

Abstract
Background:
Inherited Protein C deficiency (PCD) caused by mutations in Protein C gene (PROC) increases the risk of thrombosis. Missense mutations in PC’s signal peptide and propeptide have been reported in PCD patients, but their pathogenic mechanisms, except mutations in R42 residue, remain unclear.
Objectives:
To investigate the pathogenic mechanisms of inherited PCD caused by 11 naturally occurring missense mutations in PC’s signal peptide and propeptide.
Methods:
Through cell-based assays, we evaluated the impact of these mutations on various aspects such as the activities and antigens of secreted PC, intracellular PC expression, subcellular localization of a reporter protein, and propeptide cleavage. Additionally, we investigated their effect on pre-mRNA splicing using a minigene splicing assay.
Results:
Our data revealed that certain missense mutations (L9P, R32C, R40C, R38W, and R42C) disrupt PC secretion by impeding co-translational translocation to the ER or by causing ER retention. Additionally, some mutations (R38W, R42L/H/S) result in abnormal propeptide cleavage. However, a few missense mutations (Q3P, W14G, and V26M) do not account for PCD. Through a minigene splicing assay, we observed that several variations (c.8A>C, c.76G>A, c.94C>T, and c.112C>T) increase the incidence of aberrant pre-mRNA splicing.
Conclusion:
Our findings suggest that the variations in PC’s signal peptide and propeptide have varying effects on the biological process of PC, including posttranscriptional pre-mRNA splicing, translation, and posttranslational processing. Additionally, one variation could affect the biological process of PC at multiple levels. Except W14G, our results provide a clear understanding of the relationship between PROC genotype and inherited PCD.
Keywords: thrombophilia, inherited Protein C deficiency (PCD), signal peptide and propeptide, pathogenic mechanism, aberrant pre-mRNA splicing, missense mutation
Introduction
Protein C (PC), an important regulatory protein of the coagulation cascade, inhibits coagulation by cleaving critical sites in the activated procoagulant factors V and VIII, thus inactivating these enzymes. PC deficiency (PCD) is a heritable or acquired risk factor for thrombophilia, with clinical presentations varying from asymptomatic individuals, through venous thromboembolism (VTE), to acute life-threatening complications, such as purpura fulminans and disseminated intravascular coagulation (DIC) [1]. Mutations in PC gene (PROC) cause inherited PCD. Monoallelic mutations of PROC are much more frequent (1 in 200 to 1 in 500 births) [2] and usually cause partial deficiency, with presentations varying from asymptomatic individuals to VTE or pulmonary embolism [3]. Biallelic mutations of PROC are extremely rare (1 in 500 000 to 1 in 750 000 births) [4] and generally cause severe PCD, with striking clinical presentations occurring in infants, such as purpura fulminans, renal vein thrombosis, and vitreal vein thrombosis [5]. Typically, these mutations cause either quantitative PCD (type I) or functional PCD (type II) [6]. Type I PCD is characterized by concordant decreases in both PC activity and antigen concentration. Type II PCD is defined by decreased PC activity but normal PC antigen values. Most PC mutations result in type I deficiency, and type II deficiency accounts for about 15% of symptomatic deficiency [7].
The PROC gene is located on chromosome 2q14.3 and contains 9 exons spanning 11.6 kb [8, 9]. The mature mRNA encodes a precursor protein containing 461 amino acids (aa), which contains an 18-residue signal peptide and a 24-residue propeptide, followed by a 419-residue mature protein that is composed of a γ-carboxy-glutamic acid residue (Gla) domain, two epidermal growth factor (EGF)-like domains, a linker, an activation peptide, and a serine protease domain (Figure S1A). So far, over 500 mutations identified throughout the gene length may lead to inherited PCD by impairing protein synthesis and/or function [7]. The types of gene defects include missense mutations, nonsense mutations, splice site mutations, deletions, and insertions. Among them, missense mutations throughout the PROC coding region are the most prevalent. Eleven missense mutations related to inherited PCD have been reported in signal peptide and propeptide regions of PC [10].
PC is a vitamin K-dependent secretory glycoprotein. The signal peptide and propeptide are necessary for PC maturation. The signal peptide directs the co-translational translocation of nascent PC polypeptide into the endoplasmic reticulum (ER) lumen for secretion. The propeptide is required for vitamin K-dependent γ-carboxylation of PC’s Gla domain by γ-glutamyl carboxylase (GGCX) [11]. Proper cleavage of the propeptide between residues R42 and A43 is required to generate the functional mature PC. PCD patients with mutations in residue R42 usually cause aberrant propeptide cleavage [12–14]. However, it remains elusive about the molecular mechanism to cause inherited PCD by other missense mutations in PC’s signal peptide and propeptide regions. Here, we systemically study these mutations by multiple methods, and found that they may cause PCD by differently affecting the biological process of PC, including aberrant pre-mRNA splicing, failure of co-translational translocation to ER, ER retention and/or uncleaved propeptide. Unveiling the molecular mechanisms will help us to understand their inheritance pattern causing PCD and to evaluate their pathogenic risk, which may be benefit for the diagnosis and treatment of PCD patients.
Material and Methods
Mutations selection and nomenclature of PC
The missense mutations in PC’s signal peptide and propeptide were selected from a literature [10]. The detailed information of patients was collected in Supplementary table 1, and the mutations were described following the nomenclature system of the Human Genome Variation Society (HGVS, http://www.hgvs.org/mutnomen). The sequence NP_000303.1 is used as a reference for amino acid numbers, and numbered from the first residue of PC precursor. The NM_000312.4 is used as a reference of PROC mRNA. Sequence variations were numbered from the first nucleotide of the first codon “ATG” according to the HGVS recommendations.
Plasmid constructions
The cDNA of human PROC gene was subcloned into the EF1ɑ multicloning site of the pBud CE4.1 vector. The construct was used to detect PC’s anticoagulant and amidolytic activities, and PC’s expression and secretion.
To detect intracellular localization of PC mutants, a fusion protein (PCspg-EGFP) was designed which contains the N-terminal segment of PC, followed by mEGFP (Figure S1B, supplementary file 1). The fusion gene was subcloned into the EF1ɑ multicloning site of the pBud CE4.1 vector, and the PDI-mCherry fusion protein (ER location) or mScarlet-Giantin (Golgi location) was subcloned into the CMV multi-cloning site.
To evaluate the propeptide cleavage efficiency in mutants, a fusion protein (Flag-PCspg-MBP-His) was constructed firstly, which contains the N-terminal segment of PC, followed by MBP-His, then a flag tag was added between the signal peptide and propeptide (Figure S1C, supplementary file 1). The fusion gene was subcloned into multicloning site of pcDNA3.1.
For the minigene splicing assay, a 1928bp genomic sequence in PROC gene locus, from 200bp upstream of exon 2 to 200bp downstream of exon3 (Supplementary file 1), was synthesized and subcloned into the exon trapping vector pSPL3 between Xho I and Bam HI. All the mutants according to the variations were constructed by using a FastCloning technique [15].
Accessing anticoagulant and amidolytic activities of Protein C
HEK 293T cells were used to express PC and supernatants were used to accessing anticoagulant and amidolytic activities. Briefly, the cells were transfected with different constructs in 10cm dish. After transfection, the medium was replaced by Opti-MEM containing 10μM vitamin K. For additional 36–48 hours incubation, the mediums were collected and concentrated to 50-fold by ultrafiltration centrifugal tubes (10kD, Millipore). The concentrate was used to perform PC anticoagulant activity and amidolytic activity by the Hemoclot Protein C kit (Hyphen BioMed) and by the Biophen Protein C kit (Hyphen BioMed), respectively. The activity of the mutants was normalized by wild-type PC, which was normalized to “1”.
Detection of total secreted and Ca2+-dependent Protein C by ELISA assay
The supernatant was prepared similarly as described for PC anticoagulant activity without concentration, and CaCl2 was added to the 5mM final concentration. The sandwich-based ELISA assay was performed to detect the total and the γ-carboxylated PC antigen. To detect the total PC antigen, the ELISA plate was coated by mouse anti-human PC monoclonal antibody (GMA-067, Green Mountain Antibodies; 2μg/ml, 100μl/well) overnight at 4°C. After washed by TBST buffer for five times, the plate was blocked by bovine serum albumin (BSA). Then samples were added and incubated for 2 hours at room temperature. After washed by TBST buffer, SAPC-HRP (Affinity Biologicals, 1:5000) was added and incubated for another 1 hour at room temperature. After washed by TBST, substrate ABTS was used for color development, and the absorbance value was measured at 405nm by an ELISA plate reader.
