract
Synaptogenesis in the brain is highly organized and orchestrated by synaptic cellular adhesion molecules (CAMs) such as N-cadherin and amyloid precursor protein (APP) that contribute to the stabilization and structure of synapses. Although N-cadherin plays an integral role in synapse formation and synaptic plasticity, its function in synapse dismantling is not as well understood. Synapse weakening and loss are prominent features of neurodegenerative diseases, and can also be observed during homeostatic compensation to neuronal hyperexcitation. Previously, we have shown that during homeostatic synaptic plasticity, APP is a target for cleavage triggered by phosphorylation by Polo like kinase 2 (Plk2). Here, we found that Plk2 directly phosphorylates N-cadherin, and during neuronal hyperexcitation Plk2 promotes N-cadherin proteolytic processing, degradation, and disruption of complexes with APP. We further examined the molecular mechanisms underlying N-cadherin degradation. Loss of N-cadherin adhesive function destabilizes excitatory synapses and promotes their structural dismantling as a prerequisite to eventual synapse elimination. This pathway, which may normally help to homeostatically restrain excitability, could also shed light on the dysregulated synapse loss that occurs in cognitive disorders.
Graphical Abstract
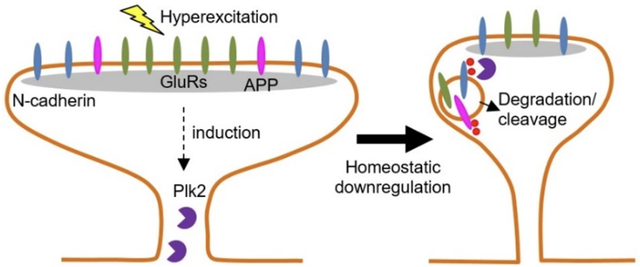
In this study, we show that neuronal hyperexcitation upregulates the activity-dependent homeostatic regulator Polo-like kinase (Plk2), which phosphorylates N-cadherin, a synaptic cellular adhesion molecule. This event triggers N-cadherin cleavage by metalloproteases and calpains and ultimate lysosomal degradation. Additionally, N-cadherin and the Alzheimer’s disease related factor amyloid precursor protein (APP) are associated at excitatory synapses. Degradation of N-cadherin accompanies downregulation of APP together with synaptic constituents PSD-95 and glutamate receptors (GluRs), leading to homeostatic synaptic destabilization and dismantling. Understanding N-cadherin metabolism will help elucidate mechanisms of synapse loss during normal learning and memory as well as under pathological conditions.
Introduction
Synapses are critical for neuronal communication as well as learning and memory. Synaptogenesis in the brain is highly organized and orchestrated by a wide variety of synaptic cellular adhesion molecules (CAMs) that contribute to the formation, stabilization, and diverse structure of synapses (Moreland & Poulain, 2022). These synaptic CAMs include cadherins, neuroligins, neurexins and amyloid precursor protein (APP), the precursor for the Alzheimer’s disease (AD)-related peptide amyloid beta (Aβ).
Classical cadherins play important roles in modulating synaptic specificity and remodeling. N-cadherin (also known as cadherin-2 or Cdh2) is a type I classical cadherin that is expressed pre- and postsynaptically in mature glutamatergic synapses (Basu et al., 2015; Stan et al., 2010a). Trans-synaptic adhesion occurs due to homophilic interactions of N-cadherin across the synaptic cleft via five extracellular calcium-binding cadherin repeats, while the cytoplasmic region of N-cadherin is primarily responsible for actin association and downstream signaling by binding to catenins (Seong et al., 2015). Although N-cadherin is known to play an integral role in the construction of synapses, its function in dismantling synapses is not understood. Studies have shown that forced asymmetrical expression of N-cadherin (loss on either pre- or postsynaptic side only) results in defects in synaptic transmission, reduced synapse formation, and increased synapse elimination (Jüngling et al., 2006; Pielarski et al., 2013). These results suggest that removal of N-cadherin from one side of the synapse could be a physiological mechanism to trigger synapse destabilization and loss.
In addition to synaptogenesis, N-cadherin has been shown to be crucial for synaptic plasticity and is required for long-term potentiation (LTP) (Bozdagi et al., 2000). Moreover, dendritic spines undergo actin-dependent changes in morphology during LTP (Harris, 2020; Hruska et al., 2018), and mutating alpha-catenin, which is required for cadherin’s actin-binding, leads to severe impairments in dendritic spine morphology (Togashi et al., 2002). Collectively, these data suggest that cadherins are important for both synaptic development and strengthening, but the role of N-cadherin in synaptic weakening is less clear.
Synapse weakening and loss are prominent features of neurodegenerative diseases, and genome-wide association studies have suggested that synaptic CAMs are impaired in AD (Bao et al., 2015). N-cadherin is widely expressed in the hippocampus, a structure which is important in learning and memory and perturbed in AD. Previous studies have shown a physical association of N-cadherin and APP using co-immunoprecipitation from transfected HEK293 cells and mouse brain lysates (Asada-Utsugi et al., 2011). N-cadherin also promotes the localization of γ-secretase to the cell surface, increasing the probability of encounter with APP and production of Aβ (Uemura et al., 2008). This enhancement can be blocked in the presence of an N-cadherin antagonist, ADH-1 (Asada-Utsugi et al., 2011). Additionally, Aβ42 synthetic peptides added to primary neuronal cultures results in p38 MAPK-mediated cell death, together with reduced N-cadherin expression, indicating that N-cadherin is downstream of Aβ (Ando et al., 2011). Collectively, these studies suggest that N-cadherin forms a complex with APP and engages in reciprocal regulation with Aβ.
Interestingly, N-cadherin and APP are proteolytically processed by a similar set of enzymes. N-cadherin is initially cleaved by the alpha-secretase ADAM 10, which results in the formation of an N-terminal fragment (NTF) and a C-terminal fragment (CTF-1) (Reiss et al., 2005). CTF-1 is further cleaved by γ-secretase component presenilin (PS1) to form C-terminal fragment 2 (CTF-2), which binds to transcriptional coactivator CREB binding protein (CBP) and promotes its proteasomal degradation, thus resulting in reduced CREB activity (Marambaud et al., 2003). These similar processing pathways further suggest that APP and N-cadherin may have some functional overlap.
Previously, we have shown that APP is a target for beta secretase processing during homeostatic synaptic plasticity, a class of compensatory mechanisms by which neurons adapt to and counteract excessive changes in network activity. Polo like kinase 2 (Plk2) is an essential regulator of homeostatic plasticity; its expression is stimulated by neuronal hyperexcitation and functions to downregulate excitatory synapses (K. J. Lee et al., 2011; Seeburg et al., 2008; Seeburg & Sheng, 2008). AD is associated with neuronal hyperactivity (Ma & Klann, 2012; Puzzo et al., 2017), which also stimulates Aβ production (Cirrito et al., 2008). Plk2 links these events by directly phosphorylating APP during hyperexcitation, resulting in APP cleavage and the generation of Aβ (Y. Lee et al., 2017). This pathway may normally help to restrain excitability through reduction of AMPARs and synapse loss (Hsieh et al., 2006). We have further shown that Plk2 inhibitors applied during hyperexcitation result in reduced Aβ generation and elevated surface expression of AMPARs (J. S. Lee et al., 2019).
Here, we examined the functional relationship between Plk2 and N-cadherin. We found that Plk2 directly phosphorylates N-cadherin, and during neuronal hyperexcitation Plk2 promotes N-cadherin proteolytic processing, degradation, and disruption of complexes with APP. Loss of N-cadherin adhesive function destabilizes excitatory synapses and promotes their structural dismantling as a prerequisite to eventual synapse elimination.
Materials and Methods
Animals
All experimental procedures using rodents were approved and performed per regulations of the Georgetown University Institutional Animal Care and Use Committee (protocol #2016–1145). Pregnant Sprague–Dawley rats (8–10-week-old females) were obtained from Charles River (Raleigh, NC). Timed-pregnant Sprague–Dawley dams were singly housed for 2 days in individually ventilated cages, with ad libitum access to food and water. This study was not pre-registered. No exclusion criteria were predetermined and no animals were excluded. A total of 24 dams were used for this study.
Primary hippocampal neurons
At embryonic day 18 (E18), pregnant Sprague Dawley dams were euthanized using a flow-regulated carbon dioxide chamber and death was verified by toe pinch and cervical dislocation. Culture preparations were obtained from two dams. An average of 7–14 embryos were excised via laparotomy and decapitated using sharp scissors immediately after euthanasia of the dam. Skulls were placed in Hank’s Buffer Saline Solution (HBSS) which is comprised of 440 mL of cell culture grade water, 50 mL of 10X HBSS, 10 mM of HEPES (Gibco cat. #15630–080) and 1% of penicillin-streptomycin (Gibco cat. #15140122). Brains were excised and the hippocampus was dissected. Hippocampal neurons were washed 3 times with 10 mL of cold HBSS. After the final wash, neurons were trypsinized for 15 minutes using 200 μl of trypsin (Gibco cat. #15090046) in 5 mL of HBSS at 37°C. Trypsin was then removed and 1 mL of pre-warmed trypsin inhibitor was added to stop trypsinization. Neurons were then washed 3 times with pre-warmed HBSS. Neurons were mechanically triturated using a 5 mL pipette 25 times, a wide flame polished glass pipette 15 times, and a narrow flame polished pipette 5 times. Neurons were filtered using a 70 μm sterile cell strainer (Fisher brand cat. #22363548) and cells were plated in 12-well plates onto nitric acid-washed borosilicate coverslips or wells pretreated with Poly-D-Lysine (Sigma, cat. #P8099) and laminin (Sigma, cat. #L2020) in 1 mL of plating media composed of Neurobasal media supplemented with 25 μM glutamate, 500 μM glutamine, and 1:500 of Primocin at a density of 75,000 cells per well for immunocytochemistry and 300,000 cells per well for biochemistry. Pooled brains from one litter of ~12 embryos provided sufficient neurons to generate approximately 96 wells for immunocytochemistry or 24 wells for biochemistry. Cells were taken from at least 2 independent culture preparations for each experimental condition.
COS-7 tissue culture
COS-7 cells were obtained from Georgetown University Lombardi Comprehensive Cancer Center Tissue Culture and Biobanking Shared Resource. Cells were grown in Dulbecco’s Modified Eagles Medium which contains 4.5 g/L of glucose, L-glutamine, and 110 mg/L of sodium pyruvate (Gibco cat. #11995065) supplemented with 10% fetal bovine serum (Gibco cat #10437028) and 1% of penicillin-streptomycin (Gibco cat #15140122).
Transfection
On DIV14, primary hippocampal neurons were transfected with a total of 2 μg of DNA using Lipofectamine 2000 (Invitrogen cat. #11688019) for 48 hrs. COS-7 cells were transfected with a total of 1 μg of DNA using Polyjet In Vitro DNA transfection reagent (SignaGen cat. # SL10068) for 48 hrs.
Constructs
Mouse N-cadherin fused to a C-terminal FLAG epitope was expressed in pcDNA3.1 (gift of Dr. Yasuji Matsuoka, Georgetown University). Myc epitope-tagged Plk2, constitutively active Plk2 (Plk2-CA, T236E), and kinase-dead Plk2 (Plk2-KD, K108M) were expressed in pGW1-CMV (British Biotechnology).
