

SUMMARY
Therapeutic neoantigen cancer vaccines have limited clinical efficacy to date. Here, we identify a heterologous prime-boost vaccination strategy using a self-assembling peptide nanoparticle TLR-7/8 agonist (SNP) vaccine prime and a chimp adenovirus (ChAdOx1) vaccine boost that elicits potent CD8 T cells and tumor regression. ChAdOx1 administered intravenously (i.v.) had 4-fold higher antigen-specific CD8 T cell responses than mice boosted by the intramuscular (i.m.) route. In the therapeutic MC38 tumor model, i.v. heterologous prime-boost vaccination enhances regression compared with ChAdOx1 alone. Remarkably, i.v. boosting with a ChAdOx1 vector encoding an irrelevant antigen also mediates tumor regression, which is dependent on type I IFN signaling. Single-cell RNA sequencing of the tumor myeloid compartment shows that i.v. ChAdOx1 reduces the frequency of immunosuppressive Chil3 monocytes and activates cross-presenting type 1 conventional dendritic cells (cDC1s). The dual effect of i.v. ChAdOx1 vaccination enhancing CD8 T cells and modulating the TME represents a translatable paradigm for enhancing anti-tumor immunity in humans.
In brief
Ramirez-Valdez et al. describe an intravenous heterologous prime-boost vaccination strategy that effectively promotes tumor regression by eliciting high-magnitude anti-tumor CD8 T cell responses and modulating the tumor microenvironment through type I IFN signaling.
Graphical abstract
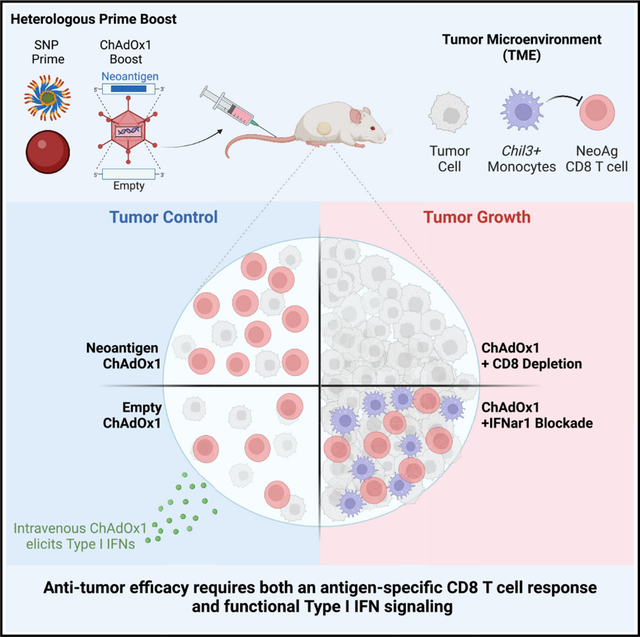
INTRODUCTION
Immunotherapies have transformed the treatment landscape for multiple tumor types. Studies using checkpoint inhibitors (CPIs) that target PD-1 or CTLA-4 have revealed a critical role for endogenous T cells that recognize tumor antigens in mediating anti-tumor efficacy.1–3 Therapeutic approaches leveraging tumor-specific adoptive T cells also exhibit tumor regression.4,5 A major goal for immunotherapy has been to develop approaches that enhance the frequency and function of tumor-specific T cells. One such approach is to develop vaccines that can induce or expand T cells against patient specific tumor antigens.6
Cancer vaccines have had limited impact on anti-tumor immunity.7 This lack of clinical efficacy is multifactorial and includes suboptimal vaccine platforms for eliciting high-magnitude T cell responses, a limited understanding of tumor antigens, and the immunosuppressive tumor microenvironment (TME) that may limit T cell function.6 Recent advances in the genomic characterization of tumors have enabled major efforts toward personalized vaccine approaches based on tumor-specific mutation-derived antigens, known as neoantigens. However, initial clinical results with neoantigen peptide-based vaccines given with a potent adjuvant have not demonstrated consistent induction of high-frequency CD8 T cell responses in humans.8 Additionally, mRNA-based neoantigen vaccines have also induced modest CD8 T cell responses.9 These data suggest that additional approaches are required to increase the magnitude of neoantigen-specific CD8 T cells. Accordingly, heterologous prime-boost vaccination (using two different vaccines) is one strategy that can significantly enhance the magnitude and alter the quality of CD8 T cell responses compared with either vaccine alone. This strategy has been used to increase immunity against ebola,10 hepatitis C virus,11 and SARS-CoV-2.12
We previously developed a self-assembling nanoparticle (SNP) vaccine that co-delivers peptide antigens with a TLR-7/8 agonist.13 The SNP vaccine was designed to be rapidly manufactured and can enable delivery of ~98% of possible neoantigen peptides as stable 20–40 nm micelles.14 The SNP vaccine elicits stem-like anti-tumor CD8 T cells when administered by the intravenous (i.v.) route in mice and mediates tumor regression.15 To expand upon these findings and to further enhance the CD8 T cell response, we modified a replication defective chimp adenovirus (ChAdOx1) to be used as a heterologous boost. The ChAdOx1 vector was selected because of the low seroprevalence in humans to avoid pre-existing neutralizing antibodies16 and can be modified to express target antigens. ChAdOx1 is safe and protective against SARS-CoV-2 in humans,17 and has demonstrated anti-tumor efficacy in multiple pre-clinical mouse tumor models as a heterologous prime-boost approach with a modified vaccinia Ankara (MVA) boost.18,19 Recent clinical trials utilizing intramuscular (i.m.) adenovirus followed by either MVA20 or a self-amplifying RNA21 were able to induce neoantigen-specific CD8 T cell responses, validating this approach for anti-tumor immunity induction.
In addition to optimizing the magnitude, breadth, and quality of tumor-specific CD8 T cell responses, an additional and important consideration is how such responses will function in the TME. The TME is a complex collection of immune and non-immune cells that can be characterized according to the degree of immune infiltration, spanning a spectrum from “immune desert” to “inflamed.”22 Recent studies have revealed that myeloid cell populations in some tumor models, such as the colorectal cancer line MC38, mirror the populations in human tumors, including monocytes, macrophages, and conventional dendritic cells (cDCs).23 Whereas cDCs can play an important role in anti-tumor functions by improving T cell activation;24 monocytes and macrophages can have either pro-tumoral or anti-tumoral functions depending on the microenvironment and associated signals.25 For example, tumor-associated macrophages (TAMs), a heterogeneous population of myeloid cells of embryonic or monocytic origin, have emerged as a promising target to promote anti-tumor immunity.26 The goal of TAM targeting therapeutics is to promote their polarization away from an anti-inflammatory (pro-tumoral) state toward a pro-inflammatory (anti-tumoral) state, sometimes referred to as an M2 phenotype to M1 phenotype polarization, respectively.27 This polarization can be induced by exposing TAMs to type I IFNs (IFNα or IFNβ), which results in the upregulation of interferon-stimulated genes (ISGs) that promote a pro-inflammatory response.28
Here, we assessed whether a heterologous prime-boost approach using SNP and ChAdOx1 could both induce high-magnitude tumor-specific CD8 T cell responses and modulate the TME. We show that i.v. ChAdOx1 elicits higher frequency tumor-specific CD8 T cell responses than i.m. ChAdOx1. Importantly, i.v. ChAdOx1 modulates the TME in a type I IFN-dependent and antigen-independent manner to promote tumor regression. These data highlight that this unique heterologous prime-boost approach with i.v. ChAdOx1 as a boost may be clinically translatable on the basis of the potency of this vaccine vector in humans.
RESULTS
Intravenous ChAdOx1 vaccination elicits high-frequency tumor-specific effector CD8 T cell responses
A key goal for therapeutic cancer vaccines is to elicit a high frequency of antigen-specific CD8 T cells. We hypothesized that a heterologous prime-boost approach could elicit higher magnitude anti-tumor CD8 T cell responses than homologous vaccination. On the basis of the previously reported data that adenoviral vectors are especially potent for eliciting CD8 T cell responses in humans, we generated a ChAdOx1 vaccine that encodes Reps1, a neoantigen expressed by the MC38 mouse colorectal cancer cell line (Figure S1A).29
We first assessed how the route of immunization altered the magnitude, phenotype, and quality of the T cell response. Mice were vaccinated i.m., the standard route used for vaccination with adenoviral vectors in the majority of infectious disease and tumor models,10,11,18,20 or i.v. with Reps1-encoding ChAdOx1. The frequency of CD8 T cell responses was assessed at peak (2 weeks) and memory (16 weeks) time points in blood and selected tissue sites by tetramer staining (Figure 1A). Two weeks post-immunization, i.v. ChAdOx1 elicited significantly higher magnitude T cell responses in the blood, spleen, liver, and popliteal lymph node (popLN) than i.m. ChAdOx1 (Figures 1B and S1B). The effector and memory phenotypes of CD8 T cells following ChAdOx1 immunization were assessed using the canonical cell surface markers CD127 (IL-7R) and KLRG1 (Figure 1C, left panel). Memory precursor effector cells (MPECs) retain CD127 expression whereas short-lived effector cells (SLECs) lose CD127 expression and upregulate KLRG1.30 Two weeks after i.v. ChAdOx1 vaccination, there was a higher frequency of SLECs and lower frequency of MPECs compared with i.m. ChAdOx1 (Figure 1C, right panel).
Figure 1. Intravenous ChAdOx1 vaccination elicits durable, higher magnitude, and more terminally differentiated CD8 T cells responses than intramuscular vaccination.
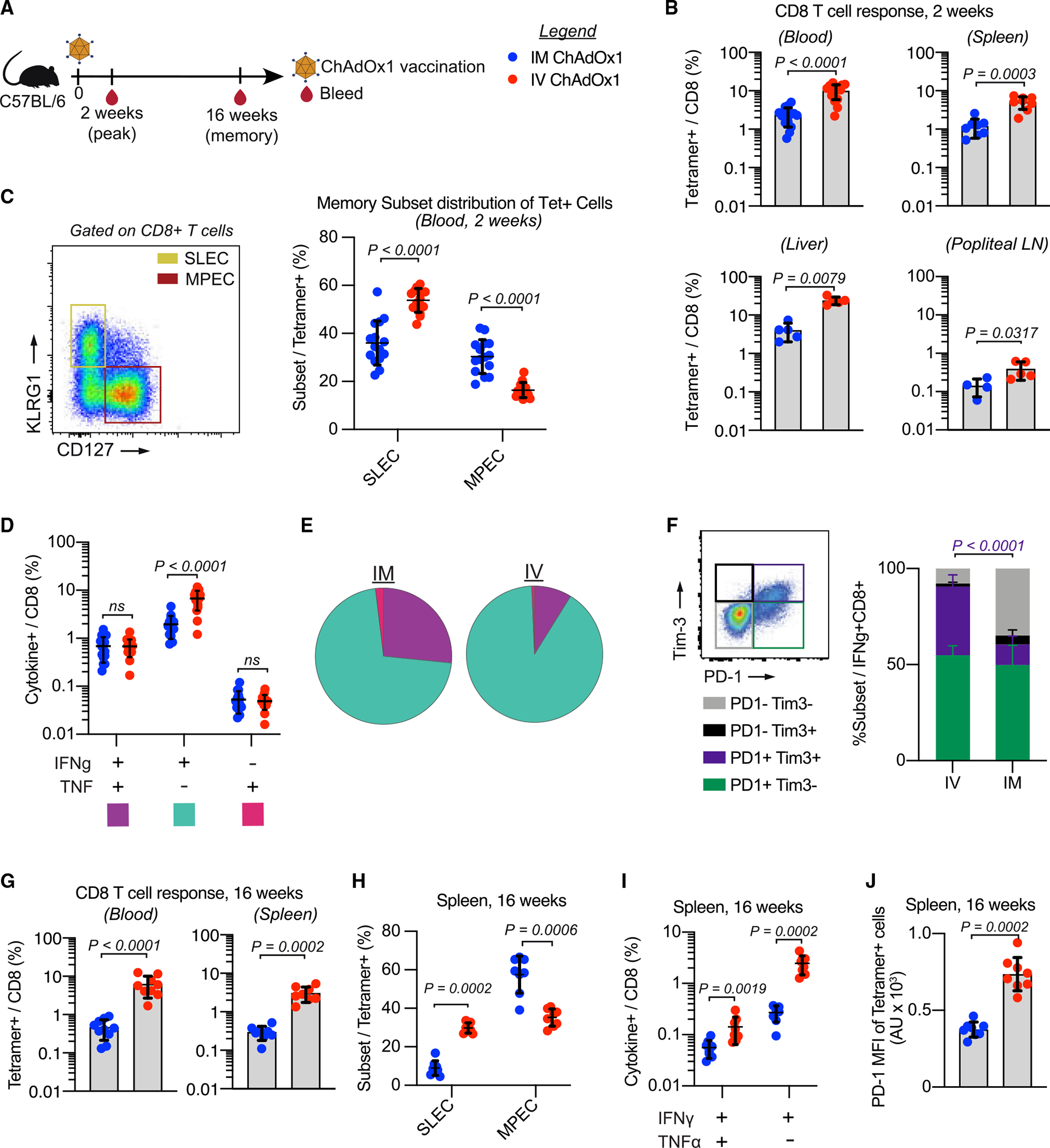
(A) Mice are vaccinated with ChAdOx1 and sampled 2 and 16 weeks post-vaccination to assess antigen-specific CD8 T cell responses. Legend is for the entire figure.
(B) Antigen-specific CD8 T cell response measured 2 weeks post-vaccination by tetramer staining single cell suspensions derived from blood (n = 15), spleen (n = 8), liver (n = 5), and popliteal lymph nodes (LNs) (n = 5). Two replicates for blood and spleen, 1 experiment for liver/popliteal LNs.
