

Abstract
Background.
Targeting short-term improvements in multicomponent risk scores for mortality in patients with pulmonary arterial hypertension (PAH) may result in improved long-term outcomes. We aimed to determine whether PAH risk scores were adequate surrogates for clinical worsening in PAH randomized clinical trials.
Methods.
We performed an individual participant data meta-analysis using three large long-term RCTs in PAH (AMBITION, GRIPHON, and SERAPHIN). We calculated predicted risk using the COMPERA Full, COMPERA 2.0, non-invasive FPHR, REVEAL 2.0, and REVEAL Lite 2 risk scores. We assessed the surrogacy of achievement of low-risk status by 16 weeks for improvement in long-term clinical worsening and survival using both mediation and meta-analysis frameworks.
Findings.
The study sample included 2508 participants. The mean age was 49 ±16 years, 78% were women, 68% were classified as white, and 11% were Hispanic/Latino. Fifty-five percent had idiopathic PAH and 31% had PAH associated with connective tissue disease. In a mediation analysis, the proportions of treatment effects explained by achievement of low-risk status ranged only from 7% to 13%. In a meta-analysis of trial-regions, the treatment effects on low-risk status were not predictive of the treatment effects on clinical worsening. A leave-one-out analysis suggested that the use of these risk scores as surrogates may lead to biased inferences regarding the effect of therapies on clinical outcomes in PAH RCTs. Results were similar when using absolute risk scores at 16 weeks as the potential surrogates.
Interpretation.
Multicomponent risk scores may have utility for the prediction of long-term outcomes in patients with PAH. Clinical surrogacy for long-term outcomes, however, is not guaranteed by results from observational studies that suggest changes in multicomponent risk scores correlate with better outcomes. Our analyses of three PAH trials with long-term follow-up suggest that further study is necessary before using these or other scores as surrogate outcomes in PAH RCTs.
Funding.
Cardiovascular Medical Research and Education Fund
Keywords: Pulmonary hypertension, surrogate outcome, risk score, pulmonary vascular disease
Introduction
Functional and hemodynamic measures have long been used to inform the diagnosis and care of patients with pulmonary arterial hypertension (PAH). More recently, there have been increasingly strong recommendations to use multicomponent risk scores that combine these and other clinical and laboratory values to guide the treatment of PAH. Popular examples include REVEAL,1 FPHR,2 and COMPERA,3,4 though several others exist or are in development.5 These scores transform several measures of disease severity, organ dysfunction, and hemodynamics into an ordinal ranking that is usually further stratified into three to four levels of mortality risk at one year.
The 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension5 advocate for “a risk-based, goal-orientated treatment approach in patients with PAH, where achieving and/or maintaining a low-risk status is favorable and recommended.” (Class I Recommendation, Level of Evidence B). This approach assumes that new (or established) therapies that improve patients to a “low-risk” score in the short term will also lead to benefits in the long term, i.e., that the risk score is a surrogate end point. However, the formal validation of these scores as surrogate outcomes remains incomplete and it is possible that the current approach to ranking and calculating these scores is suboptimal for using them as clinical surrogates to inform regulatory decisions pertaining to treatment efficacy or clinical care.
The Food and Drug Administration (FDA) defines a surrogate end point as “a laboratory measurement or physical sign that is used in therapeutic trials as a substitute for a clinically meaningful end point… and is expected to predict the effect of the therapy.”6,7 A surrogate endpoint is an alternative outcome measure that can substitute for a clinically important outcome that would happen later in time or be more difficult to measure.6,7 Such endpoints would have high utility for PAH, which is a rare disease. The low incidence of PAH necessitates long time-to-event focused trials, preventing rapid evaluation of potential therapies.
A validated surrogate endpoint can provide results faster and often more affordably than long-term or difficult-to-capture outcomes in trials.8–10 For example, if the multicomponent risk score at 4 months captured the effects of an intervention and predicted outcomes over several years, therapies could be tested in shorter and less expensive RCTs by using the risk score as a surrogate outcome.11 Prior efforts have shown that six-minute walk distance (6MWD) and hemodynamics are not adequate surrogates for outcomes in PAH,12,13 however these studies have been criticized for only including short-term follow-up and assessing single parameters at a time (6MWD, pulmonary vascular resistance, etc.). We sought to assess the surrogacy of five validated multivariable risk scores using long-term data from PAH trials.
Methods
Study population and study sample
We considered randomized clinical trials (RCTs) of any PAH therapy that were submitted to the United States (US) FDA since 2000 (Table S1) for inclusion in this study. The FDA provided us with individual participant data (IPD) from these RCTs with the goal of improving study design and quality. For this study, we included Phase III long-term event-driven trials which included data for the risk prediction scores captured at 12 or 16 weeks and follow-up of 1–4 years (Figure S1). Most trials were excluded for having a follow-up duration of less than 1 year. Three trials were included in this study. The AMBITION trial14 randomized patients to an ambrisentan 10 mg and tadalafil 40 mg combination therapy arm or one of two monotherapy arms (ambrisentan 10 mg alone and tadalafil 40 mg alone). We considered the combination therapy arm as the ‘experimental treatment arm’ for this study; monotherapy patients were combined into one ‘control arm.’ The GRIPHON trial15 randomized patients to selexipag (up to 3200 μg daily) or placebo. The SERAPHIN trial16 randomized patients to macitentan 3 mg, macitentan 10 mg, or placebo. We combined the macitentan 3 mg and macitentan 10 mg arms into one ‘experimental treatment arm’ for this study.
We harmonized the IPD obtained from the FDA as previously described.17,18 Briefly, we used the Study Data Tabulation Model (Version 1.4) to organize the data into domain datasets. Demographic, PAH etiology, World Health Organization (WHO) functional class (FC), 6MWD, laboratory, vital sign, hemodynamic values from right heart catheterization, clinical worsening, and mortality data were harmonized across the various trials. The time points at which data were captured in the individual trials were recorded. The University of Pennsylvania Institutional Review Board considered the harmonization and secondary use of these data as exempt from approval.
Description of PAH risk scores
We assessed five commonly used PAH risk scores for potential surrogacy: REVEAL 2.0,19 REVEAL Lite 2,20 COMPERA,3 COMPERA 2.0,21 and non-invasive FPHR.2 A summary of the score formulations are given in Table S2, with a full description of each score provided in the Web Supplement. The standard FPHR score (which requires right atrial pressure and cardiac index) could not be used because the included studies did not perform right heart catheterization at 12 or 16 weeks. The non-invasive FPHR score will be henceforth referred to as FPHR for brevity.
We calculated risk scores when some risk score components were missing using the standard procedures described by each respective risk score calculator; this is described in more detail in Table S3. In addition, the AMBITION and GRIPHON trials collected data for risk score calculation at 16 weeks, while SERAPHIN collected such data at 12 and 24 weeks. To harmonize the surrogate variable data, risk scores at 16 weeks in SERAPHIN were imputed using linear interpolation of the 12- and 24-week data. The 12-week data contributed two-thirds of the weighted average, and the 24-week data contributed one-third.
