

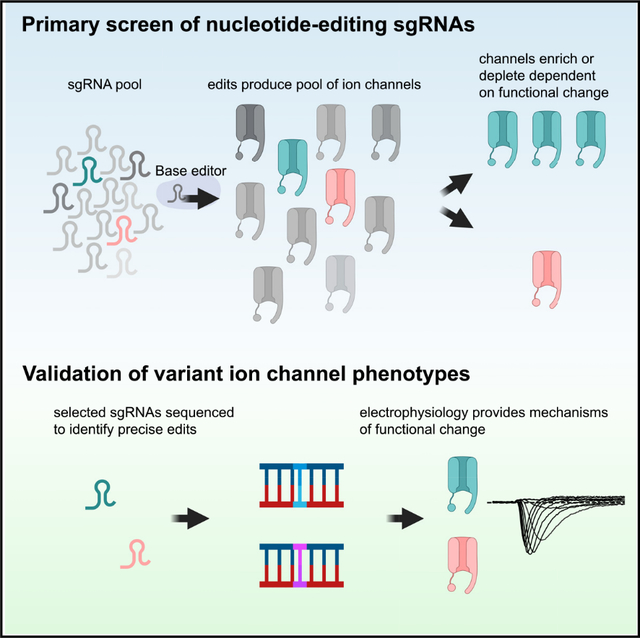
SUMMARY
It is challenging to apply traditional mutational scanning to voltage-gated sodium channels (NaVs) and functionally annotate the large number of coding variants in these genes. Using a cytosine base editor and a pooled viability assay, we screen a library of 368 guide RNAs (gRNAs) tiling NaV1.2 to identify more than 100 gRNAs that change NaV1.2 function. We sequence base edits made by a subset of these gRNAs to confirm specific variants that drive changes in channel function. Electrophysiological characterization of these channel variants validates the screen results and provides functional mechanisms of channel perturbation. Most of the changes caused by these gRNAs are classifiable as loss of function along with two missense mutations that lead to gain of function in NaV1.2 channels. This two-tiered strategy to functionally characterize ion channel protein variants at scale identifies a large set of loss-of-function mutations in NaV1.2.
In brief
Pablo et al. couple genomic base-editing technology to a pooled viability assay screening for variants that perturb the function of voltage-gated sodium channel NaV1.2. Their approach maps out a two-tiered strategy for prioritizing possible disease-causing variation in ion channels associated with neurodevelopmental disease.
Graphical Abstract
INTRODUCTION
Voltage-gated sodium channels (NaVs) are large proteins with 24 transmembrane segments1–3 responsible for the initiation of the action potential4 in most electrically excitable cells. NaV1.2 is an essential neuronal NaV encoded by the gene SCN2A. Loss of NaV1.2 in mice leads to perinatal lethality,5 and NaV1.2 haploinsufficiency produces altered axonal excitability during early development together with impaired dendritic excitability and synaptic function in mature pyramidal neurons.6 The importance of SCN2A is underscored by pathogenic variants in this gene associated with a spectrum of neurodevelopmental disorders.7,8 Two major phenotype groups have been reported for SCN2A disorders: individuals in the first group have seizures with onset before 3 months of age and typically harbor missense variants with a gain-of-function (GOF) effect. These individuals are often effectively treated with sodium channel blockers.9–11 The second group is characterized by developmental delay and seizure onset after 3 months and predominantly caused by loss-of-function (LOF) variants. Individuals in this group have a variable response to sodium channel blockers, which may even aggravate seizures in some carriers of LOF variants.9,12,13 Thus, improved understanding of coding variations that lead to LOF is essential to develop appropriate treatment strategies for patients with NaV1.2 channelopathy.
Seven-hundred and fifty-one of the 1,093 missense variants in SCN2A are listed as variants of uncertain significance or have conflicting interpretations in the ClinVar Database (as of February 2023). In addition, 509 (mostly uncharacterized) missense variants in SCN2A have been observed in at least one individual to date in the gnomAD database of more than 100,000 people. The rapid growth in the number of variants identified through clinical genetic testing of patients necessitates higher-throughput methods for determining the functional consequences of SCN2A mutations. For ion channels, high-throughput cellular assays and electrophysiology have been used to characterize the function of channel variations.14–18 These approaches invariably require the introduction of large pieces of exogenous genetic material to each cell and are not feasible for scanning mutagenesis across an entire NaV protein. Trafficking assays can assess many more variants in parallel than high-throughput electrophysiology but are focused on defects in channel localization. Deep mutational scanning is a promising approach for variant annotation at scale and has been used to comprehensively study many genes, such as p5319,20 and TDP-43.21 Recently, a deep mutational scan of the cardiac NaV gene, SCN5A, succeeded in identifying all possible mutagenic variation in a region of 12 amino acids in domain IV of the channel.22 As with previous studies, mutant channels had to be introduced and stably integrated into cells.22
The advent of CRISPR-Cas9 technology has greatly expanded the toolkit for genome-editing technology. In base editing, a nickase Cas9 molecule (nCas9) uses a guide RNA (gRNA) to target a specific genomic region without generating a double-strand break.23,24 A nucleotide base-modifying enzyme, such as cytidine deaminase, that is fused to the nCas9 then introduces a chemical modification to bases in a targeting window of approximately five nucleotides, dependent on the gRNA,23 and results in a C→T (or G→A if the edit is antisense) substitution. Coupled with a gRNA library, these genome editors allow de novo scanning of all potential C→T (or G→A) changes in the specific genomic location without the need for exogenous introduction of large pieces of pre-synthesized DNA variants. In addition, half of the known pathogenic point mutations in humans occur from the spontaneous deamination of cytosine,24 and genomic cytosine editing may capture these events effectively.
Here we apply base-editing technology to tile the NaV1.2 cDNA with cytosine-to-thymine transition, coupled with a pooled viability assay to triage variants introduced by 368 gRNAs. Most of the gRNAs that were of high interest led to guide enrichment compared with controls, consistent with LOF molecular phenotypes. Variants that were highly enriched or depleted were then confirmed with sequencing and high-throughput electrophysiological assays that established mechanistic insights. Structural analysis further corroborated the results from the screen. This approach could be widely used to build ion channel variant-to-function look-up tables.
RESULTS
Traditional mutational approaches are difficult to apply to NaV1.2 given its size, complexity, and the prohibitive nature of screening large variant libraries with electrophysiology, the gold-standard NaV phenotyping assay. Given the success of recent base-editing screens,25–27 we designed a cytosine-to-thymine base-editor gRNA library to produce C→T nucleotide changes tiling the NaV1.2 cDNA, including missense mutants, nonsense mutants, silent mutants, and controls (Figures 1A and 1D).
Figure 1. A pooled viability screen prioritizes putative gain- and loss-of-function mutations in NaV1.2.
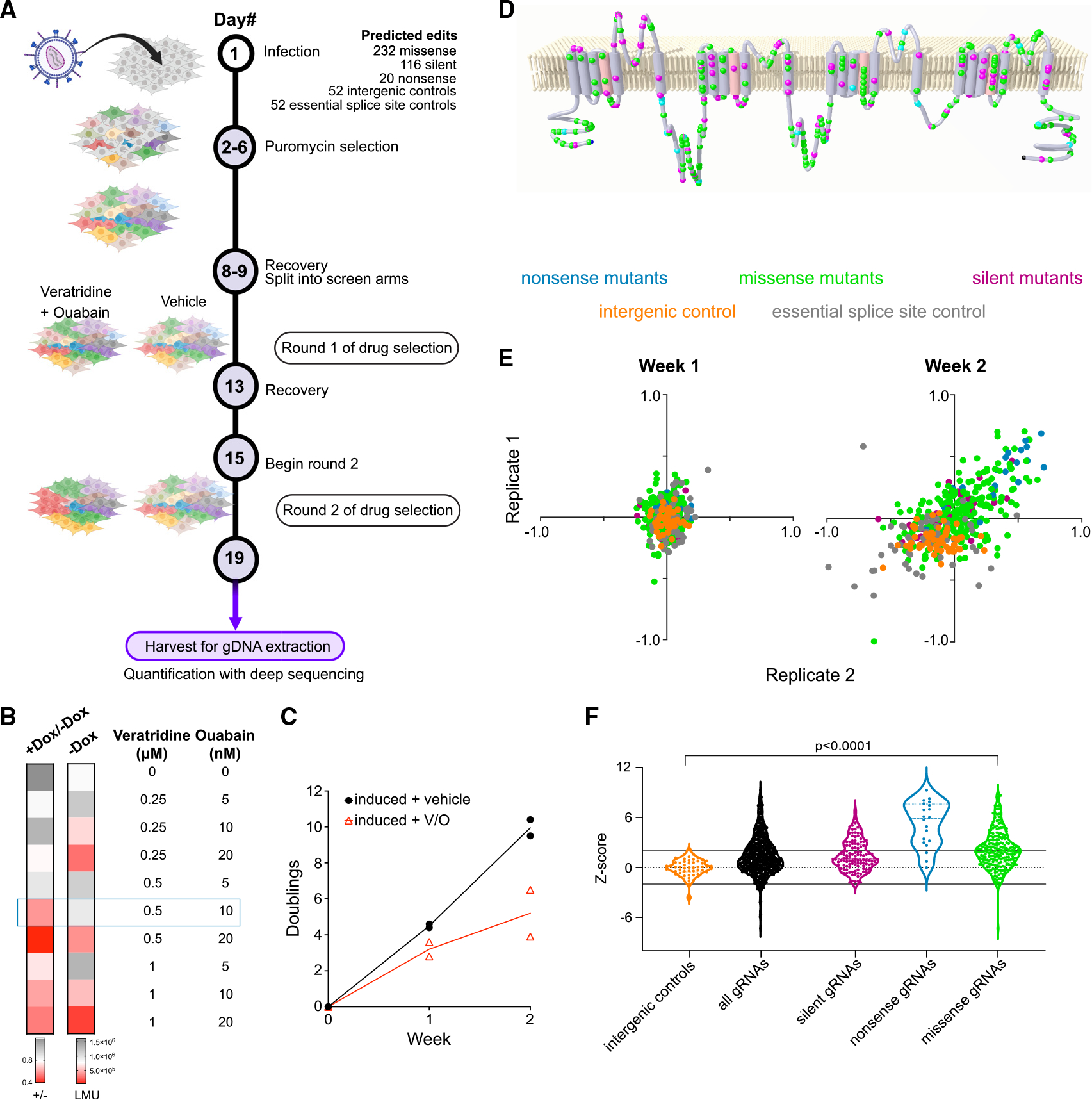
(A) Schematic of base-editing screen targeting NaV1.2. Two rounds of veratridine and ouabain treatment were applied to amplify enrichment/depletion. Figure created using BioRender.com.
(B) Heatmap showing optimization of assay conditions for a pooled viability screen in NaV1.2 cells. The cellular viability in a variety of veratridine and ouabain concentrations is assessed by luminescence using a Cell-Titer Glo assay. The first column represents the ratio of luminescence from doxycycline-induced cells to luminescence from uninduced cells as a measure of NaV1.2-dependent toxicity. The second column represents the luminescence readout (LMU) from uninduced cells and is a measure of drug toxicity independent of NaV1.2 (n = 3 wells; representative of three independent experiments).