(C) Gating strategy for SLECs/MPECs (left). Frequency of tetramer+ cells that are SLECs or MPECs.
(D) Frequency of IFNγ+ and/or TNF+ CD8 T cells.
(E) Pie charts showing proportion of IFNγ+- and/or TNF+-producing CD8 T cells.
(F) Gating strategy for Tim-3/PD-1 (left). Proportion of IFNγ+ cells that express Tim-3 and/or PD-1.
(G) Antigen-specific CD8 T cell response measured 16 weeks post-vaccination by tetramer staining single cell suspensions derived from blood (n = 10) and spleen (n = 8).
(H) Proportion of tetramer+ CD8 T cells in the spleen 16 weeks post-vaccination that fall into each of the SLEC/MPEC categories.
(I) Frequency of IFNγ- and/or TNF-producing CD8 T cells from spleens collected 16 weeks post-vaccination. Monofunctional TNF-producing cells not shown as response was not above background.
(J) MFI of PD-1 on tetramer+ CD8 T cells in the spleen 16 weeks post-vaccination.
Data represented as mean ± SD; Mann-Whitney test. In (C)–(F), n = 15, 2 experimental replicates. In (H)–(J), n = 8, 2 experimental replicates.
The frequency of CD8 T cells producing IFNγ and TNF was determined by flow cytometry following peptide restimulation to assess functionality (Figure S1C). Intravenous ChAdOx1 immunization elicited higher frequencies of CD8 T cells producing IFNγ only compared with i.m. vaccination (Figure 1D). CD8 T cells producing both IFNγ and TNF represented ~25% of the antigen-specific cells following i.m. vaccination but only ~7% following i.v. vaccination (Figure 1E). Intravenous ChAdOx1 elicited a higher frequency of PD-1 and Tim-3 co-expressing antigen-specific CD8 T cells, identified by the production of IFNγ upon peptide restimulation (Figure 1F).
Similar analyses were performed 16 weeks post-vaccination to assess the durability of the CD8 T cell response. The frequency of Reps1-specific CD8 T cells remained significantly higher after i.v. ChAdOx1 compared with i.m. immunization in blood and spleen (Figure 1G). The trends observed in memory phenotype in the blood 2 weeks post-vaccination were maintained in the spleen at 16 weeks, with i.m. ChAdOx1 eliciting a response with a lower proportion of SLECs and higher proportion of MPECs than i.v. ChAdOx1 (Figure 1H). The antigen-specific CD8 T cells were still predominantly monofunctional IFNγ producing cells (Figure 1I). The expression of PD-1 remained higher on i.v. ChAdOx1 elicited CD8 T cells compared with the i.m. route (Figure 1J). The expression of Tim-3 was no longer detectable on the antigen-specific cells at week 16, so it was not possible to compare this between the T cells elicited by either route. Collectively, these data show that i.v. vaccination with ChAdOx1 elicits higher magnitude, more terminally differentiated CD8 T cell responses that are durable over a 16 week period compared with the same dose administered by the i.m. route.
Intravenous heterologous prime boost further increases the magnitude of tumor-specific CD8 T cell responses
To determine whether the interval between prime and boost affected the magnitude of CD8 T cell responses, groups of mice were primed either 4, 2, or 1 week prior to boosting with ChAdOx1 given i.v. or i.m. and tetramer staining was performed through 16 weeks (Figure S2A). There were no significant differences in the magnitude of the CD8 T cell response on the basis of the interval between prime and boost when comparing groups that received ChAdOx1 by the i.m. or i.v. route (Figure S2B). However, i.v. ChAdOx1 boost elicited significantly higher magnitude CD8 T cell responses than i.m., regardless of interval and consistent with the differences seen when using ChAdOx1 alone (Figure S2C).
To test the protective efficacy of heterologous prime-boost vaccination in a prophylactic model, we used a 2-week interval between prime and boost, which we had previously used for homologous SNP prime-boost studies and would thus enable benchmarking.15 We tested heterologous prime boost with i.v. SNP followed by i.m. or i.v. ChAdOx1, and compared this to i.m. or i.v. ChAdOx1 alone at the time of boost, and to homologous i.v. SNP prime-boost. Mice were challenged with MC38 cells 2 weeks post-boost and received a single dose of αPD-L1 at this time (Figure 2A).
Figure 2. Intravenous heterologous prime-boost vaccination elicits high-magnitude CD8 T cell responses that protect mice from tumor challenge.
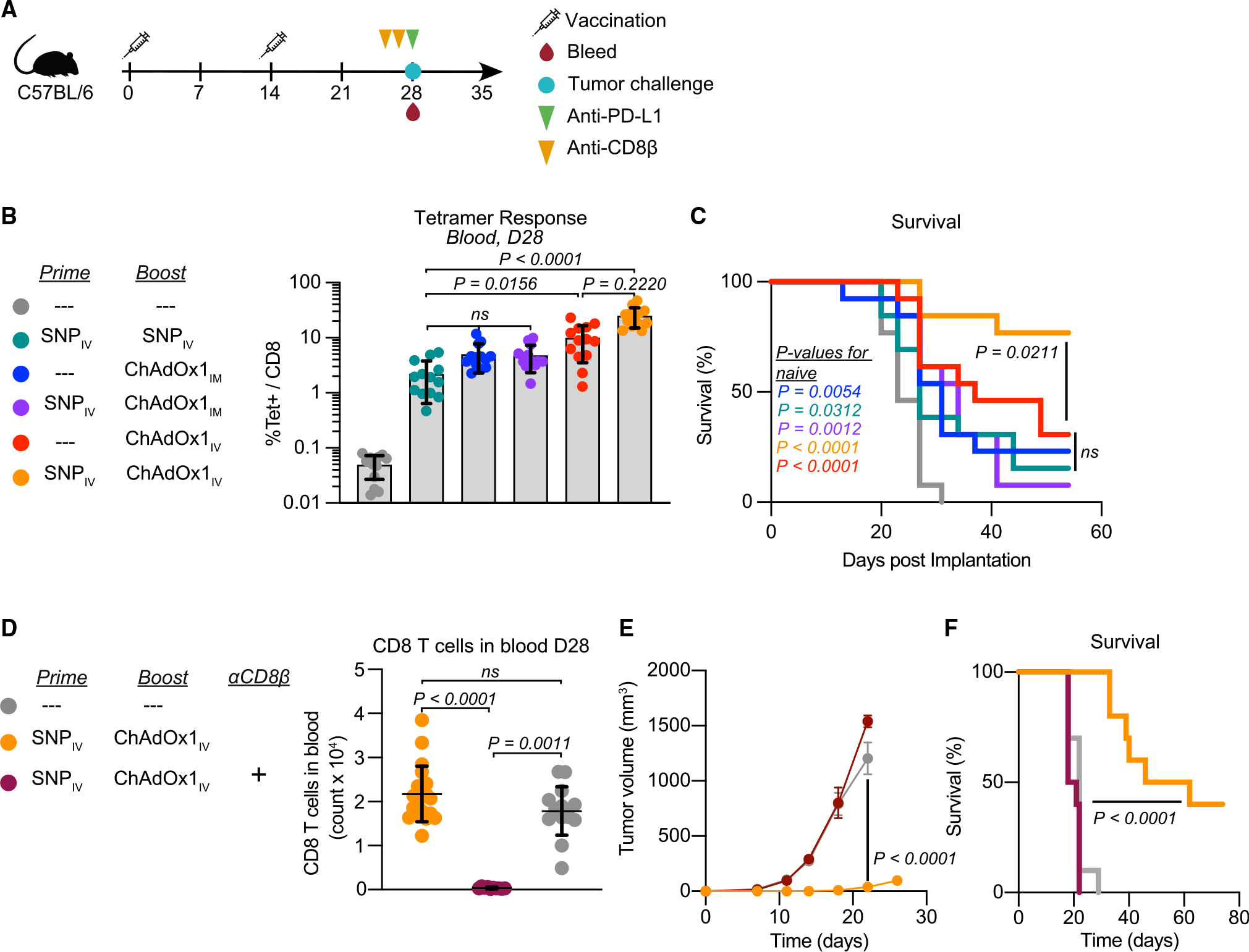
(A) Prophylactic study vaccination and sampling schedule. Mice are primed and boosted 2 weeks apart and then challenged with tumor cells 2 weeks post-boost. At the time of tumor challenge, mice are also bled to assess T cell responses and given 1 dose of αPD-L1. Some mice also received αCD8β antibody 3 days and 1 day prior to tumor challenge.
(B and C) Magnitude of Reps1-specific CD8 T cell responses in blood at the time of tumor challenge measured by tetramer staining (B). Survival curve following tumor implantation (C).
(D–F) CD8 T cell count in blood at time of challenge (D). Average tumor growth curves following MC38 tumor challenge in i.v. heterologous prime-boost group with and without CD8 T cell depletion (E). Survival curve for i.v. heterologous prime-boost group with and without CD8 T cell depletion (F).
In (B) and (D), data represented as mean ± SD; Kruskal-Wallis test with Dunn’s correction for multiple comparisons. In (C) and (F), Mantel-Cox log rank test, compared with naive mice unless otherwise indicated. In (E), two-way ANOVA with Bonferroni correction for multiple comparisons. In (B) and (C), n = 13 and representative of 2 replicates; in (D)–(F), n = 10, 1 experiment.
Heterologous prime boost with i.m. ChAdOx1 did not increase the magnitude of the Reps1-specific CD8 T cell response compared with either SNP given twice or i.m. ChAdOx1 vaccination alone (Figure 2B). In contrast, i.v. heterologous prime-boost significantly increased the magnitude of antigen-specific CD8 T cell responses compared with homologous i.v. SNP vaccination and trended toward higher magnitude relative to i.v. ChAdOx1 vaccination alone (Figure 2B). All vaccinated groups showed reduced tumor growth (Figure S3) and improved survival compared with unvaccinated animals (Figure 2C).
To confirm that protection was dependent on CD8 T cell responses, CD8β+ cells were depleted prior to challenge (Figure 2D), and this resulted in a complete loss of tumor growth control (Figure 2E) and reduced survival (Figure 2F). These data demonstrate that protection from MC38 in the prophylactic setting is dependent on the magnitude of pre-existing Reps1-specific CD8 T cell responses, which is best induced by heterologous prime boost with i.v. SNP and i.v. ChAdOx1.
Therapeutic heterologous prime-boost vaccination with i.v. ChAdOx1 controls established tumors
Protection following heterologous prime-boost immunization was next assessed in a therapeutic model with established MC38 tumors. Mice were vaccinated with SNP 1 week after tumors were implanted and became palpable, then boosted a week later with ChAdOx1. The shortened one week interval between prime and boost was necessary due to the rapid growth of the tumors. All mice received αPD-L1 weekly for a total of 3 doses starting at the time of boost (Figure 3A). Given the lack of efficacy in the prophylactic setting observed when administering ChAdOx1 i.m., all subsequent studies focused on the i.v. route of administration as a boost.
Figure 3. Intravenous ChAdOx1 vaccination promotes tumor regression when used as part of a heterologous prime-boost vaccination strategy.
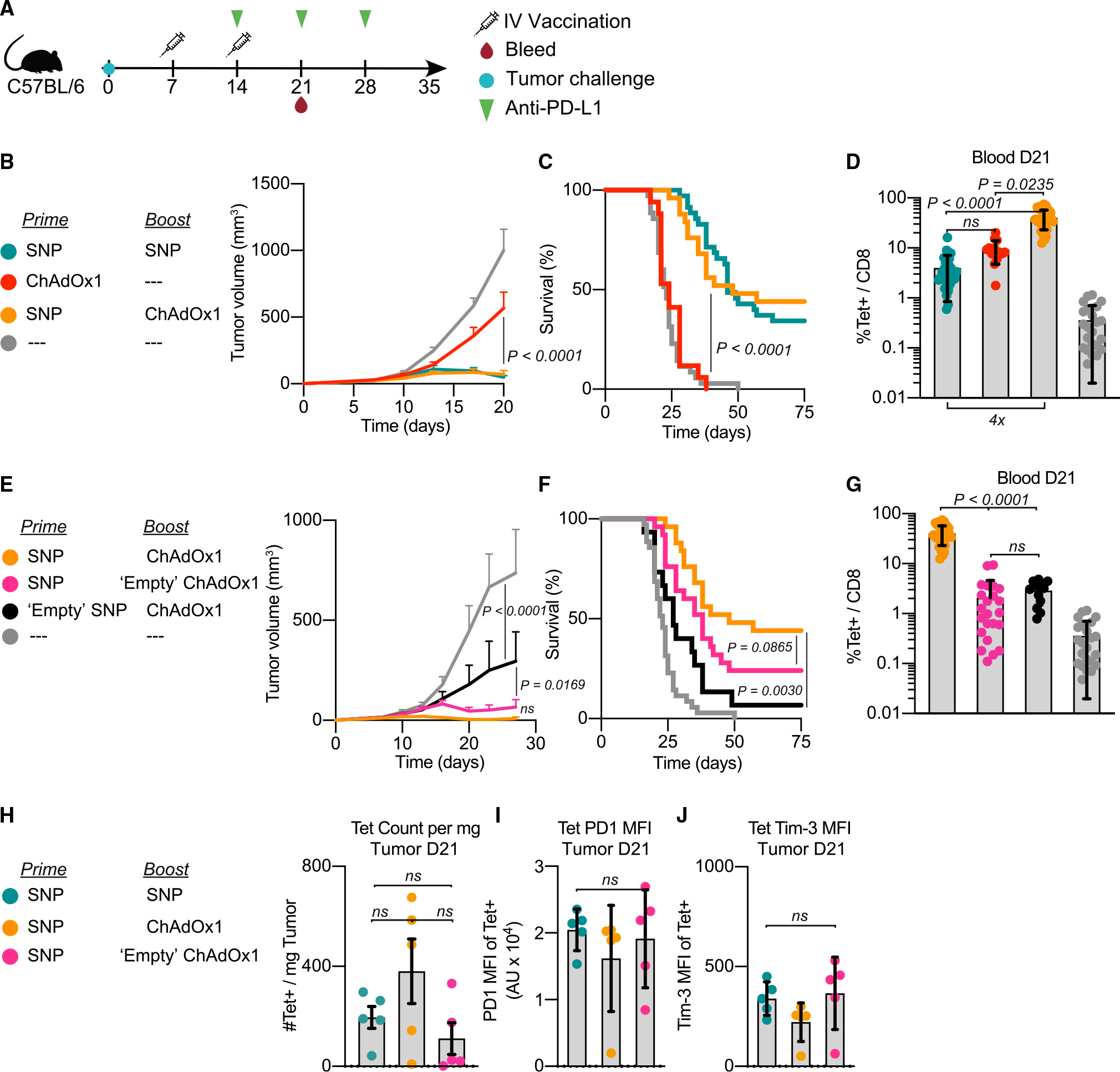
(A) Schematic of therapeutic study design. Mice were implanted with MC38 and vaccinated on days 7 and 14 with the vaccinations indicated in the legend. Mice received 3 doses of αPD-L1 administered weekly beginning on day 14. Blood, spleens, and tumors were harvested on day 21 to assess Reps1-specific CD8 T cell responses.