Description of clinical outcomes
The primary outcome of interest was time to “clinical worsening”, a composite endpoint composed of any of the following events: all-cause death, hospitalization for worsening PAH, lung transplantation, atrial septostomy, discontinuation of study treatment (or study withdrawal) for worsening PAH, initiation of parenteral prostacyclin analogue therapy, or decrease of at least 15% in 6MWD from baseline, combined with either worsening of WHO FC from baseline or the addition of an approved PAH treatment.14–16 The secondary outcome of interest was time to all-cause mortality.
Statistical Analysis
Characteristics of the study sample were shown using mean ± standard deviation (SD) or median and interquartile range (IQR). The proportion of study subjects with low-risk status for each risk score at 16 weeks was calculated for each treatment group. Corresponding risk differences were calculated along with 95% confidence intervals. The Kaplan-Meier method22 was used to estimate time-to-clinical-worsening stratified by treatment allocation and by low-risk status.
Surrogacy was assessed using three methods, (i) mediation analysis, (ii) meta-analysis, and (iii) leave-one-out meta-analysis.23 Complete-case analysis was used for all methods, as only a low proportion of surrogate outcomes were expected to be missing after performing the calculations to adjust for missing risk score components as described above. Individuals with a clinical worsening event before 16 weeks were excluded from primary analyses focused on clinical worsening to avoid conditioning on future information in models. Because ordinal variables introduce potential issues in mediation and other analyses, the primary surrogate outcome was dichotomized as 1 = low-risk status vs. 0 = not low-risk status. Figure 1 shows directed acyclic graphs corresponding to surrogate outcomes of various strengths. If a robust indirect effect of treatment exists while a direct effect is non-existent, risk status and risk scores would be considered strong surrogates. In practice, direct effects will usually be present, and the strength of surrogacy is assessed by considering the effect size of the indirect effect relative to the total effect of treatment.
Figure 1:
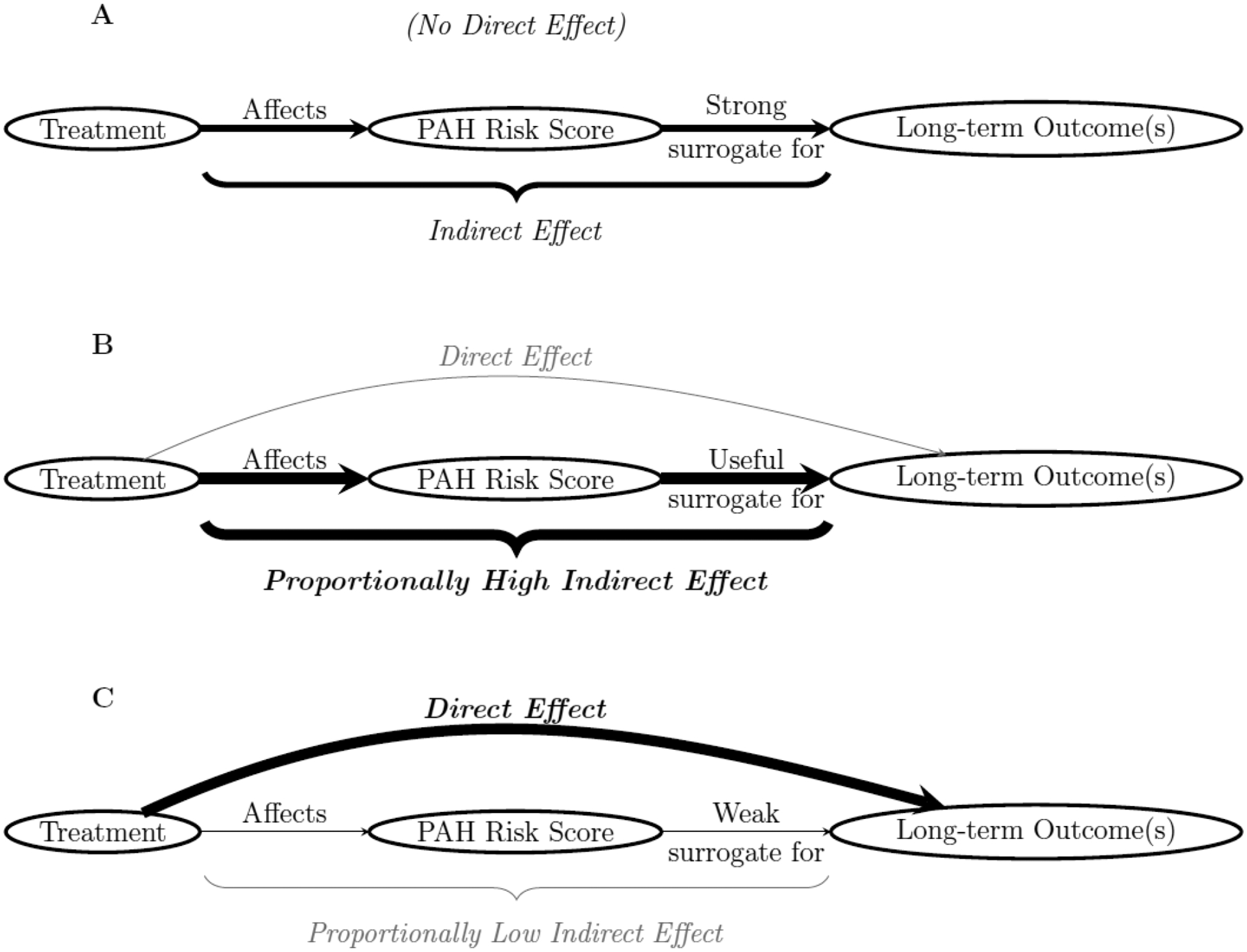
Directed acyclic graphs for A) a strong/perfect surrogate, B) a useful surrogate, and C) a weak and/or inadequate surrogate.
First, we performed a mediation analysis considering each candidate surrogate as an intermediate outcome.24,25 To avoid ‘inconsistent mediation’ (i.e., when direct and indirect effects cancel each other out, the direct effect is even larger than the total effect, or other situations that result in a negative proportion mediated), we first empirically tested four criteria to justify the mediation analysis (Table S4).26,27 Next, Cox proportional hazards models28 were fit for the clinical worsening and survival outcomes conditional on each candidate surrogate separately, as well as treatment, corresponding baseline risk score, and a fixed effect variable with three levels for trial membership (to allow for similarity within each trial/intervention). Results from these models were combined with parallel models that did not condition on the candidate surrogates, but otherwise conditioned on the same set of variables to perform the ‘difference method’ for mediation,29,30 estimating the total effect and direct effects of treatment on clinical worsening and survival, as well as the indirect effects through each candidate surrogate. The proportion of the effect mediated through each surrogate risk score was estimated, along with 95% confidence intervals via a bootstrap procedure.
Next, surrogacy was assessed using meta-analysis techniques.31,32 As recommended when the number of available trials is small,33 each geographic region within each trial was treated as its own clinical trial in the meta-analysis. For the AMBITION study, trial locations were grouped by North American and European/Australian regions. The GRIPHON and SERAPHIN studies were grouped by American, European/Australian, and Asian regions. In all trials, Israel was included in the European/Australian region. First, the effects of treatment on risk scores were estimated within each trial-region using separate logistic regression models for each risk score. Then the effects of treatment on clinical worsening in each trial-region were estimated using Cox proportional hazards models. The estimated treatment effects on the surrogates were then regressed on the corresponding estimated log hazard ratios for the outcome, weighting by the inverse variance of the estimated log hazard ratio from each trial region. The R2 metrics from the final weighted models were used to assess the strength of the potential surrogates.
