

Abstract
Background & Aims:
Glycoprotein A repetitions predominant (GARP) is a membrane protein that functions as a latent TGF-β docking molecule. While the immune regulatory properties of GARP on blood cells have been studied, the function of GARP on tissue stromal cells remains unclear. Here, we investigate the role of GARP expressed on hepatic stellate cells (HSCs) in the development of liver fibrosis.
Methods:
The function of GARP on HSCs was explored in toxin-induced and metabolic liver fibrosis models, using conditional GARP-deficient mice or a newly generated inducible system for HSC-specific gene ablation. Primary mouse and human HSCs were isolated to evaluate the contribution of GARP to the activation of latent TGF-β. Moreover, cell contraction of HSCs in the context of TGF-β activation was tested in a GARP-dependent fashion.
Results:
Mice lacking GARP in HSCs were protected from developing liver fibrosis. Therapeutically deleting GARP on HSCs alleviated the fibrotic process in established disease. Furthermore, NKT cells exacerbated hepatic fibrosis by inducing GARP expression on HSCs through IL-4 production. Mechanistically, GARP facilitated fibrogenesis by activating TGF-β and enhancing endothelin-1 (ET-1)-mediated HSC contraction. Functional GARP was expressed on human HSCs and significantly upregulated in the liver of fibrosis patients. Lastly, deletion of GARP on HSCs did not augment inflammation or liver damage.
Conclusions:
GARP expressed on HSCs drives the development of liver fibrosis via cell contraction-mediated activation of latent TGF-β. Considering that systemic blockade of TGF-β has major side effects, we highlight a therapeutic niche provided by GARP and surface-mediated TGF-β activation. Thus, our findings suggest an important role of GARP on HSCs as a promising target for the treatment of liver fibrosis.
Impact and implications:
Liver fibrosis represents a substantial and increasing public health burden globally, and specific treatments are not available. Here, we show that the protein GARP expressed on hepatic stellate cells (HSCs) drives the development of liver fibrosis. Our findings suggest GARP as a novel target for future therapies of fibrotic disease.
Keywords: GARP, hepatic stellate cells (HSCs), liver fibrosis, TGF-β, natural killer T (NKT) cells, endothelin-1 (ET-1)
Graphical abstract
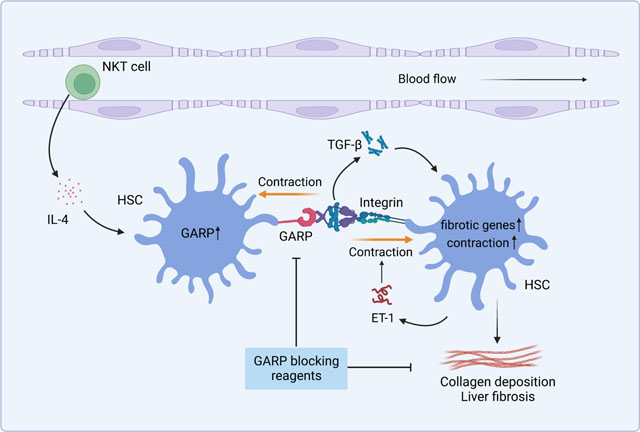
Introduction
Chronic liver disease is often caused by alcohol consumption, unhealthy diet, or persistent hepatic viral infection. Prolonged hepatocyte damage and inflammatory responses frequently lead to liver fibrosis,1,2 a state in which collagen fibers excessively accumulate in the extracellular matrix (ECM) of the liver, causing stiffness of hepatic tissue. Fibrosis is the strongest predictor of mortality,3 and if left untreated can develop into cirrhosis, a late stage and life-threatening syndrome. While cirrhosis represents a substantial and increasing public health burden globally,4 specific treatment is unfortunately not available.
Activation of hepatic stellate cells (HSCs) is a central driver of liver fibrosis.5 In healthy liver, quiescent HSCs serve as pericytes and store vitamin A. Different stimuli, such as pro-fibrotic cytokines, are able to trigger the activation of HSCs.6 Upon stimulation, they lose their vitamin A-containing lipid droplets and transdifferentiate into fibrogenic, contractile myofibroblasts that synthesize and deposit large amounts of collagen into the ECM.7
Among many pro-fibrotic cytokines, TGF-β is the one that most potently induces HSC activation.2,7 TGF-β is synthesized as a latent complex (LTGFβ), in which latency-associated peptide (LAP) encloses a TGF-β dimer. This controls the cytokine’s activity by preventing its binding to TGF-β receptor. Several factors, including some cell surface proteins, are involved in disrupting the LTGFβ complex to release mature TGF-β into the extracellular space.8
GARP (also known as LRRC32) is a transmembrane protein known to tether LTGFβ to the cell surface and is involved in TGF-β activation.9 Expression of GARP on Tregs and platelets has been reported to mediate oral tolerance10 and immune escape of cancer cells.11,12 GARP is expressed on HSCs,13 however, its role in liver fibrosis is not known. Considering its role in activation of TGF-β, we hypothesized that GARP on HSCs might contribute to the development of liver fibrosis.
Here, we found that GARP is upregulated in the liver of fibrosis patients as well as in fibrotic livers of mice. To investigate its role in fibrogenesis, we used a mouse model in which GARP was genetically deleted in HSCs. We found that GARP exacerbated liver fibrosis by activation of TGF-β, a process that was enhanced by HSC contraction. Liver natural killer T (NKT) cells induced GARP expression and augmented fibrogenesis. Moreover, therapeutically deleting GARP in established disease alleviated collagen deposition, which highlights GARP as a novel target for the treatment of liver fibrosis.