(B–D) Average tumor growth curves for the i.v. heterologous prime-boost group compared with the positive control (i.v. SNP given twice) and i.v. ChAdOx1 prime alone (B). Survival curve (C). Magnitude of Reps1-specific CD8 T cell responses in blood at day 21, measured by tetramer staining blood (D).
(E–G) Average tumor growth curves for the i.v. heterologous prime-boost group compared with antigen-free vaccination controls (E). Either SNP does not contain the Reps1 antigen (black) or ChAdOx1 does not contain the Reps1 antigen (pink). Survival curve (F). Magnitude of Reps1-specific CD8 T cell responses in blood at day 21, measured by tetramer staining blood (G).
(H–J) Number of Reps1-specific CD8 T cells per mg of tumor tissue processed in groups with equivalent efficacy (H). MFI of PD-1 (I) or Tim-3 (J) on Reps1-specific CD8 T cells in the tumor at day 21.
In (B) and (E), two-way ANOVA with Bonferroni correction for multiple comparisons, p values compared with naive mice. In (C) and (F), Mantel-Cox log-rank test, groups compared as indicated in figure by paired legend color-matched circles. In (D) and (G–J), data represented as mean ± SD; Kruskal-Wallis test with Dunn’s correction for multiple comparisons. In (B)–(G), n = 8 or 9, and data are representative of 2 or 3 experimental replicates per group. In (C), (D), (F), and (G), data from all replicates are merged. In (H)–(J), n = 5 and representative of 2 replicates.
We had previously reported that SNP given twice i.v. can provide ~40% protection in the therapeutic model with MC38 tumor challenge.15 We used this to benchmark the heterologous prime-boost group and i.v. ChAdOx1 alone group. The i.v. heterologous prime-boost vaccine group displayed similar control of tumor growth (Figure 3B) and survival (Figure 3C) as homologous prime and boost with SNP. Both prime-boost vaccine groups had significantly better tumor control than the ChAdOx1 prime alone group (Figures 3B and 3C). Interestingly, mice that received i.v. ChAdOx1 alone as a primary vaccination did not exhibit tumor regression despite having ~10% Reps1-specific CD8 T cell responses, which were comparable with the i.v. SNP prime-boost group that promoted tumor regression. (Figure 3D). These data show that homologous or heterologous prime-boost vaccination with SNP-SNP or SNP-ChAdOx1 was required for protection compared with i.v. ChAdOx1 alone. This led to further analysis of how the prime and boost vaccinations influenced the adaptive CD8 T cell response as well as a potential role of innate activation by vaccines.
Role of CD8 T cells in mediating tumor control following therapeutic prime-boost vaccination
Adenoviral vectors are potent inducers of innate cytokines.31 Thus we hypothesized that i.v. administration of ChAdOx1, in addition to generating or boosting CD8 T cell responses, could also induce systemic innate cytokine production that enhances anti-tumor efficacy when used as a boost. To delineate the requirement of the antigen versus innate activation in the prime or boost vaccinations, we used SNP and ChAdOx1 vaccines that either delivered the Reps1 antigen or an irrelevant antigen referred to as “empty.” Remarkably, priming with SNP followed by i.v. boosting with “empty” ChAdOx1 displayed significantly reduced tumor growth compared with unvaccinated control mice and were comparable with the heterologous prime boost with SNP-ChAdOx1 both containing the Reps1 antigen (Figure 3E). Moreover, the group that received Reps1-SNP prime followed by “empty” ChAdOx1 boost displayed significantly better tumor growth control than the group which received the “empty” SNP followed by Reps1-encoding ChAdOx1. These differences in tumor control were reflected in the survival curves (Figure 3F). This difference between the Reps1 SNP and the “empty” SNP group demonstrates the importance of encoding the antigen in the first vaccination to generate tumor-specific CD8 T cells.
In assessing the CD8 T cell responses in blood 7 days post-boost, the “empty” SNP prime followed by Reps1-ChAdOx1 boost group and the Reps1-SNP prime followed by “empty” ChAdOx1 boost group had equivalent CD8 T cell responses and were both significantly lower magnitude than the heterologous prime-boost the Reps1-SNP/Reps1-ChAdOx1 group (Figure 3G). This further suggests that the magnitude of CD8 T cell response one week post-boost is not the determinant factor in mediating tumor control in the therapeutic setting, thus suggesting that innate stimulus from the vaccine boost contributes to the observed efficacy of the “empty” ChAdOx1 boost.
To confirm that anti-tumor efficacy requires CD8 T cells in the therapeutic setting, anti-CD8β was administered just prior to, and immediately after, the antigen-encoding ChAdOx1 boost (Figure S4A). Depletion of CD8 T cells was verified in the spleen and tumor draining lymph node (tdLN) one day post-boost vaccination (Figures S4B and S4C) and in blood 1 week post-boost (Figure S4D). Depletion of CD8 T cells abrogated control of tumor growth (Figure S4E) and survival (Figure S4F). Thus, CD8 T cells are required for therapeutic efficacy after the boost vaccination.
Finally, we assessed the magnitude and exhaustion phenotype of Reps1-specific CD8 T cell responses in the tumors of select vaccine groups 1 week post-boost vaccination. There were no significant differences in the number of Reps1-specific CD8 T cells in the tumor (Figure 3H) or expression of PD-1 or Tim-3 on these cells (Figures 3I and 3J). Collectively the data suggest that two vaccinations are required to promote tumor regression in mice challenged with MC38. A CD8 T cell response is required, but the protection following the “empty” ChAdOx1 boost highlights a second mechanism that is independent of boosting CD8 T cell responses, given the lack of correlation with magnitude 1 week post-boost.
ChAdOx1 vaccination activates STING-dependent induction of type I IFNs required for priming CD8 T cell responses
Intravenous ChAdOx1 vaccination may promote anti-tumor efficacy through dual mechanisms, by boosting CD8 T cells and through systemic innate activation. Thus, a kinetic analysis of innate cytokine production in serum was assessed following i.v. ChAdOx1 vaccination in naive mice (Figure 4A). Intravenous vaccination with ChAdOx1 induced rapid production of IFNα, CXCL-10 (IP-10), and IL-12, canonical cytokines important for CD8 T cell priming, which are also known to have direct effects on immune activation and tumor control.32 As SNP contains the TLR-7/8a, we also assessed production of these cytokines following i.v. SNP given that this vaccine is also effective in this model (Figures 4B–4D).
Figure 4. ChAdOx1 vaccination activates STING to elicit transient systemic release of IFNα, which is required for priming CD8 T cell responses.
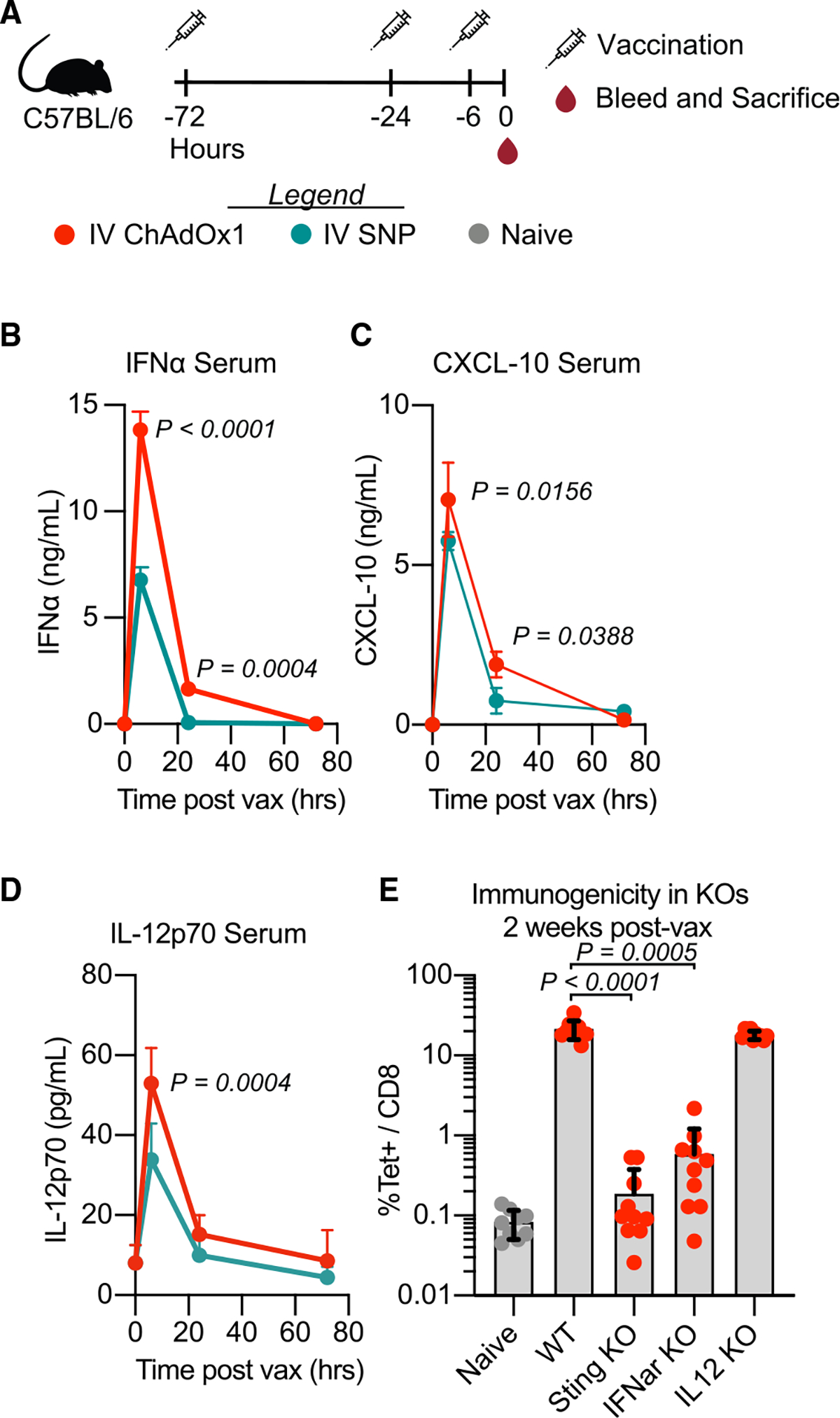
(A) Groups of mice were vaccinated with i.v. ChAdOx1 or i.v. SNP 72, 24, or 6 h prior to sampling.
(B–D) Cytokine measured in serum 6, 24, and 72 h after i.v. vaccination. (B) IFNα, (C) CXCL-10, and (D) IL-12p70. n = 3; data representative of 2 experimental replicates.
(E) Antigen-specific CD8 T cell response 2 weeks post i.v. ChAdOx1 vaccination measured by tetramer staining in WT, STING KO, IFNα receptor KO, and IL-12 KO mice in blood. n = 5; data composite of 2 experimental replicates. Data represented as mean ± SD (B–D), two-way ANOVA with Bonferroni correction for multiple comparisons. In (E), Kruskal-Wallis test with Dunn’s correction for multiple comparisons.
To assess the role of these cytokines on CD8 T cell priming following ChAdOx1 i.v. in naive mice, IFNα/β receptor subunit 1 (IFNAR1) and IL-12 knockout (KO) mice were used. IFNAR1 KO mice had a significant reduction in the magnitude of CD8 T cell responses (Figure 4E). However, there was no effect on the responses in IL-12 KO mice (Figure 4E). To determine the innate pathway by which IFNα was induced, STING deficient mice were used, as ChAdOx1 is a DNA virus and other chimp adenoviruses have been shown to activate this signaling pathway. Indeed, CD8 T cells were significantly reduced in STING deficient mice. Together these data show that ChAdOx1 induces STING-dependent type I IFNs required for CD8 T cell priming, and these cytokines potentially have an additional anti-tumor function during the boost.
Type I IFNAR signaling is required for tumor regression following i.v. ChAdOX1
We sought to understand whether type I IFNs are required for tumor control in the therapeutic setting. An IFNAR1 blocking antibody was administered intraperitoneally 1 day before and after the boost vaccination (Figure 5A). The protection mediated by i.v. ChAdOx1 boosting, with or without the Reps1 antigen, was indeed dependent on IFNAR1 signaling. The IFNAR1 blockade resulted in loss of tumor control (Figures 5B and 5D) and abrogated any survival benefits of i.v. ChAdOx1 boosting (Figures 5C and 5E). Interestingly, the magnitude of the Reps1-specific CD8 T cell response in blood 1 week post-boost was unaffected by IFNAR1 blockade (Figure 5F). The lack of tumor control in the presence of high-magnitude Reps1-specific CD8 T cell responses elicited by the i.v. heterologous prime-boost regimen observed in the presence of IFNAR1 blockade demonstrates the critical requirement of functional IFNAR signaling for anti-tumor efficacy. To validate these data in a different therapeutic tumor model, we used a B16F10 melanoma cell line that is considered a “cold tumor” as it is not immune-infiltrated. To delineate the role of CD8 T cells and the TME, we used a modified B16F10 cell line that expresses the MC38 neoantigen Adpgk. We observed the same dependence on IFNAR1 signaling using an i.v. heterologous prime-boost vaccination to treat the Adpgk-expressing B16F10 tumors (Figures S5A–S5D).
