

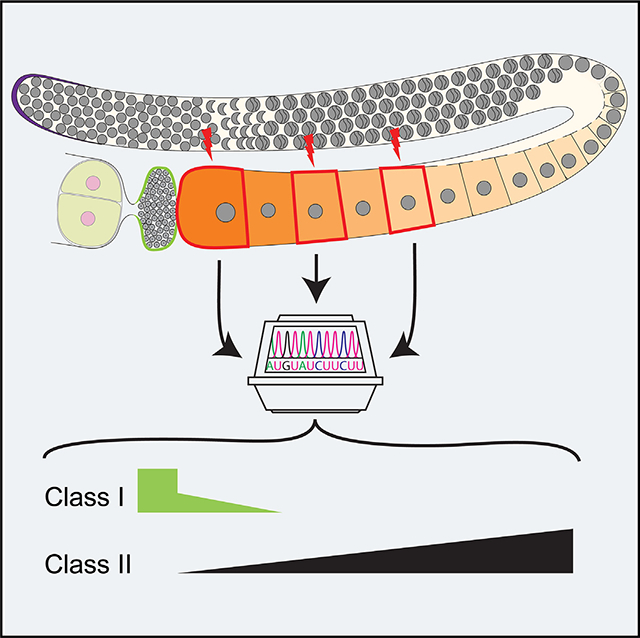
SUMMARY
Maternal RNAs are stored from minutes to decades in oocytes throughout meiosis I arrest in a transcriptionally quiescent state. Recent reports, however, propose a role for nascent transcription in arrested oocytes. Whether arrested oocytes launch nascent transcription in response to environmental or hormonal signals while maintaining the meiosis I arrest remains undetermined. We test this by integrating single-cell RNA sequencing, RNA velocity, and RNA fluorescence in situ hybridization on C. elegans meiosis I arrested oocytes. We identify transcripts that increase as the arrested meiosis I oocyte ages, but rule out extracellular signaling through ERK MAPK and nascent transcription as a mechanism for this increase. We report transcript acquisition from neighboring somatic cells as a mechanism of transcript increase during meiosis I arrest. These analyses provide a deeper view at single-cell resolution of the RNA landscape of a meiosis I arrested oocyte and as it prepares for oocyte maturation and fertilization.
In brief
Maternal transcripts, which are important for early embryogenesis, are all thought to originate exclusively from the germ cell lineage. Trimmer et al. show that a few unique transcripts are produced in the somatic cells and loaded into oocytes, suggesting other sources of maternal transcripts.
Graphical Abstract
INTRODUCTION
Oocytes arrest in prophase of meiosis I across species.1–5 In humans, this arrest often lasts for decades.6 During the arrest, oocytes grow and acquire competence (an ability to complete meiosis and undergo fertilization to produce healthy progeny). Until recently, it was believed that the oocytes go through these complex developmental transitions in a transcriptionally quiescent environment, since transcription stops prior to oocyte arrest in meiosis I.7 Thus, the coordination of oocyte arrest with oocyte growth and meiotic resumption for accurate zygotic transition was thought to be largely driven by cytoplasmic components and signaling molecules. Due to the prolonged transcriptional quiescence of this process, one key cytoplasmic component acquired by the oocyte prior to arrest is maternal RNAs. Maternal RNAs are stockpiled in the arrested oocyte until they are loaded into the developing embryo and eventually cleared in the embryo in preparation for zygotic genome activation.8–11 However, recent work from Walker et al. on Caenorhabditis elegans meiosis I arrested oocytes suggests that transcription is reactivated, as assessed through the phosphorylation of the RNA polymerase II cytoplasmic domain at serine 5, in arrested meiosis I oocytes by activation of ERK signaling due to presence of short-peptide-hormone signaling through major sperm protein in C. elegans.12 Similarly, in Drosophila, Navarro-Costa et al. observed that the oocyte epigenome is dynamic and may regulate oocyte transcription during meiotic prophase arrest,13 while in mammals, mouse oocytes arrested in meiosis I have been identified as belonging to two classes, those that launch transcription and those that do not.14 Overall, these studies question the current model that oocytes are transcriptionally inactive during meiosis I arrest. However, no study has systematically extracted oocytes arrested in meiosis I at different ages and performed a deep sequencing analysis on the mRNAs followed by analysis of active transcription to directly test the model.
C. elegans is an excellent model system to study the molecular events that occur during meiosis I of oogenesis. The C. elegans hermaphroditic germline maintains a population of continuously dividing germline stem cells (GSCs) in the progenitor zone (Figure 1A) that enter meiosis at larval stage 3 (L3); during the L3–L4 larval stages the GSCs undergo spermatogenesis.15 The hermaphroditic germline completes spermatogenesis by the end of L4, and the GSCs enter oogenesis to continuously generate oocytes during adulthood. Thus, meiosis I oocytes can be easily accessed for study during the entire adult development in C. elegans.
Figure 1. Femtosecond laser microdissection (fs-LM) method for the isolation and collection of single arrested meiosis I oocytes at different ages for scRNA seq.
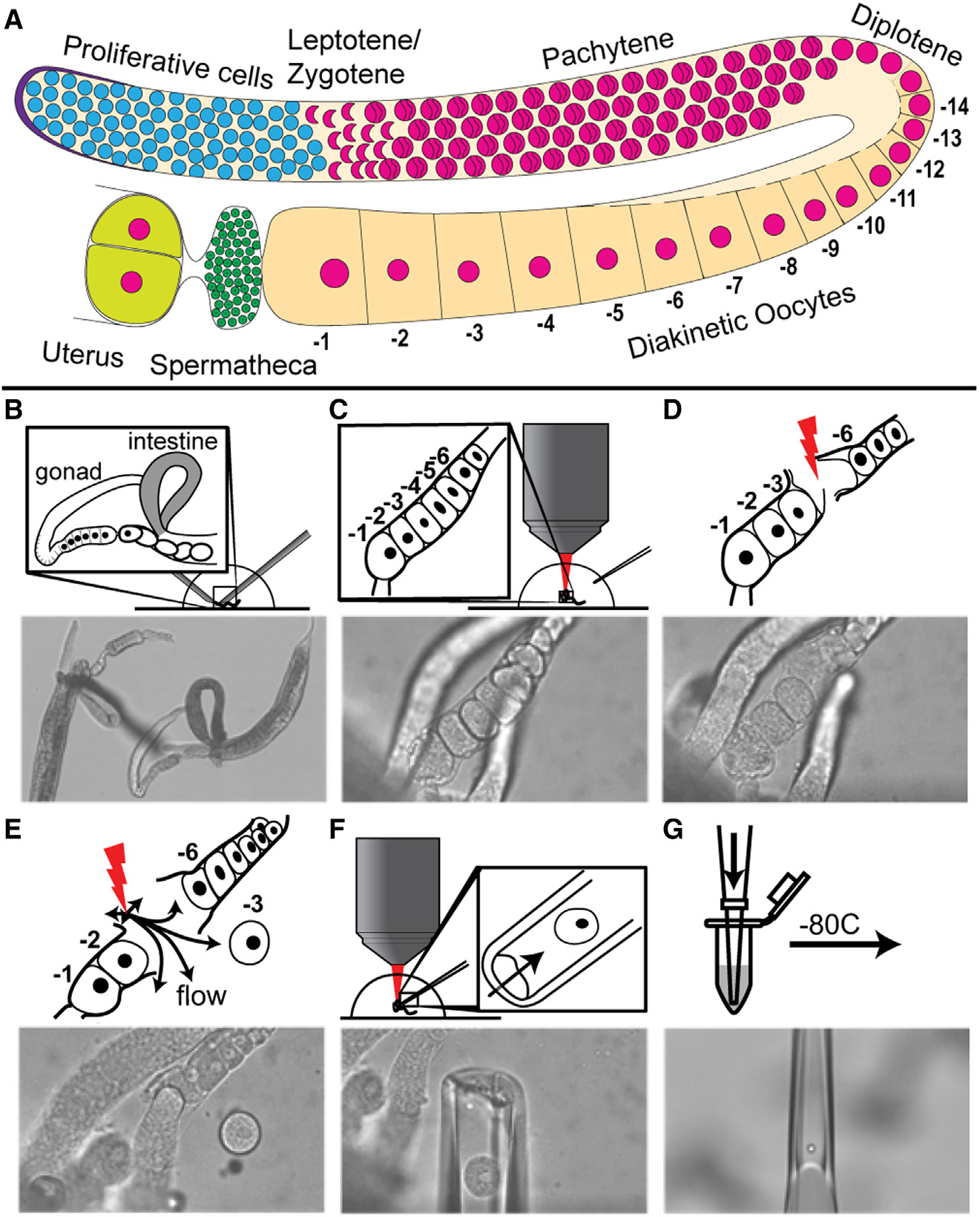
(A) Drawing of one C. elegans hermaphroditic germline arm with major cell populations labeled. (B–G) The fs-LM method16 involves the following steps. (B) Sever the animal at the pharynx with two 28G needles by hand under a stereoscope to expose the gonad and part of the uterus. (C) Transfer the buffer droplet containing the dissected animals to an upright microscope equipped with the fs-LM setup and a micropipetting system. (D) Ablate non-target oocytes (−4 and −5) closest to the target oocyte (−3) to create a passage for releasing the target oocyte. (E) Dislodge the target oocyte and move it away from the carcass by creating gentle water jets with low-energy off-target fs-laser pulses. (F) Extract the released target oocyte with a glass micropipette. (G) Deposit the collected single oocyte into the lysis buffer and freeze at −80°C. See also Figure S1.
The oogenic germ cells progress through a long pachytene stage of meiosis I and arrest in prophase of meiosis I at diplotene and diakinesis (diakinetic oocytes) (Figure 1A). The arrested prophase I oocytes release the arrest upon receiving the short-range hormonal signal mediated by major sperm protein (MSP)17 and enter meiosis II coupled with fertilization in the uterus.9 Progression through meiosis I coupled with oocyte growth requires the maternal nutrition-dependent activation of insulin-like signaling through DAF-2, which in turn regulates the activation of the RAS-ERK pathway.18 ERK is activated in arrested meiosis I oocytes by the presence of sperm,17,18 which is essential for the onset of meiotic maturation.19 Using C. elegans as a model system, we investigate the nature of the RNA dynamics and regulation of transcripts during oogenesis. Oocytes grow and move proximally in the germline and arrest at diakinesis of meiosis I. The arrested meiosis I oocytes organize in a linear fashion by age in the germline, with the oldest oocyte termed −1 (based on its proximity to the uterus and the spermatheca) and younger sibling oocytes termed −2, −3, and so on, by birth order (Figure 1A). The identity of the maternal RNAs that are stored in each of these arrested oocytes, as well as any changes in abundance as they age, remains unknown.
Active transcription occurs in germ cells from the progenitor zone until the end of pachytene stage, while the meiosis I arrested oocytes are thought to be transcriptionally silent based on cytological analyses such as nucleotide incorporation and RNA polymerase II phosphorylation.12,20,21 Thus, maternal RNAs are transcribed distally and loaded into the growing oocytes via actin-mediated cytoplasmic streaming through a syncytial structure called the rachis, which connects all the germ cells until the oocytes individualize at the −4 or −5 position (Figure 1A).22,23
Here we assess the dynamics of transcripts in arrested meiosis I oocytes at different stages of arrest. We spatially extracted oocytes at three different ages post meiosis I arrest and performed single-cell RNA sequencing followed by RNA velocity, to assess transcription, and validated using hairpin chain reaction fluorescence in situ hybridization (HCR-FISH). We found that, while most transcripts remain stable as oocytes age, there are populations of transcripts that either increase or decrease with oocyte age. Those that decrease appear to be related to metabolism or earlier germ cell processes, while those that increase do not appear to increase due to nascent transcription. Rather, on validating five transcripts that increase in the −1 oocyte by FISH analysis, we found that all five transcripts are produced in the spermatheca and likely transported into the −1 oocyte independent of sperm signal, hormone-induced signaling, and ERK activation. This provides an example of the acquisition of maternal transcripts from a somatic source, using a technically innovative method that is widely applicable to C. elegans as well as other species.