Materials and methods
The materials and methods are available under supplementary information and the supplementary CTAT table.
Results
GARP on HSCs exacerbates liver fibrosis
Primary HSCs go through spontaneous activation when cultured in vitro. Flow cytometry analysis showed that HSCs strikingly upregulate GARP expression upon activation (Fig. 1A). Since HSC activation is a hallmark feature of liver fibrosis, we analyzed the amount of GARP in fibrotic mice. C57BL/6 mice were fed with thioacetamide (TAA) for 4 months to induce hepatic fibrosis. Immunoblot showed significantly higher levels of GARP in the liver of TAA-treated animals than in control mice (Fig. 1B). To investigate how GARP affects the development of fibrosis, we crossbred Lrat-Cre mice,14 in which Cre is specifically expressed in HSCs, with GARP-floxed mice.15 After TAA treatment (Fig. 1C), LratCre+/− GARPfl/fl (GARPΔHSC) mice, in which GARP expression on HSCs was selectively deleted (Fig. 1D), were strikingly protected from liver fibrosis compared to GARPfl/fl littermates, as demonstrated by lower fibrotic gene expression in the liver, including αsmooth muscle actin (Acta2), type I and type III collagen (Col1a1 and Col3a1), as well as TIMP metallopeptidase inhibitor 1 (Timp1) (Fig. 1E). Sirius red staining of the liver revealed significantly less collagen deposition in GARPΔHSC mice (Fig. 1F). We also explored the HSC activation state by immunohistochemistry (IHC) staining of alphasmooth muscle actin (α-SMA), a marker of activated HSCs, and found that GARPΔHSC mice exhibited drastically reduced α-SMA abundance in the liver (Fig. 1G). Hydroxyproline is a major component of collagen, and its content was significantly reduced in the liver of GARP-deficient animals (Fig. 1H), again pointing to reduced collagen accumulation when GARP was lacking in HSCs. Similar results were consistently observed in the NASH diet model (Fig. S1A–E) and in the high-fat diet with high fructose and cholesterol (FPC) model (Fig. S2A–E) of liver fibrosis. These results indicate that GARP on HSCs drives fibrogenesis of the liver.
Fig. 1. GARP on HSCs exacerbates liver fibrosis.

(A) GARP expression on the surface of quiescent (1-day cultured) or activated (7-day cultured) HSCs detected by flow cytometry. (B) Immunoblot of GARP in liver lysates of mice fed with TAA for 4 months or control naïve mice. (C) Experimental design of TAA-induced liver fibrosis in GARPΔHSC mice and GARPfl/fl littermates. (D) Flow cytometry analysis of GARP expression on the surface of HSCs. HSCs were isolated from the liver of mice fed with TAA for 4 months and were gated on vitamin A-mediated autofluorescence. (E) Fibrotic gene expression in the liver of mice determined by qRT-PCR. (F) Representative images of Sirius red staining of mouse liver (left) and quantification of stained area (right). Scale bar 100 μm. (G) Representative images of IHC staining of α-SMA in the liver of mice (left) and quantification of stained area (right). Scale bar 100 μm. (H) Concentration of hydroxyproline in the liver of mice. Each data point represents one mouse. Data are presented as mean ± standard error of mean (SEM). Data are from one experiment representing at least three independent experiments with similar results. Unpaired t-test was used to compare two groups. *p<0.05;**p<0.01;***p<0.001;****p<0.0001; NS, not significant.
Therapeutic deletion of GARP in established disease alleviates fibrogenesis
Since GARP on HSCs clearly contributed to hepatic fibrosis, we next asked whether we could target GARP to curb ongoing fibrogenesis in an already diseased liver. Because an inducible model for conditional targeting of HSCs was not available, we aimed to construct an inducible Cre into the allele of an HSC-specific gene to allow for GARP deletion after induction of fibrosis. For this purpose, we screened the genes that were highly expressed in HSCs but not in other liver-resident cells, such as hepatocytes, Kupffer cells, and liver sinusoidal endothelial cells (LSEC). The top hit of the screen by microarray analysis was bone morphogenetic protein 10 (Bmp10) (Fig. 2A), a member of the TGF-β superfamily. Throughout the different tissues of the mouse organism, Bmp10 expression was limited to liver and right atrium (Fig. S3A). In the liver, Bmp10 was exclusively expressed by HSCs (Fig. 2B), making it a suitable marker for specific targeting. To this end, we generated a mouse strain in which expression of inducible Cre (CreERT2) is driven by the endogenous Bmp10 promoter (Fig. S3B,C). To examine the specificity of this mouse model, we crossed Bmp10-CreERT2 mice to Ai6 animals (that express Zsgreen fluorescence following Cre-mediated recombination) and administered tamoxifen to Cre+ offspring. Subsequent liver immunofluorescence (IF) staining showed that all Zsgreen-positive cells were also positive for desmin, an HSC marker (Fig. 2C), indicating that Bmp10-CreERT2 mice specifically express Cre in HSCs upon tamoxifen administration. Some desmin-positive cells were negative for Zsgreen, suggesting a group of Bmp10− HSCs in the liver. Using this inducible system, we crossed Bmp10-CreERT2 mice to GARPfl/fl animals and fed the offspring with NASH diet for 4 weeks to establish liver fibrosis. Subsequently, mice were given tamoxifen to induce GARP deletion in HSCs, prior to continuing the diet for another 4 weeks (Fig. 2D). As a result, GARP expression in the liver of Bmp10CreERT2+/−GARPfl/fl mice was significantly reduced compared to GARPfl/fl littermates (Fig. 2E), indicating successful targeting. In agreement with impaired GARP expression, fibrotic genes were downregulated in Bmp10CreERT2+/−GARPfl/fl mice compared to littermate controls (Fig. 2E). Before tamoxifen treatment, mice showed a similar level of collagen deposition in the liver. However, after 4 weeks of tamoxifen injection, GARPfl/fl mice progressively advanced in fibrosis, while Bmp10CreERT2+/−GARPfl/fl animals showed no increase in collagen accumulation (Fig.2F). Similarly, hydroxyproline content (Fig. S3D) was significantly diminished in the Bmp10CreERT2+/−GARPfl/fl model. IHC staining of α-SMA also indicated less HSC activation upon inducible GARP ablation when compared to GARPfl/fl animals (Fig. 2G). These results demonstrate that therapeutically targeting GARP on HSCs can alleviate liver fibrosis.