(C) The number of doublings for two independent replicates monitored over 2 weeks in the presence and absence of the veratridine-ouabain treatment (n = 2 independent infections).
(D) A membrane topology map of the NaV1.2 protein shows the distribution of targeted nonsense (cyan), missense (green), and silent (magenta) mutations included in the gDNA library.
(E) Scatterplots of the log fold change of each individual gRNA (colored dot) in the veratridine-ouabain arm compared with the vehicle control arm. Two independent replicates of the screen are plotted against each other for each week of the screen. The color code for different types of gRNAs is indicated above the plots.
(F) Violin plots of Z scores constructed for each gRNA in the screen using intergenic targeting gRNAs as negative controls (n = 2 independent infections).
In order to screen this library in a pooled fashion, we developed a NaV1.2 viability assay in a human embryonic kidney (HEK293) stable cell line with NaV1.2 cDNA expression under the control of a doxycycline-inducible promoter. We based our viability screen on a combination treatment of the compounds veratridine and ouabain previously reported to induce intracellular accumulation of Na+ ions through NaV1.2 channels.28 Veratridine leads to increased flux of sodium ions largely due to alterations in the inactivation properties of NaVs, while ouabain inhibits the Na+/K+ ATPase responsible for efflux of Na+ ions.28 The combination of these two drugs leads to cytotoxicity (Figure S1) depending on the function of NaV1.2 channels at the plasma membrane. We optimized the assay conditions by testing an array of veratridine and ouabain concentration combinations over the course of 96 h, followed by assessment of cell viability with Cell-Titer Glo, a viability assay that uses luminescence to measure ATP levels. We found that a concentration of 0.5 μM veratridine and 10 nM ouabain leads to maximal separation in luminescence (hence viability) in cells not expressing NaV1.2 (uninduced, −Dox) and cells expressing NaV1.2 (induced, +Dox; Figure 1B). Milder conditions did not induce sufficient cell death, whereas harsher conditions led to toxicity independent of NaV1.2 expression.
In this assay, cells expressing LOF variants of NaV1.2 will have a growth advantage (gRNA enrichment) over the cells expressing the wild-type (WT) reference channel, while cells with GOF variants of NaV1.2 will suffer more toxicity (gRNA depletion). After having developed the viability assay for NaV1.2, we infected our stable cell line with the library of base-editing gRNAs, selected for infected cells by puromycin selection and then divided the pool of cells into two arms. Both arms were induced to express NaV1.2, but one arm was given vehicle, while the other was treated with 0.5 μM veratridine and 10 nM ouabain for 96 h (Figure 1A). We ran the screen in duplicate (two separate infections). Across these two independent library infections, the relative representation of individual gRNAs was tightly correlated (Figure S2A). Through 2 weeks of drug treatment, there were fewer cell doublings in the arm treated with veratridine and ouabain, consistent with treatment-induced toxicity in cells expressing NaV1.2 (Figure 1C).
After treatment with veratridine and ouabain, we harvested the cells, extracted DNA, and quantified the enrichment/depletion of gRNAs compared with the untreated samples. While there were few gRNAs highly enriched/depleted at 1 week across the two replicates, gRNAs in treated samples showed greater separation from those in untreated samples after 2 weeks of treatment, indicating that enrichment/depletion amplified over longer treatment (Figure 1E). As shown, gRNAs targeting intergenic regions (orange dots), which should not affect NaV1.2 function, are generally less abundant (lower left quadrant) compared with the untreated samples, as expected from the NaV1.2-dependent toxicity in the presence of veratridine-ouabain in this assay. Consistent with previous reports25 that targeting essential splice sites reduces cell fitness, 23% of gRNAs targeting essential splice sites across the genome (gray dots) depleted significantly in our assay. Importantly, gRNAs predicted to make nonsense mutations (blue dots) were highly enriched, as should be the case when NaV1.2 function is abrogated. We constructed Z scores (see STAR Methods) using intergenic gRNAs as the baseline, effectively mean-centering our results around NaV1.2 WT function (Figure 1F; Table S1). Using this metric, the majority (17 out of 20) of the nonsense gRNAs had Z scores greater than 2, indicating the expected enrichment for LOF of Nav1.2 in this group. As discussed later, two of the three nonsense gRNAs scoring lower than 2 are likely false negatives, whereas the last nonsense gRNA introduces a stop codon toward the end of the protein, which we functionally validated to be minimally disruptive. These results showed that we are able to reliably isolate expected LOF (nonsense) variants of NaV1.2 and validated our screening approach.
As shown in Figure 1F, there is a wide spread of Z scores for gRNAs predicted to make missense mutations. Of the 232 gRNAs that we designed to introduce missense changes, 111 of them were significantly enriched (Z score >2) and likely produced NaV1.2 channels with LOF. We detected two missense gRNAs with Z scores < −2, indicating greater depletion than WT NaV1.2 and, thus, putative GOF.
We designed our screen to scan across the length of the NaV1.2 cDNA (Figure 1D) since ClinVar missense variants are also found across NaV1.2 (Figure S2B). Given the published structure of NaV1.2 (Figure 2A; PDB: 6j8e), we asked whether we could identify structure-to-function patterns of high-scoring missense gRNAs. Several gRNAs targeting the pore of NaV1.2 (S5-S6 linker of each of the transmembrane domains) were associated with significantly higher Z scores than gRNAs targeting cytoplasmic linker regions of the channel (Figure 2B; Wilcoxon rank-sum test, p = 0.018 versus D3-D4 linker, p = 0.0019 versus Other intracellular). We have previously identified regions of NaVs that are enriched for variants causing LOF,10 and gRNAs targeting these regions had significantly higher Z scores than gRNAs in other regions (Figure 2C; Wilcoxon rank-sum test, p = 1.8e−4). Focusing on the voltage-sensor and pore-forming domains, we found gRNAs targeting regions around the selectivity filter (aspartate-glutamate-lysine-alanine; DEKA motif) tended to have Z scores >4 (Figure 2D). Thus, our screen was able to detect regions of the channel that are intolerant to missense variants even if the screen did not mutate every amino acid to every other amino acid (e.g., saturation).
Figure 2. Structural analysis of predicted missense variants of NaV1.2 in the editing screen.
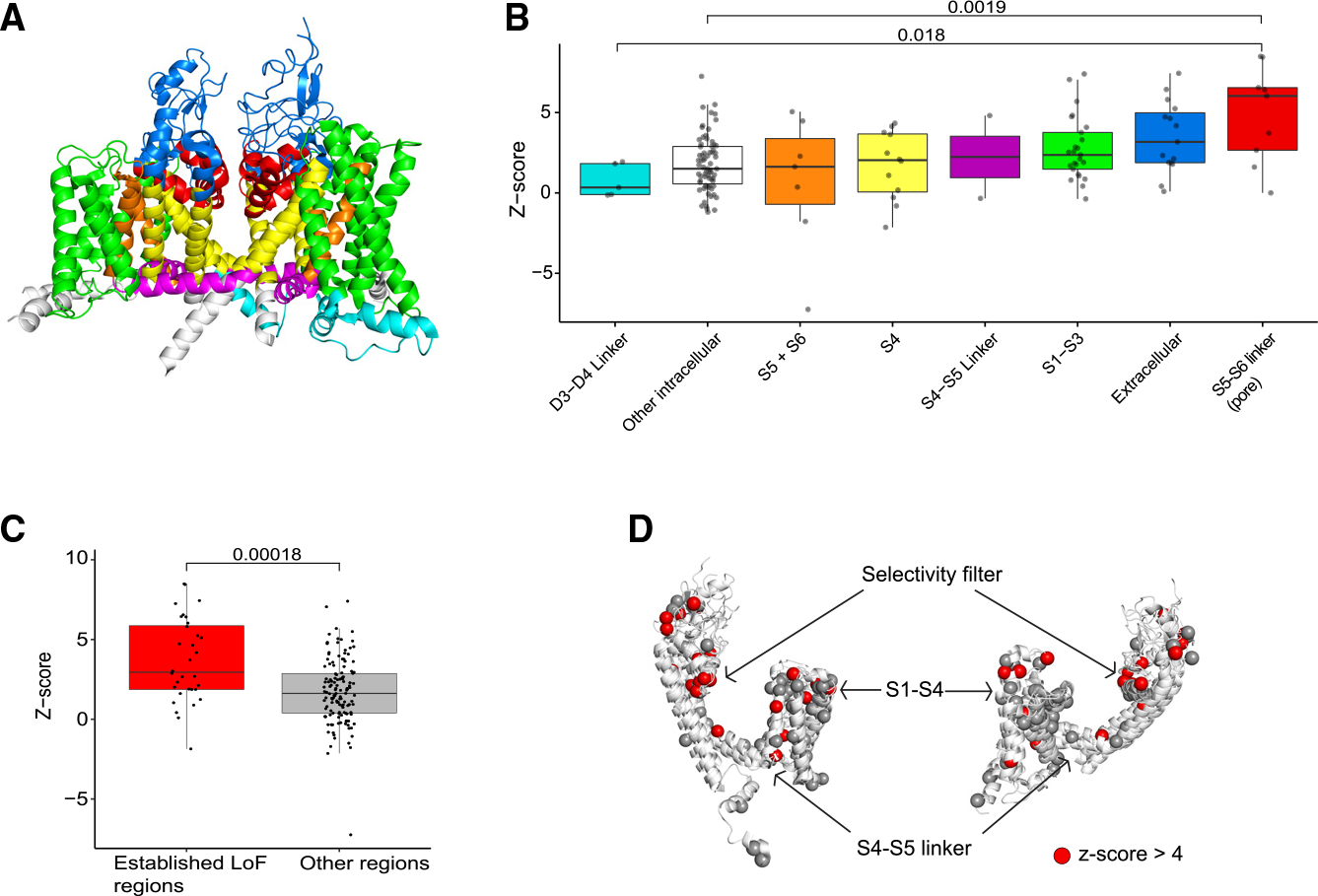
(A) Structure of the NaV1.2 channel color coded by region.
(B) gRNAs targeting the pore region (red) are more likely to have higher Z scores, indicating LOF, than gRNAs targeting D3-D4 linker and cytoplasmic domains (cyan and white).
(C) gRNAs in regions reported to be prone to LOF disruptions have significantly higher Z scores than gRNAs in other regions. The boxplots for (B) and (C) depict median and 25%/75% quartile ranges.
(D) Collapsed view of the four domains of theNaV1.2 channel aligned by homology. A preponderance of gRNAs targeting the selectivity filter have high Z scores (>4) compared with other regions.
Previous screens with base editing showed that gRNAs can introduce “collateral” mutations in addition to the predicted edits.25 To validate our screen results and to address the impact of these collateral mutations, we performed secondary confirmation. We chose 11 gRNAs to further validate either because of high absolute Z scores or because they scored below our empirical Z-score cutoff of 2 for significant change in NaV1.2 function (Table S1). We infected the NaV1.2 stable lines separately with each of the 11 gRNAs. As with the pooled screen, each individual infection was then separated into either a vehicle arm or veratridine-ouabain treatment arm over the course of 2 weeks (Figure 3A). DNA samples from each infection were then sequenced specifically in the region of the intended edit for each gRNA in order to identify and quantify the abundance of specific alleles. Each unique combination of intended and unintended edits constituted a specific allele in the cell population.
