

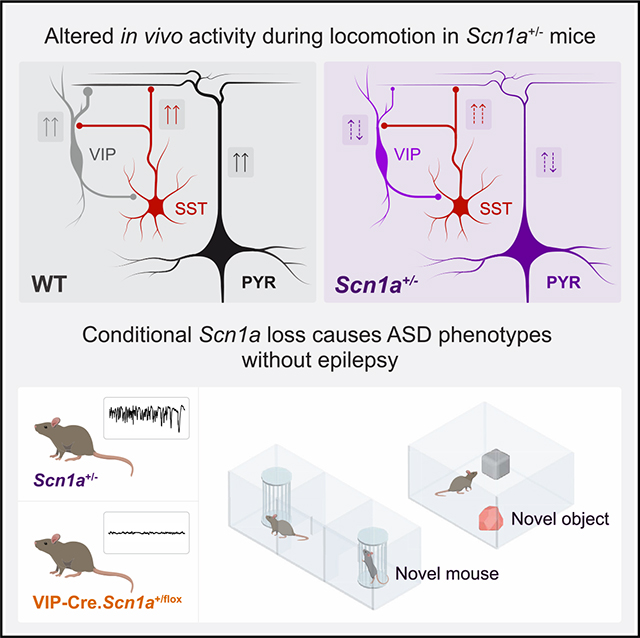
SUMMARY
Dravet syndrome (DS) is a severe neurodevelopmental disorder caused by loss-of-function variants in SCN1A, which encodes the voltage-gated sodium channel subunit Nav1.1. We recently showed that neocortical vasoactive intestinal peptide interneurons (VIP-INs) express Nav1.1 and are hypoexcitable in DS (Scn1a+/−) mice. Here, we investigate VIP-IN function at the circuit and behavioral level by performing in vivo 2-photon calcium imaging in awake wild-type (WT) and Scn1a+/− mice. VIP-IN and pyramidal neuron activation during behavioral transition from quiet wakefulness to active running is diminished in Scn1a+/− mice, and optogenetic activation of VIP-INs restores pyramidal neuron activity to WT levels during locomotion. VIP-IN selective Scn1a deletion reproduces core autism-spectrum-disorder-related behaviors in addition to cellular- and circuit-level deficits in VIP-IN function, but without epilepsy, sudden death, or avoidance behaviors seen in the global model. Hence, VIP-INs are impaired in vivo, which may underlie non-seizure cognitive and behavioral comorbidities in DS.
In brief
Goff et al. show that VIP-expressing neocortical GABAergic interneurons are dysfunctional during awake behavior in Dravet syndrome (Scn1a+/−) mice. Scn1a deletion in VIP-INs only yields behavioral abnormalities linked to autism spectrum disorder, without seizures or epilepsy. This work provides insight into the circuit basis of a severe neurodevelopmental disorder.
Graphical Abstract
INTRODUCTION
Cerebral cortex interneurons (INs) constitute a diversity of cell types defined by electrical activity, molecular profiles, anatomy, and patterns of connectivity, and are key components of the microcircuit functions that underlie behavior.1,2 IN dysfunction is also implicated in the pathogenesis of a range of neurological and psychiatric disorders including epilepsy, schizophrenia, and autism spectrum disorder (ASD).3,4 However, it remains unclear how subtype-specific IN dysfunction contributes to disorders defined by paroxysmal events (seizures) as well as durable impairments in cognitive function. Dravet syndrome (DS) is a neurodevelopmental disorder characterized by treatment-resistant epilepsy, prominent features of or formal diagnosis of ASD, cognitive impairment, and sudden unexplained death (SUDEP).5,6 DS is caused by heterozygous variants in SCN1A leading to loss of function of one copy of the voltage-gated sodium (Na+) channel α subunit Nav1.1,7,8 but a substantial gap remains between the clearly defined genetic cause of DS and a mechanistic understanding of how loss of Nav1.1 impacts brain circuits to yield this complex disorder.
At the cellular level, Nav1.1 is preferentially expressed in INs relative to excitatory cells, and data from Scn1a+/− mice demonstrates impaired IN spike generation.9,10 The three largest classes of cerebral cortex INs—those expressing parvalbumin (PV), somatostatin (SST), and vasoactive intestinal peptide (VIP-INs)—are hypoexcitable in acute brain slices prepared from young Scn1a+/− mice.10–14 Deletion of Scn1a across all INs (via Dlx1/2-Cre) largely recapitulates the global phenotype.15–18 As in human patients, pathology in Scn1a+/− mice exhibits a defined developmental trajectory such that both IN dysfunction and epilepsy/SUDEP appear at the onset of Nav1.1 expression (during the third postnatal week in mice). Subsequently, impairment of both PV-INs and SST-INs (but not VIP-INs) recovers at later developmental time points as seizure frequency decreases.10,11,19,20 However, Scn1a+/− mice exhibit long-lasting impairments shared with other models of ASD, suggesting a role of persistent VIP-IN dysfunction in the durable non-epilepsy-related impairments in cognition, sensory processing, and ASD-related behaviors characteristic of DS.
All cerebral cortex interneurons form inhibitory connections with other interneurons, and VIP-INs also inhibit pyramidal cells. However, it is the case that VIP-INs serve a prominent disinhibitory role linked to behavioral-state transitions and are regulators of stimulus gain control, synaptic plasticity, and learning.21–28 During transition to active behavioral states such as from quiet wakefulness to locomotion, VIP-INs are excited by ascending cholinergic and noradrenergic neuromodulation and in turn inhibit SST-INs, thereby contributing to disinhibition of local excitatory neurons.21,26,29–32 However, the effect of locomotion on neocortical activity is complex and influenced by more than VIP-INs, as many IN populations—including SST-INs—are strongly activated.33 Previous in vivo recording of PV and SST neurons in Scn1a+/− mice during the chronic phase of the disorder have not demonstrated the predicted reductions in activity level,13,34 and loss of Scn1a restricted to these cells only partially replicates the behavioral disease phenotype.18 Given a known disinhibitory role, we suspected that VIP-INs in fact may not contribute to the seizure phenotype of Scn1a+/− mice. As other studies have characterized a role for VIP-INs in gain modulation, plasticity, and arousal, we hypothesized that VIP-IN dysfunction in DS may underlie the non-seizure cognitive comorbidities attributable to the in vivo circuit functions of VIP-INs.
To understand how VIP-INs respond to endogenous cholinergic modulation, we first performed pharmacology experiments in acute brain slices and, surprisingly, found that application of cholinomimetics to VIP-INs in Scn1a+/− but not wild-type (WT) mice led to Na+ current downregulation that compounded existing intrinsic VIP-IN dysfunction secondary to heterozygous loss of Scn1a. Using in vivo 2-photon (2P) calcium imaging in awake mice, we found that patterns of activation of VIP-INs and pyramidal neurons associated with the onset of locomotion are attenuated in Scn1a+/− mice while SST-INs remain robustly activated. Augmenting VIP-IN recruitment using behavior-driven optogenetics led to an increase in local excitatory neuron activity to WT levels. VIP-IN specific Scn1a deletion (VIP-Cre.Scn1afl/+) replicated intrinsic physiological deficits and partially reproduced the circuit effect, particularly during pharmacologically boosted high cholinergic states. Furthermore, VIP-Cre.Scn1afl/+ mice recapitulated the core ASD endophenotypes of the global model, yet without hyperactivity, anxiety phenotypes, temperature-induced seizures, epilepsy, or SUDEP. Together, these data implicate VIP-IN dysfunction in a persistent circuit impairment linked to ASD-related disease features independent of the epilepsy phenotype of DS.
RESULTS
Cholinergic modulation exacerbates VIP-IN dysfunction in Scn1a+/− mice through Na+ current downregulation
Our previous work established that VIP-INs express Nav1.1 and are dysfunctional in Scn1a+/− mice in vitro.11 In vivo, VIP-IN activity is driven by ascending cholinergic and noradrenergic neuromodulation, which occurs during transition from quiet wakefulness to locomotion and is highly correlated with pupil dilation.29,35 In addition to direct depolarization of VIP-INs via nicotinic acetylcholine receptors, acetylcholine acts on muscarinic receptors to enhance VIP-IN excitability via downregulation of M-type potassium current.11 To link the in vitro and in vivo activity of VIP-INs in Scn1a+/− mice, we tested the response of VIP-INs to cholinergic modulation in acute brain slices. We hypothesized that VIP-INs in Scn1a+/− mice would exhibit decreased response to cholinergic modulation due to intrinsic hypoexcitability secondary to heterozygous loss of Scn1a.
Bath application of 5 μM carbachol both depolarized and modulated the firing pattern of WT VIP-INs, as we showed previously (Figure 1A). However, stimulating WT VIP-INs with slow depolarizing ramps before and after carbachol application also revealed an attenuation of the dynamic range of VIP-INs, which entered depolarization block at lower current injections (Figure 1B). VIP-INs from Scn1a+/− mice fired at lower frequencies before application of carbachol (42 ± 2.9 vs. 34 ± 2.5 Hz, WT vs. Scn1a+/−, p = 0.04) and cholinergic modulation caused a massive decrease in excitability to near silence. Many VIP-INs from Scn1a+/− mice made rapid transition to depolarization block, firing only 1–2 action potentials (APs) in response to slow depolarizing ramps (Figures 1B and 1E; carbachol AP index, WT −0.21 ± 0.06 vs. Scn1a+/− −0.63 ± 0.06, p = 0.00003). This was true for all Scn1a+/− VIP-INs regardless of their initial firing pattern11 (i.e., continuous adapting or irregular spiking; Figure S1), which indicates that VIP-IN dysfunction may be more profound in vivo (as cholinergic tone is low or absent in slice).
Figure 1. Effect of cholinergic modulation on VIP-IN excitability and Na+ current.
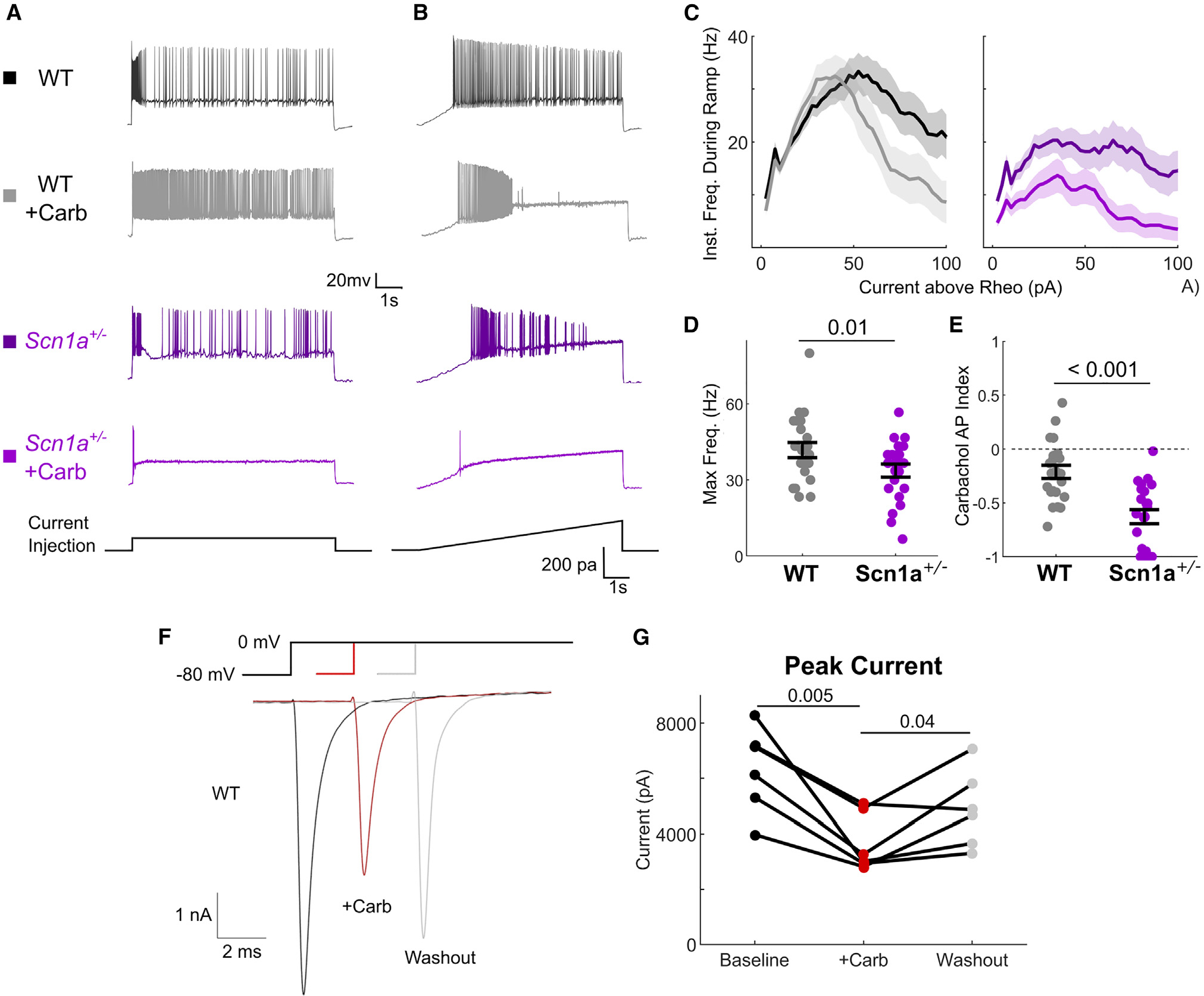
(A and B) Representative example current-clamp traces depicting responses of VIP-INs in acute brain slices prepared from either wild-type (WT) or Scn1a+/− mice. Cells were stimulated with either (A) a square 8-s-long pulse at 2× rheobase current or (B) a slow 8-s ramp current with a maximum of 200 pA at 25 pA/s. The same cells are then recorded again after bath application of 5 μM carbachol.
