

Abstract
Cyclin-dependent-kinase-4/6 inhibitor (CDK4/6i) plus endocrine therapy (ET) is standard of care for patients with hormone receptor (HR)-positive, HER2-negative metastatic breast cancer (MBC). However, resistance to CDK4/6is plus ET remains a clinical problem with limited therapeutic options following disease progression. Different CDK4/6is might have distinct mechanisms of resistance, and therefore using them sequentially or targeting their differentially altered pathways could delay disease progression. To understand pathways leading to resistance to the CDK4/6is palbociclib and abemaciclib, we generated multiple in vitro models of palbociclib-resistant (PR) and abemaciclib-resistant (AR) cell lines, as well as in vivo patient-derived xenografts (PDX) and ex vivo PDX-derived organoids from patients who progressed on CDK4/6i. PR and AR breast cancer cells exhibited distinct transcriptomic and proteomic profiles that sensitized them to different classes of inhibitors; PR cells upregulated G2/M pathways and responded to abemaciclib, while AR cells upregulated mediators of the oxidative phosphorylation pathway (OXPHOS) and responded to OXPHOS inhibitors. PDX and organoid models derived from palbociclib-resistant breast cancer patients remained responsive to abemaciclib. Resistance to palbociclib while maintaining sensitivity to abemaciclib was associated with pathway-specific transcriptional activity but was not associated with any individual genetic alterations. Lastly, data from a cohort of 52 patients indicated that patients with HR-positive/HER2-negative MBC who progressed on palbociclib-containing regimens can exhibit a meaningful overall clinical benefit from abemaciclib-based therapy when administered after palbociclib. These findings provide the rationale for clinical trials evaluating the benefit of abemaciclib treatment following progression on a prior CDK4/6i.
Significance
Palbociclib-resistant breast cancers respond to abemaciclib and express pathway-specific signatures of sensitivity, providing a biomarker-driven therapeutic option for metastatic breast cancer patients following disease progression on CDK4/6 inhibitors.
Introduction
Hormone receptor (HR)-positive, HER2-negative breast cancer subtype accounts for 70% of breast cancers (1) with endocrine therapy (ET) the mainstay of systemic therapy. While ET reduces the risk of recurrence and mortality, many patients with metastatic breast cancer (MBC) either do not respond or experience progression shortly after initiating therapy (intrinsic resistance), or experience progression over time (acquired resistance) (2). Biologically targeted therapies against mTOR, PI3K (3,4), and cyclin-dependent kinases 4/6 (CDK4/6) have successfully delayed progression in patients with MBC in combination with aromatase inhibitors (AIs) or fulvestrant, collectively called ET (5–12). Three CDK4/6 inhibitors (CDK4/6is), palbociclib, ribociclib, and abemaciclib, have been approved by the US Food and Drug Administration for use in combination with ET for MBC on the basis of improved progression-free survival (PFS) compared to PFS with ET alone (13–17). However, despite evidence demonstrating improved survival, most patients will develop resistance to CDK4/6is and progress following long-term treatment (18).
In patients with progression on treatment with CDK4/6i plus ET, there are no established guidelines for treatment selection, but strategies include: (i) changing the ET, typically from AI to fulvestrant, in combination with agents that target a collateral pathway (i.e. PI3K/Akt/mTOR) (19–22) (ii) maintaining the ET and switch to another CDK4/6i or switch both ET and CDK4/6i or (iii) chemotherapy. Systemic therapy choice often depends on the ET response and its duration.
Mechanisms of resistance to CDK4/6is have been explored by our group and others. Data from longitudinal blood and tumor samples point to upregulation of the cyclin E, IL-6/JAK/STAT3, and autophagy pathways and loss of Rb and FAT1 functions in mediating resistance to CDK4/6is (23–27). Accordingly, high cyclin E mRNA was also associated with resistance to palbociclib in the PALOMA-3 trial (23). Others have shown that activating genomic alterations in AKT1, RAS, AURKA, CCNE2, ERBB2, and FGFR2 along with loss of estrogen receptor (ER) expression or PTEN can drive resistance to CDK4/6is (28,29). However, each of these mechanisms account for a small percentage of resistant phenotypes and not all resistance can be attributed to any one of these driving alterations as evolution of resistance-driving mutations appear infrequently at the genomic level (28,30–32). In addition, resistance to ET may render the combination with CDK4/6is less effective (33).
Because the three CDK4/6is share the same nominal targets, they might be assumed to have similar mechanisms of resistance. However, there is evidence from preclinical and clinical studies suggesting that mechanisms of resistance may differ between the three agents. For example, abemaciclib can inhibit kinases that are not inhibited by palbociclib and ribociclib, such as CDK9/7/2/1, GSK3α/β, and CAMK2γ/δ, and as such, abemaciclib has been shown to inhibit cell proliferation in Rb-deficient cells, against which palbociclib and ribociclib are ineffective (34). This suggests that abemaciclib’s multi-kinase inhibitory activity may render it more therapeutically beneficial following treatment with other CDK4/6is (34–40). Moreover, clinical data have shown that some patients continue to derive meaningful clinical benefit from abemaciclib after progression on prior palbociclib (41,42).
In the study reported here, we tested the hypotheses that palbociclib and abemaciclib have distinct pathways of resistance and that using these drugs sequentially or targeting their differentially altered pathways could delay progression and/or improve survival in breast cancer. Our findings provide rationale for clinical trials evaluating the benefit of abemaciclib treatment following progression on a prior CDK4/6i.
Materials and Methods
Cell Lines and Culture Conditions
Several different pools (n=3) of MCF7(ATCC-HTB-22 RRID:CVCL_0031) and T47D (ATCC HTB-133 RRID:CVCL_0553) PR cell lines were generated by culturing each of the MCF7 and T47D pools in complete medium supplemented with increasing concentrations of palbociclib, 1.2 μM, 2.4 μM, 3.6 μM, and 4.8 μM, in a stepwise manner over a 6–7-month period. Four PR cell lines were generated corresponding to cells resistant to each concentration of palbociclib. Likewise, MCF7 AR cell lines were generated by culturing several pools (n=3) of MCF7 cells in complete medium supplemented with increasing concentrations of abemaciclib, 0.5 μM, 1 μM, and 1.5 μM, in a stepwise manner over a 7-month period. Three AR cell lines were generated corresponding to cells resistant to each concentration of abemaciclib. Each resistant cell line was maintained in medium supplemented with the respective concentration of the drug used to generate resistance. Cell lines were regularly tested for Mycoplasma and authenticated every 6 months by karyotype and short tandem repeat analysis at the MDACC Characterized Cell Line Core Facility. The detailed procedures for in vitro assays: comet, RNA sequencing, GSEA, and RPPA were described previously (27,43–49) and are included in Supplementary Materials and Methods.
Development and Treatment of PDX Models
Breast tumor tissues were obtained from patients during surgery after written informed consent was obtained under protocols approved by the MDACC Institutional Review Board and in accordance with recognized ethical guidelines. PDX model ET-R was previously developed (46). PDX model PR-4 (originally named T141-004) was kindly provided by the laboratory of Dr. Funda Meric-Bernstam’s at MDACC and was generated as previously described (50,51). PDX models PR-1, PR-2, and PR-3, originally named CTG-3277, CTG-3266, and CTG-2308, respectively, were obtained from Champions Oncology, Inc. through a Material Transfer Agreement with MDACC. In this study, the PDX models were re-established from the liquid nitrogen tumor bank and transplanted into the T4 mammary fat pad of 8-week-old female NSG mice given drinking water supplemented with 8 μg/mL estradiol (E2), except for the PR-4 model, which was not grown in the presence of E2. Tumors were allowed to grow until they reached 1000 to 1500 mm3 and were then harvested and used for further PDX expansion and drug response studies, as well as organoid generation (PDxOs). The procedures performed for these studies are included in an Institutional Animal Care and Use Committee (IACUC) approved protocol. Detailed descriptions of organoid generation, maintenance, passaging, and drug treatment are included in Supplementary Materials and Methods.
For the drug treatment experiments, 4×4-mm fragments of the PDX line were transplanted into the fat pad of the fourth mammary gland of female nude mice. Tumors were allowed to grow until they reached 150–200 mm3 in the presence of drinking water supplemented with E2 (8 μg/mL). Once tumors reached an average volume of 200 mm3, mice were randomized to the indicated treatment arms and treated with vehicle (0.5% methylcellulose), palbociclib (50 mg/kg daily by mouth or 75 mg/kg 21days on /7 days off by mouth), or abemaciclib (50 mg/kg daily by mouth). Once treatments started, the E2 supplementation was removed, mimicking the effect of ET. Drugs were administered via oral gavage. The length and width of tumor xenografts were measured by caliper 2–3 times per week, and tumor volume was calculated by the formula volume = (length × (width)2)/2. The mice were euthanized when average tumor volume in the vehicle-treatment arm reached 1000–1300 mm3. Following euthanasia, all tumors were harvested, random fragments were frozen and stored at −80 °C for RNA/protein isolation, and one random piece was fixed in formalin for histopathological and immunohistochemistry analysis. Abemaciclib methanesulfonate and palbociclib isethionate were purchased from MedChem Express, Manmouth Junction, NJ.
Patient Cohort
To identify patients for this study, after obtaining approval from the MDACC Institutional Review Board, we queried MDACC’s Department of Breast Medical Oncology database (>53,000 patients treated since the 1980s) for the following criteria: age ≥18 years, referred to MDACC during 2015–2022, ER-positive and/or PgR-positive and HER2-negative advanced breast cancer, abemaciclib-based therapy at MDACC at any time through December 2022 (Supplementary Fig. S9). Within this cohort, patients whose disease progressed on palbociclib or ribociclib in the metastatic setting and subsequently receive abemaciclib-based therapy were identified. We excluded patients who discontinued the initial CDK4/6i because of toxicity or received abemaciclib therapy as the initial CDK4/6i. Patients could receive abemaciclib sequentially, i.e., immediately after disease progression on prior CDK4/6i, or nonsequentially, i.e., after other systemic therapies after progression on the first CDK4/6i. Abemaciclib could be administrated as monotherapy or in combination with ET. Patients with HER2− positive disease or TNBC were excluded.
Endpoints included PFS and OS, with PFS defined as the length of time from start of abemaciclib treatment to progression or death. Patients who were receiving treatment or for whom no information on end date of treatment was available and patients without documentation of progression were right censored at the time of last follow-up. OS was defined as the length of time from start of abemaciclib treatment to death due to any cause; surviving patients were right censored at the time of last follow-up. The distributions of PFS and OS and the 95% CIs were estimated by the Kaplan-Meier method (52), and the conventional log-rank test (53) was performed to evaluate the differences in PFS and OS between the indicated groups.
Genomic testing on patient samples
Tumor NGS was either performed on tumor tissue, using one of five CLIA (Clinical Laboratory Improvement Amendment) -platforms with a range of 50–400 cancer-related genes assessed, or circulating tumor DNA (ctDNA), using Guardant360. Genomic testing results were retrieved from a prospectively maintained database. This data is accessible through a protocol that is approved by the Institutional Review Board and has waiver of informed consent (54–57).Genomic alterations presented represent alterations that were determined to be of somatic origin by the clinical laboratory performing the testing. The germline variants were excluded based on matched tumor-normal analysis in some labs and by using database-driven computation approaches by the others. Both publicly available and locally curated knowledge-bases were utilized to determine clinical actionability of somatic variants using laboratory-specific practices.
Study Approvals
Animal studies were performed in accordance with the animal protocol procedures approved by the Institutional Animal Care and Use Committee. Patient related studies were approved by the MD Anderson Cancer Center Institutional Review Board committee.
Data Availability
The RNA-seq data sets from the cell line, organoids cultures and PDX models generated in this study have been deposited at the Gene Expression Omnibus repository: Accession numbers GSE222367(cell lines) GSE229146 (organoids derived from PDX models) and GSE229235 (PDX models) which will be publicly available at the time of publication. The human sequence data generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. The human genomic data, listed in SNP databases analyzed in this study were obtained from gnomAD (https://gnomad.broadinstitute.org). All other raw data are available upon request from the corresponding author.
Results
Palbociclib-and abemaciclib-resistant cells exhibit distinct transcriptomic and proteomic profiles
Palbociclib was the first CDK4/6i approved by the FDA in February 2015 and has been the most frequently prescribed first CDK4/6i for any line of treatment (28,42,58). For the time periods of 2015–2020, 2021 and 2022, palbociclib remains the predominantly prescribed first CDK4/6i at The University of Texas M.D. Anderson Cancer Center (MDACC) with 92%, 80% and 60% of all CDK4/6is, respectively. There is a transition toward increased abemaciclib use (4.5% to 16.7% to 32%), while ribociclib remains less utilized in the clinic (3.6% to 4% to 8%). The change in CDK4/6i use over the past 8 years at MDACC is shown in Supplemental Fig. S1A. This data supports the immediate need to understand the differential pathways regulating the efficacy of palbociclib and abemaciclib in our patient population, and cell lines. Therefore, we sought to assess whether the pathways leading to acquired resistance to palbociclib and abemaciclib are distinct, by generating in vitro models of resistance to each agent in a stepwise manner using MCF7 and T47D estrogen receptor (ER)-positive/HER2-negative breast cancer cell lines. Parental cells were treated over a 6-month period with increasing concentrations of palbociclib starting at 1.2 μM and increasing by 2-fold at each step, up to 4.8 μM. These concentrations were based on continuous treatment of patients with MBC from two different studies (n=12 and n=13) being treated with 125 mg palbociclib QD for 21 days to generate steady state plasma levels at the end of one cycle (59). The summary of palbociclib pharmacokinetic parameters revealed that the area under the curve ranged from 1633–1982 ng.hr/ml (3646–4425 nM). Consequently, for our in vitro studies we used 3.6 and 4.8 μM palbociclib as our high dose concentrations, which reflects the highest steady state plasma levels as reported (59). For each MCF7 and T47D, four palbociclib-resistant (PR) cell lines were generated corresponding to cells resistant at each concentration and called 1.2PR, 2.4PR, 3.6PR, and 4.8PR. Dose-response analysis corroborated that PR cells were 8- to 20-fold (MCF7) and 5- to 9-fold (T47D) less sensitive to palbociclib than the parental cells were. However, the doubling times of the resistant cells were similar to those of the parental cells (Fig. 1A; Supplementary Fig. S1B). Similarly, three abemaciclib-resistant (AR) MCF7 cell lines (0.5 AR, 1.0AR, and 1.5AR) were generated over a 7-month period. These concentrations were selected on the basis of plasma levels (area under the curve, 3844–4570 ng.hr/ml) reported in patients following a single 200 mg oral dose of abemaciclib (60). AR cells were 5-fold less sensitive to abemaciclib compared to parental cells and had doubling times similar to those of the parental cells (Fig. 1B). During acquisition of resistance, the PR cells became smaller and lost the ability to form domes, indicative of epithelial-to-mesenchymal transition (EMT). No morphological changes were observed in the AR cells (Fig. 1A and B; Supplementary Fig. S1B).