Figure 5. Interferon alpha is required for mediating anti-tumor efficacy after ChAdOx1-i.v. treatment.
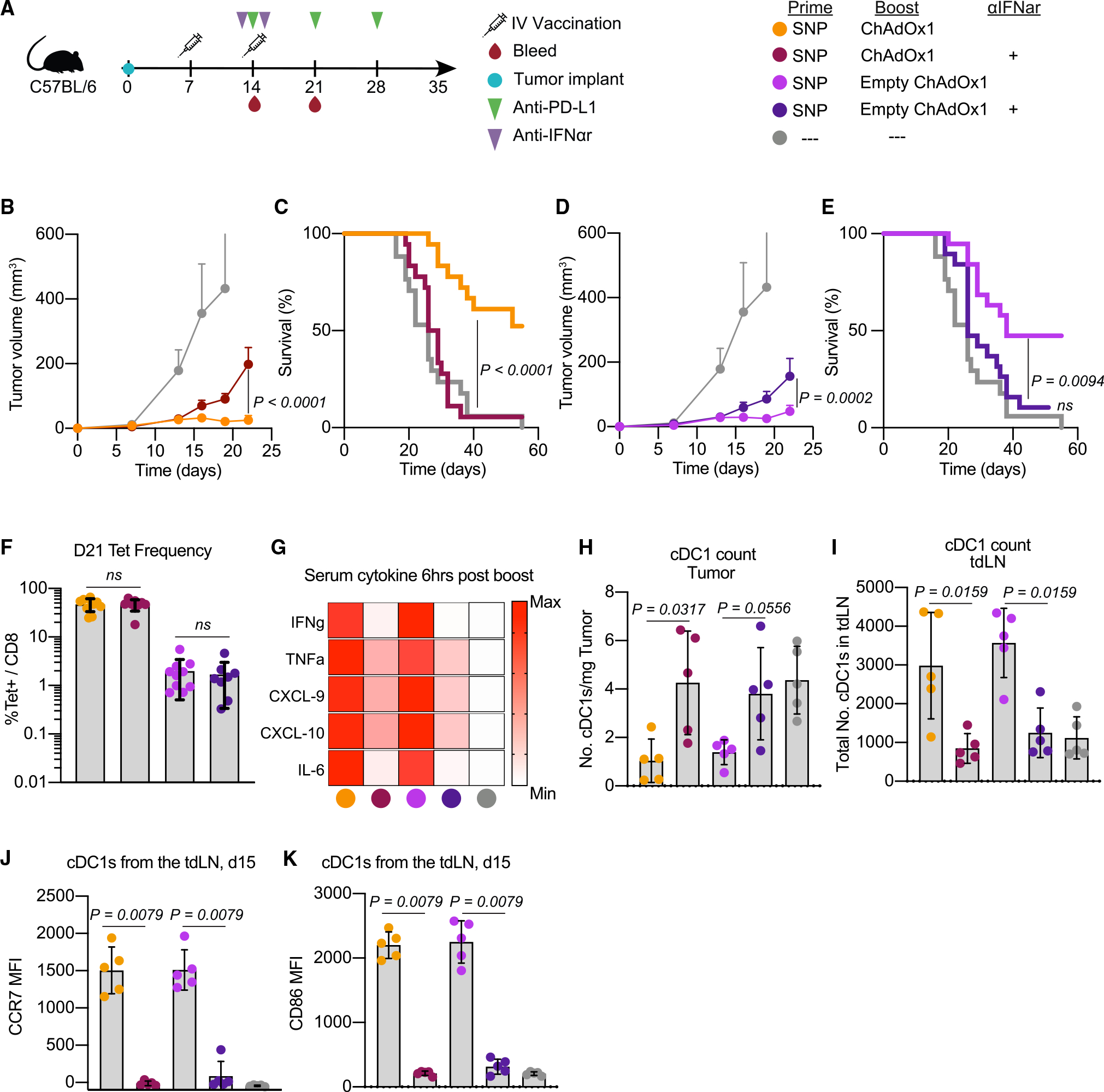
(A) Schematic of therapeutic study design as described in Figure 3. Some groups received saturating doses of IFNα receptor blocking antibody one day prior to and one day after the boost vaccination. Blood was collected on day 21 to assess Reps1-specific CD8 T cell responses. Tumor and tumor-draining lymph node (tdLN) samples were collected 1 day post-boost to assess cDC1s.
(B and D) Average tumor growth curves for the heterologous prime-boost vaccinations with either the (B) Reps1-encoding ChAdOx1 or (D) empty ChAdOx1.
(C and E) Survival curve for the heterologous prime-boost vaccination groups with either the (C) Reps1-encoding ChAdOx1 or (E) empty ChAdOx1.
(F) Magnitude of the Reps1-specific CD8 T cell response 1 week post-boost vaccination measured by tetramer staining blood.
(G) Heatmap plot of the average amount of a subset of cytokines assayed by Luminex present in serum 6 h post-boost. Scale is relative to the range for each individual cytokine.
(H) Number of cDC1s per mg of tumor found 1 day post-boost.
(I) Number of cDC1s in the tdLN 1 day post-boost.
(J and K) Expression of the maturation and migration marker (J) CCR7 and activation marker (K) CD86.
In (B) and (D), two-way ANOVA with Bonferroni correction for multiple comparisons, p values compared with naive mice unless otherwise indicated. In (C) and (E), Mantel-Cox log rank test. In (F) and (H)–(K), data represented as mean ± SD, Mann-Whitney test. In (B)–(E), n = 8 or 9, 2 experimental replicates, data for both panel sets is from the same experiments and separated into the Reps-1 encoding ChAdOx1 and empty ChAdOx1 boost for presentation purposes. In (F), n = 8–10, data representative of 2 experimental replicates. In (G)–(K), n = 5, data representative of 2 experimental replicates.
To better understand how ChAdOx1-induced type I IFN signaling may be enhancing tumor regression, we initially examined the cytokine milieu following boosting. Type I IFNs have pleotropic effects on multiple cell types, thus affecting production of cytokines, chemokines, and DC activation which could all have a role affecting tumor control. IFNAR1 blockade at the time of the boost resulted in a decrease of multiple cytokines including the pro-inflammatory cytokines IFNγ, TNF, and IL-6. In addition, there was a decrease in the chemokines CXCL-9 and CXCL-10, which promote T cell infiltration into the tumor24 (Figures 5G and S6A–S6F).
One additional and notable effect of type I IFN signaling is activation of dendritic cells which are essential for effective anti-tumor immunity.24,33 We thus focused on the effects of IFNAR1 blockade on type 1 cDCs (cDC1s) in the tumor and the tdLN 1 day post-boost, based on their critical role in CD8 T cell priming and function.34 In the tumor, i.v. ChAdOx1 vaccination was associated with a reduction in the number of cDC1s, and this was dependent on IFNAR1 signaling (Figure 5H). In contrast, the number of cDC1s in the tdLN increased in the i.v. heterologous prime-boost groups and this was also dependent on IFNα receptor signaling (Figure 5I). This was associated with increased expression of CCR7 on these cDC1s (Figure 5J), which is known to be required for migration of cDC1s from the tumor to the tdLN to prime anti-tumor CD8 T cell responses.33 In addition, type I IFNs were also required for the upregulation of the co-stimulatory molecule CD86 on cDC1s (Figure 5K). Collectively, these data suggest that type I IFNs induced by i.v. vaccination with ChAdOx1 promote the activation, maturation, and migration of cDC1s to the tdLN, which may support the anti-tumor efficacy of i.v. vaccination.
Intravenous vaccination with ChAdOx1 remodels the TME
On the basis of the heterogeneity of myeloid cells and their regulatory properties within the TME, we performed singe-cell RNA sequencing (scRNA-seq) on myeloid cells from tumors and spleens 24 h post-boost with ChAdOx1 and compared these with cells from unvaccinated mice (Figure 6A). To obtain sufficient cells for this analysis from each tissue, CD45+, lineage−, CD11b+, and/or CD11c+ cells were stained, sorted, and processed using the 10X Genomics 5′ sequencing protocol.
Figure 6. Intravenous ChAdOx1 elicits Type I IFNs that increase the ratio of pro-inflammatory to anti-inflammatory monocytes at the tumor site.
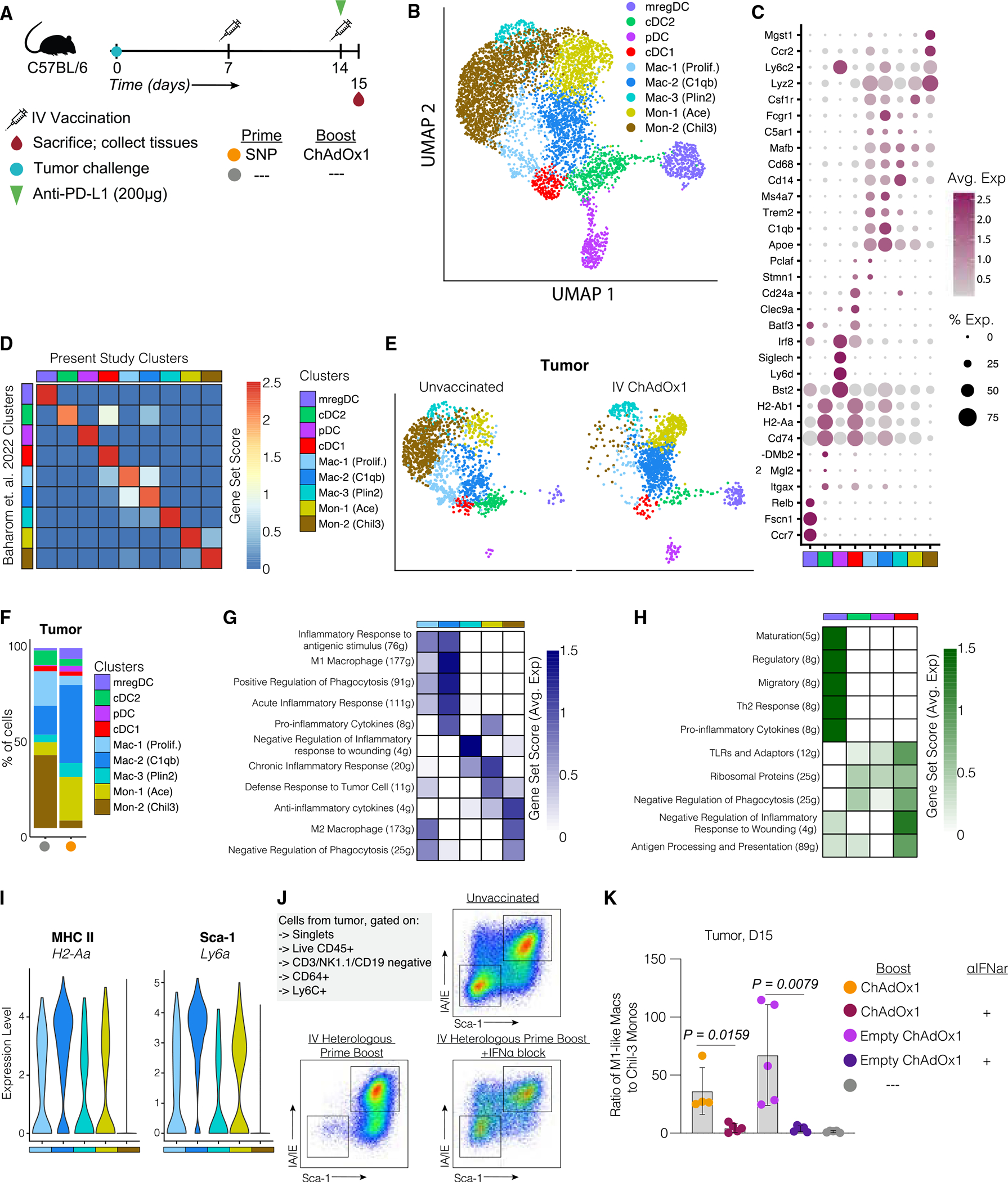
(A) Mice were implanted with MC38 and vaccinated with i.v. heterologous prime boost using the antigen-encoding ChAdOx1. Spleens and tumors were harvested 1 day post-boost vaccination. Myeloid cells were sorted by fluorescence-activated cell sorting (FACS) and used for scRNA sequencing.
(B) UMAP visualization of scRNA sequencing data from spleen and tumor isolated monocytes, macrophages, and DCs. Classified according to their metaclusters identity.
(C) Dot plot of canonical markers identifying specific DC, monocyte, and macrophage subsets.
(D) Correlation matrix of the metaclusters identified in the present study and the metaclusters identified in a published dataset (Baharom et al.).35
(E) Uniform manifold approximation and projections (UMAPs) of tumor MNPs separated by treatment group.
(F) Bar graphs summarize frequencies of monocyte and macrophage metaclusters for each animal across different treatment groups in the tumor.
(G) Heatmap of gene set score analysis results focused on mono/mac subsets.
(H) Heatmap of gene set score analysis results focused on DC subsets.
(I) Violin plots of 2 differentially expressed genes (H2–2a, Ly6A) between the Chil-3 monocytes and the remaining mono/mac metaclusters.