Inhibitors
To induce hyperexcitation, primary hippocampal neurons (DIV20) were treated with 100 μM of picrotoxin (PTX), a GABAA antagonist for 24 hrs. To inhibit Plk2 upregulation in response to hyperexcitation, 50 nM of pan-Plk inhibitor volasertib (BI-6727, Chemietek) or selective Plk2 inhibitor TC S-7005 (Tocris cat. #4459) was applied to primary hippocampal cultures at DIV21 for 4 hrs before collection. The following inhibitors were used to test pathways implicated in the cleavage of N-cadherin: 10 μM of lactacystin (Adipogen cat. #AGCN20104C500) and 1 μM of MG-132 (Tocris cat. #1748) against proteasomes; 10 nM of bafilomycin (EMD Millipore cat. #196000) and 40 μM of chloroquine for 4 hrs (Acros cat. #50-63-5) against lysosomes; 5 μM of GM-6001 (EMD Millipore cat. #364205), a pan inhibitor of MMP; 3 μM of GI254023X (Sigma cat. #SML0789) for 5 hrs, a selective ADAM-10 inhibitor; 25 μM and 50 μM of Calpain I and Calpain III (Sigma cat. #20-872-225) inhibitors; 1 μM of BSI (EMD Millipore cat. #565749) for beta secretase.
Antibodies
The following antibodies were used: mouse anti-N-cadherin (ICC 1:100, WB 1:1000, cat. #610921, RRID: AB_398236, BD Biosciences), rabbit anti-N-cadherin clone EPR1792Y (ICC 1:150, WB 1:1000, cat. #04–1126, RRID: AB_1977064, Millipore), rabbit anti-N-cadherin (ICC 1:150, cat. #363 003, RRID: AB_2620123, Synaptic Systems), N-terminal mouse anti-N-cadherin clone GC-4 (ICC 1:100, cat. #C3865, RRID: AB_262097, Sigma Aldrich), rabbit N-terminal anti-APP (1:500, cat. #A8967, RRID: AB_258427, Sigma), rabbit anti-APP Y188 (ICC: 1:500, WB 1:1000, cat. #TA300496, RRID: AB_2056556, Origene), rabbit anti-APP Y188 (WB 1:1000 cat. #ab32136, RRID: AB_2289606, Abcam), anti-mouse PSD-95 (ICC 1:200, cat. #75–028, AB_2292909, Neuromab), guinea pig anti-PSD-95 (ICC 1:250, cat. #124014, RRID: AB_2619800, Synaptic Systems), rabbit anti-PSD-95 (ICC 1:200, cat. #3450, RRID: AB_2292883, Cell Signaling Technology), mouse anti-GluA2 (1:100, cat. #556341, RRID: AB_396373, BD Biosciences), mouse anti-FLAG (ICC 1:100, WB 1:1000, cat. #F1804, RRID: AB_262044, Sigma), rabbit anti-myc (ICC 1:100, WB 1:1000, cat. # 2272, RRID: AB_10692100, Cell Signaling Technology), chicken anti-MAP2 (ICC 1:1000, cat. #1100-MAP2, RRID: AB_2492141, PhosphoSolutions).
Immunocytochemistry
Primary hippocampal neurons were fixed with 1% paraformaldehyde (PFA)/ 4% sucrose for 5 minutes followed by a methanol fix for 5 minutes at −20°C. Primary hippocampal neurons were washed 3 times with phosphate buffer solution (PBS) for 5 minutes. Coverslips were blocked with 5% normal goat serum (NGS) and 0.3% Triton X-100 in PBS and then incubated in primary antibodies in 3% NGS and 0.3% Triton X-100 in PBS overnight at 4°C. Neurons were washed 3 times in PBS with 0.1% Triton X-100 for 5 mins on the shaker before adding secondary antibodies (Alexa Fluor 647, Alexa Fluor 555, or Alexa Fluor 488; Invitrogen). Coverslips were incubated with secondary antibodies for 1 hr in the dark at room temperature then washed 3 times for 5 mins in 1X PBS with 0.1% Triton X-100 on the shaker. Coverslips were mounted with VectaShield or Prolong Diamond Antifade Mountant (Invitrogen cat. #P36970). Coverslips were then sealed with CoverGrip coverslip sealant (Biotium cat. #23005). Primary hippocampal neurons were imaged using a Zeiss Axiovert fluorescence microscope with 63X objective using Zen Blue software or a Leica SP8 confocal microscope with the same intensity across all samples
Immunoprecipitation
Primary hippocampal neurons are collected at DIV20–21. Media was aspirated and cells were washed with cold PBS. Cells were then lysed with non-denaturing lysis buffer which is composed of 25 mM Tris HCl pH 8, 150 mM NaCl, 1% Nonidet P-40 (NP-40), 1 mM EDTA, cOmplete mini protease inhibitor cocktail (Roche cat. #1836153001), AEBSF (Thermofisher Scientific cat. #PI78431), NaF, and Na3VO4. Cells were scraped and placed in a pre-cooled microcentrifuge tube and homogenized using a plastic dounce. Lysates were nutated at 4°C for 30 mins and centrifuged at 4°C for 30 mins at 16,000g. The supernatant was saved as the input fraction. To ensure equivalent protein concentrations per each treatment group, a BCA assay (Thermo Scientific Pierce cat. #2325) was performed. For 1 mg of input protein, 2 μg of antibody was added to lysates. Following an overnight incubation at 4°C, 110 μl of either Sepharose A or G beads was added to lysates for 4 hrs at 4°C under rotary agitation. After 4 hrs, the tubes were centrifuged at 2,000g and the supernatant was saved as the unbound fraction. Beads were washed 3 times with cold PBS containing protease inhibitor and centrifuged at 2,000g between washes. The wash buffer was removed and the bound fraction eluted from beads with 30 μl of 2X SDS loading buffer with beta-mercaptoethanol. Samples were boiled at 95°C for 5 mins and run on an 8% SDS page gel.
Immunoblotting
SDS-PAGE gels were soaked in 20% ethanol for 7 minutes and transferred using the iBlot2 Dry Blotting System (Thermo Fisher). Proteins were transferred using a nitrocellulose membrane with a pore size of 0.2 μm (Invitrogen cat. #IB23001). After transfer, blots were rinsed with Tris buffered saline with 0.1% Tween-20 (TBST) and incubated in blocking solution (5% skim milk/TBST) for 1 hour. Blots were incubated in primary antibody in blocking solution on the shaker overnight at 4°C. Blots were washed 3 times with TBST for 10 minutes each and incubated with either mouse or rabbit horseradish peroxidase-conjugated secondary antibodies (Sigma) in blocking solution for 1 hour on the shaker at room temperature. The blots were visualized using enhanced chemiluminescence (ECL) solution (Thermo Scientific Pierce cat. #P132106) using the Perkin Elmer Pierce system. Full western blot images are included in Supplementary Figure 4.
In vitro kinase assay
Immunoprecipitated N-cadherin from COS-7 cells was bound to Sepharose (Invitrogen). Beads were washed twice with PBS followed by a final wash with kinase buffer (50 mM Tris pH 7.5, 10 mM MgCl2, 5 mM DTT, and 2 mM EGTA). N-cadherin was combined with purified Plk2 in baculovirus (Active Motif) with 200 μM 32P-γ-ATP for 30 mins at 30°C. The reaction was terminated by eluting with 2X LSB at 50°C for 10 mins. Samples were then separated on an 8% Tris-tricine SDS PAGE gel. The gel was dried and exposed to film for autoradiography.
Image Analysis
Image J (Fiji) and Metamorph software (Molecular Dynamics) were used to measure fluorescent signals. Images were taken with the same intensity and gain across samples for a given experiment and region of interest (ROI). For dendritic ROIs, we selected at least 3 secondary or primary dendrites per neuron, thresholded images to the same level to subtract background, and measured mean fluorescent intensity. The fluorescent intensities were then averaged to obtain a neuronal mean. Secondary dendrites were defined as any branch protruding from a primary dendrite. For somatic ROIs, gain was lowered in order to avoid saturation of the signal. For colocalization analyses, Image J coloc2 plugin was used to determine Mander’s coefficient which measures the degree of overlap between N-cadherin and APP and the overlap between APP and N-cadherin in dendritic spines (marked by PSD-95 labeling to delineate ROIs). The colocalization scale is 0–1 where 0 represents no colocalization and 1 is 100% colocalization.
Statistical Analysis
Data were analyzed using Graphpad Prism 9. All values are expressed as mean±SEM from 2 or more independent experiments. The sample number represents neurons, the number of cells, or independent cultures as indicated in figure legends. For these in vitro studies, no a priori sample size calculations were performed. The sample sizes used were based on previous studies of a similar nature (Lee et al., 2011). For immunocytochemistry experiments, neurons were imaged from separate cultures and proximal and secondary dendrites were averaged per neuron. For comparison between two independent groups, two-tailed unpaired Student’s t-tests were used. For multiple group comparisons, a one-way analysis of variance (ANOVA) was used with Tukey’s post hoc test. Statistically significant differences were determined at P<0.05. No randomization was used to allocate cultured cells to treatment groups as all material was relatively homogeneous within each culture preparation. All experimentation, image acquisition, and analysis/quantification were performed blind to experimental condition to avoid bias.
Results
We hypothesized that hyperexcitation-induced Plk2 causes downregulation of synapses by phosphorylating synaptic structural components, leading to their degradation and ultimately synapse destabilization and elimination. To test whether synaptic CAMs are potential targets for Plk2, we performed a kinase-dependent molecular degradation screen of a panel of candidate synaptic CAMs. COS-7 cells were co-transfected with either myc-Plk2 constitutively active (Plk2-CA, T236E) or myc-Plk2 kinase-dead (Plk2-KD, K108M mutant), together with one of the following synaptic CAMs: β3-integrin, SynCAM1, tropomyosin receptor kinase B (TrkB), cadherin 9 (Cdh9), cadherin 8 (Cdh8), cadherin 10 (Cdh10), cadherin 13 (Cdh13), N-cadherin, Ephrin A4 (EphA4), and Ephrin B2 (EphB2) (Figure 1A). The results indicate that Plk2-CA resulted in loss of N-cadherin expression, with little effect on other synaptic CAMs (Figure 1B). Quantification indicated that N-cadherin levels were reduced by ~90% in the presence of Plk2-CA, as assayed by both immunoblotting and immunocytochemistry (Figure 1B,D). N-cadherin was not affected in the presence of an equivalent amount of Plk2-KD, suggesting that Plk2 promotes elimination of N-cadherin in a kinase-dependent manner.
Figure 1. (A) Plk2-dependent degradation screen of synaptic cellular adhesion molecules.