RESULTS
Some oocyte maternal transcripts increase with age
To investigate the mRNA dynamics of arrested oocytes, we extracted single oocytes at defined positions, indicative of the specific state of the arrest or maturation, in the germline and performed single-cell deep RNA sequencing (scRNA-seq) at a depth of >10 million reads per oocyte. We isolated and collected individual oocytes that had recently entered meiosis I arrest (n = 4), or were already in meiosis I arrest (n = 4), or just before meiotic maturation (n = 4) (positions −5, −3, and −1, respectively), using the femtosecond laser microdissection (fs-LM) method that we recently developed (Figures 1B–1G).16 The collected oocytes were prepared for single-cell RNA sequencing (STAR Methods). The oocytes were sequenced at a depth of >10 million (M) reads. We then tested for sufficient depth by comparing the number of genes detected at multiple thresholds in each oocyte to the total number of reads in that oocyte (Figures S1A–S1C; STAR Methods).16 Since the number of genes detected was not correlated with the read count of each oocyte, we determined that the read depth used was sufficient to identify any transcriptomic changes between the oocytes.
Overall, at a cutoff of 1 count per million, we detected ~8,000 genes per oocyte (Figure S1B). We compared this number with a previously published single-cell transcriptome database where Tintori et al. extracted and sequenced embryonic blastomeres from C. elegans.24 We reasoned that, since all early embryonic transcripts come from maternal deposition, this comparison is reasonable. Tintori et al. identified 8,575 genes during the analysis of the embryonic blastomeres.24 Focusing on the one-cell embryo analysis from Tintori et al., we found that 5,035 genes were above the published threshold (RPKM >25). We then compared this dataset of 5,035 genes with the genes detected in the current study and observed a ~99% overlap between the respective gene lists (Figure S2A). This is a very high degree of overlap, suggesting that many of the transcripts loaded into the one-cell embryos derive from the oocyte and are stable at this stage of zygotic initiation. We next compared our dataset with bulk sequencing of oocytes and one-cell embryos from Stoeckius et al.25 Since Stoeckius et al. had performed bulk sequencing on both oocytes and embryos, we generated two distinct gene lists: one of the oocyte dataset (6,714 genes) and the second for the one-cell embryo (7,294 genes) using the published threshold (RPKM ≥2). We then compared the gene lists in oocytes derived from the current study with this analysis and found an overlap of ~91% and ~88% for oocytes and one-cell embryos, respectively, between the two datasets (Figures S2B and S2C). Overall, these analyses demonstrate that our study has a high degree of consistency with other studies.
To determine if the mRNA abundance changes as the age of an arrested oocyte increases, we performed differential expression analyses and compared −1 with −3 and −1 with −5 oocytes using DESeq2 with a threshold of 2-fold change (STAR Methods) (Figure 2A). Overall, upon comparing the different stages of oocyte arrest, we find that the majority of the mRNA transcripts do not display any change in abundance as the arrested oocyte ages, suggesting that most transcripts are stable in the arrested oocyte once loaded. To validate this observation in situ, we selected several genes that did not significantly change in expression (pgl-1, mpk-1, cyn-7, and spn-4) and used HCR-FISH. Specificity of the FISH probes was tested by RNAi (Figure S3). We found that pgl-1, mpk-1, and cyn-7 showed no obvious change in expression as oocytes age (Figure 2B). However, spn-4 mRNA, which was identified as being stable across the age of the oocytes, appeared to decrease in the −1 oocyte by FISH analysis (Figures 2B and S3E), and we found that spn-4 expression decreased slightly from the −5 oocyte to the −3 oocyte to the −1 oocyte. This suggests that the DESeq2 analysis may identify some false negatives. We next turned our attention to transcripts that either increased or decreased as the arrested oocyte aged.
Figure 2. Differential accumulation of maternal transcripts in arrested oocytes as they age.
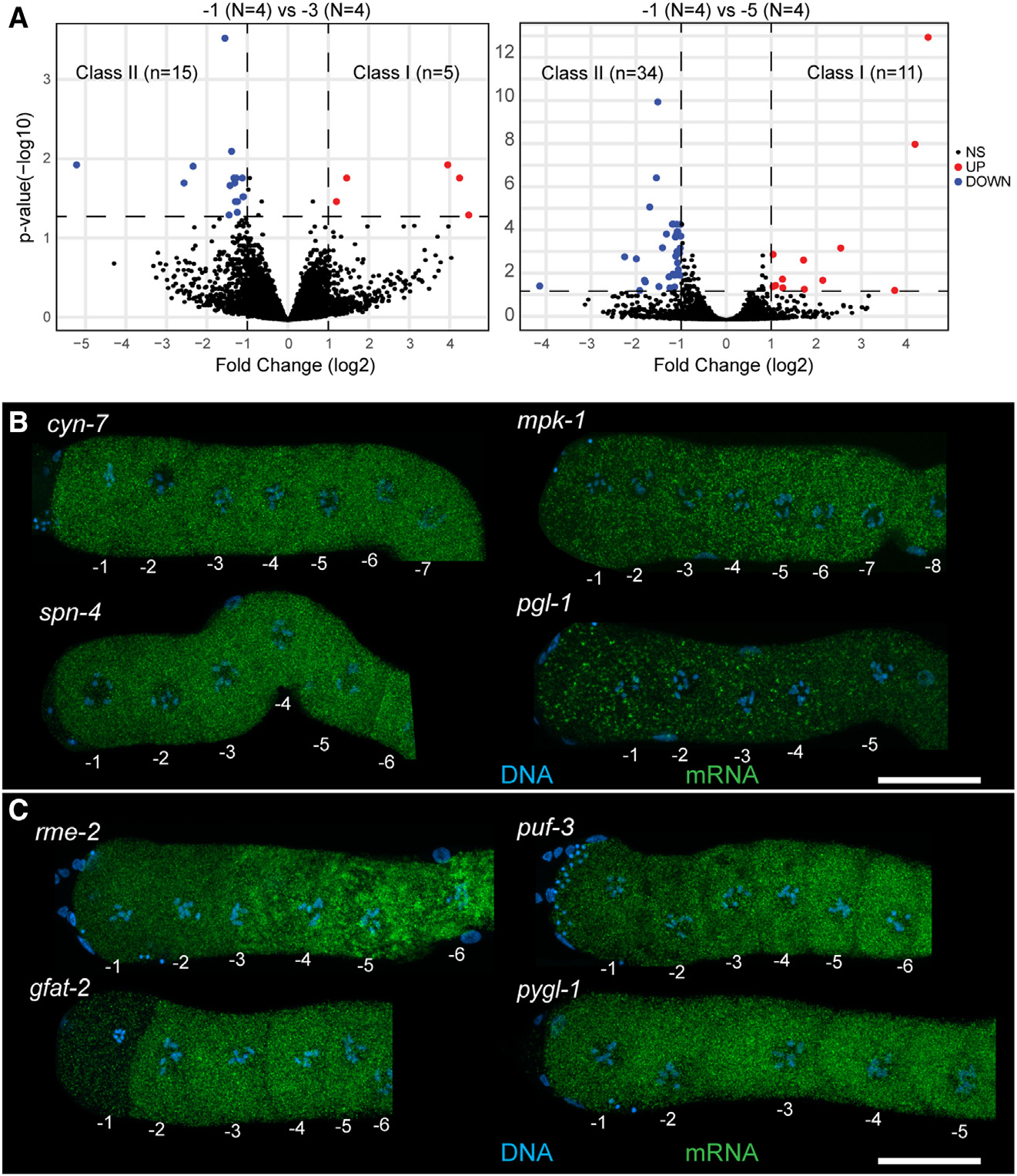
(A) DESeq2 analysis of −1 oocytes compared with −3 and −5 oocytes. Downregulated transcripts are in blue, upregulated transcripts are in red. Significance cutoff was set to 23 fold change (vertical dashed lines) and p < 0.05 (horizontal dashed line).
(B) Dissected and stained germlines showing representative expression patterns for cyn-7, mpk-1, spn-4, and pgl-1. Shown are maximum-intensity projection images through the full z stack with DNA (blue) and mRNA target (green).
(C) Dissected and stained germlines showing representative expression patterns for the class II transcripts rme-2, puf-3, gfat-2, and pygl-1. Shown are maximum-intensity projection images through the full z stack DNA (blue) and mRNA target (green). (B, C) Scale bars, 20 mm. See also Figures S3 and S4 and Tables S1 and S2.
Of the transcripts that were differentially regulated between the different ages of the arrested oocytes, using the −1 oocyte as our point of comparison, we identified two classes: (1) class I transcripts increased in abundance as the oocyte aged (thus more abundant in −1 relative to −3 or −5) (Figure 2A, red). We identified 16 unique genes in this class (Table S1). (2) Class II transcripts decreased in abundance as the oocyte aged (thus more abundant in −5 or −3 relative to −1) (Figure 2A, blue). We identified 48 unique genes in this class (Table S2). To determine if a specific cell biological process was enriched among the class I and II transcripts, we used PANTHER and assessed for gene ontology (GO) term enrichment between the pairs of oocytes (STAR Methods). While most pairwise comparisons did not contain sufficient genes to generate significant GO term enrichment, we observed that the class II mRNAs between the −5 and the −1 oocytes were enriched for transcripts involved in organic substance metabolic processes (~2.5-fold, p = 0.00753), suggesting that metabolic processes were likely downregulated as the age of the arrested oocyte increased. We speculate that a decrease in metabolic transcripts in an arrested oocyte as it ages may be similar to a phenomenon observed in mammals, wherein transcripts related to protein synthesis, metabolism, and energy production are removed upon completion of oocyte growth.27 Within class II transcripts, we also observed a reduction in RNAs that promote GSC fate (puf-3 and puf-11)28 as well as yolk transport during oocyte growth (rme-2).29 This observation suggests that some class II RNAs may decrease during the removal of germline-specific transcripts in preparation for embryogenesis. To validate these findings, we assayed four class II transcripts by HCR-FISH: two transcripts with GO terms relating to metabolic organic metabolism (gfat-2 and pygl-1), along with puf-3 and rme-2. Probes were validated by RNAi (Figure S4). We observed that rme-2, puf-3, and gfat-2 show a consistent visible decrease in the −1 oocyte relative to the −5 oocyte as determined by the DESeq2 analysis (Figure 2C). However, while pygl-1 sometimes shows a visible decrease in signal between the −5 and the −1 oocytes (Figure S3G), this decrease is not always obvious (Figure 2C), suggesting that in some events sequencing-based analysis may be more sensitive to changes in transcript abundance relative to FISH-based methods. The decrease in transcript expression levels as an arrested oocyte ages is not unexpected and has been reported, since some transcripts are destabilized by various deadenylating enzymes.25
The presence of class I RNAs, however, was unexpected, as an increase in RNA abundance in arrested oocytes has not been previously reported. If transcripts are loaded into oocytes through cytoplasmic streaming from the rachis,23 and active transcription is shut down at the end of pachytene long before formation of oocytes, then we reasoned that the transcript levels should not increase once an oocyte individualizes and undergoes arrest. It was therefore surprising to us to discover RNA populations that increased as the oocyte ages, since the oocytes have generally closed their connection with the rachis by the −4 or −5 position.22 Because a role for MPK-1 ERK signaling through the presence of MSP and sperm has been proposed in the field to lead to phosphorylation of RNA polymerase II (Pol II), leading to transcriptional activation, we assayed for de novo transcription in each of the oocytes sequenced.