(J) Example staining of Chil-3 monocytes and activated monocytes found in the tumor of different treatment groups 1 day post-boost.
K) Ratio of activated monocytes to Chil-3 monocytes found in the tumor 1 day post-boost.
In (A)–(H), 2 mice in the unvaccinated group, 3 mice in the i.v. heterologous prime-boost group, scRNA sequencing experiment performed once. In (K), n = 5, and data representative of 2 experimental replicates.
After excluding contaminating T cells, B cells, natural killer (NK) cells, and granulocytes, the remaining populations of monocytes, macrophages, and DCs were subjected to a second round of dimensionality reduction and clustering. This analysis revealed thirteen unique cell clusters/states visualized on the UMAP space (Figure S7A). These clusters were manually reclassified into nine “metaclusters” (Figure 6B), based on their hierarchical ordering using their Euclidean distance (Figure S7B) as well as the focal points of the compacted cells on a density plot (Figure S7C). The new metaclusters were annotated on the basis of the mRNA expression levels of a curated list of canonical markers (Figure 6C), and gene set scores of signature genes (Figure 6D), previously described by Baharom et al.35 The meta-clusters included four DC populations including migratory/regulatory DCs (mregDCs; Ccr7, Fscn1, and Relb), pDCs (Siglech, Ly6d, and Irf8), cDC1s (Batf3, Clec9a, and Cd24a), type 2 cDCs (cDC2s) (Mgl2, H2-Dmb2, and Itgax), three macrophage subpopulations (Stmn1, Apoe, Mafb, C1qb, and Plin2), and two monocyte populations (Csf1r, Ace, Ccr2, Mgst1, and Chil3) (Figure 6C). All nine metaclusters were present in both spleen and tumor (Figures S7D and S7E).
The reduction in Chil3 monocytes in the tumor following i.v. vaccination with ChAdOx1 was striking (Figures 6E and 6F). There was a trend toward an increase in both Ace monocytes and C1qb macrophages in the tumor of ChAdOx1-vaccinated mice compared with the unvaccinated mice (Figure 6F). Supervised functional analysis using gene sets to score the expression of specific pathways revealed that the C1qb macrophages expressed pro-inflammatory cytokines and an M1 macrophage-like transcriptional profile, which suggests an anti-tumoral function (Figure 6G). In contrast, the Chil3 monocytes highly expressed anti-inflammatory cytokines and were characterized by an M2 macrophage-like transcriptional profile, representing their immunosuppressive and pro-tumoral function.35 Thus, i.v. ChAdOx1 vaccination alters the monocyte/macrophage compartment by decreasing the frequency of immunosuppressive Chil3 monocytes and increasing the frequency of pro-inflammatory C1qb macrophages.
In addition, analysis of the DCs in the tumor revealed an increase in the frequency of mregDCs in the tumor of i.v. ChAdOx1-vaccinated mice (Figures 6E and 6F). As depicted in Figure 6H, these cells presented higher gene set scores for maturation, migratory, regulatory, and Th2 response functions,36 and pro-inflammatory cytokines, which may represent their higher maturation state. Also, cDC1 and mregDC showed higher score for negative regulation of inflammatory response to wound healing, similar to C1qb macrophages (Figure 6H). Collectively, these data suggest that i.v. vaccination increases the presence of pro-inflammatory myeloid cells in the tumor that can contribute to tumor regression.
Intravenous vaccination with ChAdOx1 results in a type I IFN-dependent decrease of Chil3+ monocytes in the TME
Testing differentially expressed genes among different monocyte/macrophage metaclusters identified two genes encoding cell surface proteins (Figure 6I), including MHC class II (H2-Aa) and stem cell antigen gene 1 (Sca-1, Ly6A). By sequential gating of antigen-presenting cells (APCs), we were able to distinguish cells that are likely the M2-like Chil3 monocytes (Sca-1−, MHC-I−) and activated M1-like macrophages (Sca-1+, MHC-II+) and used this staining panel to assess monocyte/macrophage populations in tumors 1 day post-boost (Figure 6J). There was a significant reduction in the frequency of Chil3 monocytes in the i.v. heterologous prime boost compared with the unvaccinated mice and those that received both vaccinations and the IFNAR1 blocking antibody (Figure 6J). We quantified this by calculating the ratio of activated macrophages to Chil3 monocytes and found that IFNα signaling shifts this ratio in favor of the activated and likely pro-inflammatory M1-like macrophages (Figure 6K). These data suggest that type I IFNs elicited by i.v. ChAdOx1 vaccination are altering the balance of myeloid cells at the tumor site in favor of pro-inflammatory states that may enable CD8 T cell function.
DISCUSSION
A primary goal of cancer vaccines is to prime or expand tumor-specific T cell responses that can mediate tumor regression.6 The majority of therapeutic vaccine studies in humans against cancer use one vaccine platform. This may be a suboptimal strategy to elicit high-magnitude and high-quality T cell responses, which will be required for effective anti-tumor immunity. Heterologous prime-boost vaccination strategies have been effective in generating potent adaptive immunity against multiple infectious diseases. This approach may also be optimal for generating anti-tumor immunity and has been tested in multiple pre-clinical models18,19 and clinical studies.20,21 Of note, most studies have focused on administering vaccines i.m. Here, by using the ChAdOx1 as an i.v. boost we have demonstrated a striking increase in CD8 T cell responses compared with i.m. vaccination. Such responses remained elevated over a prolonged period, remaining at ~10% of the CD8 T cells in blood 16 weeks post-vaccination in mice. This improvement in magnitude and durability following i.v. ChAdOx1 vaccination may be broadly applicable for diseases in which high-frequency systemic T cells are required for protection.
In these studies, we focused on the role of CD8 T cells, as we used defined neoantigen epitopes that induce such responses. Thus, there were low to undetectable CD4 T cell responses in these studies. Nevertheless, it is clear that CD4 T cells can have a demonstrable anti-tumor effect through a number of mechanisms and inducing such responses could be beneficial.21 Both the SNP vaccine formulation which uses long peptides linked to TLR 7/8a or adenoviral vaccines are capable of eliciting CD4 T cell immunity and could be used if neoantigens containing MHC class II epitopes were delivered by either of these platforms.
Although it is clear that T cells are required for efficacy of CPIs, in many instances this may be insufficient as the newly primed CD8 T cells may encounter an immunosuppressive TME that inhibits their function through multiple mechanisms beyond checkpoints.37 Here we demonstrated that i.v. vaccination with ChAdOx1 had a secondary effect in reducing the frequency of immunosuppressive Chil3 monocytes and a concomitant increase in the frequencies of M1 macrophages (pro-inflammatory) through an IFNAR1 signaling-dependent mechanism. We also found that systemic type I IFNs were associated with an increase in the number of cDC1s in the tdLN expressing the migratory marker CCR7 and co-stimulatory molecule CD86. We hypothesize that these may be priming de novo anti-tumor T cell responses to support anti-tumor immunity.38
The anti-tumoral effect of type I IFNs has been known for decades; despite a lack of understanding of the underlying mechanism of action, recombinant IFNα2 was used in the 1980s as a treatment for cancer.39,40 This was associated with toxicity and subsequently discontinued with the development of more well-tolerated cancer therapeutics for which the mechanism of action was better understood. The mechanisms underpinning the anti-tumor effects of IFNs on myeloid, lymphoid, and tumor cells have only begun to be understood more recently.41 We focused predominantly on the effects of systemic type I IFNs on cDC1s, monocytes, and macrophages as these were the most readily detectable. However, IFNs are pleotropic and can also increase the expression of tumor cell antigen processing and presentation machinery, increase proliferation and activation of NK cells, reduce regulatory T cell (Treg) activity, and increase T cell effector function, among other effects.28 It is possible that the vaccine elicited Type I IFNs are orchestrating a systemic enhancement of the immune response through the activation of many of these pathways to promote anti-tumor immunity. One important aspect to note is that the induction of type I IFN from vaccine stimulation in vivo may be more physiological than providing exogenous cytokine systemically.
We have previously referred to the dual role of using vaccines to increase CD8 T cells and mediate innate immunity to modulate the TME as “vax-innate” in the context of the SNP vaccine.35 Here, the use of ChAdOx1 as a boost further substantiates this concept but provides an approach to substantially enhance the magnitude of CD8 T cells that can be generated. Indeed, adenoviral vectors are among the most potent vaccines for eliciting T cells in humans for a number of infectious disease indications and it is likely that the data reported here will translate into humans. Ongoing efforts are focused on determining whether the increased boosting effect of i.v. ChAdOx1 observed in mice is also observed in non-human primates and will provide important data for the safety and translatability in humans.
Limitations of the study
There are some limitations to the studies presented herein. The studies were performed primarily with the MC38 tumor model, though the requirement of IFNAR1 signaling is confirmed in a second model of B16F10 melanoma expressing the neoantigen Adpgk. In addition, the significant improvement in protection elicited by i.v. heterologous prime boost relative to homologous SNP vaccination is most clear in the prophylactic setting (Figure 2). By contrast in the therapeutic setting, control of tumor growth is comparable between homologous and heterologous prime-boost vaccination (Figure 3). This is likely due to the inherent difference between the prophylactic setting, in which a larger neoantigen-specific CD8 T cell response present at the time of the tumor challenge enhances protection, as there is no role for innate immune activation as the tumor is not established. In the therapeutic setting both CD8 T cells and innate immunity are required. As both SNP and ChAdOx1 elicit systemic type I IFNs, which are critical for modulating the TME, the increased magnitude of CD8 T cells with the heterologous prime boost is not required for improved tumor control. Nevertheless, on the basis of the potency of adenoviral vectors in humans for inducing CD8 T cell responses, it is likely that the heterologous prime boost with ChAdOx1 will provide higher magnitude responses than homologous prime boost with SNP, which may be important across different tumors in humans.
Finally, the translatability of the IFNα-mediated TME modulation from mice to humans has not yet been demonstrated. We recently demonstrated that stratifying human tumors on the basis of the expression of the immunosuppressive Chil3 monocyte metacluster gene set, that certain cancer patients with a “high” Chil3 score had a worse clinical prognosis than those with a low Chil3 score.35 However, the data regarding the effects of type I IFNs on myeloid cells is correlational, and quantifying the specific contributions of any singular effect of type I IFNs was beyond the scope of the present studies. Nevertheless, the ability to enhance DC activation in the tdLN and the potential to limit inhibitory macrophages is possible and likely to have application across various tumors and should be assessed prospectively in humans.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by lead author, Robert Seder (rseder@mail.nih.gov).
Materials availability
Requests for SNP vaccine formulations or ChAdOx1 viral vector vaccines can be directed to Vaccitech North America.