Figure 1. PR and AR cells exhibit distinct transcriptomic profiles and differential altered pathways.
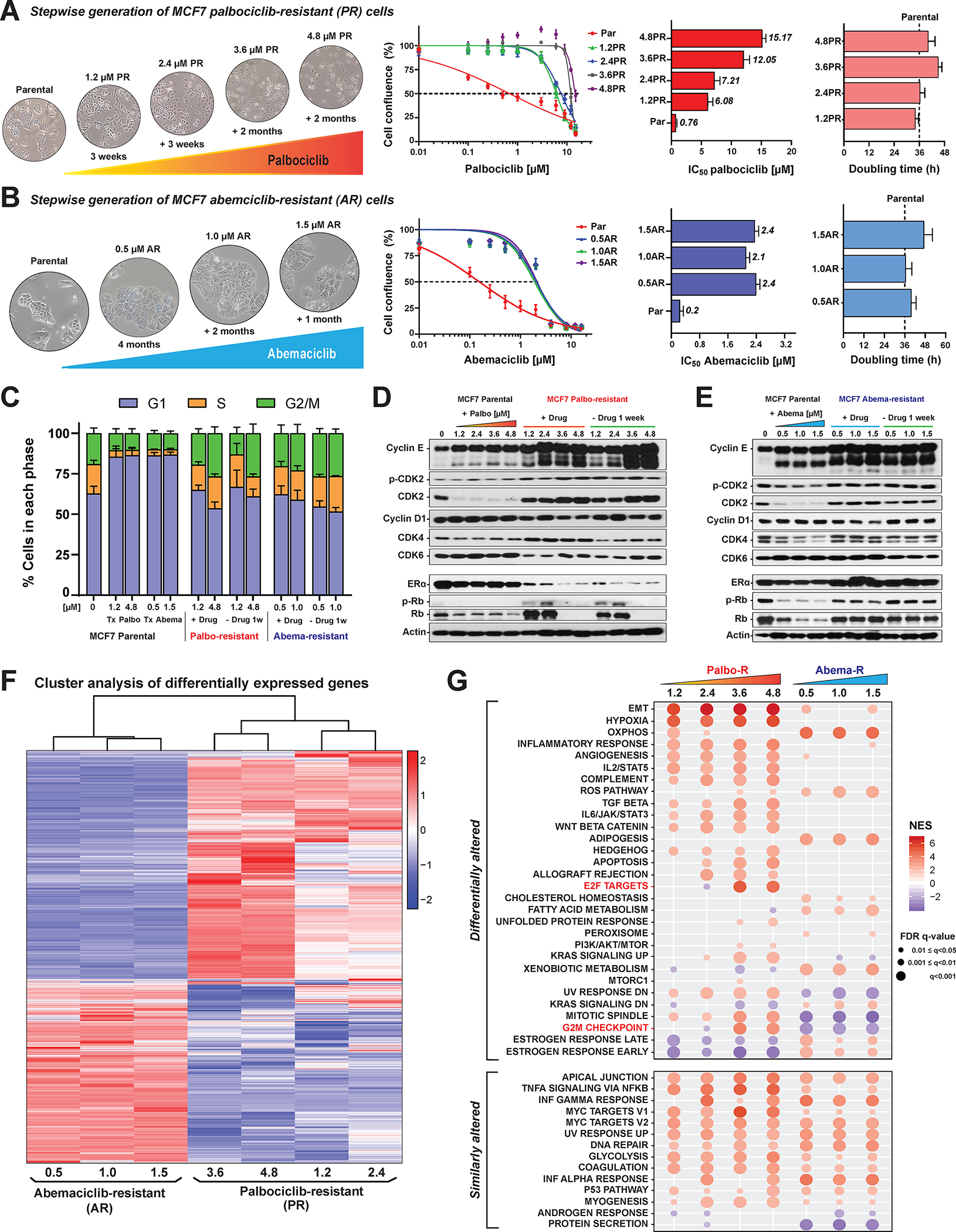
A, Left, Schematic representation of the generation of the PR breast cancer cell lines, with representative bright-field images (10× magnification) at each concentration. MCF7 PR cell lines were generated by culturing cells in medium supplemented with increasing concentrations of palbociclib from 1.2 to 4.8 μM in a stepwise manner over a 6-month period. Four cell lines resistant to palbociclib were generated: 1.2PR, 2.4PR, 3.6PR, and 4.8PR. Second from left: Dose-response curves for MCF7 parental and PR cells depicting the effect of treatment with 0.01–16 μM palbociclib for 6 days, followed by 6 days of recovery in drug-free medium. As a readout of cell proliferation, cell confluence (whole well) was assessed using Incucyte. Data were normalized to DMSO (100%), plotted, and analyzed by nonlinear regression using GraphPad Prism 9 software. The dashed line indicates IC50 values. Right graphs: horizontal bar graphs depicting the IC50 values and doubling times of PR cells. Each experiment included eight technical replicates per concentration. Data represent the mean ± SEM; n ≥ 3 independent experiments. B, Schematic representation, dose-response curves, IC50 values, and cell doubling times for MCF7 AR cells. The cells were generated using increasing concentrations of abemaciclib from 0.5 to 1.5 μM in a stepwise manner over a 7-month period. Three resistant cell lines were generated: 0.5AR, 1.0AR, and 1.5AR. Results are presented as described in A. C, Cell cycle analysis comparing the effect of palbociclib or abemaciclib treatment on MCF7 parental cells (first five bars), PR cell lines (middle four bars), and AR cell lines (last four bars), in the continuous presence of their respective drug (+Drug) and after 1 week of drug removal (−Drug 1w). Stacked bars represent the percentage of cells in each cell cycle phase, by color. D and E, Western blot analysis with the indicated antibodies for MCF7 parental and resistant cells under the conditions described in C. F, Heatmap depicting the hierarchical cluster analysis of differentially expressed genes in each MCF7 PR and AR cell line. Data represent the average expression of each gene for each cell line derived from three independent biological replicates. Red denotes genes with high expression levels; blue, genes with low expression levels. The color ranges from red to blue indicates the standardized log2 normalized expression from large to small. G, GSEA of cancer Hallmark gene sets in MCF7 PR and AR cells using the Molecular Signatures Database. Genes were ranked according to the significance (P values) in combination with the sign of log2 fold change values resulting from two-group comparisons (each resistant cell line at each concentration vs parental cells). The GSEA was performed for each comparison between resistant cells at each concentration of PR or AR vs parental cells. FDR (false discovery rate) cutoff of 5% was used to select significant gene sets, which were then compared across all resistant cells to analyze differential alteration between each concentration and each CDK4/6i. The upper dot plot depicts the pathways that showed a differentially altered pattern between PR and AR cells, while the bottom dot plot depicts the pathways that were similarly altered across all the resistant cell lines. The two pathways highlighted in red were downregulated in cells resistant to low concentrations and upregulated in cells resistant to high concentrations of palbociclib. NES, normalized enrichment score.
Analysis of cell cycle distribution (Supplementary Fig. S1C–S1E) and proliferation rate (Supplementary Fig. S1F–S1G) confirmed that in contrast to the parental cells, none of the PR or AR cells arrested in G1 following CDK4/6i treatment, demonstrating their complete proliferative adaptation to the CDK4/6is.
To determine if long-term culture in drug-free medium could re-sensitize resistant cells to palbociclib or abemaciclib, the resistant cells were cultured for up to 3 weeks in the presence or absence of the drug and then assessed for response to palbociclib or abemaciclib (Supplementary Fig. S2A–F). Dose-response curves showed similar sensitivity to the CDK4/6is in cells cultured in the presence or absence of the drug for 3 weeks (Supplementary Fig. S2B–S2C, and S2F), suggesting that the resistant phenotype was stable. Likewise, the cell cycle distribution was similar for resistant cells cultured in the presence or absence of the CDK4/6i for 7 days (Fig. 1C; Supplementary Fig. S2D). Cell cycle protein levels remained similar for each resistant panel, independent of being cultured in the presence or absence of each CDK4/6i for 7 days (Fig. 1D and E; Supplementary Fig. S2E). Interestingly, cyclin E, pCDK2, and CDK2 were similarly upregulated in PR and AR cells; however, ER, Rb, and pRb were differentially altered depending on which CDK4/6i was used to generate resistance (Fig. 1D and 1E; Supplementary Fig. S2E), suggesting that while PR and AR cells share some of the cell cycle bypass mechanisms of acquired resistance, they may also have distinct pathways of resistance.
Next, we examined the molecular signatures associated with resistance to palbociclib and abemaciclib through genome-wide RNA expression profiles of the PR and AR cell lines by RNA sequencing. First, PR and AR cells were analyzed as a single group and compared to parental cells in terms of differentially expressed genes (DEGs). Using a false discovery rate (FDR) cutoff of 5%, we identified 9099 significant DEGs in PR cells (Supplementary Fig. S3A, left panel) and 10,779 significant DEGs in AR cells (Supplementary Fig. S3A, middle panel) compared to parental cells. We also identified 12,576 DEGs in PR cells compared to AR cells (Supplementary Fig. S3A, right panel). Unsupervised hierarchical clustering comparing each resistant cell line to parental cells revealed that PR and AR cells harbored distinct transcriptomic profiles, as indicated by two distinct clusters (Fig. 1F). Furthermore, the PR cell cluster was bifurcated in subclusters corresponding to cells resistant to low (1.2 and 2.4 μM) and high (3.6 and 4.8 μM) concentrations of palbociclib. Principal components analysis (PCA) in MCF7 (Supplementary Fig. S3B) and T47D (Supplementary Fig. S3C) PR cells confirmed that the cells resistant to low concentrations (1.2 and 2.4 μM) mapped close to each other, while the cells resistant to high concentrations (3.6 μM and 4.8 μM) mapped in a different cluster. A PCA map of the AR cells revealed only two well-defined clusters corresponding to resistant vs parental cells; no distinct clusters related to different drug concentrations were observed (Supplementary Fig. 3D). These gene expression patterns suggest that acquisition of resistance to palbociclib is a multistep process, with changes gradually accumulating as the drug dose increases, while acquisition of resistance to abemaciclib is a monophasic process, with no additional changes as the drug dose increases. The three independent pools that were used to generate each of the PR and AR resistant cells have a high degree of concordance at the transcriptomic level. A heatmap of spearman correlation coefficient values of the RNA expression profiles among the different resistant cell lines shows that the three independent pools of each PR MCF7 and T47D cell lines (1.2, 2.4, 3.6 and 4.8), have high correlation (0.94 for T47D, 0.75 for MCF-7 and >0.6 for both; Supplemental Fig. 4A). Similarly, the spearman correlation coefficient heatmap from the RNA expression of the three independent pools of each abemaciclib resistant MCF7 cell lines (0.5, 1.0 and 1.5) shows a very high degree of correlation between the pools (>0.94, Supplemental Fig. S4B).
GSEA and RPPA reveal differentially altered pathways in palbociclib resistant compared to abemaciclib resistant cells
Since PR cells and AR cells have distinct transcriptomic profiles, we next examined the key pathways differentially altered between them by Gene Set Enrichment Analysis (GSEA). This analysis unveiled 30 significantly differentially altered and 14 similarly enriched pathways (Fig. 1G). EMT and hypoxia were the top pathways significantly upregulated in PR but not AR cells. OXPHOS, reactive oxygen species, adipogenesis, fatty acid metabolism, and cholesterol homeostasis were the top pathways significantly upregulated in AR but not PR cells. Glycolysis, DNA repair, and inflammatory-response-related pathways were similarly upregulated in PR and AR cells compared to parental cells.
In PR cells, several pathways exhibited higher enrichment with higher concentration of palbociclib to which cells were resistant (Fig. 1G). Two related pathways, E2F targets and G2/M checkpoint, were downregulated in cells resistant to low concentrations (1.2PR and 2.4PR) but upregulated in cells resistant to high concentrations (3.6PR and 4.8PR). Comparison of MCF7 and T47D PR cells (Supplementary Fig. S5) confirmed that most pathways were similarly altered in the two types of cells. In contrast, in AR cells, most of the pathways were similarly enriched across the cells resistant to different concentrations of abemaciclib (Fig. 1G), suggesting that resistance to abemaciclib occurs as a switch (i.e., is monophasic) and not in a stepwise manner.
To determine if the pathways enriched at the mRNA level (GSEA) were also induced at the protein level, we evaluated the proteomic profiles of PR and AR cells by reverse-phase protein array (RPPA) of 483 antibodies that recognize key proteins in multiple signaling pathways. Heatmap depiction of the resulting proteomic profiles revealed distinct protein expression patterns in PR and AR cells (Fig. 2A). To identify the key altered proteomic pathways, we performed a protein pathway enrichment analysis (PPEA) (see Supplemental Materials and Methods) (Fig. 2B; Supplementary Table S1). The results were highly concordant with the GSEA results (Fig. 1G), showing that EMT and cell cycle pathways (including several G2/M proteins) were the main pathways induced in PR but not AR cells, while OXPHOS was induced in AR but not PR cells. Taken together, the changes observed at the transcriptomic and proteomic levels suggest that pathways driving resistance to palbociclib and abemaciclib are distinct (Fig. 2B).
Figure 2. Divergent pathways are activated in PR and AR cells.