Oocyte maternal transcript increase is not due to de novo transcription
For transcription to occur, RNA Pol II must be activated via phosphorylation at two phospho-residues, serine 5 for initiation and serine 2 for extension.30,31 To determine whether the increase in transcripts in the aged oocytes was due to an activation of MPK-1 ERK signaling by the presence of MSP and sperm, we performed staining for pSer5 RNA Pol II (p5RNAP2) and pSer2 RNA Pol II (p2RNAP2) with dpMPK-1 (to assess dually phosphorylated active state of MPK-1 ERK) in mated and unmated female (fog-2) germlines (STAR Methods). After 2 h of mating, dpMPK-1 signal was clearly detectable in the proximal oocytes; however, we failed to observe any p5RNAP2 or p2RNAP2 signal in the most proximal oocytes (Figure 3). These data demonstrate that the presence of sperm signal and activation of MPK-1 ERK does not lead to phosphorylation of RNA Pol II at Ser5 or Ser2. Because, upon mating, we observe activation of MPK-1 ERK and presence of sperm in the oocytes, but not p5RNAP2 or p2RNAP2, we conclude that transcription is neither initiated nor extended by the presence of sperm and dpMPK-1 in the oocytes, although we cannot rule out a situation where some p5RNAP2 or p2RNAP2 was present below the detection limit. To directly assess for nascent transcription in the arrested oocytes, we performed RNA velocity analysis on the scRNA-seq data from the −5, −3, and −1 oocytes.
Figure 3. Sperm signal and MPK-1 ERK activation do not induce activation and phosphorylation of RNA Pol II Ser5 or Ser2.
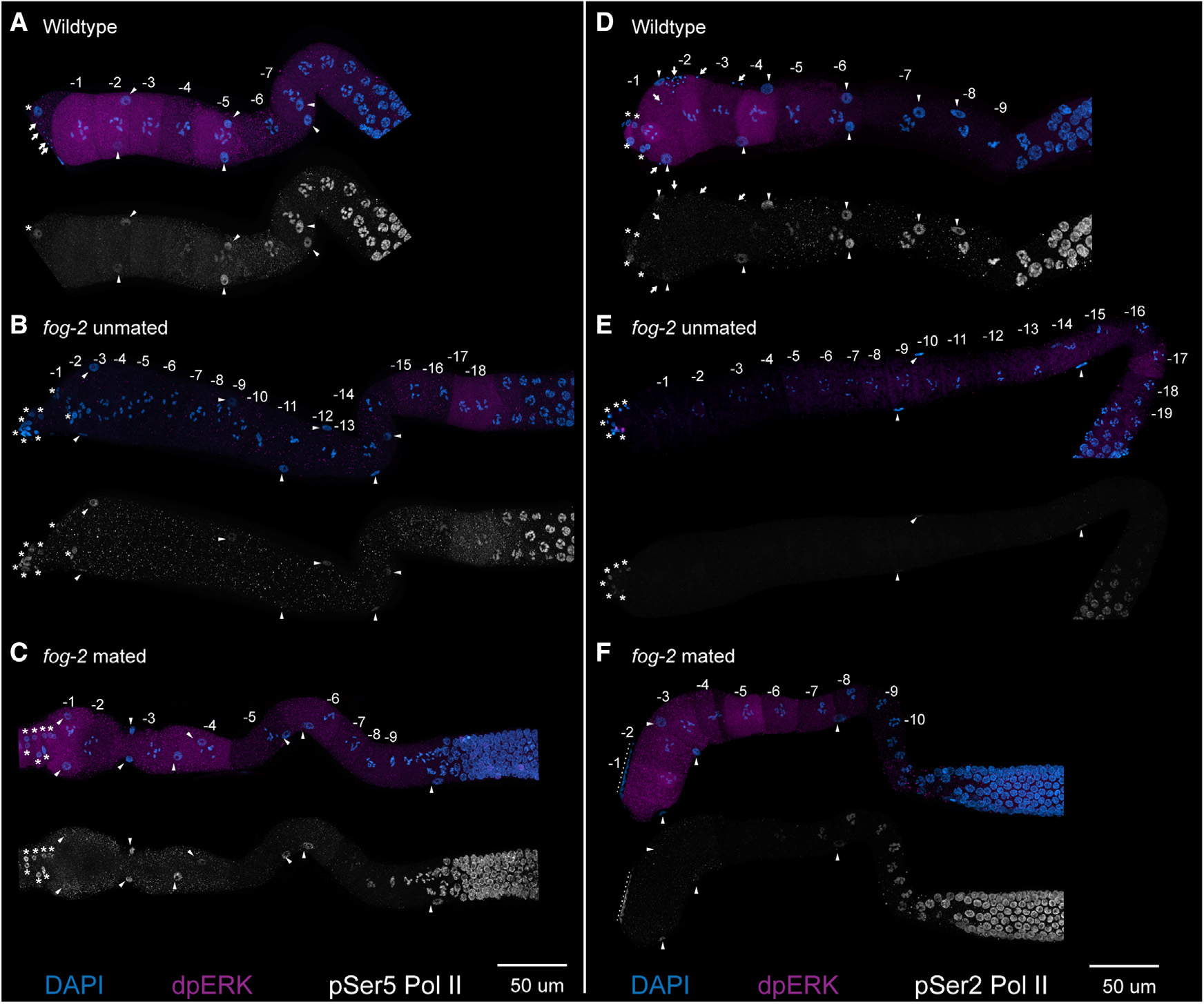
(A–F) Maximum-intensity projections from dissected germlines from (A and D) wild type, (B and E) feminized (fog-2) unmated, and (C and F) fog-2 mated for 2 h are shown with numbered oocytes. Germlines are stained with DAPI (blue), dpMPK-1 (magenta), and either pSer5 Pol II (white, A–C) or pSer2 Pol II (white, D–F). The nuclei of sheath cells (triangles), spermathecal cells (asterisks), and sperm (arrows) are labeled. Nuclei near the dotted lines in (F) are from a neighboring germline. Scale bars, 50 μm.
RNA velocity analysis (STAR Methods) was performed using reads that mapped to introns.32 Overall, we observed the presence of intronic reads to be ~0.35% of the total reads (Figure 4A) in each of the oocytes sequenced (−5, −3, and −1). However, 0.35% is an extremely low number compared with the 15%–25% of intronic reads in somatic cells.32–34 These data suggest that there is a drastic reduction in overall nascent transcription in meiosis I arrested C. elegans oocytes. Nevertheless, to determine whether the 0.35% of the genes that displayed intronic reads were differentially changed between oocytes, we compared pairs of arrested oocytes at different ages and observed that, as an oocyte progresses from −5 to −3 to −1, there is a very slight increase in the number of genes that carry intronic signatures (Figures 4B–4D). However, none of the genes with intronic reads correlated with significant differential expression of the exonic reads from the same gene (Figures 4E and 4F), suggesting that if there is a minimal nascent transcription, it does not significantly contribute to the transcriptome of the arrested oocyte.
Figure 4. RNA velocity to assess nascent transcription in arrested meiosis I oocytes.
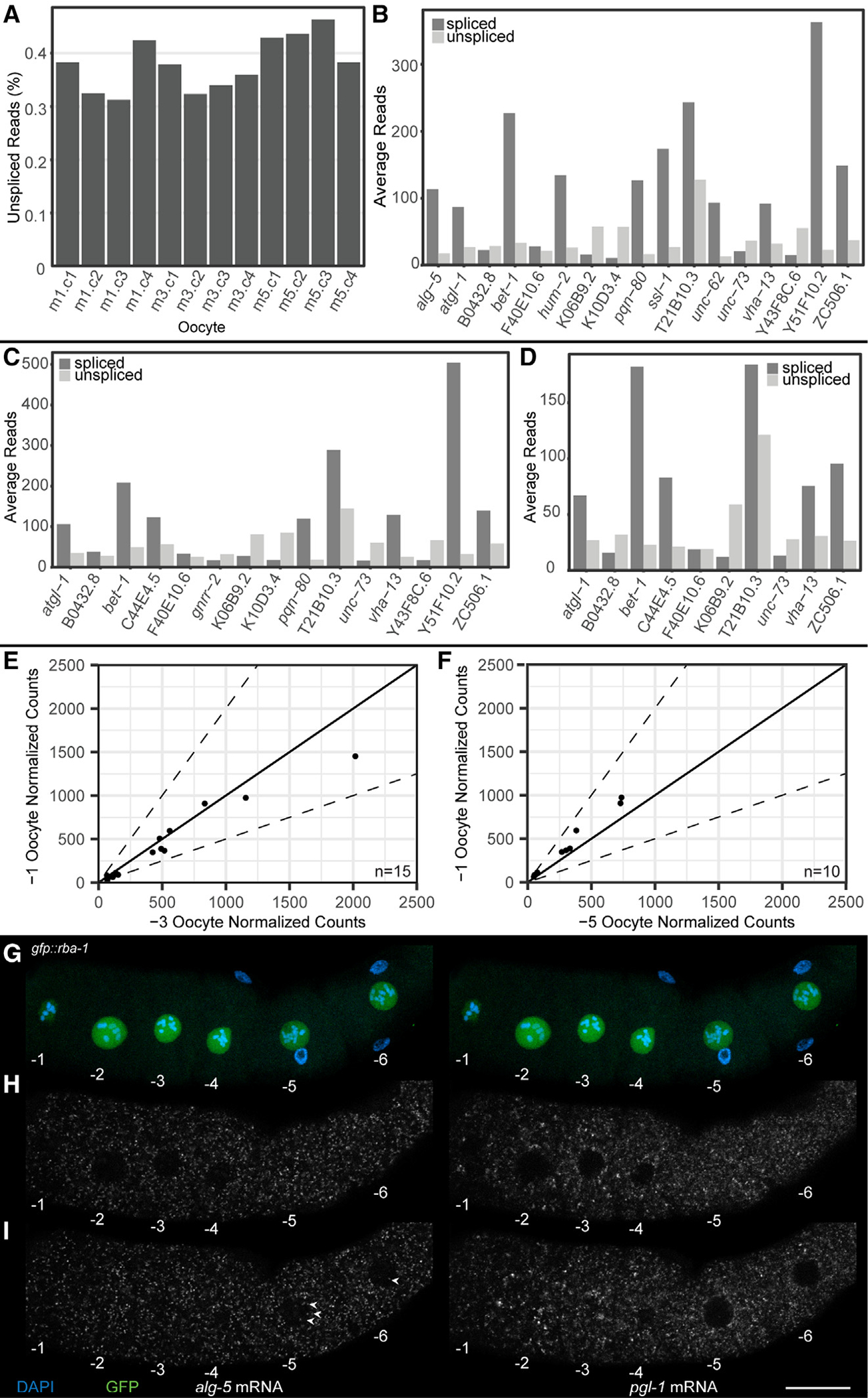
(A) Percentage intronic reads vs. total RNA reads. Oocyte positions are labeled m1 (−1 oocyte), m3 (−3 oocyte), and m5 (−5 oocyte).
(B–D) Intronic read count vs. exonic read count for genes detected as expressing intronic reads in (B) −1 oocyte, (C) −3 oocyte, and (D) −5 oocyte.
(E and F) Comparison of normalized exonic reads from DESeq2 of genes containing intronic reads in the −1 oocyte. Comparisons are between (E) the −1 and the −3 oocytes and (F) the −1 and the −5 oocytes. Dotted line depicts ±2-fold difference.
(G–I) Oocytes from a dissected gfp::rba-1 germline with DAPI (blue), GFP::RBA-1 (green), alg-5 mRNA (white, left side), and pgl-1 mRNA (white, right side). Oocytes are numbered decreasing from oldest to youngest. (G) Maximum-intensity projections of DAPI and GFP showing the positions and sizes of the oocyte nuclei. (H and I) Individual slices from a z stack of the dissected germline, one (H) at the medial plane of oocytes −1 through −4 and another (I) through the medial plane of oocytes −5 and −6. Nuclear alg-5 puncta are labeled (arrowheads). Scale bar, 20 μm. See also Figure S5 and Table S3.