Data and code availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-mouse B220 (PE-Cy7); clone RA3-6B2 | BD Biosciences | Cat# 552772; RRID:AB_394458 |
| Anti-mouse CCR7 (BV421); clone 4B12 | Biolegend | Cat# 120120; RRID:AB_2561446 |
| Anti-mouse CD3 (BUV395); clone145-2C11 | BD Biosciences | Cat# 563565; RRID:AB_2738278 |
| Anti-mouse CD3 (Alexa 700); clone 17A2 | Biolegend | Cat# 100216; RRID:AB_493697 |
| Anti-mouse CD4 (BUV395); clone RM4-4 | BD Biosciences | Cat# 740209; RRID:AB_2739958 |
| Anti-mouse CD8 (APCeFluor780); clone 53–6.7 | eBioscience | Cat# 47-0081-82; RRID:AB_1272185 |
| Anti-mouse CD11b (AF700); clone M1/70 | BD Biosciences | Cat# 557960; RRID:AB_396960 |
| Anti-mouse CD11c (PE); clone HL3 | BD Biosciences | Cat# 553802; RRID:AB_395061 |
| Anti-mouse CD16/32; clone 2.4G2 | BD Biosciences | Cat# 553142; RRID:AB_394657 |
| Anti-mouse CD19 (BUV395); clone 1D3 | BD Biosciences | Cat# 563557; RRID:AB_2722495 |
| Anti-mouse CD44 (BUV737); clone IM7 | BD Biosciences | Cat# 564392; RRID:AB_2738785 |
| Anti-mouse CD45 (BUV661); clone 30-F11 | BD Biosciences | Cat# 565079; RRID:AB_2739057 |
| Anti-mouse CD64 (BV785); clone X54-5/7.1 | BD Biosciences | Cat# 741024; RRID:AB_2740644 |
| Anti-mouse CD86 (BV711); clone GL1 | BD Biosciences | Cat# 740688; RRID:AB_2734766 |
| Anti-mouse CD127 (PE-Cy5); clone A7R34 | Biolegend | Cat# 135016; RRID:AB_1937261 |
| Anti-mouse CD172α (PerCP-eF710); clone P84 | Life Technologies | Cat# 46-1721-82; RRID:AB_10804639 |
| Anti-mouse Eomes (PerCP-eF710); clone Dan11mag | Invitrogen | Cat# 46-4875-82; RRID:AB_10597455 |
| Anti-mouse F4/80 (PE-Cy5); clone BM8 | eBioscience | Cat# 15-4801-82; RRID:AB_468798 |
| Anti-mouse FoxP3 (PE-Cy7); clone FJK-16S | Invitrogen | Cat# 25-5773-82; RRID:AB_891552 |
| Anti-mouse IA/IE (AF488); clone M5/114.15.2 | Biolegend | Cat# 107616; RRID:AB_493523 |
| Anti-mouse IFNγ (APC); clone XMG1.2 | BD Biosciences | Cat# 554413; RRID:AB_398551 |
| Anti-mouse KLRG1 (BV785); clone 2F1 | BD Biosciences | Cat# 565477; RRID:AB_2739256 |
| Anti-mouse Ly6A/E (PE-CF594); clone D7 | BD Biosciences | Cat# 562730; RRID:AB_2737751 |
| Anti-mouse Ly6C (APC-eF780); clone AL-21 | BD Biosciences | Cat# 560596; RRID:AB_1727555 |
| Anti-mouse Ly6G (BUV563); clone 1A8 | BD Biosciences | Cat# 565707; RRID:AB_2739334 |
| Anti-mouse NK1.1 (BUV395); clone PK136 | BD Biosciences | Cat# 564144; RRID:AB_2738618 |
| Anti-mouse PD-1 (BV421); clone 29F.A12 | Biolegend | Cat# 135218; RRID:AB_2561447 |
| Anti-mouse SiglecH (BUV805); clone 440C | BD Biosciences | Cat# 748291; RRID:AB_2872718 |
| Anti-mouse TCF-1 (AF647); clone C63D9 | Cell Signaling | Cat# 6709S; RRID:AB_2797631 |
| Anti-mouse Tim-3 (BV605); clone RMT3-23 | Biolegend | Cat# 119721; RRID:AB_2616907 |
| Anti-mouse TNFα (BV650); clone MP6-XT22 | Biolegend | Cat# 506333; RRID:AB_2562450 |
| Anti-mouse XCR1 (BV650); clone ZET | Biolegend | Cat# 148220; RRID:AB_2566410 |
| Anti-mouse IFNα receptor-1; clone MAR1-5A3 | BioXCell | Cat# BE0241; RRID:AB_2687723 |
| Anti-mouse PD-L1; clone 10F.9G2 | BioXCell | Cat# BE0101; RRID:AB_10949073 |
| TotalSeq-C0301 anti-mouse Hashtag 1; clones M1/42, 30-F11 | Biolegend | Cat# 155861; RRID:AB_2800693 |
| TotalSeq-C0302 anti-mouse Hashtag 2; clones M1/42, 30-F11 | Biolegend | Cat# 155863; RRID:AB_2800694 |
| TotalSeq-C0303 anti-mouse Hashtag 3; clones M1/42, 30-F11 | Biolegend | Cat# 155865; RRID:AB_2800695 |
| TotalSeq-C0304 anti-mouse Hashtag 4; clones M1/42, 30-F11 | Biolegend | Cat# 155867; RRID:AB_2800696 |
| TotalSeq-C0305 anti-mouse Hashtag 5; clones M1/42, 30-F11 | Biolegend | Cat# 155869; RRID:AB_2800697 |
| TotalSeq-C0306 anti-mouse Hashtag 6; clones M1/42, 30-F11 | Biolegend | Cat# 155871; RRID:AB_2819910 |
| TotalSeq-C0307 anti-mouse Hashtag 7; clones M1/42, 30-F11 | Biolegend | Cat# 155873; RRID:AB_2819911 |
| TotalSeq-C0308 anti-mouse Hashtag 8; clones M1/42, 30-F11 | Biolegend | Cat# 155875; RRID:AB_2819912 |
| TotalSeq-C0309 anti-mouse Hashtag 9; clones M1/42, 30-F11 | Biolegend | Cat# 155877; RRID:AB_2819913 |
| TotalSeq-C0310 anti-mouse Hashtag 10; clones M1/42, 30-F11 | Biolegend | Cat# 155879; RRID:AB_2819914 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| ChAdOx1-Reps1 | Viral Vector Core Faciliy, Jenner Institute, Oxford | N/A |
| ChAdOx1-Adpgk | Viral Vector Core Faciliy, Jenner Institute, Oxford | N/A |
| ChAdOx1-M39 (empty) | Viral Vector Core Faciliy, Jenner Institute, Oxford | N/A |
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Dasatinib | Selleckchem | S1021 |
| Isoflurane, USP | Baxter Healthcare Corp. | NDC 10019-360-60 |
| Paraformaldehyde (PFA, 16%) | Electron Microscopy Sciences | 15710 |
| Tween 20 | Sigma | P-7949 |
| Heparin | Fresenius Kabi | NDC 63323-540-05 |
| Dimethylsulfoxide (DMSO) | Sigma-Aldrich | 276855-100mL |
| Reps1 SNP vaccine “GRVLELFRAAQLANDVVLQIMELCGATR” | Vaccitech North America | N/A |
| Adpgk SNP vaccine “GIPVHLELASMTNMELMSSIVHQQVFPT” | Vaccitech North America | N/A |
| ABTS (2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)) | Sigma | A3219 |
| Murine IL-12 ABTS ELISA Kit | Peprotech | 900-K97 |
| Reps1 Tetramer (H-2Db, AQLANDVVL) | Gift from J. Finnigan | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse IFN Alpha All Subtype ELISA Kit, High Sensitivity | PBL Assay Science | Cat# 42115-1 |
| eBioscience™ Foxp3/Transcription Factor Staining Set | Invitrogen | Cat# 00-5523-00 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit | ThermoFisher Scientific | Cat# L34962 |
| ArC™ Amine Reactive Compensation Bead Kit | ThermoFisher Scientific | Cat# A10346 |
| BD Horizon Brilliant Stain Buffer | BD Biosciences | 566385 |
| Streptavidin PE (SaPE) | BD Biosciences | S866 |
| Trypsin-EDTA (0.25%) | Gibco, Life Tech | 25200–056 |
| ViaStain AOPI Staining Solution | Nexcelom Bioscience | CS2-0106 |
| Milliplex MAP mouse cytokine/chemokine Magnetic Kit | Millipore Sigma | MCYTMAG-70K-PX32 |
| Phosphate buffered saline (PBS) | Gibco | 10010–023 |
| RPMI 1640 | Cytiva, HyClone Labs | SH30027.02 |
| FBS | Gibco | 10438–026 |
| Penicillin/Streptomycin/Glutamine (100X) | Gibco, Life Tech | 10378–016 |
| Non-essential Amino Acids (100X) | Cytiva, HyClone Labs | SH30238.01 |
| Sodium Pyruvate (100mM) | Cytiva, HyClone Labs | SH30239.01 |
| Collagenase D | Roche | 11088882001 |
| DNase I | Roche | 04536282001 |
| ACK (Ammonium-Chloride-Potassium) lysing buffer | Quality Biological | 118-156-101 |
| Chromium Single Cell 50 Reagent Kit | 10X Genomics | https://support.10xgenomics.com/single-cell-vdj/library-prep |
| Dynabead MyOne Silane | Thermo Fisher Scientific | Cat#37002D |
| SPRIselect for Size Selection | Beckman Coulter | Cat#B23319 |
|
| ||
| Deposited data | ||
|
| ||
| scRNA Sequencing data | GEO | GSE214741 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MC38 Murine colorectal cancer cell line | Genentech, L. Delamarre | N/A |
| B16-F10-Adpgk | Gift from J. Finnegan | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: wild-type C57BL/6J mice | Jackson Labs | 000664 |
| Mouse: IL-12 KO C57BL/6J mice | Jackson Labs | 002693 |
| Mouse: IFNαr KO C57BL/6J mice | Jackson Labs | 028288 |
| Mouse: STING KO C57BL/6J mice | Jackson Labs | 025805 |
|
| ||
| Software and algorithms | ||
|
| ||
| Flowjo v10 | Tree Star | N/A |
| GraphPad Prism v8 | GraphPad software | N/A |
| xPONENT software | Luminex | N/A |
| R (V 4.2.1) | R Core Team | https://www.r-project.org/ |
| Cellranger (V 6.0.1) | 10X Genomics | https://support.10xgenomics.com |
| Seurat (V 4.1.1) | Hao and Hao et al.42 | https://cran.r-project.org/web/packages/Seurat/index.html |
| DoubletFinder (V 2.0.3) | McGinnis et al.43 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Pheatmap (V 1.0.12) | Pheatmap | https://cran.r-project.org/web/packages/pheatmap/index.html |
| dittoSeq (V 1.8.1) | DittoSeq | https://bioconductor.org/packages/release/bioc/html/dittoSeq.html |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
Wild-type (WT) C57BL/6J, mice were purchased from The Jackson Laboratory and housed in specific-pathogen-free conditions. Upon arrival, mice were given 1 week to adjust to the new animal facility prior to being used. Mice used in studies were between 8–10 weeks old. All mice used were females. All animal experiments were performed at the Vaccine Research Center at the National Institutes of Health (NIH) with the approval of the Institutional Animal Care and Use Committee at the NIH. Experiments complied with the ethical guidelines set by the Institutional Animal Care and Use Committee and animals were humanely killed at defined end points.
Tumor cell lines
The MC38 cell line was a kind gift from L. Delamarre (Genentech). The MC38 cells were grown in media comprised of DMEM + 10% FBS + 1% penicillin/streptomycin/glutamine + 1% non-essential amino acids + 1 mM sodium pyruvate. Stocks of MC38 were generated upon receipt of the cells and used for tumor experiments.
The B16F10 cell line expressing Adpgk was a gift from John Finnigan at the Icahn School of Medicine at Mount Sinai. The B16F10 cells were grown in media comprised of RPMI 1640 + 10% FBS + 1% penicillin/streptomycin/glutamine + 1% non-essential amino acids + 1 mM sodium pyruvate. Stocks of B16F10-Adpgk were generated upon receipt of cells and used for tumor experiments.
METHOD DETAILS
SNP vaccine
SNP vaccines were produced as described previously.14 Peptide antigens modified to form nanoparticles as part of an SNP vaccine were produced by GenScript. These peptides were linked to hydrophobic blocks containing an imidazoquinoline-based TLR-7/8 agonist (Vaccitech North America) using a click chemistry reaction.
ChAdOx1 vaccines
Transgenes encoding either Adpgk, Reps1, or an irrelevant antigen (M39) were used to create a transgene for insertion into ChAdOx1. The constructs consisted of a fragment of the murine invariant chain (CD74, denoted mIi), followed by a spacer sequence (GGGPGGG) and the mouse codon optimized sequence for one of the target antigen sequences. The spacer-antigen fragment was repeated a total of 5 times to complete the transgene insert, resulting in a pentamer repeat. Stop codons were added at the end of the pentamer repeat sequence. This resulted in the creation of 3 unique ChAdOx1 vectors, 1 encoding each of Reps1, Adpgk, and M39. The transgenes were produced as plasmids by Geneart, and were flanked by NotI and KpnI restriction enzyme sites.
Transgenes were excised by NotI and KpnI restriction enzymes and cloned into a shuttle vector that contained gateway recombination sites. Gateway recombination was performed to introduce the transgenes into the ChAdOx1 bacterial artificial chromosome (BAC). Successful recombination was confirmed by sequencing the insert within the ChAdOx1 BAC. Positive clones were grown in E. coli (DH5α) cells for MIDI prep. Purified ChAdOx1 BAC sequences isolated through the MIDI prep were linearized by incubating with restriction enzyme PmeI. Linearized BACs were sent to the Jenner Institute Viral Vector Core Facility (VVCF) for ChAdOx1 production.
Upon receiving ChAdOx1 stocks, these were thawed on ice and aliquoted into 100μl single use aliquots and stored at −80°C.
Immunizations and treatments
SNP vaccines were prepared in sterile PBS (Gibco) and administered intravenously by tail vein injection (100 μl). For ChAdOx1 vaccinations, viral stocks were thawed on ice and then resuspended in PBS at a concentration that yielded 1 × 108 IU per 100 μl IV dose or 1 × 108 IU per 50μl IM dose. For IFNα receptor blockade, mice were treated 500μg of anti-IFNα receptor 1 antibody (MAR1–5A3; Bio X Cell) in 100μl of PBS via intraperitoneal injection. Mice were also treated with 200μg of anti-PD-L1 antibody in 100μl of PBS via intraperitoneal injection (10F.9G2; BioXcell).
Tumor implantation
For each tumor implantation, a frozen cell aliquot was thawed and cultured in MC38 or B16F10 media at 37°C and 5% CO2, passaged once and collected using trypsin EDTA (Gibco). Then, 105 cells in sterile PBS per mouse were implanted subcutaneously on the right flank. Tumors were measured twice a week using digital calipers. Tumor volume was estimated using the formula: (tumor volume = short × short × long/2). Animals were killed when tumors surpassed 1,000 mm3.
Blood and tissue processing
Heparin-treated blood was collected and lysed with ACK lysis buffer (Quality Biological) to isolate PBMCs. Lungs, liver, kidneys and tumors were collected in digestion media containing Roswell Park Memorial Institute (RPMI) 1640, 10% FCS, 50 U ml−1 DNase I (Sigma-Aldrich) and 0.2 mg ml−1 collagenase D (Sigma-Aldrich). Tissues were mechanically disrupted using the respective programs on the gentleMACS dissociator (Miltenyi Biotec) and incubated at 37°C for 30–45 min in a shaking incubator. Spleens were mechanically disrupted and lysed with ACK lysis buffer. Lymph nodes were mechanically disrupted in BioMasher tubes (Nippi). All single-cell suspensions were filtered through a 70-μm cell strainer and resuspended in PBS for flow cytometry staining.
Flow cytometry
For T cell tetramer analysis, cells were assessed for viability with LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen) in PBS containing 50 nM dasatinib (STEMCELL Technologies) for 30 min at room temperature. Samples were then washed and blocked with anti-CD16/CD32 (BD Biosciences). Cells were then stained with fluorescently conjugated tetramer in cell staining buffer (PBS and 2% FCS) containing 50nM dasatinib to enhance staining. Cells were simultaneously stained with the following surface antibodies to: CD8 (clone 53–6.7), PD-1 (clone 29F.A12), Tim-3 (clone RMT3–23), and CD44 (clone IM7) purchased from BioLegend and CD4 (clone RM4–4) purchased from BD Biosciences. After a 1-h incubation at 4°C, cells were washed twice in cell staining buffer, fixed and permeabilized using the FoxP3 transcription factor staining buffer set (eBioscience). Cells were stained overnight at 4°C with CD3 (clone 17A2) from BD Biosciences. Stain was washed off the following morning and samples resuspended in eBioscience FoxP3 transcription factor permeabilization wash buffer after which samples were acquired on an LSRFortessa X50 (BD Biosciences) using the FACSDiva software v8.0.1 (BD Biosciences).