(C) Current-frequency relationships for WT and Scn1a+/− VIP-INs elicited from 8-s ramp depolarizations. Data were aligned to the rheobase current for each cell, and instantaneous firing frequency was calculated using a 300-ms sliding window. Solid lines and shaded areas represent mean ± SEM.
(D and E) (D) Maximum firing frequency for each WT and Scn1a+/− VIP-IN during the 8-s ramp at baseline and (E) the effect of carbachol on the total number of spikes generated during an 8-s ramp depolarization calculated as an index value.
(F) Inward Na+ currents recorded in voltage clamp from WT VIP-INs in acute slices from a single depolarizing step from −80 to 0 mV. Individual sweeps are shown for each condition (no averaging). Peak current was measured, repeated during bath application of 5 μM carbachol, and a full or partial washout was observed in most cells.
(G) Summary of data from the experiment in (F), where connected lines show individual cells (n = 6 VIP-INs from N = 5 WT mice).
Comparisons and p values were calculated using a paired Student’s t test. There was no difference between baseline and washout conditions. p values in (D) and (E) were determined by Mann-Whitney U test. Dots represent individual data points (WT, n = 22 cells; Scn1a+/−, n = 22 cells). All line and bar graphs show mean ± bootstrapped 95% confidence interval (CI) of the mean unless noted otherwise. See also Figure S1.
This paradoxical deleterious effect of carbachol on VIP-INs in Scn1a+/− mice is potentially explained by action of cholinergic neuromodulation via second messengers to downregulate Na+ current.36,37 To test this, we performed voltage-clamp recordings of VIP-INs and confirmed that bath application of carbachol led to a reversible decrease in Na+ current (Figures 1F and 1G). This reveals a mechanism whereby VIP-IN hypofunction in Scn1a+/− mice may be more dramatic during periods of high cholinergic tone in vivo.
Dravet syndrome mice exhibit impaired activation of VIP-INs and local pyramidal neurons in V1 during active behavior states
Based on VIP-IN dysfunction in Scn1a+/− mice in vitro, we hypothesized that VIP-INs may be impaired in Scn1a+/− mice in vivo. Furthermore, these circuit abnormalities may be most prominent during periods of increased arousal, given the paradoxical response to cholinergic modulation of VIP-INs in Scn1a+/− mice. Behavioral transition from quiet wakefulness to locomotion in mice is correlated with recruitment of both VIP-IN activity and pyramidal neurons in primary visual cortex (V1) as well as with fluctuations in pupil diameter.29,35 Using 2P calcium imaging of GCaMP7s expressed in tdTomato+ (tdT+) VIP-INs and tdT− non-VIP cells (mainly putative pyramidal neurons) in V1, we recorded activity in awake head-fixed WT and Scn1a+/− mice that were free to run or rest and compared activity across behavioral epochs (Figure 2A).
Figure 2. Impaired patterns of behaviorally linked neocortical activity in Scn1a+/− mice.
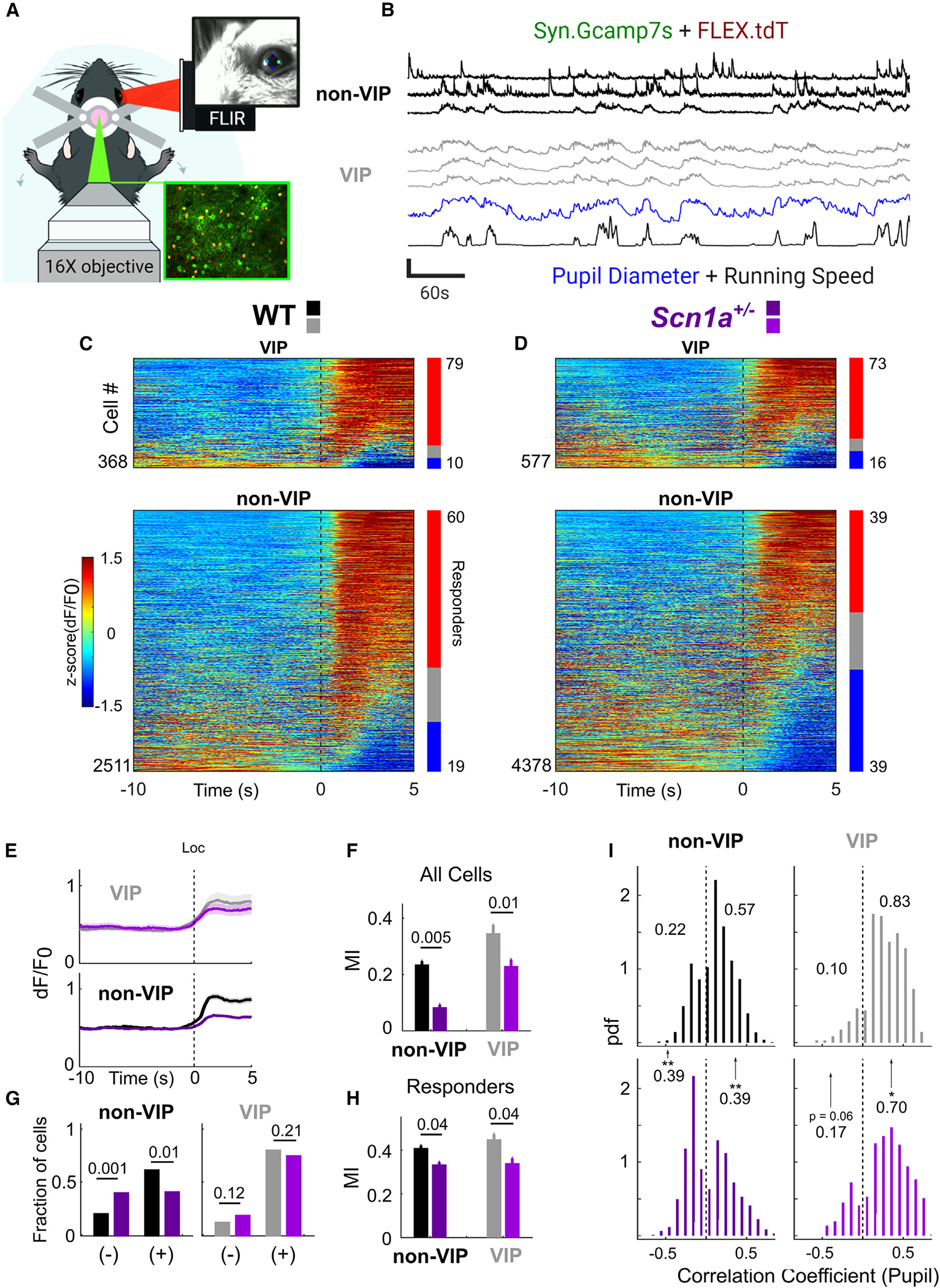
(A) Experimental design. Adult WT and Scn1a+/− mice implanted with a cranial window over V1 were head-fixed and allowed to rest or run freely. In vivo 2P imaging was synchronized to locomotion and video of the mouse’s pupil and whisker (extracted post hoc using DeepLabCut).
(B) GCaMP7s calcium transients, calculated as dF/F0, of non-VIP neurons (tdT−) and VIP-INs (tdT+), were extracted from each field of view (FOV) and aligned to behavioral data. Traces of three individual VIP-INs and non-VIP neurons (the top two of which are presumptive pyramidal neurons; Figure S3), are shown, along with pupil diameter and running speed. All traces are Z-scored for clarity (vertical scale bar represents Z score of 4).
(C) Heatmap showing activity for each cell at the onset of >5 s locomotion bouts, averaged from multiple running bouts and Z-scored within each cell. Bars indicate percentage activated (red) or inhibited (blue) at onset of locomotion. Black dotted line represents onset of locomotion.
(D) As in (C), but for Scn1a+/− mice. Black dotted line represents onset of locomotion.
(E) Average dF/F0 at onset of locomotion across all VIP-INs and non-VIP neurons. Lines and shaded area represent mean ± 95% CI.
(F) Modulation of dF/F0 during locomotion measured as an index value (modulation index [MI]; see STAR Methods).
(G) Fraction of cells activated (+) or inhibited (−) at onset of locomotion as in (C) and(D) by genotype. Responders were cells that showed significant positive (+) or negative (−) correlation to locomotion vs. shuffled behavioral data. The numbers of positively and negatively correlated neurons were compared across genotype.
(H) MI as in (F) but for those cells activated during locomotion (responders).
(I) Histograms of the time-zero correlation coefficient between the activity of individual cells vs. pupil diameter, normalized as a probability density (where the size of each bar is equal to the probability of a cell populating that bin divided by bin width). Shown are cells with significant positive or negative correlation (as in G), and the proportion of cells with positive or negative correlation are indicated. Significant differences across genotype are indicated. *p < 0.05; **p < 0.01 (see STAR Methods).
For all experiments, n = 2,511 tdT− and 368 VIP-INs from N = 5 WT mice (3 male, 2 female); n = 4,378 tdT− and 577 VIP-INs from N = 6 Scn1a+/− mice (2 male, 4 female). See also Figures S2–S7.
The increase of VIP-IN and pyramidal neuron activity during locomotion reported by GCaMP7s fluorescence was quantified as a locomotion modulation index (MI) (STAR Methods),33 which was reduced in Scn1a+/− mice compared with age-matched WT littermates (Figures 2C–2F; VIP-INs −0.11 ± 0.04, p = 0.01; non-VIP neurons −0.16 ± 0.05, p = 0.005). There was reduced locomotion MI in Scn1a+/− mice when restricting analysis to only responding cells (Figures 2G and 2H; −0.09 ± 0.04, p = 0.04), and a trend toward fewer responding VIP-INs in Scn1a+/− mice relative to WT during locomotion. Additionally, a smaller percentage of pyramidal neurons were responders (−0.23 ± 0.08, p = 0.009), and more pyramidal neurons were negatively responsive to locomotion (+0.20 ± 0.06, p = 0.001). There was also a lower locomotion MI in responding pyramidal neurons (−0.082 ± 0.04, p = 0.04). Similarly, fewer VIP-IN activity profiles were positively correlated with pupil diameter in Scn1a+/− mice relative to WT (−0.15 ± 0.07, p = 0.04). Fewer pyramidal neurons exhibited an activity profile that was positively correlated with pupil diameter (−0.19 ± 0.07, p = 0.01), with more pyramidal neurons showing anticorrelation (Figure 2I; +0.16 ± 0.06, p = 0.008). As an additional readout of activity, we found both reduced locomotion MI of the background neuropil signal in Scn1a+/− mice relative to WT and reduced correlation of the neuropil signal with pupil diameter (Figure S2).
Baseline activity levels during quiet rest measured by deconvolved event frequency were similar between genotypes (Figure S3), indicating that VIP-IN dysfunction in Scn1a+/− mice is state dependent. As we reported previously,34 WT and Scn1a+/− mice had a similar number of running bouts, ran at similar speed, and had identical pupillomotor activity associated with locomotion in this experimental setup (Figure S4). Therefore, impairments in Scn1a+/− mice were not an artifact of genotype differences in behavior or locomotion. Finally, there was decreased correlation between the fluorescence signals of VIP-INs and non-VIP neurons relative to WT mice during rest (Figure S5), which may indicate subtle impairment of VIP-IN-mediated circuit function that was not reflected in overall changes in activity levels at rest.
One important caveat of our experimental setup is that while tdT− non-VIP neurons predominantly represent excitatory neurons in layer 2/3, this may also include other non-VIP-IN populations including SST-INs, which may be differentially affected relative to pyramidal neurons by both loss of Scn1a and impairment of VIP-IN excitability. To validate that our results indeed reflected the activity of excitatory pyramidal neurons, we recorded from pyramidal cell-specific Slc17a7-Cre.WT mice injected with Cre-dependent GCaMP7s under otherwise identical conditions, thereby limiting GCaMP7s expression to a genetically identified excitatory neuron population. We found that unlabeled WT neurons from VIP-Cre.WT mice had levels of modulation by locomotion similar to those of Slc17a7+ neurons. We additionally paired this with analysis of skew in calcium traces, which can reliably be used to define pyramidal neurons without genetic identification (Figure S5).33 An optimal receiver-operating characteristic separation between pyramidal cells and INs (SST and PV) at a skew value of 0.9 captured 90% of pyramidal neurons. We nevertheless took a strict skew cutoff of 2.35, which weighted inclusion of INs 2.5-fold and captured 36% of pyramidal neurons while excluding 93% of INs. When limiting analysis to high-skew putative pyramidal neurons, we found that differences in WT and Scn1a+/− mice were preserved, indicating that our results from tdT− non-VIP neurons accurately reflect changes in the pyramidal neuron population (Figure S5).