Finally, we carried out a trial-region leave-one-out meta-analysis. Specifically, the meta-regression described above was performed for all but one trial-region. Then, the effect of the treatment on the surrogate was used to predict the hazard ratio in the left-out trial region, using the fitted meta-regression. This full procedure was repeated for each trial-region and the predicted hazard ratios were compared to the observed hazard ratios.
We performed sensitivity analyses, including specifying accelerated failure time (AFT) models rather than Cox proportional hazards models within the mediation and meta-analyses. We also repeated the analyses using the raw absolute risk scores at 16 weeks as surrogates, which utilized ordinal logistic regression when modeling the risk scores. Finally, a post-hoc sensitivity analysis incorporating events before 16 weeks was performed. All analyses were carried out in R 4.1.2 statistical software.34 The statistical code used to perform all analyses is available at https://github.com/harhay-lab/PAH-surrogates.
Role of the funder
The funder(s) had no role in data collection, analysis, interpretation, writing of the manuscript, or decision to submit.
Results
Of the 28 PAH trials received from the FDA that were considered for inclusion, we included three Phase III event driven trials in this study (Figure S1). The characteristics of the included studies are shown in Table S5. The study sample included 2508 subjects. The mean age was 49 ± 16 years, 1956 (78%) were women, 1704 (68%) were white, and 280 (11%) were Hispanic/Latino. Of these, 1388 (55%) had idiopathic PAH, and 776 (31%) had PAH associated with connective tissue disease. Table 1 shows the baseline characteristics of the study sample stratified by COMPERA risk score.
Table 1.
Baseline characteristics of study participants stratified by COMPERA risk score
| Characteristic | N | Overall N = 2,508 | Low risk N = 617 | Intermediate risk N = 1,766 | High risk N = 125 |
|---|---|---|---|---|---|
| Age (years), mean ± SD | 2,508 | 49.3 ± 15.9 | 46.3 ± 16.0 | 50.5 ± 15.6 | 47.8 ± 16.3 |
| Sex (Female), n (%) | 2,508 | 1,956 (78.0) | 497 (80.6) | 1,361 (77.1) | 98 (78.4) |
| Race, n (%) | 2,508 | ||||
| White | 1,704 (67.9) | 388 (62.9) | 1,226 (69.4) | 90 (72.0) | |
| Asian | 463 (18.5) | 148 (24.0) | 294 (16.6) | 21 (16.8) | |
| Black | 86 (3.4) | 19 (3.1) | 63 (3.6) | 4 (3.2) | |
| Multiple / Other | 31 (1.2) | 18 (2.9) | 13 (0.7) | 0 (0.0) | |
| Unknown | 224 (8.9) | 44 (7.1) | 170 (9.6) | 10 (8.0) | |
| Ethnicity, n (%) | 2,508 | ||||
| Not Hispanic/Latino | 2,227 (88.8) | 561 (90.9) | 1,551 (87.8) | 115 (92.0) | |
| Hispanic/Latino | 280 (11.2) | 56 (9.1) | 214 (12.1) | 10 (8.0) | |
| Unknown | 1 (0.0) | 0 (0.0) | 1 (0.1) | 0 (0.0) | |
| Body mass index (kg/m 2 ), mean ± SD | 2,508 | 26.9 ± 5.7 | 26.0 ± 5.0 | 27.2 ± 5.9 | 27.1 ± 5.3 |
| PAH etiology, n (%) | 2,503 | ||||
| Idiopathic | 1,388 (55.5) | 292 (47.5) | 1,013 (57.5) | 83 (66.4) | |
| Associated with connective tissue disease | 776 (31.0) | 233 (37.9) | 509 (28.9) | 34 (27.2) | |
| Associated with congenital heart disease | 181 (7.2) | 56 (9.1) | 122 (6.9) | 3 (2.4) | |
| Drug and toxin-induced | 72 (2.9) | 15 (2.4) | 56 (3.2) | 1 (0.8) | |
| Heritable/familial | 56 (2.2) | 13 (2.1) | 40 (2.3) | 3 (2.4) | |
| Associated with HIV infection | 30 (1.2) | 6 (1.0) | 23 (1.3) | 1 (0.8) | |
| Background PAH therapy, n (%) | 2,508 | ||||
| None | 1,348 (53.7) | 344 (55.8) | 946 (53.6) | 58 (46.4) | |
| Phosphodiesterase type 5 | 736 (29.3) | 184 (29.8) | 510 (28.9) | 42 (33.6) | |
| inhibitor alone | |||||
| Phosphodiesterase type 5 | 227 (9.1) | 43 (7.0) | 171 (9.7) | 13 (10.4) | |
| inhibitor and Endothelin receptor | |||||
| antagonist | |||||
| Endothelin receptor antagonist alone | 156 (6.2) | 33 (5.3) | 113 (6.4) | 10 (8.0) | |
| Phosphodiesterase type 5 inhibitor and Prostacyclin analogue | 24 (1.0) | 9 (1.5) | 13 (0.7) | 2 (1.6) | |
| Prostacyclin analogue alone | 17 (0.7) | 4 (0.6) | 13 (0.7) | 0 (0.0) | |
| WHO functional class, n (%) | 2,507 | ||||
| I | 10 (0.4) | 5 (0.8) | 5 (0.3) | 0 (0.0) | |
| II | 1,110 (44.3) | 485 (78.6) | 622 (35.2) | 3 (2.4) | |
| III | 1,361 (54.3) | 127 (20.6) | 1,125 (63.7) | 109 (87.9) | |
| IV | 26 (1.0) | 0 (0.0) | 14 (0.8) | 12 (9.7) | |
| Six-minute walk distance (m), mean ± SD | 2,506 | 353.3 ± 89.5 | 400.0 ± 78.2 | 342.5 ± 84.7 | 275.0 ± 103.5 |
| NT-proBNP (pg/mL) | 1,744 | 365 [208, 1,750] | 156 [81, 268] | 939 [365, 1,924] | 2,527 [1,880, 3,486] |
| Mean right atrial pressure (mmHg), median [IQR] | 2,332 | 8.0 [5.0, 11.0] | 5.0 [4.0, 7.0] | 8.0 [6.0, 12.0] | 16.0 [15.0, 20.0] |
| Mean pulmonary artery pressure (mmHg), median [IQR] | 2,503 | 51 [41, 60] | 44 [36, 54] | 52 [43, 62] | 58 [51, 65] |
| Cardiac output (L/min), median [IQR] | 2,423 | 4.2 [3.4, 5.1] | 5.0 [4.4, 6.0] | 4.0 [3.2, 4.8] | 2.9 [2.4, 3.4] |
| Cardi ac index (L/min/m 2 ), median [IQR] | 2,431 | 2.4 [1.9, 2.9] | 2.9 [2.6, 3.4] | 2.2 [1.9, 2.6] | 1.7 [1.4, 1.9] |
| Pulmonary vascular resistance (Wood units), median [IQR] | 2,445 | 10 [7, 14] | 7 [5, 9] | 11 [7, 15] | 17 [13, 21] |
| Pulmonary artery wedge pressure (mmHg), median [IQR] | 2,387 | 9.0 [7.0, 12.0] | 9.0 [6.0, 11.0] | 10.0 [7.0, 12.0] | 10.0 [7.0, 11.5] |
| Treatment assignment | 2,508 | ||||
| Experimental | 1,372 (54.7) | 326 (52.8) | 982 (55.6) | 64 (51.2) | |
| Control | 1,136 (45.3) | 291 (47.2) | 784 (44.4) | 61 (48.8) |
After calculations adjusting for missing risk score components (Table S3), 37–55 (2%) of enrolled individuals had missing risk scores at 16 weeks, with slight variation by risk score. With similar variation, 56–81 individuals (2–3%) had clinical worsening before 16 weeks but were still enrolled for risk score assessment; these were excluded from relevant primary analyses. Of those at risk for events after surrogate measurement, 717 (32%) experienced clinical worsening and 138 (6%) died. Allocation to the experimental arm increased the probability of achieving low-risk status for each risk score at 16 weeks compared to allocation to the control/placebo arm (Table S6). Randomization to the experimental arm was also associated with increased time to clinical worsening (Figure 2A), although no effect on long-term survival was found (Figure 2B). Achievement of low-risk status was associated with both a longer time to clinical worsening (Figure 2C and Figure S2) and a longer time to death (Figure 2D and Figure S3) for all risk scores considered.