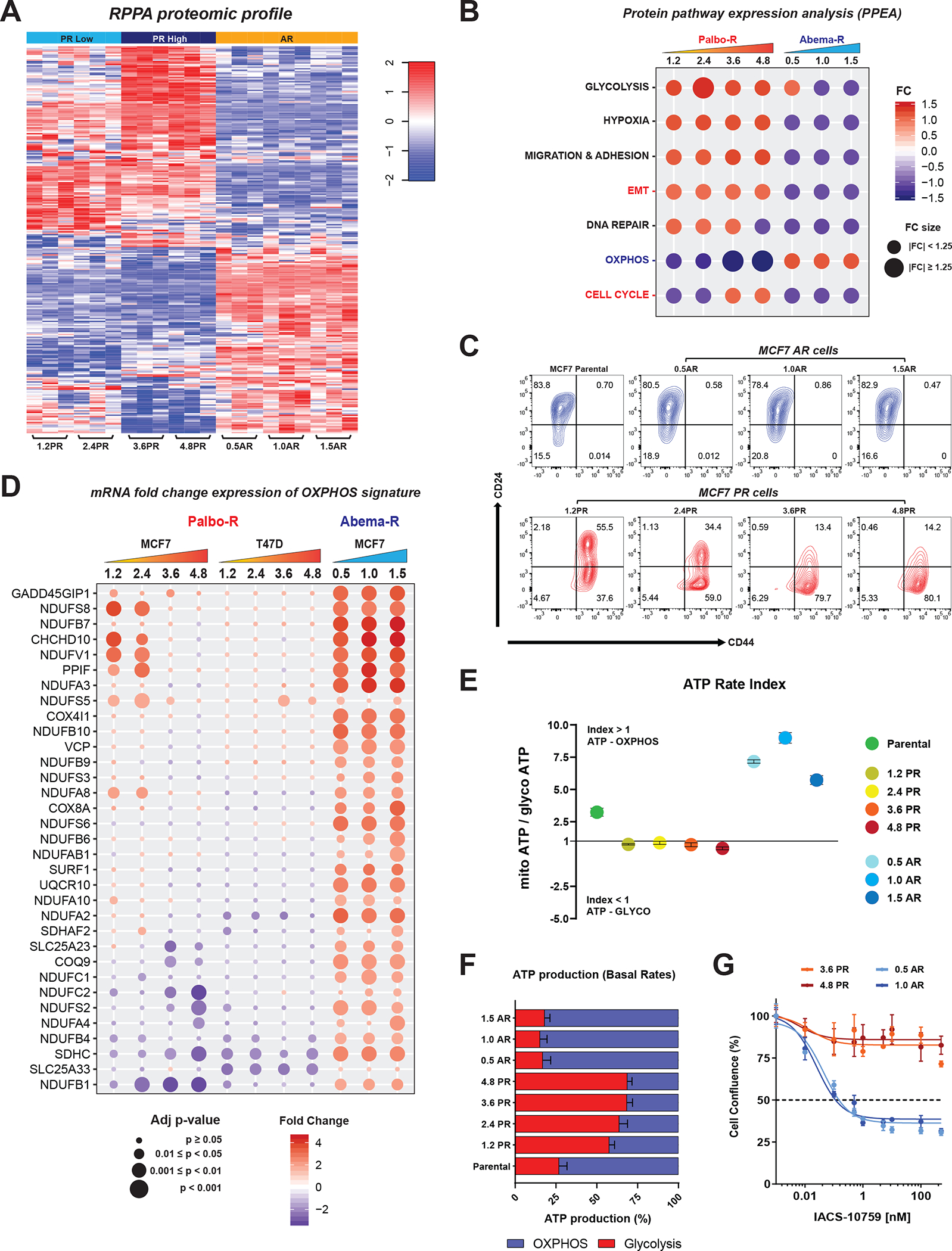
A, Heatmap depicting the changes in protein expression levels in PR and AR cells assayed by RPPA. Significant differences were tested with one-way ANOVA and the Tukey’s honest significance test, FDR=0.05. Three replicates for each cell line are shown. RPPA data was generated by the RPPA core facility and analyzed by the Department of Bioinformatics and Computational Biology at MDACC. B, Dot plot depicting the protein pathway enrichment analysis (PPEA) using the RPPA data. The fold change (FC) for each pathway corresponds to the average of the individual fold change values for the proteins that contribute to that pathway. C, Flow cytometry analysis of CD44 and CD24 markers showing the enrichment of the CSCs population (CD44+/high/CD24−/low) in PR and AR cells. Contour plots are representative of at least three independent experiments. D, mRNA fold change expression of an OXPHOS gene signature generated from the literature (see Supplementary Methods) across MCF7 and T47D PR cells and MCF7 AR cells. Fold change gene expression values correspond to the average of three biological replicates per cell line. E, Real-time ATP rate assay in MCF7 parental, PR, and AR cells. The ATP rate index is the ratio of oxygen consumption rate to extracellular acidification rate. A high index value represents a more oxidative, less glycolytic phenotype, and vice versa. Graphed values correspond to one representative experiment. Data represent the mean ± SEM; n = 3 biological replicates. F, Percentage of ATP production from OXPHOS and glycolysis in MCF7 parental, PR, and AR cells. Bars represent the mean ± SEM; n = 3 independent experiments. G, Dose-response curves in MCF7 PR and AR cells depicting the effect of treatment with increasing concentrations of the OXPHOS inhibitor IACS-10759 (0.001–500 nM) for 72 hours. Cell confluence (whole well) was assessed using Incucyte. Data were normalized to DMSO (100%), plotted, and analyzed by nonlinear regression using GraphPad Prism 9 software. The dashed line indicates IC50 values. Data represent the mean ± SEM; n = 4 biological replicates.
We next validated the key enriched pathways in the PR and AR cell lines through analyzing expression of a chronological EMT signature generated from the literature (61–65) across both the AR and PR cells. Assessment of key genes and regulators of the EMT pathway in these cell lines divulged an increase in the mRNA expression of specific early, intermediate, and late EMT genes in both MCF7 and T47D PR cells but not AR cells relative to parental cells. Downregulation of epithelial late EMT genes such as those encoding claudins was observed in PR cells, while upregulation of these genes was observed in AR cells (Supplementary Fig. S6A). PR but not AR cells exhibited increased protein expression of mesenchymal markers vimentin, N-cadherin, and the EMT transcription factor snail. In contrast, AR but not PR cells, expressed the epithelial marker E-cadherin (Supplementary Fig. S6B). These findings suggested that unlike PR cells, AR cells do not exhibit upregulation of the EMT pathway. In accordance with EMT upregulation, PR (but not AR cells) exhibited a stem-like phenotype, harboring a high population of CD44high/CD24low cells, identified as breast cancer stem cell-like (B-CSC-L) (27,28,66,67) (Fig. 2C; Supplementary Fig. S6C).
Consistent with significant enrichment of the IL-6/STAT3 pathway in PR but not AR cells (Fig. 1G), we found significantly lower levels of secreted IL-6 in AR cells than in PR cells (Supplementary Fig. S6D), suggesting a nonpivotal role of IL-6 as a driver of abemaciclib resistance. Although the ERα response pathway (Fig. 1G) and ERα protein levels (Supplementary Fig. S6E) were downregulated in PR cells and upregulated in AR cells, ERα was functionally compromised in both resistant models. Dose-response analysis in cells treated with fulvestrant showed that both PR and AR cells were resistant to fulvestrant compared to parental cells (Supplementary Fig. S6F).
The finding that OXPHOS was the major pathway upregulated at the RNA and protein levels in AR but not PR cells (Figs. 1G and 2B), suggested a role of this pathway in acquisition of resistance to abemaciclib. To further investigate the role of the OXPHOS pathway in resistance to abemaciclib, we interrogated the expression of a literature derived OXPHOS gene signature (68) in PR and AR cells. The expression pattern across the resistant cell lines showed that the majority of the genes in the OXPHOS signature were significantly upregulated in AR cells but not in PR cells (Fig. 2D). To assess the dependency of AR and PR cells on OXPHOS, we measured the kinetic activity of the two main ATP-producing pathways, mitochondrial respiration and glycolysis. Results showed that AR cells relied mainly on mitochondrial activity for energy production, as indicated by a high ATP rate index and a greater production of ATP from OXPHOS than from glycolysis, whereas PR cells relied mainly on glycolysis (Fig. 2E and 2F; Supplementary Fig. S6G). To investigate whether the OXPHOS dependency could be a vulnerability specifically targeted in AR cells versus PR cells, we evaluated response of PR and AR cells to the OXPHOS inhibitor IACS-010759, a clinical-grade small-molecule inhibitor of complex I of the mitochondrial electron transport chain (69). Treatment with IACS-010759 inhibited the proliferation of AR cells, but not PR cells (Fig. 2G), suggesting that the distinct pathway alterations during the acquisition of resistance to palbociclib and abemaciclib can identify specific vulnerabilities and dependencies.
Collectively, our results suggest that PR and AR cells have different mechanisms of resistance and provide a rationale for investigating the responsiveness of PR models to abemaciclib.
Abemaciclib inhibits entry into mitosis in palbociclib resistant cells
Consistent with the hypothesis that PR cells are sensitive to abemaciclib, PR cells had decreased inhibitory concentration (IC50) values for abemaciclib with increasing concentrations of palbociclib. For example, while the 1.2 μM PR cells responded to 2.0 μM abemaciclib, at the 4.8 μM PR dose, these cells responded to a much lower dose of abemaciclib (0.765μM), which showed 2.7 times more sensitivity to abemaciclib than the 1.2 μM PR (Fig 3A). This inverse relationship between response to palbociclib and abemaciclib can be seen by the shift in the IC50 values (IC50 palbociclib – IC50 abemaciclib) with the ΔIC50 difference ranging from 4.97 to 10.46 between the 1.2PR and 4.8PR cells (Fig. 3A, Supplemental Fig. S7A). The cell lines resistant to palbociclib at concentrations of 3.6 μM and 4.8 μM were the most sensitive to abemaciclib, with 9.47 and 10.46-unit shift in IC50 values. Conversely, AR cells displayed full cross-resistance to palbociclib, with higher IC50 values for palbociclib than for abemaciclib, as depicted by negative ΔIC50 values (Fig. 3B Supplemental Fig. S7A). Although the PR cells retained sensitivity to abemaciclib, the IC50 of abemaciclib in the PR cells was higher than that of the parental cells. This was not unexpected because the parental cells are untreated and the PR cells have undergone long-term treatment with palbociclib; However, we next assessed whether this observed response to abemaciclib was also accompanied by functional changes in the cells.
Figure 3. Abemaciclib induces G2 arrest and overrides entry into mitosis in PR cells.
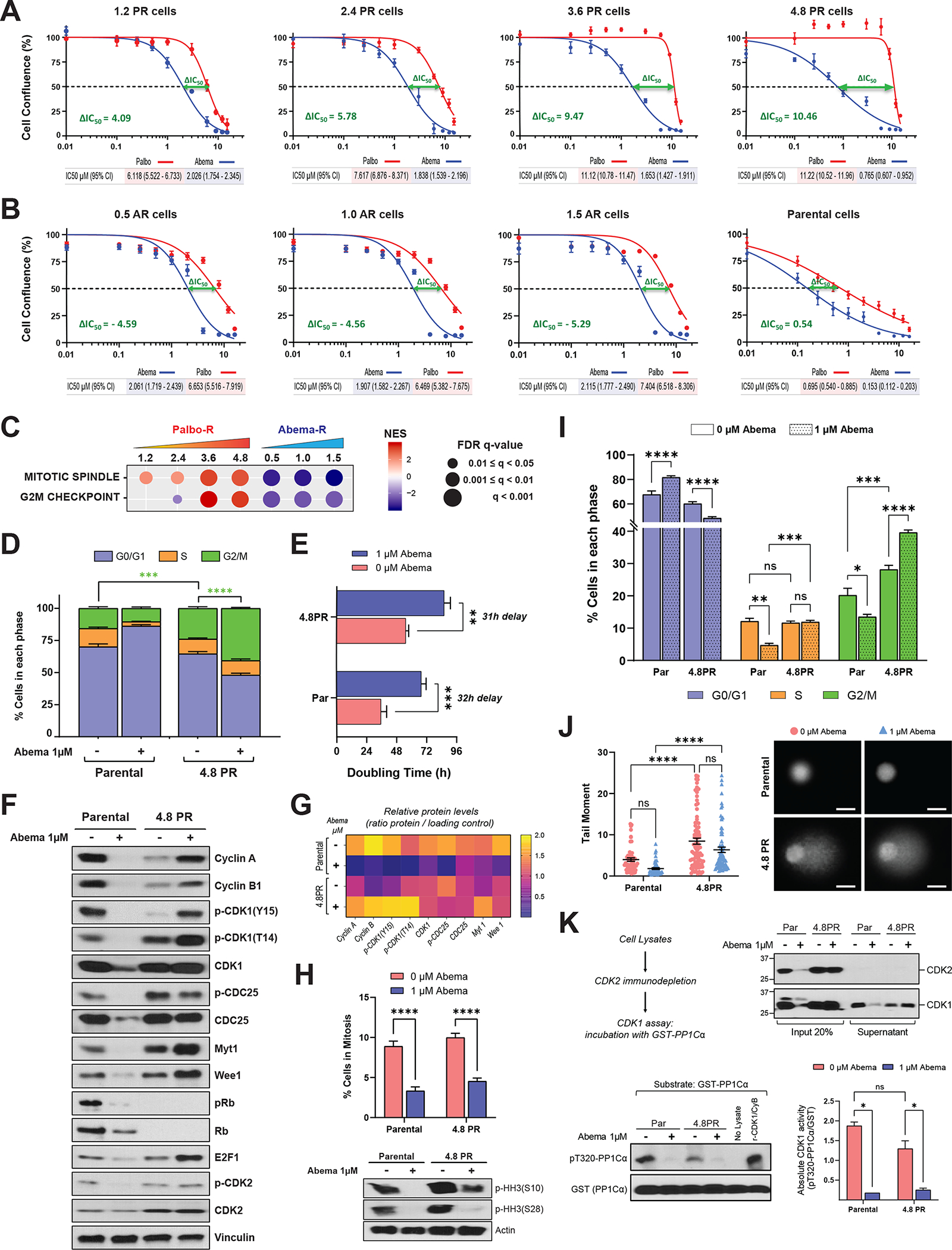
A and B, Dose-response curves in MCF7 PR (A), AR (B), and parental (B) cells depicting the effect of treatment with increasing concentrations of abemaciclib or palbociclib (0.01–16 μM) for 6 days, followed by 6 days of recovery in drug–free medium. As a readout of cell proliferation, cell confluence (whole well) was assessed using Incucyte. Data were normalized to DMSO (100%), plotted, and analyzed by nonlinear regression using GraphPad Prism 9 software. Each experiment included eight biological replicates per concentration and was repeated at least 3 times. Data represent the mean ± SEM; n ≥ 3 independent experiments. The IC50 values are indicated by the dashed line and specified below each plot including the 95% CI. The difference between the IC50 values for palbociclib and abemaciclib, or ΔIC50, was calculated by subtracting the IC50 of the tested drug from the IC50 of the drug to which the cells were resistant: For PR cells, ΔIC50 = (IC50 palbociclib − IC50 abemaciclib); for AR cells, ΔIC50 = (IC50 abemaciclib − IC50 palbociclib). The ΔIC50 is indicated in each plot. C, GSEA of two cancer hallmark gene sets, mitotic spindle and G2/M, in MCF7 PR and AR cells (see Fig. 1G). FDR, false discovery rate; NES, normalized enrichment score. D, Cell cycle analysis in MCF7 parental and 4.8PR cells comparing the effect of treatment with 1 μM abemaciclib for 6 days. Stacked bars represent the percentage of cells in each cell cycle phase. The difference in the percentages of cells in G2/M phase between the indicated groups was evaluated by two-way ANOVA, Tukey’s multiple comparisons test. *** P = 0.0003; **** P < 0.0001. Data represent the mean ± SEM; n ≥ 4 independent experiments. E, Bar graph depicting the effect of 1 μM abemaciclib treatment for 6 days on the doubling time of MCF7 parental and 4.8PR cells. Cells were counted on day 0 and on day 6, after treatment. The difference between groups was evaluated by two-way ANOVA, Šídák’s multiple comparisons test. ** P = 0.0010; *** P = 0.0004. Data represent the mean ± SEM; n ≥ 4 independent experiments. F, Western blot analysis of the indicated proteins in MCF7 parental and 4.8PR cells treated with 1 μM abemaciclib for 6 days. G, Densitometry analysis for the western blots in F showing the average protein levels (ratio for each protein/loading control) of three independent experiments. H, Top: Percentage of MCF7 parental and 4.8PR cells in mitosis after treatment with 1 μM abemaciclib for 6 days evaluated by flow cytometry. The percentage of cells in mitosis was determined by quantitation of histone H3 phosphorylated at S28 (pHH3-S28) in parallel with DNA content (propidium iodide) to evaluate the cell cycle. Percentage of cells in mitosis = (cells in mitosis / cells in G2/M) × 100. The difference between groups was evaluated by two-way ANOVA, Dunnett’s multiple comparisons test. **** P ≤ 0.0001. Bottom: Western blot analysis of pHH3 (phosphorylated at S28 and S10) using cell lysates from two independent representative experiments with MCF7 parental and 4.8PR cells treated under the same conditions. I, Percentages of cells gated in each cell cycle phase according to the DNA content (measured by propidium iodide) from the experiments in H. Multiple comparisons were evaluated by two-way ANOVA, Tukey’s multiple comparisons test. * P = 0.0139; ** P = 0.0052; *** P ≤ 0.0007; **** P ≤ 0.0001; ns: not significant. Data represent the mean ± SEM; n ≥ 4 independent experiments. Cell cycle experiments in I are independent from those in D. J, DNA damage in MCF7 parental and 4.8PR cells upon 1μM abemaciclib treatment for 6 days evaluated by comet assay. Representative images of different conditions are shown. Images were captured using 20× magnification. Bars = 25 μm. Cells were scored using the Comet Assay Software Project (CASP) tool; 50–100 cells were scored per condition per independent experiment. Each experiment included two biological replicates per concentration and was repeated twice. Data are representative of one independent experiment and depict the tail moment mean ± SEM. Tail moment = percentage of DNA in the tail × tail length. The difference between groups was evaluated by two-way ANOVA, Šídák’s multiple comparisons test. **** P ≤ 0.0001; ns: not significant. K, In vitro CDK1 kinase activity measured by quantitation of the phosphorylation levels on T320 of the substrate protein phosphatase 1Cα (pT320-PP1Cα). Top left: Experimental flow chart. Top right: Western blot analysis with both CDK2 and CDK1 antibodies showing the presence of CDK1 after immunodepletion of CDK2. Bottom left: CDK2-immunodepleted lysates were incubated with recombinant GST-PP1Cα and assayed for pT320-PP1Cα by western blot analysis. Recombinant CDK1/cyclin B (CyB) complex was used as a positive control. Bottom right: Bar graph shows absolute in vitro CDK1 activity quantified as the ratio of pT320-PP1Cα to GST from two independent experiments. Densitometry analysis was performed using ImageJ software. The difference between groups was evaluated by two-way ANOVA, Tukey’s multiple comparisons test. * P < 0.05; ns: not significant.