While VIP-INs form direct synaptic connections with pyramidal neurons, a disinhibitory action in the cortex has been attributed to VIP-IN influence on surrounding IN populations including SST-INs. We recorded from SST-Cre WT and Scn1a+/− mice expressing GCaMP7s in SST-INs to characterize the effect of loss of Scn1a on SST-IN activity (Figure S6). SST-INs from these mice showed a reduced locomotion MI compared with WT mice that was present only when analyzing responding cells. However, unlike VIP-INs and pyramidal neurons, SST-INs in WT and Scn1a+/− mice were robustly activated, consistent with prior results in WT mice,33,38 with no significant changes in overall locomotion MI, percentage of responding cells, or correlation with pupil diameter.
Optogenetic VIP-IN stimulation normalizes neocortical dynamics in Scn1a+/− mice
Given the above results, we tested whether enhancing VIP-IN recruitment during locomotion could increase the activity of local pyramidal neurons in Scn1a+/− mice by performing 2P imaging with simultaneous optogenetic activation of VIP-INs using selective expression of a red shifted opsin.39 Photostimulation reliably activated VIP-INs at rest, resulting in a broad increase in the GCaMP7s signal of non-VIP cells (Figures 3A–3C). A small population of non-VIP cells showed a strong decrease in activity in response to VIP-IN stimulation; this population of cells had high baseline activity with low-skew calcium traces and were considered as putative INs (p-INs) that are directly inhibited by VIP-INs (Figure S7), although a subset could be excitatory pyramidal neurons directly inhibited by VIP-INs. Optogenetic photoactivation of VIP-INs during locomotion in Scn1a+/− mice restored overall activity of all non-VIP neurons to WT levels (Figures 3D–3F; MI +0.07 ± 0.006, p = 0.001), while activity of p-INs was dramatically reduced (−0.23 ± 0.04, p = 0.004). Therefore, direct activation of VIP-INs was sufficient to normalize the modulation of non-VIP neurons in Scn1a+/− mice during locomotion to WT levels.
Figure 3. Optogenetic activation of VIP-INs normalizes neocortical dynamics associated with the transition to locomotion in Scn1a+/− mice.
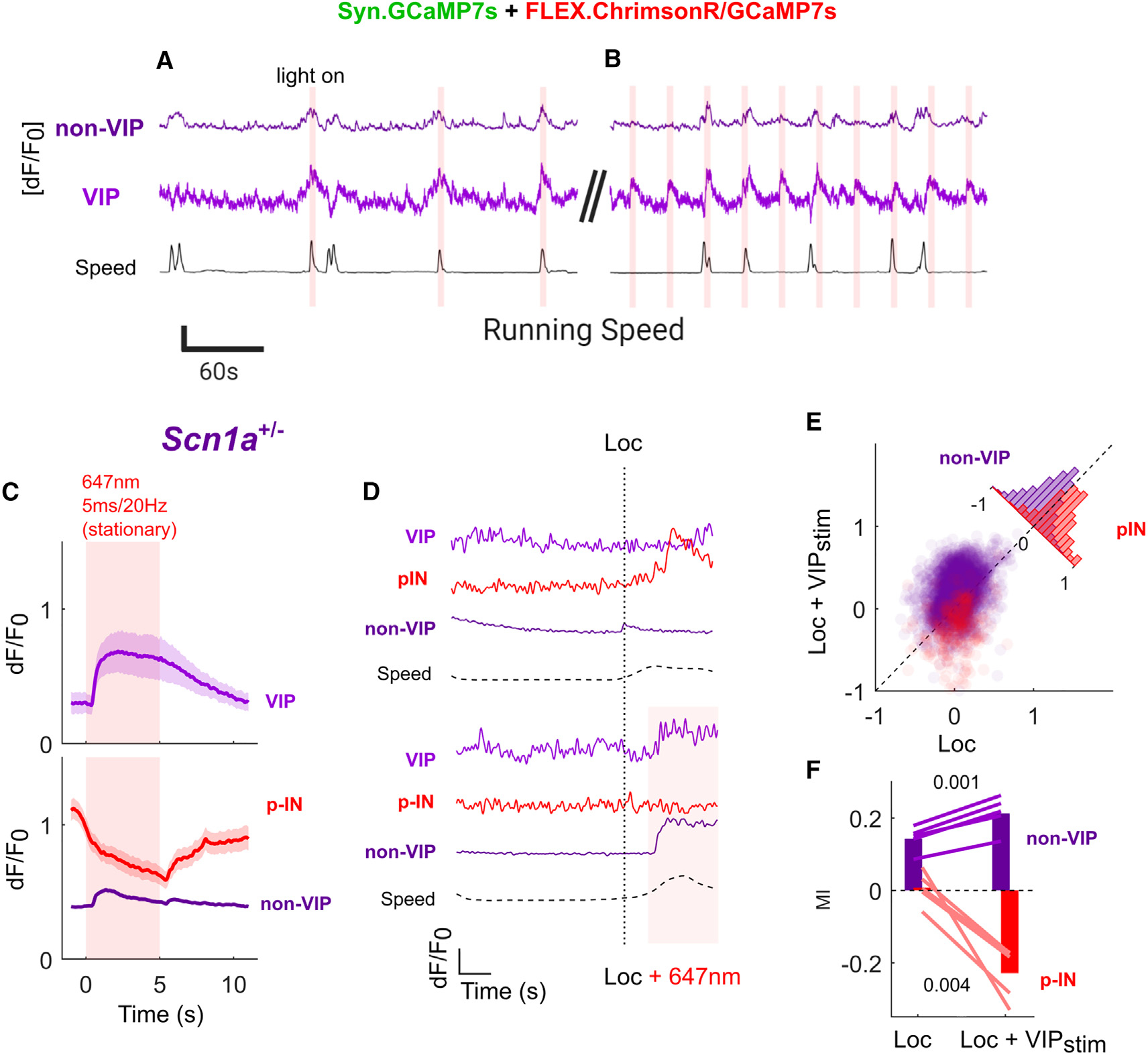
(A and B) Example trial of simultaneous imaging:optogenetics in Scn1a+/− mice in vivo. VIP-INs expressing ChrimsonR were stimulated for 5 s with 20-Hz/5-ms red-light pulses either (A) triggered by onset of locomotion with a 60-s time-out period between stimulations or (B) every 30 s. This ensured that stimulation occurred during both locomotion and quiet resting periods for each mouse. Neural data are an average of all cells (VIP or non-VIP) within a single FOV − [dF/F0]. Scale bars: y axis, 0.5 dF/F0 or 100 mm/s; x axis, 60 s.
(C) Average VIP-IN and non-VIP dF/F0 elicited by 5-s laser stimulation during stationary epochs. VIP-INs are uniformly activated, while non-VIP neurons show a heterogeneous response (Figure S7) with an average disinhibitory response across most cells and strong inhibition of approximately ~10% of non-VIP cells, which are likely the population of INs directly targeted by VIP-INs (p-INs) based on low-skew traces of dF/F0. Lines and shaded area represent mean ± 95% CI. (D) Example dF/F0 traces from a single VIP-IN, p-IN, and putative pyramidal (non-VIP) neuron aligned to an individual locomotion bout with and without laser stimulation. Horizontal dotted lines represent running speed (Speed), the vertical dotted line is detected locomotion onset (Loc), and the solid line and shaded area represent laser stimulation (+647). Scale bars: y axis, 1 dF/F0 or 100 mm/s; x axis, 2 s.
(E) Locomotion MI was calculated for each non-VIP neuron separately for epochs either with optogenetic VIP-IN activation (Loc + VIPstim) or without (Loc) compared with quiet wakefulness with no VIP-IN activation. Seventy-six percent of non-VIP neurons show a greater locomotion MI with concurrent VIP-IN activation, while 78% of p-INs have a lower (more negative) MI (inset histogram).
(F) Average locomotion MI for all non-VIP neurons and p-IN with or without VIP-IN stimulation. Lines represent the average response of all cells in individualScn1a+/− mice, and bars represent the grand average across mice.
n = 3,246 non-VIP and 69 VIP cells from N = 5 Scn1a+/− mice (2 male, 3 female). Within-group comparisons were made with a paired Student’s t test using the average of each mouse. See also Figure S8.
Conditional deletion of Scn1a in VIP-INs recapitulates physiology and circuit deficits
DS is a multifaceted disease with a complex circuit basis and many cell types affected by loss of Scn1a. To ask how VIP-Ins specifically contribute to the overall disease phenotype of DS, we generated VIP-Cre.Scn1a+/fl mice in which heterozygous deletion of Scn1a was restricted to VIP-INs. We confirmed the intrinsic electrophysiological deficit of VIP-INs to be present in VIP-Cre.Scn1a+/fl mice including deficits in repetitive firing and AP generation, which closely resemble VIP-IN dysfunction in Scn1a+/− mice (Figures 4A–4C and S8). We hypothesized that in vivo activity of VIP-INs would be similarly disrupted in VIP-Cre.Scn1a+/fl mice. When we performed the same 2P imaging experiment as in the global model, we unexpectedly found no genotype difference in the locomotion MI of VIP-IN or pyramidal neurons (Figures 4D–4I). However, we found that the deconvolved event frequencies of non-VIP cells were slightly decreased during quiet wakefulness in VIP-Cre.Scn1a+/fl mice (Figure 4J; arbitrary units, −0.03 ± 0.003; p = 0.04), suggesting decreased overall excitability of V1 in VIP-Cre.Scn1a+/fl mice in vivo.
Figure 4. Loss of Scn1a in VIP-INs reproduces in vitro physiology with no overt circuit deficit.
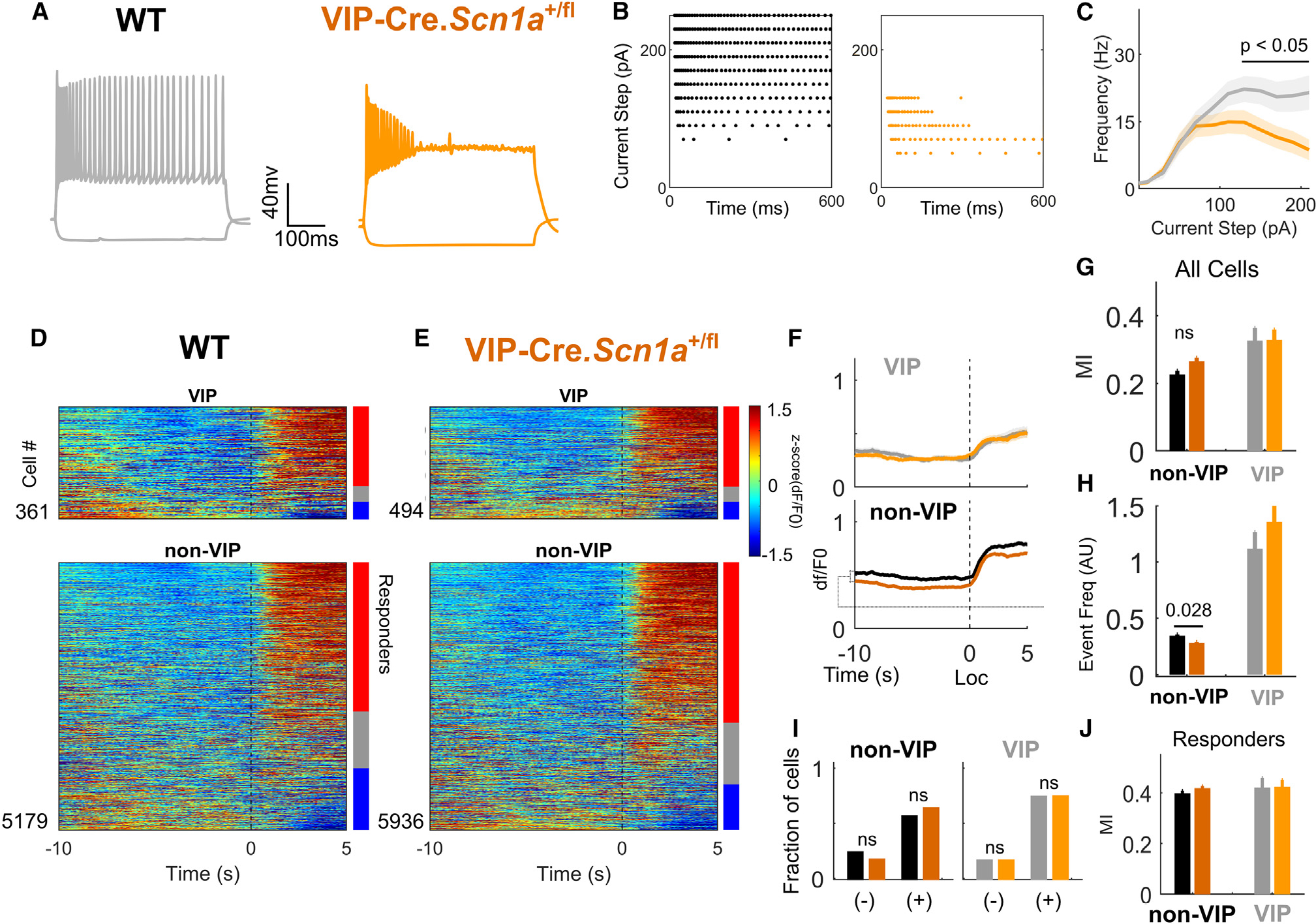
(A) Representative traces of the voltage response of an individual VIP-IN to a 600-ms square depolarizing current at 2× rheobase. VIP-INs from Scn1a+/fl mice show action potential failure and depolarization block.