Figure 2:
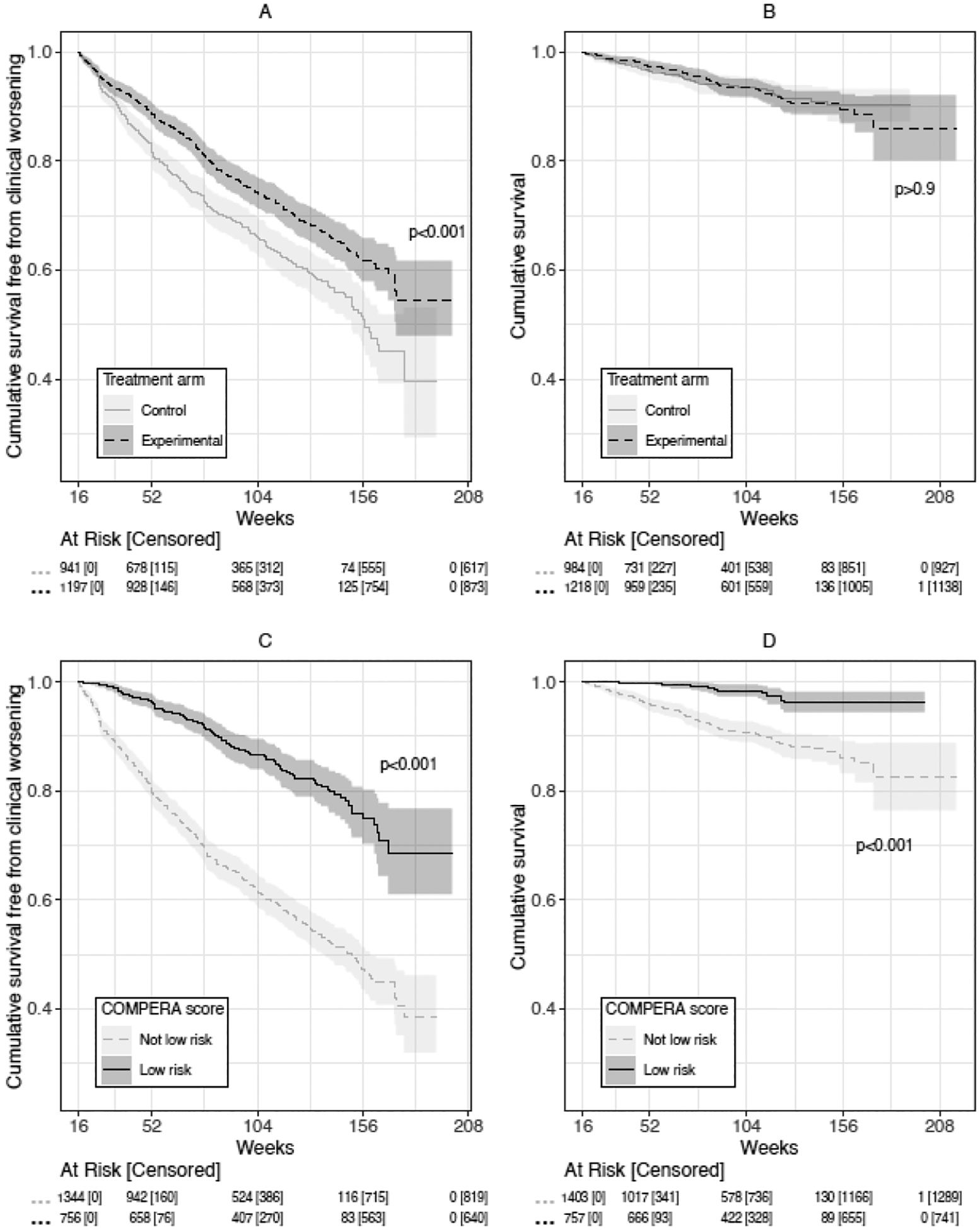
Kaplan Meier curves for A) cumulative survival free from clinical worsening, stratified by experimental or control arm, B) cumulative survival, stratified by experimental or control arm, C) cumulative survival free from clinical worsening, stratified by COMPERA low-risk status at 16 weeks vs other status, and D) cumulative survival, stratified by COMPERA low-risk status at 16 weeks vs other status.
The four standard criteria for carrying out mediation analysis were assessed (Table S4). All criteria were met for the clinical worsening outcome, but they were not met for mortality, as treatment group was not significantly associated with survival in the combined trial sample (logrank test p=0.6; HR = 0.97 [95% CI: 0.72, 1.31]; Figure 2B). Figure 3 shows the key results of the mediation analysis for clinical worsening (Table S7 shows the full results). Indirect and direct effects are reported in the figure, where the indirect effect is the part of the treatment effect which is mediated through the risk status, while the direct effect summarizes the portion of the treatment effect which is not mediated through the risk status. Attainment of low-risk status was not a strong mediator for the effect of treatment on time to clinical worsening for any of the risk scores, with proportionally small and only marginally significant indirect treatment effects. The proportions of the effects mediated through the risk scores were at best modest, ranging from 0.07 to 0.13.