To detect the γ-carboxylated PC, the ELISA plate was coated by a Ca2+-dependent monoclonal antibody (provided by Dr. Paul Bajaj) [16, 17] and detected by SAPC-HRP. The ELISA was performed similarly to the above protocol. The antigen was normalized by wild-type, which was normalized to “1”.
Subcellular localization of reporter protein by fluorescence confocal imaging
The subcellular localization of PCspg-EGFP reporter of wild-type and mutants were examined by fluorescence confocal microscope. Briefly, the coverslips were placed in a 6-well plate before seeding the HEK 293T cells, and the reporter was transiently expressed. Transfected cells were cultured in medium with 10μM vitamin K for 36–48 hours, then fixed with 4% paraformaldehyde. The coverslip was mounted to glass slide with DAPI-Fluoromont G medium. The fluorescent image was collected by the Zeiss LSM 880 II Airyscan FAST confocal microscope. Fluorescent signal was quantified by ImageJ.
Western blot for detecting cell lysate and secreted protein
HEK 293T cells were treated in a 6-well plate similarly as described for PC anticoagulant activity. The cell lysate was collected as previous described [18]. The medium were precipitated by trichloroacetic acid (TCA) and re-suspended to perform western blot [19]. For the western blot, the samples were subjected to reduced SDS-PAGE and transferred to a PVDF membrane. The membrane was blocked by 5% skim milk in TBST, and then incubated at 4°C overnight with the primary antibodies, including SAPC-HRP, anti-Gla antibody (Biomedica diagnostics), β-actin (Proteintech), anti-Flag M2 antibody (Sigma), and anti-His antibody (Proteintech). Corresponding HRP-conjugated secondary antibodies were used to detect the primary antibodies. Signals were detected by an ECL kit (Millipore).
Propeptide cleavage assay
HEK 293T cells were transfected with the constructs for propeptide cleavage assay. After transfection, the medium was replaced by Opti-MEM containing 10μM warfarin. After 36–48 hours incubation, the medium was collected divided into two parts, one for ELISA assay directly and the other for western blot after TCA precipitation. The anti-His and anti-Flag M2 primary antibody was used to detect the total secreted reporter protein (TSRP) and the propeptide uncleaved reporter protein (PURP), respectively. ELISA assay was performed to quantify the TSRP and PURP. Briefly, the 96-well plate was coated by rabbit polyclonal MBP antibody (Proteintech), and then detected by anti-His-HRP or by anti-Flag M2-HRP to detect the TSRP or the PURP, respectively. The ELISA assay was performed similarly to detect the total PC antigen.
Minigene splicing assay
The constructs for the minigene splicing assay were transfected into HEK293 cells. Total RNAs were extracted and were reverse transcripted to cDNAs for PCR amplification. The PCR products were used for electrophoresis by 1% agarose gel, and the target bands were collected by gel purification kit and the DNA fragments were ligated into the pMD18-T vector (Clontech). The transformation was performed in DH5α competent E.coli cells. The positive colonies were selected and identified for sequencing. The sequences were analyzed by Snapgene.
Statistical analysis
Statistical analysis was calculated using Graphpad Prism7. Data for ELISA and activity results were presented as mean SD, and differences between groups were determined by Student’s t-test with significance at p<0.05. Differences in minigene splicing results were determined by Chi-square test with significance at p<0.05.
Results
1. Characterizing activities and antigens of secreted Protein C of missense mutations in signal peptide and propeptide
In a reported database, there were 27 PCD patients carrying eleven missense mutations in signal peptide and propeptide region of PC (Figure S2, Supplementary table 1) [10]. About half patients (14 in 27) have mutations in residue R42 with four different mutations (R42C/S/H/L), and the other mutations are distributed sporadically in seven residues of signal peptide and propeptide (Figure S2A). So far, mutations in R42 have been reported to cause PCD by abnormal propeptide cleavage [12–14]. However, it remains unknown how other mutations cause PCD. To elucidate the molecular basis of PCD in these mutations, we investigated the mutation effect on the anticoagulant activity, amidolytic activity, total antigen, and γ-carboxylated antigen of secreted PC. Our results showed that anticoagulant activity (Figure 1A) dramatically decreased in nine mutations, but not in Q3P and W14G. Among these nine mutations, however, amidolytic activity, total antigen, and γ-carboxylated antigen (Figure 1B, C and D) consistently decreased only in five mutations, including mutations of L9P, R32C, R38W, R40C, and R42C.
Figure 1. Relative activities and antigens of secreted Protein C with missense mutations in signal peptide and propeptide.
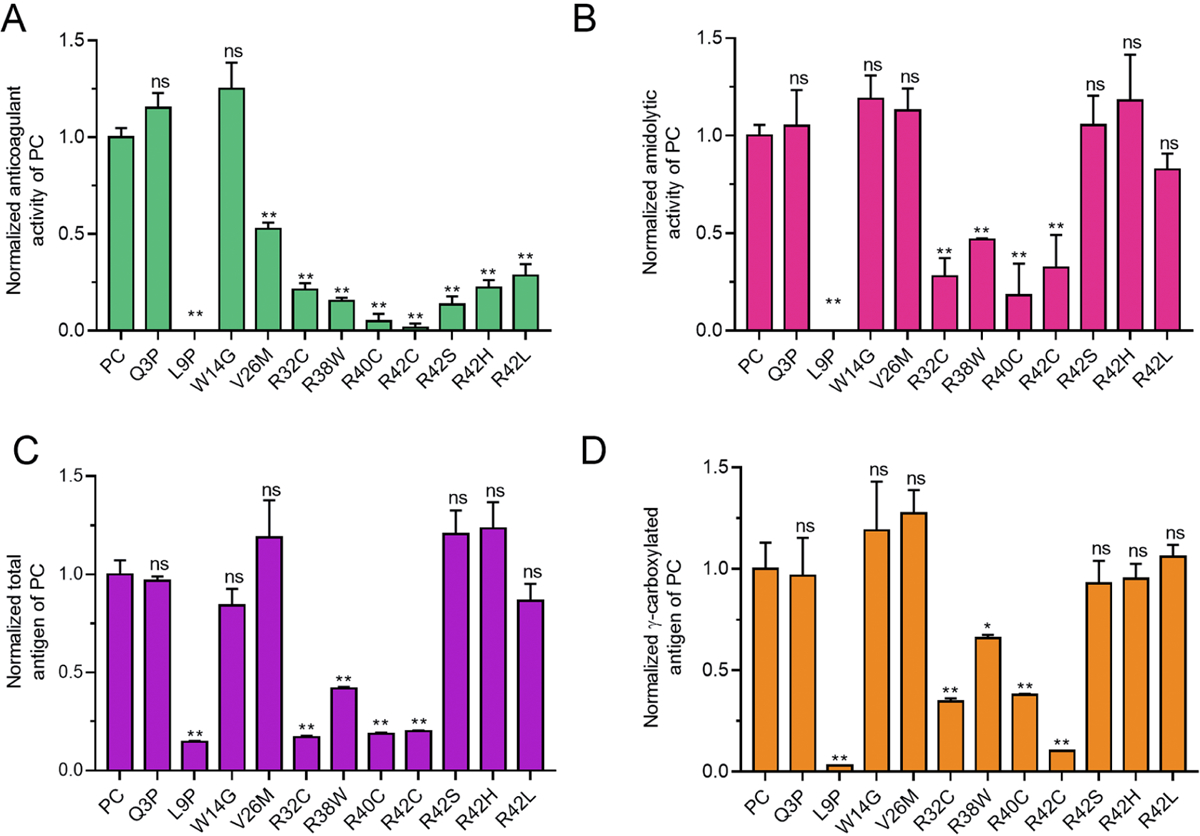
The constructs with missense mutations were overexpressed in HEK 293T cells in the medium with 10μM vitamin K, and anticoagulant activity, amidolytic activity, total antigens and γ-carboxylated antigens of the secreted PC were determined by corresponding methods (see methods), respectively. Relative values were normalized by wild-type PC which value is defined as “1”. A. Relative PC’s anticoagulant activity. B. Relative PC’s chromogenic activity. C. Relative total antigen of PC. D. Relative γ-carboxylated PC. The t-test is performed by Graphpad Prism 7. n.s. indicates no significant difference. * indicates p<0.05, and ** indicates p<0.01.