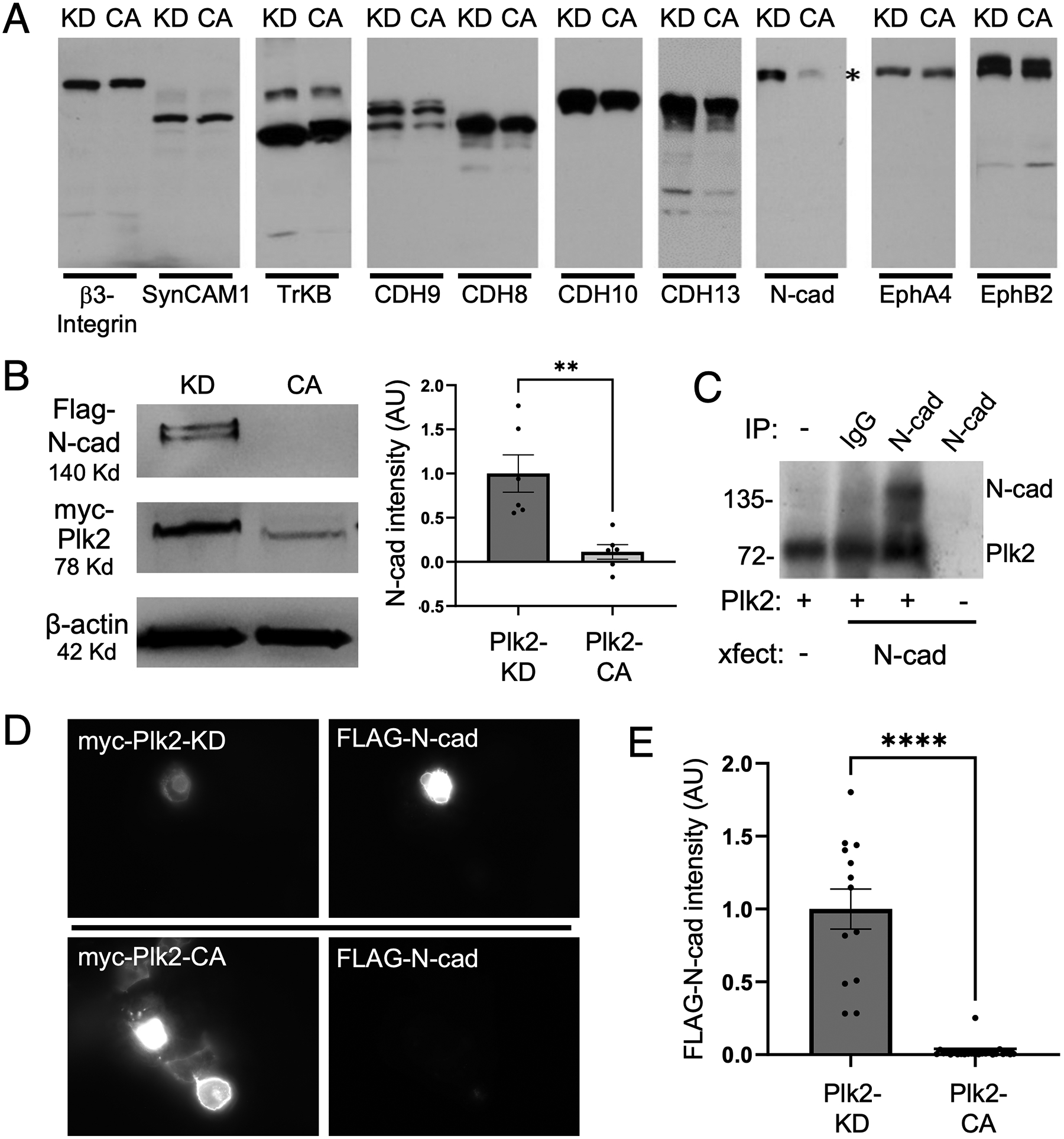
Candidate cellular adhesion molecules (CAMs) as indicated above the blots were transfected into COS-7 cells together with either kinase dead (KD) or constitutively active (CA) Plk2. Immunoblotting for each CAM demonstrated that only N-cadherin levels (highlighted by asterisk) were decreased by active Plk2. (B) Immunoblots of FLAG-N-cadherin and myc-tagged Plk2-KD or -CA co-transfected in COS-7 cells. Loading control is β-actin. Quantification is normalized to Plk2-KD; **p=0.0030, t=3.902, DF=10, unpaired two-tailed student’s t-test (n=6 cell culture preparations). (C) Direct phosphorylation of N-cadherin by Plk2. Autoradiogram of in vitro kinase assays using N-cadherin immunopurified from transfected COS-7 cells added to recombinant Plk2. Nonimmune IgG negative control to demonstrate immunoprecipitation specificity. Note Plk2 is autophosphorylated (lower band). (D) Immunostaining of COS-7 co-transfected with FLAG-N-cadherin and either myc-Plk2-KD (upper panels) or -CA (lower panels). (E) Quantification of images in (D) normalized to Plk2-KD; ***p<0.0001, t=9.702, DF=35, unpaired student’s t-test (n=13–24 cells).
To test whether Plk2 directly phosphorylates N-cadherin, an in vitro kinase assay was performed using N-cadherin immunopurified from COS-7 cells and purified full-length Plk2 produced in baculovirus (Active Motif) in the presence of 32P-γ-ATP. The autoradiogram showed that Plk2 indeed can phosphorylate N-cadherin, as well as exhibiting autophosphorylation (Figure 1C). These results demonstrate that N-cadherin is a bona fide and direct phosphorylation substrate of Plk2.
To determine whether Plk2 is capable of eliminating N-cadherin in neurons, we transfected Plk2-KD, Plk2-CA, and Plk2-WT into primary hippocampal neurons and measured the expression of endogenous N-cadherin. Neurons transfected with Plk2-CA exhibited ~60% loss of N-cadherin in secondary dendrites as well as in the soma, as compared to inactive Plk2-KD (Figure 2). Additionally, a significant decrease in N-cadherin somatic expression was observed in Plk2-WT neurons compared to Plk2-KD (Figure 2). No significant difference in Plk2 expression in the soma was observed among any of the constructs (Figure 2). These data further support that N-cadherin loss is kinase dependent and occurs in neuronal dendrites.
Figure 2. Loss of endogenous N-cadherin is dependent on Plk2 kinase activity in primary hippocampal neurons.
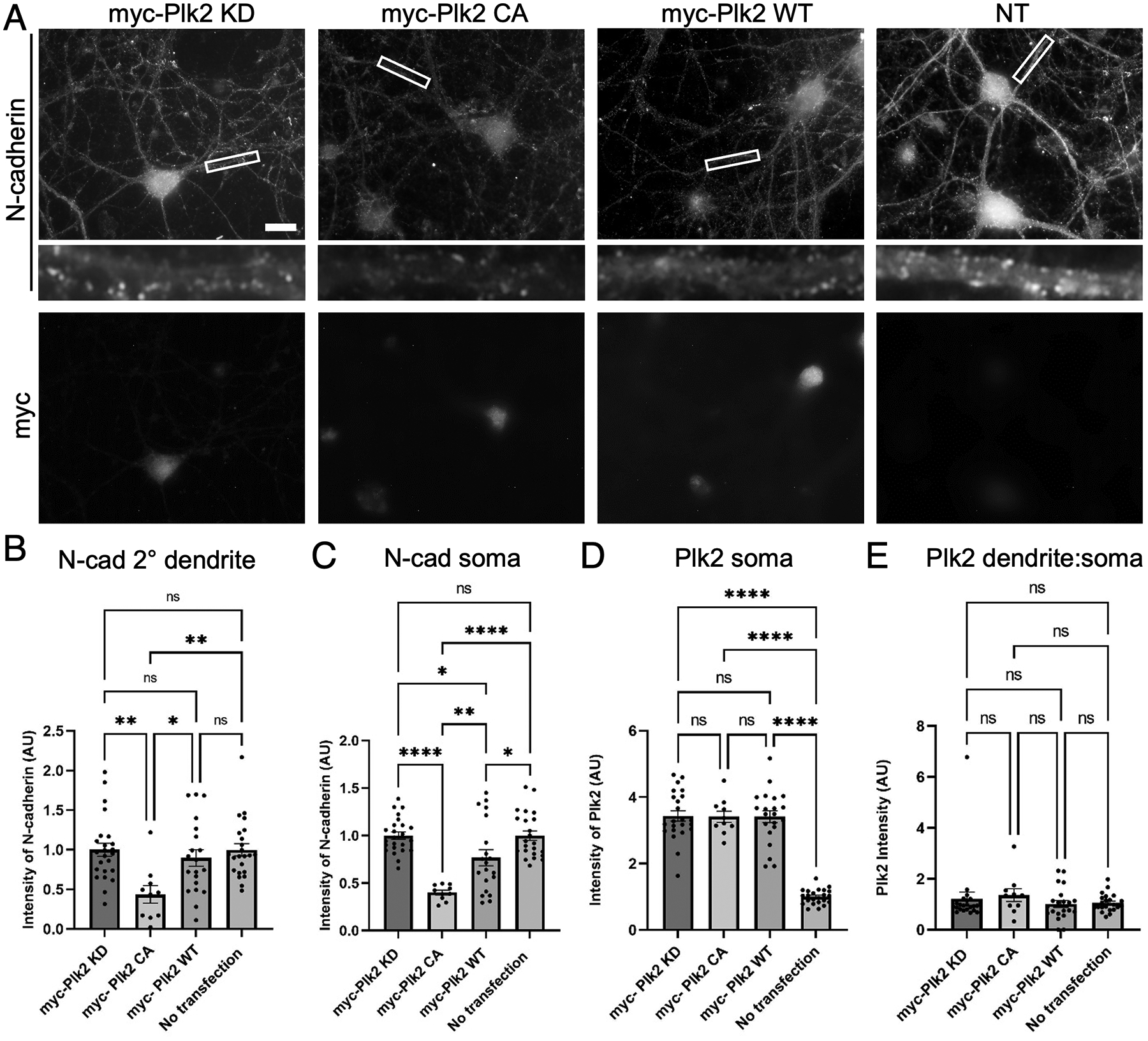
(A) Hippocampal neurons were transfected with myc-Plk2 constructs as indicated above images and immunostained for endogenous N-cadherin (upper panels) or for myc epitope (lower panels). Higher magnification views of representative dendrites (boxed region) are shown below each neuron for N-cadherin. Scale, 10 μm. (B-C) Quantification of N-cadherin intensity across transfection conditions measured (B) in secondary dendrites (P= 0.0024, F=5.281, DF=3) and (C) in soma (P< 0.0001, F=16.14, DF=3). (D-E) Plk2 intensity in (D) soma (p< 0.0001, F=79.52, DF=3) and (E) proximal dendrites across the various transfection conditions (p=0.6680, F=0.5228, DF=3) One-way ANOVA and Tukey’s multiple comparison test, n=10–24 neurons; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns=nonsignificant.
Next, we examined if N-cadherin loss occurs physiologically in neurons during hyperexcitation conditions that induce Plk2 expression. For this purpose, we used picrotoxin (PTX) to block inhibitory GABAA receptors, which causes global stimulation of excitatory synaptic activity and triggers homeostatic adaptation. Under basal conditions, N-cadherin is distributed in a highly punctate pattern in dendrites. Upon chronic stimulation with PTX for 24 hrs, N-cadherin was substantially diminished in secondary dendrites and soma in comparison to unstimulated control (Figure 3). PTX had no significant effect on levels of the dendritic protein MAP2 (Supplementary Figure 1), indicating specificity of the N-cadherin reduction. To test if PTX-induced N-cadherin loss is mediated by Plk2, we used two different antagonists (pan Plk-family inhibitor BI-6727, or selective Plk2 inhibitor TCS-7005; 50 nM each) for 4 hours prior to collection. Both Plk2 inhibitors blocked the removal of N-cadherin during hyperexcitation in secondary dendrites and soma (Figure 3). Thus, hyperexcitation-induced loss of native N-cadherin in neurons requires endogenous Plk2 activity.
Figure 3. Hyperactivity-induced loss of endogenous N-cadherin in primary hippocampal neurons requires Plk2 kinase activity.