(B) Raster plots of the action potentials fired by the cells in (A) during the full series of 600-ms current injections from 0 to 250 pA. Each dot represents an action potential. The Scn1a+/fl cell enters depolarization block starting at 100 pA, while the WT VIP-IN continues firing past 250 pA.
(C) Current-frequency relationship for all cells recorded as in (A) and (B). n = 30 VIP-INs from four VIP-Cre.Scn1a+/fl mice, and n = 27 VIP-INs from four WT mice. See Figure S7 for additional parameters of repetitive firing and single action potential generation.
(D) Heatmap showing activity for each cell at the onset of locomotion bouts, as in Figure 2C. Bars show percentage of activated (red, responders) and inhibited (blue) cells.
(E) Same as (D), for Scn1a+/fl mice.
(F) Average dF/F0 for all VIP-INs (light shades) and non-VIP neurons (dark shades) aligned to locomotion onset (Loc) in WT (grayscale) and Scn1a+/fl mice (orange) (as in Figure 2E). Lines and shaded area represent mean ± 95% CI.
(G) Locomotion MI as in Figure 2F.
(H) Deconvolved event frequency during stationary epochs (as in Figure S3C).
(I) Fraction of cells either activated (+) or inhibited (−) at the onset of locomotion by genotype. Responders were determined compared with shuffled data as in Figure 2G and STAR Methods.
(J) MI of cells activated during locomotion (responders), as in Figure 2H.
For (D) to (J), n = 5,179 tdT− and 361 VIP-INs from N = 6 WT (4 male, 2 female) mice; n = 5,936 tdT− and 494 VIP-INs from N = 7 Scn1a+/fl mice (3 male, 4 female). ns, not significant. See also Figure S9.
VIP-INs constitute one component of the complex changes in neural circuit dynamics that occur at the onset of locomotion. As VIP-IN impairment in Scn1a+/− mice was exacerbated by cholinomimetics (Figure 1), we considered that the circuit impact of VIP-IN dysfunction might be amplified in the context of increased cholinergic tone in vivo. We tested this by injecting a low dose of the muscarinic agonist pilocarpine (10 mg/kg; typical convulsant doses are 300–400 mg/kg) and measured activity of individual cells within the same imaging field during both resting and locomotion epochs before and after pilocarpine injection (Figure 5A).
Figure 5. Circuit deficits from VIP-Cre.Scn1afl/+ mice closely resemble the global model following pharmacologically enhanced muscarinic activation.
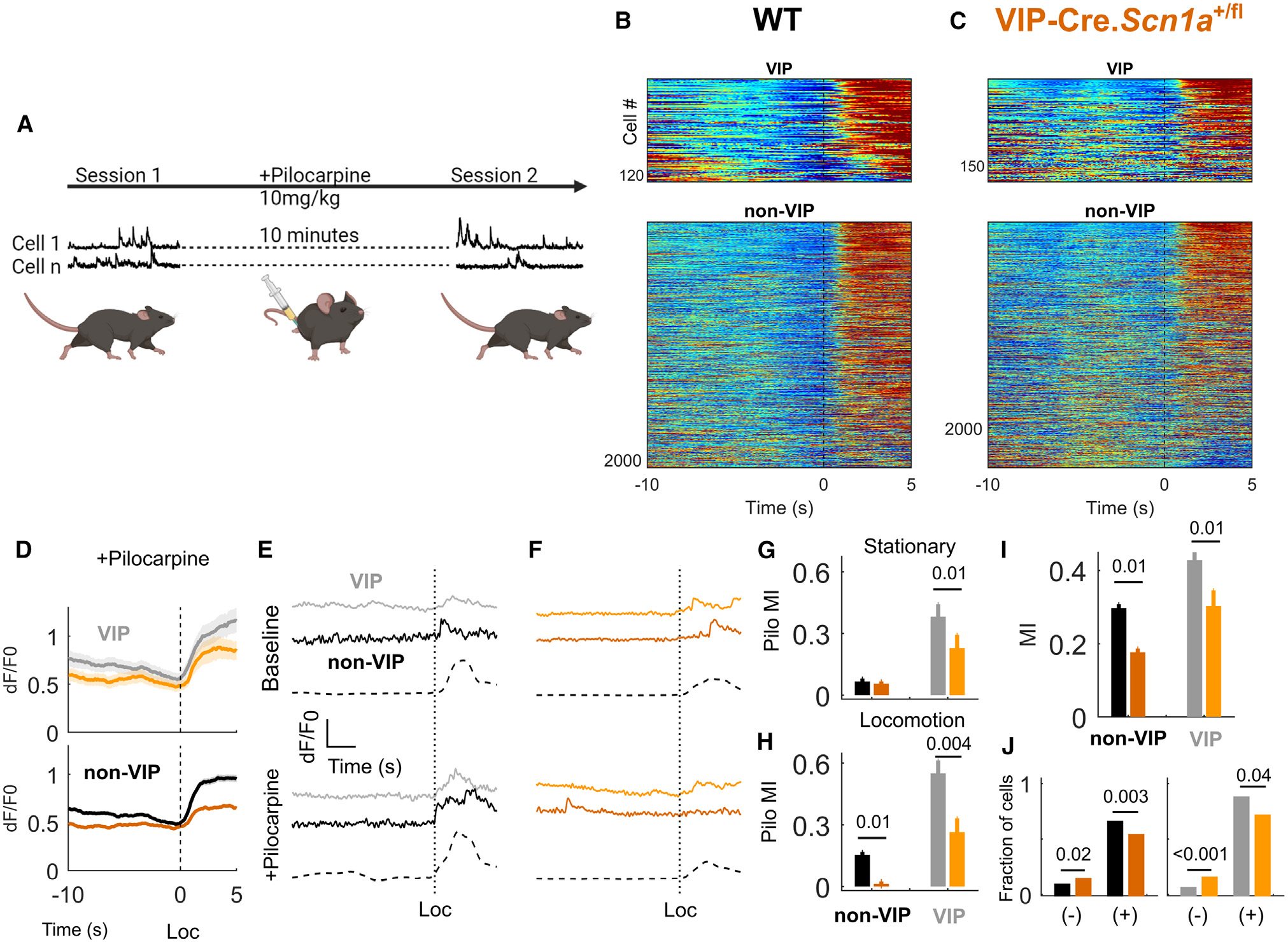
(A) Diagram of experimental design for (B) to (H). After a single FOV was recorded as above, the mouse was injected with low subconvulsive 10 mg/kg pilocarpine, and the same FOV was recorded 10 min later.
(B and C) Heatmap showing activity for each cell at the onset of locomotion bouts, as in Figures 4D and 4E, following the application of pilocarpine.
(D) Averaged dF/F0 response, as in (C), aligned to locomotion bouts, after the injection pilocarpine.
(E) Example of the dF/F0 trace from a single VIP and non-VIP neuron aligned to a single locomotion bout onset, with the same cells shown below after intraperitoneal injection of pilocarpine.
(F) Same as (E) but for example cells from a VIP-Cre.Scn1a+/fl mouse.
(G) The effect of pilocarpine on deconvolved event frequencies during stationary epochs calculated as an index value.
(H) The effect of pilocarpine on deconvolved event frequencies during locomotion epochs calculated as an index value.
(I) Locomotion MI for VIP and non-VIP cells after injection of pilocarpine.
(J) Fraction of cells either activated (+) or inhibited (−) during locomotion after the injection of pilocarpine, compared across genotypes as above.
For (B) to (H) and (I), n = 2,068 tdT− and 119 VIP-INs from N = 6 WT (4 male, 2 female) mice; n = 2,384 tdT− and 178 VIP-INs from N = 7 Scn1a+/fl mice (3 male, 4 female).
Pilocarpine caused a rise in the deconvolved event rate recorded in VIP-INs. However, this effect was much smaller in VIP-Cre.Scn1a+/fl mice compared with WT littermates at rest (Figure 5G; pilocarpine index −0.1 ± 0.06, p = 0.01) and during locomotion (Figure 5H; −0.3± 0.1, p = 0.004). There was a similar increase in non-VIP-IN activity that was again reduced in VIP-Cre.Scn1a+/fl mice during locomotion epochs (−0.15 ± 0.06, p = 0.01). The presence of pilocarpine recapitulated the genotype differences in locomotion MI of VIP and non-VIP neurons seen in the global model (Figures 5B, 5C, 5I, and 5J). Our data indicate that VIP-IN dysfunction in DS mice is partially responsible for decreased neocortical excitatory neuron activity during active behaviors, and manipulation of VIP-IN activity—as in our behaviorally driven optogenetic experiment—is sufficient to normalize neocortical excitatory neuron activity in the global Scn1a+/− mice to WT levels.
Selective loss of Scn1a in VIP-INs yields features of ASD without seizures
Finally, dysfunction of VIP-INs in DS in vivo could give rise to pathological behaviors linked to circuit dysfunction. VIP-INs have been shown to influence spatial memory, sensory processing, and sociability. Dysregulation of these behaviors is common across models of ASD, including in Scn1a+/− mice. We found that none of 60 VIP-Cre.Scn1a+/fl mice had seizures, and none had SUDEP prior to postnatal day 150; none of eight mice tested had temperature-induced seizures. Compared with age- and sex-matched WT littermates, VIP-Cre.Scn1a+/fl mice interacted less with a novel mouse in a 3-chamber task (Figure 6A; social index −0.14 ± 0.05, p = 0.009), buried more marbles in a marble burying assay (Figure 6B; +16% ± 5%. p = 0.005), and showed no object preference in a novel object recognition (NOR) task (Figure 6C; preference index −0.16 ± 0.06, p = 0.01). These results recapitulated the task-specific deficits observed in global VIP-Cre.Scn1a+/− mice (Figures 6A–6C). However, VIP-Cre.Scn1a+/fl mice and VIP-Cre.Scn1a+/− mice showed divergent phenotypes in the open-field test: VIP-Cre.Scn1a+/fl mice showed no evidence of hyperactivity compared with WT littermates whereas VIP-Cre.Scn1a+/− mice ran further and faster, and spent less time in the center of the open field compared with both WT and VIP-Cre.Scn1a+/fl mice (Figure 6D). As an independent test of anxiety-related phenotypes, VIP-Cre.Scn1a+/fl mice performed no differently from WT mice in the elevated plus maze (Figure S8). In summary, VIP-Cre.Scn1a+/fl mice show several consistent abnormal behaviors that are conserved across models of ASD.17,40,41 However, VIP-Cre.Scn1a+/fl mice differ in important ways from the global DS model, as they do not exhibit hyperactivity, temperature-sensitive seizures, chronic epilepsy, or SUDEP characteristic of Scn1a+/− mice and attributed to dysfunction of PV- and SST-INs,18 thereby dissociating the epilepsy-related and non-epilepsy behavioral phenotypes of this disorder.
Figure 6. Loss of Scn1a in VIP-INs replicates core cognitive impairments and ASD endophenotypes of Scn1a+/− mice.
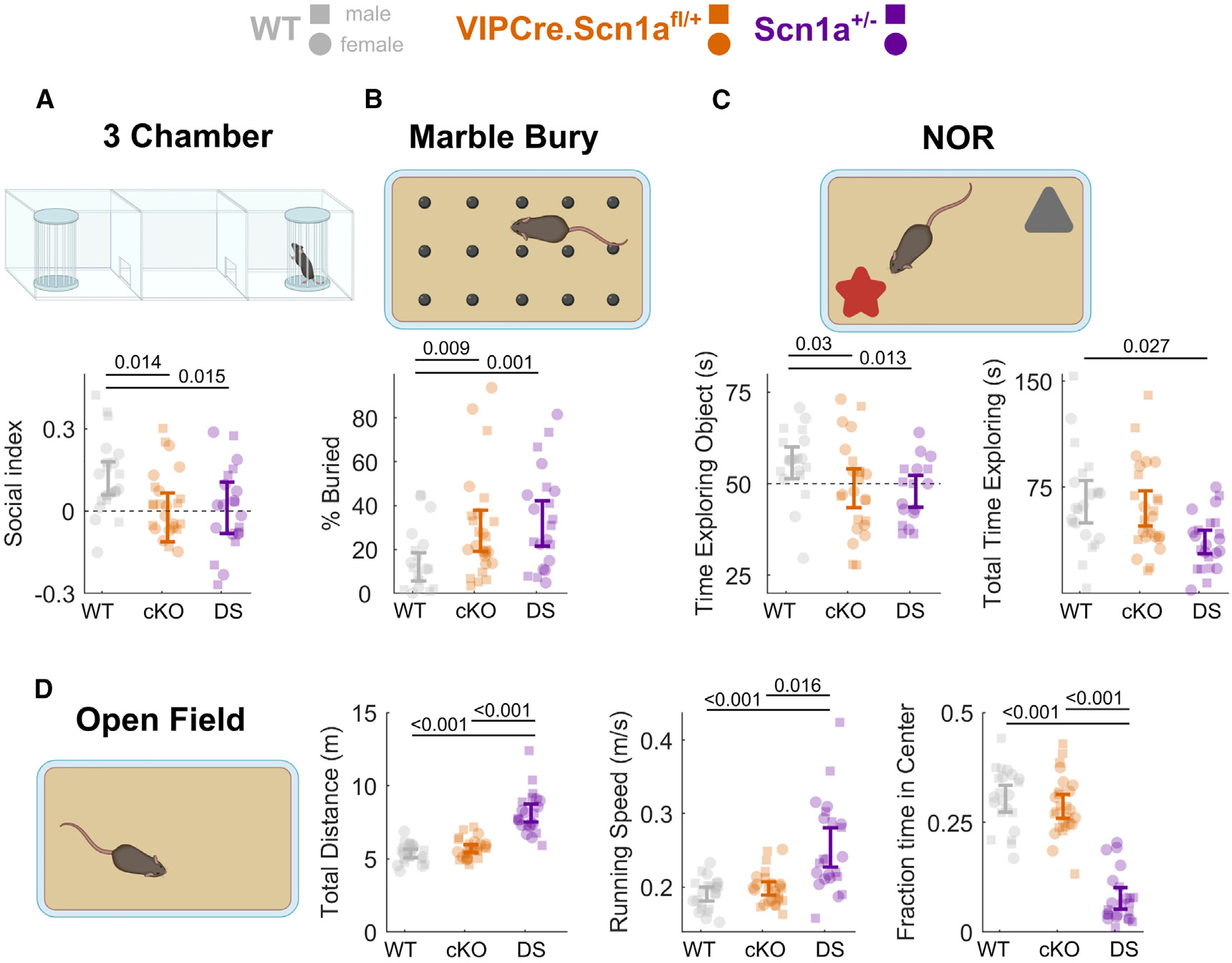
(A) Social interaction index for time spent with a novel conspecific vs. an empty enclosure in the opposite chamber (higher values indicate more time spent with the novel mouse, and index of 0 [dotted line] indicates equal time with each). Scn1afl/+ (cKO) and Scn1a+/− (DS). This result remained when instead comparing the raw time spent interacting with the novel mouse between experimental groups (Figure S10).