The previous study demonstrated that mutants of R42S/H/L lead to type II PCD due to abnormal propeptide cleavage that disrupts the function of PC’s Gla domain [12–14]. This mechanism can well explain their discrepancy between PC’s anticoagulant activity and amidolytic activity, which is also consistent with clinical data in patients (Figure S2B). Our data also indicates that PCD can be partially explained by the decreased PC secretion in mutations of L9P, R32C, R38W, R40C and R42C, as the decreased anticoagulant activity is associated with the decreased PC antigen (Figure 1C). However, the data cannot explain how the mutations of Q3P, W14G and V26M resulted in PCD. In the following sections, we will explore their mutation effect on the PC’s biological process.
2. L9P mutation causes abnormal subcellular localization of Protein C
To investigate how these mutations cause PCD, we performed western blot to access the mutation effect on PC’s expression and secretion. The data obviously showed that L9P mutation significantly decreases intracellular PC expression and secretion (Figure 2A). However, mutations of R32C, R40C and R42C decrease PC secretion significantly, but not intracellular PC expression or γ-carboxylation (Figure 2A). The secreted PC detected by western blot is consistent with that from ELISA (Figure 1C). Additionally, the intracellular L9P expression shows a lower molecular weight band than wild-type PC, which indicates that this mutation should have a distinct mechanism to cause PCD.
Figure 2. L9P mutation affects the secretion of Protein C due to abnormal subcellular localization.
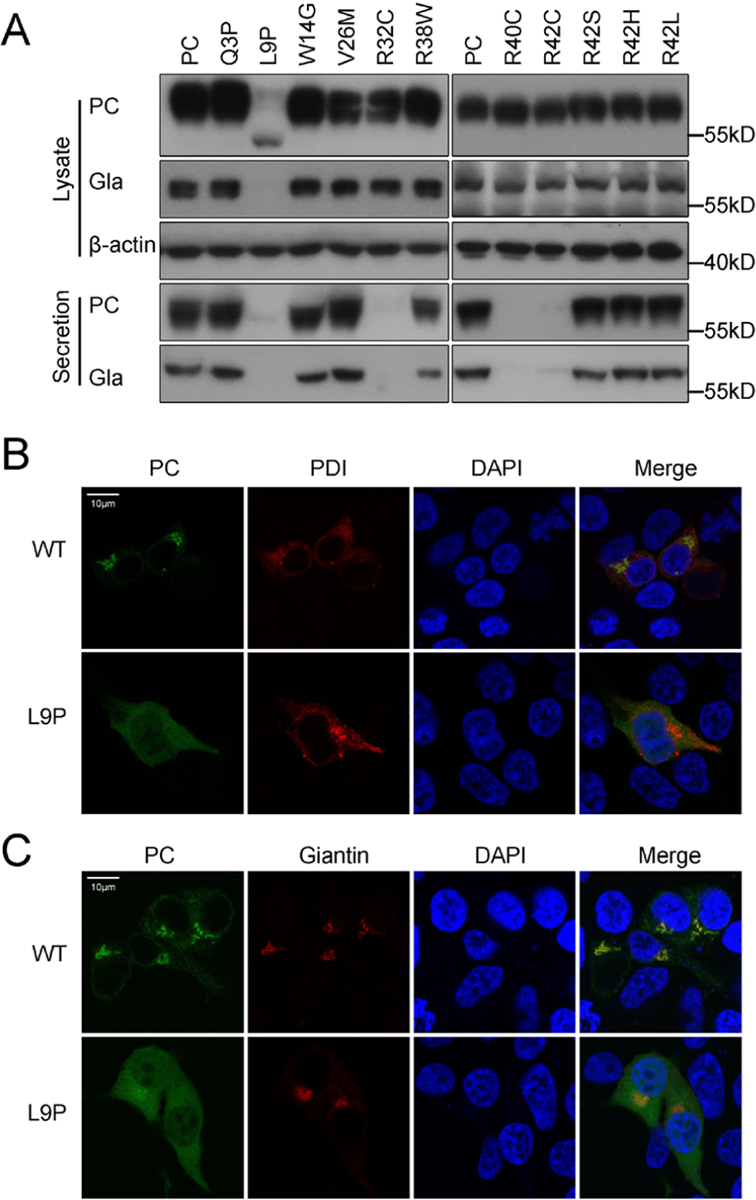
A. Expression and γ-carboxylation of intracellular expressed and secreted protein C was detected by western blot using SAPC-HRP antibody and anti-Gla antibody, respectively. The β-actin was used as a loading control for cell lysate samples. B and C. Subcellular localization of the PCspg-EGFP reporter with L9P mutation. The PDI-mCherry and mScarlet-Giantin were used as ER marker and Golgi marker, respectively. The nucleus was stained by DAPI. The cells were culture in medium with 10μM vitamin K.
The residue L9 belongs to the hydrophobic region of PC’s signal peptide (Figure S2A), which is recognized by signal recognition particle (SRP). As the SRP facilitates co-translational translocation of the secretory protein to ER, we assumed that L9P mutation may disrupt the function of signal peptide and cannot translocate the translation initiation complex from the cytosol to ER. Using an online server (https://services.healthtech.dtu.dk/service.php?SignalP-5.0) [20], we found that L9P mutation decreases the probability of signal peptide from 97.8% to 78.6%.
To confirm L9P mutation destroying the function of PC’s signal peptide, we used the reporter protein PCspg-EGFP, which also affects secretion (Figure S3A), to explore the mutation effect on subcellular localization (see supplementary file 1 for more explanation). Our data showed that the reporter protein with L9P mutation spreads throughout the cell, whereas the wild-type signal peptide is co-localized with ER marker (PDI, Figure 2B) and Golgi marker (Giantin, Figure 2C). Collectively, our data suggested that L9P mutation may cause PCD by disrupting the co-translational translocation function of signal peptide.
3. Three mutations of arginine to cysteine in the propeptide region result in ER retention of Protein C
PC is a secretory protein that is synthesized in ER, then transfers to the Golgi complex, which is the sorting center of secretory proteins. Our data (Figure 1C and 2A) indicates that the mutants of R32C, R40C and R42C affect PC secretion. So we checked their effect on subcellular localization by PCspg-EGFP reporter, as three mutants of reporter affect secretion similarly to corresponding PC mutants (Figure S3B, see supplementary file 1 for more explanation).
Our results showed that the reporter protein of wild-type or mutants could be generally located in ER (Figure 3A) and Golgi complex (Figure 3B). However, by quantifying the fluorescent signal of reporter protein, the signal in Golgi is significantly stronger in wild-type than mutations (Figure 3C). That is to say, relatively more reporter of mutants is retained in ER than that in wild-type. To further confirm that, the constructs with full-length PC were expressed in HEK293T cells, the data from immuno-fluorescent assay also indicated that three mutants cause ER retention of PC (Figure S4). So ER retention caused by these mutations should be the reason of decreased PC secretion.
Figure 3. Mutations of R32C, R40C and R42C result in ER retention of the PCspg-EGFP reporter.
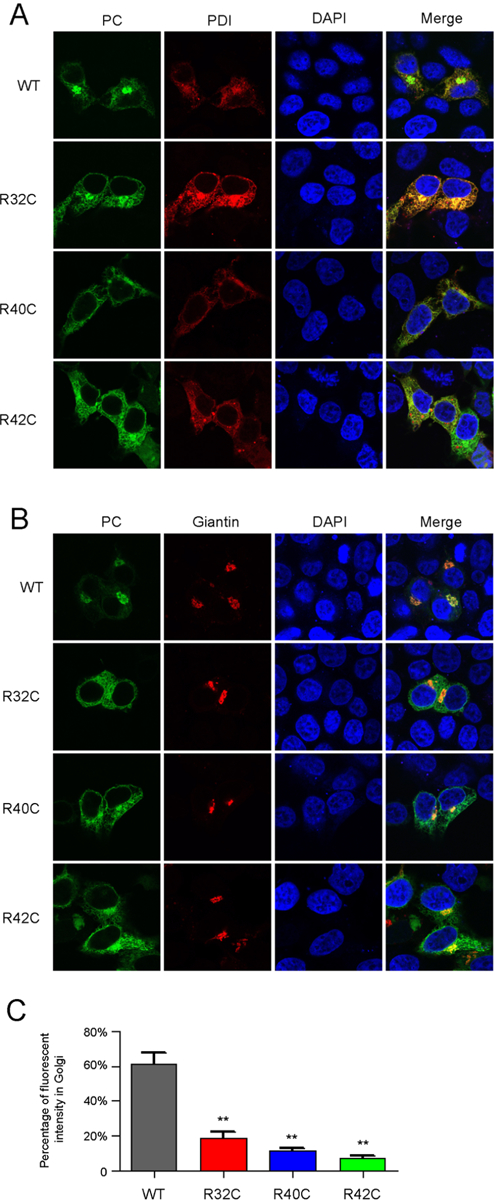
A and B. Subcellular localization of the PCspg-EGFP reporter protein in ER (A) or Golgi (B). The PDI-mCherry (A) and mScarlet-Giantin (B) were used as ER marker and Golgi marker, respectively. The nucleus was stained by DAPI. The cells were culture in medium with 10μM vitamin K. C. Quantification of fluorescent intensity. Green fluorescent intensity of the whole cell and Golgi in Figure 3B was quantified by ImageJ. The percentage of reporter’s fluorescent intensity in Golgi was calculated by dividing the fluorescent intensity in Golgi to that in the whole cell. Eight to ten cells were analyzed for each construct, and the t-test was performed between wild-type and mutants by Graphpad Prism 7. ** indicates p<0.01.