Figure 3. Sequence validation of select gRNAs uncovers mutations that drive GOF or LOF.
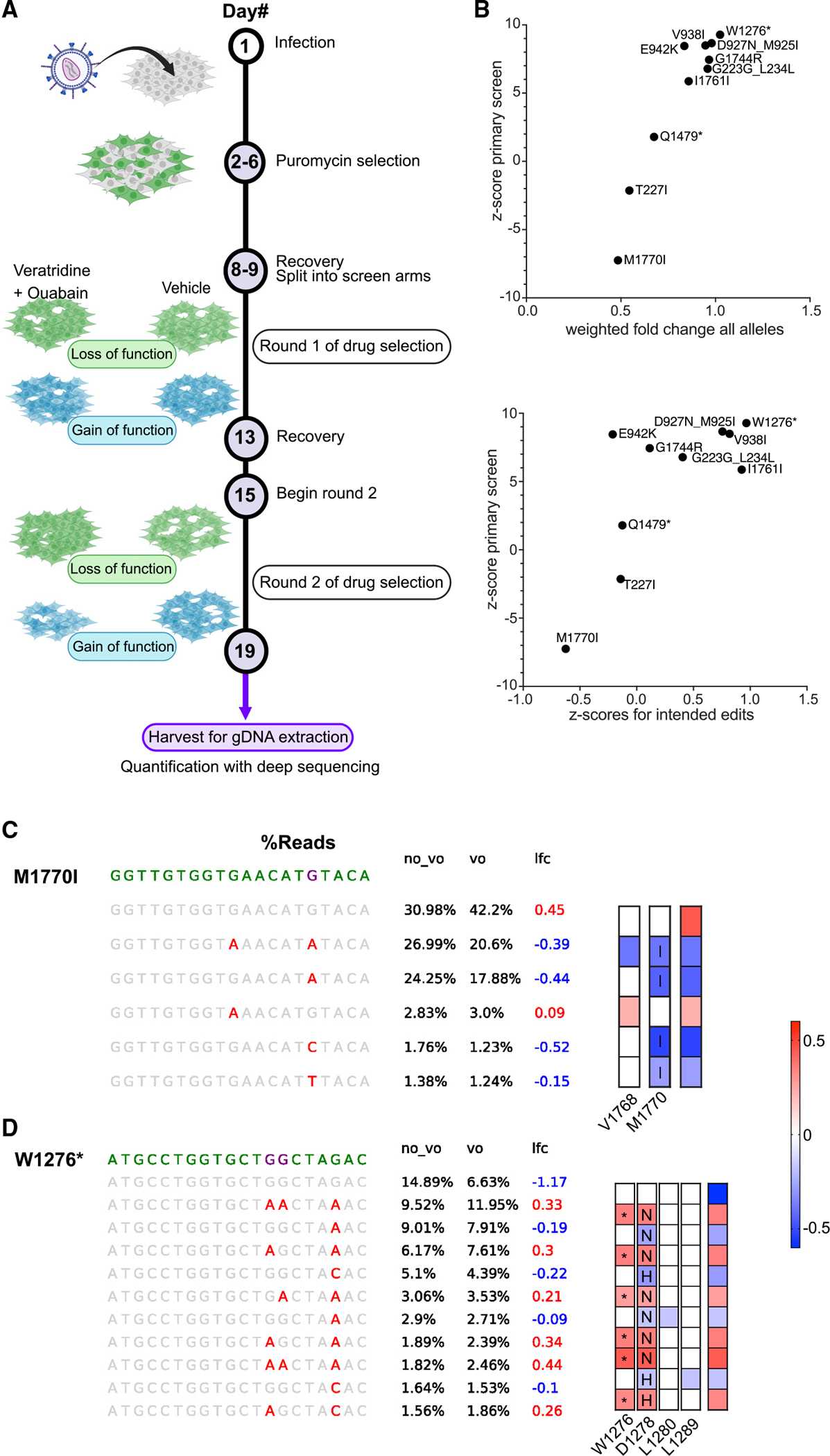
(A) Schematic of arrayed base-editing screens using individual gRNAs. Figure created using BioRender.com.
(B) Correlations of weighted fold change of all alleles (top), or Z scores of specific alleles (bottom), for each indicated gRNA compared with Z scores from the primary screen (see section “results” and STAR Methods for details).
(C and D) (C) Sequence analysis of the M1770I locus and (D) sequence analysis of the W1276* locus. (Left) Purple letters indicate the predicted nucleotide edits, while red letters indicate actual edits detected. (Right) Heatmap colors indicate the level of enrichment or depletion in log2 (fold change). Letters in boxes signify amino acid changes caused by the nucleotide mutations while asterisk (*) indicates a stop codon. Colored boxes with no letter indicate silent mutations. n = 3 sequencing replicates.
For each gRNA, we assessed the enrichment or depletion of all alleles (weighted by allele abundance) in the veratridine-ouabain arm compared with the vehicle arm. We reasoned that the cumulative enrichment/depletion of the alleles produced by a gRNA would be reflected in the enrichment/depletion of the same gRNA in the primary screen. For example, if a gRNA in the primary screen was enriched, then the majority of the alleles produced by base editing would be expected to be enriched. We found significant correlation between the weighted fold change of all alleles created by a gRNA and the Z score of the same gRNA from the primary screen (r2 = 0.91, p < 0.0001) (Figure 3B, top panel). This result suggests that the individual gRNA secondary confirmation analyses were highly consistent with the primary screen.
We then constructed Z scores of the specific alleles induced by each gRNA (relative to the unedited allele) to further assess cumulative enrichment/depletion. We found that the enrichment/depletion of only the alleles containing the predicted edits (Figure 3B, bottom panel) also correlated well (r2 = 0.54, p < 0.01) with the Z scores from the primary screen, although taking all alleles (including collateral edits) into account led to better correlation (Figure 3B, top panel). Taken together, this suggests that intended edits explain a significant portion of the effects of the gRNAs we tested.
We examined the sequences of alleles for each gRNA and confirmed that the cellular phenotype (LOF or GOF) could be explained by the predicted edits. For example, M1770I was the gRNA with the most negative Z score in the primary screen (most depleted; Figure 3C). Any allele including the methionine to isoleucine amino acid change was depleted in the veratridine-ouabain condition relative to the unedited allele, validating the gRNA depletion from the primary screen. In contrast, a collateral silent mutation at position 1768 was not depleted by itself and only depleted together with p.M1770I mutation, suggesting this silent mutation did not change channel function (Figure 3C).
On the other end of the spectrum, W1276* was the gRNA with the highest Z score. Alleles with the nonsense mutation reliably enriched, whereas two other collateral missense mutations were not enriched when uncoupled from the intended nonsense mutation (Figure 3D). The allele analysis for all gRNAs is shown in Figure S3.
Having thus established that our screen was capable of identifying mutations and residues prone to functional disruption, we characterized selected mutations using a high-throughput electrophysiology assay. We assessed sodium current density, the voltage dependence of channel activation, and the voltage dependence of channel inactivation of NaV1.2 variant channels transiently expressed in HEK293 cells (Figure 4A; Table S2) (see STAR Methods). A decrease in current density, less activation at more depolarized voltages (right shift in activation curve), or greater inactivation at more hyperpolarized voltages (left shift in inactivation curve) could all be construed as LOF effects. Conversely, an increase in current density, greater activation at more hyperpolarized voltages (left shift in activation curve), or less inactivation at more depolarized voltages (right shift in inactivation curve) may all be interpreted as GOF changes that allow more sodium influx through NaV1.2.7 The only two mutations that were putative GOF (T227I, M1770I, Z score < −2) showed greater channel activation at more hyperpolarized voltages (Figures 4A and 4B, see representative blue traces upon depolarization to −10 mV). Although the inactivation of M1770I seemed unchanged relative to WT NaV1.2, T227I also left shifted inactivation, indicating mixed GOF and LOF effects (Figures 4A and 4B). Aside from the greater editing efficiency of the M1770I gRNA (Figures 3C and S3), the mixed effects of the T227I mutation may also explain its less negative Z score (M1770I = −7.25, T227I = −2.14). Residues targeted by high-Z-scoring gRNAs were clear LOF as expected. D927N and G1744R led to a dramatic decrease in current density to the point where a significant proportion of transfected cells did not express discernible NaV1.2 current (Figures 4A, 4C, and 4D). As further validation of our approach, mutations predicted by low-Z-scoring gRNAs (Z score <2) had little functional consequence (G839S, A1631T, P77S; Figure 4D). The Q1923* variant had little functional disruption (Figure 4D), demonstrating that a nonsense mutation toward the end of the protein is relatively benign and explaining why the Z score for this gRNA fell below 2 (Figure 1F). In contrast, Q1479* truncates NaV1.2 in the middle of the protein and, as expected, has severe LOF (Figure 4D). In this case, the reason the gRNA had a Z score below 2 was because of editing inefficiency (see Figure S3 and section “discussion”). When we introduced silent mutations predicted by high-Z-scoring “silent-mutation” gRNAs (I1761I, S1392S, T365T) (Figures 1D–1F), we found only small changes in the measured electrophysiological parameters, unlikely to explain the phenotype in the cytotoxicity assay (Figure 4D). As confirmed by our sequence validation (Figure S3 and section “discussion”), it is likely that the high Z scores of these gRNAs were driven by collateral missense mutations. The benefit of sequence validation of these silent mutation gRNAs is that they yielded likely LOF missense variants that could be tested in the future.
Figure 4. Electrophysiological characterization of NaV1.2 variants identifies mechanisms of altered channel function.