(B) Percentage of marbles buried during a 30-min period.
(C) Left: percent time exploring a novel vs. familiar object during the 20-s evaluation period. Right: total time spent exploring both objects during the 5-min test period.
(D) Left: total distance traveled during a 15-min open-field period. Center: maximum running speed (90th percentile of running speed during the open-field period). Right: percent time spent in the center, more than 4 cm from any edge of the arena.
Data from male (squares) and female (circles) mice are plotted separately, with error bars representing the bootstrapped 95% CI of the mean; p values represent comparisons between genotypes using a mixed-effects model with sex as a random effect. N = 26 (13 male, 13 female) VIPCre.Scn1afl/+, N = 23 (11 male, 12 female) VIPCre.Scn1a+/−, and N = 22 (12 male, 10 female) WT mice. p values indicate differences between genotype determined by linear mixed-effects modeling considering sex as a random effect. Sex had no statistically significant impact on performance of any test (STAR Methods). Diagrams created using BioRender. See also Figures S10 and S11.
DISCUSSION
Here, we assessed the activity of VIP-IN impairment in vivo in a model of DS, a prominent neurodevelopmental disorder characterized by epilepsy as well as durable and untreatable non-seizure comorbidities including ASD and intellectual disability. Our main questions concerned (1) how impaired VIP-IN cellular excitability in Scn1a+/− mice alters their activity and circuit function in vivo, and (2) how VIP-IN dysfunction is related to abnormal non-seizure/epilepsy ASD-linked behaviors characteristic of DS. We found that loss of Scn1a drives profound circuit-level defects in neocortex that can be restored by enhancing VIP-IN activity at behavioral-state transitions. These results are partially replicated by selective deletion in VIP-INs, and VIP-IN-specific Scn1a deletion dissociates features of ASD from the characteristic epilepsy phenotype of DS. This also indicates that the presence of seizures and SUDEP in early life is not required for developmental impairment in this model of DS.
2P imaging reveals impaired activation of VIP-INs and excitatory neurons during locomotion in Scn1a+/− mice
We used an established 2P calcium imaging paradigm to simultaneously assess the activity of VIP-INs and pyramidal neurons during behavioral-state transition in vivo. We found that activity during transition to active states was markedly reduced in Scn1a+/− mice. Using pupillometry as a correlate of neuromodulatory input,35 we found the correlation structure of both VIP-INs and local pyramidal neurons with pupil diameter to be disrupted in Scn1a+/− mice, with fewer neurons displaying activity that was positively correlated with pupil diameter as well as decreased correlation between VIP-IN and pyramidal neuron activity. In acute brain slices, other IN types (i.e., not VIP-INs) regain excitability in Scn1a+/− by young adulthood.10,11,19,20 Interestingly, SST-INs had a lower amplitude of response during locomotion in responding cells but as a population had a preserved proportion of cells activated during behavior. Overall, SST-INs showed no differences in locomotion MI and, in contrast to VIP-INs and pyramidal neurons, trended toward having a higher fraction of cells positively responding to locomotion. One way to interpret these results is that SST-INs remain hypoexcitable in V1 of adult Scn1a+/− mice; however, their overall activity is not reduced relative to WT because of decreased VIP-IN activity in Scn1a+/− mice. If VIP-INs are under-recruited during active behaviors relative to other inhibitory INs in Scn1a+/− mice, this would result in relatively higher levels of inhibitory neuron activity and lower levels of excitatory neuron activity. This idea is supported by our finding that optogenetic stimulation of VIP-INs during behavior restores normal activity levels in excitatory neurons, at least in part by inhibiting other INs. While VIP-IN impairment likely also affects neocortical development,42 our data suggest that boosting VIP-IN activity could be an avenue toward addressing reversible circuit deficits.
Limitations of the study: Loss of Scn1a specifically in VIP-INs partially recapitulates Scn1a+/− circuit deficits
While the conditional deletion of Scn1a in VIP-INs replicated the intrinsic electrophysiological phenotype found in VIP-INs in Scn1a+/− mice, it partially reproduced the altered in vivo activity and neocortical dynamics of the global model. We did not observe deficits in recruitment at onset of locomotion, yet baseline pyramidal neuron activity was reduced in VIP-Cre.Scn1a+/fl mice (Figures 5C and 5D). However, VIP-IN activity was similar between WT and VIP-Cre.Scn1a+/fl mice. One explanation for this apparent disconnect is that circuit deficits in both Scn1a+/− and VIP-Cre.Scn1a+/fl mice may be influenced by impaired VIP-IN spike propagation and/or synaptic release, which are not reflected by somatic calcium transients.19,43 Nevertheless, the activity of VIP-INs in Scn1a+/− mice was exquisitely sensitive to increased cholinergic tone in vivo. Low (subconvulsive) doses of pilocarpine boosted the activity of VIP-INs in WT mice in vivo yet had a markedly reduced effect in VIP-Cre.Scn1a+/fl mice, resulting in markedly decreased VIP-IN and excitatory neuron activity relative to WT. Using pharmacology in the acute brain slice preparation, we found that VIP-IN physiology is paradoxically impaired in Scn1a+/− mice by increased cholinergic tone, likely due to a differential sensitivity to the downregulation of Na+ current induced by cholinergic muscarinic modulation, reported previously in other cell types (Figure 1).36,37 This finding may explain how low subconvulsant doses of the cholinergic muscarinic agonist pilocarpine reproduced circuit deficits in VIP-Cre.Scn1a+/fl mice in vivo. The divergence between Scn1a+/− and VIP-Cre.Scn1a+/fl mice raises the interesting possibility of a differential or impaired cholinergic outflow from subcortical structures in DS that was not explored here.
The overall effect of locomotion on activity in sensory neocortex is complex and involves increased activity of both VIP-IN and SST-IN populations,33,38 which we replicate here and study in the context of a neurodevelopmental disorder. Furthermore, VIP-INs have many well-conserved features including participation in disinhibitory circuit motifs, but are a diverse cell population and provide direct inhibition of pyramidal cells as well as co-release of acetylcholine.25–27,44–47 Similarly, we previously found that a subpopulation of ~50% of VIP-INs which are irregular spiking are the most impaired by loss of Scn1a+/− in vitro. While there are some genetic strategies for targeting VIP-IN subsets, they do not correspond neatly to those VIP-INs that are dysfunctional in Scn1a+/− mice.11 In the future, a comprehensive understanding of VIP-IN diversity and improved tools for targeting subsets of VIP-INs could allow us to link hypoexcitability of VIP-INs in Scn1a+/− to the observed abnormalities in circuit dynamics during locomotion, clarifying a potential role of impaired VIP-IN-mediated disinhibition vs. other VIP-IN-mediated circuit functions. However, our data do support the conclusion that decreased pyramidal neuron activity in DS mice during transitions to locomotion is correlated with and partially due to VIP-IN dysfunction, and augmenting VIP-IN function is sufficient to recover WT activity levels. Such limitations highlight the challenges of linking cellular and circuit function to behavior in the context of disease.
VIP-IN dysfunction underlies non-seizure ASD-linked endophenotypes of Dravet syndrome
VIP-Cre.Scn1a+/fl mice also show a subset of the behavioral abnormalities identified in the global DS model and other models of ASD40,42,48 yet without hyperactivity, temperature-sensitive seizures, epilepsy, or premature lethality. This suggests that VIP-IN dysfunction is not driven by ongoing seizure activity and does not contribute to the epilepsy phenotype of DS, but instead gives rise to non-seizure disease features including ASD and intellectual disability. The circuit function of VIP-INs is conserved across various neocortical regions as well as the hippocampus and amygdala,23,45,49 and VIP-INs are similarly impaired in Scn1a+/− mice across multiple sensory neocortical regions.11 Future work will identify the specific VIP-IN population(s) and brain region(s) that contribute to the cognitive abnormalities in DS. For instance, VIP-INs in the hippocampus appear to play a critical role in the remapping of place cells,23 while VIP-INs in the anterior insular cortex respond strongly to social interaction and are required for social preference.50,51 VIP-INs also shape sensory processing in complex ways, boosting gain of small but salient stimuli: for example, in V1 these cells act to enhance discrimination between novel and familiar images.22,24,38,52
VIP-INs as a locus of dysfunction in neurodevelopmental disorders
While many studies have examined the effect of loss of Nav1.1 on the intrinsic excitability of various IN subtypes in vitro, we demonstrate here a subtype-specific IN dysfunction at the circuit level in awake, behaving DS mice in vivo. Our data indicate that VIP-IN dysfunction may underlie the prominent comorbid ASD endophenotypes of DS independent of epilepsy. Indeed, genes implicated in ASD but not epilepsy are enriched in VIP-INs.53 Manipulation of VIP-IN activity may represent a therapeutic strategy for cognitive impairment in DS and across a range of neurodevelopmental disorders. The durable and severe cognitive comorbidities that persist throughout life in DS patients could potentially be addressed during the chronic phase of disease via enhanced VIP-IN function. Further work is needed to understand how ASD-related behaviors are directly tied to specific VIP-IN circuit functions that may vary across neocortical regions. Overall, our work demonstrates that VIP-INs represent a targetable nexus of dysfunction in DS and perhaps across neurodevelopmental disorders, highlighting the importance of developing new cell-type-specific tools and therapies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ethan M. Goldberg (goldberge@chop.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Preprocessed 2P, electrophysiological, and behavioral data will be deposited at G-node and be publicly available as of the date of publication. DOIs are listed in the key resources table. Additional raw data reported in this paper will be shared by the lead contact upon request.
All original code will be deposited at G-node and will be publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| AAV9.pCAG.Flex.tdTomato.WPRE | Penn Vector Core | AV-9-ALL864 |
| AAV9.pGP.AAV.syn.FLEX.jGCaMP7s.WPRE | GENIE Project/Addgene | 104491 |
| AAV9.pGP.AAV.syn.jGCaMP7s.WPRE | GENIE Project/Addgene | 104487 |
| AAV9.syn.FLEX.ChrimsonR.tdT | E. Boyden/Addgene | 62723 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Carbamoylcholine chloride | Sigma | C4382 |
| Pilocarpine hydrochloride | Sigma | P6503 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| 129S-Scn1atm1Kea/Mmjax | Jax | RRID:MMRRC_037107-JAX |
| Viptm1(cre)Zjh/J | Jax | RRID:IMSR_JAX:010908 |
| B6; 129P2-Pvalbtm1(cre)Arbr/J | Jax | RRID:IMSR_JAX:008069 |
| B6.Cg-Slc17a7tm1.1(cre)Hze/J | Jax | RRID:IMSR_JAX:037512 |
| B6J.Cg-Ssttm2.1(cre)Zjh/MwarJ | Jax | RRID:IMSR_JAX: 028864 |
| B6.129X1(SJL)-Scn1atm2.1Wac/Mmucd | UC Davis | RRID:MMRRC_041829-UCD |
| 129S6.SvEvTac | Taconic | RRID:IMSR_TAC: 129sve |
| C57BL/6J | Jax | RRID:IMSR_JAX:000664 |
|
| ||
| Software and algorithms | ||
|
| ||
| Pclamp 10 | Clampfit | |
| MATLAB | Mathworks | 2021a |
| Gramm | (Morel et al.)54 | https://doi.org/10.21105/joss.00568 |
| Deeplabcut (Deeplabcutcore) | (Nath et al.)55 | https://doi.org/10.1038/s41593-018-0209-y |
| FlyCapture2 | Point Gray (now FLIR) | |
| Suite2P | (Pachitariu et al.)56 | https://doi.org/10.1101/061507 |
| Python | Anaconda | v3.7.4 |
| Biorender | Biorender.com | Agreement number: XZ249HQ8GU |
| Original Code and data | G-Node:https://doi.org/10.12751/g-node.8s1yed | |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental animals
All procedures and experiments were approved by the Institutional Animal Care and Use Committee at the Children’s Hospital of Philadelphia and were conducted in accordance with the ethical guidelines of the National Institutes of Health. For 2P imaging experiments and slice electrophysiology, male and female mice were used in equal proportions. For mouse behavior experiments, both sexes were used, and sex was considered in our statistical design (see below); no sex-specific differences were identified unless otherwise noted, in which case the data were pooled. After weaning at P21, mice were group-housed with up to 5 mice per cage and maintained on a 12-h light/dark cycle with ad libitum access to food and water. In a subset of cases, extended weaning at P25 was allowed if pups appeared small at P21.