For the mononuclear phagocyte uptake analysis, cells were assessed for viability with the LIVE/DEAD Fixable Blue Dead Cell Stain Kit for 10 min at room temperature. After FcR blocking, cells were stained for 20 minutes at room temperature with the following surface antibodies: NK1.1 (clone PK136), CD19 (clone 1D3), CD3 (clone 145–2C11), Ly6G (clone 1A8), CD45 (clone 30-F11), Siglec-H (clone 440c), CD86 (clone GL1), CD11c (clone HL3), CD80 (clone 16–10A1), B220 (clone RA3–6B2), CD64 (clone X54–5/7.1), CD11b (clone MI/70), Ly6A/E (clone D7), and Ly6C (clone AL-21) purchased from BD Biosciences, CCR7 (clone 4B12), MHC class II (I-A/I-E, clone M5/114.15.2), CD169 (clone 3D6.112) and XCR1 (clone ZET) purchased from BioLegend, and CD172a (clone P84) from Thermo Fisher Scientific. The stain was washed off and cells were then fixed in 0.5% PFA in PBS until they were acquired on an LSRFortessa X50 (BD Biosciences) using the FACSDiva software v8.0.1 (BD Biosciences).
In vivo imaging
Whole-body imaging of mice after immunization with Alexa Fluor 647-labeled vaccines was performed using the IVIS Spectrum In Vivo Imaging System (PerkinElmer).
ELISA and Luminex
Serum from whole blood was collected at specified time points after vaccination. Commercially available ELISA kits were used to measure IL-12 subunit p40 (PeproTech) and all subtypes of IFNα (PBL Assay Science) according to the manufacturer’s protocols. A commercially available Luminex kit (Millipore Sigma) was used according to the manufacturer’s protocols to assess multiple analytes from serum samples.
Cell sorting for scRNA-seq
Spleens and tumors from mice that had been boosted one day prior were collected and processed into single cell suspensions by mechanical dissociation. Samples were stained with LIVE/DEAD Fixable Blue Dead Cell Stain Kit for 10 min at room temperature. Then samples were washed with FACS buffer (2% FBS in PBS) and stained with Fc block (Anti-mouse CD16/32, BD Biosciences) prior to addition of a surface stain. The surface stain antibody master mix contained: CD3 BUV395, CD19 BUV395, CD45 BUV661, CD11c PE, and CD11b AF700. Each sample was also stained with a unique hashtag antibody. Samples were incubated in surface stain for 20 minutes at room temperature after which all surface stain antibodies were washed off. Samples were resuspended in FACS buffer and sorted by fluorescence activated cell sorting to isolate the live CD45+ CD11b+ CD11c+ cells. Sorted samples were pooled together by tissue prior to loading in duplicate into a Chromium single cell sorting system (10x Genomics). Expression and hashtag library construction was performed following the Chromium Single Cell VDJ Library protocol with a loading target of 1 × 104 per lane. At the conclusion, there were 4 expression and hash tag libraries from spleen samples and another 4 from tumor samples. The libraries were sequenced on a NovaSeq 6000 S2 chip.
Pre-processing of scRNA-seq data
The FASTQ files containing raw scRNA-seq data were mapped to mm10 mouse reference genome using the Cell Ranger Single-Cell software v6.0.1 from 10x Genomics. The output count matrices were analyzed using Seurat R package v4.1.1.42 Only singlet cells were chosen during demultiplexing, and also doublet cells removed using DoubletFinder R package (v2.0.3)43 by assuming doublet formation rate of 12%.
Integration, dimension reductions and cell clustering by Seurat
According to Seurat integration approach, the filtered count matrices were log-normalized using NormalizeData function, the anchors were identified using FindIntegrationAnchors function (dims = 1:20, k.anchor= 5, k.filter= 30, anchor.features = 3000, reduction = “cca”), and the cell-to-cell distance (batch-corrected) matrix was imputed using IntegrateData function (dims= 1:20).
Next, following a linear-dimensionality reduction (PCA) on the whole distance matrix using RunPCA function (npcs = 50) and finding nearest neighbours using FindNeighbors function (dims = 1:10), a non-linear dimensionality reduction by means of Uniform Manifold Approximation and Projection (UMAP) was performed using RunUMAP function and umap-learn method (dims = 1:10, min.dist = 0.5) and cell clusters/states were identified using FindClusters function (resolution = 0.8) in an unsupervised manner. These clusters were characterised based on their expression of canonical markers, and clustering representing monocyte, macrophages and dendritic cell types were passed through a second round of RunPCA (npcs = 50), FindNeighbors (dims = 1:8), RunUMAP (dims = 1:8, min.dist = 0.6) and FindClusters (resolution = 0.8) functions. The latter clusters (original clusters) were manually combined into meta-clusters by considering the focal points of the compacted cells on a density plot and also based on their hierarchical ordering using their Euclidean distance (calculated by mean expression of the 3000 anchor genes) using pheatmap R package v1.0.12.
The identity of meta-clusters were characterised based on their expression of canonical markers (Figure 6C) as well as by calculating the average expression of signature genes as we have previously described (top-10 differentially expression genes, Baharom et al.35) for each meta-clusters (Figure 6C).
Cluster distribution
The distribution of each Mon/Mac/DC meta-cluster within each experimental group or tissue was calculated using dittoSeq R package (v1.8.1)44 according to the following formula:
The statistical cross-condition or cross-tissue comparison of the meta-cluster distribution was done in GraphPad Prism software v9.3.1 using non-parametric unpaired two-samples Wilcoxon test. (Mann-Whitney test). A P value < 0.05 was considered as statistically significant.
Biological pathways analysis
To provide a vision on biological pathways differentially present among Mon/Mac or DC meta-clusters, a curated gene list of each pathway was prepared from literature and public databases including MGI, Maier et al.,45 Orecchioni et al.,46 and KEGG, and the average expression of the genes was calculated in each cell type, and visualised using pheatmap R package.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of biological data
All results are presented as the mean with SD unless otherwise stated. Statistics were assessed using a Kruskal-Wallis test with Dunn’s correction for multiple comparisons [immunogenicity], two-way ANOVA with Bonferroni correction [tumor growth curves], log rank test [survival curves], Mann-Whitney U-test [cytokines].
Supplementary Material
Highlights.
Intravenous heterologous prime boost with SNP-ChAdOx1 elicits potent CD8 T cell responses
Antigen-specific T cells are necessary but insufficient for therapeutic efficacy
Intravenous ChAdOx1 promotes tumor regression through type I IFN-dependent TME remodeling
Type I IFN reduces the frequency of immunosuppressive Chil3 monocytes in the TME
ACKNOWLEDGMENTS
We thank members of the Seder lab for scientific discussions and members of the Translational Research Program (Vaccine Research Center) for their valuable support with the animal studies, especially Dr. Ruth Woodward, Dr. Diana Scorpio, Nina Callaham, Marqui Dorsey, Sienna Rush, Gloria Salbador, Oscar Hernandez, and Arelis Island. This work was supported by the National Institutes of Health Intramural Research Program of the U.S. (R.A.S.), the NIH-Oxford-Cambridge Scholars Program (R.A.R.-V.), and EMBO YIP and the Singapore Immunology Network core funding (F.G.). C.S.K.L. was supported by a fellowship from the Swiss National Science Foundation (P300P3_155374). The graphical abstract was created with BioRender.com.
Footnotes
DECLARATION OF INTERESTS
A.S.I., G.M.L., and R.A.S. are listed as inventors on patents describing polymer-based vaccines. A.S.I. and G.M.L. are employees of Vaccitech North America, which is commercializing polymer-based drug delivery technologies for immunotherapeutic applications. C.S.K.L. and B.J.V.d.E. are inventors on a patent that covers viral vectors and methods for the prevention and treatment of cancer. F.B. is an employee of Genentech, a member of the Roche group, which develops and markets drugs for profit.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112599.
REFERENCES
- 1.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. (2015). Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 348, 124–128. 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J, et al. (2014). Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581. 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. (2016). T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 375, 2255–2262. 10.1056/nejmoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, et al. (2014). Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645. 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saxena M, Van Der Burg SH, Melief CJM, and Bhardwaj N (2021). Therapeutic cancer vaccines. Nat. Rev. Cancer 21, 360–378. 10.1038/s41568-021-00346-0. [DOI] [PubMed] [Google Scholar]
- 7.Blass E, and Ott PA (2021). Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 18, 215–229. 10.1038/s41571-020-00460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. (2017). Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226. 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 10.Stanley DA, Honko AN, Asiedu C, Trefry JC, Lau-Kilby AW, Johnson JC, Hensley L, Ammendola V, Abbate A, Grazioli F, et al. (2014). Chimpanzee adenovirus vaccine generates acute and durable protective immunity against ebolavirus challenge. Nat. Med. 20, 1126–1129. 10.1038/nm.3702. [DOI] [PubMed] [Google Scholar]
- 11.Swadling L, Capone S, Antrobus RD, Brown A, Richardson R, Newell EW, Halliday J, Kelly C, Bowen D, Fergusson J, et al. (2014). A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci. Transl. Med. 6, 261ra153. 10.1126/scitranslmed.3009185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X, Shaw RH, Stuart ASV, Greenland M, Aley PK, Andrews NJ, Cameron JC, Charlton S, Clutterbuck EA, Collins AM, et al. (2021). Safety and immunogenicity of heterologous versus homologous prime-boost schedules with an adenoviral vectored and mRNA COVID-19 vaccine (Com-COV): a single-blind, randomised, non-inferiority trial. Lancet 398, 856–869. 10.1016/s0140-6736(21)01694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lynn GM, Laga R, Darrah PA, Ishizuka AS, Balaci AJ, Dulcey AE, Pechar M, Pola R, Gerner MY, Yamamoto A, et al. (2015). In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat. Biotechnol. 33, 1201–1210. 10.1038/nbt.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lynn GM, Sedlik C, Baharom F, Zhu Y, Ramirez-Valdez RA, Coble VL, Tobin K, Nichols SR, Itzkowitz Y, Zaidi N, et al. (2020). Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat. Biotechnol. 38, 320–332. 10.1038/s41587-019-0390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baharom F, Ramirez-Valdez RA, Tobin KKS, Yamane H, Dutertre CA, Khalilnezhad A, Reynoso GV, Coble VL, Lynn GM, Mulè MP, et al. (2021). Intravenous nanoparticle vaccination generates stem-like TCF1+ neoantigen-specific CD8+ T cells. Nat. Immunol. 22, 41–52. 10.1038/s41590-020-00810-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dicks MDJ, Spencer AJ, Edwards NJ, Wadell G, Bojang K, Gilbert SC, Hill AVS, and Cottingham MG (2012). A novel chimpanzee adenovirus vector with low human seroprevalence: improved systems for vector derivation and comparative immunogenicity. PLoS One 7, e40385. 10.1371/journal.pone.0040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voysey M, Clemens SAC, Madhi SA, Weckx LY, Folegatti PM, Aley PK, Angus B, Baillie VL, Barnabas SL, Bhorat QE, et al. (2021). Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 397, 99–111. 10.1016/s0140-6736(20)32661-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cappuccini F, Stribbling S, Pollock E, Hill AVS, and Redchenko I (2016). Immunogenicity and efficacy of the novel cancer vaccine based on simian adenovirus and MVA vectors alone and in combination with PD-1 mAb in a mouse model of prostate cancer. Cancer Immunol. Immunother. 65, 701–713. 10.1007/s00262-016-1831-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McAuliffe J, Chan HF, Noblecourt L, Ramirez-Valdez RA, Pereira-Almeida V, Zhou Y, Pollock E, Cappuccini F, Redchenko I, Hill AV, et al. (2021). Heterologous prime-boost vaccination targeting MAGE-type antigens promotes tumor T-cell infiltration and improves checkpoint blockade therapy. J. Immunother. Cancer 9, e003218. 10.1136/jitc-2021-003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.D’Alise AM, Brasu N, De Intinis C, Leoni G, Russo V, Langone F, Baev D, Micarelli E, Petiti L, Picelli S, et al. (2022). Adenoviral-based vaccine promotes neoantigen-specific CD8+ T cell stemness and tumor rejection. Sci. Transl. Med. 14, eabo7604. 10.1126/scitranslmed.abo7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palmer CD, Rappaport AR, Davis MJ, Hart MG, Scallan CD, Hong S-J, Gitlin L, Kraemer LD, Kounlavouth S, Yang A, et al. (2022). Individualized, heterologous chimpanzee adenovirus and self-amplifying mRNA neoantigen vaccine for advanced metastatic solid tumors: phase 1 trial interim results. Nat. Med. 28, 1619–1629. 10.1038/s41591-022-01937-6. [DOI] [PubMed] [Google Scholar]