4. R38W mutation leads to aberrant propeptide cleavage
The decreased PC secretion in R38W mutation could only partially explain patients’ PCD, as the anticoagulant activity of R38W (~15% of WT) decreased more than its amidolytic activity and antigen(~46% of WT, Figure 1). It indicates that there may be another mechanism to cause PCD in R38W. Residue R38 is near the C-terminal of propeptide cleavage site, in which mutations usually cause abnormal propeptide cleavage [12–14]. So we assumed that R38W mutation may lead to abnormal propeptide cleavage.
Firstly, we used a Flag-PC construct to detect the mutation effect on propeptide cleavage, and the results indicated that R38W mutation should perturb the propeptide cleavage (Figure S5, see supplementary file 1 for more explanation). To simplify the western blot results, we used the reporter Flag-PCspg-MBP-His to detect the propeptide cleavage efficiency. In theory, if the propeptide is not cleaved, the PURP can be detected by anti-Flag antibody, while both the propeptide cleaved reporter protein (PCRP) and the PURP can be detected by anti-His (Figure 4A). Here, the secreted reporter protein in the medium was detected to evaluate the propeptide cleavage efficiency by western blot and ELISA. To exclude the interference of γ-carboxylation on analyzing propeptide cleavage, the cells were cultured in the medium with 10μM warfarin. R42H mutation was used as a positive control, because it had been reported to cause abnormal propeptide cleavage.
Figure 4. R38W mutation leads to aberrant propeptide cleavage of Protein C.
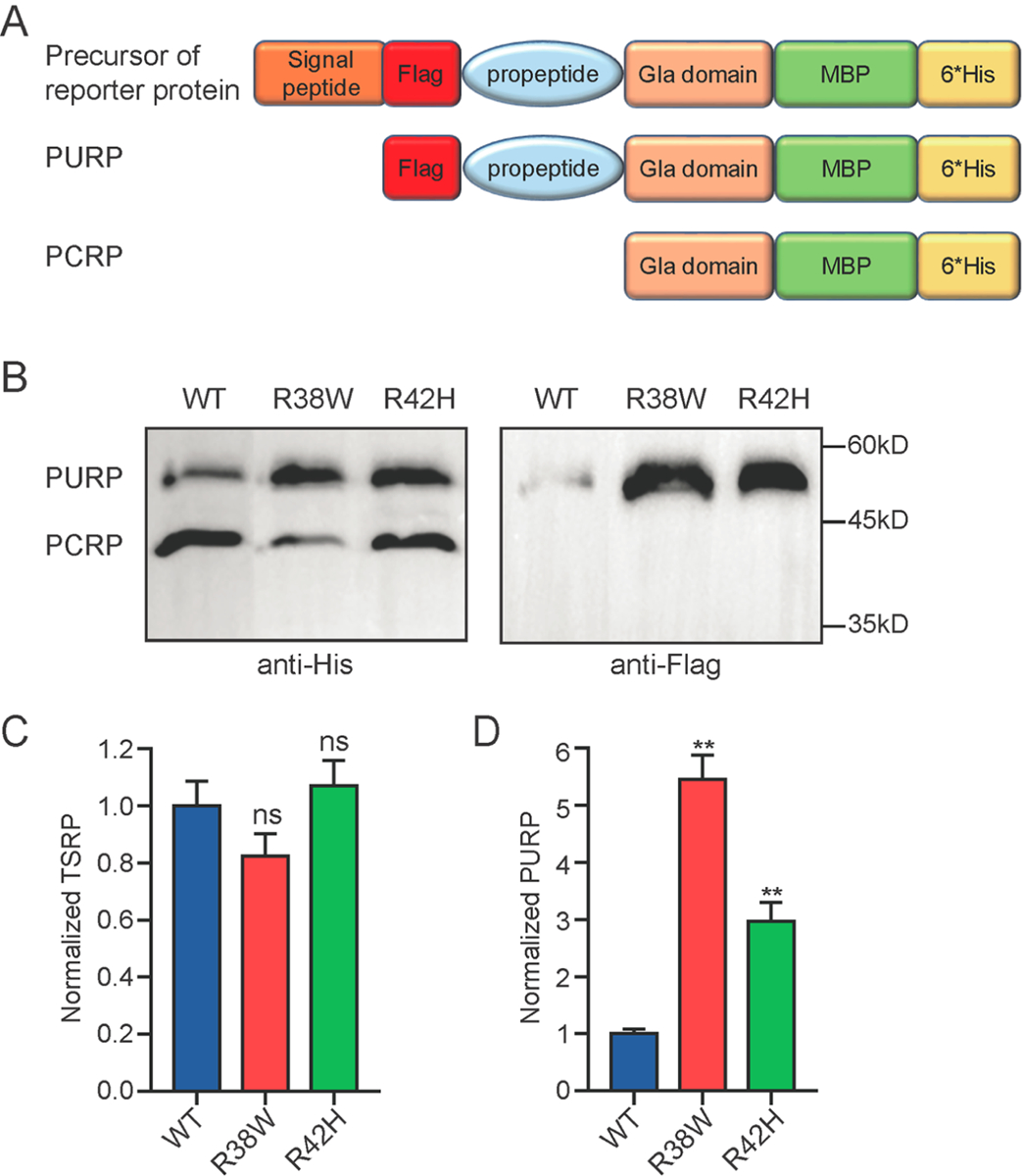
A. Scheme of the Flag-PCspg-MBP-His reporter protein precursor, PURP and PCRP. The reporter protein precursor represents the full sequence of the designed Flag-PCspg-MBP-His reporter protein. PURP represents the propeptide uncleaved reporter protein, and PCRP represents the propetide cleaved reporter protein. B. Analyzing the propeptide cleavage by western blot. The anti-His antibody was used to detect total secreted reporter protein (TSRP, including both PURP and PCRP). The anti-Flag antibody was used to detect the secreted PURP. C and D. Analyzing of secreted TSRP (C) and PURP (D) by ELISA. The MBP polyclonal antibody was coated to an ELISA plate to catch the antigen, then anti-His-HRP was used to detect the TSRP, and anti-Flag-HRP was used to detect the PURP. The cells were culture in medium with 10μM warfarin. The t-test was performed by Graphpad Prism 7. n.s. indicates no significant difference. * indicates p<0.05, and ** indicates p<0.01.
As expected, two bands are detected by anti-His antibody, the upper band with larger molecular weight is the PURP which is further confirmed by anti-Flag antibody and the lower band should be PCRP (Figure 4B). Comparison of the PCRP and PURP in different constructs, the data showed that the PCRP in wild-type is more than that in R38W, R42H, instead, the PURP in the mutants is more than that in wild-type (Figure 4B). Comparison of the PCRP and PURP in each construct, the data showed that there is more PCRP than PURP in wild-type, and there is more PURP than PCRP in mutant of R38W (Figure 4B, left). To quantify the propeptide cleavage efficiency in wild-type and mutations, the ELISA assay was performed. The TSRP (including PURP and PCRP) is no significant difference between mutants and wildtype (Figure 4C), but the PURP is significantly increased in both mutants (Figure 4D). Compared to wild-type, the PURP increased about 5.5-fold and 3-fold in R38W and R42H, respectively. Overall, our data showed that the R38W mutation should affect the propeptide cleavage efficiency, which could well explain that its amidolytic activity is about three-fold of its anticoagulant activity in Figure 1.