Mouse strains used in this study included: Scn1a+/− mice on a 129S6.SvEvTac background (RRID:MMRRC_037107-JAX) generated by a targeted deletion of exon 1 of the Scn1a gene, VIP-Cre mice (Viptm1(cre)Zjh/J; RRID:IMSR_JAX:010908 on a mixed C57BL/6; 129S4 background), PV-Cre mice (B6; 129P2-Pvalbtm1(cre)Arbr/J; RRID:IMSR_JAX:008069), SST-Cre mice (B6J.Cg-Ssttm2.1(cre)Zjh/MwarJ; RRID:IMSR_JAX: 028864), wild-type 129S6.SvEvTac (Taconic Biosciences model #129SVE; RRID:IMSR_TAC: 129sve), and wild-type C57BL/6J (RRID:IMSR_JAX:000664). Scn1a+/fl mice (RRID:MMRRC_041829-UCD) were rederived from frozen embryos in the CHOP mouse genetics core.
Female VIP-Cre homozygous mice were crossed to male 129S6.Scn1a+/− mice to generate Scn1a.VIP-Cre mice and WT VIP-Cre littermate controls. The genotype of all mice was determined via PCR of tail snips obtained before P10. All mice used for experiments were on a near 50:50 129S6:B6J background, and Scn1a+/− mice on this background have been shown to replicate the core phenotype of Dravet syndrome.57,58 We previously confirmed that progeny from this breeding configuration exhibited similar rates of SUDEP and epilepsy as well as threshold for temperature-induced seizures as Scn1a+/− mice without Cre.11 The same approach was used to generate VIP-Cre.Scn1a+/fl mice and WT VIP-Cre littermate controls; however, these mice are on a pure B6J background and thus WT control groups were not combined. Additionally, both male and female Scn1a+/fl were used for breeding.
METHOD DETAILS
AAV injections and cranial window implantation
Mice to be used for slice electrophysiology recordings were injected at P0 with AAV9.pCAG.Flex.tdTomato.WPRE as described previously.59 Briefly, P0 mice were anesthetized on ice, and then injected through the skull with 1.5 μL of AAV9 divided between both hemispheres. For 2P imaging experiments, VIP-INs from double transgenic Scn1a.VIP-Cre and WT.VIP-Cre littermates from the cross described above were labeled via stereotaxic injection of AAV9.pCAG.Flex.tdTomato.WPRE. Mice age > P35 were anesthetized with isoflurane (induction, 3–4%; maintenance, 1–1.5%) and body temperature and breathing were continuously monitored. A craniotomy 1 mm anterior and 3 mm lateral to lambda was made and virus was injected with a 50–75 μm beveled tip diameter glass pipette driven by a Nanoject III (Drummond Scientific). 60 nL of a mixture of AAV9.pGP.AAV.syn.jGCaMP7s.WPRE, AAV9.pGP.AAV.syn.FLEX.jGCaMP7s.WPRE, and AAV9.pCAG.Flex.tdTomato.WPRE diluted in sterile PBS to approximately 2.5 X E12 was injected at 20 nL/min across 4–5 locations separated by at least 0.5 mm centered around primary visual cortex V1 (+1.0 mm AP, −3.0 mm ML). The pipette remained in place for 5 min to allow the virus to spread at each injection location. Following virus injection, a cranial window made of a 3mm circular coverslip glued to a 5mm circular coverslip was affixed in place over the craniotomy, and a titanium headbar was cemented on top of the coverslip. All mice were given buprenorphine-SR 0.5 mg/kg, cefazolin 500 mg/kg, and dexamethasone 5 mg/kg perioperatively and monitored for recovery and infection periodically for 48 h following surgery.
Acute slice preparation
Mice were anesthetized with isoflurane and transcardially perfused with ice-cold artificial cerebral spinal fluid (ACSF) containing (in mM): NaCl, 87; sucrose, 75; KCl, 2.5; CaCl2, 1.0; MgSO4, 6.0; NaHCO3, 26; NaH2PO4, 1.25; glucose, 10, and equilibrated with 95% O2 and 5% CO2. The brain was removed and mounted on a holder of the Leica VT-1200S vibratome and sliced at 300–350 μm thickness. Slices were allowed to recover for 30 min in ACSF warmed to 30°C, then maintained at room temperature before recording. Slices were transferred to a recording chamber on the stage of a BX-61 upright microscope and perfused with recording solution at 30–32°C and 2–3 mL/min containing, in mM: NaCl, 125; KCl, 2.5; CaCl2, 2.0; MgSO4, 1.0; NaHCO3, 26; NaH2PO4, 1.25; glucose, 25.
Slice recordings
VIP-INs were identified by tdTomato expression. Whole-cell recordings were obtained from superficial (layer 2/3) primary somatosensory cortex (S1; “barrel”) and visual cortex (V1) as described previously. Patch pipettes were pulled from borosilicate glass using a Narashige PC-100 puller and filled with intracellular solution containing (in mM): K-gluconate, 130; KCl, 6.3; EGTA, 0.5; MgCl2, 1.0; HEPES, 10; Mg-ATP, 4.0; Na-GTP, 0.3; pH was adjusted to 7.30 with KOH, and osmolarity adjusted to 285 mOsm with 30% sucrose. Pipettes had a resistance of 4–6 MΩ when filled and placed in recording solution. Voltage was sampled at 50 kHz with a MultiClamp 700B amplifier (Molecular Devices), filtered at 10 kHz, digitized using a DigiData 1550A, and acquired using pClamp10 software. Recordings were discarded if the cell had an unstable resting membrane potential and/or a membrane potential greater than −50 mV, or if access resistance increased by > 20% during the recording. We did not correct for liquid junction potential. For experiments using carbachol, repeat measurements were made 5 min after wash-in and up to 30 min after wash-out of drug.
Electrophysiology data analysis
All analysis was performed blind to genotype using custom written MATLAB (Mathworks) code. Resting membrane potential was the average value of a 1 s sweep with no direct current injection. Input resistance was calculated using the responses to −10 to −50 pA hyperpolarizing current injections near rest using for each sweep. AP threshold was the value at which the derivative of the voltage reached 10 mV/ms. Spike peak refers to the maximum voltage value of an individual AP, while spike amplitude is the difference between spike peak and AP threshold. Maximum rise slope was taken as the max value of during a single AP. Unless indicated, all quantification of single spike properties was done using the first AP elicited at rheobase.
Rheobase was determined as the minimum current injection that elicited APs using sequential 600 ms sweeps at 10 pA intervals. Maximal instantaneous firing was calculated using the smallest inter-spike interval (ISI) elicited at near-maximal current injection (before entering depolarization block). Maximal steady-state firing was defined as the maximum number of spikes generated in a single 600ms sweep, with a minimum requirement for a spike being an amplitude of 40 mV and height overshooting at least 0 mV. I-f relationships were generated from 600ms current steps as well as from 8 s ramp depolarizations (Figure 1) as described previously. For Figure 1, the maximum instantaneous firing rate was calculated from the shortest ISI during a single 8 s ramp depolarization. The effect of carbachol on firing was calculated as an index value from the total number of spikes generated before and after bath application of carbachol during an 8 s ramp depolarization:
In vivo 2-P calcium imaging
Imaging was performed using a mode-locked pulsed infrared laser (18–30 mW, InSight, SpectraPhysics) controlled by a Pockels cell (Conoptics) on a commercial 2P microscope (Bruker) equipped with a resonant scanner (Cambridge Technology). A 16/0.8 NA water-immersion objective (Nikon) was used. GCaMP7s was imaged at 950 nm with a gallium arsenide phosphide photodetector (H7422–40, Hamamatsu). Simultaneously, tdT was imaged with a multi-alkali detector (R3896, Hamamatsu). For experiments using ChrimsonR.tdT, a single reference image (average of 100 frames) was taken before and after each imaging field at 1020 nm to isolate and verify tdT signal. Data were acquired and converted to .tif stacks for analysis. Fluorescence data were acquired at 30 Hz.
Approximately 3 weeks after head bar implantation, mice were allowed to habituate to head fixation in a floating Mobile Home Cage (Neurotar) in the imaging apparatus for 20–30 min daily for 5 days prior to the first imaging experiment. By the end of habituation, all mice showed spontaneous running bouts, occasional grooming, and the absence of escape or prolonged freezing behaviors. For test sessions, mice were allowed to freely “move” in the floating chamber for 12–15 min. The chamber had a transparent siding, and a 7-inch LCD screen (resolution: 1024X600, refresh: 60Hz) displaying a greyscale image (mean luminance 50 cd/m2) positioned 15 cm away from and perpendicular to the contralateral eye. Airflow into the Mobile Home Cage stage provided approximately pink noise during the experiment, measured at ~40–45 db. An infrared (IR) CCD camera (Grasshopper 3, FLIR Teledyne) was positioned 15 cm away from the ipsilateral eye, and the image field was adjusted to include the entirety of the pupil, whisker pad, and nose, illuminated by an IR light source (850nm). Pupillometry videos were acquired at 100Hz, and locomotion speed was tracked by the mobile home cage locomotion tracking software (Neurotar). Neural and behavioral data acquisition was synchronized by a TTL pulse at the start and end of each recording session, and all behavioral data were decimated to match the sample frequency of the fluorescence data prior to analysis. For in vivo optogenetics experiments, a separate TTL output from either a Master-9 (A.M.P.I.; for q30s stimulation) or the mobile home cage interface (running triggered stimulation) was used to trigger a red laser routed through the imaging objective (10 mW/mm2, 647 nm; OBIS, Coherent). This combination of stimulation patterns was used to ensure that we collected sufficient epochs where there was: 1) laser stimulation at rest; 2) laser stimulation concurrent to locomotion; and 3) locomotion epochs without laser stimulation.
For experiments with pilocarpine, a single FOV was recorded per mouse as above, then acquisition was stopped, and the mouse was given an intraperitoneal injection of 10 mg/kg pilocarpine. After 10 min, the injection was verified by an increased pupil diameter and increased salivation by the mouse. Imaging and behavioral data acquisition was restarted such that the same cells were recorded before and after pilocarpine injection.
Cell detection and extraction of neuron activity
The Suite2p package was used for preprocessing of the acquired 2P calcium imaging data followed by cell detection and extraction of neural activity.56 A non-rigid registration was implemented for the dataset in which each image was divided into sub-blocks for more accurate movement correction. Once the images were registered, alignment metrics were checked for quality of data; a small number of sessions were excluded if z drift was detected. After registration, cells were detected as ROIs spatially located around the peaks of the smoothened principal components, and a blinded experimenter manually rejected non-cell ROIs that were erroneously assigned as cells. Fluorescence values were extracted from the detected cells and neuropil values are derived from the area around the cells across the imaging session. In cases where two imaging sessions on the same field of view were recorded in series (before and after pilocarpine injection, or using different optogenetic stimulation patters), the resulting fluorescence data were aligned and combined prior to cell detection in suite2P.
Normalization and background correction
Because background fluorescence is correlated with locomotion, we adopted the method of diPoppa et al. (2018) for estimating the adjacent neuropil signal for each cell and finding the correct scaling factor for background subtraction. Suite2P was used to output a neuropil mask for each cell extending 30 μm from each ROI, excluding regions belonging to other detected ROIs. Then, we determined the minimum cellular fluorescence that corresponded to the surrounding neuropil signal by binning the neuropil signal into 20 segments and calculating the fifth percentile of the cellular signal that corresponded to each bin. We then used linear regression to calculate the correction factor for each cell. To avoid overestimating with highly active cells which are strongly correlated with , we averaged from sparsely firing cells with high skew values (>4) for each FOV to give a correction value for that experiment (Figure S2). For a minority of FOVs that did not have at least 20 high skew cells, we used an average correction factor from all other FOVs .
Analysis of neural activity
Fluorescence values in all figures are reported as , where is the 10th percentile of each neuron’s fluorescence trace. was adjusted for long (>6 min) recording sessions by using a linear interpolation between the average values for the first and second half of the recording.
To measure how neural activity is modulated by locomotion, we calculated a modulation index MI for each cell by separating the entire neural trace into 1 s chunks and computing the average activity for each. These chunks were separated based on the average running speed into locomotion and quiescent epochs. MI was calculated for each cell as:
For Figure 2, MI was calculated independently for chunks which also had concurrent red-light stimulation during at least 15 of 30 sample points within the chunk length. Chunks with between 1 and 15 sample points where there was red light stimulation were discarded. Using this method, we were able to combine the q30s and triggered datasets, although our results were nearly identical if we separated our analysis based on stimulation pattern (not shown).