- 22.Chen DS, and Mellman I (2013). Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10. 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, et al. (2020). Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 181, 442–459.e29. 10.1016/j.cell.2020.03.048. [DOI] [PubMed] [Google Scholar]
- 24.Spranger S, Dai D, Horton B, and Gajewski TF (2017). Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31, 711–723.e4. 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engblom C, Pfirschke C, and Pittet MJ (2016). The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 16, 447–462. 10.1038/nrc.2016.54. [DOI] [PubMed] [Google Scholar]
- 26.Hourani T, Holden JA, Li W, Lenzo JC, Hadjigol S, and O’Brien-Simpson NM (2021). Tumor associated macrophages: origin, recruitment, phenotypic diversity, and targeting. Front. Oncol. 11, 788365. 10.3389/fonc.2021.788365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang Y, Guo J, and Huang L (2020). Tackling TAMs for cancer immunotherapy: it’s nano time. Trends Pharmacol. Sci. 41, 701–714. 10.1016/j.tips.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parker BS, Rautela J, and Hertzog PJ (2016). Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer 16, 131–144. 10.1038/nrc.2016.14. [DOI] [PubMed] [Google Scholar]
- 29.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. (2014). Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576. 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 30.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295. 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quinn KM, Zak DE, Costa A, Yamamoto A, Kastenmuller K, Hill BJ, Lynn GM, Darrah PA, Lindsay RWB, Wang L, et al. (2015). Antigen expression determines adenoviral vaccine potency independent of IFN and STING signaling. J. Clin. Invest. 125, 1129–1146. 10.1172/jci78280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Propper DJ, and Balkwill FR (2022). Harnessing cytokines and chemokines for cancer therapy. Nat. Rev. Clin. Oncol. 19, 237–253. 10.1038/s41571-021-00588-9. [DOI] [PubMed] [Google Scholar]
- 33.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 30, 324–336. 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Böttcher JP, and Reis e Sousa C (2018). The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer 4, 784–792. 10.1016/j.trecan.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baharom F, Ramirez-Valdez RA, Khalilnezhad A, Khalilnezhad S, Dillon M, Hermans D, Fussell S, Tobin KKS, Dutertre C-A, Lynn GM, et al. (2022). Systemic vaccination induces CD8+ T cells and remodels the tumor microenvironment. Cell 185, 4317–4332.e15. 10.1016/j.cell.2022.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, et al. (2020). A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262. 10.1038/s41586-020-2134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tie Y, Tang F, Wei Y. q., and Wei X. w. (2022). Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J. Hematol. Oncol. 15, 61. 10.1186/s13045-022-01282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy TL, and Murphy KM (2022). Dendritic cells in cancer immunology. Cell. Mol. Immunol. 19, 3–13. 10.1038/s41423-021-00741-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, and Blum RH (1996). Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J. Clin. Oncol. 14, 7–17. 10.1200/jco.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- 40.Rai KR, Davey F, Peterson B, Schiffer C, Silver RT, Ozer H, Golomb H, and Bloomfield CD (1995). Recombinant alpha-2b-interferon in therapy of previously untreated hairy cell leukemia: long-term follow-up results of study by Cancer and Leukemia Group B. Leukemia 9, 1116–1120. [PubMed] [Google Scholar]
- 41.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, and Kroemer G (2015). Type I interferons in anticancer immunity. Nat. Rev. Immunol. 15, 405–414. 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 42.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bunis DG, Andrews J, Fragiadakis GK, Burt TD, and Sirota M (2020). dittoSeq: universal user-friendly single-cell and bulk RNA sequencing visualization toolkit. Bioinforma. Oxf. Engl. 36, 5535–5536. btaa1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, et al. (2020). A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orecchioni M, Ghosheh Y, Pramod AB, and Ley K (2019). Macrophage polarization: different gene signatures in M1(LPS+) vs. Classically and M2(LPS−) vs. Alternatively activated macrophages. Front. Immunol. 10, 1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-mouse B220 (PE-Cy7); clone RA3-6B2 | BD Biosciences | Cat# 552772; RRID:AB_394458 |
| Anti-mouse CCR7 (BV421); clone 4B12 | Biolegend | Cat# 120120; RRID:AB_2561446 |
| Anti-mouse CD3 (BUV395); clone145-2C11 | BD Biosciences | Cat# 563565; RRID:AB_2738278 |
| Anti-mouse CD3 (Alexa 700); clone 17A2 | Biolegend | Cat# 100216; RRID:AB_493697 |
| Anti-mouse CD4 (BUV395); clone RM4-4 | BD Biosciences | Cat# 740209; RRID:AB_2739958 |
| Anti-mouse CD8 (APCeFluor780); clone 53–6.7 | eBioscience | Cat# 47-0081-82; RRID:AB_1272185 |
| Anti-mouse CD11b (AF700); clone M1/70 | BD Biosciences | Cat# 557960; RRID:AB_396960 |
| Anti-mouse CD11c (PE); clone HL3 | BD Biosciences | Cat# 553802; RRID:AB_395061 |
| Anti-mouse CD16/32; clone 2.4G2 | BD Biosciences | Cat# 553142; RRID:AB_394657 |
| Anti-mouse CD19 (BUV395); clone 1D3 | BD Biosciences | Cat# 563557; RRID:AB_2722495 |
| Anti-mouse CD44 (BUV737); clone IM7 | BD Biosciences | Cat# 564392; RRID:AB_2738785 |
| Anti-mouse CD45 (BUV661); clone 30-F11 | BD Biosciences | Cat# 565079; RRID:AB_2739057 |
| Anti-mouse CD64 (BV785); clone X54-5/7.1 | BD Biosciences | Cat# 741024; RRID:AB_2740644 |
| Anti-mouse CD86 (BV711); clone GL1 | BD Biosciences | Cat# 740688; RRID:AB_2734766 |
| Anti-mouse CD127 (PE-Cy5); clone A7R34 | Biolegend | Cat# 135016; RRID:AB_1937261 |
| Anti-mouse CD172α (PerCP-eF710); clone P84 | Life Technologies | Cat# 46-1721-82; RRID:AB_10804639 |
| Anti-mouse Eomes (PerCP-eF710); clone Dan11mag | Invitrogen | Cat# 46-4875-82; RRID:AB_10597455 |
| Anti-mouse F4/80 (PE-Cy5); clone BM8 | eBioscience | Cat# 15-4801-82; RRID:AB_468798 |
| Anti-mouse FoxP3 (PE-Cy7); clone FJK-16S | Invitrogen | Cat# 25-5773-82; RRID:AB_891552 |
| Anti-mouse IA/IE (AF488); clone M5/114.15.2 | Biolegend | Cat# 107616; RRID:AB_493523 |
| Anti-mouse IFNγ (APC); clone XMG1.2 | BD Biosciences | Cat# 554413; RRID:AB_398551 |
| Anti-mouse KLRG1 (BV785); clone 2F1 | BD Biosciences | Cat# 565477; RRID:AB_2739256 |
| Anti-mouse Ly6A/E (PE-CF594); clone D7 | BD Biosciences | Cat# 562730; RRID:AB_2737751 |
| Anti-mouse Ly6C (APC-eF780); clone AL-21 | BD Biosciences | Cat# 560596; RRID:AB_1727555 |
| Anti-mouse Ly6G (BUV563); clone 1A8 | BD Biosciences | Cat# 565707; RRID:AB_2739334 |
| Anti-mouse NK1.1 (BUV395); clone PK136 | BD Biosciences | Cat# 564144; RRID:AB_2738618 |
| Anti-mouse PD-1 (BV421); clone 29F.A12 | Biolegend | Cat# 135218; RRID:AB_2561447 |
| Anti-mouse SiglecH (BUV805); clone 440C | BD Biosciences | Cat# 748291; RRID:AB_2872718 |
| Anti-mouse TCF-1 (AF647); clone C63D9 | Cell Signaling | Cat# 6709S; RRID:AB_2797631 |
| Anti-mouse Tim-3 (BV605); clone RMT3-23 | Biolegend | Cat# 119721; RRID:AB_2616907 |
| Anti-mouse TNFα (BV650); clone MP6-XT22 | Biolegend | Cat# 506333; RRID:AB_2562450 |
| Anti-mouse XCR1 (BV650); clone ZET | Biolegend | Cat# 148220; RRID:AB_2566410 |
| Anti-mouse IFNα receptor-1; clone MAR1-5A3 | BioXCell | Cat# BE0241; RRID:AB_2687723 |
| Anti-mouse PD-L1; clone 10F.9G2 | BioXCell | Cat# BE0101; RRID:AB_10949073 |
| TotalSeq-C0301 anti-mouse Hashtag 1; clones M1/42, 30-F11 | Biolegend | Cat# 155861; RRID:AB_2800693 |
| TotalSeq-C0302 anti-mouse Hashtag 2; clones M1/42, 30-F11 | Biolegend | Cat# 155863; RRID:AB_2800694 |
| TotalSeq-C0303 anti-mouse Hashtag 3; clones M1/42, 30-F11 | Biolegend | Cat# 155865; RRID:AB_2800695 |
| TotalSeq-C0304 anti-mouse Hashtag 4; clones M1/42, 30-F11 | Biolegend | Cat# 155867; RRID:AB_2800696 |
| TotalSeq-C0305 anti-mouse Hashtag 5; clones M1/42, 30-F11 | Biolegend | Cat# 155869; RRID:AB_2800697 |
| TotalSeq-C0306 anti-mouse Hashtag 6; clones M1/42, 30-F11 | Biolegend | Cat# 155871; RRID:AB_2819910 |
| TotalSeq-C0307 anti-mouse Hashtag 7; clones M1/42, 30-F11 | Biolegend | Cat# 155873; RRID:AB_2819911 |
| TotalSeq-C0308 anti-mouse Hashtag 8; clones M1/42, 30-F11 | Biolegend | Cat# 155875; RRID:AB_2819912 |
| TotalSeq-C0309 anti-mouse Hashtag 9; clones M1/42, 30-F11 | Biolegend | Cat# 155877; RRID:AB_2819913 |
| TotalSeq-C0310 anti-mouse Hashtag 10; clones M1/42, 30-F11 | Biolegend | Cat# 155879; RRID:AB_2819914 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| ChAdOx1-Reps1 | Viral Vector Core Faciliy, Jenner Institute, Oxford | N/A |
| ChAdOx1-Adpgk | Viral Vector Core Faciliy, Jenner Institute, Oxford | N/A |
| ChAdOx1-M39 (empty) | Viral Vector Core Faciliy, Jenner Institute, Oxford | N/A |
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Dasatinib | Selleckchem | S1021 |
| Isoflurane, USP | Baxter Healthcare Corp. | NDC 10019-360-60 |
| Paraformaldehyde (PFA, 16%) | Electron Microscopy Sciences | 15710 |
| Tween 20 | Sigma | P-7949 |
| Heparin | Fresenius Kabi | NDC 63323-540-05 |
| Dimethylsulfoxide (DMSO) | Sigma-Aldrich | 276855-100mL |
| Reps1 SNP vaccine “GRVLELFRAAQLANDVVLQIMELCGATR” | Vaccitech North America | N/A |
| Adpgk SNP vaccine “GIPVHLELASMTNMELMSSIVHQQVFPT” | Vaccitech North America | N/A |
| ABTS (2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)) | Sigma | A3219 |
| Murine IL-12 ABTS ELISA Kit | Peprotech | 900-K97 |
| Reps1 Tetramer (H-2Db, AQLANDVVL) | Gift from J. Finnigan | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Mouse IFN Alpha All Subtype ELISA Kit, High Sensitivity | PBL Assay Science | Cat# 42115-1 |
| eBioscience™ Foxp3/Transcription Factor Staining Set | Invitrogen | Cat# 00-5523-00 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit | ThermoFisher Scientific | Cat# L34962 |
| ArC™ Amine Reactive Compensation Bead Kit | ThermoFisher Scientific | Cat# A10346 |
| BD Horizon Brilliant Stain Buffer | BD Biosciences | 566385 |
| Streptavidin PE (SaPE) | BD Biosciences | S866 |
| Trypsin-EDTA (0.25%) | Gibco, Life Tech | 25200–056 |
| ViaStain AOPI Staining Solution | Nexcelom Bioscience | CS2-0106 |
| Milliplex MAP mouse cytokine/chemokine Magnetic Kit | Millipore Sigma | MCYTMAG-70K-PX32 |
| Phosphate buffered saline (PBS) | Gibco | 10010–023 |
| RPMI 1640 | Cytiva, HyClone Labs | SH30027.02 |
| FBS | Gibco | 10438–026 |
| Penicillin/Streptomycin/Glutamine (100X) | Gibco, Life Tech | 10378–016 |
| Non-essential Amino Acids (100X) | Cytiva, HyClone Labs | SH30238.01 |
| Sodium Pyruvate (100mM) | Cytiva, HyClone Labs | SH30239.01 |
| Collagenase D | Roche | 11088882001 |
| DNase I | Roche | 04536282001 |
| ACK (Ammonium-Chloride-Potassium) lysing buffer | Quality Biological | 118-156-101 |
| Chromium Single Cell 50 Reagent Kit | 10X Genomics | https://support.10xgenomics.com/single-cell-vdj/library-prep |
| Dynabead MyOne Silane | Thermo Fisher Scientific | Cat#37002D |
| SPRIselect for Size Selection | Beckman Coulter | Cat#B23319 |
|
| ||
| Deposited data | ||
|
| ||
| scRNA Sequencing data | GEO | GSE214741 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MC38 Murine colorectal cancer cell line | Genentech, L. Delamarre | N/A |
| B16-F10-Adpgk | Gift from J. Finnegan | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: wild-type C57BL/6J mice | Jackson Labs | 000664 |
| Mouse: IL-12 KO C57BL/6J mice | Jackson Labs | 002693 |
| Mouse: IFNαr KO C57BL/6J mice | Jackson Labs | 028288 |
| Mouse: STING KO C57BL/6J mice | Jackson Labs | 025805 |
|
| ||
| Software and algorithms | ||
|
| ||
| Flowjo v10 | Tree Star | N/A |
| GraphPad Prism v8 | GraphPad software | N/A |
| xPONENT software | Luminex | N/A |
| R (V 4.2.1) | R Core Team | https://www.r-project.org/ |
| Cellranger (V 6.0.1) | 10X Genomics | https://support.10xgenomics.com |
| Seurat (V 4.1.1) | Hao and Hao et al.42 | https://cran.r-project.org/web/packages/Seurat/index.html |
| DoubletFinder (V 2.0.3) | McGinnis et al.43 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Pheatmap (V 1.0.12) | Pheatmap | https://cran.r-project.org/web/packages/pheatmap/index.html |
| dittoSeq (V 1.8.1) | DittoSeq | https://bioconductor.org/packages/release/bioc/html/dittoSeq.html |