To validate whether the intronic reads detected by RNA velocity were “nascent transcripts” in vivo, we performed a FISH analysis on three candidate RNAs. We limited the RNAs to be validated to those with more than 10 intronic reads and ranked them using the following criteria: (1) intronic reads were found in more than one intron, (2) at least one intronic read crossed an intron-exon boundary, and (3) intronic reads did not correlate with a misannotated 3′ UTR, using data from a recent in-depth 3′ UTR analysis35 (STAR Methods) (Table S3). We performed FISH analysis on the top three identified from these criteria (alg-5, pqn-80, and bet-1) and assayed the level of nascent transcription (signal in the nucleus) and its potential impact on the total abundance of the RNA (signal in the cytoplasm) using HCR-FISH.36 While targeting the introns directly would be the best way to assess this transcription, HCR-FISH requires at least 20 intronic probes against each target, and the intronic lengths for the targets being tested were insufficient to generate unique probes.36 Thus, we used the presence of a nuclear signal from the exonic probes as a readout of nascent transcription. We generated probes against alg-5, bet-1, and pqn-80 and used the stable transcript pgl-1, an abundant germ cell marker that did not show any changes in either intronic or exonic RNA abundance in the analysis, as a positive control for FISH and a negative control for intronic signal. Each probe was tested for specificity by RNAi-mediated depletion of the target gene, although only alg-5 was confirmed to be specific (Figure S5) (STAR Methods). To assay for nuclear signal, we used a nuclear marker, GFP::RBA-1 (STAR Methods), which localizes to the oocyte nucleoplasm and could thus be used to determine the boundaries of the nucleus. Confocal images from the HCR-FISH analysis of alg-5 were acquired and assayed together with nucleoplasmic GFP signal (which was used as a marker of nuclear area; STAR Methods). The HCR-FISH signal specifically contained within the GFP nucleoplasmic area was marked as a “positive” nuclear signal. We assayed both the cytoplasmic signal (marker for mature mRNA) from the probes and the nuclear signal. Overall, we observed very few nuclear puncta compared with the large amount of cytoplasmic signal (Figures 4G–4I). This suggests that if there is any nascent transcription in the oocytes, it is likely not biologically relevant, given the large amount of cytoplasmic transcript already loaded into the oocyte from the immature oogenic pachytene-stage cells. This observation is consistent with the RNA velocity analysis that showed that none of the genes with intronic signatures displayed any significant increase in exonic expression levels in the oocytes (Figures 4E and 4F). Thus, we conclude that (1) arrested meiosis I oocytes do not undergo significant nascent transcription and (2) small levels of nascent transcription do not contribute to the increase in transcript abundance as an arrested oocyte ages.
The highest fold-change class I transcripts in the −1 oocyte are acquired from the spermatheca
The increase in abundance of transcripts in oocytes in the absence of any transcription, however, is intriguing. This led us to investigate the potential mechanisms that may lead to an increase in transcripts in an arrested oocyte as it ages. To determine the nature of the transcripts that increased in abundance as the age of an arrested oocyte increased and to validate this phenomenon in vivo, we focused on the mRNAs that were most abundant in the −1 oocyte compared with either the −3 or the −5 oocyte. From the two separate comparisons, we identified five mRNAs that increased as an oocytes aged. Of these, one mRNA (ule-5) was significantly increased in the −1 oocyte compared with both the −3 and the −5 oocytes (Figure 5A); ZK813.1 and F17E9.4 were higher in the −1 vs. the −3 oocyte comparison, and ZC373.2 and D1054.10 were higher in the −1 vs. the −5 comparison (Table S1). We assayed all five for their expression using HCR-FISH probes and confirmed the specificity of the probes by RNAi (Figures S6 and S8). We observed that, as detected by scRNA-seq analysis, there was signal in the cytoplasm from the −1 oocyte for each of the five genes tested relative to the −3 and −5 oocytes (Figure 5). We were surprised to detect a very strong signal for each of the five genes, which was magnitudes of order higher in abundance, in the spermathecal cells relative to the −1 oocyte (Figures 5B–5K). This observation suggested that the expression of each of the genes tested likely originated from the spermatheca and was somatic in nature. Compared with the FISH signal in the spermatheca for these genes, the signal in the −1 oocyte appeared relatively low. Thus, to clearly detect this signal and ensure that it was present in the oocyte cytoplasm, we enhanced the contrast in the −1 oocyte, as shown for ule-5 (Figure 5C). The z sections from a central portion of the −1 oocyte (Figures 5E–5G) clearly demonstrate the presence of the ule-5 FISH puncta in the −1 oocyte cytoplasm, and these puncta are reduced or absent in the −3 and −5 oocytes, consistent with the scRNA-seq data (Figures 5C and S7). Strikingly, in the oocytes undergoing fertilization within the spermatheca, the number of puncta corresponding to the transcripts increased dramatically relative to the arrested −1 oocyte (Figure 5D). The strong signal in the spermathecal cells and oocytes undergoing fertilization relative to an arrested −1 oocyte suggested that the transcripts are formed in the spermatheca and then transported into the arrested or fertilizing oocyte. This was intriguing and exciting, since mRNAs have not been previously described in any system that may be taken up from neighboring cells prior to maturation, much less during ovulation and fertilization. However, because we observed this pattern with all five genes tested, the data suggest that there may be a general phenomenon present where the spermatheca provides maternal RNAs to the arrested oocyte.
Figure 5. Class I transcripts are present in the arrested −1 oocyte and the spermatheca.
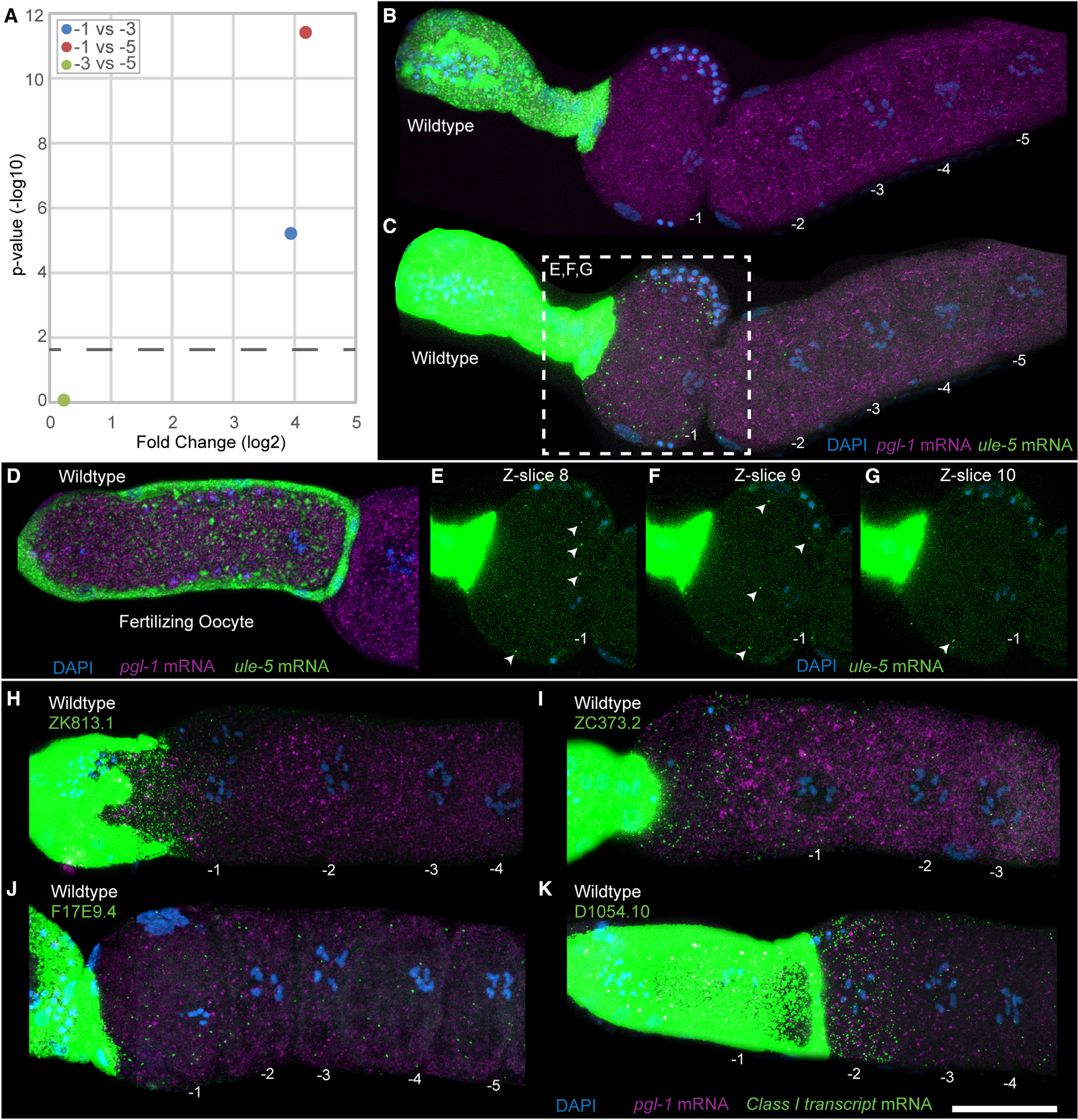
(A) Plot showing the fold change in ule-5 between different oocyte positions. The p value cutoff (horizontal dashed line) was p < 0.05.
(B–G) HCR-FISH staining of dissected wild-type germlines with DAPI (blue), ule-5 mRNA (green), and pgl-1 mRNA (magenta) with oocytes numbered, acquired as a z stack with 14 slices with a step size of 1.5 μm. (B and C) Maximum-intensity projection images through the full z stack with (B) normal acquisition intensity for ule-5 mRNA and (C) enhanced intensity for ule-5 mRNA. (D) Dissected wild-type germline with oocyte in the spermatheca. (E–G) Individual slices from central planes (−1 nucleus visible) of the z stack with enhanced intensity, ule-5 FISH signal (arrowheads) and slice number labeled. For −2, −3, −4, and −5 oocytes and end slices 1 and 14, see Figure S7.
(H–K) Dissected and stained germlines showing representative expression patterns for the class II transcripts (H) ZK813.1, (I) ZC373.2, (J) F17E9.4, and (K) D1054.10. Shown are maximum intensity projection images through the full z stack DNA (blue), pgl-1 mRNA (purple), and target mRNA (green). Scale bar, 25 μm. See also Figures S7, S8, and S9.
Since the spermatheca harbors the sperm, and only the −1 oocytes (along with fertilizing oocytes) were positive for the ule-5 mRNA signal, we wondered whether ule-5 transcription in the spermatheca was induced by the sperm signal. To test this, we assayed for ule-5 expression, using HCR-FISH in mated and unmated females (fog-2) (STAR Methods). We observed ule-5 signal in the −1 oocyte and spermatheca of both mated and unmated females, suggesting that ule-5 is not induced by the sperm signal but is instead generated in the spermatheca and likely acquired by the oocyte from the neighboring spermathecal cells (Figures 6A–6D). The expression of ule-5 RNA mirrors that of the ULE-5 protein, which is expressed in the spermatheca and secreted via COPII secretory vesicles, localizing to the eggshell of the fertilized embryo.37 However, the experiments to assess ULE-5 protein expression were performed using C. elegans transgenic lines generated through extrachromosomal arrays and high-copy-number transgenes, which are silenced in the C. elegans germline.37,38 To more directly test that the transcripts were being transported from the spermatheca to the −1 oocyte, we used CRISPR-Cas9 (STAR Methods) to replace the promoter of one of these class I transcripts, ZK813.1, with a spermatheca-specific promoter.
Figure 6. Class I transcripts are transferred from the spermatheca into the arrested −1 oocyte.