Figure 3:
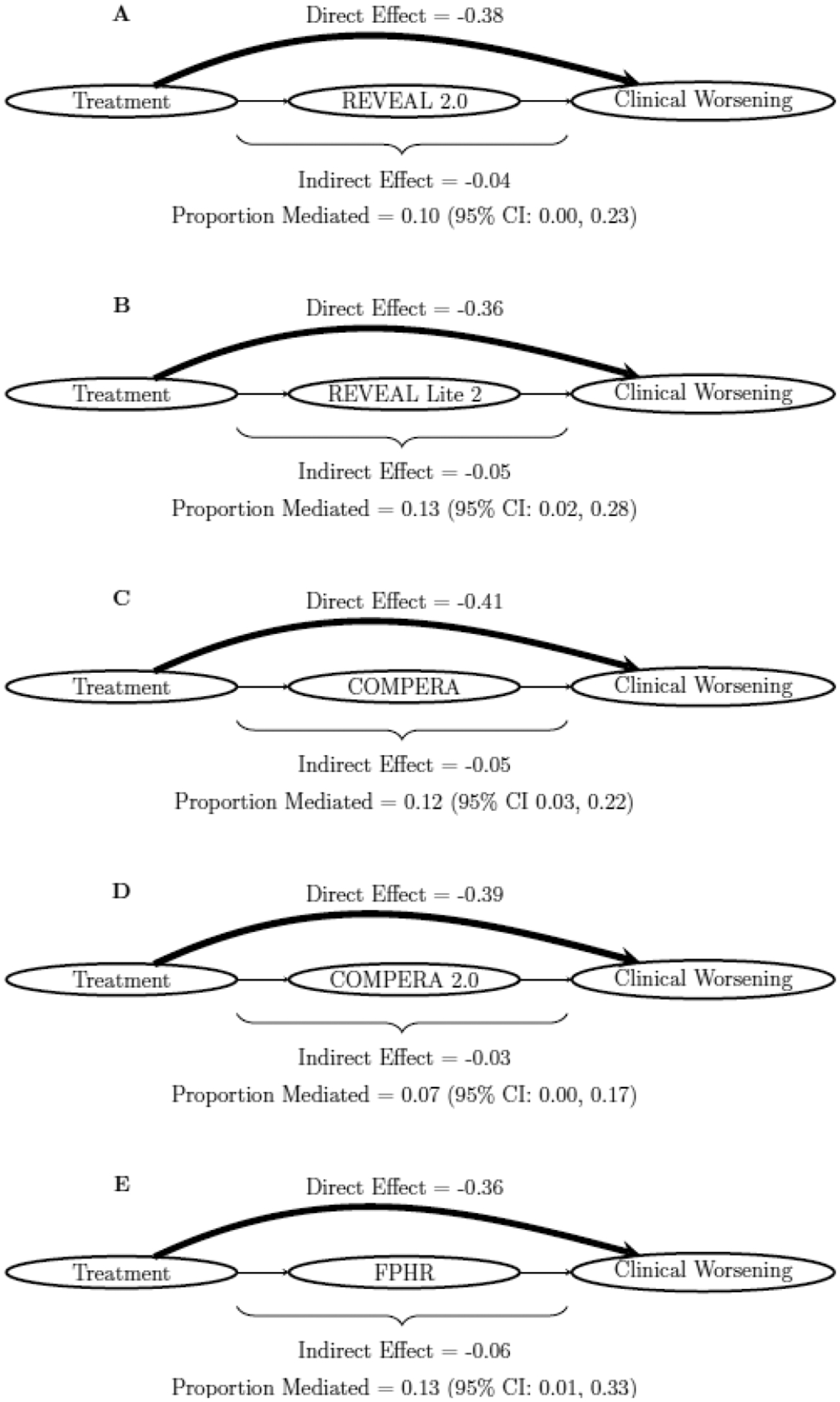
Visualization of mediation results for clinical worsening for A) REVEAL 2.0, B) REVEAL Lite 2, C) COMPERA, D) COMPERA 2.0, and E) FPHR risk score mediators. Size of direct and indirect effect lines were weighted according to the relative size of the effects.
Figure 4 shows the meta-regression results for clinical worsening. The meta-regression was weighted by the inverse variance of the effect estimate on clinical worsening, which is proportional to the sample of each trial-region. For each risk score, R2 values were between 0.01 and 0.19, indicating no or weak correlations between the treatment effect on achieving low-risk status at 16 weeks and the treatment effect on long-term clinical worsening. The meta-regression for mortality also showed low correlations, ranging from 0 to 0.2. Thus, the treatment effects on risk status do not seem to predict the eventual treatment effects on clinical worsening or mortality.
Figure 4:
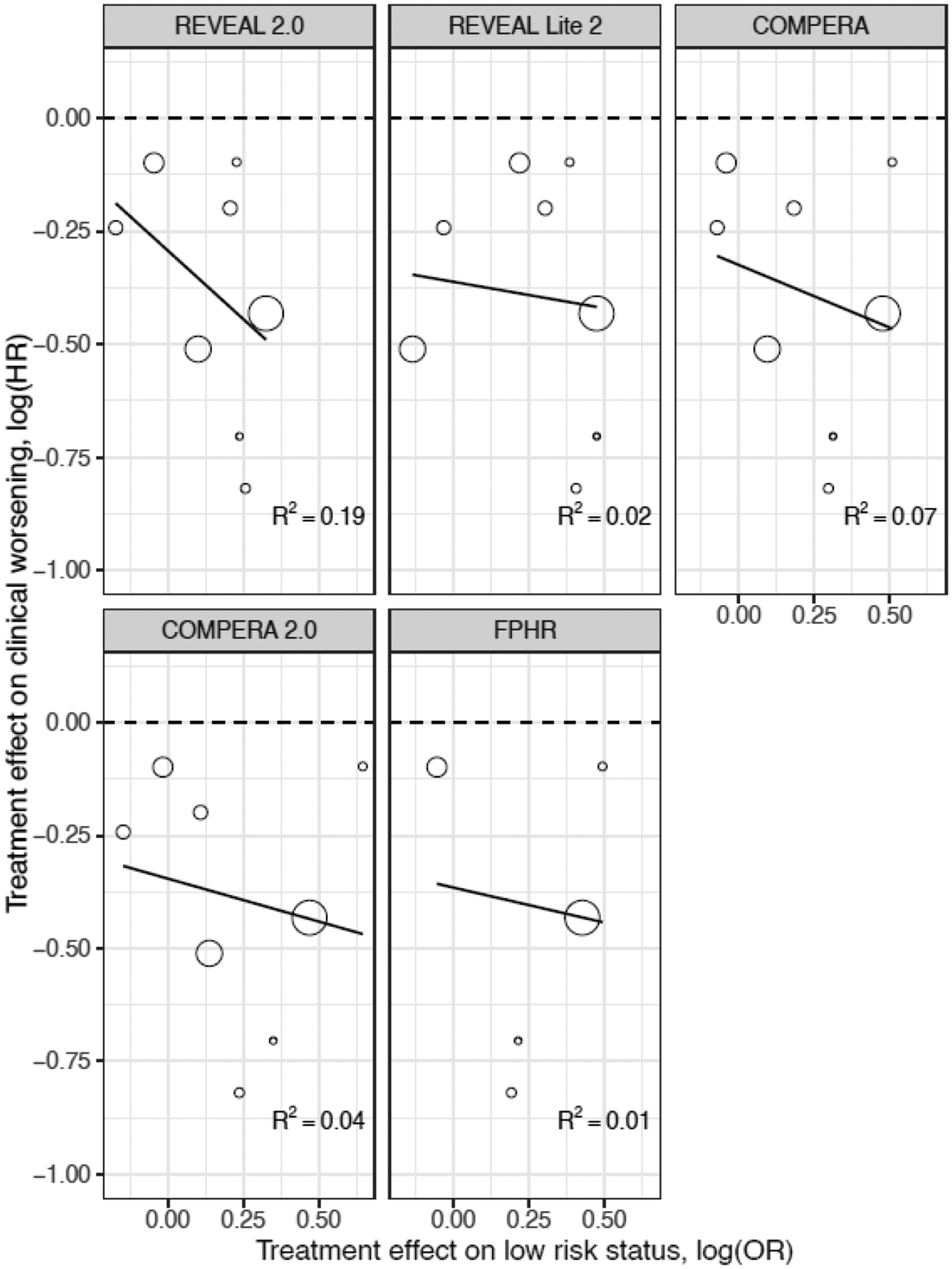
Meta-regression of the treatment effects on clinical worsening and on the risk status at the trial-region level.