Fig. 2. Therapeutic deletion of GARP in established disease alleviates fibrogenesis.

(A) Heat map of the genes with high expression in HSCs (signal intensity >500) and low expression in other liver-resident cells (signal intensity <100). Gene expression was determined by microarray analysis. (B) Bmp10 mRNA expression in liver-resident cells detected by qRT-PCR. (C) IF staining in the liver of Bmp10CreERT2+/−Ai6 mice 12 days after last tamoxifen administration as analyzed by confocal microscopy. Zsgreen (green), desmin (red), nucleus (blue). Scale bar 100 μm. (D) Experimental design of induced GARP deletion during NASH diet-mediated liver fibrosis. (E) GARP and fibrotic gene expression in the liver of mice detected by qRT-PCR. (F) Representative images of Sirius red staining of mouse liver (left) and quantification of stained area (right). Scale bar 100 μm. (G) Representative images of IHC staining of α-SMA in the liver of mice (left) 8 weeks after NASH diet and quantification of stained area (right). Each data point represents a single mouse. Data are presented as mean ± SEM. Data in E-G are from one experiment representing at least two independent experiments with similar results. Unpaired t-test was used to compare two groups. *p<0.05;**p<0.01. NS, not significant.
Natural killer T (NKT) cells facilitate fibrotic scarring by inducing GARP expression on HSCs through IL-4 production
Next, we explored the mechanism of GARP regulation on HSCs. We hypothesized that NKT cells, one of the most abundant lymphocytes in the liver with a key role in hepatic fibrogenesis,16 may be involved in the regulation of GARP. To investigate this, we fed wild-type (WT) or Traj18-knockout mice, which lack invariant NKT (iNKT) cells, with NASH diet (Fig. 3A). After 8 weeks, GARP and fibrotic gene expression in the liver were downregulated in iNKT-deficient compared to WT animals (Fig. 3B). Collagen deposition in mice lacking iNKT cells was also reduced (Fig. 3C and Fig. S4A), corroborating the contribution of iNKT cells to the development of liver fibrosis. Since GARP expression in iNKT-deficient mice was reduced, we sought to test the direct impact of iNKT cells on GARP regulation by HSCs. For this purpose, we first isolated iNKT cells from the liver of naïve mice and stimulated them in vitro by TCR cross-linking. Subsequently, incubation with iNKT cell-conditioned medium significantly induced GARP expression on HSCs, an effect largely inhibited in the presence of anti-IL-4 but not isotype control antibody (Fig. 3D). This result implies that IL-4 produced by iNKT cells directly triggered HSCs to express GARP. Indeed, activated HSCs are equipped with the necessary machinery to sense this cytokine as they expressed IL-4 receptor (Fig. S4B), and recombinant IL-4 strongly induced GARP upregulation (Fig. 3E). Moreover, induction of GARP on HSCs was prevented by AS1517499 (Fig. 3E), a specific inhibitor of the IL-4 downstream signaling molecule STAT6. To translate these findings in vivo and to assess whether iNKT cells produce IL-4 to promote GARP-mediated liver fibrosis, we injected OCH, an αgalactosylceramide (α-GalCer) analog that strongly biases iNKT cells to preferentially produce IL-4,17,18 into NASH diet-fed GARPΔHSC mice or GARPfl/fl littermates (Fig. 3F). OCH injection strongly induced IL-4 expression in murine liver, and GARP production was significantly amplified in OCH-treated mice compared to DMSO controls (Fig. S4C). Consequently, GARPfl/fl mice that received OCH showed augmented liver fibrosis as reflected by increased fibrotic gene expression (Fig. S4C), collagen deposition (Fig. 3G), and α-SMA staining (Fig. 3H). Interestingly, despite elevated IL-4 abundance in the liver, GARPΔHSC mice showed no obvious change in magnitude of fibrosis (Fig. 3G, H, and S4C). Taken together, these data suggest that iNKT cells produce IL-4 to promote liver fibrosis by inducing GARP expression on HSCs.
Fig. 3. NKT cells promote fibrotic scarring by inducing GARP expression on HSCs through IL-4 production.