5. Effects of several PROC variations on the pre-mRNA splicing
Our data from the cell-based assay could not well explain the PCD in patients with missense mutations of Q3P, W14G, and V26M. V26M mutation had been reported causing type I PCD [10]. However, our data showed that V26M mutation does not affect PC antigen secretion, which is inconsistent with its clinical phenotype. Several studies had reported that nucleotide variations could lead to aberrant pre-mRNA splicing [21–23]. So we assumed that these three variations (c.8A>C, c.40T>G, and c.76G>A) may affect the PROC pre-mRNA splicing. Here, we used a minigene splicing assay to examine the variation effect on the PROC pre-mRNA splicing. The c.8A and c.40T are located in exon 2 and c.76G is located in exon 3, so the corresponding PROC genomic sequence was sub-cloned into pSPL3 vector (Figure 5A). For comparison, the splicing effect was also examined in the variations of c.26T>C (L9P), c.94C>T (R32C), c.112C>T (R38W), and c.118C>T (R40C).
Figure 5. The effects of the PROC variations on the pre-mRNA splicing.
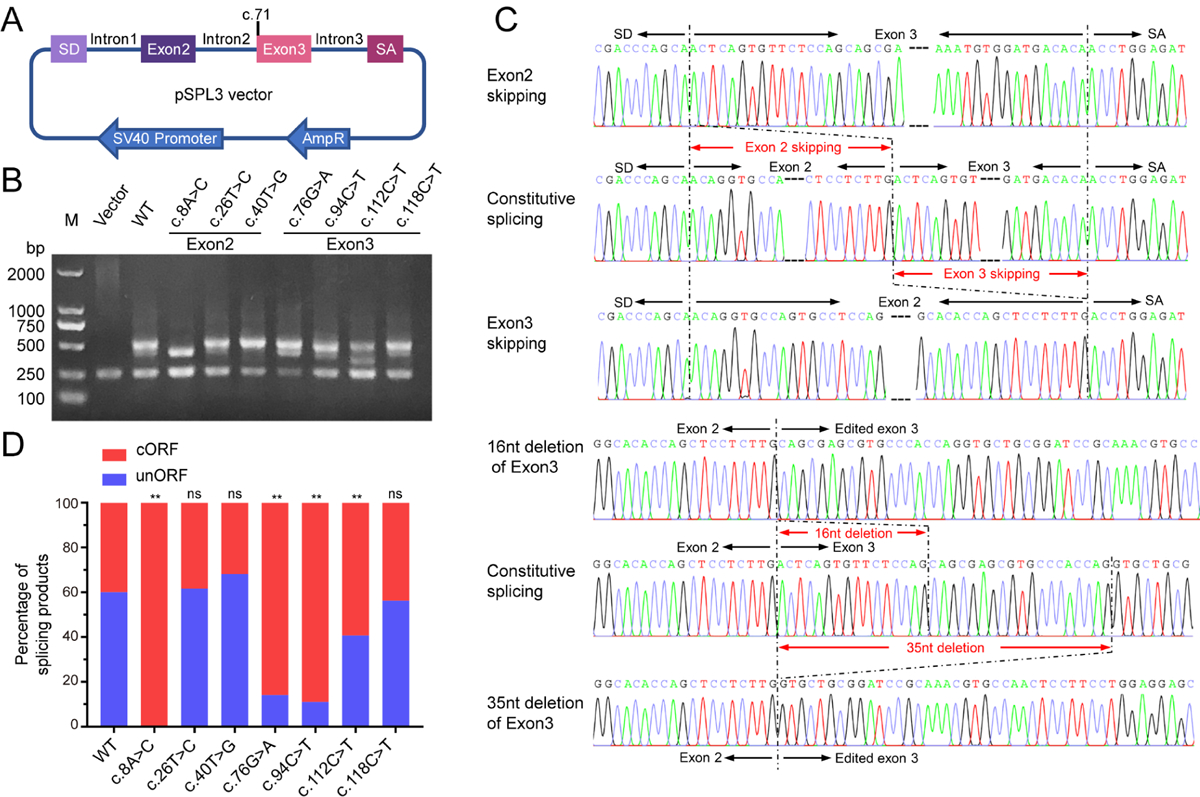
A. Scheme of the construct for minigene splicing assay. The genome sequence of PROC gene was sub-cloned into multi-cloning site (MCS) of pSPL3 vector. The c.71 indicates the position of the first nucleotide of exon 3. B. Agarose gel electrophoresis to analyzing the splicing products of PROC variations. The constructs were transiently transfected in HEK 293T cells. The splicing products were amplified by reverse transcriptase-PCR and analyzed by agarose gel electrophoresis. M indicates DNA ladder marker. Vector indicates splicing product from the empty pSPL3 vector. C. Comparison of sequencing chromatograms between constitutive and aberrant splicing products. Sequence changes were indicated by red arrow lines. D. Statistics of splicing products. About 100 sequencing results of each construct were analyzed. The splicing products were classified into two groups, changed ORF (cORF) and unchanged ORF (unORF). Percentage of each group was calculated. The cORF transcript cause non-functional protein product due to frame shift, and the unORF splicing products encode the normal PC. The Chi-square analysis is performed by Graphpad Prism 7. n.s. indicates no significant difference. * indicates p<0.05, and ** indicates p<0.01.
Based on the gel analysis of PCR products, the data showed that aberrant splicing was obviously observed in c.8A>C and c.112C>T (Figure 5B). To confirm this observation, the PCR products (except the 250bp band) of each construct were ligated to a T-vector. Surprisingly, complex alternative splicing was found in the wild-type and the variations, and total 22 alternative splicing products are detected (Supplementary table 2), including 6 splicing products with unchanged open-reading frame (unORF) and 16 aberrant splicing products with changed ORF (cORF). There are multiple patterns to cause aberrant splicing, including exon skipping, exon deletion and intron insertion (Figure S6). The aberrant splicing products with cORF due to deletion or insertion will cause frameshift, thereby leading to PCD. Among the aberrant splicing products, there are five major basic types, including exon 2 skipping, exon 3 skipping, 16nt deletion of exon 3, 19nt deletion of exon 3, and 35nt deletion of exon3 (Figure 5C, Figure S7). Additionally, 4 types of aberrant splicing products are also detected in the construct with wild-type PROC genomic sequence, and exon 2 skipping and 35nt deletion of exon3 are two major aberrant types (Supplementary table 2).
To investigate if the variations change the translational outcomes of the splicing products, we categorized the splicing products into two groups, the unORF and cORF. In wild-type, there are about 60.2% splicing products with unORF and 39.8% with cORF (Figure 5D). Compared with wild-type, variations of c.8A>C, c.76G>A, c.94C>T, and c.112C>T significantly increase the aberrant splicing products with cORF (Figure 5D). Complete aberrant splicing was only observed in the c.8A>C variation, which suggests that this variation cause PCD by inducing complete aberrant pre-mRNA splicing. Variations of c.76G>A, c.94C>T, and c.112C>T significantly increase the aberrant pre-mRNA splicing products to 85.9%, 89% and 59.2%, respectively, which indicates that these three variations leading to PCD should be partially dependent on aberrant splicing.
To show how these variations lead to aberrant splicing, we separately analyze three major splicing types in exon 2 (including canonical exon 2, 4nt deletion of exon2, and exon 2 skipping, Figure 6A–C) or five major splicing types in exon 3 (include normal exon 3, exon 3 skipping, 16nt deletion of exon3, 19nt deletion of exon3 and 35nt deletion of exon3, Figure 6D–H), because the combination of these major splicing types in exon 2 and exon 3 constituted the most alternative splicing products. Among variations in exon 2, the c.8A>C variation significantly causes exon 2 skipping. Although the c.40T>G variation does not significantly affect the unORF splicing products, but it significantly decreases exon 2 skipping (Figure 6C) and increases aberrant splicing of exon 3 (Figure 6D). For the variations in exon 3, c.76G>A, c.94C>T, and c.112C>T significantly increase aberrant splicing of exon 3 (Figure 6D). In detail, c.112C>T significantly increases the exon 3 skipping (Figure 6E), the c.76G>A significantly increases 16nt deletion of exon 3 (Figure 6F) and 19nt deletion of exon 3 (Figure 6G), and c.94C>T increases 35nt deletion of exon 3 (Figure 6H). Additionally, c.76G>A and c.112C>T also change the splicing pattern of exon 2 (Figure 6C). The c.76G>A increases the exon 2 skipping (Figure 6C), and the c.112C>T decreases the exon 2 skipping (Figure 6C) and increases canonical exon 2 splicing type (Figure 6A).