The leave-one-out meta-regression results for clinical worsening are presented in Table 2. The predicted hazard ratios for the association of the surrogate with time to clinical worsening were generally biased for most trial-regions, with both overestimation and underestimation for different regions. The meta-regression results predicted a large effect on increased time to clinical worsening in SERAPHIN: Asia, GRIPHON: Europe/Australia, and GRIPHON: Asia (predicted HR 0.53–0.71) which was not observed in the trial-regions (estimated HR 0.82–0.91). The leave-one-out meta-regression results for mortality were similarly biased (Table S8).
Table 2.
Leave-one-out meta-regression results and observed hazard ratio for the left-out trial-region for clinical worsening
| Predicted trial-region hazard ratio (95% CI) using surrogate in meta-regression | ||||||
|---|---|---|---|---|---|---|
| Trial-region left out of meta-analysis | Observed trial-region hazard ratio | REVEAL 2.0 | REVEAL Lite 2 | COMPERA | COMPERA 2.0 | FPHR |
| AMBITION: North America | 0.49 | 0.66 (0.52, 0.84) | 0.69 (0.49, 0.97) | 0.68 (0.53, 0.87) | 0.68 (0.53, 0.88) | 0.71 (0.33, 1.52) |
| AMBITION: Europe/Australia | 0.44 | 0.67 (0.53, 0.84) | 0.70 (0.54, 0.90) | 0.69 (0.56, 0.86) | 0.70 (0.57, 0.87) | 0.73 (0.37, 1.45) |
| SERAPHIN: Americas | 0.60 | 0.73 (0.57, 0.94) | 1.00 (0.48, 2.06) | 0.75 (0.54, 1.03) | 0.72 (0.53, 0.96) | -- |
| SERAPHIN: Europe/Australia | 0.78 | 0.87 (0.41, 1.84) | 0.67 (0.43, 1.06) | 0.72 (0.43, 1.21) | 0.70 (0.38, 1.28) | -- |
| SERAPHIN: Asia | 0.82 | 0.64 (0.51, 0.81) | 0.66 (0.51, 0.85) | 0.67 (0.52, 0.87) | 0.68 (0.50, 0.91) | -- |
| GRIPHON: Americas | 0.65 | 0.57 (0.35, 0.91) | 0.67 (0.41, 1.08) | 0.61 (0.34, 1.09) | 0.65 (0.39, 1.06) | 0.65 (0.13, 3.18) |
| GRIPHON: Europe/Australia | 0.91 | 0.71 (0.46, 1.09) | 0.65 (0.51, 0.83) | 0.65 (0.41, 1.03) | 0.65 (0.44, 0.96) | 0.28 (0.11, 0.71) |
| GRIPHON: Asia | 0.91 | 0.63 (0.50, 0.78) | 0.64 (0.49, 0.84) | 0.57 (0.40, 0.82) | 0.53 (0.33, 0.85) | 0.57 (0.20, 1.62) |
Each row displays the predicted hazard ratio (95% CI) corresponding to the treatment effect on clinical worsening, had the trial-region listed in the first column been excluded from the meta-regressions. These predictions can then be compared to the observed trial-region hazard ratio displayed in the second column to assess how well each surrogate would have worked if they had been used in each trial-region at 16 weeks rather than waiting to observe the long-term clinical worsening outcome.
A post-hoc sensitivity analysis including patients with clinical worsening before 16 weeks who were still enrolled for surrogate measurement at 16 weeks (rather than excluding them as above) yielded the same conclusions (Table S9). Planned sensitivity analyses using AFT models are provided in Tables S10 and S11. For the mediation analysis, these yielded slightly higher proportions of treatment effect mediated by low-risk status for each risk score, although all were less than 0.2, which is not consistent with strong mediation/surrogacy (Table S10). Meta regression results using AFT models are displayed in Figure S5, yielding improved (but still weak) R2 values ranging from 0.02 to 0.28. The leave-one-out meta-regression results for clinical worsening using AFT models are presented in Table S11, showing biases in predicting trial-region effects. The magnitude of these biases cannot be easily compared to those from the primary analysis as the estimands are on a different scale.
We also performed sensitivity analyses using raw ordinal risk score values at 16 weeks as candidate surrogates rather than binary attainment of low-risk status (Table S12). The estimated proportions mediated were marginally higher than those found using binary low-risk status, but there were no particularly strong mediators identified. Meta-regression using ordinal mediators (Figure S6) and leave-one-out meta-regression using ordinal mediators (Table S13) yielded similar conclusions to the meta-regressions using binary low-risk status.
Discussion
We used IPD from three large RCTs in PAH to assess whether established multicomponent risk scores (both “low-risk status” and the ordinal scores themselves) were adequate surrogates for time to clinical worsening. Using mediation analysis, we showed that achieving low-risk status explained about 7–13% of the effect of experimental arm allocation on time to clinical worsening. Sensitivity analyses showed that the proportion mediated could be higher when considering ordinal risk scores or using accelerated failure time models. The ordinal score results were similar to those presented in a recent mediation analysis of the FREEDOM-EV trial (which was not included in this analysis).35 Meta-regression of trial-regions showed weak associations between the treatment effect on the prediction rules and the treatment effect on the outcomes. Leave-one-out regressions did not predict the actual treatment effect on time to clinical worsening for the trial-region left out with substantial differences both towards and away from a null treatment effect in several instances. Sensitivity analyses for the meta-analyses did not yield substantially different results.
Counter to recent guidelines and proposals to use established multicomponent risks scores as surrogates in RCTs or as targets in clinical management, our results suggest that this approach could lead to erroneous conclusions about the effects of new PAH treatments on long-term clinical worsening. While the threshold to determine adequate surrogacy is not clearly established in any disease state, most set a high bar for surrogacy, with 50% or more proportion of treatment effect often stated as the goal. Targeting a surrogate outcome that is not a valid surrogate could have negative consequences, such as finding a “positive” result for a surrogate (when there is actually no impact on long-term outcomes) or a “negative” result for a surrogate, when the intervention is actually effective in improving long-term outcomes. The strength of the surrogate effect could also differ greatly from the strength of the effect on the outcome, even if in the same direction.