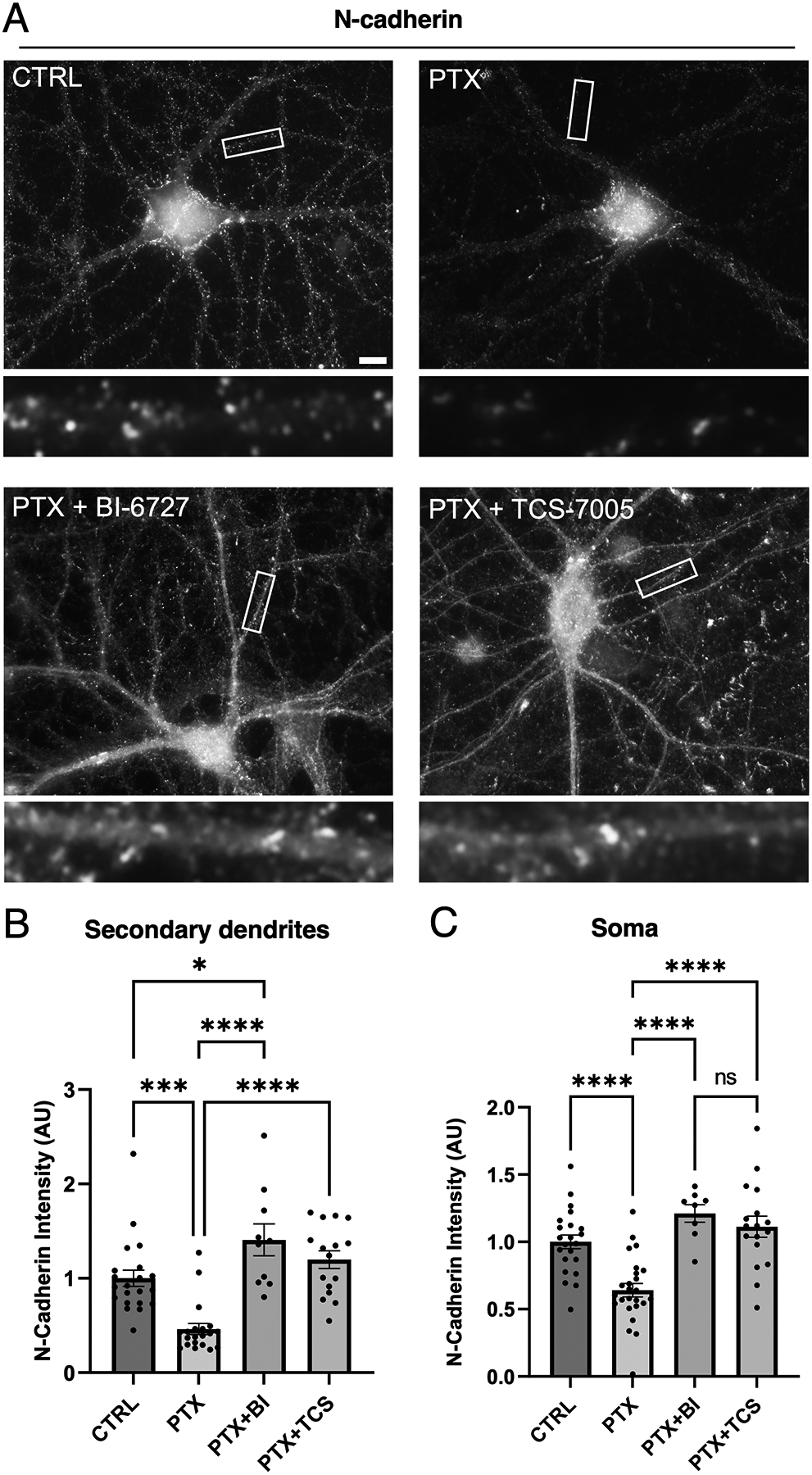
(A) Neurons were unstimulated (CTRL), treated with PTX alone, or treated with PTX in combination with Plk2 inhibitors BI-6727 or TCS-7005, as indicated, and immunostained for native N-cadherin. Higher magnification views of representative secondary dendrites (boxed regions) are shown below each neuron. Scale, 10 μm. (B-C) Quantification of N-cadherin intensity in (B) secondary dendrites (p<0.0001, F=18.20, DF=3) and (C) soma (p<0.0001, F=16.75, DF=3) (n=10–21 neurons per group, one-way ANOVA and Tukey’s multiple comparison test; *p<0.05, ***p<0.001, ****p<0.0001, ns=nonsignificant).
Accumulating evidence points to a protein complex of N-cadherin and APP in stabilizing synapses. To examine whether hyperexcitation alters this complex association, we immunostained for N-cadherin and APP. We found that both synaptic CAMs were lost following PTX treatment, which was rescued by BI6727 or TCS-7005 (Figure 4). Additionally, we found that APP co-immunoprecipitated with N-cadherin from primary hippocampal neurons (Figure 5A) and transfected heterologous cells (Figure 5B). When neurons were treated with PTX for 24 hrs, we found that hyperexcitation abolished the N-cadherin and APP association (Figure 5A). BI-6727 or TC S-7005 inhibition of Plk2 restored the interaction of these CAMs, and indeed rescued to a level higher than baseline. This finding confirms that APP and N-cadherin form a physiological assembly, and that during hyperexcitation Plk2 causes the dismantling of this complex.
Figure 4. APP and N-cadherin are co-diminished by hyperactivity in a Plk2-dependent manner.
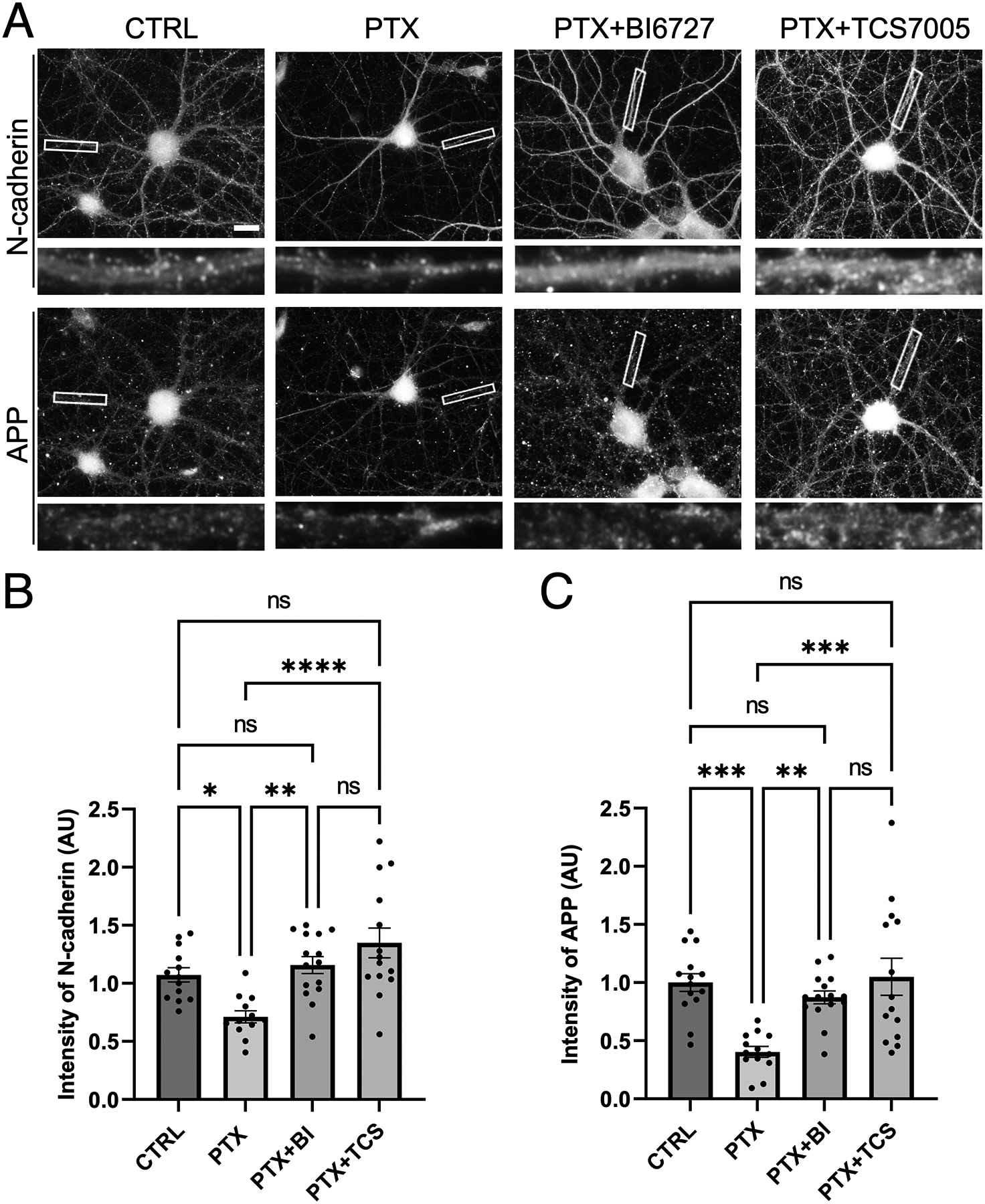
(A) Immunostaining of endogenous APP and N-cadherin in cultured hippocampal neurons left unstimulated (CTRL) or treated with PTX, PTX+BI 6727, or PTX+TCS-7005 (n=12–15 neurons). Higher magnification views of representative secondary dendrites (boxed regions) are shown below each neuron. Scale, 10 μm. (B-C) Quantification of images in (A) for (B) N-cadherin (p<0.0001, F=8.913, DF=3) or (C) APP (p< 0.0001, F=9.047, DF=3); (n=12–15 neurons for N-cadherin, 13–15 neurons for APP; one-way ANOVA and Tukey’s multiple comparison test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns=nonsignificant).
Figure 5. Association of APP and N-cadherin is disrupted by hyperactivity in a Plk2-dependent manner.
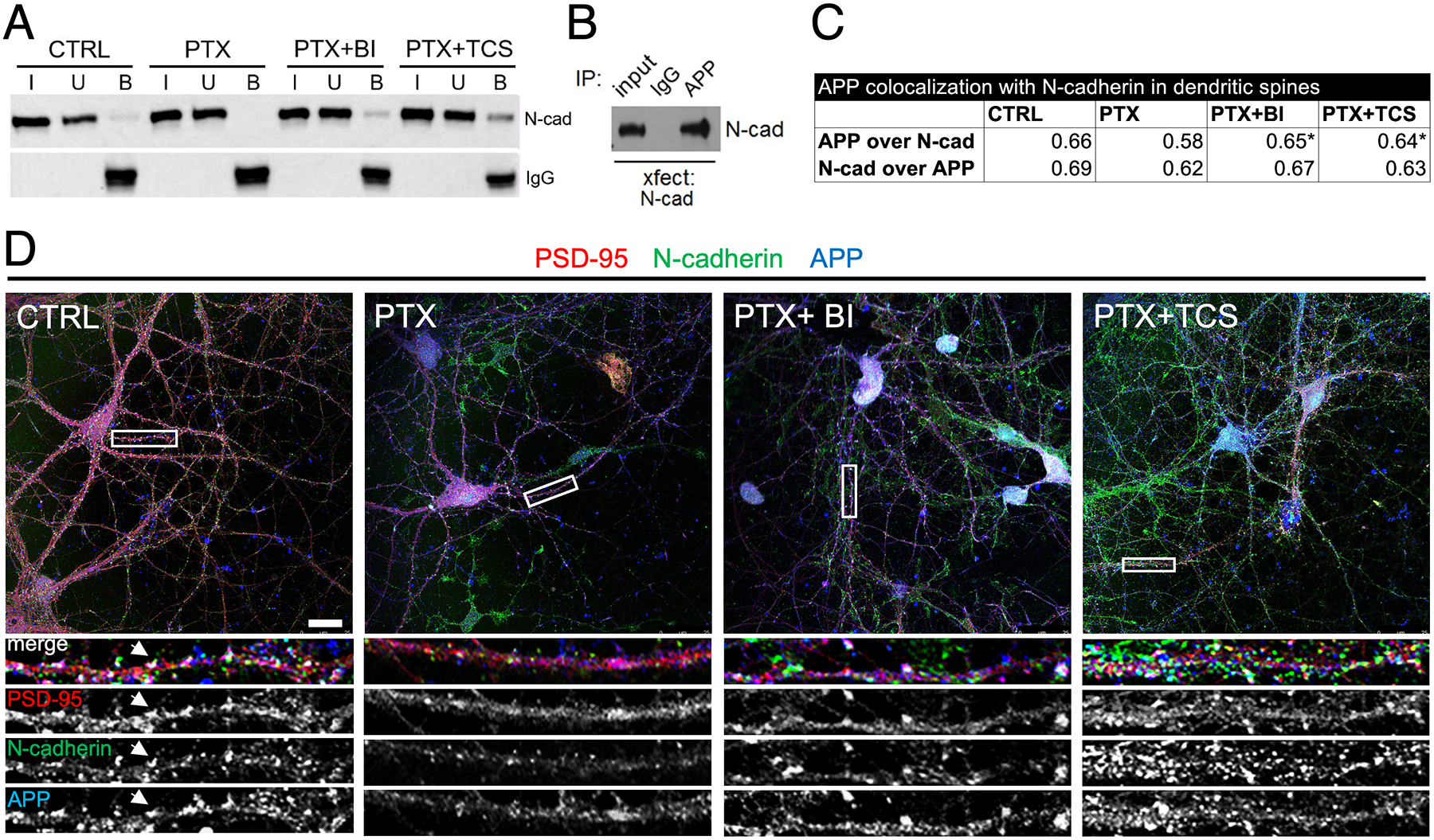
(A) Cultured hippocampal neuron lysates were prepared following treatments as shown at top, immunoprecipitated for APP, and probed for N-cadherin. I, input; U, unbound; B, bound. CTRL, unstimulated; BI, BI-6727; TCS, TCS-7005. (B) COS-7 cells were transfected with N-cadherin, immunoprecipitated with APP antibodies or nonimmune IgG, and probed for N-cadherin. (C) Quantification of mean overlap using Mander’s coefficient between APP and N-cadherin (upper line) (p= 0.0127, F=3.638, DF=3), or between N-cadherin and APP (lower line) (p= 0.0076, F=4.021, DF=3), within PSD-95 puncta across the different treatments. One-way ANOVA and Tukey’s multiple comparison test; *p<0.05 vs. PTX. (D) Triple label immunostaining of PSD-95 (red), N-cadherin (green), and APP (blue) to label dendritic spines under the different treatment conditions as indicated. Arrowheads highlight white puncta indicating triple co-localization at synapses. Higher magnification views of representative secondary dendrites (boxed in full neuron image) are shown below each neuron in merged color as well as in gray scale of separate channels for better visualization of individual distributions. Scale, 10 μm.