Figure 6. The effect of variations on the splicing patterns of exon 2 or exon 3.
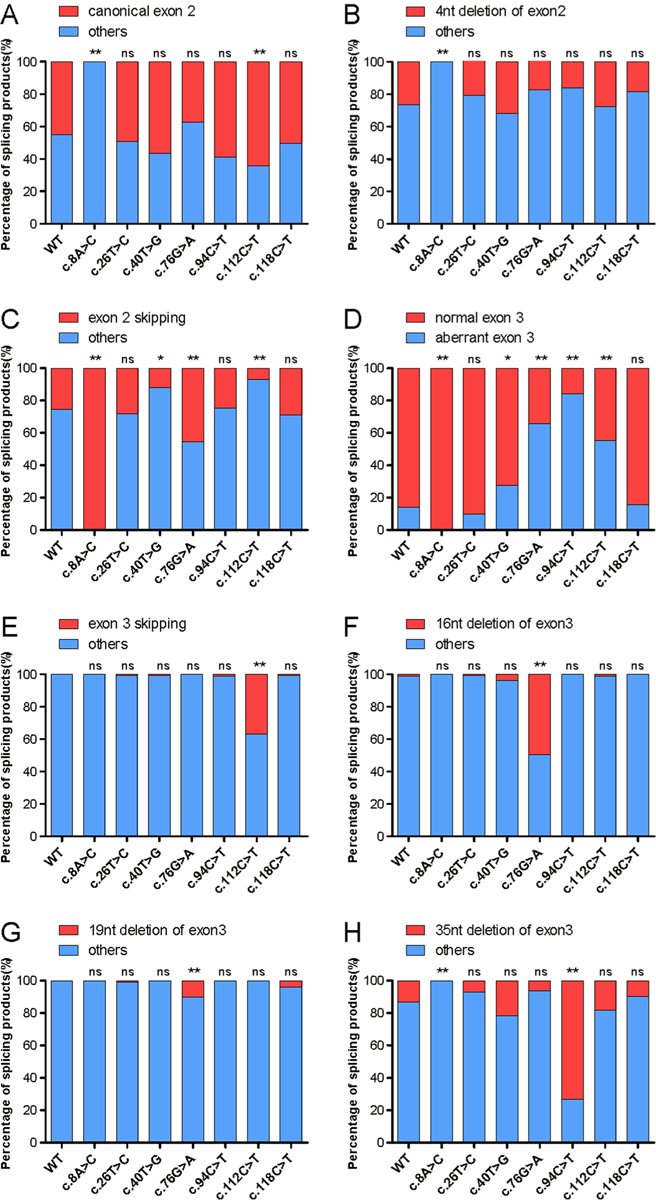
Three major splicing types in exon 2 (A-C) and five major splicing types in exon 3(D-H) are selected, ant they are analyzed separately about how the variations affect the splicing patterns of exon 2 or exon 3. For each graph, the splicing products were divided into two groups, a specific splicing form (shown in red color) and others (shown in blue color) which refer to the splicing forms other than the specific splicing form. In exon 2, the selected splicing types include canonical exon 2 (A), 4nt deletion of exon 2 (B), and exon 2 skipping (C). In exon 3, the selected splicing types include normal exon 3 (D), exon 3 skipping (E), 16nt deletion of exon 3 (F), 19nt deletion of exon 3 (G) and 35nt deletion of exon 3 (H). The percentage of splicing products derives from sequencing of about 100 colonies for each variant in Supplementary Table 2. The Chi-square analysis was performed between wild-type and mutants by Graphpad Prism 7. n.s. indicates no significant difference. * indicates p<0.05, and ** indicates p<0.01.
Discussion
Inherited PCD is caused by PROC mutation that is a risk factor for thromboembolism. Heterozygous PROC mutation is more common and leads to a four to seven fold increased risk for VTE as well as an increased risk of recurrent thrombosis [3, 24, 25]. The inheritance pattern of PCD may be autosomal recessive or dominant with incomplete penetrance, and phenotypes of inherited PCD vary largely among individuals [7, 24], however, the reasons remain largely unknown. So far, more than 500 mutations in PROC gene locus have been identified in human gene mutation database (HGMD, available at www.hgmd.cf.ac.uk), and most of them are missense mutations [26]. However, it is largely unknown if and how missense mutations are pathogenic. Elucidating the molecular pathogenic mechanism of these variations will benefit to evaluate their pathogenic risk of inherited PCD, and to understand their inheritance patterns and clinical presentation variations.
In patients with missense mutations in signal peptide and propeptide, PCD usually appears as a partial deficiency and segregates as an autosomal dominant trait [10]. It had been reported that mutations in residue R42 caused PCD due to abnormal propeptide cleavage in mature PC [12–14]. As R42 mutations did not affect the catalytic domain, so patients with these mutations show the type II PCD with normal PC’s amidolytic activity. Here, the data from cell-based assay about R42S/L/H mutations is consistent with the clinical observations and could well explain their autosomal dominant inheritance pattern. However, it was still unknown about the pathogenic mechanism of other mutations in signal peptide and propeptide regions.
Using a cell-based assay to characterize 11 reported missense mutations, we found that only 8 missense mutations could explained PCD, but not the mutations of Q3P, W14G and V26M (Figure 1). As patients carrying these three variations were type I deficiency, and previous studies showed that missense mutations may induce aberrant splicing to cause nonfunctional protein [21–23, 27], so we assumed that these three variants may induce aberrant pre-mRNA splicing. The most suitable method to identify splicing aberrations is based on the study of patient’s RNA of the affected tissue. As this sort of sample is not available for us, so we used a minigene splicing assay to test whether a specific DNA variant affects pre-mRNA splicing.
On the basis of minigene splicing assay, all the constructs showed the complex alternative splicing, and the aberrant splicing products were also detected in wild-type construct. It has been reported that aberrant splicing of the PROC gene occurs in healthy individuals [28]. To rationalize the complex alternative splicing, we analyzed the twelve naturally occurring alternative splicing events of the PROC gene reported in NCBI database. We found that exon 2 skipping and 35nt deletion of exon 3 exist in naturally occurring alternative splicing events, which are also two major types of aberrant splicing in wild-type construct of the minigene splicing assay. Additionally, the natural alternative splicing events with unORF, including 52nt retention of intron 1 and 4nt deletion of exon 2, are also detected in wild-type construct by the minigene splicing assay. It indicates that the minigene splicing assay is reliable and reflects the natural alternative splicing events of PROC gene. The multiple alternative splicing events in PROC gene also indicate that there are several potential splicing sites or elements spreading around exon 2 and exon 3, and the variations near the region may increase the risk of inducing aberrant splicing.
To elucidate if and how the variations affect the splicing events, statistics analysis was performed on the basis of about 100 sequencing results of T-vector colonies. Our data showed that four variants could lead to aberrant pre-mRNA splicing at different extent (Figure 5). Variation of c.8A>C leads to complete aberrant splicing by inducing exon 2 skipping. Variations of c.76G>A, c.94C>T, and c.112C>T significantly increase aberrant splicing, however, there are still about 15%, 11%, and 40% splicing products with unORF, respectively. It indicates that these three variations are also affected by missense mutations. Their unORF splicing products account for about 1/4, 1/5, and 2/3 that of wild-type. As V26M (c.76G>A) missense mutation has 50% PC’s anticoagulant activity of wild-type (Figure 1A), so the c.76G>A should theoretically generate about 12.5% PC’s activity (1/4*50%) of wild-type by combining the effect of aberrant splicing and missense mutation. Similarly, missense mutations of R32C (c.94C>T) and R38W (c.112C>T) reduce PC’s anticoagulant activity to about 20% and 15% of wild-type (Figure 1A), so the variation of c.94C>T and c.112C>T should theoretically reduce PC’s activity to about 4% (1/5*20%) and 10% (2/3*15%) of wild-type, respectively. Interestingly, in an Italian family, thrombotic events were observed in adolescents in two patients with homozygous variation of c.94C>T, and their PC’s activity ranged from 5% to 9%, and PC antigen levels were about 5% [29], which is well consistent with our experiment prediction.