These results may seem surprising. These prediction rules are well-established and validated approaches to predicting mortality in PAH patients. It has also been shown that allocation to experimental arms in RCTs in PAH is associated with improved time to clinical worsening. We confirmed both of these results in our study, however, these findings are insufficient to infer surrogacy.9 There is a pervasive fallacy that showing an impact of treatment on the potential surrogate and an association between the surrogate and outcome is sufficient to validate a surrogate end point. The treatment of ventricular ectopy with anti-arrhythmics and of systolic heart failure with chronic milrinone infusion are classic examples of targeting what were thought to be surrogate endpoints which ended up failing.36 In PAH, one study (COMPASS-3) tried using a “targeted” treatment approach based on a surrogate which did not translate into improved longer-term outcome.37 We believe that early-phase clinical trials should only use validated surrogates as primary outcomes (or include possible surrogates as secondary outcomes for hypothesis generation). Any goal-oriented treatment approaches (like targeting “low-risk status”) require testing in RCTs comparing them to usual care before making evidence-based recommendations. The risks of using such unvalidated surrogates or treatment targets include erroneous inferences about treatment effects and potential over-treatment of patients with multiple expensive drugs with significant respective side effects.
Strengths of this study include the relatively large number of subjects with representation from various international regions, randomization of allocation to study intervention or control arms (required to validate surrogates), harmonization of the clinical worsening definition to be consistent between studies, and the IPD meta-analysis. There were also several limitations to this study. First, the mediation method may be overly liberal for determining surrogacy;38 a strong mediator doesn’t necessarily make for a good surrogate. However, we did not identify any metric (achieving low-risk status or ordinal risk score) as a strong mediator of the treatment effect on outcome, mitigating this concern. Another limitation is that only 3 trials met the analysis inclusion criteria, which is less than typically used to assess surrogacy in a meta-analytic framework. We tried to partially address this limitation by considering each trial-region as its own trial for analysis, as has been previously proposed and validated.33 More large long-term trials would increase confidence in the findings. FREEDOM-EV was a long-term trial that was not included in the analysis because it concluded after we had received the data from the FDA. This study found similar estimates for the proportion of treatment effect explained by the REVEAL Lite 2 prediction rule (15–33%).35
As all trials were submitted to the FDA for regulatory approval, this is likely a biased sample of trials; external validity (especially for new therapies and patient populations not included in the current set of trials) would be important to establish. Relatedly, the population represented in these trials is somewhat different from that represented in risk score registries and the population as a whole (in particular, our sample was younger on average and a higher proportion female). We were unable to assess whether these rules were adequate for surrogacy for mortality using mediation analysis due to the lack of overall treatment effect on mortality in the combined sample. This was likely due to opposing benefit and harm effects in the randomized phases of AMBITION and GRIPHON, respectively. There was little evidence of surrogacy in the meta-regression analyses for mortality. Overall, the inclusion of more trials with greater variability of treatment effects on survival (and more mortality events) would improve this analysis and allow for further investigation of surrogacy. Because these prediction rules were derived to predict mortality at one-year, new prediction rules derived to predict clinical worsening might have better performance as surrogates. The time to clinical worsening outcome has not been comprehensively evaluated in the risk score registries, despite becoming a common primary endpoint for late-phase approval trials for PAH therapies.
There were inevitably missing data for the risk scores and risk score components (e.g., hemodynamic data was not collected at baseline in two of the included trials), and data needed to be interpolated for one trial due to measurement at different time points compared to the other two trials. These factors could have contributed to the poor performance of risk scores as surrogates, but the data completeness likely reflects other Phase III studies. While the main analysis focused on maintaining low-risk status at 16 weeks after randomization as the surrogate marker, sensitivity analyses incorporating the actual risk scores themselves showed slightly better results. These results can only be applied early in treatment and assessment of patients with PAH, as we do not know whether these prediction rules applied later in the course of disease may be surrogates for longer-term outcomes.
In summary, these data do not support the use of current prediction rules as surrogates of time to clinical worsening in clinical trials. While the risk scores remain strong predictive tools and can be used as such, this approach should not be conflated with use as a validated surrogate end point. Future work could consider alternative clinical outcomes, such as composite clinical improvement endpoints. Studies should try to refine risk scores as surrogate endpoints and leverage large, harmonized sets of data and use machine-learning tools to derive variables specifically designed for surrogacy rather than risk prediction. Such a priori approaches are essential for all stakeholders in PAH, as validated surrogate outcomes could improve clinical care and expedite clinical research for this disease.
Data Sharing Statement
The data used for this paper were provided by the FDA for secondary analyses of the included clinical trials. This data and related study documents will not be made available with this manuscript.
Supplementary Material
Research in context.
Evidence before this study
Many have advocated for the use of multicomponent risk scores as surrogate outcomes in pulmonary arterial hypertension (PAH) trials and clinical care. While studies have shown that these scores are predictive of later mortality, this correlation is not sufficient in isolation to support the use of changes in multicomponent risk scores as surrogate outcomes. We completed a search of Medline to identify literature on the assessment of surrogate outcomes for PAH on April 14, 2023, including all languages, using these search terms: ((pulmonary hypertension) OR (pulmonary arterial hypertension)) AND (surrogate). The search identified older papers that examined hemodynamic values and six-minute walk distance as potential surrogate outcomes for PAH, and a mediation analysis of the FREEDOM-EV trial in which the REVEAL Lite 2 score had a proportion of treatment effect explained of 15% to 33%.
Added value of this study
In this paper, data from three large PAH trials with long-term follow-up were analyzed using both mediation and meta-analysis frameworks to assess the potential surrogacy of five popular multicomponent PAH risk scores (COMPERA, COMPERA 2.0, REVEAL 2.0, REVEAL Lite 2, and non-invasive FPHR).
Implications of all the available evidence
Evidence for surrogacy for five popular multicomponent PAH risk scores is weak to moderate, and using these risk scores as surrogates in future trials may lead to erroneous conclusions.
Acknowledgments
We thank the Cardiovascular Medical Research and Education Fund for supporting this project.