We further analyzed the association of APP and N-cadherin at synapses using triple immunostaining of these proteins along with PSD-95 as a marker of excitatory synapses. We observed considerable overlap of the three proteins in dendritic spines (seen as white puncta due to triple co-localization) (Figure 5C,D). APP colocalization with N-cadherin decreased (albeit in a nonsignificant manner) with PTX, but that Plk2 inhibitors restored the colocalization to a level significantly different from PTX alone (Fig. 5C,D). N-cadherin, however, did not change its colocalization extent with APP under any conditions (Fig. 5C,D), suggesting that N-cadherin association with APP is strictly maintained even under dynamically changing protein levels.
To examine the molecular mechanisms that underlie the activity- and Plk2-dependent loss of N-cadherin, we hypothesized that this pathway may involve proteolytic cleavage and degradation. N-cadherin is a substrate of multiple proteases including calpains, matrix metalloproteinases (MMPs), and the proteasome. Pharmacological inhibition was utilized to identify proteolytic pathways involved in N-cadherin loss in hyperexcitation. First, we tested GM6001 (a pan-MMP inhibitor) and GI254023X (a specific inhibitor of ADAM10) and found that these inhibitors rescued the loss of N-cadherin normally observed with chronic PTX (Figure 6). This observation suggests hyperexcitation induces processing of N-cadherin through upregulation of ADAM10 (Figure 6).
Figure 6. Metalloproteases and ADAM10 proteolytic cleave N-cadherin during hyperexcitation.
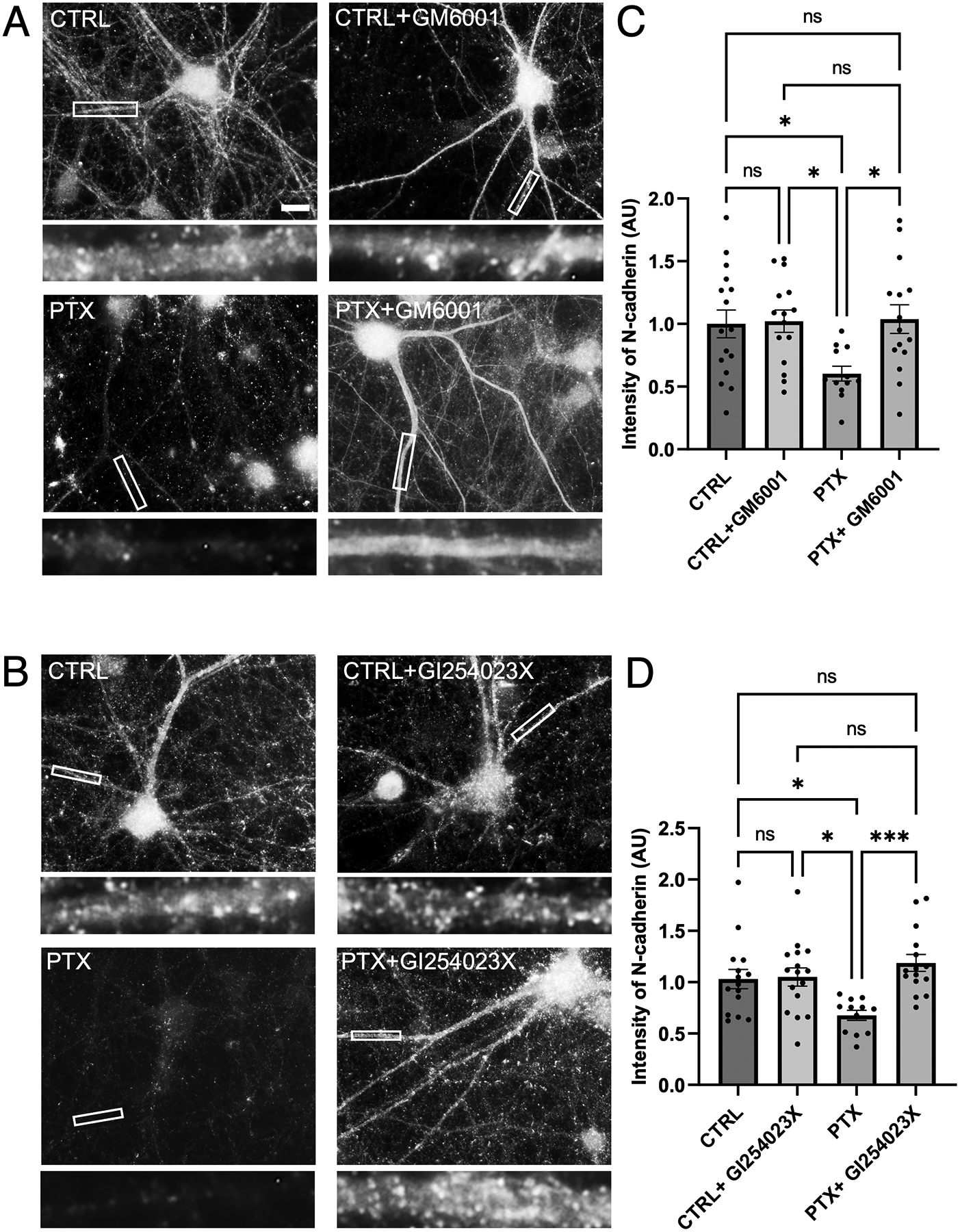
(A-B) Immunostaining of endogenous N-cadherin in cultured hippocampal neurons left unstimulated (CTRL) or treated with PTX, either with or without broad spectrum MMP inhibitor GM6001 or specific ADAM10 inhibitor GI254023X. Higher magnification views of representative secondary dendrites (boxed regions) are shown below each neuron. Scale, 10 μm. (C-D) Quantification of (C) GM6001 (p= 0.0143, F=3.856, DF=3) and (D) GI254023X treatments (p=0.0012, F= 6.059, DF=3) (n=12–16 neurons for GM6001, 12–26 neurons for GI254023X; one-way ANOVA and Tukey’s multiple comparison test; *p<0.05, ***p<0.001, ns=nonsignificant).
Calpains, which are calcium-dependent cysteine proteases (Goll et al., 2003), are also upregulated by increased intracellular calcium during hyperexcitation (Lynch & Baudry, 1984). Thus we wanted to examine if calpains are involved in proteolytic cleavage of N-cadherin. We tested two different concentrations of calpain I and calpain III inhibitors (25 μM and 50 μM.) We found that 25 μM of Calpain I inhibitor did not rescue N-cadherin loss during hyperexcitation; however, 25 μM of Calpain III inhibitor did prevent the proteolytic cleavage of N-cadherin. With 50 μM, both inhibitors rescued N-cadherin loss in comparison to PTX-treated control neurons (Figure 7). This result suggests that calpains are upregulated during hyperexcitation and also play a role in the proteolytic cleavage of N-cadherin (Figure 7).
Figure 7. Calpain proteolytically cleaves N-cadherin during hyperexcitation.
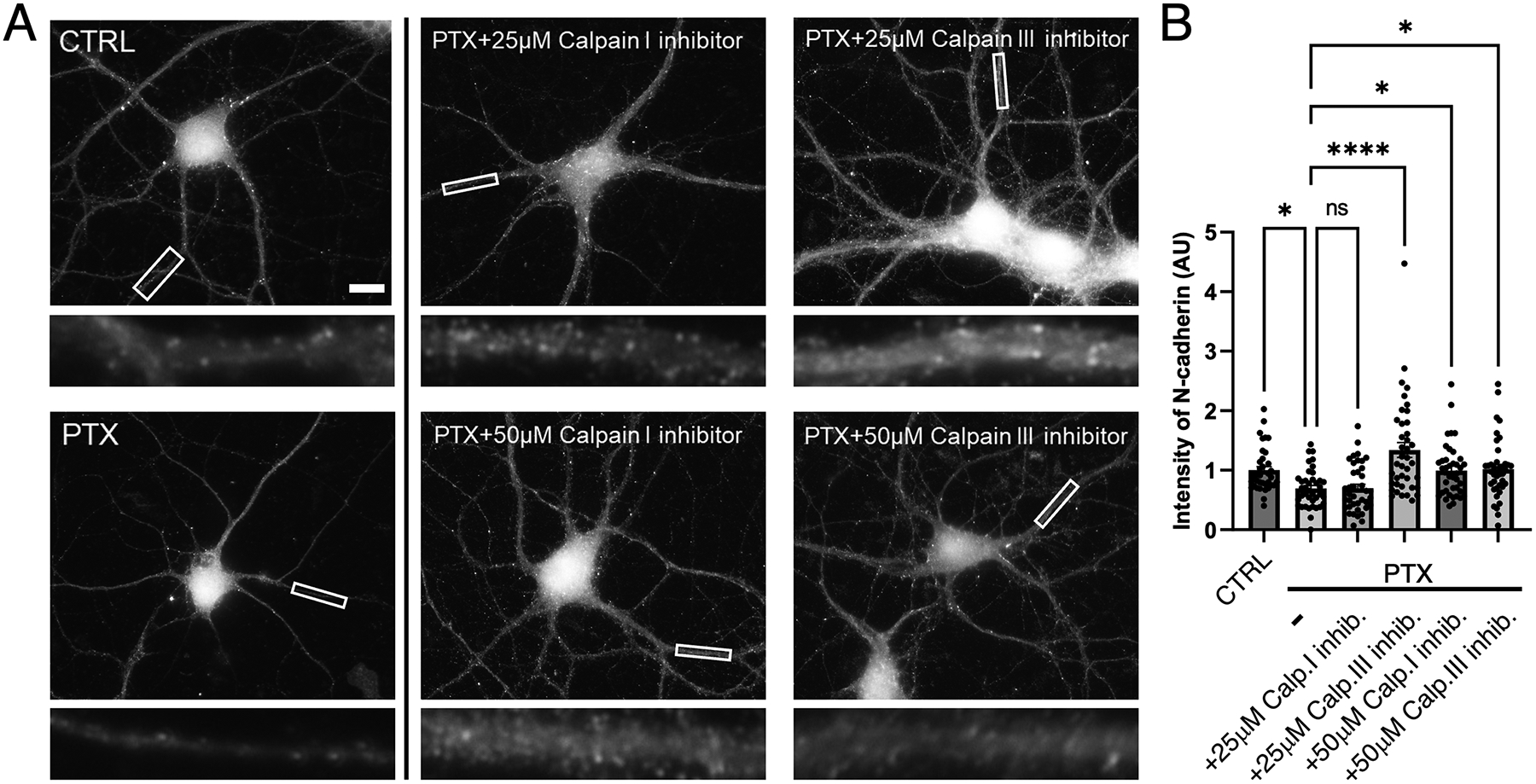
(A) Immunostaining of endogenous N-cadherin in cultured hippocampal neurons left unstimulated (CTRL), treated with PTX by itself, or treated with PTX in combination with calpain inhibitors as indicated. Higher magnification views of representative secondary dendrites (boxed regions) are shown below each neuron. Scale, 10 μm. (B) Quantification of images from (A) (p < 0.0001, F=9.159, DF=5) (n=36–40 neurons; one-way ANOVA and Sidak’s post-hoc test, *p<0.05, ****p<0.0001, ns=nonsignificant).