To further investigate the mechanisms mediating the response of PR cells to abemaciclib, we focused on the G2/M pathway since it exhibited an opposite enrichment pattern in PR and AR cells (Fig. 3C), suggesting that it may be targeted by abemaciclib. Cell cycle analysis revealed an increased accumulation of untreated PR cells in the G2/M phase compared to parental cells that was exacerbated by abemaciclib treatment (Fig. 3D; Supplementary Fig. S7B), resulting in a G2/M-mediated 31-hour delay in their doubling time compared to untreated PR cells (Fig. 3E). Parental cells, in contrast, were arrested in the G1 phase with a concomitant G1-mediated 32-hour delay in their doubling time in response to abemaciclib treatment. Western blot analysis of key G2/M proteins in PR cells treated with abemaciclib showed higher accumulation of cyclin B, inhibitory pCDK1 (Y15 and T14), and Myt1, while levels of p-CDC25C were lower in PR cells as compared to untreated cells (Fig. 3F and 3G). These results suggest that in response to abemaciclib treatment, the cyclin B/CDK1 complex is inactivated, resulting in abemaciclib-mediated G2 arrest of PR cells. In contrast, in parental cells treated with abemaciclib, the levels of these proteins decreased, along with the levels of cyclin A, p-CDK2, and Wee1, consistent with G1 arrest, as corroborated by low levels of Rb, p-Rb, and E2F1 (Fig. 3F and G).
To evaluate if abemaciclib inhibits the entry of PR cells into mitosis, we assessed phosphorylation of histone H3 (pHH3, a mitotic marker) in parallel to cell cycle changes. Flow cytometry (Fig. 3H top) and western blot analyses (Fig. 3H bottom) indicated a significant reduction in the mitotic index of both parental and PR cells following abemaciclib treatment as compared to untreated cells. In PR cells, this reduction was mediated through a G2 cell cycle arrest as evidenced by increased accumulation of cells in G2/M phase and decreased accumulation of cells in G1 phase without changes in S phase (Fig. 3I). On the other hand, in parental cells, the reduction in mitotic index was mediated by G1 arrest as evidenced by reduced accumulation of cells in S and G2/M and increased accumulation of cells in G1 (Fig. 3I). The abemaciclib-induced G2 arrest in PR cells did not appear to be due to increased DNA damage as the comet tail length was similar in abemaciclib-treated and untreated PR cells (Fig. 3J). Next, we evaluated if abemaciclib can inhibit CDK1 kinase activity in PR cells. To this end, we examined phosphorylation of the T320 site in the human protein phosphatase PP1Cα by CDK1 as described (70,71). Since this substrate can also be phosphorylated by CDK2, we first immunodepleted all cell lysates with an anti-CDK2 antibody and then assessed the CDK1 activity (Fig. 3K top). We found that CDK1 activity was significantly attenuated following abemaciclib treatment in both parental and 4.8PR cells (Fig. 3K bottom). Taken together, our results suggest that PR models exhibiting upregulation of the G2/M pathway could be sensitive to abemaciclib.
Patient-derived models of palbociclib resistant breast cancer retain abemaciclib sensitivity
We next assessed the response to abemaciclib in patient-derived models of PR breast cancer. We used three patient-derived xenografts (PDXs; termed PR-1, PR-2, and PR-3) previously established from patients whose disease progressed during first-line treatment with palbociclib plus ET (Supplementary Fig. S8A) to generate long-term organoid cultures (PDxOs) (Fig. 4A). We used an additional PDX model derived from a patient whose disease progressed during ET alone and was CDK4/6i treatment naïve (ET-R) as a model of breast cancer sensitive to both palbociclib and abemaciclib (Supplementary Fig. S8A–S8B). PDxOs were morphologically heterogeneous (Fig. 4B, bright field) and recapitulated the histopathological features of the parent PDX tumors: high-grade (grade 3) adenocarcinoma, pleomorphic with a high percentage of mitotic figures (Fig. 4B, H&E). PDxOs also retained the hormone receptor and Ki67 status of the parental PDX tumors (Fig. 4B) and the original patient tumor biopsy (Supplementary Fig. S8A). RNA-sequencing analysis showed that PDxOs had gene expression profiles concordant with those of the parent PDX tumors, as evidenced by high Pearson correlation coefficients between organoids and PDX tumors. Inter-model correlation was higher between PR models, which clustered in a separate group from the ET-R model. All models showed high intra-model correlation (Fig. 4C).
Figure 4. Patient-derived palbociclib resistant breast cancer models retain abemaciclib sensitivity.
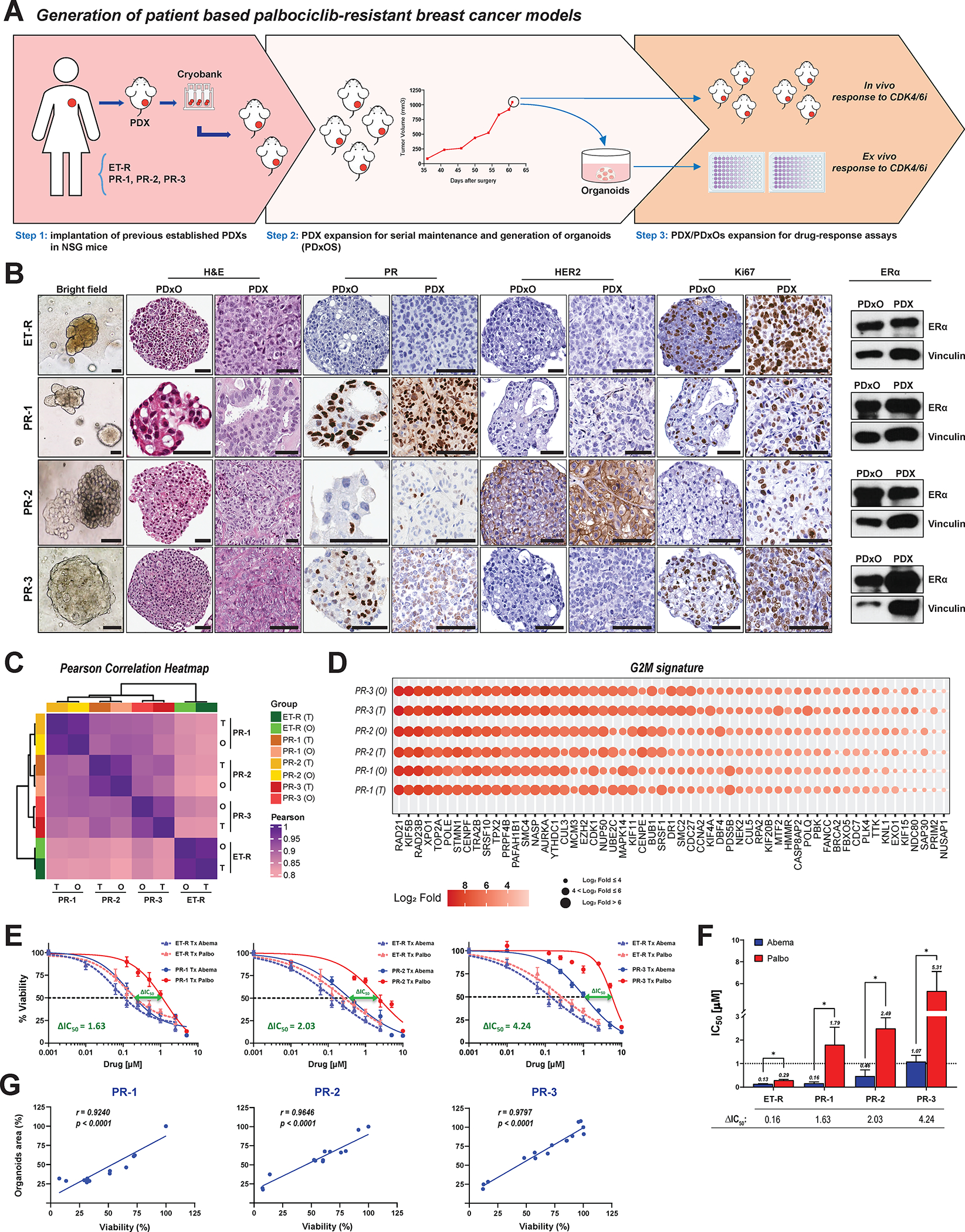
A, Schema for establishing human PDXs and long-term organoid cultures from PDXs (PDxOs). See Supplementary Fig. S8A for additional details on the PDX models. B, Morphological and histological characterization of matched PDxOs and PDXs. First column: Bright-field images (20× magnification) from the four PDxO models depicting the organoid phenotypes. ET-R and PR-3 had cohesive, dense/solid organoids; PR-1 had cohesive organoids, with a mix of cystic/hollow and dense/solid organoids; and PR-2 had discohesive organoids. Second and third columns: H&E staining comparing the histopathological features of PDxOs and their matched PDXs. PDxOs show a tissue architecture/organization reminiscent of the original PDX lesions, high-grade adenocarcinoma. ET-R, PR-1, and PR-2 exhibited some areas forming tubular gland structures, while PR-3 exhibited marked nuclear atypia and mitotic activity with no glandular/tubular differentiation. Fourth to ninth columns and ERα column: IHC staining for progesterone receptor (PgR), HER2, and Ki67 and western blot analysis of ERα showing the concordance of the hormone receptor status and proliferation status of the PDX/PDxO pairs with the originating patient tumors (details in Supplementary Fig. S8A). Scale bars: 50μm. C, Hierarchical clustering correlation heatmap illustrating transcriptome fidelity between PDX tumors (T) and their derived organoids (O). The color scale within the heatmap indicates the Pearson correlation coefficient. Hierarchical clustering on the top and left side depicts the distance between models. D, G2/M gene signature expression in PR PDX tumors (T) and their derived PDxOs (O). Color scale indicates the extent of gene expression levels (on log2 scale). E, Dose-response curves for PDxO models depicting the effect of treatment with 0.001–10 μM concentrations of abemaciclib or palbociclib for 6 days, followed by 6 days of recovery in drug-free medium. Organoid viability was assessed by Cell Titer-Blue assay. Data were normalized to DMSO (100%), plotted, and analyzed by nonlinear regression using GraphPad Prism 9 software. The dashed lines indicate IC50 values. The difference between the IC50 values for palbociclib and abemaciclib treatment, or ΔIC50, was calculated as IC50 palbociclib – IC50 abemaciclib. Each experiment included three biological replicates per concentration. Data represent the mean ± SEM; n ≥ 3 independent experiments. F, Bar graph summarizing differences in IC50 values for each PDxO model. G, Pearson correlation between the PDxO drug response assessed by Cell Titer-Blue viability assay and by measuring the organoid area.