A Pearson correlation coefficient was used to describe the relationship between neural activity and pupil diameter . We then used a shuffle test to determine whether an individual cell was either positively or negatively correlated with either locomotion or pupil diameter. Briefly, neural data were divided into 1s chunks and randomly shuffled times, then was recalculated for each cell in each shuffle to give a distribution of values . A cell was positively or negatively correlated with pupil diameter if or after correcting for multiple comparisons. For demonstration purposes, we extracted individual running bouts defined by a duration of running of at least 5s preceded by 10s of rest. We then averaged the trace for each cell across running bouts and plotted the grand averaged and bootstrapped 95% CI across all cells. Inter-population correlation (Figure S4) was calculated as described previously33 independently for locomotion and quiescent epochs.
We additionally extracted deconvolved ‘spiking’ event trains for some analyses. Briefly, after calculating for each cell as described above, we used the OASIS deconvolution algorithm60 to extract . To tune the values for the transient decay constant (tau) and noise floor, we randomly selected a high skew (>2.5) non-VIP neuron and a moderately high skew (>1) VIP-IN from each FOV and used a simple threshold to extract a template kernel from each trace. We then fit the decay time of the kernel as well as the noise floor and averaged across all FOVs and used these values across all experiments. We then used a de-noising algorithm to remove low amplitude events that are near the noise floor. Briefly, the maximum of was scaled to the maximum of for each cell and we calculated the median absolute deviation of . We then binned into 100ms intervals and set the value of each bin equal to zero if it fell below a multiple of the noise level where .
For calculating the modulation index based on ne-noised deconvolved events from each cell, we segmented the chunked data in the same way into locomotion and stationary epochs, and used the simplified formula:
We found that calculation of locomotion MI using either event traces or yielded equivalent results. We included analyses using event trains primarily as a tool to describe the baseline activity of neurons independent of locomotion or any external stimulus, or to compare activity within the same cell across conditions (e.g., +/− pilocarpine). However, because the exact relationship between spiking and calcium fluorescence likely differs between cell types, we did not attempt to estimate exact firing rates or compare firing rates between or across cell types.
Mouse behavior
All animal behavior was filmed through a glass bottom chamber (25 × 15 inches) with opaque sides and lid using a Google Pixel 5 phone camera (1920×1080 30hz). All mice performed the same panel of behavioral experiments on consecutive days in the order described below. Mice were maintained on a regular 12h light dark schedule, and all experiments were performed in a 6-h window around the transition from light to dark in an isolated, noise-proof, dimly lit room. The blinded experimenter exited the room after initializing all tests. Mice were handled daily for one week prior to performing behavioral experiments.
Three chamber social task
The test chamber was divided into three equal sections with plexiglass dividers. A novel mouse that was age, genotype, and sex-matched to the test animal was placed under a small clear square cage that had gaps large enough for nose-to-nose interactions. An identical cage was placed in the opposite chamber, and then the test mouse was placed in the central compartment and allowed to habituate for 5 min. After this time, a portion of the Plexiglas divider was removed exposing a path into each side chamber, and the test animal was allowed to explore for 10 min. After this time, both mice were returned to their home cages, and the apparatus was thoroughly cleaned with 70% EtOH. Mouse, divider, and holding cage position were extracted from behavioral videos using DeepLabCut (DLC; see below) and social preferences were calculated as index values:
where denotes the total time the test mouse spent on the side with the novel mouse and on the side with the empty chamber.48,50
Novel object recognition and open field task
Prior to the familiarization phase of the NOR task, mice were allowed to freely explore and habituate to the empty test chamber for 15 min. This open field period was recorded, and the mouse and chamber positions were extracted using DLC. We calculated the total distance traveled, average running speed, and percent of time spent in the center (defined as > 3 inches from the walls). After habituation, the test animal was briefly removed, and two identical objects were placed diagonally from one another, 3 inches away from the side wall. The mouse was returned to the chamber and allowed to explore the objects for 7 min. 24 h later, each mouse was returned to the chamber and allowed to explore a novel object placed in a new symmetrical location, and a familiar object used on the day before for a total of 5 min. Mouse and object location were extracted using DLC. As preliminary analysis indicated that interaction times based solely on proximity using these videos were heavily affected by non-exploratory behaviors near each object, videos were scored by an independent blind experimenter for time spent interacting with each object based on previously reported guidelines61 (Figure S11). The time spent interacting with the novel and familiar objects was recorded during the first 20 s of total interaction time. was compared across groups for genotype differences, and we tested the null hypothesis within each experimental group with a paired Student’s t-test to determine if there was any preference toward the novel object. Eight mice ) were excluded for not spending at least 20 s interacting with either object61 and hence did not meet a priori criteria for inclusion. Total time spent exploring both objects was also reported.
Marble burying
Compulsive and repetitive behaviors were assessed with a marble burying test. Twelve equally sized marbles were placed in a uniform grid in a standard housing cage with 2 inches of bedding. A single mouse was allowed to explore the cage for 30 min and, following this period, a single en face photo was taken from 1 ft away from the surface of the bedding and was scored for % marbles buried by a blinded experimenter.
Elevated plus maze (EPM)
Two identical EPM apparatus (2 ft elevation, arm length of 12 in.) with two closed and two open arms were placed in the test room and separated by a white curtain. Videos were taken from 6 feet above the apparatus, and two mice were tested simultaneously if they were from the same housing cage. Mice were placed in the center of the apparatus and allowed to freely explore for 5 min. Mouse position and location of the apparatus arm were extracted with DLC, and both the time and distance traveled on the open ( and ) and closed ( and ) arm were calculated. Willingness to explore the open arm was calculated as an index for both time and distance.
DLC markerless pose estimation
DLC55 was used on a Windows system equipped with an Nvidia RTX 3090 GPU. We used separately trained networks for the following applications: A) Pupil and whisk-ometry during 2P imaging; B) Three chamber social task; C) Open field and NOR task; and D) EPM task. In all cases, we extracted 5 frames per animal (10 for pupillometry) using the k-means algorithm and manually labeled the desired body parts and/or apparatus markers (below). For training, we used the resnet 50 pre-trained model, and otherwise default parameters. We trained each network for 300,000 iterations, and then evaluated the networks for accuracy and generalization, and in some cases extracted additional outlier frames for labeling and retrained the network to improve results. Details for the various networks are listed in Table S1.
All marker locations were exported as a csv and analyzed in MATLAB. Where indicated, ‘mouse position’ refers to an average of the nose, neck, and tail base position. For pupillometry, the four markers identifying the bounds of the pupil were fit with a circle, and the diameter was calculated. Saccades, closed eyelids, and grooming were detected and excluded from analysis. We also extracted a motion index for whisking and found this to be correlated with pupil diameter as previously reported, but we did not include this in our analysis of neural activity. All behavioral data extracted from DLC was filtered with a 300ms rolling average.
QUANTIFICATION AND STATISTICAL ANALYSIS
Because of the highly nested structure of our data, unless otherwise noted, we primarily relied on linear mixed effects models to determine significant differences between genotypes. For 2P datasets, where many hundreds of cells are recorded from each animal, we treated individual mice as random effects and genotype as the fixed effect. For a given measured parameter (e.g., MI) we constructed models with default settings in MATLAB using the following formula:
accounting for different baseline values for each mouse (random intercept model). Slope coefficients and errors (normalized as effect sizes in relation to WT values), and p values representing the estimated difference between genotypes, are reported in the main text.
For mouse behavior experiments, we included both male and female mice in our analysis in accordance with guidance from the NIH. We used mixed effects models to test for differences between genotypes
and tested whether sex influenced behavior by comparing the above model to a simplified one:
Among all experiments, sex only had a trend toward an effect on distance traveled in the open arm of the EPM with males spending less time in the open arm (Effect size: −0.18; Error: 0.06; p value: 0.1). Otherwise, for all differences we observed between genotypes, sex did not significantly affect performance.
In some indicated cases, within group comparisons are made using a paired Student’s t-test. For slice electrophysiology experiments, we treated each cell as and made all comparisons using the Kruskal-Wallis test as several parameters did not have normal distributions. Sample sizes were not estimated with power analyses but were based on previous studies examining similar phenomenon in models of ASD and epilepsy. All line and bar graphs show mean +/− bootstrapped 95% CI of the mean unless noted otherwise. Wherever possible, values for individual cells/mice are shown.
Supplementary Material
Highlights.
VIP interneurons are dysfunctional in Dravet syndrome (Scn1a+/−) mice in vivo
Cholinergic modulation exacerbates VIP-IN impairment during behavior
Scn1a deletion in VIP-INs only results in ASD-linked behaviors, without seizures
ACKNOWLEDGMENTS
We would like to thank Xiaohong Zhang for expert technical support and mouse colony maintenance, Jennifer Kearney for the gift of Scn1a+/− mice, Edward Boyden for the distribution of ChrimsonR, and the GENIE Project at the Howard Hughes Medical Institute Janelia Research Campus for GCaMP7s. We thank Bernardo Rudy, Eddie Zagha, and Benjamin Scholl for comments on a previous version of the manuscript. This work was supported by the National Institutes of Neurological Disorders and Stroke of the National Institutes of Health under F31 NS111803 (to K.M.G.) and R01 NS110869 (to E.M.G.), by the Dana Foundation David Mahoney Neuroimaging Program and Burroughs Wellcome Career Award for Medical Scientists (to E.M.G.), and a Postdoctoral Fellowship award from the Dravet Syndrome Foundation (to A.S.).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112628.
REFERENCES
- 1.Gouwens NW, Sorensen SA, Baftizadeh F, Budzillo A, Lee BR, Jarsky T, Alfiler L, Baker K, Barkan E, Berry K, et al. (2020). Integrated morphoelectric and transcriptomic classification of cortical GABAergic cells. Cell 183, 935–953. 10.1016/j.cell.2020.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yao Z, van Velthoven CTJ, Nguyen TN, Goldy J, Sedeno-Cortes AE, Baftizadeh F, Bertagnolli D, Casper T, Chiang M, Crichton K, et al. (2021). A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 184, 3222–3241. 10.1016/j.cell.2021.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Contractor A, Ethell IM, and Portera-Cailliau C (2021). Cortical interneurons in autism. Nat. Neurosci. 24, 1648–1659. 10.1038/s41593-021-00967-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dienel SJ, and Lewis DA (2019). Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol. Dis. 131, 104208. 10.1016/j.nbd.2018.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dravet C (2011). Dravet syndrome history. Dev. Med. Child Neurol. 53, 1–6. 10.1111/j.1469-8749.2011.03964.x. [DOI] [PubMed] [Google Scholar]
- 6.Dravet C, and Oguni H (2013). Dravet syndrome (severe myoclonic epilepsy in infancy). Handb. Clin. Neurol. 111, 627–633. 10.1016/B978-0-444-52891-9.00065-8. [DOI] [PubMed] [Google Scholar]