Next, we wanted to determine if these proteolytic cleavage products are ultimately degraded by the proteasome or lysosome. We utilized two pharmacological inhibitors of the proteasome (MG-132 and lactacystin), but neither inhibitor rescued the loss of N-cadherin induced by PTX (1 μM or 10 μM for 5 hrs), suggesting that N-cadherin is not degraded by the proteasome. We then tested two pharmacological inhibitors of the lysosome (10 nM bafilomycin and 40 μM chloroquine), and found that lysosome inhibition for 4 hours resulted in rescue of N-cadherin during hyperexcitation (Figure 8). Collectively, these data suggest that hyperexcitation results in an upregulation of Plk2, which signals proteolytic cleavage of N-cadherin by ADAM10 and calpains, followed by degradation of C-terminal cleavage products by the lysosome.
Figure 8. N-cadherin is degraded by the lysosome.
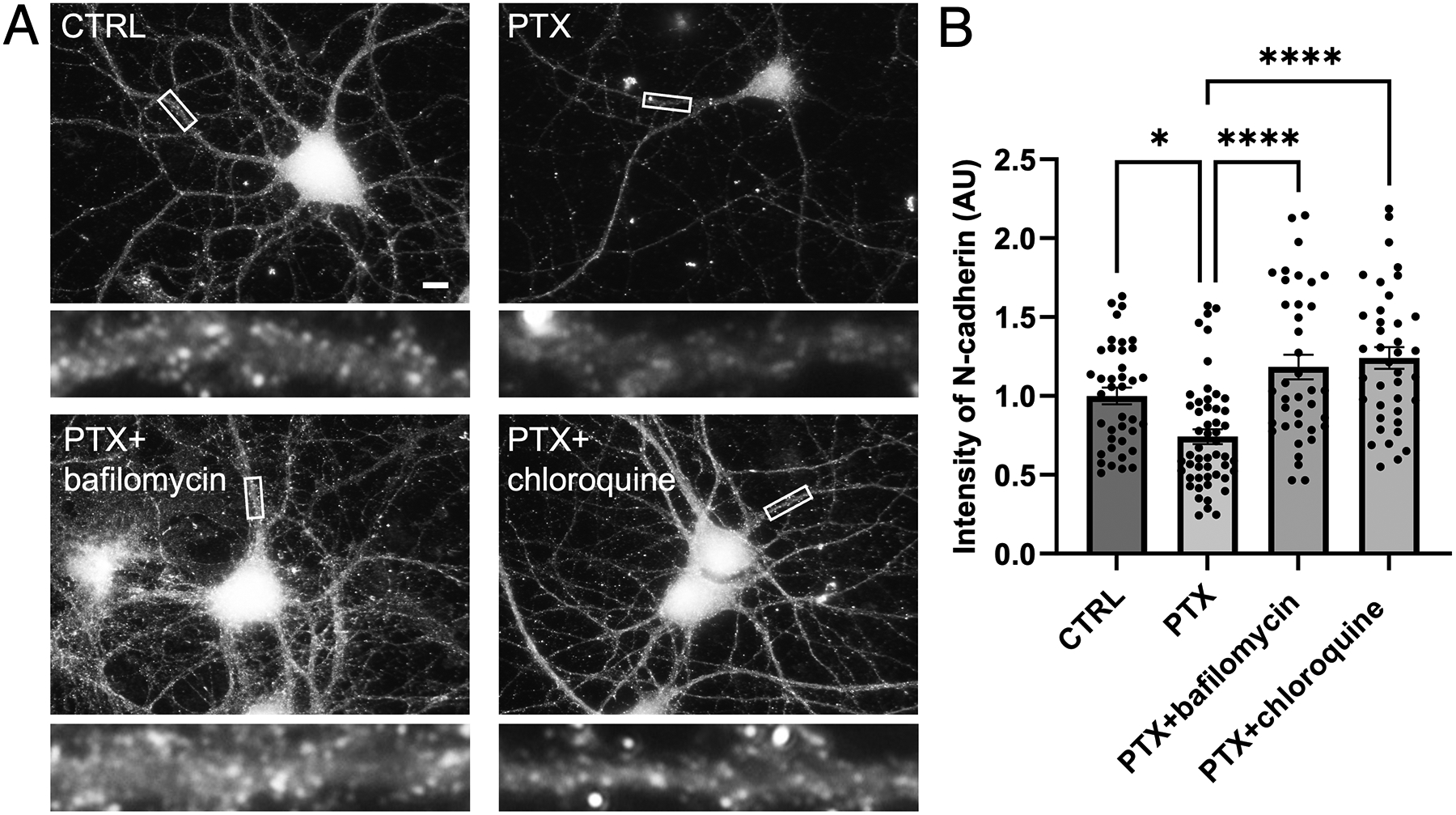
(A) Immunostaining of endogenous N-cadherin in cultured hippocampal neurons left unstimulated (CTRL), treated with PTX alone, or treated with PTX in combination with lysosome inhibitor bafilomycin or chloroquine. Higher magnification views of representative secondary dendrites (boxed regions) are shown below each neuron. Scale, 10 μm. (B) Quantification of images in (A) (p< 0.0001, F=14.87, DF=3) (n=39–52 neurons; one-way ANOVA and Tukey’s multiple comparison test, *p<0.05, ****p<0.0001, ns=nonsignificant).
To understand the functional role of the loss of N-cadherin on the synapse, we examined the expression of the postsynaptic marker PSD-95 as a readout of synapse stability. Global stimulation of synaptic activity by PTX resulted in the loss of both N-cadherin and PSD-95 after 24 hours (Supplementary Figure 2). To exclude the possibility that the loss of N-cadherin is simply a secondary effect of synapse elimination, neurons were stimulated with PTX over a time course of 4, 8, and 16 hours and immunostained for N-cadherin and PSD-95. The results indicate that at 4 hours there is no change yet in the expression of N-cadherin and PSD-95 with PTX (Supplementary Figure 3A). At the 8-hour PTX timepoint, there was no significant change in PSD-95, however, unexpectedly there was increased expression of N-cadherin (Supplementary Figure 3B). After 16 hours of PTX treatment, both PSD95 and N-cadherin were reduced in secondary dendrites and the soma, but to different degrees (Supplementary Figure 3C). These results support the idea that N-cadherin levels are not simply changing as a result of altered synapse size or density.
Previous findings demonstrated functional and physical interactions among APP, N-cadherin, and GluA2 (K. J. Lee et al., 2010; Saglietti et al., 2007b). Therefore, we wished to examine changes in AMPAR surface expression during hyperexcitation, and whether such regulation required the cleavage of N-cadherin and APP. During hyperexcitation, there is a reduction of surface GluA2 (sGluA2) receptors in comparison to unstimulated control. In the presence of beta secretase inhibitor (BSI) to block APP beta processing, sGluA2 levels were still reduced with PTX. However, PTX with the addition of GI254023X alone, or GI254023X in combination with BSI, did rescued sGluA2 levels (Figure 9), suggesting that N-cadherin cleavage may be important for removal of surface AMPARs during homeostatic adaptation to hyperexcitation.
Figure 9. Inhibition of APP and N-cadherin cleavage rescues loss of surface GluA2 during hyperexcitation.
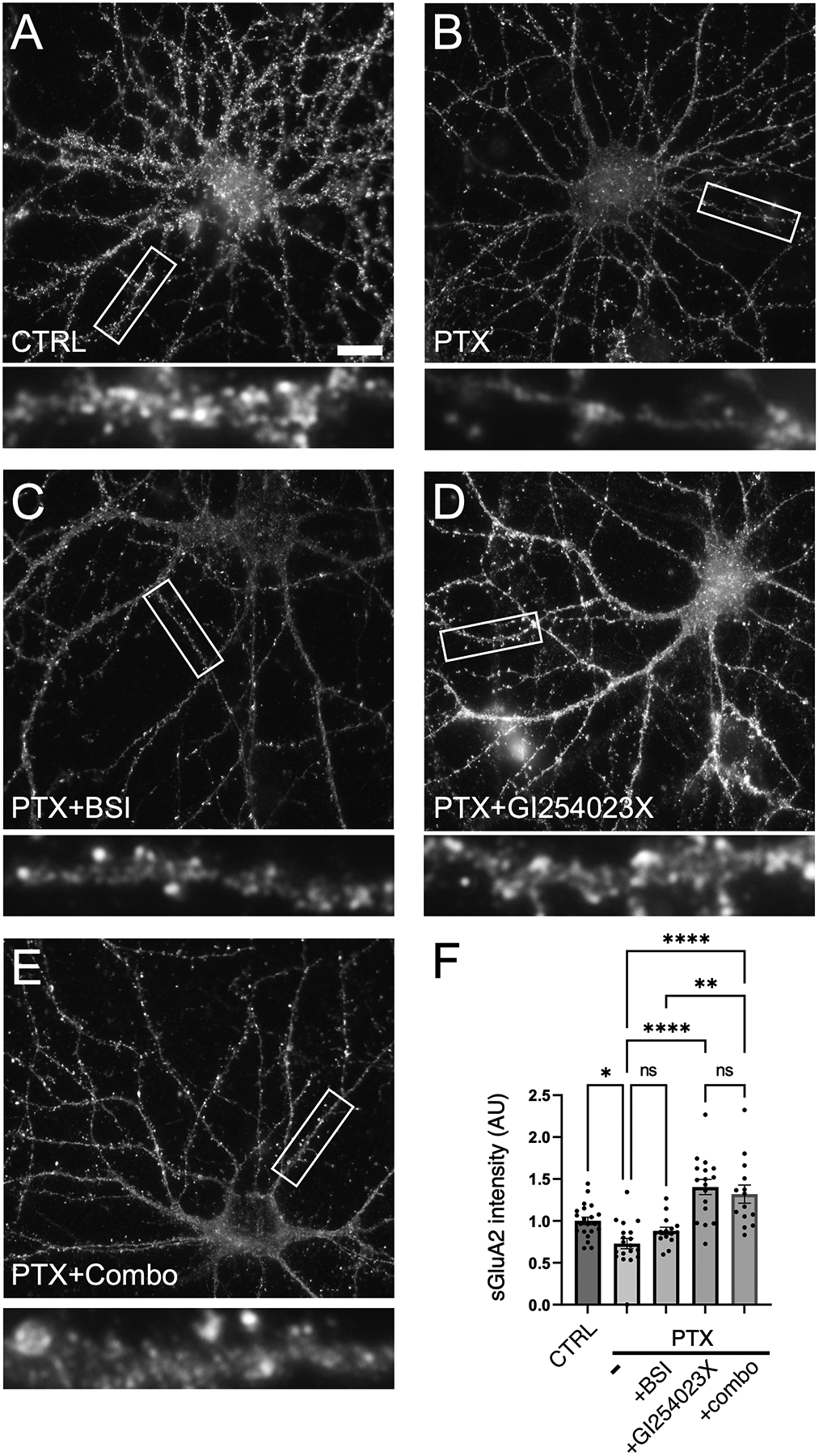
(A-E) Immunostaining of hippocampal neurons for surface GluA2 (sGluA2) (A) left unstimulated (CTRL), or treated with (B) PTX alone, (C) PTX and beta secretase inhibitor (BSI), (D) PTX and ADAM10 inhibitor GI254023X, or (E) PTX and both BSI+GI254023X (combo). Higher magnification views of representative secondary dendrites (boxed regions) are shown below each neuron. Scale, 10 μm. (F) Quantification of images in (A-E) (p< 0.0001, F= 17.29, DF=3) n=14–21; one way ANOVA and Tukey’s multiple comparison test, *p<0.05, **p<0.001, ****p<0.0001, ns=nonsignificant).