To investigate if the G2/M pathway enrichment observed in PR cells was conserved in both PDX and PDxO models of palbociclib resistant breast cancer, we generated a G2/M signature by selecting the overlapping genes contributing to the leading-edge of the G2/M pathway derived from both MCF7 and T47D PR cells. The G2/M signature expression pattern was concordant with the results of the GSEA and highly expressed in MCF7 and T47D cells resistant to palbociclib at concentrations of 3.6 and 4.8 μM (Supplementary Fig. S8C). All three PR breast cancer PDX and PDxO models also expressed the G2/M gene signature, with high fidelity between tumors and organoids (Fig. 4D). To determine whether genomic alterations could be identified that are driving the gene expression changes of the resistant models, we performed mutational analysis on our PDX samples and the CDK4/6i resistant MCF-7 and T47D cells. Genes (n=34) previously shown to drive resistance to CDK4/6is were observed to be mainly amplified or deleted, in agreement with previous reports (25,28,42,72–77). However, the mutational landscape of the patient derived samples was heterogenous and lacked concordance with genomic alterations of the cell lines. There were only three mutational variants, two of which were in TP53, that do not account for the transcriptional changes observed with palbociclib resistance (Supplementary Fig. S8D) nor have been shown to alter response to palbociclib in sensitive cells (47). Next, we interrogated the most frequently occurring mutations (n=17 genes), rather than selecting those that have been reported to be drivers of CDK4/6i resistance (Supplementary Fig. S8E). Again, the mutational landscape of these 17 genes was heterogeneous, especially in the PDX samples with minimal concordance with the resistant cell lines.
All three PR PDxOs were refractory to palbociclib, but responded to abemaciclib treatment, with IC50 values ≤ 1 μM (Fig. 4E and 4F). In contrast, the ET-R (CDK4/6i naïve) PDxOs were sensitive to both CDK4/6is, exhibiting low IC50 values and only a 0.16-unit shift between the palbociclib and abemaciclib IC50 values (Fig. 4E and 4F), consistent with the in vivo responses of parent PDXs to these CDK4/6is (Supplementary Fig. 8B). Comparison of the response to abemaciclib between the sensitive PDxOs (ET-R) and each PR PDxO model showed that PR PDxOs retained sensitivity to abemaciclib, as indicated by dose-response curves with similar slopes and a 1.6- to 4-unit shift in the IC50 value between palbociclib and abemaciclib (Fig. 4E and F). PR PDxO drug responses were reproducible when the organoid size (area) was used as the treatment response readout, as evidenced by a significant Pearson correlation between organoid viability and organoid area (Fig. 4G). These results suggest that patient-derived models refractory to palbociclib treatment with an upregulated G2/M pathway are responsive to abemaciclib.
Abemaciclib inhibits tumor growth and prolongs survival after progression on palbociclib
To evaluate whether palbociclib resistant breast cancer PDX models are also sensitive to abemaciclib, we assessed their in vivo response to abemaciclib and palbociclib treatments. Abemaciclib treatment of the PR-3 PDX model (Supplementary Fig. S8A), significantly inhibited its tumor growth (Fig. 5A) and prolonged survival (Fig. 5B) compared to palbociclib-treated mice, which were refractory to palbociclib, recapitulating the clinical response of the patient from whom the PDX was derived, and had a similar tumor growth to that in vehicle-treated mice. A similar growth-inhibitory response to abemaciclib was observed in the PR-2 PDX model (Supplementary Fig. S9A–S9B). The PR-3 and PR-2 PDXs exhibited a drug response concordant with that of their matched organoids as demonstrated by inhibition of tumor growth and organoid viability after abemaciclib treatment versus no inhibition after palbociclib treatment (Fig. 5C; Supplementary Fig. S9C). There was a significant positive linear correlation between tumor volume and organoid viability (Fig. 5D; Supplementary Fig. S9D). The therapeutic response to abemaciclib was further confirmed by significant reductions in proliferation markers Ki67 and pHH3, to <8% and <1%, respectively, for abemaciclib-treated PDX tumors compared to >37% and >2%, respectively, for palbociclib and vehicle-treated PDX tumors (Fig. 5E and 5F). Collectively, the results from these pre-clinical ex vivo and in vivo models suggest that tumors which have progressed on palbociclib can respond to abemaciclib.
Figure 5. Abemaciclib inhibits tumor growth and prolongs survival after progression on palbociclib therapy.
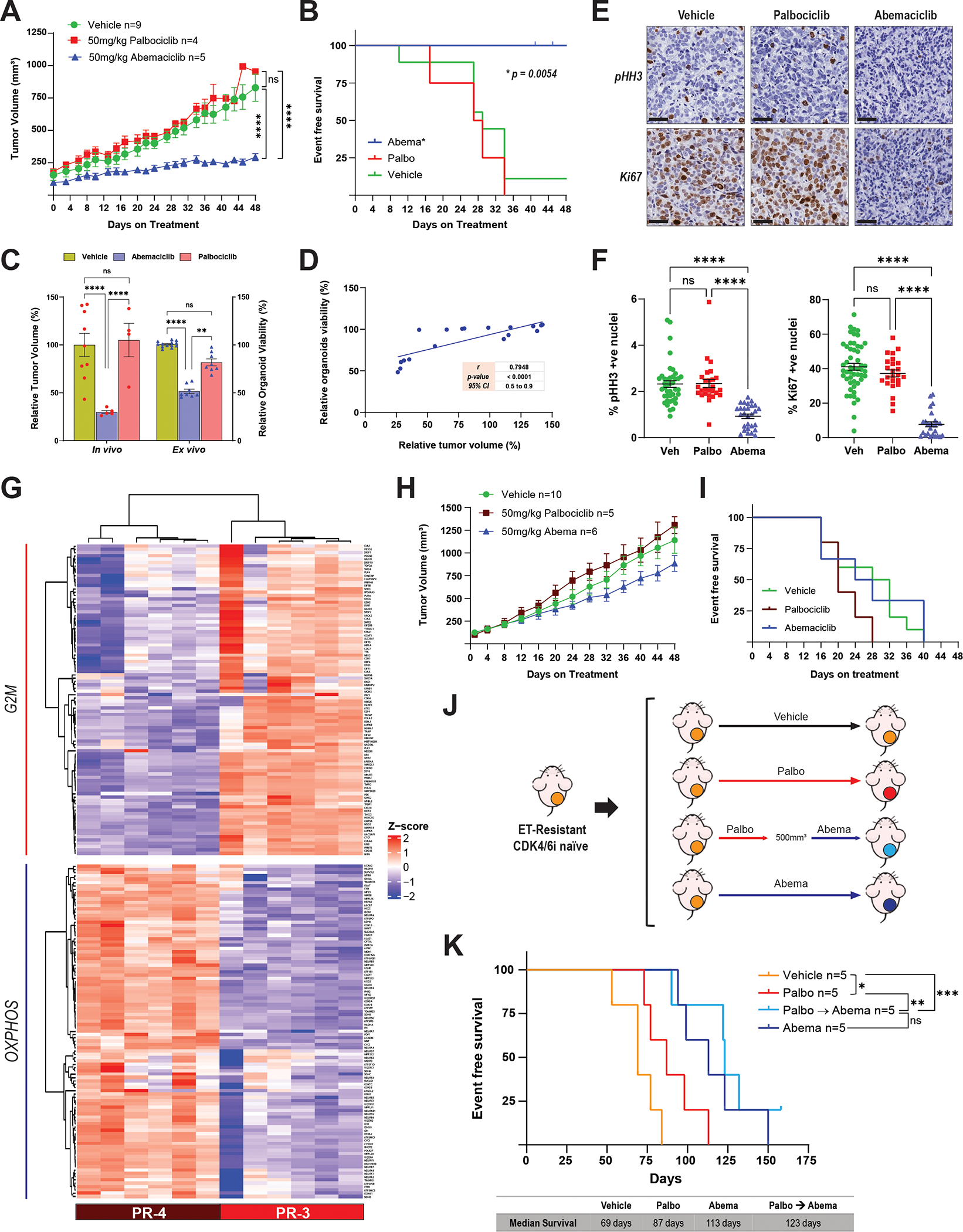
A, Tumor growth curves of female nude mice bearing the PR-3 PDX model and treated with abemaciclib (50 mg/kg PO, every day), palbociclib (50 mg/kg PO, 21-day cycle), or vehicle for 48 days. Before treatment, tumors were allowed to grow until they reached a volume of 100–200 mm3 in the presence of E2 (8 μg/mL) supplementation in the drinking water. Once treatment started, the E2 supplementation was removed to mimic the effect of ET. The length and width of tumor xenografts were measured by calipers 2–3 times per week, and the tumor volume was calculated as (length × (width)2)/2. All tumors were collected at the same time and processed for biomarker analysis. The differences between groups were evaluated by two-way ANOVA, Tukey’s multiple comparisons test. **** P ≤ 0.0001; ns: not significant. Data represent the tumor volume mean ± SEM; n = 9 mice for vehicle, n = 4 mice for palbociclib, n = 5 mice for abemaciclib. B, Survival analysis of female nude mice bearing the PR-3 PDX model and treated as described in A. Event-free survival was calculated based on the time on treatment when tumor volume reached 500 mm3. The difference between survival curves was calculated using the log-rank (Mantel-Cox) test. C, Comparison of the response to abemaciclib and palbociclib treatment in the PR-3 PDX model (in vivo) and its matched organoids (ex vivo). Relative tumor volume (%) was calculated from the experiment shown in A, and relative organoid viability (%) was calculated from the experiments shown in Fig. 4E. The difference between groups was evaluated by two-way ANOVA, Tukey’s multiple comparisons test. ** P ≤ 0.0085; **** P ≤ 0.0001; ns: not significant. D, Pearson correlation between the tumor volume and the organoid viability from experiments in A and Fig. 4E, respectively. E and F, pHH3 and Ki67 immunohistochemistry analysis of PR-3 PDX tumor tissues from the experiment shown in A. Representative images (40× magnification, Scale bars: 50μm) are shown in E. Quantification of Ki67- and pHH3-positive nuclei was performed using QuPath software (and independently by a pathologist) by assessing multiple areas in each individual tumor for each treatment to cover the entire tumor, and data are presented as mean ± SEM. The difference between groups was evaluated by two-way ANOVA, Tukey’s multiple comparisons test. **** P ≤ 0.0001; ns: not significant. G, Hierarchical clustering heatmap depicting the leading-edge gene expression of G2/M and OXPHOS pathways in PR-3 (n = 6 mice) compared to PR-4 (n = 6 mice) models. H, Tumor growth curves of female nude mice bearing the PR-4 PDX model and treated as described in A. The difference between groups was evaluated by two-way ANOVA, Tukey’s multiple comparisons test; no significant differences were observed. Data represent the tumor volume mean ± SEM; n = 10 mice for vehicle, n = 5 mice for palbociclib, n = 6 mice for abemaciclib. I, Survival analysis of female nude mice bearing the PR-4 PDX model and treated as described in A. Event-free survival was calculated based on the time on treatment when tumor volume reached 500 mm3. The difference between survival curves was calculated using the log-rank (Mantel-Cox) test. No significant differences were observed. J, Preclinical experimental design to assess the response to abemaciclib following lack of response to palbociclib in an endocrine therapy–resistant (ET-R) and CDK4/6i-naive PDX model. Female nude mice bearing the ET-R PDX model were randomized into four arms as indicated and treated with vehicle; palbociclib (75 mg/kg PO, 21-day cycle); palbociclib as the first CDK4/6i followed by sequential treatment with abemaciclib once the tumors reached 500 mm3; or abemaciclib (50 mg/kg PO, every day). E2 supplementation was given as described in A. K, Survival analysis of mice treated as described in J. Event-free survival was calculated based on the duration of treatment prior to the tumor burden reaching the ethical endpoint. The difference between survival curves was calculated using the log-rank (Mantel-Cox) test. * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; ns: not significant.
To determine which palbociclib resistant tumors could benefit from a switch to abemaciclib, we evaluated the response of an additional model (PR-4 PDX) to abemaciclib. PR-4 was derived from a patient with metastatic ER-positive/PgR-negative/HER2-negative breast cancer whose disease progressed after 3 months of second-line treatment with palbociclib plus fulvestrant (50) (Supplementary Fig. S8A). Compared to the PR-3 abemaciclib-sensitive model, PR-4 had upregulated expression of genes in the OXPHOS pathway (leading edge) and downregulated expression of genes in the G2/M pathway (leading edge) (Fig. 5G), which we had identified as differential pathways of resistance to abemaciclib (OXPHOS high) and palbociclib (G2/M high) (Figs. 1–4). Similar expression patterns were observed in PR-3 and PR-4, when we interrogated our previous cell line-derived G2/M and literature-derived OXPHOS gene signatures (Supplementary Fig. S9E). As predicted by high OXPHOS and low G2/M gene signature expression, PR-4 did not respond to abemaciclib treatment, resulting in lack of tumor growth inhibition (Fig. 5H) or survival benefit (Fig. 5I). PR-4 was previously reported to respond to the OXPHOS inhibitor IACS-010759 (50). Taken together, these results suggest that tumors refractory to palbociclib therapy that are likely to benefit from abemaciclib treatment could be selected on the basis of a G2/M high and OXPHOS low gene set enrichment pattern.
Lastly, we tested if the abemaciclib switching treatment strategy had therapeutic benefit in a preclinical model that mimics the clinical treatment course of patients with ER-positive/HER2-negative MBC whose disease initially responds to CDK4/6 inhibition but becomes resistant during long-term treatment. In these experiments, we used ET-R, a CDK4/6i-naïve PDX model that was derived from a patient with ER-positive/HER2-negative MBC who progressed on AI treatment (Supplementary Fig. S8A) and was responsive to both palbociclib and abemaciclib (Supplementary Fig. S8B). Mice bearing ET-R tumors were treated with (i) vehicle; (ii) palbociclib alone; (iii) abemaciclib alone; or (iv) palbociclib followed by abemaciclib once the tumors reached 500 mm3 during palbociclib treatment, indicating lack of response (Fig. 5J). Kaplan-Meier analysis revealed that mice switched to abemaciclib after no response on palbociclib had significantly longer median survival (123 days) than mice receiving only palbociclib (87 days) or vehicle (69 days). The median survival in the palbociclib followed by abemaciclib group was similar to that in mice receiving abemaciclib continuously (Fig. 5K). These results suggest that treatment of palbociclib-refractory tumors with abemaciclib could significantly improve outcome.