(A) Experimental design of NASH diet-induced liver fibrosis in wild-type (WT) or Traj18-knockout (iNKT-deficient) mice. (B) GARP and fibrotic gene expression in the liver of mice detected by qRT-PCR. (C) Representative images of Sirius red staining of mouse liver (left) and quantification of stained area (right). Scale bar 100 μm. (D) GARP mRNA expression in HSCs treated with complete RPMI (cRPMI) or iNKT-conditioned medium in the presence or absence of anti-IL-4 or isotype control antibody. (E) Representative histogram plot (left) and mean fluorescent intensity (MFI) (right) of GARP staining on the surface of HSCs after incubation with PBS, recombinant IL-4, or IL-4 plus STAT6 inhibitor AS1517499. (F) Scheme of OCH injection during NASH diet-induced liver fibrosis. (G) Representative images of Sirius red staining of mouse liver (left) and quantification of stained area (right). Scale bar 100 μm. (H) Representative images of IHC staining of α-SMA in the liver of mice (left) and quantification of stained area (right). Scale bar 100 μm. Each data point represents a single mouse in A-C and F-H or one well in D-E. Data are presented as mean ± SEM and are from one experiment representative of at least two independent experiments with similar results. Unpaired t-test in B-C, G-H and one-way ANOVA in D-E were used to compare two groups. *p<0.05;**p<0.01;***p<0.001;****p<0.0001; NS, not significant.
GARP on HSCs drives fibrogenesis by activating TGF-β
TGF-β is a cytokine that strongly activates HSCs and is well known for its ability to promote liver fibrosis.2 TGF-β is produced in its latent form in which latency-associated peptide (LAP) encloses a TGF-β dimer to prevent it from binding to its receptor.8 GARP is known to bind and activate LTGFβ in several blood cell populations.9,11 We observed that GARP is co-expressed with LTGFβ on the cell surface of activated HSCs (Fig. 4A). However, it was not clear whether HSCs utilize GARP for TGF-β activation. To address this question, we used a reporter system where HSCs isolated from mouse liver were cocultured with active TGF-β reporter cells to detect TGF-β release (Fig. 4B). In contrast to GARPfl/fl, HSCs isolated from GARP-deficient mice failed to activate TGF-β (Fig. 4C). The respective isoform released from HSCs was mainly TGF-β1, as demonstrated by blocking of luciferase activity using an anti-TGF-β1 antibody (Fig. 4D). Thus, HSCs activate TGF-β1 using GARP. To exclude the possibility that HSCs from GARPΔHSC mice exhibit off-target effects regarding TGF-β activation, we knocked down GARP in WT HSCs and subjected them to our TGF-β reporter assay. Accordingly, knockdown confirmed the requirement of GARP for HSCs to release mature TGF-β (Fig. 4E). In line with previous reports,19 an integrin αV inhibitor abolished TGF-β activation by HSCs (Fig. 4E). Therefore, GARP and integrin αV are both indispensable for HSC-mediated TGF-β release. In order to investigate TGF-β activation during the development of liver fibrosis, we analyzed the level of phosphorylated Smad2 (p-Smad2), an important downstream signaling molecule of TGF-β receptor, in the liver of fibrotic mice. Immunoblot of whole liver lysate and IHC staining of liver sections revealed increased amounts of p-Smad2 in fibrotic GARPfl/fl mice compared to naïve animals (Fig. 4F, G). In sharp contrast, fibrosisresistant GARP-deficient mice exhibited similar levels of p-Smad2 as untreated control animals (Fig. 4F, G). Altogether, these results indicate that GARP on HSCs facilitates liver fibrosis by activating latent TGF-β.
Fig. 4. GARP on HSCs facilitates fibrogenesis by activating TGF-β.

(A) Flow cytometry analysis of GARP and LAP expression on the surface of HSCs isolated from GARPfl/fl or GARPΔHSC mice and culture-activated for 7 days. (B) Schematic of TGF-β reporter assay using isolated HSCs and active TGF-β reporter cells. (C) TGF-β activation by HSCs from GARPfl/fl or GARPΔHSC mice. (D) TGF-β activation by WT HSCs in the presence of anti-TGF-β1 or isotype antibody. (E) TGF-β activation by WT HSCs treated with scramble siRNA, or GARP siRNA, or incubated with PBS, or integrin inhibitor CWHM12. (F) Immunoblot of p-Smad2 or total Smad2 in the liver lysates of mice fed with TAA for 4 months or control naïve mice. (G) Representative images of IHC staining for p-Smad2 in the liver of mice fed with NASH diet for 8 weeks, or TAA for 4 months, or control naïve mice (left), and quantification of stained area (right). Scale bar 50 μm. Each data point represents one well in C-E or a single mouse in G. Data are presented as mean ± SEM. One-way ANOVA in C-D and unpaired t-test in E-G were used to compare two groups. ***p<0.001;****p<0.0001; NS, not significant.
GARP fosters a vicious cycle involving ET-1 and TGF-β
Mechanical pulling force has been proposed to be involved in TGF-β release from its latent complex.20,21 Activated HSCs transdifferentiate into highly contractile myofibroblasts. As cell contraction inevitably generates pulling force, we then asked whether contraction of HSCs contributes to TGF-β activation. To explore this, we first evaluated the contraction of HSCs using a collagen gel assay (Fig. 5A), in which cells are seeded into a collagen matrix to measure the subsequent contraction-mediated gel shrinkage. In this context, HSCs showed clear contractile properties, which were enhanced by the potent vasoconstrictor endothelin-1 (ET-1), and abolished by blebbistatin, a myosin inhibitor (Fig. 5B). Correspondingly, our TGF-β reporter assay demonstrated that ET-1 strikingly increased TGF-β activation by HSCs, an increase specifically abolished by bosentan, an endothelin receptor antagonist (Fig. 5C). Notably, while blebbistatin abolished cell contraction, it also diminished active TGF-β release from HSCs (Fig. 5C). These data indicate that cell contraction is needed by HSCs to activate TGF-β.