Discussion
In this study, we examined molecular mechanisms contributing to the homeostatic loss of synapses that occurs as a compensatory response to chronic neuronal hyperexcitation. During overactivity of neurons, expression of the homeostatic regulator Plk2 is known to be induced, resulting in the phosphorylation of synaptic substrates and downregulation of excitatory synapses. By screening several synaptic CAMs, here we identified N-cadherin as a novel and selective Plk2 target. Plk2 caused the dramatic loss of N-cadherin expression, both in heterologous cells and in hippocampal neurons. This effect was dependent on Plk2 kinase activity, and indeed N-cadherin could be directly phosphorylated by Plk2 in a purified system. Moreover, hyperexcitation induced the removal of N-cadherin puncta from excitatory synapses in a manner requiring Plk2 kinase activity. Examination of MAP2 and previous studies of other synaptic proteins such as liprin (Lee et al., 2011) further underscored the selectivity of the effect on N-cadherin. Together, these findings indicate that this mechanism constitutes a physiological pathway of specific CAM disassembly during homeostatic synaptic plasticity.
Using various pharmacological inhibitors, we identified proteases responsible for the loss of N-cadherin in hyperexcitation. MMPs have been shown to cleave N-cadherin resulting in an N-terminal fragment and CTF-1 that is further cleaved by presenilin and ultimately degraded (Marambaud et al., 2003; Reiss et al., 2005). This cleavage is activity dependent, as it is enhanced by bicuculline, a GABAA receptor inhibitor similar to PTX (Malinverno et al., 2010). Our findings confirmed that the selective ADAM10 inhibitor GI254023X rescued N-cadherin loss during hyperexcitation. ADAM10 is known to localize to excitatory synapses, and inhibition of ADAM10 cleavage of N-cadherin leads to increased dendritic spine size (Malinverno et al., 2010) as well as improvement in spatial working memory (Asada-Utsugi et al., 2021). Our data build upon this idea that proteolytic cleavage of N-cadherin results in reductions in synaptic efficacy, expanding such function to govern homeostatic weakening and loss of synapses in hyperexcitation.
In addition to MMPs, we found that calpains also contribute to N-cadherin degradation during neuronal overactivity. In the CNS, calpains are widely expressed and play roles in cell death, proliferation, and synaptic plasticity (Baudry et al., 2013), while several calpain substrates are important in neurodegeneration affecting Aβ production, tau phosphorylation, and disrupted neuronal structure (Atherton et al., 2014; Jin et al., 2015; Lynch & Baudry, 1987). Inhibiting calpain I and calpain II through gene knockout caused decreased dendritic spine number, reduced expression of NMDAR and AMPAR subunits, lowered levels of PSD-95, and impairment in LTPs and learning and memory (Amini et al., 2013). Furthermore, a reduction of calpains in vitro (Bednarski et al., 1995) or in vivo ameliorates cell death in glutamate- or NMDA-induced excitotoxicity (Amini et al., 2013). Conversely, activation of calpain results in the degradation of several synaptic scaffolding proteins such as GRIP-1 (Lu et al., 2000)(Jourdi et al., 2005; Lu et al., 2001). GRIP-1 is responsible for trafficking of sGluA2 receptors to control synaptic scaling (Liang et al., 2017; Tan et al., 2015), and GRIP-1 also links N-cadherin to AMPARs (Heisler et al., 2014). Degradation of GRIP-1 in response to hyperexcitation results in the reduction of sGluA2 and internalization of receptors (Tan et al., 2015). Thus, both inhibition and activation of calpains appears to perturb synapse structure and function, and the precise balance of calpain levels may be needed to preserve N-cadherin integrity and synapse stability.
To address the functional role of N-cadherin on the synapse in response to hyperexcitation, we analyzed the distribution and levels of PSD-95 as a marker of excitatory synapses. PSD-95 is a scaffold protein that creates slots for stabilization of AMPARs (Malinow & Malenka, 2002; Newpher & Ehlers, 2008; Schnell et al., 2002), and as such the amount of PSD-95 at synapses is an index of AMPAR content. During homeostatic synaptic scaling, hyperexcitation reduces synaptic markers such as PSD-95 and PSD-93 (Sun & Turrigiano, 2011). N-cadherin knockdown in vitro (Aiga et al., 2011) or knockout in vivo (Stan et al., 2010b) also decreases PSD-95. Our results showed that a selective inhibitor of Plk2 rescues both PSD-95 and N-cadherin in response to hyperexcitation.
To test whether N-cadherin degradation led to loss of PSD-95, or whether loss of excitatory synapses led to the disappearance of N-cadherin as a secondary consequence, we performed a time course from 4–24 hrs. N-cadherin and PSD-95 generally displayed similar changes at all time points tested except at 8 hrs, at which there was no change in PSD-95 intensity but an unexpected increase in the expression of N-cadherin with PTX stimulation. These findings demonstrate that N-cadherin dynamics does not simply follow PSD-95 levels passively. However, after the 8 hr time point Plk2 is known to be induced, which leads to a dramatic decrease in both N-cadherin and PSD-95. It may be that at early time points, a relatively brief period of heightened synaptic activity upregulates N-cadherin expression to increase adhesive properties and maintain synapse number. In contrast, more prolonged overactivity prompts a more severe response necessitating synapse elimination.
Lastly, we and others have shown functional and physical interactions between N-cadherin, APP, and AMPARs (Okuda et al., 2007; Saglietti et al., 2007a; Tai et al., 2008; Xie et al., 2008). Our colocalization data in particular argue that APP and N-cadherin are tightly coregulated during plasticity, maintaining relatively consistent co-localization at synapses while undergoing dramatic changes in synaptic protein levels. To examine the relative importance of the degradation of these synaptic CAMs to changes in surface AMPAR subunit GluA2, we blocked the cleavage of both N-cadherin and APP using a combination of ADAM10 and beta secretase inhibitors. During hyperexcitation, sGluA2 levels were reduced but BSI treatment did not block this AMPAR downregulation. However, ADAM10 inhibitor rescued sGluA2 levels, and a combination of both drugs further increased sGluA2, consistent with a synergistic effect between N-cadherin and APP on determining AMPAR levels.
Collectively, these studies suggest that Plk2 initiates a signaling cascade beginning with N-cadherin and APP processing, which triggers downstream removal of PSD-95 that, in conjunction with other destabilizing events such as GRIP-1 degradation, culminates in the synaptic scaling of AMPARs. Further understanding the molecular underpinnings of the synapse provided by synaptic CAMs such as N-cadherin and APP and their remodeling in synaptic plasticity is essential for dissecting abnormalities in synapse function that underlie neurodegenerative diseases and other cognitive disorders.
Supplementary Material
FUNDING
This work was supported by NIH grant RF1 AG056603 (to DTSP).
Abbreviations
- Aβ
Amyloid beta
- AD
Alzheimer’s disease
- ANOVA
Analysis of variance
- APP
Amyloid precursor protein
- BSI
Beta secretase inhibitor
- Cdh8
Cadherin 8
- Cdh9
Cadherin 9
- Cdh10
Cadherin 10
- Cdh13
Cadherin 13
- CAMs
Cellular adhesion molecules
- CBP
CREB binding protein
- CTF-1
C-terminal fragment 1
- CTF-2
C-terminal fragment 2
- ECL
Enhanced chemiluminescence
- EphA4
Ephrin A4
- EphB2
Ephrin B2
- HBSS
Hank’s Buffer Saline Solution
- LTP
Long term potentiation
- MMPs
Matrix metalloproteinases
- NGS
Normal goat serum
- NTF
N-terminal fragment
- PBS
Phosphate buffer solution
- Plk2
Polo like kinase 2
- Plk2-CA
Constitutively active Plk2
- Plk2-KD
Kinase-dead Plk2
- PTX
Picrotoxin
- PS1
Presenilin
- RRID
Research Resource Identifier
- sGluA2
Surface GluA2
- TrkB
Tropomyosin receptor kinase B
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Aiga M, Levinson JN, & Bamji SX (2011). N-cadherin and neuroligins cooperate to regulate synapse formation in hippocampal cultures. The Journal of Biological Chemistry, 286(1), 851–858. 10.1074/jbc.M110.176305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amini M, Ma C-L, Farazifard R, Zhu G, Zhang Y, Vanderluit J, Zoltewicz JS, Hage F, Savitt JM, Lagace DC, Slack RS, Beique J-C, Baudry M, Greer PA, Bergeron R, & Park DS (2013). Cellular/Molecular Conditional Disruption of Calpain in the CNS Alters Dendrite Morphology, Impairs LTP, and Promotes Neuronal Survival following Injury. 10.1523/JNEUROSCI.4247-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Uemura K, Kuzuya A, Maesako M, Asada-Utsugi M, Kubota M, Aoyagi N, Yoshioka K, Okawa K, Inoue H, Kawamata J, Shimohama S, Arai T, Takahashi R, & Kinoshita A (2011). N-cadherin regulates p38 MAPK signaling via association with JNK-associated leucine zipper protein: implications for neurodegeneration in Alzheimer disease. The Journal of Biological Chemistry, 286(9), 7619–7628. 10.1074/jbc.M110.158477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arikkath J, & Reichardt LF (2008). Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends in Neurosciences, 31(9), 487. 10.1016/J.TINS.2008.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada-Utsugi M, Uemura K, Kubota M, Noda Y, Tashiro Y, Uemura TM, Yamakado H, Urushitani M, Takahashi R, Hattori S, Miyakawa T, Ageta-Ishihara N, Kobayashi K, Kinoshita M, & Kinoshita A (2021). Mice with cleavage-resistant N-cadherin exhibit synapse anomaly in the hippocampus and outperformance in spatial learning tasks. Molecular Brain, 14(1), 1–16. 10.1186/S13041-021-00738-1/FIGURES/8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada-Utsugi M, Uemura K, Noda Y, Kuzuya A, Maesako M, Ando K, Kubota M, Watanabe K, Takahashi M, Kihara T, Shimohama S, Takahashi R, Berezovska O, & Kinoshita A (2011). N-cadherin enhances APP dimerization at the extracellular domain and modulates Aβ production. Journal of Neurochemistry, 119(2), 354–363. 10.1111/j.1471-4159.2011.07364.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton J, Kurbatskaya K, Bondulich M, Croft CL, Garwood CJ, Chhabra R, Wray S, Jeromin A, Hanger DP, & Noble W (2014). Calpain cleavage and inactivation of the sodium calcium exchanger-3 occur downstream of Aβ in Alzheimer’s disease. Aging Cell, 13(1), 49–59. 10.1111/ACEL.12148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Liu G, Jiang Y, Jiang Q, Liao M, Feng R, Zhang L, Ma G, Zhang S, Chen Z, Zhao B, Wang R, Li K, & Liu G (2015). Cell adhesion molecule pathway genes are regulated by cis-regulatory SNPs and show significantly altered expression in Alzheimer’s disease brains. Neurobiology of Aging, 36(10), 2904.e1–2904.e7. 10.1016/j.neurobiolaging.2015.06.006 [DOI] [PubMed] [Google Scholar]
- Basu R, Taylor MR, & Williams ME (2015). The classic cadherins in synaptic specificity. Cell Adhesion & Migration, 9(3), 193–201. 10.1080/19336918.2014.1000072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M, Chou MM, & Bi X (2013). Targeting calpain in synaptic plasticity. Expert Opinion on Therapeutic Targets, 17(5), 579. 10.1517/14728222.2013.766169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarski E, Vanderklish P, Gall C, Saido TC, Bahr BA, & Lynch G (1995). Translational suppression of calpain I reduces NMDA-induced spectrin proteolysis and pathophysiology in cultured hippocampal slices. Brain Research, 694(1–2), 147–157. 10.1016/0006-8993(95)00851-G [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, & Huntley GW (2000). Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron, 28(1), 245–259. http://www.ncbi.nlm.nih.gov/pubmed/11086998 [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang J-E, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, & Holtzman DM (2008). Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron, 58(1), 42–51. 10.1016/j.neuron.2008.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, & Cong J (2003). The calpain system. Physiological Reviews, 83(3), 731–801. 10.1152/PHYSREV.00029.2002 [DOI] [PubMed] [Google Scholar]