Abemaciclib following progression on palbociclib results in clinical benefit in patients with HR-positive/HER2-negative MBC
To evaluate the potential clinical benefit of abemaciclib after disease progression during treatment with a prior CDK4/6i, we conducted a retrospective analysis of patients treated at MDACC. We first identified patients with HR-positive/HER2-negative MBC who had received abemaciclib (n=242). From this cohort, we excluded patients who discontinued the prior CDK4/6i because of toxicity (n=39) or received abemaciclib as the initial CDK4/6i (n=151) (Supplementary Fig. S10). Patients were included (n= 52) if they progressed on palbociclib (n=51) or ribociclib (n=1) and were subsequently treated with abemaciclib either “sequentially,” i.e., upon progression with the first CDK4/6i (n=18), or “non-sequentially,” i.e., following other systemic therapies (n=34) (Supplementary Fig. S10). Twenty-six of the 34 patients who received abemaciclib non-sequentially had received chemotherapy (Table 1). The first CDK4/6i was given either in combination with an AI (n=34) or an oral selective estrogen degrader (SERD, n=18) (Table 1 and Fig. 6A). Fifteen patients (28.8%) received abemaciclib as monotherapy, and 37 (71.2%) received abemaciclib plus ET. Of these 37 patients, 10 (19.2%) continued with the same class of ET plus abemaciclib, and 27 (52%) were switched to a different ET plus abemaciclib; specifically, 25 (48.1%) were switched from palbociclib plus AI to abemaciclib plus fulvestrant, and 2 (3.9%) were switched from palbociclib plus fulvestrant to abemaciclib plus AI. At the data cutoff of December 2022, 13 patients (25%) were still receiving abemaciclib, while 39 (75%) had discontinued abemaciclib because of disease progression (Fig. 6A). Twenty patients (38%) received abemaciclib for more than 6 months, and 13 (25%) received abemaciclib for more than 1 year (Fig. 6A). Mutational analysis of 98 genes in 28 of the patient tumor samples did not show any consistent alterations with time to progression on abemaciclib (Fig 6B). As with the PDX models (Supplementary Figs. S8D. and S8E), the mutational landscape of the patient samples was highly heterogeneous. ESR1 was the most frequently altered gene with missense mutations and/or fusions in 39% of the patients, followed by PIK3CA, TP53, and FGFR1 in 36%, 32% and 25% of the patients, respectively. These data are in agreement with the data published by Wander et al. (42) and again shows that the genomic landscape of CDK4/6i resistance is heterogeneous with no particular genetic mutation being observed in a majority of cases that could account for the CDK4/6i resistance, stratified according to time on abemaciclib, sequential vs non-sequential treatment or death events (Fig. 6B, Supplementary Fig. S11, Supplementary Fig S12, respectively).
Table 1.
Characteristics of patients with MBC who received abemaciclib after disease progression on a prior CDK4/6i (N=52)
| Parameter | n (%)* |
|---|---|
| Mean age (range), y | |
| Original diagnosis | 47.5 (29–75) |
| Diagnosis of metastasis | 53 (35–75) |
| Sex | |
| Female | 50 (96) |
| Male | 2 (4) |
| Race/ethnicity | |
| White | 38 (73) |
| Black | 4 (8) |
| Hispanic | 2 (4) |
| Asian/Pacific Islander | 5 (10) |
| Other | 3 (6) |
| Abemaciclib sequence following prior CDK4/6i | |
| Sequential | 18 (35) |
| Non-sequential | 34 (65) |
| Abemaciclib treatment course | |
| Monotherapy | 15 (29) |
| Endocrine partner | 37 (71) |
| Aromatase inhibitor | 3 (6) |
| Fulvestrant | 34 (65) |
| Clinical outcome on abemaciclib at data cutoff (July 1, 2022) | |
| On treatment | 13 (25) |
| Discontinued, progressive disease | 39 (75) |
| Prior CDK4/6i treatment course | |
| Ribociclib | 1 (2) |
| Palbociclib | 51 (98) |
| Endocrine partner | |
| Aromatase inhibitor | 34 (65) |
| SERD | |
| Fulvestrant | 17 (33) |
| other | 1 (2) |
| Mean duration of CDK4/6i treatment, months (range) | |
| First CDK4/6i (palbociclib or ribociclib) | 11.1 (1 – 43.6) |
| Second CDK4/6i (abemaciclib) | 7.6 (0.9 – 29.3) |
| Other treatment lines prior to abemaciclib, metastatic setting (nonsequential cohort) | |
| Chemotherapy | 26 (50) |
| Everolimus | 11 (21) |
| Tamoxifen | 7 (14) |
| Alpelisib | 2 (4) |
| Bevacizumab | 1 (2) |
| Olaparib | 1 (2) |
| Megestrol | 1 (2) |
Data are presented as n(%) unless otherwise specified
Figure 6. Clinical outcomes in patients with HR+/HER2-negative metastatic breast cancer treated with abemaciclib after progression on prior CDK4/6i treatment.
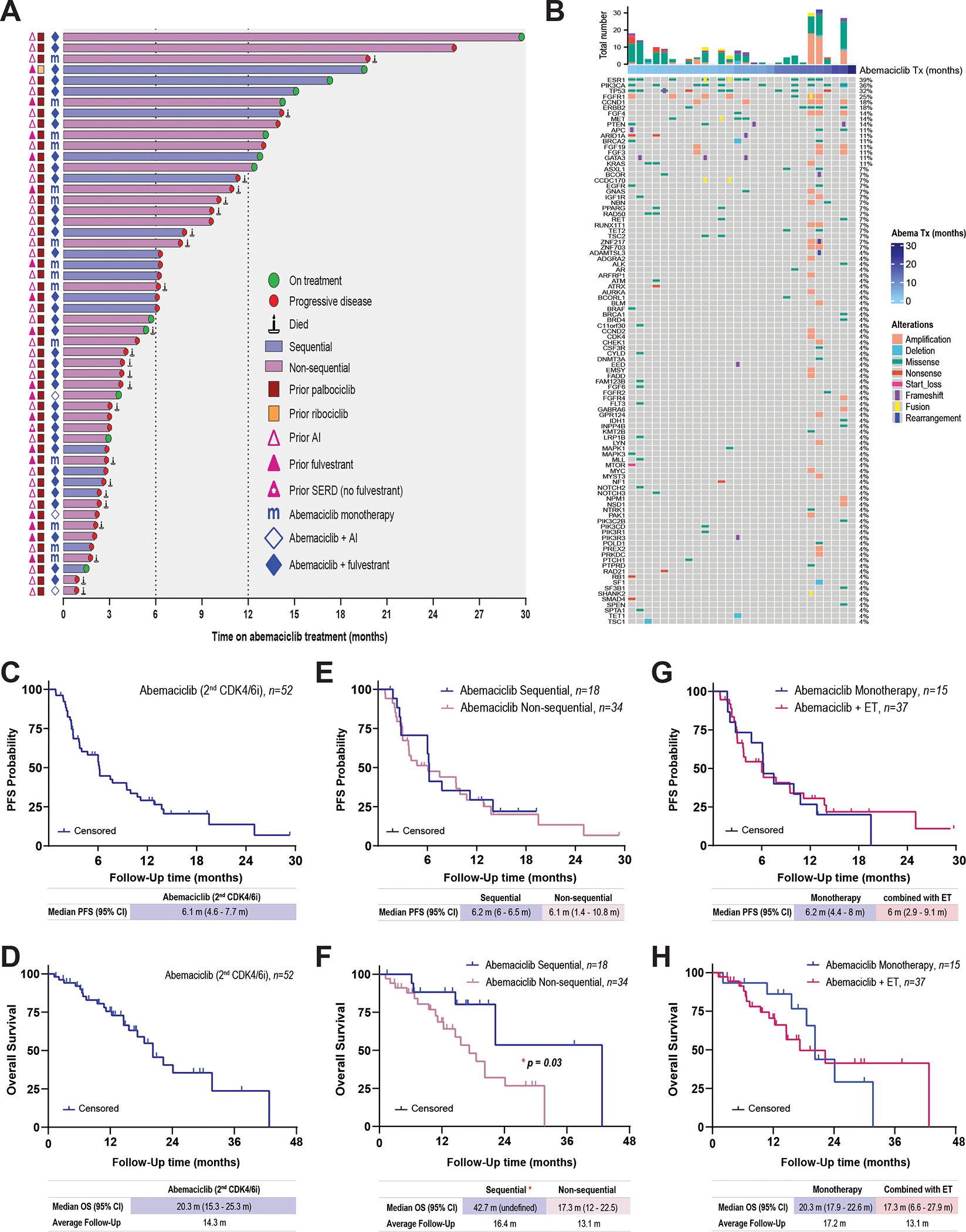
A, Swimmer plot depicting the prior CDK4/6i treatment course, duration of abemaciclib-based therapy, and subsequent clinical outcome. From left to right: pink triangles represent prior ET. Red and yellow squares represent the initial CDK4/6i agent received. Blue “m” and diamonds represent the abemaciclib treatment regimen received after progression on the first CDK4/6i: blue “m”, monotherapy; blue diamonds, combinations with ET. The bars indicate the abemaciclib treatment sequence after the prior CDK4/6i: blue, sequential; purple, non-sequential. The symbols at the ends of the bars represent the clinical outcome on abemaciclib treatment: green circle, still receiving abemaciclib treatment at data cutoff in December 2022; red circle, disease progressed; candle, patient died. B. Distribution of alterations on the gene panel of the patient baseline biopsies (n = 28). Oncoprint plot shows all the genes of interest sorted by mutation frequency form high to low (see supplemental Materials and Methods). The percentage of samples with any gene alteration is indicated on the right side. Each column represents a patient sample. Samples are ordered by abemaciclib treatment time in months. Total number of alterations for each sample is indicated in the bar plot on the top. Colors and shapes refer to different variant types. C and D, Kaplan–Meier analysis of PFS (C) and OS (D) in patients receiving abemaciclib therapy after prior disease progression on palbociclib or ribociclib. E and F, PFS (E) and OS (F) in patients stratified based on whether they received abemaciclib treatment sequentially or non-sequentially after the prior CDK4/6i. G and H, PFS (G) and OS (H) in patients stratified based on whether they received abemaciclib as monotherapy or combined with ET. Survival curves were compared using the log-rank (Mantel-Cox) test.
For the overall group of 52 patients who received abemaciclib, the median PFS was 6.1 months (Fig. 6C), and the median overall survival (OS) was 20 months (Fig. 6D). PFS was similar in the patients who received abemaciclib sequentially (6.2 months) and nonsequentially (6.1 months) (Fig. 6E), but OS was significantly longer in patients who received abemaciclib sequentially (42.7 months versus 17.3 months) (Fig. 6F). To assess the contribution of ET to the observed clinical benefit, we stratified the 52 patients into those who received abemaciclib in combination with ET (n=37) and those who received abemaciclib as monotherapy (n=15). PFS and OS were similar between these two cohorts (Fig. 6G and H), suggesting that the clinical benefit from abemaciclib is independent of ET. Collectively, these results suggest that patients with HR-positive/HER2-negative MBC may receive a meaningful clinical benefit from abemaciclib after progression on palbociclib.
Discussion
The combination of CDK4/6i with ET is considered the first line standard treatment for HR-positive/HER2-negative MBC patients (22). However, since most patients progress following long-term treatment, resistance to CDK4/6i remains a significant unmet clinical challenge and the question “what is effective beyond progression on CDK4/6i” is still unmet. Due to the predominance of palbociclib as the first ordered CDK4/6i over the past eight years, we focused on understanding whether mechanisms leading to resistance provide biomarkers for identifying subsequent response to abemaciclib. It is assumed that the mechanisms of resistance to palbociclib and abemaciclib are similar, suggesting potential cross-resistance if they are used sequentially. In this study, we directly tested the hypothesis that the mechanisms of resistance are distinct between palbociclib and abemaciclib and using them sequentially or targeting their differentially altered pathways may be effective in delaying progression and/or improving survival. We provide evidence from cell lines, organoid cultures, PDXs, and patient data that PR models and clinical cases that progress on palbociclib are responsive to abemaciclib. We show that PR and AR cells exhibit distinct transcriptomic and proteomic profiles sensitizing them to different classes of inhibitors, whereas genomic alterations are heterogeneous and cannot consistently predict response to palbociclib. PR cells respond to abemaciclib, while AR cells respond to OXPHOS inhibitors. Similarly, PDX and PDxO models derived from patients with HR-positive/HER2-negative MBC with progression on palbociclib responded to abemaciclib treatment. Clinically, patients with ER-positive/HR-positive/HER2-negative MBC with progression on palbociclib-containing therapy obtained a meaningful PFS and OS benefit from abemaciclib-based therapy, more so if administered sequentially after palbociclib.
The purpose of the current study was to differentiate pathways of resistance to palbociclib versus abemaciclib in order to determine whether switching to abemaciclib is a feasible option for patients who have progressed on palbociclib. There is previously published data from our team on the drivers of CDK4/6i resistance, including our 2019 publication by Kettner et al. (27), where we show that IL-6 by itself is sufficient to confer resistance to palbociclib in parental cells through upregulation of STAT3 and EMT. In an earlier paper, we show that knockdown of Rb and overexpression of the low molecular weight forms of cyclin E (LMW-E), but not knock down of p53, were sufficient to mediate resistance to palbociclib (47). In yet another study we showed that induced overexpression of LMW-E rendered cells resistant to palbociclib through activation of CDK2 (26). Our goal in the current manuscript, was to identify pathways of resistance and how to circumvent these pathways by additional therapies.
Using multiomics analysis, we identified 14 similarly and 30 differentially altered pathways between PR and AR in vitro models. Interestingly, Rb/pRb were differentially altered depending on which CDK4/6i was used to generate resistance. Given the importance of Rb during cell cycle progression, it is hypothesized that loss of RB1 function is associated with resistance to palbociclib (23,24,78–82). Consistent with this hypothesis, PR cells exhibited down-regulation of Rb/pRb levels and enrichment of the E2F targets pathway. In contrast, AR cells exhibited no significant changes in the levels of Rb/pRb and no enrichment of the E2F targets pathway, consistent with studies showing abemaciclib has potential activity in Rb-deficient models (34,35,83).
EMT and hypoxia were the main pathways enriched in PR cells but not in AR cells, while OXPHOS and reactive oxygen species were the main pathways enriched in AR cells but not PR cells. We previously reported that IL-6/STAT3-mediated upregulation of the EMT and CSCs pathways, and IL-6/STAT3-mediated downregulation of the ER pathway can drive development of palbociclib resistance (27). Moreover, matched tumor samples from breast cancer patients who progressed on palbociclib exhibited deregulation in IL-6/STAT3 compared with their pretreatment biopsy samples, suggesting that the IL-6/STAT3 pathway is a key driver of palbociclib resistance (27). These findings led to a phase 1b/2 clinical trial investigating the addition of STAT3 inhibitor TTI-101 to reverse resistance in HR-positive/HER2-negative MBC that progresses after at least 6 months on first line palbociclib plus AI therapy (NCT05384119). In the current study, we found that PR cells were enriched in the CD44+/high/CD24−/low population and had increased levels of secreted IL-6 protein, along with upregulation of the EMT pathway and significant enrichment of the IL-6/STAT3, TGF-β, Wnt, Hedgehog, and TNF-α/NF-κB signaling pathways. In contrast, AR cells did not exhibit CD44+/high/CD24−/low population enrichment, EMT pathway upregulation, increased levels of secreted IL-6 protein, or significant enrichment of the IL-6/STAT3, TGF-β, Wnt, or Hedgehog signaling pathways, suggesting that these pathways are not significant drivers in the acquisition of resistance to abemaciclib.