Fig. 5. GARP fosters a vicious cycle involving endothelin-1 (ET-1) and TGF-β.

(A) Schematic of collagen gel assay to measure contraction of HSCs. (B) Representative images of collagen gel mixed with WT HSCs in the presence of PBS, ET-1, or blebbistatin, at time point 24 and 48 hours after gel release from the well side (left), and statistical analysis of change in gel area (right). (C) TGF-β activation by WT HSCs in the presence of PBS, ET-1, ET-1 plus bosentan, or blebbistatin. (D) ELISA of ET-1 concentration in the supernatant of GARPΔHSC or GARPfl/fl HSCs cultured in the presence of recombinant TGF-β1 (rTGF-β1) or anti-TGF-β1 antibody. (E) ET-1 mRNA level in the liver of mice fed with NASH diet for 8 weeks determined by qRT-PCR. (F) Representative images of collagen gel mixed with HSCs from GARPΔHSC or GARPfl/fl mice in the presence of PBS, or bosentan at 24 and 48 hours after gel release from the well side (left), and statistical analysis of change in gel area (right). (G) A graph illustrating GARP-mediated TGF-β activation, ET-1 production, and contraction by HSCs. Each data point represents one well in B-D and F or one mouse in E. Data are presented as mean ± SEM. One-way ANOVA in B-D, F and unpaired t-test in E were used to compare two groups. **p<0.01;***p<0.001;****p<0.0001; NS, not significant.
In fibrotic liver, HSCs are a main source of ET-1.22,23 Therefore, we assessed ET-1 production by HSCs isolated from GARPfl/fl or GARPΔHSC mice. GARPfl/fl HSCs produced significantly higher amounts of ET-1 than GARP-deficient cells (Fig. 5D). Recombinant TGF-β (rTGF-β) further increased ET-1 production in HSCs, while TGF-β blocking had the opposite effect (Fig. 5D). Consistent with this observation, ET-1 expression was significantly higher in the liver of GARPfl/fl control mice than in GARP-deficient animals fed with NASH diet (Fig. 5E). These results suggest that ET-1 enhances contraction-mediated TGF-β activation, while released TGF-β in turn increases ET-1 production by HSCs.
To determine the consequences of dampened ET-1 production by GARP-deficient HSCs, we tested their ability to shrink collagen gel. Strikingly, GARP-deficient HSCs were much less contractile compared to GARPfl/fl, and bosentan clearly inhibited the contraction of GARPfl/fl HSCs (Fig. 5F). This illustrates the important role of autocrine ET-1 in the regulation of HSC contraction. In conclusion, these observations indicate that GARP facilitates contraction-mediated TGF-β release from HSCs. Released TGF-β works in an autocrine manner, inducing ET-1 production to amplify HSC contraction (Fig. 5G). Supporting this notion, blocking integrin αv, which contributes to TGF-β release from HSCs, impaired cell contraction (Fig. S5). Thus, we describe a vicious cycle formed by the crosstalk between ET-1 and TGF-β to drive liver fibrosis. Of note, targeting GARP provides an opportunity to break this loop.
Expression of GARP is upregulated in the liver of fibrosis patients
Next, we performed experiments in the human system, culture-activated human primary HSCs, and found expression of GARP on the cell surface as analyzed by flow cytometry (Fig. 6A). To explore the involvement of human GARP in the activation of TGF-β, we knocked down GARP in human HSCs (Fig. 6B), prior to subjecting them to our TGF-β reporter assay. Human HSCs were able to activate TGF-β, and similar to our experiments in the murine model, GARP proved to be indispensable for this activity (Fig. 6C). We then retrieved and analyzed transcriptional data from public databases (Gene Expression Omnibus accession code: GSE83452,24 GSE84044,25 and GSE1432326) to evaluate GARP expression in liver biopsies of three large cohorts of patients with chronic liver disease. GARP expression in the liver of NASH patients was significantly higher than in non-NASH individuals. Furthermore, the expression of GARP was even more strikingly increased in the liver of HBV/HCV-infected fibrosis/cirrhosis patients than in non-fibrosis patients or healthy donors (Fig. 6D). These results strongly indicate that GARP is expressed on human HSCs and contributes to the development of liver fibrosis of different etiology.
Fig. 6. Functional GARP is present on human HSCs and upregulated in the liver of fibrosis patients.

(A) Flow cytometry analysis of GARP expression on the surface of human primary HSCs after 7 days culture. (B) FACS analysis of GARP expression on the surface of human HSCs treated with or without scramble siRNA or GARP siRNA. (C) TGF-β activation by human HSCs treated with or without scramble siRNA or GARP siRNA. Data are presented as mean ± SEM. (D) Level of GARP mRNA expression in liver biopsies of patients with NASH (left), HBV-induced advanced fibrosis (middle), or HCV-induced cirrhosis (right) compared to corresponding non-fibrosis or healthy individuals. Solid black line in the violin plots represents median. The upper and lower dotted black lines represent 75th and 25th percentile. Each data point indicates one well in C or one patient in D. One-way ANOVA in C and unpaired t-test in D were used to compare two groups. ***p<0.001; ****p<0.001;NS, not significant.