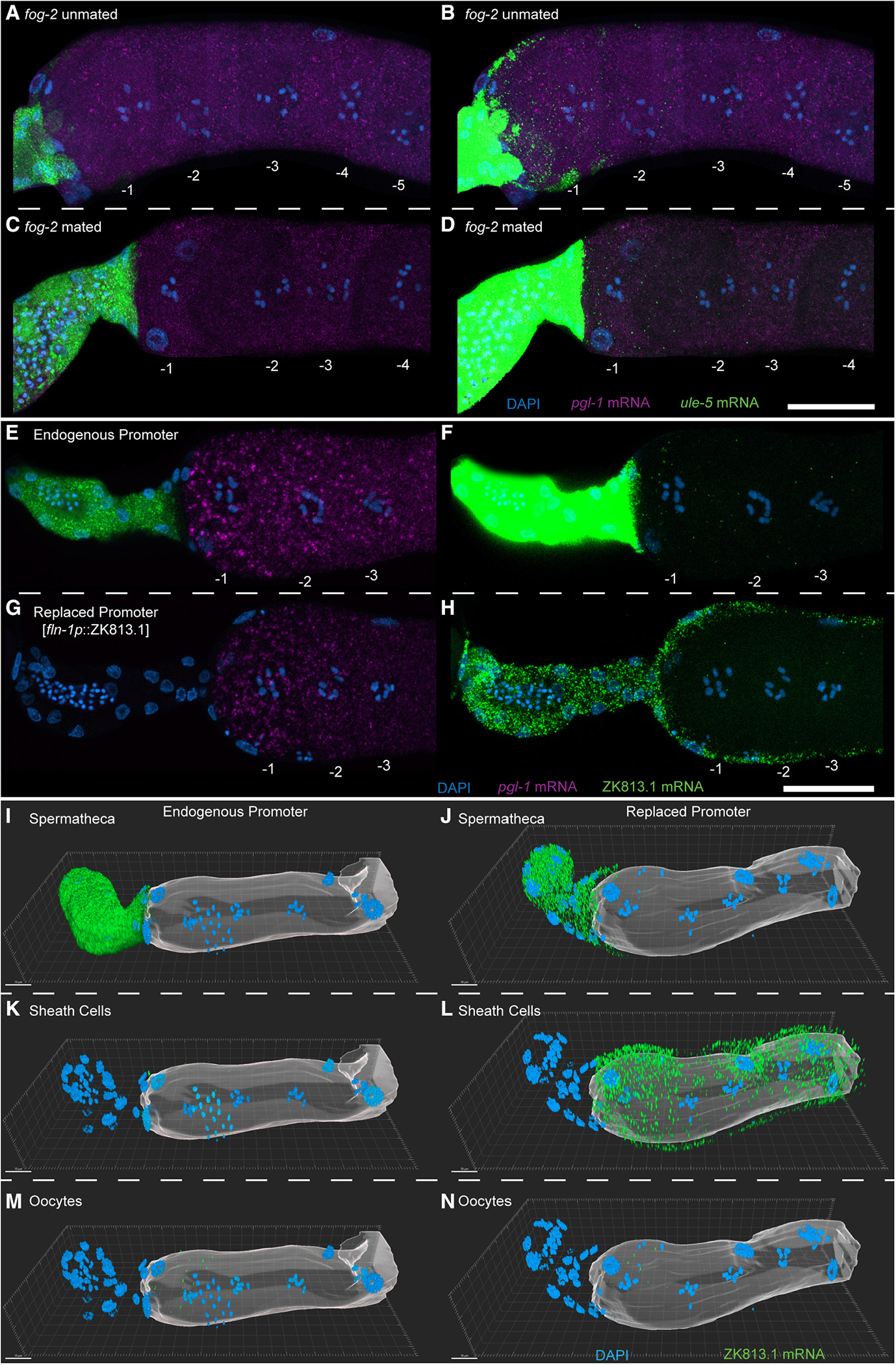
(A–N) HCR-FISH staining of dissected fog-2 (A and B) unmated and (C and D) mated germlines with DAPI (blue), ule-5 mRNA (green), and pgl-1 mRNA (magenta) with oocytes numbered. Shown are maximum-intensity projection images through the full z stack with (H and J) normal acquisition intensity for ule-5 mRNA and (I and K) enhanced intensity for ule-5 mRNA. (E–H) HCR-FISH staining of dissected and stained germlines from animals with (E and F) the endogenous ZK813.1 promoter or with (G and H) the ZK813.1 promoter replaced with the fln-1 promoter. Shown are maximum intensity projections of slices containing oocyte chromatin with DAPI (blue), ZK813.1 mRNA (green), and pgl-1 mRNA (magenta) with oocytes numbered. (I–N) Three-dimensional reconstruction of germlines with either (I, K, and M) the endogenous ZK813.1 promoter or (J, L, and N) the ZK813.1 promoter replaced with the fln-1 promoter. ZK813.1 mRNA signal has been separated by cell type: (I and J) spermatheca, (K and L) sheath cells, and (M and N) oocytes. Oocytes are represented by a translucent surface. Scale bars: (A–H) 25 μm, (I–N) 10 μm. See also Figure S10.
We used ZK813.1 and not ule-5 for this analysis because we reasoned that (1) the phenomenon seems general, so the identity of the gene itself that is being transported should not matter, and (2) the promoter of ule-5 is only 200 bp, with a predicted gene on the reverse strand which might share this promoter sequence. Since ZK813.1 has 1.1 kb upstream until the next gene and this region is not shared with any other genes, we replaced the putative promoter region upstream of ZK813.1 with a previously reported spermatheca promoter, a 957 bp region 5′ of the transcription start site (TSS) for fln-1a (Y66H1B.2a.1)39 (Figure S9A). Using two independent lines, we performed HCR-FISH to compare the expression pattern of wild-type ZK813.1 mRNA with that driven by the fln-1 promoter. We find that ZK813.1 mRNA is orders of magnitude less abundant in the spermatheca when driven by the fln-1 promoter compared with its endogenous promoter (Figures 6E and 6G). By enhancing the ZK813.1 signal, we found that not only is ZK813.1 mRNA still expressed in the spermatheca when driven by the fln-1 promoter, albeit at a very low level, it is now expressed in the sheath cells as well (Figures 6F, 6H, and 6I–6L, Videos S1 and S2). In addition, we found that ZK813.1 mRNA is still present in the oocytes, including the −1 oocyte, when driven by the fln-1 promoter (Figures 6H and 6N, Video S2), with no visible increase in puncta compared with the endogenous promoter (Figures 6F and 6M, Video S1). While the expression in the sheath cells increased drastically when driven by the fln-1 promoter, expression in the −1 oocyte did not reflect this increase in the sheath cells, suggesting that the ZK813.1 transcript is not transported from the sheath cells. Furthermore, we found that, while the ZK813.1 mRNA is still present in fertilizing oocytes when driven by the fln-1 promoter, it is orders of magnitude lower than when driven by the endogenous promoter (Figure S10). This change in expression levels mirrors that found in the spermatheca, suggesting that it is the spermatheca that provides the transcript. Together, these results provide an example of a somatic tissue providing mRNA transcripts to a maturing or fertilizing oocyte.
DISCUSSION
In this study, we report deep sequencing analysis of meiosis I arrested oocytes at different ages of meiosis I arrest. Currently, very little is known of these events in arrested meiosis I oocytes because oocytes have not been profiled at a single-cell resolution as a function of their spatial and temporal developmental state and response to hormonal signals. This is largely due to three technical challenges posed by this analysis, which we address through technical innovation in this study. (1) Currently there is no reliable biomarker for differentiating the oocytes’ birth order outside the gonad. Therefore, it is imperative that the identities of individual oocytes be accurately tracked throughout the extraction process to retain the knowledge of their spatial location in the gonad. (2) The oocytes need to be extracted from live animals, which is critical for capturing the RNA content closest to in vivo. Speed of extraction is another critical aspect for obtaining high-quality RNA sequencing. Oocytes ovulate every 20 min in an adult animal, and hormone-signaling through MSP rapidly induces changes to oocyte maturation within 15 min of mating.40 Since a majority of stimuli take only ~10–20 min to perturb RNA content in cells,41,42 the extraction procedure of oocytes needs to be rapid enough (<10 min per procedure) to capture RNA dynamics of interest and avoid undesired transcriptomic noise.
The existing techniques are inadequate to satisfy these requirements. While traditional laser capture microdissection (LCM) can maintain spatial information, it has poor resolution (~1/10 of the length of gonadal arms, ~50 μm) and cannot isolate single oocytes without damaging them.41 In addition, LCM requires sample fixing/freezing that incurs RNA degradation and impedes library preparation.43,44 The prevalent method for acquiring single C. elegans cells involves chemomechanical dissociation and fluorescence-activated cell sorting, which not only cannot distinguish individual oocytes due to lack of fluorescent markers, but also is a lengthy procedure (>2 h) and disruptive, while inducing undesirable shifts in RNA content of collected cells.45,46
This study addresses these challenges through the development of an image-guided, fs-LM method to enable rapid, contamination-free oocyte extraction. This method was based on our previous work where we used fs-laser axotomy in C. elegans to cut single axons with nanoscale resolution and minimal collateral damage to surrounding tissue.47–49 In fs-LM, we take advantage of the precision of fs-laser ablation to extract intact single oocytes from the gonads of living animals with the exact knowledge of their original location through imaging (Figure 1). By precisely ablating around a target oocyte in a 3D pattern, we resect the target oocyte from the gonad and release it with laser-induced microbubbles. Prior to collection with a micropipette, contamination such as attached somatic gonad debris is further removed by laser. The whole process takes 8 min to extract a single oocyte, which satisfies the speed requirement (<10 min) set forward by cellular signal transduction. By eliminating the need for slicing, freezing, or fixing, fs-LM al lows us to acquire healthy, intact cells, which is critical to the quality of downstream library construction for RNA sequencing. This technical innovation allowed us to characterize fundamental principles that govern RNA dynamics during this distinct and crucial developmental phase, with implications across evolution.
We observed that mRNA molecules are taken up by the oldest meiosis I arrested oocyte and fertilizing oocytes from the adjacent spermatheca. While it is possible that each of these transcripts in the maturing and fertilizing oocyte can be translated in the −1 arrested and fertilizing oocyte, it is likely that these class I RNAs contribute to the developing zygote as a maternal contribution for embryonic development. Regardless, it is intriguing that arrested meiosis I oocytes acquire mRNAs from somatic neighboring cells, since the implications for this in cases such as mammalian oocytes that are arrested for decades before ovulation suggests that the somatic microenvironment may greatly influence oocyte quality over time due to the acquisition of various RNAs from the soma. In worms, examples of somatic cells contributing mRNA to the germ cells have not been identified. Although somatic gonadal cells have been shown to regulate the intake of metabolites to support germ cell development through gap junctions,50 gap junctions are not expressed between spermathecal cells and oocytes in C. elegans,51–54 thus ruling this out as a method of transport. It is likely the five class I transcripts that were validated are transported through exosome-based communication, such as extracellular vesicles (EVs), or some as yet undiscovered mechanism. Somatic granulosa cells in mammals, which surround the oocyte, have been shown to secrete EVs into the follicular fluid that potentially transport small microRNA species as well as mRNAs and lipids to the oocyte, inducing gene expression changes in vitro,55 although in vivo evidence for these phenomena is currently lacking. To investigate EVs as a potential transfer mechanism, we screened through the available C. elegans EV datasets for the ule-5 transcript and found that it is present in EVs collected from C. elegans.56 However, we also discovered a number of germline-specific genes such as pgl-1 in these EVs, which we did not detect in the somatic cells of our scRNA-seq analysis or FISH analysis, thus suggesting that at this time, the specificity of the mRNAs contained in EVs as a mode of extracellular communication needs further investigation.56
Together, these data provide in vivo support of a model of maternal RNA accumulation wherein the oocytes take up RNA from neighboring somatic cells during meiosis I arrest (Figure 7). We speculate that similar mechanisms may work in mammalian oocytes to acquire mRNAs from neighboring cells. If so, the implications for this acquisition are numerous, since oocytes arrest for decades, leaving a long period of time during which oocytes could receive external mRNA transcripts with potential far-reaching effects on organismal development and fitness.
Figure 7. Model depicting the transfer of mRNA transcripts from the spermatheca into the −1 oocyte.