- 7.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, and De Jonghe P (2001). De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 68, 1327–1332. 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catterall WA (2017). Forty years of sodium channels: structure, function, pharmacology, and epilepsy. Neurochem. Res. 42, 2495–2504. 10.1007/s11064-017-2314-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, and Catterall WA (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 10.Favero M, Sotuyo NP, Lopez E, Kearney JA, and Goldberg EM (2018). A transient developmental window of fast-spiking interneuron dysfunction in a mouse model of dravet syndrome. J. Neurosci. 38, 7912–7927. 10.1523/JNEUROSCI.0193-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goff KM, and Goldberg EM (2019). Vasoactive intestinal peptide-expressing interneurons are impaired in a mouse model of Dravet syndrome. Elife 8, e46846. 10.7554/eLife.46846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tai C, Abe Y, Westenbroek RE, Scheuer T, and Catterall WA (2014). Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 111, 3139–3148. 10.1073/pnas.1411131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Stasi AM, Farisello P, Marcon I, Cavallari S, Forli A, Vecchia D, Losi G, Mantegazza M, Panzeri S, Carmignoto G, et al. (2016). Unaltered network activity and interneuronal firing during spontaneous cortical dynamics in vivo in a mouse model of severe myoclonic epilepsy of infancy. Cerebr. Cortex 26, 1778–1794. 10.1093/cercor/bhw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rudy B, Fishell G, Lee S, and Hjerling-Leffler J (2011). Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 71, 45–61. 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, Yanagawa Y, Tamamaki N, Hensch TK, Itohara S, and Yamakawa K (2013). Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of dravet syndrome. Hum. Mol. Genet. 22, 4784–4804. 10.1093/hmg/ddt331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oakley JC, Kalume F, Yu FH, Scheuer T, and Catterall WA (2009). Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc. Natl. Acad. Sci. USA 106, 3994–3999. 10.1073/pnas.0813330106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tatsukawa T, Ogiwara I, Mazaki E, Shimohata A, and Yamakawa K (2018). Impairments in social novelty recognition and spatial memory in mice with conditional deletion of Scn1a in parvalbumin-expressing cells. Neurobiol. Dis. 112, 24–34. 10.1016/j.nbd.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 18.Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, and Catterall WA (2015). Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain 138, 2219–2233. 10.1093/brain/awv142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaneko K, Currin CB, Goff KM, Wengert ER, Somarowthu A, Vogels TP, and Goldberg EM (2022). Developmentally regulated impairment of parvalbumin interneuron synaptic transmission in an experimental model of Dravet syndrome. Cell Rep. 38, 110580. 10.1016/j.celrep.2022.110580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Almog Y, Fadila S, Brusel M, Mavashov A, Anderson K, and Rubinstein M (2021). Developmental alterations in firing properties of hippocampal CA1 inhibitory and excitatory neurons in a mouse model of Dravet syndrome. Neurobiol. Dis. 148, 105209. 10.1016/j.nbd.2020.105209. [DOI] [PubMed] [Google Scholar]
- 21.Fu Y, Kaneko M, Tang Y, Alvarez-Buylla A, and Stryker MP (2015). A cortical disinhibitory circuit for enhancing adult plasticity. Elife 4, e05558. 10.7554/eLife.05558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garrett M, Manavi S, Roll K, Ollerenshaw DR, Groblewski PA, Ponvert ND, Kiggins JT, Casal L, Mace K, Williford A, et al. (2020). Experience shapes activity dynamics and stimulus coding of VIP inhibitory cells. Elife 9, e50340. 10.7554/eLife.50340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turi GF, Li WK, Chavlis S, Pandi I, O’Hare J, Priestley JB, Grosmark AD, Liao Z, Ladow M, Zhang JF, et al. (2019). Vasoactive intestinal polypeptide-expressing interneurons in the Hippocampus support goal-oriented spatial learning. Neuron 101, 1150–1165.e8. 10.1016/j.neuron.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan AG, Poort J, Chadwick A, Blot A, Sahani M, Mrsic-Flogel TD, and Hofer SB (2018). Distinct learning-induced changes in stimulus selectivity and interactions of GABAergic interneuron classes in visual cortex. Nat. Neurosci. 21, 851–859. 10.1038/s41593-018-0143-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfeffer CK, Xue M, He M, Huang ZJ, and Scanziani M (2013). Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat. Neurosci. 16, 1068–1076. 10.1038/nn.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S, Kruglikov I, Huang ZJ, Fishell G, and Rudy B (2013). A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat. Neurosci. 16, 1662–1670. 10.1038/nn.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang X, Shen S, Cadwell CR, Berens P, Sinz F, Ecker AS, Patel S, and Tolias AS (2015). Principles of connectivity among morphologically defined cell types in adult neocortex. Science 350, aac9462. 10.1126/science.aac9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kullander K, and Topolnik L (2021). Cortical disinhibitory circuits: cell types, connectivity and function. Trends Neurosci. 44, 643–657. 10.1016/j.tins.2021.04.009. [DOI] [PubMed] [Google Scholar]
- 29.McGinley MJ, Vinck M, Reimer J, Batista-Brito R, Zagha E, Cadwell CR, Tolias AS, Cardin JA, and McCormick DA (2015). Waking state: rapid variations modulate neural and behavioral responses. Neuron 87, 1143–1161. 10.1016/j.neuron.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson J, Ayzenshtat I, Karnani MM, and Yuste R (2016). VIP+ interneurons control neocortical activity across brain states. J. Neurophysiol. 115, 3008–3017. 10.1152/jn.01124.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munoz W, Tremblay R, Levenstein D & Rudy B Layer-specific modulation of neocortical dendritic inhibition during active wakefulness. Science. 355, 954–959. [DOI] [PubMed] [Google Scholar]
- 32.Karnani MM, Jackson J, Ayzenshtat I, Hamzehei Sichani A, Manoocheri K, Kim S, and Yuste R (2016). Opening holes in the blanket of inhibition: localized lateral disinhibition by vip interneurons. J. Neurosci. 36, 3471–3480. 10.1523/JNEUROSCI.3646-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dipoppa M, Ranson A, Krumin M, Pachitariu M, Carandini M, and Harris KD (2018). Vision and locomotion shape the interactions between neuron types in mouse visual cortex. Neuron 98, 602–615.e8. 10.1016/j.neuron.2018.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran CH, Vaiana M, Nakuci J, Somarowthu A, Goff KM, Goldstein N, Murthy P, Muldoon SF, and Goldberg EM (2020). Interneuron desynchronization precedes seizures in a mouse model of Dravet syndrome. J. Neurosci. 40, 2764–2775. 10.1523/JNEUROSCI.2370-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reimer J, Froudarakis E, Cadwell CR, Yatsenko D, Denfield GH, and Tolias AS (2014). Pupil fluctuations track fast switching of cortical states during quiet wakefulness. Neuron 84, 355–362. 10.1016/j.neuron.2014.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cantrell AR, Ma JY, Scheuer T, and Catterall WA (1996). Muscarinic modulation of sodium current by activation of protein kinase C in rat hippocampal neurons. Neuron 16, 1019–1026. 10.1016/S0896-6273(00)80125-7. [DOI] [PubMed] [Google Scholar]
- 37.Cantrell AR, and Catterall WA (2001). Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat. Rev. Neurosci. 2, 397–407. 10.1038/35077553. [DOI] [PubMed] [Google Scholar]
- 38.Millman DJ, Ocker GK, Caldejon S, Kato I, Larkin JD, Lee EK, Luviano J, Nayan C, Nguyen TV, North K, et al. (2020). VIP interneurons in mouse primary visual cortex selectively enhance responses to weak but specific stimuli. Elife 9, e55130. 10.7554/eLife.55130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. (2014). Independent optical excitation of distinct neural populations. Nat. Methods 11, 338–346. 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, and Catterall WA (2012). Autistic behavior in Scn1a +/− mice and rescue by enhanced GABAergic transmission. Nature 489, 385–390. 10.1038/nature11356.Autistic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ Jr., Clarkson RL, Sanders SJ, and Bender KJ (2019). The autism-associated gene Scn2a contributes to dendritic excitability and synaptic function in the prefrontal cortex. Neuron 103, 673–685. 10.1016/j.neuron.2019.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Batista-Brito R, Vinck M, Ferguson KA, Chang JT, Laubender D, Lur G, Mossner JM, Hernandez VG, Ramakrishnan C, Deisseroth K, et al. (2017). Developmental dysfunction of VIP interneurons impairs cortical circuits. Neuron 95, 884–895. 10.1016/j.neuron.2017.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uchino K, Kawano H, Tanaka Y, Adaniya Y, Asahara A, Deshimaru M, Kubota K, Watanabe T, Katsurabayashi S, Iwasaki K, and Hirose S (2021). Inhibitory synaptic transmission is impaired at higher extracellular Ca2+ concentrations in Scn1a+/− mouse model of Dravet syndrome. Sci. Rep. 11, 10634. 10.1038/s41598-021-90224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dávid C, Schleicher A, Zuschratter W & Staiger JF The innervation of parvalbumin-containing interneurons by VIP-immunopositive interneurons in the primary somatosensory cortex of the adult rat. Eur. J. Neurosci. 25, 2329–2340 DOI: 10.1111/j.1460-9568.2007.05496.x. [DOI] [PubMed] [Google Scholar]
- 45.Guet-McCreight A, Skinner FK, and Topolnik L (2020). Common principles in functional organization of VIP/calretinin cell-driven disinhibitory circuits across cortical areas. Front. Neural Circ. 14, 32–14. 10.3389/fncir.2020.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obermayer J, Luchicchi A, Heistek TS, de Kloet SF, Terra H, Bruinsma B, Mnie-Filali O, Kortleven C, Galakhova AA, Khalil AJ, et al. (2019). Prefrontal cortical ChAT-VIP interneurons provide local excitation by cholinergic synaptic transmission and control attention. Nat. Commun. 10, 5280. 10.1038/s41467-019-13244-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Granger AJ, Wang W, Robertson K, El-Rifai M, Zanello AF, Bistrong K, Saunders A, Chow BW, Nuñez V, Turrero García M, et al. (2020). Cortical ChAT+ neurons co-transmit acetylcholine and GABA in a target-and brain-region-specific manner. Elife 9, e57749. 10.7554/eLife.57749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mossner JM, Batista-Brito R, Pant R, and Cardin JA (2020). Developmental loss of MeCP2 from VIP interneurons impairs cortical function and behavior. Elife 9, e55639. 10.7554/eLife.55639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krabbe S, Paradiso E, d’Aquin S, Bitterman Y, Courtin J, Xu C, Yonehara K, Markovic M, Muller C, Eichlisberger T, et al. (2019). Adap-€ tive disinhibitory gating by VIP interneurons permits associative learning. Nat. Neurosci. 22, 1834–1843. 10.1038/s41593-019-0508-y. [DOI] [PubMed] [Google Scholar]
- 50.Ramos-Prats A, Paradiso E, Castaldi F, Sadeghi M, Mir MY, Hörtnagl H, Göbel G, and Ferraguti F (2022). VIP-expressing interneurons in the anterior insular cortex contribute to sensory processing to regulate adaptive behavior. Cell Rep. 39, 110893. 10.1016/j.celrep.2022.110893. [DOI] [PubMed] [Google Scholar]
- 51.Miura I, Sato M, Overton ETN, Kunori N, Nakai J, Kawamata T, Nakai N, and Takumi T (2020). Encoding of social exploration by neural ensembles in the insular cortex. PLoS Biol. 18, e3000584. 10.1371/journal.pbio.3000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Vries SEJ, Lecoq JA, Buice MA, Groblewski PA, Ocker GK, Oliver M, Feng D, Cain N, Ledochowitsch P, Millman D, et al. (2020). A large-scale standardized physiological survey reveals functional organization of the mouse visual cortex. Nat. Neurosci. 23, 138–151. 10.1038/s41593-019-0550-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goff KM, and Goldberg EM (2021). A role for vasoactive intestinal peptide interneurons in neurodevelopmental disorders. Dev. Neurosci. 43, 168–180. 10.1159/000515264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morel P (2018). Gramm: grammar of graphics plotting in Matlab. J. Open Source Softw. 3, 568. 10.21105/joss.00568. [DOI] [Google Scholar]
- 55.Nath T, Mathis A, Chen AC, Patel A, Bethge M, and Mathis MW (2019). Using DeepLabCut for 3D markerless pose estimation across species and behaviors. Nat. Protoc. 14, 2152–2176. 10.1038/s41596-019-0176-0. [DOI] [PubMed] [Google Scholar]
- 56.Pachitariu M, Stringer C, Dipoppa M, Schröder S, Rossi LF, Dalgleish H, Carandini M, Harris KD. Suite2p: beyond 10,000 neurons with standard two-photon microscopy. bioRxiv 10.1101/061507. [DOI] [Google Scholar]
- 57.Miller AR, Hawkins NA, McCollom CE, and Kearney JA (2014). Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Gene Brain Behav. 13, 163–172. 10.1111/gbb.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mistry AM, Thompson CH, Miller AR, Vanoye CG, George AL Jr., and Kearney JA (2014). Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol. Dis. 65, 1–11. 10.1016/j.nbd.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao Y, Hisey E, Bradshaw TWA, Erata E, Brown WE, Courtland JL, Uezu A, Xiang Y, Diao Y, and Soderling SH (2019). Plug-and-Play protein modification using homology-independent universal genome engineering. Neuron 103, 583–597.e8. 10.1016/j.neuron.2019.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Friedrich J, Zhou P, and Paninski L (2017). Fast online deconvolution of calcium imaging data. PLoS Comput. Biol. 13, e1005423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P, and Freret T (2013). Object recognition test in mice. Nat. Protoc. 8, 2531–2537. 10.1038/nprot.2013.155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Preprocessed 2P, electrophysiological, and behavioral data will be deposited at G-node and be publicly available as of the date of publication. DOIs are listed in the key resources table. Additional raw data reported in this paper will be shared by the lead contact upon request.
All original code will be deposited at G-node and will be publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| AAV9.pCAG.Flex.tdTomato.WPRE | Penn Vector Core | AV-9-ALL864 |
| AAV9.pGP.AAV.syn.FLEX.jGCaMP7s.WPRE | GENIE Project/Addgene | 104491 |
| AAV9.pGP.AAV.syn.jGCaMP7s.WPRE | GENIE Project/Addgene | 104487 |
| AAV9.syn.FLEX.ChrimsonR.tdT | E. Boyden/Addgene | 62723 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Carbamoylcholine chloride | Sigma | C4382 |
| Pilocarpine hydrochloride | Sigma | P6503 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| 129S-Scn1atm1Kea/Mmjax | Jax | RRID:MMRRC_037107-JAX |
| Viptm1(cre)Zjh/J | Jax | RRID:IMSR_JAX:010908 |
| B6; 129P2-Pvalbtm1(cre)Arbr/J | Jax | RRID:IMSR_JAX:008069 |
| B6.Cg-Slc17a7tm1.1(cre)Hze/J | Jax | RRID:IMSR_JAX:037512 |
| B6J.Cg-Ssttm2.1(cre)Zjh/MwarJ | Jax | RRID:IMSR_JAX: 028864 |
| B6.129X1(SJL)-Scn1atm2.1Wac/Mmucd | UC Davis | RRID:MMRRC_041829-UCD |
| 129S6.SvEvTac | Taconic | RRID:IMSR_TAC: 129sve |
| C57BL/6J | Jax | RRID:IMSR_JAX:000664 |
|
| ||
| Software and algorithms | ||
|
| ||
| Pclamp 10 | Clampfit | |
| MATLAB | Mathworks | 2021a |
| Gramm | (Morel et al.)54 | https://doi.org/10.21105/joss.00568 |
| Deeplabcut (Deeplabcutcore) | (Nath et al.)55 | https://doi.org/10.1038/s41593-018-0209-y |
| FlyCapture2 | Point Gray (now FLIR) | |
| Suite2P | (Pachitariu et al.)56 | https://doi.org/10.1101/061507 |
| Python | Anaconda | v3.7.4 |
| Biorender | Biorender.com | Agreement number: XZ249HQ8GU |
| Original Code and data | G-Node:https://doi.org/10.12751/g-node.8s1yed | |