Targeting GARP on HSCs does not augment liver damage or inflammation
Systemically neutralizing TGF-β or its receptor may induce uncontrollable inflammation.27,28 However, deletion of GARP on HSCs did not lead to an increase of inflammatory cell infiltration into the liver in both NASH diet and TAA model (Fig. 7A, B), nor did it augment the expression of pro-inflammatory genes (Fig. 7C). As a consequence, deletion of GARP did not enhance liver damage as demonstrated by serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels (Fig. 7D). Natural killer (NK) cells are known to kill activated HSCs and are important for the control of liver fibrosis.29.30 The frequency, activation phenotype, and cytokine production of liver NK cells as well as their cytotoxicity toward activated HSCs were well preserved in GARPΔHSC mice compared to GARPfl/fl littermates (Fig. 7E, F). Kupffer cells and monocyte-derived macrophages (MDMs) are important TGF-β producers in the liver and promote activation of HSCs by TGF-β and other cytokines.31 We found that GARP is not expressed on Kupffer cells or MDMs (Fig. S6A), thus not mediating TGF-β activation in these cell types. Flow cytometry and IHC analysis showed that targeting GARP on HSCs did not change the frequency of Kupffer cells or MDMs in the liver (Fig. S6B–C), nor did it affect the TGFβ production in macrophages (Fig. S6D). Moreover, conditioned medium from liver macrophages did not significantly modulate GARP expression in HSCs (Fig. S6E). These results show that, although curbing fibrogenesis, targeting GARP on HSCs does not augment liver damage or inflammation, nor does it affect the homeostasis of liver macrophages or the clearance of activated HSCs by NK cells.
Fig. 7. Targeting GARP on HSCs does not augment liver damage or inflammation.

(A) Representative FACS plots of granulocytes in the liver of mice (left) and quantification of its frequency (right). (B) Representative images of hematoxylin and eosin staining on mouse liver sections after 4 months of TAA treatment (left) and quantification of inflammation score (right). Black arrow indicates foci of infiltrating inflammatory cells. Scale bar 50 μm. (C) Expression of pro-inflammatory genes in the liver of mice quantified by qRT-PCR. (D) ALT and AST activity in serum of mice after 8 weeks of NASH or normal diet. (E) Frequency of NK cells and level of activating receptor NKG2D on NK cells in the liver of mice after 8 weeks of NASH diet. (F) Frequency of IFN-γ producing cells among total NK cells isolated from the liver of mice, and cytotoxicity of isolated liver NK cells against HSCs. Each data point represents a single mouse. Data are presented as mean ± SEM. Unpaired t-test was used to compare two groups. *p<0.05; NS, not significant.
Discussion
Previous research on GARP mainly focused on its expression on blood cells32–35 and its role in anti-tumor immune responses.36–38 GARP expression in stromal cells, especially its role in tissue pathology is, however, not well characterized. Here, using three different disease models, we show that GARP expressed on HSCs is a critical driver of liver fibrosis. By activating the pro-fibrotic cytokine TGF-β, GARP facilitates HSC activation and TGF-β-mediated fibrogenesis. We demonstrate that GARP and αV integrin are both indispensable for HSCs to mediate mature TGF-β release. Importantly, expression of GARP is enhanced on activated HSCs and in fibrotic liver, making it an appropriate disease biomarker and a promising therapeutic target. Indeed, by generating Bmp10-CreERT2 mice, a novel mouse tool with inducible Cre expression in HSCs, we show that deleting GARP after establishment of disease alleviates liver fibrosis, despite an ongoing fibrotic process due to continued NASH diet. Blocking of TGF-β can be beneficial for the treatment of liver fibrosis and cancer, yet systemically neutralizing TGF-β or blockade of its receptor induced unacceptable side effects due to its ubiquitous distribution and pleiotropic biological functions.27,28 By contrast, GARP expression is confined to a limited group of cells, thus, we propose that targeting GARP to block TGF-β activation could avoid systemic adverse effects. As a proof of principle, our results show that, while curbing fibrogenesis, ablation of GARP on HSCs does not exacerbate liver damage or inflammation, nor does it affect NK cell-mediated clearance of activated HSCs. This highlights the targeting of GARP as a safe and feasible approach for treating liver fibrosis. The role of NKT cells in liver fibrosis has been gradually acknowledged.16 Increased numbers of activated NKT cells were found in patients with NASH.39,40 Murine studies showed that the CXCR6-CXCL16 axis mediates accumulation of NKT cells during the development of hepatic fibrosis,41 and NKT cells exacerbated fibrosis of the liver in different mouse models.39,40,42 However, the molecular mechanism of how NKT cells contribute to fibrogenesis is not well understood. In this study, we show that iNKT cells promote liver fibrosis by inducing GARP expression on activated HSCs, and IL-4 produced by iNKT cells mainly mediates the induction of GARP. iNKT cell-derived IL-4 likely represents an important mechanism to mediate wound healing in the liver, as it has been implicated to induce wound repair in sterile liver injury.43 However, in the case of chronic liver disease, persistent damage to the liver could trigger and amplify this healing mechanism for too long, eventually shifting the balance from a beneficial effect to a devastating process.