- Harris KM (2020). Structural LTP: from synaptogenesis to regulated synapse enlargement and clustering. Current Opinion in Neurobiology, 63, 189. 10.1016/J.CONB.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisler FF, Lee HK, Gromova K. v., Pechmann Y, Schurek B, Ruschkies L, Schroeder M, Schweizer M, & Kneussel M (2014). GRIP1 interlinks N-cadherin and AMPA receptors at vesicles to promote combined cargo transport into dendrites. Proceedings of the National Academy of Sciences of the United States of America, 111(13), 5030–5035. 10.1073/PNAS.1304301111/-/DCSUPPLEMENTAL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruska M, Henderson N, le Marchand SJ, Jafri H, & Dalva MB (2018). Synaptic nanomodules underlie the organization and plasticity of spine synapses. Nature Neuroscience, 21(5), 671. 10.1038/S41593-018-0138-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, & Malinow R (2006). AMPAR Removal Underlies Aβ-Induced Synaptic Depression and Dendritic Spine Loss. Neuron, 52(5), 831–843. 10.1016/j.neuron.2006.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YN, Jung YS, Soo HL, Moon CH, Kim CH, & Eun JB (2009). Calpain-mediated N-cadherin proteolytic processing in brain injury. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 29(18), 5974–5984. 10.1523/JNEUROSCI.6178-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin N, Yin X, Yu D, Cao M, Gong CX, Iqbal K, Ding F, Gu X, & Liu F (2015). Truncation and activation of GSK-3β by calpain I: a molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Scientific Reports, 5. 10.1038/SREP08187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdi H, Lu X, Yanagihara T, Lauterborn JC, Bi X, Gall CM, & Baudry M (2005). Prolonged Positive Modulation of α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA) Receptors Induces Calpain-Mediated PSD-95/Dlg/ZO-1 Protein Degradation and AMPA Receptor Down-Regulation in Cultured Hippocampal Slices. Journal of Pharmacology and Experimental Therapeutics, 314(1), 16–26. 10.1124/JPET.105.083873 [DOI] [PubMed] [Google Scholar]
- Jüngling K, Eulenburg V, Moore R, Kemler R, Lessmann V, & Gottmann K (2006). N-Cadherin Transsynaptically Regulates Short-Term Plasticity at Glutamatergic Synapses in Embryonic Stem Cell-Derived Neurons. Journal of Neuroscience, 26(26), 6968–6978. 10.1523/JNEUROSCI.1013-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Lee Y, André EA, Lee KJ, Nguyen T, Feng Y, Jia N, Harris BT, Burns MP, & Pak DTS (2019). Inhibition of Polo-like kinase 2 ameliorates pathogenesis in Alzheimer’s disease model mice. PLoS ONE, 14(7). 10.1371/JOURNAL.PONE.0219691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Lee Y, Rozeboom A, Lee JY, Udagawa N, Hoe HS, & Pak DTS (2011). Requirement for Plk2 in orchestrated Ras and Rap signaling, homeostatic structural plasticity, and memory. Neuron, 69(5), 957. 10.1016/J.NEURON.2011.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Moussa CEH, Lee Y, Sung Y, Howell BW, Turner RS, Pak DTS, & Hoe HS (2010). Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience, 169(1), 344–356. 10.1016/j.neuroscience.2010.04.078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Lee JS, Lee KJ, Turner RS, Hoe H-S, & Pak DTS (2017). Polo-like kinase 2 phosphorylation of amyloid precursor protein regulates activity-dependent amyloidogenic processing. Neuropharmacology, 117, 387–400. 10.1016/j.neuropharm.2017.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Li JL, Han Y, Luo YX, Xue YX, Zhang Y, Zhang Y, Zhang LB, Chen ML, Lu L, & Shi J (2017). Calpain-GRIP Signaling in Nucleus Accumbens Core Mediates the Reconsolidation of Drug Reward Memory. The Journal of Neuroscience, 37(37), 8938. 10.1523/JNEUROSCI.0703-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Rong Y, & Baudry M (2000). Calpain-mediated degradation of PSD-95 in developing and adult rat brain. Neuroscience Letters, 286(2), 149–153. 10.1016/S0304-3940(00)01101-0 [DOI] [PubMed] [Google Scholar]
- Lu X, Wyszynski M, Sheng M, & Baudry M (2001). Proteolysis of glutamate receptor-interacting protein by calpain in rat brain: implications for synaptic plasticity. Journal of Neurochemistry, 77(6), 1553–1560. 10.1046/J.1471-4159.2001.00359.X [DOI] [PubMed] [Google Scholar]
- Lynch G, & Baudry M (1984). The Biochemistry of Memory: A New and Specific Hypothesis. Science, 224(4653), 1057–1063. 10.1126/SCIENCE.6144182 [DOI] [PubMed] [Google Scholar]
- Lynch G, & Baudry M (1987). Brain spectrin, calpain and long-term changes in synaptic efficacy. Brain Research Bulletin, 18(6), 809–815. 10.1016/0361-9230(87)90220-6 [DOI] [PubMed] [Google Scholar]
- Malinow R, & Malenka RC (2002). AMPA receptor trafficking and synaptic plasticity. Annual Review of Neuroscience, 25, 103–126. 10.1146/ANNUREV.NEURO.25.112701.142758 [DOI] [PubMed] [Google Scholar]
- Malinverno M, Carta M, Epis R, Marcello E, Verpelli C, Cattabeni F, Sala C, Mulle C, di Luca M, & Gardoni F (2010). Synaptic Localization and Activity of ADAM10 Regulate Excitatory Synapses through N-Cadherin Cleavage. The Journal of Neuroscience, 30(48), 16343. 10.1523/JNEUROSCI.1984-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, & Robakis NK (2003). A CBP Binding Transcriptional Repressor Produced by the PS1/ϵ-Cleavage of N-Cadherin Is Inhibited by PS1 FAD Mutations. Cell, 114(5), 635–645. 10.1016/J.CELL.2003.08.008 [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GCR, Nemoto T, Miyashita Y, Iino M, & Kasai H (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nature Neuroscience 2001 4:11, 4(11), 1086–1092. 10.1038/nn736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreland T, & Poulain FE (2022). To Stick or Not to Stick: The Multiple Roles of Cell Adhesion Molecules in Neural Circuit Assembly. Frontiers in Neuroscience, 16. 10.3389/FNINS.2022.889155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newpher TM, & Ehlers MD (2008). Glutamate receptor dynamics in dendritic microdomains. Neuron, 58(4), 472–497. 10.1016/J.NEURON.2008.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Lujan R, Laube G, Roberts JDB, Molnar E, & Somogyi P (1998). Cell Type and Pathway Dependence of Synaptic AMPA Receptor Number and Variability in the Hippocampus. Neuron, 21(3), 545–559. 10.1016/S0896-6273(00)80565-6 [DOI] [PubMed] [Google Scholar]
- Okuda T, Yu LMY, Cingolani LA, Kemler R, & Goda Y (2007). β-Catenin regulates excitatory postsynaptic strength at hippocampal synapses. Proceedings of the National Academy of Sciences of the United States of America, 104(33), 13479–13484. 10.1073/PNAS.0702334104/ASSET/223235A3-5C78-439A-9E55-390540361F29/ASSETS/GRAPHIC/ZPQ0330772850005.JPEG [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielarski KN, van Stegen B, Andreyeva A, Nieweg K, Jüngling K, Redies C, & Gottmann K (2013). Asymmetric N-cadherin expression results in synapse dysfunction, synapse elimination, and axon retraction in cultured mouse neurons. PloS One, 8(1), e54105. 10.1371/journal.pone.0054105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss K, Maretzky T, Ludwig A, Tousseyn T, de Strooper B, Hartmann D, & Saftig P (2005). ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. The EMBO Journal, 24(4), 742–752. 10.1038/sj.emboj.7600548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, & Passafaro M (2007a). Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron, 54(3), 461–477. 10.1016/J.NEURON.2007.04.012 [DOI] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset M-C, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, & Passafaro M (2007b). Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron, 54(3), 461–477. 10.1016/j.neuron.2007.04.012 [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, & Nicoll RA (2002). Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proceedings of the National Academy of Sciences of the United States of America, 99(21), 13902–13907. 10.1073/PNAS.172511199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Feliu-Mojer M, Gaiottino J, Pak DTS, & Sheng M (2008). Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron, 58(4), 571. 10.1016/J.NEURON.2008.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, & Sheng M (2008). Activity-Induced Polo-Like Kinase 2 Is Required for Homeostatic Plasticity of Hippocampal Neurons during Epileptiform Activity. Journal of Neuroscience, 28(26), 6583–6591. 10.1523/JNEUROSCI.1853-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong E, Yuan L, & Arikkath J (2015). Cadherins and catenins in dendrite and synapse morphogenesis. In Cell Adhesion and Migration (Vol. 9, Issue 3, pp. 202–213). Taylor and Francis Inc. 10.4161/19336918.2014.994919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan A, Pielarski KN, Brigadski T, Wittenmayer N, Fedorchenko O, Gohla A, Lessmann V, Dresbach T, & Gottmann K (2010a). Essential cooperation of N-cadherin and neuroligin-1 in the transsynaptic control of vesicle accumulation. Proceedings of the National Academy of Sciences of the United States of America, 107(24), 11116–11121. 10.1073/pnas.0914233107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan A, Pielarski KN, Brigadski T, Wittenmayer N, Fedorchenko O, Gohla A, Lessmann V, Dresbach T, & Gottmann K (2010b). Essential cooperation of N-cadherin and neuroligin-1 in the transsynaptic control of vesicle accumulation. Proceedings of the National Academy of Sciences of the United States of America, 107(24), 11116–11121. 10.1073/PNAS.0914233107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, & Turrigiano GG (2011). PSD-95 and PSD-93 Play Critical But Distinct Roles in Synaptic Scaling Up and Down. The Journal of Neuroscience, 31(18), 6800. 10.1523/JNEUROSCI.5616-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai CY, Kim SA, & Schuman EM (2008). Cadherins and synaptic plasticity. Current Opinion in Cell Biology, 20(5), 567–575. 10.1016/J.CEB.2008.06.003 [DOI] [PubMed] [Google Scholar]
- Tan HL, Queenan BN, & Huganir RL (2015). GRIP1 is required for homeostatic regulation of AMPAR trafficking. Proceedings of the National Academy of Sciences of the United States of America, 112(32), 10026–10031. 10.1073/PNAS.1512786112/-/DCSUPPLEMENTAL [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, & Takeichi M (2002). Cadherin regulates dendritic spine morphogenesis. Neuron, 35(1), 77–89. 10.1016/S0896-6273(02)00748-1 [DOI] [PubMed] [Google Scholar]
- Uemura K, Ando K, Aoyagi N, Shimohama S, Takahashi R, & Kinoshita A (2008). P4–262: N-cadherin–based adhesion enhances Abeta release and decreases Abeta42/40 ratio. Alzheimer’s & Dementia, 4(4), T748. 10.1016/j.jalz.2008.05.2331 [DOI] [Google Scholar]
- Xie Z, Photowala H, Cahill ME, Srivastava DP, Woolfrey KM, Shum CY, Huganir RL, & Penzes P (2008). Coordination of synaptic adhesion with dendritic spine remodeling by AF-6 and kalirin-7. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 28(24), 6079–6091. 10.1523/JNEUROSCI.1170-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.