Although the variation of c.40T>G could change the splicing pattern of wild-type by reducing exon 2 skipping (Figure 6C) and increasing aberrant splicing of exon3 (Figure 6D), but it does not change the splicing products with unORF significantly (Figure 5D). So it could not explain the PCD with this variation by the minigene splicing assay, which may be caused by limitation of this assay. Previous studies showed that a single mutation could affect splicing away from the natural splicing site [23, 30]. Here, the splicing assay only uses partial exon and intron of the PROC genomic sequence, but not the full genome of the gene. So the minigene assay might hide the mutation effect on the splicing of the whole PROC gene.
For the molecular pathogenic mechanisms of the variants resulting in missense mutations, our data showed that they cause PCD by differently affecting the biological process of PC from translation to maturation (Figure 7). In signal peptide region, L9P missense mutation reduces PC expression by disrupting the co-translational translocation function of signal peptide (Figure 7). It is similar to our previous study in hemophilia B with mutations in signal peptide of FIX [18]. The propeptide mediated the γ-carboxylation of PC’s Gla domain [11]. Interestingly, our data showed that missense mutations of R32C, R40C, and R42C decrease PC secretion due to these mutations inducing PC’s ER retention. So we proposed that the propeptide may be important for PC’s sorting from ER to Golgi complex, and the propeptide residue mutating to cysteine lead to PC’s ER retention by interfering with the sorting function of propeptide (Figure 7). However, the underlying mechanism should be further studied. For the missense mutation of V26M, we checked the EC50 in response to vitamin K. We found that the EC50 of V26M is about 2-fold that of wild-type (Figure S8), which could partially explain its reduced anticoagulant activity.
Figure 7. Summary of mutation effects on biological process of Protein C.
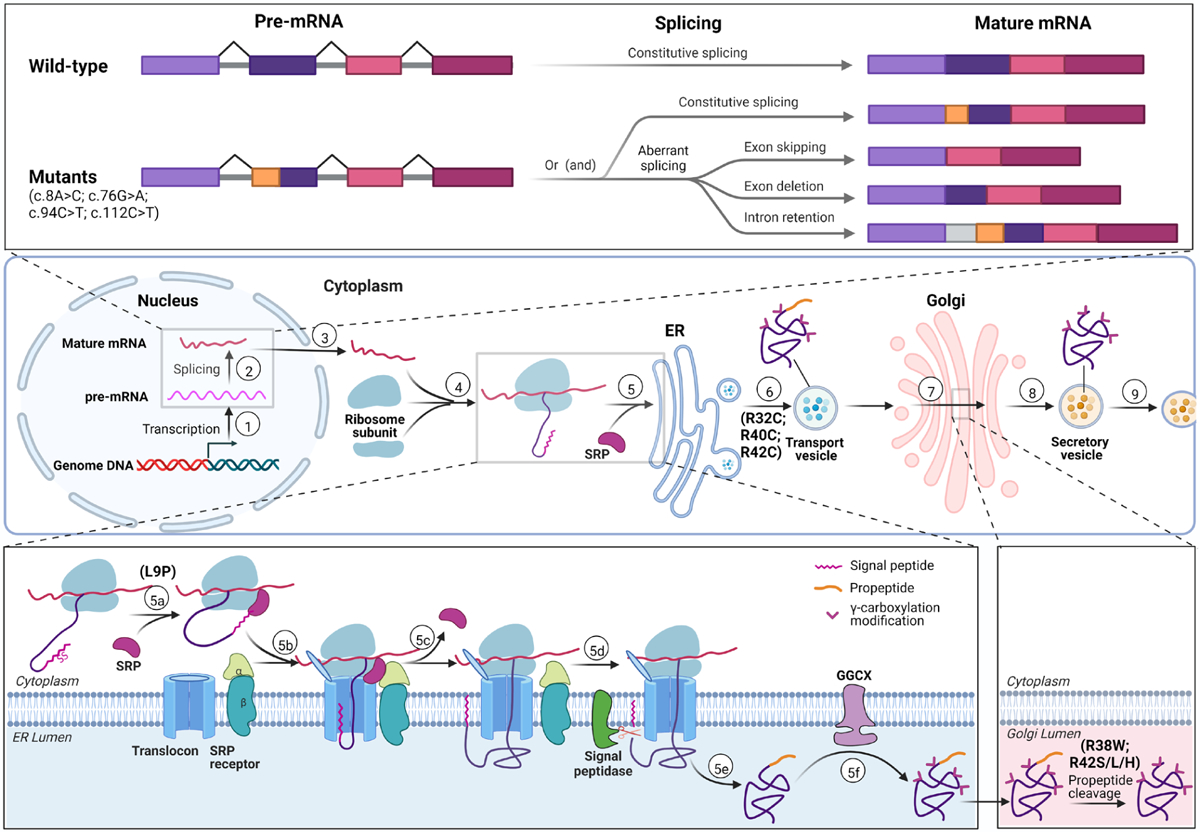
Top panel: Diagram of variants affecting pre-mRNA splicing. Mutations (c.8A>C, c.76G>A, c.94C>T, c.112C>T) can lead to aberrant splicing, including exon skipping, exon deletion and intron retention. The orange color indicates the region contains the variation. Middle panel: Scheme of PC biological process from gene transcription to protein secretion. In nucleus, the PROC gene is transcribed (①) into preliminary mRNA (pre-mRNA), which is spliced into mature mRNA (②); Mature mRNA transfers to cytosol (③) and forms translation initial complex with ribosome subunits (④). Directed by SRP, the translation initial complex is translocated to ER to finish translation, and then the protein is modified in ER lumen (⑤). The protein is transferred to Golgi complex by transport vesicle (⑥), and further modification (including propeptide cleavage) is processed to generate mature PC in Golgi (⑦). Mature PC is coated into secretory vesicles (⑧) and secreted into extracellular space (⑨). Mutations of R32C, R40C, R42C may interfere the step ⑥ and lead to ER retention. Bottom panel: Biological process of protein C in ER and Golgi. Bottom left: Biological process of PC from cytosol to ER. SRP recognizes the signal peptide (5a), directs the translation complex to translocon in ER membrane to continue translation (5b), and then is released (5c). The signal peptide is cleaved by signal peptidase (5d). After translation is finished (5e), PC is translocated into ER lumen, in which γ-carboxylation is carried by GGCX. L9P mutation destroys the signal peptide function which translocates protein translation from cytosol into ER by binding SRP. Bottom right: PC propeptide is cleaved by propeptidase in Golgi complex. Mutations of R38W and R42S/L/H lead to mis-cleaved or uncleaved propeptide in mature PC.
The C-terminal of propeptide is propeptidase recognition site in all vitamin K-dependent clotting factors (VKCFs) [16, 18]. Usually, the −1 and −4 positions of the propeptide are the arginine, and −3 position is arginine or lysine in the motif of propeptidase recognition site. However, the −4 position of PC’s propeptide is isoleucine, but not arginine. While the −5 position of PC’s propeptide is an arginine residue, R38. So far, it is unclear if residue R38 is required for propeptide cleavage. Here we developed a propeptide cleavage assay, and the data showed that R38W mutation significantly affects the PC’s propeptide cleavage. It indicates that the sequence RIRKR should be the motif of PC’s propeptidase recognition site. However, there is a shortage of the propeptide cleavage assay. The assay could only detect the propeptide uncleaved products of aberrant propeptide cleavage, but not the products with changed cleavage site. Previous studies showed that R42S/L/H mutations shift the cleavage site to the −1 and −2 position of the propeptide, which resulted in remaining an additional residue in the N-terminal of mature PC. The −1 residue remained product could not be detected by anti-Flag antibody, and might not be distinguished from propeptide normally cleaved species by SDS-PAGE. That may be the reason why R38W mutation appears to affect propeptide cleavage more than R42H (Figure 4).
In summary, our data indicates that the nucleotide variations in signal peptide and propeptide of PC cause PCD by differently affecting the biological process of PC, including posttranscriptional pre-mRNA splicing, translation, posttranslational modification and process. Moreover, our data indicated that one variation could affect different biological process of PC at multiple levels, such as the variants of c.76G>A, c.94C>T, and c.112C>T lead to PCD by aberrant splicing and missense mutation simultaneously. Nevertheless, except for W14G, our data showed that these variants could significantly decrease PC’s activity by different mechanisms, so their inheritance pattern should be autosomal dominant theoretically. So the individuals carrying these heterozygous variations should significantly increase the risk of venous thromboembolism. These homozygous mutations may be life threatening. In one word, our study clarifies these variations should be pathogenic.