Our results show that GARP and integrins are both required for HSCs to activate LTGFβ. However, previous studies showed that binding of GARP and integrins to the LTGFβ complex was not sufficient to release active TGF-β.44 Based on the structure of the GARP-LTGFβ complex,45 it is postulated that mechanical tension force is required to open the LTGFβ prodomain. In agreement with this notion, here we show that contraction is necessary for HSCs to activate TGF-β. In this context, activated HSCs express abundant α-SMA, thus are well equipped to impose tension force to the LTGFβ complex through GARP, integrins, and the cytoskeletal network. In addition to the upregulation of α-SMA, activated HSCs are also the major source of ET-1 in fibrotic liver.22,23 ET-1 is the most potent vasoconstrictor that induces HSC contraction and activation.46,47 It has been reported to contribute to fibrogenesis and portal hypertension in chronic liver disease.47–50 Our results demonstrate that GARP promotes ET-1 production in activated HSCs through TGF-β release. Putting together these findings, we propose that GARP fosters a vicious cycle involving TGF-β activation, ET-1 production, and cell contraction in HSCs that drives the development of liver fibrosis.
Supplementary Material
Highlights:
GARP on HSCs contributes to the development of liver fibrosis through TGF-β activation.
NKT cells induce GARP expression on HSCs by producing IL-4.
GARP-mediated cell contraction forms a vicious cycle driving fibrogenesis.
Patients with liver fibrosis exhibit a high level of GARP expression.
Targeting GARP on HSCs provides a safe approach to control liver fibrosis.
Financial support:
This study was supported by NIH grants R01AI136939 and R01AI083426 to F.W.
Abbreviations
- GARP
Glycoprotein A repetitions predominant
- TGF-β
Transforming growth factor beta
- HSC
hepatic stellate cell
- NKT cell
Natural killer T cell
- ET-1
endothelin-1
- ECM
extracellular matrix
- LAP
latency-associated peptide
- Tregs
Regulatory T cells
- TAA
thioacetamide
- Lrat
Lecithin retinol acyltransferase
- IHC
immunohistochemistry
- NASH
Nonalcoholic Steatohepatitis
- FPC
fructose-palmitate-cholesterol
- LSEC
liver sinusoidal endothelial cells
- Bmp10
bone morphogenetic protein 10
- IF
immunofluorescence
- TCR
T cell receptor
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- MDM
monocyte-derived macrophage
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability statement
All data are available in the manuscript, supplementary materials, and supplementary CTAT Table. Microarray data have been deposited to Gene Expression Omnibus under accession number GSE226103.
References
Author names in bold designate shared co-first authorship
- [1].Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest 2017;127:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schwabe RF, Tabas I, Pajvani UB. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020;158:1913–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015;61:1547–1554. [DOI] [PubMed] [Google Scholar]
- [4].Sepanlou SG, Safiri S, Bisignano C, Ikuta KS, Merat S, Saberifiroozi M, et al. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. The Lancet Gastroenterology & Hepatology 2020;5:245–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008;88:125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017;14:397–411. [DOI] [PubMed] [Google Scholar]
- [7].Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol 2021;18:151–166. [DOI] [PubMed] [Google Scholar]
- [8].Robertson IB, Rifkin DB. Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb Perspect Biol 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stockis J, Dedobbeleer O, Lucas S. Role of GARP in the activation of latent TGF-beta1. Mol Biosyst 2017;13:1925–1935. [DOI] [PubMed] [Google Scholar]
- [10].Edwards JP, Hand TW, Morais da Fonseca D, Glass DD, Belkaid Y, Shevach EM. The GARP/Latent TGF-beta1 complex on Treg cells modulates the induction of peripherally derived Treg cells during oral tolerance. Eur J Immunol 2016;46:1480–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Metelli A, Salem M, Wallace CH, Wu BX, Li A, Li X, et al. Immunoregulatory functions and the therapeutic implications of GARP-TGF-beta in inflammation and cancer. J Hematol Oncol 2018;11:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lecomte S, Devreux J, de Streel G, van Baren N, Havelange V, Schroder D, et al. Therapeutic activity of GARP:TGF-beta1 blockade in murine primary myelofibrosis. Blood 2022. [DOI] [PubMed] [Google Scholar]
- [13].Li Y, Kim BG, Qian S, Letterio JJ, Fung JJ, Lu L, et al. Hepatic Stellate Cells Inhibit T Cells through Active TGF-beta1 from a Cell Surface-Bound Latent TGF-beta1/GARP Complex. J Immunol 2015;195:2648–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Edwards JP, Fujii H, Zhou AX, Creemers J, Unutmaz D, Shevach EM. Regulation of the expression of GARP/latent TGF-beta1 complexes on mouse T cells and their role in regulatory T cell and Th17 differentiation. J Immunol 2013;190:5506–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Crosby CM, Kronenberg M. Tissue-specific functions of invariant natural killer T cells. Nat Rev Immunol 2018;18:559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Miyamoto K, Miyake S, Yamamura T. A synthetic glycolipid prevents autoimmune encephalomyelitis by inducing TH2 bias of natural killer T cells. Nature 2001;413:531–534. [DOI] [PubMed] [Google Scholar]
- [18].Oki S, Chiba A, Yamamura T, Miyake S. The clinical implication and molecular mechanism of preferential IL-4 production by modified glycolipid-stimulated NKT cells. J Clin Invest 2004;113:1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 2013;19:1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF-beta structure and activation. Nature 2011;474:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Buscemi L, Ramonet D, Klingberg F, Formey A, Smith-Clerc J, Meister JJ, et al. The single-molecule mechanics of the latent TGF-beta1 complex. Curr Biol 2011;21:2046–2054. [DOI] [PubMed] [Google Scholar]