Another pathway that differed between PR and AR cells was the G2/M pathway, which was upregulated in PR cells but downregulated in AR cells. Using both organoids and PDX models derived from patients with HR-positive/HER2-negative MBC that progressed on palbociclib plus AI therapy, we confirmed that tumors harboring high expression of G2/M pathway gene sets had a good response to abemaciclib. Mechanistically, treatment of PR cells with abemaciclib results in G2-phase accumulation and increased inhibitory phosphorylation of CDK1 at Y15/T14, concomitant with a lack of progression of cells into mitosis. These findings are consistent with a previous study showing that a high G2/M pathway score in metastatic tumors was associated with worse survival in ER-positive/HER2-negative breast cancer and correlated with expression of other cell-proliferation-related gene sets, such as mitotic spindle and E2F targets (84), which were also highly enriched in the PR cells but not AR cells in our study. Taken together, our results suggest that during the acquisition of resistance to palbociclib, the upregulation of gene sets related to G2M checkpoint pathway, sensitizes PR tumor cells to abemaciclib.
Both transcriptomic and proteomic analyses identified OXPHOS as the major upregulated pathway in AR cells and showed that PR cells relied mainly on glycolysis. Consistent with these results, PR cells and AR cells responded differentially to the OXPHOS inhibitor IACS-010759 (69). PR cells were resistant to OXPHOS inhibition while AR cells were sensitive, suggesting their dependence on OXPHOS for survival and a role in the acquisition of resistance to abemaciclib. Moreover, PR-4 PDX model, derived from a patient with HR-positive/MBC that progressed on palbociclib therapy, had high OXPHOS pathway gene set expression and consequently did not respond to abemaciclib. Interestingly, the same PDX model (originally named T141-004) was previously reported to respond to IACS-010759 (50). Collectively, our in vitro and in vivo results suggest that high OXPHOS pathway gene set expression could be used as a predictor of resistance while enrichment of the G2/M pathway is likely a predictor of sensitivity to abemaciclib.
The retrospective analysis of patients with HR-positive/HER2-negative MBC who received abemaciclib following progression on a prior CDK4/6i showed a meaningful clinical benefit with abemaciclib-based therapy. The median PFS (6.1 months) was similar to that observed in heavily pretreated patients in both the MONARCH-1 trial of single agent abemaciclib (6 months) (85) and the study from Wander et al. (5.3 months) (42). Interestingly, when we stratified our patient cohort according to whether abemaciclib was administered sequentially or non-sequentially, we found significantly longer median OS for patients treated sequentially (42.7 months vs 17.3 months).
Of note, the median OS in the heavily pretreated nonsequential cohort (17.3 months) was similar to that observed in the MONARCH-1 trial (17.7 months) and Wander et al. study (17.2 months)(42). Similarly, no difference in median PFS or OS was observed among patients who received abemaciclib monotherapy compared to patients who received abemaciclib plus ET. These findings, along with data from MONARCH-1 and nextMONARCH studies, suggest that single agent abemaciclib has meaningful clinical activity.
Our clinical data adds to the previous studies (42,85) by showing that there is an overall survival benefit to the sequential administration of abemaciclib after progression on palbociclib, rather than after other systemic strategies (non-sequential). Moreover, the genomic alterations that are associated with each patient (Figure 6B) did not show a clear concordance with response to abemaciclib, suggesting that the drivers of progression on palbociclib are most likely non-genomic in nature. Hence, our laboratory-based studies provide a framework for understanding how palbociclib resistance can leverage the abemaciclib response. Specifically, we propose that high G2/M and low OXPHOS pathways could serve as biomarkers to help determine whether a patient could derive a benefit from abemaciclib treatment following progression on palbociclib.
A limitation to the laboratory-based studies is that the concentrations used to generate resistance in the cell models were higher than the Cmax reported in patient plasma (60). However, we selected our doses based on the AUC reported after the administration of multiple oral doses of palbociclib (125 mg), over a 21-day period which represents the total drug exposure across time (59). MBC patients who develop resistance to palbociclib receive multiple cycles of the drug. Using these concentrations, we were able to generate cell models that recapitulate the transcriptomic changes observed in PDX models that were generated from patients who progressed following long term (e.g. 16 months/cycles, PR-1) treatment with palbociclib. It is unknown what the levels of CDK4/6i are in the patient’s tumor after prolonged drug treatment and it is possible that they accumulate to higher concentrations than those measured in pharmacokinetic studies using plasma after one bolus dose and could be closer to the concentrations used in this study.
Another limitation of our study has to do with the small clinical sample size and its retrospective nature. The clinical question regarding the optimal use of abemaciclib after progression on previous CDK4/6i should be addressed in a large, prospective, randomized clinical trial. An ongoing clinical trial, postMONARCH (NCT05169567) is evaluating the activity of abemaciclib plus fulvestrant compared to fulvestrant alone for the treatment of HR+/HER2-negative breast cancer who progressed on or recurred after prior treatment with a CDK4/6i plus ET. Likewise, other studies aimed to evaluate the combination of abemaciclib with novel SERDs or SERMs are clinically relevant (NCT04432454) (86).
Palbociclib and ribociclib have also been investigated in trials evaluating the maintenance of CDK4/6i blockade with a switch in ET after initial progression on CDK4/6i, with mixed outcomes. Results from the randomized phase 2 MAINTAIN trial (NCT02632045) showed that treatment with ribociclib in combination with an alternate ET after progression on a prior CDK4/6i, reduced the risk of progression or death compared to the placebo plus ET in HR-positive/HER2-negative MBC patients (87). The phase III trial of ribociclib and ET, NATALEE, met the primary endpoint of invasive DFS (88). Conversely, in the recently reported PACE trial (NCT03147287), designed to explore the activity of continuation of palbociclib beyond progression with change of ET to fulvestrant, no significant improvement in PFS was observed with continuing palbociclib following progression. However, a longer PFS was observed when a PD-L1 inhibitor was added (89), suggesting the need to better understand the mechanisms driving disease progression on CDK4/6i therapy. There will likely be a transition toward increased use of ribociclib as the CDK4/6i of choice and thus future studies will need to investigate biomarkers for decision making upon ribociclib resistance.
In conclusion, the results presented here provide evidence that HR-positive/HER2-negative MBC patients whose disease had become refractory to palbociclib could be responsive to abemaciclib. Transcriptomic and proteomic, but not genomic, analyses revealed differentially altered pathways in PR and AR models, we propose that patients with enrichment of the G2/M pathway are most likely to benefit from abemaciclib treatment post progression on palbociclib, while patients with enrichment of the OXPHOS pathway are most likely to be refractory. Our results provide a rationale for prospective trials evaluating the benefit of abemaciclib treatment following progression on a prior CDK4/6i.
Supplementary Material
Acknowledgements:
We thank Dr. Taiwo Adesoye, Dr. Linjie Luo and Dr. Anthony J. Caruso for the critical reading of this manuscript and Ms. Stephanie Deming and Ms. Sunita Patterson, Research Medical Library, MD Anderson Cancer Center, for editing the manuscript and Champions Oncology for sharing Whole Exome Sequencing information for the PDX models. This work was supported by NIH grant P30CA016672 to The University of Texas MD Anderson Cancer Center, Department of Defense Breakthrough Post-Doctoral Fellowship BC170615 (N.M. Kettner), NIH/NCI grants CA255960 and CA223772 (K. Keyomarsi), Cancer Prevention and Research Institute of Texas (CPRIT) Multi-Investigator grant RP180712 (K.K. Hunt and K. Keyomarsi), and CPRIT Research Training Program grants RP170067 and RP210028 (N.M. Kettner and C.S. Bishop).
Footnotes
Conflict of interest:
J. Navarro-Yepes: None.
N.M. Kettner: None.
X. Rao: None.
C.S. Bishop: None
T. Bui: None.
H.F. Wingate: None.
A.S. Raghavendra: None.
Y. Wang: None.
J. Wang: None.
A. Sahin: None.
F. Meric-Bernstam: AbbVie, Aduro BioTech Inc., Aileron Therapeutics Inc., Alkermes, AstraZeneca, Black Diamond, Bayer Healthcare Pharmaceutical, Biovica, Calithera Biosciences Inc., Curis Inc., CytomX Therapeutics Inc., Daiichi Sankyo Co. Ltd., DebioPharm, Ecor1 Capital, eFFECTOR Therapeutics, Eisai, F. Hoffman-La Roche Ltd., FogPharma, GT Apeiron, Genentech Inc., Guardant Health Inc., Harbinger Health, IBM Watson, Immunomedics, Infinity Pharmaceuticals, Inflection Biosciences, Jackson Laboratory, Karyopharm Therapeutics, Kolon Life Science, Klus Pharma, Lengo Therapeutics, Loxo Oncology, Menarini Group, Mersana Therapeutics, Novartis, OnCusp Therapeutics, OrigiMed, PACT Pharma, Parexel International, Pfizer Inc., Protai Bio Ltd, Puma Biotechnology Inc., Samsung Bioepis, Sanofi, Seattle Genetics Inc., Silverback Therapeutics, Spectrum Pharmaceuticals, Taiho Pharmaceutical Co., Takeda Pharmaceutical, Tallac Therapeutics, Tyra Biosciences, Xencor, Zentalis, Zymeworks
K.K. Hunt: Armada Health, AstraZeneca, Cairn Surgical, Eli Lilly & Co, Lumicell.
S. Damodaran: EMD Serono, Guardant Health, Novartis, Pfizer, Sermonix, Taiho.
D. Tripathy: AstraZeneca, GlaxoSmithKline, Gilead, Novartis, OncoPep, Pfizer, Polyphor, Personalis, Puma Biotechnology, Sermonix, Stemline-Menarini.
K. Keyomarsi: Apeiron, BluePrint, REPARE, Schrodinger and Novartis.
References
- 1.Noone AM, Cronin KA, Altekruse SF, Howlader N, Lewis DR, Petkov VI, et al. Cancer Incidence and Survival Trends by Subtype Using Data from the Surveillance Epidemiology and End Results Program, 1992–2013. Cancer Epidemiol Biomarkers Prev 2017;26:632–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mariotto AB, Etzioni R, Hurlbert M, Penberthy L, Mayer M. Estimation of the Number of Women Living with Metastatic Breast Cancer in the United States. Cancer Epidemiol Biomarkers Prev 2017;26:809–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baselga J, Campone M, Piccart M, Burris HA, 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012;366:520–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andre F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med 2019;380:1929–40 [DOI] [PubMed] [Google Scholar]
- 5.Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol 2016;17:425–39 [DOI] [PubMed] [Google Scholar]
- 6.Finn RS, Martin M, Rugo HS, Jones S, Im SA, Gelmon K, et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 2016;375:1925–36 [DOI] [PubMed] [Google Scholar]
- 7.Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med 2016;375:1738–48 [DOI] [PubMed] [Google Scholar]
- 8.Preusser M, De Mattos-Arruda L, Thill M, Criscitiello C, Bartsch R, Ruhstaller T, et al. CDK4/6 inhibitors in the treatment of patients with breast cancer: summary of a multidisciplinary round-table discussion. ESMO Open 2018;3:e000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tripathy D, Im SA, Colleoni M, Franke F, Bardia A, Harbeck N, et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol 2018;19:904–15 [DOI] [PubMed] [Google Scholar]
- 10.Turner NC, Huang Bartlett C, Cristofanilli M. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N Engl J Med 2015;373:1672–3 [DOI] [PubMed] [Google Scholar]
- 11.Vijayaraghavan S, Moulder S, Keyomarsi K, Layman RM. Inhibiting CDK in Cancer Therapy: Current Evidence and Future Directions. Target Oncol 2018;13:21–38 [DOI] [PubMed] [Google Scholar]
- 12.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 2015;16:25–35 [DOI] [PubMed] [Google Scholar]
- 13.Im SA, Lu YS, Bardia A, Harbeck N, Colleoni M, Franke F, et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N Engl J Med 2019;381:307–16 [DOI] [PubMed] [Google Scholar]
- 14.Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N Engl J Med 2019 [DOI] [PubMed] [Google Scholar]
- 15.Sledge GW Jr, Toi M Neven P, Sohn J, Inoue K, Pivot X, et al. The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor-Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy-MONARCH 2: A Randomized Clinical Trial. JAMA Oncol 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rugo HS, Brufsky A, Liu X, Li B, McRoy L, Chen C, et al. Real-world study of overall survival with palbociclib plus aromatase inhibitor in HR+/HER2− metastatic breast cancer. NPJ Breast Cancer 2022;8:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ha MJ, Singareeka Raghavendra A, Kettner NM, Qiao W, Damodaran S, Layman RM, et al. Palbociclib plus endocrine therapy significantly enhances overall survival of HR+/HER2− metastatic breast cancer patients compared to endocrine therapy alone in the second-line setting: A large institutional study. Int J Cancer 2022;150:2025–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N Engl J Med 2017;377:1836–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberto M, Astone A, Botticelli A, Carbognin L, Cassano A, D’Auria G, et al. CDK4/6 Inhibitor Treatments in Patients with Hormone Receptor Positive, Her2 Negative Advanced Breast Cancer: Potential Molecular Mechanisms, Clinical Implications and Future Perspectives. Cancers (Basel) 2021;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cardoso F, Paluch-Shimon S, Senkus E, Curigliano G, Aapro MS, Andre F, et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann Oncol 2020;31:1623–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gennari A, Andre F, Barrios CH, Cortes J, de Azambuja E, DeMichele A, et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann Oncol 2021;32:1475–95 [DOI] [PubMed] [Google Scholar]
- 22.Burstein HJ, Somerfield MR, Barton DL, Dorris A, Fallowfield LJ, Jain D, et al. Endocrine Treatment and Targeted Therapy for Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer: ASCO Guideline Update. J Clin Oncol 2021;39:3959–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J Clin Oncol 2019;37:1169–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 2018;29:640–5 [DOI] [PubMed] [Google Scholar]