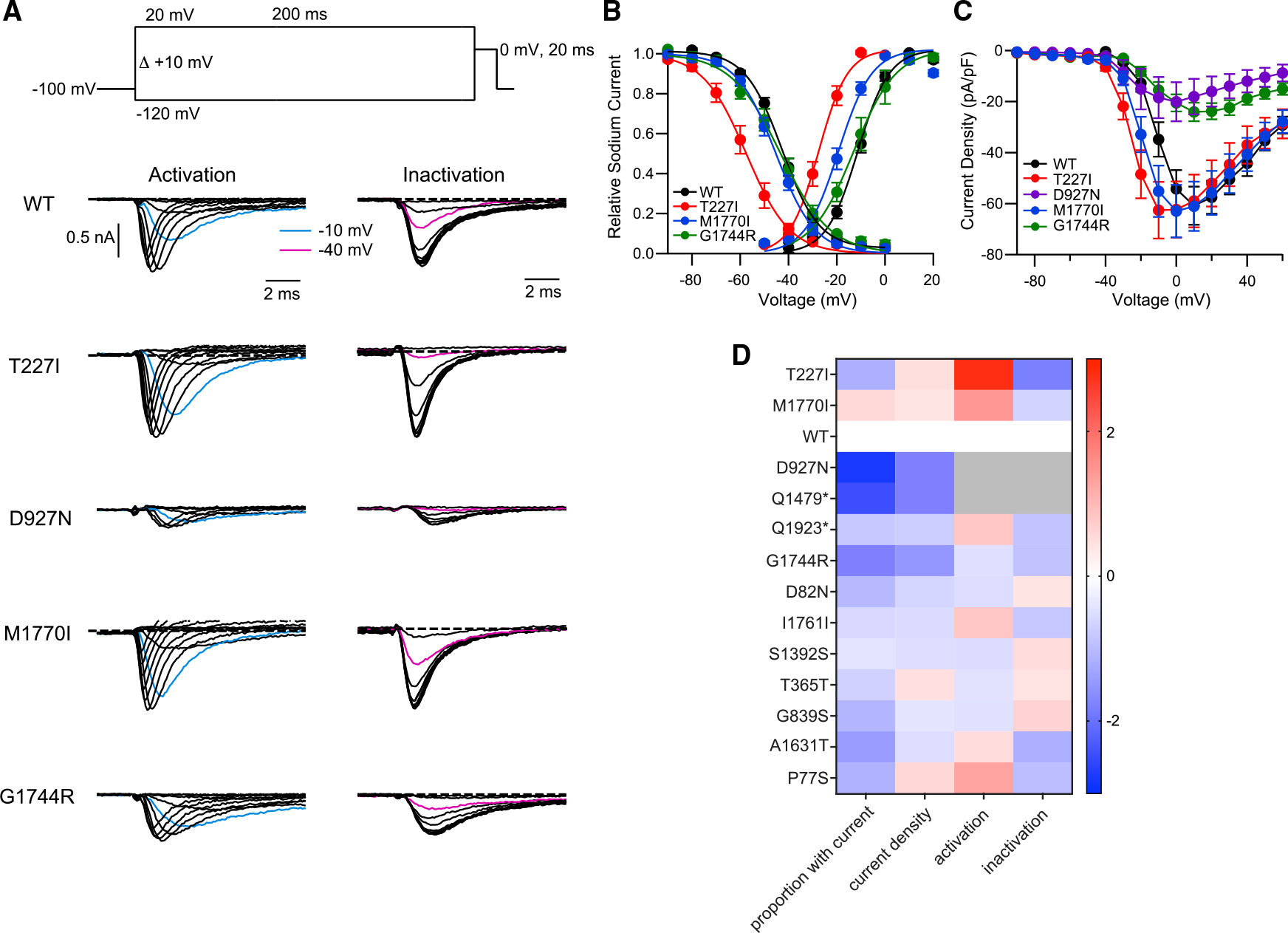
(A) Biophysical properties of WT, T227I, D927N, M1770I, and G1744R variants. (Top) The stimulus protocol to determine the voltage dependence of activation and inactivation. Sodium currents were evoked by various voltage steps from a holding potential of −100 mV to test voltages from −120 mV to +20 mV for 200 ms with 10-mV increments (pulse 1), then stepped to 0 mV for 20 ms to measure the available sodium currents (pulse 2), which were stimulated once every 10 s. Voltage-dependent activation and inactivation were assessed from the peak currents of pulse 1 and pulse 2, respectively. (Middle to bottom) Representative current traces of WT, T227I, D927N, M1770I, and G1744R from pulse 1 (activation) and pulse 2 (inactivation).
(B) Voltage dependence of activation and inactivation determined from the recorded currents. Data were normalized to the maximum peak current and fit by the Boltzmann equation (for activation) and (for inactivation), where V is test potential, Vh1 and Vh2 are the half-maximal voltage activation and inactivation, and k1 and k2 are the slope factor of activation and inactivation, respectively.
(C) Current-voltage relationships plotted for WT, T227I, D927N, M1770I, and G1744R variants. Current is normalized to capacitance for each recorded cell.
(D) Heatmap of functional characteristics for the variant NaV1.2 channels. The first column is the log2 fold change compared with WT in proportion of cells recorded that passed our threshold for current density. The next three columns represent Z scores calculated between WT and variant NaV1.2 channels, indicating the distance from the WT channel measured by standard deviations. Warm colors indicate shifts toward GOF, while cool colors denote LOF changes. Since debilitating loss of currents would supersede any changes in biophysical properties, no other parameters are reported (grayed out) for D927N and Q1479* as the currents are too small to reliably measure these parameters. Datapoints are represented by mean and error by SEM; n = 192–1,920 cells recorded per channel variant, which are then filtered for parameters such as currents that are larger than background, leading to 4–275 cells brought forward for further analysis.
DISCUSSION
NaV-encoding genes, including SCN1A, SCN2A, SCN3A, and SCN8A, are associated with a range of neurodevelopmental disorders, including benign or devastating forms of epilepsy, and autism spectrum disorder (ASD).7,8 More than 2,000 likely pathogenic and pathogenic variants in neurodevelopmental disorder (NDD)-associated NaV genes have been listed in ClinVar29 (database accessed 12/12/2022). Together, these NDD-associated NaV genes SCN1A, SCN2A, SCN3A, and SCN8A represent one of the most studied gene families across all epilepsy-associated genes.30 In particular, variants of SCN2A have been associated with epilepsy syndromes, ASD, and other neurodevelopmental disorders with diverse clinical severity. To date, SCN2A is routinely sequenced in people with neurodevelopmental disorders and early-onset epilepsies. However, diagnostic genetic tests usually report variants with unclear functional consequences. As a result, ~70% of clinically identified SCN2A missense variants are classified as variants of uncertain significance (VUSs).31 Distinguishing benign variants from disease-causing mutations remains a huge challenge for NaVs, and traditional scanning mutagenesis across the entire cDNA is often not applicable for NaVs because of their size and the difficulty in manipulating their molecular biology. Here, utilizing a CRISPR-Cas9-based C→T editor, we changed cytosines to thymines in the cDNA of NaV1.2. Many of the gRNAs that we designed to introduce missense changes (111 of 232) were enriched in our assay and produced NaV1.2 channels with significant LOF (Z score >2), compared with two gRNAs (of 232) that are depleted and produced NaV1.2 channels with GOF (Z score < −2). There are two potential explanations for this difference in number between LOF and GOF. First, we optimized our veratridine-ouabain assay for LOF. Specifically, we compared the effects of veratridine-ouabain on our stable line where NaV1.2 cDNA expression is either induced or not induced (i.e., doxycycline or no doxycycline treatment). We did not develop this assay using a cell line containing a known NaV1.2 GOF mutation. Second, it is likely easier to disrupt rather than enhance channel function.
We validated the primary base-editor screen using two orthogonal assays, one with select individual gRNAs and one with site-directed mutagenesis coupled with electrophysiology. This study serves as a proof of concept for utilizing DNA-editing technology to identify a large set of single-nucleotide variants that alter the function of NaV1.2 channels, particularly in the LOF direction, followed by validation to identify specific variant-to-function relationships.
The CRISPR-Cas9 cytosine base editor has been utilized in pooled screens to evaluate the impact of endogenous genomic variants on cell survival.25–27,32 Our report assesses the impact of C/G to A/T base pair transitions tiling an entire channel involved in neurodevelopmental disorders and identified 111 putative LOF mutations and two GOF mutations. In the two replicates of our screen, LOF variants were associated with enriched gRNAs and the two confirmed GOF variants were associated with depleted gRNAs (Figures 1 and S1). We confirmed that two significantly depleted gRNAs produced predicted nucleotide changes (T227I and M1770I) that led to NaV1.2 channels with enhanced sodium influx upon stimulation (Figures 3 and 4). A different change at 1770 (M1770L) was found in patients with epileptic encephalopathy but the variant is of undetermined functional consequence.33 T227I was previously reported by a study that sequenced patients with epilepsy but was not functionally characterized.9 Our data confirmed that mutations M1770I and T227I alter the voltage dependence of activation in a GOF direction. We validated that LOF variants typically produced NaV1.2 channels with lower current density (Figure 4) and, even at sub-saturation, our screen identified regions of the channel that are prone to disruption consistent with structural analysis (Figure 2).
Not surprisingly, our validation assays pointed to editing efficiency and editing accuracy as major considerations in interpreting the editing screen results. The frequency of unedited WT alleles ranged from 10% to 60% across the 11 gRNAs that we validated individually, and every gRNA produced collateral mutations in addition to predicted alterations in the editing window. When the intended editing efficiency is low, unintended C→T edits may influence the results. For example, silent mutation of I1761I did not significantly alter channel function (Figure 4). However, the gRNA was significantly enriched, producing a strong LOF signal (Z = 5.9). Sequence analysis identified a nearby missense mutation S1763F created by an unintended edit of a C nucleotide five bases away from the intended target site that drove the LOF phenotype (Figure S3). While this underscores the need for validation in base-editing screens, as emphasized before,25 it also nominates S1763F for future validation as a likely mutation that disrupts channel function. Another example is Q1479*, a nonsense mutation that produces strong LOF (Figure 4) and yet the gRNA has a Z score of 1.79 in our primary screen. This can be explained by the inefficiency of this guide at introducing edits of the target locus, with 60% of the alleles remaining unedited (Figure S3), consistent with recent reports where the editing efficiency was examined using a surrogate.26,27
More broadly, the secondary screens using individual gRNAs were consistent with the performance of these gRNAs when they were pooled in the primary screen (Figure 3B), supporting data that the cytotoxicity assay is reliable for determining the consequence of base edits. Despite the limitations discussed, this approach overall identified the precise nucleotide changes underlying both GOF gRNAs and six of seven of the precise nucleotide edits underlying LOF. As the technology matures in the future, improved editing efficiency and accuracy will make collateral edits less prevalent and interpretation of intended edits more direct. Furthermore, the development of A→G editors34 and Cas proteins with an expanded range of protospacer adjacent motif (PAM) targeting would greatly diversify possible gRNAs and edits in a library35 to make this approach closer to saturation.
Twenty-six missense variants in the ClinVar Database (as of February 2023) are exact matches of missense mutations that gRNAs in our screen were designed to make. Of these, 17 mutations are annotated as VUSs or conflicting interpretation. These variants could be benign, GOF, or LOF. Ten of them cross-referenced to gRNAs with Z scores higher than 2, making them putative LOF. The highest-scoring gRNA in this set (G1744R, Z score = 7.4) was included in our secondary characterization (Figure 4), which supported an LOF classification for G1744R (Figure 4). The other nine putative LOF variants are candidates for future electrophysiological validations. In total, our screen identified >100 putative disruptive missense variants of SCN2A, providing a means to prioritize functional validation of mutants likely to significantly disrupt channel function. Our ability to perform mutational scanning across NaV1.2 utilizing CRISPR-Cas9-based editing technology paves a path for saturated mutational scanning in NaV1.2 using genome-editing technology. Identified sensitive residues/regions will further facilitate computational prediction of the functional consequences of variants. The confluence of these and future efforts will be crucial in determining treatment options for patients harboring specific mutations.