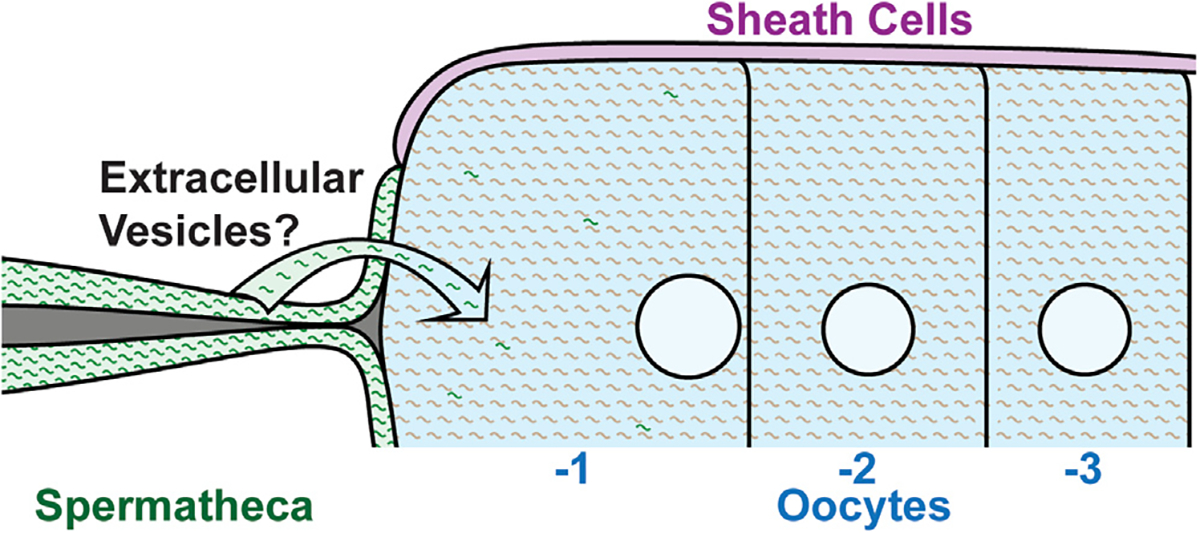
mRNA transcripts are transported from the spermatheca to the −1 oocyte by an unknown mechanism, although extracellular vesicles are likely.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Swathi Arur (sarur@mdanderson.org).
Materials availability
Plasmids generated during this study are freely available upon request.
C. elegans strains generated during this study have been deposited to the CGC.
Data and code availability
Sequencing files and selected processed data are available from the NCBI Gene Expression Omnibus. DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this work is available from the lead contact on request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Primary: anti-RNA Polymerase II (pSer 5) | Active Motif | Cat. 61085; RRID: AB_2687451 |
| Primary: anti-RNA Polymerase II (pSer 2) | Active Motif | Cat. 61083; RRID: AB_2687450 |
| Primary: anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling | Cat. 9101; RRID: AB_331646 |
| Secondary: goat anti-rat 488 | Molecular Probes | Cat. A11006; RRID: AB_141373 |
| Secondary: donkey anti-rabbit 594 | Invitrogen | Cat. A21207; RRID: AB_141637 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| pgl-1 RNAi | Vidal RNAi Library | Plate 10022: C4 |
| cyn-7 RNAi | Vidal RNAi Library | Plate 11052: C11 |
| mpk-1 RNAi (pSA001) | Arur Lab | |
| spn-4 RNAi | Vidal RNAi Library | Plate 11069: E9 |
| puf-3 RNAi | Vidal RNAi Library | Plate 10027: A4 |
| rme-2 RNAi | Vidal RNAi Library | Plate 10022: B9 |
| gfat-2 RNAi | Vidal RNAi Library | Plate 10028: B8 |
| pygl-1 RNAi | Vidal RNAi Library | Plate 10028: G7 |
| bet-1 RNAi | Vidal RNAi Library | Plate 11023: D10 |
| pqn-80 RNAi | Vidal RNAi Library | Plate 11206: H7 |
| alg-5 RNAi | Ahringer RNAi Library | Plate 1–5-1: B01 |
| ule-5 RNAi | Vidal RNAi Library | Plate 10008: E8 |
| ZK813.1 RNAi | Vidal RNAi Library | Plate 11207: F7 |
| ZC373.2 RNAi | Ahringer RNAi Library | Plate 6–4-4: H03 |
| F17E9.4 RNAi | Vidal RNAi Library | Plate 11040: B11 |
| D1054.10 RNAi | Vidal RNAi Library | Plate 10192: H11 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| RNase inhibitor | Takara Bio | Cat. 2313B |
|
| ||
| Critical commercial assays | ||
|
| ||
| SMART-seq v4 3’ DE Kit | Takara Bio | Cat. 635040 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed RNAseq data | This paper | GEO: GSE209988 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C. elegans: Strain LW4502: rba-1(jj188 [GFP::3xFLAG::RBA-1]) | Gift from Kelly Jun Liu | WB Strain: LW4502 |
| C. elegans: Strain BS553: fog-2(oz40) | CGC | WB Strain: BS553; WormBase: WBStrain00003934 |
| C. elegans: Strain AUM1870: ZK813.1(viz166) | This study | |
|
| ||
| Oligonucleotides | ||
|
| ||
| HCR FISH Probes | Molecular Instruments | See Table S4 |
| CRISPR-related oligos | This Study, Paix et al.57 | See Table S5 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | Schneider et al.58 | https://imagej.nih.gov/ij/ |
| Cutadapt v1.18 | Martin59 | https://cutadapt.readthedocs.io/en/v1.18/ |
| FastQC v0.11.5 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR aligner v2.4.0 | Dobin et al.60 | https://github.com/alexdobin/STAR |
| featureCounts v1.5.3 | Liao et al.61 | http://subread.sourceforge.net/ |
| DESeq2 | Love et al.26 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Velocyto | La Manno et al.32 | http://velocyto.org/ |
| Imaris | Oxford Instruments | https://imaris.oxinst.com/ |
| Adobe Photoshop | Adobe | https://www.adobe.com/products/photoshop/ |
|
| ||
| Other | ||
|
| ||
| RNAseq analysis and figure code | This paper | https://doi.org/10.5281/zenodo.7847503 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans
Worms were maintained at 20°C using standard culture conditions.62 Briefly, worms were maintained with Nematode Growth Media (NGM) plates seeded with of E. coli (OP50) as a food source. Worms were passaged under a stereomicroscope using a platinum wire pick. Strains used in this study: N2, BS553 fog-2(oz40), LW4502 rba-1(jj188 [GFP::3xFLAG::RBA-1]) (gift from Kelly Jun Liu at Cornell University). Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
METHOD DETAILS
Femtosecond laser microdissection (fs-LM) and RNA-sequencing
The femtosecond laser microdissection (fs-LM) method for the isolation of single neurons has been described in our previous paper.16 For the isolation of oocytes, we have slightly modified this method. In brief, healthy adult-stage animals were washed in M9 solution to remove bacteria, then transferred to a droplet of egg buffer containing ~5 mM of levamisole for up to 10 minutes. Using two 28G needles, the anesthetized animals were severed at the pharynx to expose their gonad and uterus under a stereomicroscope (Figure 1B). The buffer droplet containing the animals was then transferred to an upright microscope (BX51, Olympus) with a water dipping objective (60X, 1.0NA, LUMPLFLN60XW, Olympus) for fs-LM (Figure 1C) using 805nm wavelength, 1kHz repetition rate, 250 fs pulse width laser (Spitfire, Spectra Physics). The fs-LM process involved 4 main steps: 1) First, the gonadal sheath was punctured at 1–2 oocytes distal to the target oocyte using 150 fs-laser pulses with 50 nJ pulse energies to create an opening through which the target oocyte could be released (Figure 1D). 2) Next, lower energy fs-laser pulses (20 nJ) were used to generate gentle liquid flows to clear the debris, dislodge the target oocyte, and move it away from the carcass (Figure 1E). 3) The released oocyte was inspected for integrity prior to collection, for which we used pneumatically controlled (IM-11–2, Narishige) 40 μm inner diameter glass micropipettes coated with silicone (Sigmacote, Sigma-Aldrich) and prefilled with L15 solution. When approaching the released oocyte, we applied positive pressure to the micropipette and kept a constant outflow to avoid contamination and blew the released oocyte from the surrounding debris. 4) The target oocyte was collected through aspiration (Figure 1F) and washed twice in a fresh ice-cold L15 medium to further remove potential contaminants. All collected oocytes were lysed in 5.25 μl of lysis buffer with 2 U/μl of RNase inhibitor (Takara Bio) and then promptly frozen at −80 °C (Figure 1G). For RNA-sequencing, we used SMART-seq v4 3’ DE Kit (Takara Bio) following the manufacturer’s manual. Four oocytes were collected from each desired position (−1, −3, and −5). Maturity of the −1 oocyte is unknown. All collected single oocyte samples showed uniform amplification, which indicates high sample quality. We constructed Next-generation sequencing (NGS) libraries using the Nextera XT Kit (Illumina). All libraries were sequenced on Hi-seq 4000 (Illumina) PE150 mode with 1% PhiX control at a depth of 10 million reads per oocyte.
RNAi
RNAi was performed by feeding as described previously.63 pgl-1, bet-1, alg-5, and ule-5 RNAi clones were sequence verified and grown overnight at 37°C on LB plates containing 100ug/mL of ampicillin and 50 ug/ml of tetracycline. They were then grown in liquid LB containing 100ug/mL of ampicillin to an optical density of 0.6 – 0.7. Cultures were then seeded onto NGM agar plates supplemented with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and containing 100 μg/ml of ampicillin and 50 μg/ml of tetracycline. pgl-1 RNAi was performed by transferring L4 stage wildtype animals onto pgl-1 RNAi. alg-5 and bet-1 RNAi was performed by allowing wildtype animals to lay progeny on the RNAi plates for 4 hours, after which the mothers were removed from the plate. The animals were then synchronized at the mid-L4 stage for dissection and analysis at 24 hours past mid-L4.
Brood size and viability
To determine brood size, individual L4 animals which had been treated with RNAi from hatch were transferred to separate RNAi plates. They were transferred twice a day, with their embryos counted on the previous plate after each transfer. This continued until adult animals laid no more embryos. To determine viability, adults were counted from each F1 plate. Percent survival was determined as the number of embryos counted divided by the number of adults counted multiplied by 100.
ZK813.1 promoter replacement
Primers JHS 51 and JHS 52, each containing 30bp homology arms to an intergenic region 5’ of ZK813.1 to ZK813.1 Exon 1 respectively, were used to amplify a 975bp fragment containing the fln-1 promoter sequence39 from N2 bristol genomic DNA. This fragment was cloned into the TOPO vector pCR2.1 to create pSYC196. This plasmid was then used to generate a hybrid ssDNA repair template as previously described64 using primers JHS 49, JHS 50, JHS 51, and JHS 52. The repair template was amplified via PCR and gel purified. Co-CRISPR was adapted from Paix et. al 2017.57 Repair template was injected at a concentration of 188 ng/μL into late L4/Young Adult N2 worms along with 2 crRNAs (ZK813.1_pro_5’, ZK813.1_pro_3’), along with a dpy-10 co-CRISPR marker to delete 1046 bp upstream of ZK813.1 and replace it with the 975 bp fln-1 promoter sequence (Figure S9A). F1 rollers and dumpy rollers were cloned out and edits were screened via PCR and partial sequencing of the 3’ junction of the insertion on F1 animals using primers JHS 53, JHS 54, and JHS 55. Homozygous F2s were then screened using primers JHS 53, JHS 54, JHS 57. Sequencing was then performed on homozygous animals to verify the insertion sequence, leading to the verification of 5 independent lines (Figure S9B). In lieu of backcrossing, HCR-FISH was performed on 3 independent CRISPR lines alongside N2 as described below.
Dissection and Immunostaining and HCR-FISH
All animals were dissected as adults at 24 hours past mid L4 unless otherwise mentioned. Germlines were dissected and immunostained as described previously65 with the following alterations. Fixation was performed in 3% PFA for 15 min. Methanol treatment was performed overnight. For HCR-FISH, dissections were performed as above. After methanol treatment, germlines were processed as previously described36 with the following alterations. Processing was performed in the same tubes as immunostaining (6mm tube) and all wash incubation volumes were halved and DAPI was added during the final washes. Additionally, amplification hairpins were added at ½ of the recommended concentration. In the case that there were two treatments for a particular experiment, probe hybridization was performed on one of the treatments using probes against either mpk-1 or cyb-1 to differentiate between treatments. That sample was then washed with probe wash buffer as in the protocol, and then two conditions were combined and probe hybridization was performed for the experimental probes.