- [22].Pinzani M, Milani S, De Franco R, Grappone C, Caligiuri A, Gentilini A, et al. Endothelin 1 is overexpressed in human cirrhotic liver and exerts multiple effects on activated hepatic stellate cells. Gastroenterology 1996;110:534–548. [DOI] [PubMed] [Google Scholar]
- [23].Shao R, Yan W, Rockey DC. Regulation of endothelin-1 synthesis by endothelin-converting enzyme-1 during wound healing. J Biol Chem 1999;274:3228–3234. [DOI] [PubMed] [Google Scholar]
- [24].Lefebvre P, Lalloyer F, Bauge E, Pawlak M, Gheeraert C, Dehondt H, et al. Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARalpha-regulated dermatopontin. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang M, Gong Q, Zhang J, Chen L, Zhang Z, Lu L, et al. Characterization of gene expression profiles in HBV-related liver fibrosis patients and identification of ITGBL1 as a key regulator of fibrogenesis. Sci Rep 2017;7:43446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mas VR, Maluf DG, Archer KJ, Yanek K, Kong X, Kulik L, et al. Genes involved in viral carcinogenesis and tumor initiation in hepatitis C virus-induced hepatocellular carcinoma. Mol Med 2009;15:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tolcher AW, Berlin JD, Cosaert J, Kauh J, Chan E, Piha-Paul SA, et al. A phase 1 study of anti-TGFbeta receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother Pharmacol 2017;79:673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Akhurst RJ. Targeting TGF-beta Signaling for Therapeutic Gain. Cold Spring Harb Perspect Biol 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Melhem A, Muhanna N, Bishara A, Alvarez CE, Ilan Y, Bishara T, et al. Antifibrotic activity of NK cells in experimental liver injury through killing of activated HSC. J Hepatol 2006;45:60–71. [DOI] [PubMed] [Google Scholar]
- [30].Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006;130:435–452. [DOI] [PubMed] [Google Scholar]
- [31].Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol 2021;18:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLoS One 2008;3:e2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 2009;106:13439–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 2009;106:13445–13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wallace CH, Wu BX, Salem M, Ansa-Addo EA, Metelli A, Sun S, et al. B lymphocytes confer immune tolerance via cell surface GARP-TGF-beta complex. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cuende J, Lienart S, Dedobbeleer O, van der Woning B, De Boeck G, Stockis J, et al. Monoclonal antibodies against GARP/TGF-beta1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci Transl Med 2015;7:284ra256. [DOI] [PubMed] [Google Scholar]
- [37].Salem M, Wallace C, Velegraki M, Li A, Ansa-Addo E, Metelli A, et al. GARP Dampens Cancer Immunity by Sustaining Function and Accumulation of Regulatory T Cells in the Colon. Cancer Res 2019;79:1178–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rachidi S, Metelli A, Riesenberg B, Wu BX, Nelson MH, Wallace C, et al. Platelets subvert T cell immunity against cancer via GARP-TGFbeta axis. Sci Immunol 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010;51:1998–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Maricic I, Marrero I, Eguchi A, Nakamura R, Johnson CD, Dasgupta S, et al. Differential Activation of Hepatic Invariant NKT Cell Subsets Plays a Key Role in Progression of Nonalcoholic Steatohepatitis. J Immunol 2018;201:3017–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wehr A, Baeck C, Heymann F, Niemietz PM, Hammerich L, Martin C, et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J Immunol 2013;190:5226–5236. [DOI] [PubMed] [Google Scholar]
- [42].Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, et al. NKT associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 2012;61:1323–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liew PX, Lee WY, Kubes P. iNKT Cells Orchestrate a Switch from Inflammation to Resolution of Sterile Liver Injury. Immunity 2017;47:752–765 e755. [DOI] [PubMed] [Google Scholar]
- [44].Wang R, Zhu J, Dong X, Shi M, Lu C, Springer TA. GARP regulates the bioavailability and activation of TGFbeta. Mol Biol Cell 2012;23:1129–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lienart S, Merceron R, Vanderaa C, Lambert F, Colau D, Stockis J, et al. Structural basis of latent TGF-beta1 presentation and activation by GARP on human regulatory T cells. Science 2018;362:952–956. [DOI] [PubMed] [Google Scholar]
- [46].Rockey DC. Characterization of endothelin receptors mediating rat hepatic stellate cell contraction. Biochem Biophys Res Commun 1995;207:725–731. [DOI] [PubMed] [Google Scholar]
- [47].Rockey DC, Chung JJ. Endothelin antagonism in experimental hepatic fibrosis. Implications for endothelin in the pathogenesis of wound healing. J Clin Invest 1996;98:1381–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sogni P, Moreau R, Gomola A, Gadano A, Cailmail S, Calmus Y, et al. Beneficial hemodynamic effects of bosentan, a mixed ET(A) and ET(B) receptor antagonist, in portal hypertensive rats. Hepatology 1998;28:655–659. [DOI] [PubMed] [Google Scholar]
- [49].Kojima H, Yamao J, Tsujimoto T, Uemura M, Takaya A, Fukui H. Mixed endothelin receptor antagonist, SB209670, decreases portal pressure in biliary cirrhotic rats in vivo by reducing portal venous system resistance. J Hepatol 2000;32:43–50. [DOI] [PubMed] [Google Scholar]
- [50].Rockey DC. Endothelial dysfunction in advanced liver disease. Am J Med Sci 2015;349:6–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the manuscript, supplementary materials, and supplementary CTAT Table. Microarray data have been deposited to Gene Expression Omnibus under accession number GSE226103.