Limitations of the study
As with any other screen, our results will be affected by variables particular to the screening assay. The editing efficiency and accuracy of the specific gRNAs coupled to the CRISPR editor in this screen may lead to unedited alleles or collateral edits that may confound the results of certain gRNAs. In addition, mutations that affect the binding of veratridine either directly or allosterically will potentially be counted as “hits” that may or may not reflect disruptions in channel functions that would occur without the drug. Our screen yielded many more enriched gRNAs than depleted guides, of which there were only two. This may be due to the optimization of our assay of Nav1.2 function against no channel expression, but not a GOF mutation. Future development of the viability assay using known GOF mutations may yield more depleted gRNAs. In addition, it may be easier to disrupt than to enhance channel function with randomly introduced nucleotide edits. The secondary functional validation we outline in our approach will be able to determine the effects of specific gRNAs and mutations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jen Pan (jpan@broadinstitute.org).
Materials availability
All general-use vectors and plasmid sequence files are available from the lead contact upon request. The SCN2A plasmid used for electroporation is available on Addgene (Addgene Plasmid #162279).
Data and code availability
Access to sequence validation data can be found at https://doi.org/10.5061/dryad.zkh1893fc. Access to original cell culture and electrophysiological data is available upon request.
Access to custom code is available in a GitHub repository. The DOI is 10.5281/zenodo.7901798.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture
NaV1.2 stable cell line
Cells expressing NaV1.2 channels were established by transfecting parental FlpIn TREx HEK293 cells (Thermo Fisher, ref: R78007) with pcDNA5/pFRT/TO vectors containing the cDNA encoding the WT human NaV1.2 channel (SCN2A, NCBI Accession: NM_021007), and pOG44 vector encoding FLP recombinase (Thermo Fisher, ref: K650001) in 1:9 ratio using Lipofectamine 2000 (Life Technologies; ref. 11668019).38 Single cDNA insertion events were selected after two weeks in 200 μg/mL hygromycin B (Invitrogen); resistant polyclonal cell lines (10–20 colonies) were expanded and maintained with 180 μg/mL hygromycin B (50 mg/mL, Gibco Life Technologies, ref. 10687010) and 15 μg/mL blasticidin S (10 mg/mL, Gibco Life Technologies, ref: A1113903). cDNA integration was validated with Sanger sequencing of the insertion locus. The expression of NaV1.2 channels was induced by doxycycline (1 μg/mL, MilliporeSigma, ref: D9891–10G). The original HEK293T cell line was derived from a human, female host.
We cultured our cells in DMEM/F-12, supplemented with GlutaMAX (Life Technologies, ref. 10565042) and tetracycline-free fetal bovine serum (10% final concentration; Takara Bio, ref. 631106), referred to as “media” below.
We maintained NaV1.2-WT stable cells in antibiotic-treated (Blasticidin S and Hygromycin B) media in a T75 Tissue Culture flask (VWR International LLC, ref: BD353136). To dissociate cells from the flask, we added TrypLE (TrypLE Express Enzyme (1X), Gibco Life Technologies, ref. 12604039) to the flask, rocked the flask to coat all the cells, then promptly removed the TrypLE from the flask. We incubated the cells at 37°C for 3–5 min, then quenched the dissociation reaction using media (10 mL for T75, 15 mL for T175). Next we determined the density (cells/mL) of the homogenized cell suspension (Cellometer Auto T4, Nexcelom Bioscience LLC) to seed the appropriate number of cells. Approximately 72 h before each infection, we seeded six to nine million NaV1.2-WT cells into a T175 Falcon Tissue Culture flask (VWR International LLC, ref. 29185–308) in 25 mL of media treated with antibiotics, 37.5μL 10 mg/mL Blasticidin S and 90 μL 50 mg/mL Hygromycin B. At this time, we also seeded 1–1.5e6 cells into a T75 flask as a maintenance flask. Media was kept at 4°C and warmed to room temperature (RT) for use; PBS, trypan blue, and TrypLE were all kept at RT. We used this cell line for the Drug Enrichment Screens and Genomic DNA Extractions.
HEK293T cell line
We maintained HEK293T cells in the antibiotic-free media in a T75 tissue-culture flask. To dissociate cells from the flask, we added TrypLE to the flask, rocked the flask to coat all the cells, then promptly removed the TrypLE from the flask. We incubated the cells at 37°C for 3–5 min, then quenched the dissociation reaction using media (10 mL for T75, 15 mL for T175). Next we determined the density (cells/mL) of the homogenized cell suspension (Cellometer Auto T4, Nexcelom Bioscience LLC) to seed the appropriate number of cells. For maintenance flasks, we seeded 1–1.5e6 cells in 10mL media. The original HEK293T cell line was derived from a human, female host. We used this cell line for Lentivirus Production, Electroporation for transient transfections, and Automated Patch Clamp.
Plasmid vectors
pRDA_256 (Addgene #158581): U6 promoter expresses customizable guide RNA with a 10x guide capture sequence at the 3′ end of tracrRNA to facilitate future use with direct capture, single-cell RNA sequencing; core EF1a (EFS) expresses codon-optimized BE3 with 2xSV40NLS and 2A site provides puromycin resistance.25
pIR-CMV-SCN2A-Variant-1-IRES-mScarlet (Addgene #162279): Eukaryotic expression of human SCN2A variant 1 isoform, stabilized with IRES intron and with a mScarlet marker for visual confirmation of plasmid expression in cell culture.36
Lentivirus vectors
For both the pooled library and subsequent validation screens, we used pRDA_256 vector (BE3, Addgene #158581) as our gRNA vector.25 Additionally, to produce lentivirus containing our gRNAs for the pooled library and validation screens, we used psPAX2 (Addgene #12260) and pCMV_VSVG (Addgene #8454).25
METHOD DETAILS
Lentivirus production
To produce lentivirus, we followed methods described previously by Hanna et al.25 In brief, for large-scale production of the pooled library virus, we seeded 18e6 HEK293T cells into a T175 flask with 25 mL of media the day before transfection to ensure the cells would be at 80% confluence. The following morning, we prepared two master mixes. The first included the following: 6 mL OptiMEM (Reduced-Serum Medium, Gibco Life Technologies, ref. 31985088) and 305 μL TransIT-LT1 transfection reagent (Mirus Bio LLC, ref: MIR 2306). We homogenized this reagent mix by inverting the tube, then incubated it at RT for 3 min. We made the DNA mastermix with RT reagents in a 1.5 mL microcentrifuge tube (Eppendorf, DNA/RNA LoBind, VWR International LLC, ref. 80077-230): 50 μL OptiMEM, 40 μg of the lentivirus plasmid DNA, 50 μg psPAX2, and 5 μg VSV-G. We pipetted the DNA mix to the reagent mix, then homogenized it. We incubated the mixture at RT for 30 min before adding the solution dropwise to the prepared T175 of HEK293T cells. We incubated the transfected cells at 37°C (5% CO2) for 6–8 h, then replaced the transfection media with a fresh 40 mL of media. Thirty-six hours post-transfection, we transferred the virus-containing supernatant media from the T175 flask to a 50 mL centrifuge tube. We centrifuged this tube at 230 ×g for 1 min to pellet any cell debris. We then prepared 1 mL aliquots of virus-containing supernatant. Prior to storing at −80°C, we flash-froze the tubes by incubating them in a dry-ice and 70% ethanol slurry for several minutes.
To produce lentivirus for the secondary validation screens, we used a small-scale version of the above procedure. We seeded HEK293T cells in a Falcon 6-well plate (VWR International, ref. 62406–161) at 5.5e5 cells/well in 2.5 mL media (1 well per virus plasmid); the next morning, we prepared two master mixes for each lentivirus plasmid DNA (pRDA_256 containing virus gRNA). We prepared the reagent master mix in a 1.5 mL microcentrifuge tube: 12 μL Opti-MEM and 3 μL TransIT-LT1 transfection reagent, then incubated at RT for 5 min. We made the DNA mastermix in a separate 1.5 mL microcentrifuge tube: 500 ng of the lentivirus plasmid DNA, 500 ng psPAX2, 50 ng VSV-G, and enough RT OptiMEM to bring the final volume to 37.5 μL. We transferred the reagent mix to the plasmid mix by pipette, then carefully homogenized the transfection solution by stirring with the pipette tip. We then incubated this at RT in the tissue culture hood for 30 min before adding the solution dropwise to the respective well in the prepared plate of HEK293T. We incubated the transfected cells at 37°C (5% CO2) for 18 h, then replaced the transfection media in each well with a fresh 2.5 mL of media. The next day (24 h-post media change), we harvested the first batch of virus-containing supernatant media from each well. For each well, we transferred the supernatant media to a conical tube, added a fresh 2.5 mL media to the well, then returned the plate to 37°C (5% CO2) for 24 h. We centrifuged the tube of virus-containing media for 5 min at 1256 rpm to pellet any cell debris, then aliquoted the virus-containing media into labeled tubes for freezing. Prior to storing at −80°C, we flash-froze the tubes by incubating them in a dry-ice and 70% ethanol slurry for several minutes. The next day (24 h-post harvest 2), we repeated this virus harvesting process, then discarded the transfected cells. OptiMEM and TransIT-LT were stored at 4°C and warmed to room temperature before use. All the plasmids were stored at −20°C and thawed to room temperature before use.
Tiling library guide design
All possible guides with NGG PAM targeting the NaV1.2 cDNA were designed using an in-house design tool. The predicted C→T edits in the 4–8 nucleotide window for every guide were annotated. The library was filtered to exclude any promiscuous guides and guides with BsmBI sites or a poly-T sequence.25 The library also contained 52 guides targeting intergenic regions and 52 guides targeting splice sites of essential genes as controls.
Growth rate determination
Over-confluence of cells in each well could alter their response to compound treatment in our screen. To determine the growth rate of the NaV1.2-WT cells, we seeded NaV1.2-WT cells at different densities in each well of a 6-well plate: A) 20,000 cells; B) 40,000 cells; C) 80,000 cells; D) 160,000 cells; E) 320,000 cells; and F) 640,000 cells in 2 mL. We collected cells and counted the total cell count per well after 96 h. We calculated the doubling time for each well (i.e. each initial seeding count) using the following equation:
We chose the density ratio of 8.333e3 cells/cm2 (i.e. 80,000 cells initial seeding count to 9.6 cm2 surface area), assuming a 23.67 h doubling time. We used this ratio to guide the seeding density for all the following screen experiments.
Lentivirus titer determination
To determine the viral titer for our pooled screen, we collected uninduced NaV1.2-WT stable cells from a T175, then diluted the cell suspension to 1.5 million cells/mL in media in a 50 mL conical tube. We added Polybrene Transfection/Infection Reagent (10 mg/mL; Millipore Corporation, ref: TR-1003-G) at 0.8 μL/mL to the cell suspension, and inverted the tube gently. We added 2 mL of the polybrene-treated suspension to 5 wells of a 12-well plate (3e6 cells/well). We thawed the aliquot of virus on ice and added to each well of cells, leaving one well as a non-infected control (NIC): A) 0 μL virus (NIC), B) 250 μL virus, C) 500 μL virus, D) 750 μL virus, E) 1000 μL virus. We brought the total volume in each well up to 3 mL using polybrene-free media. We centrifuged the plate of cells with virus for 2 h (30°C, 2000 rpm), incubated the plate for two more hours at 37°C in a tissue culture incubator (5% CO2), then changed out the media on each well with 2 mL fresh media. Cells were returned to the incubator overnight.