Antibodies
The following primary antibodies were used at 1:400: anti-RNA Polymerase II (pSer 5) (cat. 61085, Active Motif), anti-RNA Polymerase II (pSer 2) (cat. 61083, Active Motif), anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (Cat. 9101, Cell Signaling). The following secondary antibodies were used at 1:800: goat anti-rat 488 (Cat. A11006, Molecular Probes), donkey anti-rabbit 594 (Cat. A21207, Invitrogen)
Image acquisition and processing
Images were taken on a Zeiss Axio Observer with an LSM 800 confocal laser scanning module using Zeiss Zen micro-imaging software. Images with more than one field were captured with overlapping boundaries either as z-stacks, or single slices while maintaining the same focal plane. Montage images were then assembled in Adobe Photoshop 2020 and processed identically apart from the enhanced intensity images for the ule-5 probe. For these images, starting with the acquisition intensity image, the channel corresponding to ule-5 (green) was altered to increase the overall intensity using the Levels adjustment in Photoshop 2020 to more prominently display the weak ule-5 signal present in the oocytes. This Levels adjustment was performed identically for sets of compared images. Maximum intensity projections were created using ImageJ.58 3D reconstruction was performed using Imaris (Oxford Instruments). Briefly, images were first deconvolved using default parameters. Surfaces were then created manually to approximately envelop 3 different regions: the spermatheca, the sheath cells, and the oocytes. ZK813.1 HCR-FISH signal was then masked using each surface to assign signal to each compartment. Finally, DAPI and ZK813.1 signal was visualized using ‘Blend’ rendering. For movie creation, outlines for the spermatheca (purple) and sheath cells (light purple) were set to opaque and clipping planes were used to create a 3um thick slice which move through the entire sample along the Z axis.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq data analysis
Raw FASTQ files were first demultiplexed according to the cellular barcode to retrieve reads for every single oocyte. On average, each oocyte had 10M read pairs. As read 2 mostly contain the cellular barcode and poly-A tail, we only used read 1 for further analysis. We trimmed the reads for adaptor and low quality (Q > 30) base pairs with Cutadapt v1.1859 and performed a quality check with FastQC v0.11.5. Alignment was performed by STAR aligner v2.4.060 to C. elegans reference genome WS259, with a unique mapping rate of ~90%. We summarized the mapping results to counts with featureCounts v1.5.3.61 Only unique mapping and unique assignments were accounted for during the process.
As only the 3’ end of the transcript was retained during library preparation, we normalized the counts into Count Per Million (CPM) for each gene. ~8000 genes were detected at a threshold of 1 CPM (Figure S1B), which is comparable to similar RNA-seq performed on single embryonic cells.66 We further subsampled the SAM files at varied ratios (10%–90%) to confirm sufficient sequencing depth for maximal gene detection. The expression level of genes displayed a smooth distribution, with most of the genes expressed at a moderate level (10 – 100 CPM, Figure S1B). DESeq226 was then used to normalize the counts and show changes in expression level. A cutoff of 2-fold change with an adjusted p-value < 0.05 was used to identify expression level changes. GO term enrichment analysis was performed using the PANTHER Overrepresentation Test using a list of genes with >1 fold decrease from the −5 to −1 oocyte using the PANTHER GO-Slim Biological Process annotation data set and was tested by a Fisher’s Test with a Bonferroni correction.67,68
RNA Velocity analysis
Reads were re-aligned using the STAR aligner with the WBcel235.97 assembly of the C. elegans genome. Following alignment and sorting of BAM files, we performed library complexity analysis using picard. The analysis showed a very high percentage of duplicate reads (over 90% in almost all cells), indicating potential library over-sequencing, which could be due to initially low cDNA load, type of cell sequenced, or could be protocol-specific. Removing duplicates from paired-end reads is generally more effective due to the lower probability of incorrectly marking non-duplicate reads as duplicates and can have significant effects on differential expression analysis. Correctly identified duplicates lead to a reduced amount of noise due to PCR amplification. Incorrect identification of duplicates can reduce the number of significantly expressed genes, especially for highly expressed genes. Therefore, we continued two analyses in parallel with filtered and unfiltered datasets, though only the filtered data are shown for the intronic analysis. Data were then combined into per-sample count matrices separately for intronic and exonic reads using velocyto.32 Genes which contained at least 10 reads in each sample for a particular oocyte age were considered to have intronic reads. For validation, genes were then ranked using the following criteria: 1) Intronic reads were found in more than one intron, 2) At least one intronic read crossed an intron-exon boundary, 3) Intronic reads did not correlate with a mis-annotated 3′ UTR, using data from a recent in-depth 3′ UTR analysis.35
Supplementary Material
Highlights.
Individual oocytes were extracted using femtosecond laser microdissection
Oocyte transcript abundance changes during meiosis I arrest
Maternal transcripts are acquired from somatic cells
ACKNOWLEDGMENTS
We thank Biomage for assistance with the RNA velocity analysis, all the members of the Arur laboratory for helpful discussions, and Kelly Jun Liu for the LW4502 strain. This work was supported by National Institutes of Health (NIH) grants R35-GM140933 (S.A.), R01-HD101269 (S.A.), and R21-NS109821 (A.B.-Y.); a grant from The University of Texas System Neuroscience and Neurotechnology Research Institute (UTS-NNRI); and a training fellowship from the Gulf Coast Consortia, on the Training In Precision Environmental Health Sciences (TPEHS) Program (NIH Grant No. T32ES027801) (K.A.T.). S.A. is the Andrew Sabin Fellow at the University of Texas MD Anderson Cancer Center.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112544.
REFERENCES
- 1.Nader N, Courjaret R, Dib M, Kulkarni RP, and Machaca K (2016). Release from Xenopus oocyte prophase I meiotic arrest is independent of a decrease in cAMP levels or PKA activity. Development 143, 1926–1936. 10.1242/dev.136168. [DOI] [PubMed] [Google Scholar]
- 2.Das D, Pal S, and Maitra S (2016). Releasing prophase arrest in zebra-fish oocyte: synergism between maturational steroid and Igf1. Reproduction 151, 59–72. 10.1530/REP-15-0389. [DOI] [PubMed] [Google Scholar]
- 3.Richard S, and Baltz JM (2014). Prophase I arrest of mouse oocytes mediated by natriuretic peptide precursor C requires GJA1 (connexin-43) and GJA4 (connexin-37) gap junctions in the antral follicle and cumulus-oocyte complex. Biol. Reprod. 90, 137. 10.1095/biolreprod.114.118505. [DOI] [PubMed] [Google Scholar]
- 4.Huber S, and Fieder M (2018). Evidence for a maximum “shelf-life” of oocytes in mammals suggests that human menopause may be an implication of meiotic arrest. Sci. Rep. 8, 14099. 10.1038/s41598-018-32502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huelgas-Morales G, and Greenstein D (2018). Control of oocyte meiotic maturation in C. elegans. Semin. Cell Dev. Biol. 84, 90–99. 10.1016/j.semcdb.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hassold T, and Hunt P (2009). Maternal age and chromosomally abnormal pregnancies: what we know and what we wish we knew. Curr. Opin. Pediatr. 21, 703–708. 10.1097/MOP.0b013e328332c6ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seydoux G, and Schedl T (2001). The germline in C. elegans: origins, proliferation, and silencing. Int. Rev. Cytol. 203, 139–185. [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto I, Kosinski ME, and Greenstein D (2006). Start me up: cell signaling and the journey from oocyte to embryo in C. elegans. Dev. Dyn. 235, 571–585. 10.1002/dvdy.20662. [DOI] [PubMed] [Google Scholar]
- 9.Robertson S, and Lin R (2013). The oocyte-to-embryo transition. Adv. Exp. Med. Biol. 757, 351–372. 10.1007/978-1-4614-4015-4_12. [DOI] [PubMed] [Google Scholar]
- 10.Schultz RM, Stein P, and Svoboda P (2018). The oocyte-to-embryo transition in mouse: past, present, and future. Biol. Reprod. 99, 160–174. 10.1093/biolre/ioy013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Svoboda P, Franke V, and Schultz RM (2015). Sculpting the transcriptome during the oocyte-to-embryo transition in mouse. Curr. Top. Dev. Biol. 113, 305–349. 10.1016/bs.ctdb.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 12.Walker AK, Boag PR, and Blackwell TK (2007). Transcription reactivation steps stimulated by oocyte maturation in C. elegans. Dev. Biol. 304, 382–393. 10.1016/j.ydbio.2006.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navarro-Costa P, McCarthy A, Prudêncio P, Greer C, Guilgur LG, Becker JD, Secombe J, Rangan P, and Martinho RG (2016). Early programming of the oocyte epigenome temporally controls late prophase I transcription and chromatin remodelling. Nat. Commun. 7, 12331. 10.1038/ncomms12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun MJ, Zhu S, Li YW, Lin J, Gong S, Jiao GZ, Chen F, and Tan JH (2016). An essential role for the intra-oocyte MAPK activity in the NSN-to-SN transition of germinal vesicle chromatin configuration in porcine oocytes. Sci. Rep. 6, 23555. 10.1038/srep23555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hubbard EJ, and Greenstein D (2000). The Caenorhabditis elegans gonad: a test tube for cell and developmental biology. Dev Dyn 218, 2–22. . [DOI] [PubMed] [Google Scholar]
- 16.Zhao P, Martin C, Ma KY, Jiang N, and Ben-Yakar A (2023). Femtosecond laser microdissection for isolation of regenerating C. elegans neurons for single-cell RNA sequencing. Nat. Methods 20, 590–599. 10.1038/s41592-023-01804-3. [DOI] [PubMed] [Google Scholar]
- 17.Miller MA, Ruest PJ, Kosinski M, Hanks SK, and Greenstein D (2003). An Eph receptor sperm-sensing control mechanism for oocyte meiotic maturation in Caenorhabditis elegans. Genes Dev. 17, 187–200. 10.1101/gad.1028303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez AL 3rd, Chen J, Joo HJ, Drake M, Shidate M, Kseib C, and Arur S (2013). DAF-2 and ERK couple nutrient availability to meiotic progression during Caenorhabditis elegans oogenesis. Dev. Cell 27, 227–240. 10.1016/j.devcel.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee MH, Ohmachi M, Arur S, Nayak S, Francis R, Church D, Lambie E, and Schedl T (2007). Multiple functions and dynamic activation of MPK-1 extracellular signal-regulated kinase signaling in Caenorhabditis elegans germline development. Genetics 177, 2039–2062. 10.1534/genetics.107.081356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly WG, Schaner CE, Dernburg AF, Lee MH, Kim SK, Villeneuve AM, and Reinke V (2002). X-chromosome silencing in the germline of C. elegans. Development 129, 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schisa JA, Pitt JN, and Priess JR (2001). Analysis of RNA associated with P granules in germ cells of C. elegans adults. Development 128, 1287–1298. 10.1242/dev.128.8.1287. [DOI] [PubMed] [Google Scholar]
- 22.Maddox AS, Habermann B, Desai A, and Oegema K (2005). Distinct roles for two C. elegans anillins in the gonad and early embryo. Development 132, 2837–2848. 10.1242/dev.01828. [DOI] [PubMed] [Google Scholar]
- 23.Wolke U, Jezuit EA, and Priess JR (2007). Actin-dependent cytoplasmic streaming in C. elegans oogenesis. Development 134, 2227–2236. 10.1242/dev.004952. [DOI] [PubMed] [Google Scholar]