Declaration of interests
CV received institutional funding for participation in a randomized trial sponsored by Altavant Sciences and personal fees from Regeneron, Merck, Janssen, and Gather-ED CME, support for travel from the American Thoracic Society, and for serving on a data safety monitoring board from Altavant Sciences and Acceleron/Merck. JF received institutional funding for participation in a randomized trial sponsored by United Therapeutics. JAM received personal fees for serving on a data safety monitoring board for United Therapeutics, Merck, and Janssen. SMK received consulting fees from Janssen, Regeneron, and Morphic, travel support from Aerovate, for serving on a data safety monitoring board from United Therapeutics, Acceleron, Vivus, Aerivate, and Proteo Biotech, editorial fees from the European Respiratory Journal, has stock or stock options in Verve Therapeutics, and has received in-kind remote monitoring equipment from PhysIQ. MOH received statistical consulting fees from Unlearn.AI and Berkeley Research Group, fees for editorial services from Elsevier and the American Thoracic Society, for serving on a data safety monitoring board from the University of California, San Francisco, and the University of Pittsburgh, and for pilot grant reviews from Brown University and New York University. No other authors have any declarations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Benza RL, Gomberg-Maitland M, Miller DP, et al. The REVEAL Registry risk score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest 2012; 141(2): 354–62. [DOI] [PubMed] [Google Scholar]
- 2.Boucly A, Weatherald J, Savale L, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J 2017; 50(2): 1700889. [DOI] [PubMed] [Google Scholar]
- 3.Hoeper MM, Kramer T, Pan Z, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. European Respiratory Journal 2017; 50(2): 1700740. [DOI] [PubMed] [Google Scholar]
- 4.Kylhammar D, Kjellström B, Hjalmarsson C, et al. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. European Heart Journal 2018; 39(47): 4175–81. [DOI] [PubMed] [Google Scholar]
- 5.Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2022. [DOI] [PubMed] [Google Scholar]
- 6.Temple R Are surrogate markers adequate to assess cardiovascular disease drugs? JAMA 1999; 282(8): 790–5. [DOI] [PubMed] [Google Scholar]
- 7.US Food Drug Administration. Surrogate endpoint resources for drug and biologic development. 2020. [Google Scholar]
- 8.Buyse M, Molenberghs G, Paoletti X, et al. Statistical evaluation of surrogate endpoints with examples from cancer clinical trials. Biom J 2016; 58(1): 104–32. [DOI] [PubMed] [Google Scholar]
- 9.Ventetuolo CE, Benza RL, Peacock AJ, Zamanian RT, Badesch DB, Kawut SM. Surrogate and combined end points in pulmonary arterial hypertension. Proc Am Thorac Soc 2008; 5(5): 617–22. [DOI] [PubMed] [Google Scholar]
- 10.Snow JL, Kawut SM. Surrogate end points in pulmonary arterial hypertension: assessing the response to therapy. Clin Chest Med 2007; 28(1): 75–89, viii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weatherald J, Boucly A, Sahay S, Humbert M, Sitbon O. The Low-Risk Profile in Pulmonary Arterial Hypertension. Time for a Paradigm Shift to Goal-oriented Clinical Trial Endpoints? Am J Respir Crit Care Med 2018; 197(7): 860–8. [DOI] [PubMed] [Google Scholar]
- 12.Ventetuolo CE, Gabler NB, Fritz JS, et al. Are hemodynamics surrogate end points in pulmonary arterial hypertension? Circulation 2014; 130(9): 768–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabler NB, French B, Strom BL, et al. Validation of 6-minute walk distance as a surrogate end point in pulmonary arterial hypertension trials. Circulation 2012; 126(3): 349–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galie N, Barbera JA, Frost AE, et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N Engl J Med 2015; 373(9): 834–44. [DOI] [PubMed] [Google Scholar]
- 15.Sitbon O, Channick R, Chin KM, et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N Engl J Med 2015; 373(26): 2522–33. [DOI] [PubMed] [Google Scholar]
- 16.Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013; 369(9): 809–18. [DOI] [PubMed] [Google Scholar]
- 17.Urbanowicz RJ, Holmes JH, Appleby D, et al. A Semi-Automated Term Harmonization Pipeline Applied to Pulmonary Arterial Hypertension Clinical Trials. Methods Inf Med 2022; 61(1–02): 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Min J, Appleby DH, McClelland RL, et al. Secular and Regional Trends among Pulmonary Arterial Hypertension Clinical Trial Participants. Ann Am Thorac Soc 2022; 19(6): 952–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benza RL, Gomberg-Maitland M, Elliott CG, et al. Predicting Survival in Patients With Pulmonary Arterial Hypertension: The REVEAL Risk Score Calculator 2.0 and Comparison With ESC/ERS-Based Risk Assessment Strategies. Chest 2019; 156(2): 323–37. [DOI] [PubMed] [Google Scholar]
- 20.Benza RL, Kanwar MK, Raina A, et al. Development and Validation of an Abridged Version of the REVEAL 2.0 Risk Score Calculator, REVEAL Lite 2, for Use in Patients With Pulmonary Arterial Hypertension. Chest 2021; 159(1): 337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoeper MM, Pausch C, Olsson KM, et al. COMPERA 2.0: a refined four-stratum risk assessment model for pulmonary arterial hypertension. Eur Respir J 2022; 60(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. Journal of the American statistical association 1958; 53(282): 457–81. [Google Scholar]
- 23.Joffe MM, Greene T. Related causal frameworks for surrogate outcomes. Biometrics 2009; 65(2): 530–8. [DOI] [PubMed] [Google Scholar]
- 24.Cheng C, Spiegelman D, Li F. Estimating the natural indirect effect and the mediation proportion via the product method. BMC Med Res Methodol 2021; 21(1): 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.VanderWeele T Explanation in causal inference: methods for mediation and interaction: Oxford University Press; 2015. [Google Scholar]
- 26.MacKinnon DP, Krull JL, Lockwood CM. Equivalence of the mediation, confounding and suppression effect. Prev Sci 2000; 1(4): 173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baron RM, Kenny DA. The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J Pers Soc Psychol 1986; 51(6): 1173–82. [DOI] [PubMed] [Google Scholar]
- 28.Cox DR. Regression models and life-tables. Journal of the Royal Statistical Society: Series B (Methodological) 1972; 34(2): 187–202. [Google Scholar]
- 29.Cheng C, Spiegelman D, Li F. Is the Product Method More Efficient Than the Difference Method for Assessing Mediation? Am J Epidemiol 2023; 192(1): 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nevo D, Liao X, Spiegelman D. Estimation and Inference for the Mediation Proportion. Int J Biostat 2017; 13(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buyse M, Molenberghs G, Burzykowski T, Renard D, Geys H. The validation of surrogate endpoints in meta-analyses of randomized experiments. Biostatistics 2000; 1(1): 49–67. [DOI] [PubMed] [Google Scholar]
- 32.Daniels MJ, Hughes MD. Meta-analysis for the evaluation of potential surrogate markers. Stat Med 1997; 16(17): 1965–82. [DOI] [PubMed] [Google Scholar]
- 33.Geybels M, Wolthers BO, Kreiner FF, Rasmussen S, Bauer R. Surrogate endpoint evaluation using data from one large global randomized controlled trial. BMC Med Inform Decis Mak 2021; 21(1): 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. . 2021. [Google Scholar]
- 35.Benza RL, Gomberg-Maitland M, Farber HW, et al. Contemporary risk scores predict clinical worsening in pulmonary arterial hypertension - An analysis of FREEDOM-EV. J Heart Lung Transplant 2022; 41(11): 1572–80. [DOI] [PubMed] [Google Scholar]
- 36.Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med 1996; 125(7): 605–13. [DOI] [PubMed] [Google Scholar]
- 37.Benza RL, Raina A, Gupta H, et al. Bosentan-based, treat-to-target therapy in patients with pulmonary arterial hypertension: results from the COMPASS-3 study. Pulm Circ 2018; 8(1): 2045893217741480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanderweele TJ. Surrogate measures and consistent surrogates. Biometrics 2013; 69(3): 561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.