The next day (day 2), we collected cells from each well (per well: 1 mL 1X PBS wash, 200 μL TrypLE, 3 min incubation at 37°C, and deactivating with 1.8 mL media), and seeded 80,000 cells to each well of two 6-well plates. We seeded cells for each virus condition (0–1000 μL virus) to 1 well per plate in 2 mL media. On day 3, we replaced the media in each well, and began the puromycin treatment, with a negative control plate for determining virus toxicity and a plate at 0.5 μg/mL puromycin dihydrochloride (+Puro; 10 mg/mL, Gibco Life Technologies, ref: A1113803) in media. We monitored the cells for 72 h. On day 6, we collected cells and counted the cells from each well. We calculated the virus toxicity percentage and infection efficiency by comparing the different virus and ±puromycin conditions to the NIC and puromycin control conditions. From this, we determined 1 mL virus achieved the greatest infection efficiency (>20%) with minimal toxicity to the infected cells (>80% cell viability).
Drug enrichment screens
Pooled library
Similar to the viral titration, on the first day we prepared uninduced NaV 1.2-WT cell suspension (1.5 million cells/mL, 0. 8μL polybrene/mL) in media in a 50 mL conical tube. We seeded 2 mL of cell suspension to four wells of a 12-well plate (3e6 cells/well), then added 1 mL of thawed pooled library virus to each of three wells of cells (9e6 total cells for infection). We added 1 mL media to a fourth well as a non-infected control (NIC). The cells were centrifuged with virus for 2 h (30°C, 2000 rpm), and incubated for two more hours at 37°C. Fresh media was added and cells were returned to the incubator overnight.
The next day (day 2), we collected cells from each well of the 12-well plate. We pooled the infected cells and seeded 1.5e6 infected cells to each of six T175 flasks from the pooled suspension in 30 mL media/flask. We prepared an in-line assay, a proxy for monitoring puromycin selection of the infected cells in the T175s, in a 6-well plate. In the in-line assay, we seeded 80,000 cells per well in 2 mL media, with 4 wells of infected cells and 2 wells of NIC cells. We prepared the surplus cells for gDNA extraction. Cells were returned to the incubator overnight. The following day (day 3), we treated cells in the T175s with 0.5 μg/mL puromycin in media. Parallel treatment was done for the in-line assay (1 well of NIC cells, 2 wells of infected cells); puromycin control wells received fresh media without puromycin. After 72 h of puromycin selection (day 6), when all the NIC cells under puromycin selection died off, we collected and counted the cell suspension from each well of in-line assay, then calculated the virus toxicity percentage and infection efficiency by comparing the infected condition and ±puromycin conditions to the NIC and puromycin control conditions. At this time, we replaced the media in the T175 flasks of infected cells with puromycin-free media, then let the cells recover for 48 h at 37°C (5% CO2).
After 48 h (day 8), we collected and pooled cells from the six T175 flasks, counted the homogenized cell suspension, then seeded 1.5e6 infected cells in 30 mL media to three T175 flasks. We added doxycycline to induce two of the three flasks. We used surplus infected cells for gDNA extraction.
The next day (day 9), we began the first round of drug enrichment using optimized concentrations of veratridine and ouabain. To prepare the drugs for use, we dissolved veratridine (≥90% (HPLC), powder; Sigma-Aldrich, CAS: 71–62-5, ref: v5754–25) in 100% ethanol, at 50 mM, and stored at −20°C. We used a working stock solution of 1 mM veratridine. We dissolved ouabain (powder; Sigma-Aldrich, CAS: 11018–89-6, ref: PHR1945–300MG) in autoclaved DI water, at 5 μM, and stored at RT. We treated the induced and uninduced infected cells with optimized drug concentrations for 96 h (0.5 μM veratridine/ethanol, 10 nM ouabain/water) (Figure 1B).
On day 13, we collected and counted cells from each flask, keeping the cell suspensions separate. Using the below formula, we calculated the number of times the cell population doubled during one round of drug enrichment (Figure 1C):
We seeded three T175 flasks with 1.5e6 cells in 30 mL, maintaining 1 flask per treatment condition and treating the two induced cell populations with doxycycline. We returned the flasks to 37°C for 48 h to allow them to recover from seeding before proceeding with the second round of VO drug enrichment. At the end of this recovery period (day 15), just as on day 9, we started the second round of drug treatment. We prepared the surplus cell suspension from each drug arm for gDNA extraction. After 96 h (day 19), we collected and counted cells from each flask and calculated the number of times the population doubled during one round of drug enrichment. Using the following equation, we calculated the cumulative number of times the cell population doubled during both rounds of drug enrichment for each drug arm (Figure 1C):
We harvested the cells for gDNA extraction. We sequenced this gDNA the same way as for the individual gRNA validation. We calculated z-scores for individual guides by taking the difference of their log fold change in the drug arm versus vehicle arm and the average log fold change of the intergenic controls in the drug arm versus vehicle arm, then dividing this by the standard deviation of the log fold changes of the intergenic controls.
Individual gRNA validation
We designed the guideRNA plasmid (pRDA_256), with one plasmid per variant of interest from the pooled library, using SnapGene (www.snapgene.com). To do this, we loaded the pRDA_256 sequence file into SnapGene, then inserted the specific guide sequence for a variant (Table S3) in the region immediately upstream of the gRNA scaffold region. The inserted gRNA was bordered immediately up- and downstream by a BsmBI restriction site. These files are available upon request. We ordered pRDA_256 custom plasmids from Genscript, and we did this for the 11 variants validated by base-editor screen (Table S3, VO SCREEN).
We received gRNA plasmids containing the specific variant guide sequence as lyophilized DNA. We reconstituted the plasmids to 100 ng/μL stocks in nuclease-free water. We confirmed the plasmid sequence using Sanger sequencing with the U6 universal primer (800 ng DNA; Genewiz). We used a 5.5 μL aliquot of the 100 ng/μL stock gRNA plasmid for small-scale lentivirus production (see Lentivirus Production above).
For individual gRNA infection, we ran a scaled-down version of the pooled library screen using lentivirus containing one gRNA at a time (Table S3). The protocol is similar to Pooled Library Screen described above, but adjusted for volumes in accordance with the seeding ratio described in Growth Rate Determination: we used 1 well of the 12-well plate per individual gRNA-containing virus on day 1, and seeded T75 flasks with 6.25e5 cells/mL in 10 mL media. Like in the pooled screen, we ran an in-line assay alongside the puromycin selection, with two wells per variant-infected cells and an NIC well (one well under puromycin selection, and the other in puromycin-free media) to determine the virus toxicity and infection efficiency. Unlike the pooled screen, we only harvested infected cells for gDNA extraction at the end of the individual gRNA validation screen (results shown in Figure 3).
Genomic DNA extraction
We extracted gDNA from cells collected at the end of the screens using the DNeasy Blood and Tissue Kit (QIAGEN, ref. 69504). Briefly, we centrifuged cells (≤5e6) at 300 ×g for 5 min at RT. The cell pellet was resuspended and washed in 1 mL 1X PBS before storage at −80°C. For processing, the cell pellets were thawed at RT for 15–30 min and suspended in 200 μL 1X PBS, with 20 μL Proteinase K (>600 mAU/mL, QIAGEN, ref. 19133) and 4 μL RNase A (100 mg/mL, QIAGEN, ref. 19101), gently mixed by vortexing on low/medium in a 5 s burst (to avoid shearing the DNA), and incubated at RT for 2 min. We added 200 μL lysis buffer to the tube and mixed gently by vortexing on low/medium for 5–10 s, and then incubated the solution at 70°C overnight (12–18 h).
The next day, we added 200 μL ethanol to the lysate after equilibration to RT for 20–30 min. We vortexed 5–10 s to mix thoroughly. The solution was then added to the DNeasy mini spin column in a 2 mL collection tube, and the column was washed with the buffers according to the manufacturer’s instructions. We eluted the genomic DNA with 150 μL elution buffer after 1 min incubation at RT. Then, we repeated this elution process with an additional 50 μL elution buffer in the same spin column and microcentrifuge tube (total elution of 200 μL).
We used the dsDNA HS Assay (Life Technologies, ref: Q33230) to measure the genomic DNA concentration following the manufacturer’s protocol, and measured the concentration with a Qubit 2.0 Fluorometer (Life Technologies, Thermo Fisher Scientific).
Genomic DNA amplification, purification, and sequencing
To sequence gRNAs for both the pooled library and the individual screens, we amplified using a two-step PCR. The first step amplified genomic DNA using custom primers (Table S4) to amplify the target site (Table S3) and adding vector-binding regions (in red in Table S4) to the ends of the amplicon. We assembled the following reaction on ice for each well of a 96-well PCR Plate: 50 μL NEBNext High Fidelity 2X PCR Master Mix (New England Biolabs, ref: M0541), 49 μL gDNA extraction, and 0.5 μL of 100 μM primers (Table S4). We ran a touchdown PCR with the following thermocycler parameters: 1) 98°C for 1 min; 2) 98°C for 30 s; 3) 68°C for 30 s (−1° per cycle); 4) 72°C for 1 min; 5) return to step 2, 15 cycles; 6) 72°C for 10 min; 7) 4°C forever.
The second step of PCR added well-specific barcodes and Illumina adapters to the ends of amplicons using the P7 “Beaker” primer for the pooled screen, P7 “Kermit” primer for the individual screens, and P5 “Argon” primer for all screens (Table S4). We assembled the following reaction for each well of a 96-well PCR plate: 55 μL molecular biology grade water (VWR International LLC, ref. 71002–726), 10 μL of 10X Titanium Taq buffer (Clontech Takara, ref. 639242), 8 μL of dNTPs (Clontech Takara, ref. 4030), 5 μL of DMSO (Sigma Aldrich, ref: D9170–5VL), 0.5 μL of P5 primer (100 μM), 10 μL of P7 primer (5 μM), 10 μL of PCR product from the first PCR step, and 1.5 μL Titanium Taq polymerase (Clontech Takara, ref. 639208). To each well, we added water, 10X buffer, dNTPs, P5 primer, and polymerase as a master mix, then added the amplified gDNA PCR product, followed by the P7 primer. We ran the second PCR with the following thermocycler parameters: 1) 95°C for 5 min; 2) 95°C for 30 s; 3) 53°C for 30 s; 4) 72°C for 20 s; 5) return to step 2, 28 cycles; 6) 72°C for 10 min; and 4°C forever.
With all screens, we cleaned the PCR product with an SPRI purification after the second PCR step. To do this, we first pooled all the PCR products together by transferring 15 μL/well (1X) from the 96-well plate to a 25 mL trough, changing tips between rows. We homogenized the pooled products by gently pipetting the solution in the trough. We added 100 mL of the homogenized pooled product to each of two wells of a round-bottom 96-well plate (Corning Costar, VWR International LLC, ref. 29445–154), and transferred the remaining pooled PCR product from the trough to a microcentrifuge tube to store at −20°C. We added 100 μL of chilled SPRI beads (Beckman Coulter, ref: B23318) to each well containing PCR product (1:1 ratio of volumes), and pipetted vigorously to mix before incubating the mixtures at RT for 5 min. We then placed the round-bottom plate atop a 96-well magnetic plate (96S Super Magnet, Alpaqua, ref: A001322), and let the beads form a circular ring at the bottom of the plate and clear the supernatant (2–3 min). We carefully removed and discarded the supernatant solution from each well without disturbing the ring. We washed each well with 100 μL 70% ethanol twice, adding the ethanol dropwise to the well. We removed the round-bottom plate from the magnet plate and let the beads dry for 1 min at RT. We eluted the purified product by adding 40 μL elution buffer (10 mM Tris$Cl, pH 8.5), resuspending the product by vigorously pipetting, and incubating the elution in its well for 30 s. Returning the round-bottom plate to the magnetic plate, we let the beads form a ring at the bottom again, then transferred and pooled the supernatant purified products to a microcentrifuge tube. We determined the concentration of the product using a Qubit 2.0 fluorometer.