- 24.Tintori SC, Osborne Nishimura E, Golden P, Lieb JD, and Goldstein B (2016). A transcriptional lineage of the early C. elegans embryo. Dev. Cell 38, 430–444. 10.1016/j.devcel.2016.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoeckius M, Grün D, Kirchner M, Ayoub S, Torti F, Piano F, Herzog M, Selbach M, and Rajewsky N (2014). Global characterization of the oocyte-to-embryo transition in Caenorhabditis elegans uncovers a novel mRNA clearance mechanism. EMBO J. 33, 1751–1766. 10.15252/embj.201488769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su YQ, Sugiura K, Woo Y, Wigglesworth K, Kamdar S, Affourtit J, and Eppig JJ (2007). Selective degradation of transcripts during meiotic maturation of mouse oocytes. Dev. Biol. 302, 104–117. 10.1016/j.ydbio.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haupt KA, Law KT, Enright AL, Kanzler CR, Shin H, Wickens M, and Kimble J (2020). A PUF hub drives self-renewal in Caenorhabditis elegans germline stem cells. Genetics 214, 147–161. 10.1534/genetics.119.302772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grant B, and Hirsh D (1999). Receptor-mediated endocytosis in the Caenorhabditis elegans oocyte. Mol. Biol. Cell 10, 4311–4326. 10.1091/mbc.10.12.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komarnitsky P, Cho EJ, and Buratowski S (2000). Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14, 2452–2460. 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schroeder SC, Schwer B, Shuman S, and Bentley D (2000). Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev. 14, 2435–2440. 10.1101/gad.836300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaidatzis D, Burger L, Florescu M, and Stadler MB (2015). Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat. Biotechnol. 33, 722–729. 10.1038/nbt.3269. [DOI] [PubMed] [Google Scholar]
- 34.Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, and Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 10, 1096–1098. 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 35.Steber HS, Gallante C, O’Brien S, Chiu PL, and Mangone M (2019). The C. elegans 3’ UTRome v2 resource for studying mRNA cleavage and polyadenylation, 3’-UTR biology, and miRNA targeting. Genome Res. 29, 2104–2116. 10.1101/gr.254839.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi HMT, Schwarzkopf M, Fornace ME, Acharya A, Artavanis G, Stegmaier J, Cunha A, and Pierce NA (2018). Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, dev165753. 10.1242/dev.165753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimmerman SM, Hinkson IV, Elias JE, and Kim SK (2015). Reproductive aging drives protein accumulation in the uterus and limits lifespan in C. elegans. PLoS Genet. 11, e1005725. 10.1371/journal.pgen.1005725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly WG, Xu S, Montgomery MK, and Fire A (1997). Distinct requirements for somatic and germline expression of a generally expressed Caernorhabditis elegans gene. Genetics 146, 227–238. 10.1093/genetics/146.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kovacevic I, and Cram EJ (2010). FLN-1/filamin is required for maintenance of actin and exit of fertilized oocytes from the spermatheca in C. elegans. Dev. Biol. 347, 247–257. 10.1016/j.ydbio.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCarter J, Bartlett B, Dang T, and Schedl T (1999). On the control of oocyte meiotic maturation and ovulation in Caenorhabditis elegans. Dev. Biol. 205, 111–128. 10.1006/dbio.1998.9109. [DOI] [PubMed] [Google Scholar]
- 41.Yosef N, and Regev A (2011). Impulse control: temporal dynamics in gene transcription. Cell 144, 886–896. 10.1016/j.cell.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uhlitz F, Sieber A, Wyler E, Fritsche-Guenther R, Meisig J, Landthaler M, Klinger B, and Blüthgen N (2017). An immediate-late gene expression module decodes ERK signal duration. Mol. Syst. Biol. 13, 944. 10.15252/msb.20177986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tzur YB, Winter E, Gao J, Hashimshony T, Yanai I, and Colaiácovo MP (2018). Spatiotemporal gene expression analysis of the Caenorhabditis elegans germline uncovers a syncytial expression switch. Genetics 210, 587–605. 10.1534/genetics.118.301315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anjam MS, Ludwig Y, Hochholdinger F, Miyaura C, Inada M, Siddique S, and Grundler FMW (2016). An improved procedure for isolation of high-quality RNA from nematode-infected Arabidopsis roots through laser capture microdissection. Plant Methods 12, 25. 10.1186/s13007-016-0123-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson-Maduro J, Baron CS, Robin C, and van Oudenaarden A (2017). Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936. 10.1038/nmeth.4437. [DOI] [PubMed] [Google Scholar]
- 46.Spencer WC, McWhirter R, Miller T, Strasbourger P, Thompson O, Hillier LW, Waterston RH, and Miller DM 3rd. (2014). Isolation of specific neurons from C. elegans larvae for gene expression profiling. PLoS One 9, e112102. 10.1371/journal.pone.0112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bourgeois F, and Ben-Yakar A (2007). Femtosecond laser nanoaxotomy properties and their effect on axonal recovery in C. elegans. Opt Express 15, 8521–8531. [DOI] [PubMed] [Google Scholar]
- 48.Guo SX, Bourgeois F, Chokshi T, Durr NJ, Hilliard MA, Chronis N, and Ben-Yakar A (2008). Femtosecond laser nanoaxotomy lab-on-a-chip for in vivo nerve regeneration studies. Nat. Methods 5, 531–533. 10.1038/nmeth.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gokce SK, Guo SX, Ghorashian N, Everett WN, Jarrell T, Kottek A, Bovik AC, and Ben-Yakar A (2014). A fully automated microfluidic femtosecond laser axotomy platform for nerve regeneration studies in C. elegans. PLoS One 9, e113917. 10.1371/journal.pone.0113917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Starich TA, Bai X, and Greenstein D (2020). Gap junctions deliver malonyl-CoA from soma to germline to support embryogenesis in Caenorhabditis elegans. Elife 9, e58619. 10.7554/eLife.58619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.El-Hayek S, and Clarke HJ (2016). Control of oocyte growth and development by intercellular communication within the follicular niche. Results Probl. Cell Differ. 58, 191–224. 10.1007/978-3-319-31973-5_8. [DOI] [PubMed] [Google Scholar]
- 52.Gilula NB, Reeves OR, and Steinbach A (1972). Metabolic coupling, ionic coupling and cell contacts. Nature 235, 262–265. 10.1038/235262a0. [DOI] [PubMed] [Google Scholar]
- 53.Starich TA, Hall DH, and Greenstein D (2014). Two classes of gap junction channels mediate soma-germline interactions essential for germline proliferation and gametogenesis in Caenorhabditis elegans. Genetics 198, 1127–1153. 10.1534/genetics.114.168815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bohrmann J, and Zimmermann J (2008). Gap junctions in the ovary of Drosophila melanogaster: localization of innexins 1, 2, 3 and 4 and evidence for intercellular communication via innexin-2 containing channels. BMC Dev. Biol. 8, 111. 10.1186/1471-213X-8-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gebremedhn S, Gad A, Aglan HS, Laurincik J, Prochazka R, Salilew-Wondim D, Hoelker M, Schellander K, and Tesfaye D (2020). Extracellular vesicles shuttle protective messages against heat stress in bovine granulosa cells. Sci. Rep. 10, 15824. 10.1038/s41598-020-72706-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Russell JC, Kim TK, Noori A, Merrihew GE, Robbins JE, Golubeva A, Wang K, MacCoss MJ, and Kaeberlein M (2020). Composition of Caenorhabditis elegans extracellular vesicles suggests roles in metabolism, immunity, and aging. Geroscience 42, 1133–1145. 10.1007/s11357-020-00204-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Paix A, Folkmann A, and Seydoux G (2017). Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods 121–122, 86–93. 10.1016/j.ymeth.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martin M (2011). Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads, 17, p. 3. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 60.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 62.Brenner S (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arur S, Ohmachi M, Nayak S, Hayes M, Miranda A, Hay A, Golden A, and Schedl T (2009). Multiple ERK substrates execute single biological processes in Caenorhabditis elegans germ-line development. Proc. Natl. Acad. Sci. USA 106, 4776–4781. 10.1073/pnas.0812285106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dokshin GA, Ghanta KS, Piscopo KM, and Mello CC (2018). Robust genome editing with short single-stranded and long, partially single-stranded DNA donors in Caenorhabditis elegans. Genetics 210, 781–787. 10.1534/genetics.118.301532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gervaise AL, and Arur S (2016). Spatial and temporal analysis of active ERK in the C. elegans germline. J. Vis. Exp. 10.3791/54901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hashimshony T, Wagner F, Sher N, and Yanai I (2012). CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2, 666–673. 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 67.Thomas PD, Ebert D, Muruganujan A, Mushayahama T, Albou LP, and Mi H (2022). PANTHER: making genome-scale phylogenetics accessible to all. Protein Sci. 31, 8–22. 10.1002/pro.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, and Thomas PD (2019). Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 14, 703–721. 10.1038/s41596-019-0128-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing files and selected processed data are available from the NCBI Gene Expression Omnibus. DOI is listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this work is available from the lead contact on request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Primary: anti-RNA Polymerase II (pSer 5) | Active Motif | Cat. 61085; RRID: AB_2687451 |
| Primary: anti-RNA Polymerase II (pSer 2) | Active Motif | Cat. 61083; RRID: AB_2687450 |
| Primary: anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling | Cat. 9101; RRID: AB_331646 |
| Secondary: goat anti-rat 488 | Molecular Probes | Cat. A11006; RRID: AB_141373 |
| Secondary: donkey anti-rabbit 594 | Invitrogen | Cat. A21207; RRID: AB_141637 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| pgl-1 RNAi | Vidal RNAi Library | Plate 10022: C4 |
| cyn-7 RNAi | Vidal RNAi Library | Plate 11052: C11 |
| mpk-1 RNAi (pSA001) | Arur Lab | |
| spn-4 RNAi | Vidal RNAi Library | Plate 11069: E9 |
| puf-3 RNAi | Vidal RNAi Library | Plate 10027: A4 |
| rme-2 RNAi | Vidal RNAi Library | Plate 10022: B9 |
| gfat-2 RNAi | Vidal RNAi Library | Plate 10028: B8 |
| pygl-1 RNAi | Vidal RNAi Library | Plate 10028: G7 |
| bet-1 RNAi | Vidal RNAi Library | Plate 11023: D10 |
| pqn-80 RNAi | Vidal RNAi Library | Plate 11206: H7 |
| alg-5 RNAi | Ahringer RNAi Library | Plate 1–5-1: B01 |
| ule-5 RNAi | Vidal RNAi Library | Plate 10008: E8 |
| ZK813.1 RNAi | Vidal RNAi Library | Plate 11207: F7 |
| ZC373.2 RNAi | Ahringer RNAi Library | Plate 6–4-4: H03 |
| F17E9.4 RNAi | Vidal RNAi Library | Plate 11040: B11 |
| D1054.10 RNAi | Vidal RNAi Library | Plate 10192: H11 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| RNase inhibitor | Takara Bio | Cat. 2313B |
|
| ||
| Critical commercial assays | ||
|
| ||
| SMART-seq v4 3’ DE Kit | Takara Bio | Cat. 635040 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed RNAseq data | This paper | GEO: GSE209988 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C. elegans: Strain LW4502: rba-1(jj188 [GFP::3xFLAG::RBA-1]) | Gift from Kelly Jun Liu | WB Strain: LW4502 |
| C. elegans: Strain BS553: fog-2(oz40) | CGC | WB Strain: BS553; WormBase: WBStrain00003934 |
| C. elegans: Strain AUM1870: ZK813.1(viz166) | This study | |
|
| ||
| Oligonucleotides | ||
|
| ||
| HCR FISH Probes | Molecular Instruments | See Table S4 |
| CRISPR-related oligos | This Study, Paix et al.57 | See Table S5 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | Schneider et al.58 | https://imagej.nih.gov/ij/ |
| Cutadapt v1.18 | Martin59 | https://cutadapt.readthedocs.io/en/v1.18/ |
| FastQC v0.11.5 | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR aligner v2.4.0 | Dobin et al.60 | https://github.com/alexdobin/STAR |
| featureCounts v1.5.3 | Liao et al.61 | http://subread.sourceforge.net/ |
| DESeq2 | Love et al.26 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Velocyto | La Manno et al.32 | http://velocyto.org/ |
| Imaris | Oxford Instruments | https://imaris.oxinst.com/ |
| Adobe Photoshop | Adobe | https://www.adobe.com/products/photoshop/ |
|
| ||
| Other | ||
|
| ||
| RNAseq analysis and figure code | This paper | https://doi.org/10.5281/zenodo.7847503 |