- 25.Li Z, Razavi P, Li Q, Toy W, Liu B, Ping C, et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018;34:893–905 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doostan I, Karakas C, Kohansal M, Low KH, Ellis MJ, Olson JA Jr, et al. Cytoplasmic Cyclin E Mediates Resistance to Aromatase Inhibitors in Breast Cancer. Clin Cancer Res 2017;23:7288–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kettner NM, Vijayaraghavan S, Durak MG, Bui T, Kohansal M, Ha MJ, et al. Combined Inhibition of STAT3 and DNA Repair in Palbociclib-Resistant ER-Positive Breast Cancer. Clin Cancer Res 2019;25:3996–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia JE, Lloyd MR, et al. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer Discov 2020;10:1174–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costa C, Wang Y, Ly A, Hosono Y, Murchie E, Walmsley CS, et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer Discov 2020;10:72–85 [DOI] [PubMed] [Google Scholar]
- 30.Schoninger SF, Blain SW. The Ongoing Search for Biomarkers of CDK4/6 Inhibitor Responsiveness in Breast Cancer. Mol Cancer Ther 2020;19:3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anurag M, Haricharan S, Ellis MJ. CDK4/6 Inhibitor Biomarker Research: Are We Barking Up the Wrong Tree? Clin Cancer Res 2020;26:3–5 [DOI] [PubMed] [Google Scholar]
- 32.O’Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov 2018;8:1390–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner NC, Neven P, Loibl S, Andre F. Advances in the treatment of advanced oestrogen-receptor-positive breast cancer. Lancet 2017;389:2403–14 [DOI] [PubMed] [Google Scholar]
- 34.Knudsen ES, Hutcheson J, Vail P, Witkiewicz AK. Biological specificity of CDK4/6 inhibitors: dose response relationship, in vivo signaling, and composite response signature. Oncotarget 2017;8:43678–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hafner M, Mills CE, Subramanian K, Chen C, Chung M, Boswell SA, et al. Multiomics Profiling Establishes the Polypharmacology of FDA-Approved CDK4/6 Inhibitors and the Potential for Differential Clinical Activity. Cell Chem Biol 2019;26:1067–80 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen P, Lee NV, Hu W, Xu M, Ferre RA, Lam H, et al. Spectrum and Degree of CDK Drug Interactions Predicts Clinical Performance. Mol Cancer Ther 2016;15:2273–81 [DOI] [PubMed] [Google Scholar]
- 37.Cousins EM, Goldfarb D, Yan F, Roques J, Darr D, Johnson GL, et al. Competitive Kinase Enrichment Proteomics Reveals that Abemaciclib Inhibits GSK3beta and Activates WNT Signaling. Mol Cancer Res 2018;16:333–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Brien N, Conklin D, Beckmann R, Luo T, Chau K, Thomas J, et al. Preclinical Activity of Abemaciclib Alone or in Combination with Antimitotic and Targeted Therapies in Breast Cancer. Mol Cancer Ther 2018;17:897–907 [DOI] [PubMed] [Google Scholar]
- 39.Gelbert LM, Cai S, Lin X, Sanchez-Martinez C, Del Prado M, Lallena MJ, et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs 2014;32:825–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Torres-Guzman R, Calsina B, Hermoso A, Baquero C, Alvarez B, Amat J, et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 2017;8:69493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mariotti V, Khong HT, Soliman HH, Costa RL, Fisher S, Boulware D, et al. Efficacy of abemaciclib (abema) after palbociclib (palbo) in patients (pts) with metastatic breast cancer (MBC). Journal of Clinical Oncology 2019;37 [Google Scholar]
- 42.Wander SA, Han HS, Zangardi ML, Niemierko A, Mariotti V, Kim LSL, et al. Clinical Outcomes With Abemaciclib After Prior CDK4/6 Inhibitor Progression in Breast Cancer: A Multicenter Experience. J Natl Compr Canc Netw 2021:1–8 [DOI] [PubMed] [Google Scholar]
- 43.Luo L, Keyomarsi K. PARP inhibitors as single agents and in combination therapy: the most promising treatment strategies in clinical trials for BRCA-mutant ovarian and triple-negative breast cancers. Expert Opin Investig Drugs 2022:1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lulla AR, Akli S, Karakas C, Ha MJ, Fowlkes NW, Mitani Y, et al. LMW cyclin E and its novel catalytic partner CDK5 are therapeutic targets and prognostic biomarkers in salivary gland cancers. Oncogenesis 2021;10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen X, Yang D, Carey JPW, Karakas C, Albarracin C, Sahin AA, et al. Targeting Replicative Stress and DNA Repair by Combining PARP and Wee1 Kinase Inhibitors Is Synergistic in Triple Negative Breast Cancers with Cyclin E or BRCA1 Alteration. Cancers (Basel) 2021;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, Low KH, Alexander A, Jiang Y, Karakas C, Hess KR, et al. Cyclin E Overexpression Sensitizes Triple-Negative Breast Cancer to Wee1 Kinase Inhibition. Clin Cancer Res 2018;24:6594–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun 2017;8:15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Francis AM, Alexander A, Liu Y, Vijayaraghavan S, Low KH, Yang D, et al. CDK4/6 Inhibitors Sensitize Rb-positive Sarcoma Cells to Wee1 Kinase Inhibition through Reversible Cell-Cycle Arrest. Mol Cancer Ther 2017;16:1751–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duong MT, Akli S, Wei C, Wingate HF, Liu W, Lu Y, et al. LMW-E/CDK2 deregulates acinar morphogenesis, induces tumorigenesis, and associates with the activated b-Raf-ERK1/2-mTOR pathway in breast cancer patients. PLoS Genet 2012;8:e1002538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans KW, Yuca E, Scott SS, Zhao M, Paez Arango N, Cruz Pico CX, et al. Oxidative Phosphorylation Is a Metabolic Vulnerability in Chemotherapy-Resistant Triple-Negative Breast Cancer. Cancer Res 2021;81:5572–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evans KW, Yuca E, Akcakanat A, Scott SM, Arango NP, Zheng X, et al. A Population of Heterogeneous Breast Cancer Patient-Derived Xenografts Demonstrate Broad Activity of PARP Inhibitor in BRCA1/2 Wild-Type Tumors. Clin Cancer Res 2017;23:6468–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. Journal of the American Statistical Association 1958;53:457–81 [Google Scholar]
- 53.Mantel N Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep 1966;50:163–70 [PubMed] [Google Scholar]
- 54.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luthra R, Patel KP, Routbort MJ, Broaddus RR, Yau J, Simien C, et al. A Targeted High-Throughput Next-Generation Sequencing Panel for Clinical Screening of Mutations, Gene Amplifications, and Fusions in Solid Tumors. J Mol Diagn 2017;19:255–64 [DOI] [PubMed] [Google Scholar]
- 56.Williamson JB, Solano L, Yuki A, Burkhart VD, Chitwood J, Cao R, et al. Analytical validation of the Oncomine Pan-Cancer Cell-Free Assay in a CLIA-and CAP-regulated laboratory for detection of solid tumor-derived variants in blood plasma. American Society of Clinical Oncology; 2019. [Google Scholar]
- 57.Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PloS one 2015;10:e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burne R, Balu S, Guerin A, Bungay R, Sin R, Paul ML. Comparison of healthcare resource utilization and costs of patients with HR+/HER2− advanced breast cancer treated with ribociclib versus other CDK4/6 inhibitors. J Med Econ 2021;24:806–15 [DOI] [PubMed] [Google Scholar]
- 59.Zirkelback J Center for Drug Evaluation and Research. Clinical Phamracology and Biopharmaceutic Review(s) For Ibrance. Volume 207103Orig1s000: FDA; 2016. [Google Scholar]
- 60.Beaver J Center for Drug Evaluation and Research: Multi-Discipline Review for Abemaciclib. Volume 208716Orig1s000: FDA; 2017. [Google Scholar]
- 61.Kwon MJ. Emerging roles of claudins in human cancer. Int J Mol Sci 2013;14:18148–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Savci-Heijink CD, Halfwerk H, Hooijer GKJ, Koster J, Horlings HM, Meijer SL, et al. Epithelial-to-mesenchymal transition status of primary breast carcinomas and its correlation with metastatic behavior. Breast Cancer Res Treat 2019;174:649–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin X, Shang X, Manorek G, Howell SB. Regulation of the Epithelial-Mesenchymal Transition by Claudin-3 and Claudin-4. PLoS One 2013;8:e67496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 2019;20:69–84 [DOI] [PubMed] [Google Scholar]
- 66.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003;100:3983–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu XQ, Pan XH, Wang TT, Wang J, Yang B, He QJ, et al. Intrinsic and acquired resistance to CDK4/6 inhibitors and potential overcoming strategies. Acta Pharmacol Sin 2021;42:171–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frederick M, Skinner HD, Kazi SA, Sikora AG, Sandulache VC. High expression of oxidative phosphorylation genes predicts improved survival in squamous cell carcinomas of the head and neck and lung. Sci Rep 2020;10:6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 2018;24:1036–46 [DOI] [PubMed] [Google Scholar]
- 70.Lewis CW, Taylor RG, Kubara PM, Marshall K, Meijer L, Golsteyn RM. A western blot assay to measure cyclin dependent kinase activity in cells or in vitro without the use of radioisotopes. FEBS Lett 2013;587:3089–95 [DOI] [PubMed] [Google Scholar]
- 71.Lewis CW, Taylor RG, Golsteyn RM. Measurement of Cdk1/Cyclin B Kinase Activity by Specific Antibodies and Western Blotting. Methods Mol Biol 2016;1342:337–48 [DOI] [PubMed] [Google Scholar]
- 72.Asghar US, Kanani R, Roylance R, Mittnacht S. Systematic Review of Molecular Biomarkers Predictive of Resistance to CDK4/6 Inhibition in Metastatic Breast Cancer. JCO Precis Oncol 2022;6:e2100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Esteban-Burgos L, Wang H, Nieto P, Zheng J, Blanco-Aparicio C, Varela C, et al. Tumor regression and resistance mechanisms upon CDK4 and RAF1 inactivation in KRAS/P53 mutant lung adenocarcinomas. Proceedings of the National Academy of Sciences of the United States of America 2020;117:24415–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jansen VM, Bhola NE, Bauer JA, Formisano L, Lee KM, Hutchinson KE, et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res 2017;77:2488–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee JS, Yost SE, Li SM, Cui Y, Frankel PH, Yuan YC, et al. Genomic Markers of CDK 4/6 Inhibitor Resistance in Hormone Receptor Positive Metastatic Breast Cancer. Cancers (Basel) 2022;14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.The I, Ruijtenberg S, Bouchet BP, Cristobal A, Prinsen MB, van Mourik T, et al. Rb and FZR1/Cdh1 determine CDK4/6-cyclin D requirement in C. elegans and human cancer cells. Nat Commun 2015;6:5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rodriguez MJ, Perrone MC, Riggio M, Palafox M, Salinas V, Elia A, et al. Targeting mTOR to overcome resistance to hormone and CDK4/6 inhibitors in ER-positive breast cancer models. Sci Rep 2023;13:2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bui TBV, Burgering BMT, Goga A, Rugo HS, van ‘t Veer LJ. Biomarkers for Cyclin-Dependent Kinase 4/6 Inhibitors in the Treatment of Hormone Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Advanced/Metastatic Breast Cancer: Translation to Clinical Practice. JCO Precis Oncol 2022;6:e2100473. [DOI] [PubMed] [Google Scholar]
- 79.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res 2016;76:2301–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taylor-Harding B, Aspuria PJ, Agadjanian H, Cheon DJ, Mizuno T, Greenberg D, et al. Cyclin E1 and RTK/RAS signaling drive CDK inhibitor resistance via activation of E2F and ETS. Oncotarget 2015;6:696–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Finn RS, Liu Y, Zhu Z, Martin M, Rugo HS, Dieras V, et al. Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naive Metastatic Breast Cancer. Clin Cancer Res 2020;26:110–21 [DOI] [PubMed] [Google Scholar]
- 82.Malorni L, Piazza S, Ciani Y, Guarducci C, Bonechi M, Biagioni C, et al. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 2016;7:68012–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chong QY, Kok ZH, Bui NL, Xiang X, Wong AL, Yong WP, et al. A unique CDK4/6 inhibitor: Current and future therapeutic strategies of abemaciclib. Pharmacol Res 2020;156:104686. [DOI] [PubMed] [Google Scholar]
- 84.Oshi M, Takahashi H, Tokumaru Y, Yan L, Rashid OM, Matsuyama R, et al. G2M Cell Cycle Pathway Score as a Prognostic Biomarker of Metastasis in Estrogen Receptor (ER)-Positive Breast Cancer. International Journal of Molecular Sciences 2020;21:2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dickler MN, Tolaney SM, Rugo HS, Cortes J, Dieras V, Patt D, et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR(+)/HER2(−) Metastatic Breast Cancer. Clin Cancer Res 2017;23:5218–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Damodaran S, Plourde PV, Moore HCF, Anderson IC, Portman DJ. Open-label, phase 2, multicenter study of lasofoxifene (LAS) combined with abemaciclib (Abema) for treating pre- and postmenopausal women with locally advanced or metastatic ER+/HER2− breast cancer and an ESR1 mutation after progression on prior therapies. Journal of Clinical Oncology 2022;40:1022- [Google Scholar]
- 87.Kalinsky K, Accordino MK, Chiuzan C, Mundi PS, Trivedi MS, Novik Y, et al. A randomized, phase II trial of fulvestrant or exemestane with or without ribociclib after progression on anti-estrogen therapy plus cyclin-dependent kinase 4/6 inhibition (CDK 4/6i) in patients (pts) with unresectable or hormone receptor–positive (HR+), HER2-negative metastatic breast cancer (MBC): MAINTAIN trial. Journal of Clinical Oncology 2022;40:LBA1004–LBA [Google Scholar]
- 88.Novartis. Novartis Kisqali® Phase III NATALEE trial meets primary endpoint at interim analysis demonstrating clinically meaningful benefit in broad population of patients with early breast cancer. Press Release-Basel, Switzerland: 2023 [Google Scholar]
- 89.Mayer EL, Wander SA, Regan MM, DeMichele A, Forero-Torres A, Rimawi MF, et al. Palbociclib after CDK and endocrine therapy (PACE): A randomized phase II study of fulvestrant, palbociclib, and avelumab for endocrine pre-treated ER+/HER2− metastatic breast cancer. Journal of Clinical Oncology 2018;36:TPS1104–TPS [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data sets from the cell line, organoids cultures and PDX models generated in this study have been deposited at the Gene Expression Omnibus repository: Accession numbers GSE222367(cell lines) GSE229146 (organoids derived from PDX models) and GSE229235 (PDX models) which will be publicly available at the time of publication. The human sequence data generated in this study are not publicly available due to patient privacy requirements but are available upon reasonable request from the corresponding author. The human genomic data, listed in SNP databases analyzed in this study were obtained from gnomAD (https://gnomad.broadinstitute.org). All other raw data are available upon request from the corresponding author.