We submitted the purified PCR product for MiSeq (300 cycles) Illumina sequencing with the following parameters: fragment size of 350 bp, single read, read 1 length of 300, i7 primer index length of 8, 10% Phix, and loading samples at 60%.
Plasmid mutagenesis for SCN2A variant electrophysiology validation
Full-length WT and variant cDNAs encoding an intron-stabilized human NaV1.236 corresponding to NM_021007 were engineered into a modified pIRES2 vector with a high efficiency IRES element followed by the monomeric red fluorescent protein mScarlet (pIRES2-mScarlet; Addgene Plasmid #162279). Variants were introduced into NaV1.2 by site-directed mutagenesis using Q5 high-fidelity DNA polymerase Master Mix (New England Biolabs, Ipswich, MA; ref: M0492S). Mutagenic primers (Table S5) were designed using custom software (available upon request). We validated these variants using electrophysiology (see Automated Patch Clamp) after introducing them into cells with transient transfections (see Electroporation for Transient Transfection).
Electroporation for transient transfection
We used the pIR-CMV-IRES-mScarlet plasmid vector containing the human SCN2A adult sequence (NCBI Accession: NM_021007) for electroporation (Addgene #162279).
Electroporation of cells on the MaxCyte system was carried out following manufacturer’s procedures. Typically, for 8 batches of electroporation, we used a total of 2e7 cells, seeded at a density of 1.5–2e6 cells per T175 flask in 30 mL of media. On the day of the electroporation, we aliquoted 5 μg/μL stocks of DNAs into corresponding DNase-free tubes along with the electroporation buffer (MaxCyte, Gaitherbug, MD; ref: EPB-1) so the final volume would be 13 μL per tube. We used cells no more than 85% confluent for electroporations. We washed cells with 5 mL of 1X PBS, then incubated them in 3 mL of TrypLE dissociation reagent. We added 7 mL of DMEM/F12+ GlutaMAX media to quench the dissociation, transferred the cell suspension into 50 mL conical tubes, then spun down the cells at 1000 rpm for 5 min. We counted the cells, then reconstituted them in electroporation buffer to a concentration of 1.20e8 cells/mL in a DNAse free tube.
We mixed 87 μL of the cell suspension with the premade DNA/electroporation buffer solution, then pipetted this gently into a 100×2 cassette. We ran this cassette on the MaxCyte electroporator, using the built-in, standard HEK293 protocol. Upon completing the electroporation, we added 10 μL of stock concentration DNase I, and we incubated the electroporated cells in a 37ΰC, 5% CO2 incubator for 30 min for recovery. We then resuspended the cells in 1 mL DMEM/F12+ GlutaMAX media, and transferred them to two T175 flasks (30mL media total per flask).
Automated patch clamp
Automated patch clamp recording was performed using a SyncroPatch 384PE (Nanion Technologies, Munich, Germany).38,39 In brief, 48 h after electroporation, cells were harvested and suspended with DMEM/F12 media Divalent-Free solution without FBS (mM, 145 NaCl, 4 KCl, 10 HEPES, 8 Glucose, pH 7.4, 300 mmol/L; 1:1 ratio). For electrophysiological recording, we used external solution (mM, 80 NaCl, 60 NMDG, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 5 Glucose, pH 7.4, 289 mmol/L) and internal solution (mM, 110 CsF, 10 CsCl, 20 EGTA, 10 HEPES, pH7.2, 285 mmol/L). Before addition of cells, the fast capacitive component was compensated and slow capacitive component was canceled. The liquid junction potential (~12 mV) was not corrected. For each 384-well plate, blank HEK293 cells and cells expressing WT hNaV1.2 were used as a negative control and positive control (reference), respectively. The data were acquired at 20 kHz and filtered at 10 kHz using the software PatchControl 384 (Nanion Technologies) and were exported to and analyzed with the software DataControl384 for additional biophysical analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of deep sequencing data from validation experiments
We used CRISPResso2 (version 2.1.3) to process sequencing reads from the validation experiment (Figure 3).37 CRISPResso2 was run in base editor mode using the default settings. All samples but one (V354M_C353Y) had >40% aligned reads. V354M_C353Y was removed from the subsequent analysis due to its low alignment rate (<5%).
We used the “Alleles_frequency_table_around_sgRNA” file from the CRISPResso2 output to calculate the weighted fold change of the percentage of the modified alleles (weighted_fc_%modified) and the z-scores for the intended edits. No filters for allele frequency were applied when calculating the weighted_fc_%modified value and the z-scores for the intended edits. When visualizing individual alleles, we filtered out any alleles that comprised <1% of the total reads in all the samples for that sgRNA.
The value is the fold change of the values in the samples treated with veratridine and ouabain compared to the samples without veratridine and ouabain. The value is the average value in the samples treated with veratridine ouabain.
To get the z-scores for the intended edits, we first calculated the weighted_fc_%modified values for all alleles:
The and values are the mean and the variance of the values in all the alleles containing the intended edits. The and values are the mean and the variance of the values in all the alleles that do not contain the intended edits.
Electrophysiology data analysis
Cells were selected for seal resistance >0.5 GΩ and series resistance <20 MΩ and further excluded if the maximum peak current at 0 mV from −100 mV was less than 300 pA, because 3% of naive HEK293 cells (4 of 147 cells) displayed up to 300 pA current likely due to endogenous channels. Voltage-dependence of activation and inactivation was fitted by Boltzmann equation: (for activation) and (for inactivation)) and the value of and was collected from the software DataControl384 (Nanion; Munich, Germany).
SCN2A structural analysis
We conducted our analysis of gRNA z-scores in R (version 4.1.2) using the basic statistical package, {stats} (Version 4.1.2), available in the base version of R. Multiple group comparisons were conducted with 1-way ANOVA followed by post-hoc pairwise t-tests, Bonferroni corrected for multiple comparisons. We applied the Wilcoxon rank-sum test to evaluate if variants located in close proximity to the pore of NaV1.2 (S5-S6-Linker) have significantly higher z-scores compared to variants at the D3-D4 linker or other intracellular regions of the channel.
Group details
The data from Figures 1C, 1E, and 1F are taken from two independent infections with the pooled library as also described in the figure caption. Data for the sequence validation in Figure 3 is from 3 sequencing replicates. Data in Figure 4 is recorded from n = 192–1920 cells recorded per channel variant which are then filtered for parameters such as currents that are larger than background, leading to 4–275 cells brought forward for further analysis. Further details can be found in the relevant figure captions.
ADDITIONAL RESOURCES
The authors have no additional resources to report.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| DMEM/F-12, supplemented with GlutaMAX™ | Life Technologies | Cat#: 10565042 |
| tetracycline-free fetal bovine serum | Takara Bio | Cat#: 631106 |
| TrypLE™ Express Enzyme (1X) | Gibco Life Technologies | Cat#: 12604039 |
| Hygromycin B (50 mg/mL) | Gibco Life Technologies | Cat# 10687010 |
| Blasticidin S (10 mg/mL) | Gibco Life Technologies | Cat# A1113903 |
| Doxycycline (1 μg/mL) | Millipore Sigma | Cat# D9891-10G |
| TransIT-LT1 (lentivirus production) | Mirus Bio LCC | Cat# MIR 2306 |
| Polybrene (10 mg/mL) | Millipore Corporation | Cat# TR-1003-G |
| Puromycin dihydrochloride (10 mg/mL) | Gibco Life Technologies | Cat# A1113803 |
| Veratridine (CAS: 71-62-5) | Sigma-Aldrich | Cat# v5754-25 |
| Ouabain (CAS: 11018-89-6) | Sigma-Aldrich | Cat# PHR1945-300MG |
| Lipofectamine 2000 (cell line generation) | Life Technologies | Cat# 11668019 |
| Proteinase K (>600 mAU/mL) (gDNA extraction) | QIAGEN | Cat# 19133 |
| RNase A (100 mg/mL) (gDNA extraction) | QIAGEN | Cat# 19101 |
| DMSO (PCR) | Sigma Aldrich | Cat# D9170-5VL |
| Electroporation Buffer (electroporation) | Maxcyte | Cat#: EPB-1 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Cell Titer Glo | Promega | Cat# G7571 |
| Titanium Taq DNA Polymerase | Clontech Takara | Cat# 639208 |
| 10X Titanium Taq buffer | Clontech Takara | Cat# 639242 |
| PureLink HiPure Plasmid DNA Purification Kit (lentivirus production) | Invitrogen | Cat# K2100-04 |
| DNeasy Blood and Tissue Kit (gDNA extraction) | QIAGEN | Cat# 69504 |
| dsDNA HS Assay | Life Technologies | Cat# Q33230 |
| NEBNext High Fidelity 2X PCR Master Mix | New England Biolabs | Cat# M0541 |
| dNTPs | Clontech Takara | Cat# 4030 |
| SPRI beads | Beckman Coulter | Cat# B23318 |
|
| ||
| Deposited data | ||
|
| ||
| Sequencing data from gRNA validation | This paper | https://doi.org/10.5061/dryad.zkh1893fc |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Stable Nav1.2 WT Cell Line (background FlpIn TREx HEK293 cells) | This paper (hSCN2A: NM_021007) | N/A |
| HEK293T | ATCC | Cat# CRL-3216 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Tables S4 and S5 | N/A | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pRDA_256 (for library and variant guides) | Hanna et al.25 (The Broad Institute) | Addgene #158581 |
| psPAX2 (for lentivirus production) | Hanna et al.25 (The Broad Institute) | Addgene #12260 |
| pCMV_VSVG (for lentivirus production) | Hanna et al.25 (The Broad Institute) | Addgene #8454 |
| pIR-CMV-SCN2A-Variant-1-IRES-mScarlet | DeKeyser et al.36 (SCN2A: NM_021007) | Addgene #162279 |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism7 | GraphPad Software | https://www.graphpad.com/ |
| CRISPResso2 (version 2.1.3) | Clement et al.37 | https://github.com/pinellolab/CRISPResso2 |
| Biorender | Biorender | https://app.biorender.com/ |
| DataControl384 (Syncropatch) | Nanion | https://www.nanion.de/products/syncropatch-384/software/ |
| SnapGene software | SnapGene by Dotmatics | www.snapgene.com |
| Our Code | GitHub | https://doi.org/10.5281/zenodo.7901798 |
Highlights.