Supplementary Material
Essentials.
Mechanisms of PCD caused by mutations in PC’s signal peptide and propeptide remain unclear.
We systemically studied the pathogenic mechanisms of these mutations by cell-based assays.
These variations have varying effects on the biological process of PC to cause PCD.
Understanding the mechanisms helps to evaluate pathogenic risk and inheritance patterns.
Acknowledgements
This work was supported by grant 82170133 and 81770140 (to G. S.), and 31900412(to M. G.) from National Natural Science Foundation of China, grant 212102310629 (to Y. S.) and 212102310877 (to G. S.) from Henan Department of Science & Technology, and grant HL121718 (to W. L.), EY028705 (to W. L.), (HL131690 to J.K.T. and D.W.S.) from National Institutes of Health, and Jiangsu Specially-Appointed Professor Start-up Funds grant (to Z. H.), and grant 2022M712691 from China Postdoctoral Science Foundation (to N. J.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict-of-Interest Disclosure The authors declare no competing financial interest.
References
- 1.Dinarvand P, Moser KA. Protein C Deficiency. Arch Pathol Lab Med. 2019;143:1281–1285. [DOI] [PubMed] [Google Scholar]
- 2.Goldenberg NA, Manco-Johnson MJ. Protein C deficiency. Haemophilia. 2008;14:1214–1221. [DOI] [PubMed] [Google Scholar]
- 3.Mahmoodi BK, Brouwer JL, Ten Kate MK, et al. A prospective cohort study on the absolute risks of venous thromboembolism and predictive value of screening asymptomatic relatives of patients with hereditary deficiencies of protein S, protein C or antithrombin. J Thromb Haemost. 2010;8:1193–1200. [DOI] [PubMed] [Google Scholar]
- 4.Millar DS, Johansen B, Berntorp E, et al. Molecular genetic analysis of severe protein C deficiency. Hum Genet. 2000;106:646–653. [DOI] [PubMed] [Google Scholar]
- 5.Minford A, Brandao LR, Othman M, et al. Diagnosis and management of severe congenital protein C deficiency (SCPCD): Communication from the SSC of the ISTH. J Thromb Haemost. 2022;20:1735–1743. [DOI] [PubMed] [Google Scholar]
- 6.Seidel H, Haracska B, Naumann J, Westhofen P, Hass MS, Kruppenbacher JP. Laboratory Limitations of Excluding Hereditary Protein C Deficiency by Chromogenic Assay: Discrepancies of Phenotype and Genotype. Clin Appl Thromb Hemost. 2020;26:1076029620912028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alhenc-Gelas M, Plu-Bureau G, Mauge L, Gandrille S, Presot I. Genotype-Phenotype Relationships in a Large French Cohort of Subjects with Inherited Protein C Deficiency. Thromb Haemost. 2020;120:1270–1281. [DOI] [PubMed] [Google Scholar]
- 8.Foster DC, Yoshitake S, Davie EW. The nucleotide sequence of the gene for human protein C. Proc Natl Acad Sci U S A. 1985;82:4673–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plutzky J, Hoskins JA, Long GL, Crabtree GR. Evolution and organization of the human protein C gene. Proc Natl Acad Sci U S A. 1986;83:546–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reitsma PH, Bernardi F, Doig RG, et al. Protein C deficiency: a database of mutations, 1995 update. On behalf of the Subcommittee on Plasma Coagulation Inhibitors of the Scientific and Standardization Committee of the ISTH. Thromb Haemost. 1995;73:876–889. [PubMed] [Google Scholar]
- 11.Foster DC, Rudinski MS, Schach BG, et al. Propeptide of human protein C is necessary for gamma-carboxylation. Biochemistry. 1987;26:7003–7011. [DOI] [PubMed] [Google Scholar]
- 12.Lind B, Johnsen AH, Thorsen S. Naturally occurring Arg(−1) to His mutation in human protein C leads to aberrant propeptide processing and secretion of dysfunctional protein C. Blood. 1997;89:2807–2816. [PubMed] [Google Scholar]
- 13.Miyata T, Zheng YZ, Sakata T, Kato H. Protein C Osaka 10 with aberrant propeptide processing: loss of anticoagulant activity due to an amino acid substitution in the protein C precursor. Thromb Haemost. 1995;74:1003–1008. [PubMed] [Google Scholar]
- 14.Simioni P, Kalafatis M, Tormene D, et al. Abnormal propeptide processing resulting in the presence of two abnormal species of protein C in plasma: characterization of the dysfunctional protein C Padua3 (protein C(R-1L/propeptide)). Thromb Haemost. 2001;86:1017–1022. [PubMed] [Google Scholar]
- 15.Li C, Wen A, Shen B, Lu J, Huang Y, Chang Y. FastCloning: a highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 2011;11:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao Z, Jin DY, Stafford DW, Tie JK. Vitamin K-dependent carboxylation of coagulation factors: insights from a cell-based functional study. Haematologica. 2020;105:2164–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ndonwi M, Broze GJ Jr., Agah S, Schmidt AE, Bajaj SP. Substitution of the Gla domain in factor X with that of protein C impairs its interaction with factor VIIa/tissue factor: lack of comparable effect by similar substitution in factor IX. J Biol Chem. 2007;282:15632–15644. [DOI] [PubMed] [Google Scholar]
- 18.Gao W, Xu Y, Liu H, et al. Characterization of missense mutations in the signal peptide and propeptide of FIX in hemophilia B by a cell-based assay. Blood Adv. 2020;4:3659–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen G, Cui W, Zhang H, et al. Warfarin traps human vitamin K epoxide reductase in an intermediate state during electron transfer. Nat Struct Mol Biol. 2017;24:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen H, Tsirigos KD, Brunak S, von Heijne G. A Brief History of Protein Sorting Prediction. Protein J. 2019;38:200–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fraile-Bethencourt E, Diez-Gomez B, Velasquez-Zapata V, Acedo A, Sanz DJ, Velasco EA. Functional classification of DNA variants by hybrid minigenes: Identification of 30 spliceogenic variants of BRCA2 exons 17 and 18. PLoS Genet. 2017;13:e1006691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hao Z, Jin DY, Chen X, Schurgers LJ, Stafford DW, Tie JK. gamma-Glutamyl carboxylase mutations differentially affect the biological function of vitamin K-dependent proteins. Blood. 2021;137:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buratti E, Baralle M, Baralle FE. Defective splicing, disease and therapy: searching for master checkpoints in exon definition. Nucleic Acids Res. 2006;34:3494–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Minno MN, Ambrosino P, Ageno W, Rosendaal F, Di Minno G, Dentali F. Natural anticoagulants deficiency and the risk of venous thromboembolism: a meta-analysis of observational studies. Thromb Res. 2015;135:923–932. [DOI] [PubMed] [Google Scholar]
- 25.Koster T, Rosendaal FR, Briet E, et al. Protein C deficiency in a controlled series of unselected outpatients: an infrequent but clear risk factor for venous thrombosis (Leiden Thrombophilia Study). Blood. 1995;85:2756–2761. [PubMed] [Google Scholar]
- 26.Weronska A, Potaczek DP, Oto J, Medina P, Undas A, Wypasek E. A Series of 14 Polish Patients with Thrombotic Events and PC Deficiency-Novel c.401–1G>A PROC Gene Splice Site Mutation in a Patient with Aneurysms. Genes (Basel). 2022;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borras N, Orriols G, Batlle J, et al. Unraveling the effect of silent, intronic and missense mutations on VWF splicing: contribution of next generation sequencing in the study of mRNA. Haematologica. 2019;104:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berg LP, Soria JM, Formstone CJ, et al. Aberrant RNA splicing of the protein C and protein S genes in healthy individuals. Blood Coagul Fibrinolysis. 1996;7:625–631. [DOI] [PubMed] [Google Scholar]
- 29.Cafolla A, D’Andrea G, Baldacci E, Margaglione M, Mazzucconi MG, Foa R. Hereditary protein C deficiency and thrombosis risk: genotype and phenotype relation in a large Italian family. Eur J Haematol. 2012;88:336–339. [DOI] [PubMed] [Google Scholar]
- 30.Gabut M, Mine M, Marsac C, Brivet M, Tazi J, Soret J. The SR protein SC35 is responsible for aberrant splicing of the E1alpha pyruvate dehydrogenase mRNA in a case of mental retardation with lactic acidosis. Mol Cell Biol. 2005;25:3286–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.