Genomic base editing enables screening of a library of NaV1.2 variants
Tiling the entire NaV1.2 coding sequence highlights regions intolerant to variation
Follow-up sequencing identifies variants likely to perturb NaV1.2 function
Electrophysiology of NaV1.2 variants reveals mechanisms of functional disruption
ACKNOWLEDGMENTS
J.Q.P. was funded by NS108874 and NS110355, and A.L.G. was funded by NS108874. Schematics were created with BioRender.com. We thank everyone at the Broad Institute Genetic Perturbation Platform and the Genomics Platform, especially David Root and Amy Goodale for generating our lentivirus library and for their support and guidance throughout this project. We thank Estefania Bricio, Lingling Yang, David Baez-Nieto, and the rest of the Pan Lab for their support and guidance on this project.
Footnotes
DECLARATION OF INTERESTS
A.L.G. consults for and receives sponsored research funding from Praxis Precision Medicines and Neurocrine Biosciences for unrelated work.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper self-identifies as living with a disability.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112563.
REFERENCES
- 1.Pan X, Li Z, Huang X, Huang G, Gao S, Shen H, Liu L, Lei J, and Yan N (2019). Molecular basis for pore blockade of human Na+ channel Nav1.2 by the μ-conotoxin KIIIA. Science 363, 1309–1313. 10.1126/science.aaw2999. [DOI] [PubMed] [Google Scholar]
- 2.Payandeh J, Scheuer T, Zheng N, and Catterall WA (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catterall WA, Lenaeus MJ, and Gamal El-Din TM (2020). Structure and pharmacology of voltage-gated sodium and calcium channels. Annu. Rev. Pharmacol. Toxicol. 60, 133–154. 10.1146/annurev-pharmtox-010818-021757. [DOI] [PubMed] [Google Scholar]
- 4.Bean BP (2007). The action potential in mammalian central neurons. Nat. Rev. Neurosci. 8, 451–465. 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- 5.Planells-Cases R, Caprini M, Zhang J, Rockenstein EM, Rivera RR, Murre C, Masliah E, and Montal M (2000). Neuronal death and perinatal lethality in voltage-gated sodium channel αII-deficient mice. Biophys. J. 78, 2878–2891. 10.1016/S0006-3495(00)76829-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ Jr., Clarkson RL, Sanders SJ, and Bender KJ (2019). The autism-associated gene Scn2a contributes to dendritic excitability and synaptic function in the prefrontal cortex. Neuron 103, 673–685.e5. 10.1016/j.neuron.2019.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meisler MH, Hill SF, and Yu W (2021). Sodium channelopathies in neurodevelopmental disorders. Nat. Rev. Neurosci. 22, 152–166. 10.1038/s41583-020-00418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reynolds C, King MD, and Gorman KM (2020). The phenotypic spectrum of SCN2A-related epilepsy. Eur. J. Paediatr. Neurol. 24, 117–122. 10.1016/j.ejpn.2019.12.016. [DOI] [PubMed] [Google Scholar]
- 9.Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, Lesca G, Ville D, Milh M, Villard L, et al. (2017). Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain 140, 1316–1336. 10.1093/brain/awx054. [DOI] [PubMed] [Google Scholar]
- 10.Brunklaus A, Feng T, Brünger T, Perez-Palma E, Heyne H, Matthews E, Semsarian C, Symonds JD, Zuberi SM, Lal D, and Schorge S (2022). Gene variant effects across sodium channelopathies predict function and guide precision therapy. Brain 145, 4275–4286. 10.1093/brain/awac006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunklaus A, Du J, Steckler F, Ghanty II, Johannesen KM, Fenger CD, Schorge S, Baez-Nieto D, Wang H-R, Allen A, et al. (2020). Biological concepts in human sodium channel epilepsies and their relevance in clinical practice. Epilepsia 61, 387–399. 10.1111/epi.16438. [DOI] [PubMed] [Google Scholar]
- 12.Hedrich UBS, Lauxmann S, and Lerche H (2019). SCN2A channelopathies: mechanisms and models. Epilepsia 60, S68–S76. 10.1111/epi.14731. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Shalom R, Keeshen CM, Berrios KN, An JY, Sanders SJ, and Bender KJ (2017). Opposing effects on Na V 1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol. Psychiatr. 82, 224–232. 10.1016/j.biopsych.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng C-A, Ullah R, Farr J, Hill AP, Kozek KA, Vanags LR, Mitchell DW, Kroncke BM, and Vandenberg JI (2022). A massively parallel assay accurately discriminates between functionally normal and abnormal variants in a hotspot domain of KCNH2. Am. J. Hum. Genet. 109, 1208–1216. 10.1016/j.ajhg.2022.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozek KA, Glazer AM, Ng C-A, Blackwell D, Egly CL, Vanags LR, Blair M, Mitchell D, Matreyek KA, Fowler DM, et al. (2020). High-throughput discovery of trafficking-deficient variants in the cardiac potassium channel KV11.1. Heart Rhythm 17, 2180–2189. 10.1016/j.hrthm.2020.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coyote-Maestas W, Nedrud D, He Y, and Schmidt D (2022). Determinants of trafficking, conduction, and disease within a K+ channel revealed through multiparametric deep mutational scanning. Elife 11, e76903. 10.7554/eLife.76903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glazer AM, Wada Y, Li B, Muhammad A, Kalash OR, O’Neill MJ, Shields T, Hall L, Short L, Blair MA, et al. (2020). High-throughput reclassification of SCN5A variants. Am. J. Hum. Genet. 107, 111–123. 10.1016/j.ajhg.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanoye CG, Desai RR, Fabre KL, Gallagher SL, Potet F, DeKeyser J-M, Macaya D, Meiler J, Sanders CR, and George AL (2018). High-throughput functional evaluation of KCNQ1 decrypts variants of unknown significance. Circ. Genom. Precis. Med. 11, e002345. 10.1161/circgen.118.002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boettcher S, Miller PG, Sharma R, McConkey M, Leventhal M, Krivtsov AV, Giacomelli AO, Wong W, Kim J, Chao S, et al. (2019). A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604. 10.1126/science.aax3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, Howard TP, Takeda DY, Ly SH, Kim E, et al. (2018). Mutational processes shape the landscape of TP53 mutations in human cancer. Nat. Genet. 50, 1381–1387. 10.1038/s41588-018-0204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolognesi B, Faure AJ, Seuma M, Schmiedel JM, Tartaglia GG, and Lehner B (2019). The mutational landscape of a prion-like domain. Nat. Commun. 10, 4162. 10.1038/s41467-019-12101-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glazer AM, Kroncke BM, Matreyek KA, Yang T, Wada Y, Shields T, Salem J-E, Fowler DM, and Roden DM (2020). Deep mutational scan of an SCN5A voltage sensor. Circ. Genom. Precis. Med. 13, e002786. 10.1161/CIRCGEN.119.002786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komor AC, Kim YB, Packer MS, Zuris JA, and Liu DR (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, and Liu DR (2017). Programmable base editing of A$T to G$C in genomic DNA without DNA cleavage. Nature 551, 464–471. 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanna RE, Hegde M, Fagre CR, DeWeirdt PC, Sangree AK, Szegletes Z, Griffith A, Feeley MN, Sanson KR, Baidi Y, et al. (2021). Massively parallel assessment of human variants with base editor screens. Cell 184, 1064–1080.e20. 10.1016/j.cell.2021.01.012. [DOI] [PubMed] [Google Scholar]
- 26.Kim Y, Lee S, Cho S, Park J, Chae D, Park T, Minna JD, and Kim HH (2022). High-throughput functional evaluation of human cancer-associated mutations using base. Nat. Biotechnol. 40, 874–884. 10.1038/s41587-022-01276-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sánchez-Rivera FJ, Diaz BJ, Kastenhuber ER, Schmidt H, Katti A, Kennedy M, Tem V, Ho Y-J, Leibold J, Paffenholz SV, et al. (2022). Base editing sensor libraries for high-throughput engineering and functional analysis of cancer-associated single nucleotide variants. Nat. Biotechnol. 40, 862–873. 10.1038/s41587-021-01172-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiser T (2004). A novel toxicity-based assay for the identification of modulators of voltage-gated Na+ channels. J. Neurosci. Methods 137, 79–85. 10.1016/j.jneumeth.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 29.Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. 10.1093/nar/gkx1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macnee M, Pérez-Palma E, López-Rivera JA, Ivaniuk A, May P, Møller RS, and Lal D (2022). Data-driven historical characterization of epilepsy-associated genes. Eur. J. Paediatr. Neurol. 10.1016/j.ejpn.2022.12.005. [DOI] [PubMed] [Google Scholar]
- 31.Pérez-Palma E, Gramm M, Nürnberg P, May P, and Lal D (2019). Simple ClinVar: an interactive web server to explore and retrieve gene and disease variants aggregated in ClinVar database. Nucleic Acids Res. 47, W99–W105. 10.1093/nar/gkz411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cuella-Martin R, Hayward SB, Fan X, Chen X, Huang J-W, Taglialatela A, Leuzzi G, Zhao J, Rabadan R, Lu C, et al. (2021). Functional interrogation of DNA damage response variants with base editing screens. Cell 184, 1081–1097.e19. 10.1016/j.cell.2021.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He N, Lin Z-J, Wang J, Wei F, Meng H, Liu X-R, Chen Q, Su T, Shi Y-W, Yi Y-H, and Liao WP (2019). Evaluating the pathogenic potential of genes with de novo variants in epileptic encephalopathies. Genet. Med. 21, 17–27. 10.1038/s41436-018-0011-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richter MF, Zhao KT, Eton E, Lapinaite A, Newby GA, Thuronyi BW, Wilson C, Koblan LW, Zeng J, Bauer DE, et al. (2020). Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 38, 883–891. 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sangree AK, Griffith AL, Szegletes ZM, Roy P, DeWeirdt PC, Hegde M, McGee AV, Hanna RE, and Doench JG (2022). Benchmarking of SpCas9 variants enables deeper base editor screens of BRCA1 and BCL2. Nat. Commun. 13, 1318. 10.1038/s41467-022-28884-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeKeyser J-M, Thompson CH, and George AL Jr. (2021). Cryptic prokaryotic promoters explain instability of recombinant neuronal sodium channels in bacteria. J. Biol. Chem. 296, 100298. 10.1016/j.jbc.2021.100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, and Pinello L (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226. 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan JQ, Baez-Nieto D, Allen A, Wang H-R, and Cottrell JR (2018). Developing high-throughput assays to analyze and screen electrophysiological phenotypes. Methods Mol. Biol. 1787, 235–252. 10.1007/978-1-4939-7847-2_18. [DOI] [PubMed] [Google Scholar]
- 39.Baez-Nieto D, Allen A, Akers-Campbell S, Yang L, Budnik N, Pupo A, Shin Y-C, Genovese G, Liao M, Pérez-Palma E, et al. (2022). Analysing an allelic series of rare missense variants of CACNA1I in a Swedish schizophrenia cohort. Brain 145, 1839–1853. 10.1093/brain/awab443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Access to sequence validation data can be found at https://doi.org/10.5061/dryad.zkh1893fc. Access to original cell culture and electrophysiological data is available upon request.
Access to custom code is available in a GitHub repository. The DOI is 10.5281/zenodo.7901798.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.