

Abstract
Psoriasis is a common, debilitating immune-mediated skin disease. Genetic studies have identified biological mechanisms of psoriasis risk, including those targeted by effective therapies. However, the genetic liability to psoriasis is not fully explained by variation at robustly identified risk loci. To move towards a saturation map of psoriasis susceptibility we meta-analysed 18 GWAS comprising 36,466 cases and 458,078 controls and identified 109 distinct psoriasis susceptibility loci, including 45 that have not been previously reported. These include susceptibility variants at loci in which the therapeutic targets IL17RA and AHR are encoded, and deleterious coding variants supporting potential new drug targets (including in STAP2, CPVL and POU2F3). We conducted a transcriptome-wide association study to identify regulatory effects of psoriasis susceptibility variants and cross-referenced these against single cell expression profiles in psoriasis-affected skin, highlighting roles for the transcriptional regulation of haematopoietic cell development and epigenetic modulation of interferon signalling in psoriasis pathobiology.
Introduction
Psoriasis is a common immune-mediated skin disease with significant impact on psychosocial wellbeing, lifelong morbidity, and mortality.1, 2 With an estimated 60 million people affected worldwide,3 it represents a substantial economic burden.4, 5
Psoriasis has a strong genetic component, with heritability estimated at 66%.6 Previous genome-wide association study (GWAS) meta-analyses have identified 65 genomic loci at which genetic variation is associated with psoriasis susceptibility in European ancestry populations,7–9 and 17 more reported in Asian ancestry populations.10 A substantial fraction of genetic risk is attributed to the Major Histocompatibility Complex (MHC) class I allele HLA-C*06:02 and related antigen processing and presentation functions, while pathogenic roles for the IL-23/IL-17 immune axis, type I interferons and NF-κB have also been established.7, 11–13 Evidence that genetic variation influences psoriasis risk through these pathways underscores the remarkable consistency between genetic perturbations and the effectiveness of biologic therapies targeting IL-23 and IL-17.1
While they have greatly influenced current models of psoriasis pathobiology, previous GWAS meta-analyses of psoriasis have been modest in size compared to those of other common diseases in the current era of population-based bioresources.14 Larger studies offer enhanced ability to detect genetic associations with small effects and discriminate independent association signals within loci. There remain open questions around how, and in which cell types, the presence of risk-increasing alleles can lead to the dysregulated immune processes that characterise psoriasis. Unpacking these causal mechanisms and better understanding their heterogeneity across individuals should help inform how existing targeted therapies can be deployed to specifically disrupt the inflammatory loop underlying psoriasis while limiting unintended adverse consequences,15, 16 and suggest new therapeutic targets for patients in whom current treatments are ineffective or response is transient.17 It will also begin to explain the mechanistic basis for the high burden of co-morbidities suffered by individuals with psoriasis.18, 19
Here we report a meta-analysis of 18 case-control genome-wide association studies conducted by an international consortium to increase the statistical power for genetic discovery in psoriasis, and to characterise causal variants, genes, pathways, and cell types.
Results
Discovery of new psoriasis susceptibility regions
To identify genomic loci at which genetic variation is associated with psoriasis susceptibility, we performed fixed-effect standard-error-weighted GWAS meta-analysis for a total of 11,808,957 autosomal variants across 18 studies comprising a total of 494,544 unrelated European ancestry individuals. The genomic inflation factor20 (λGC) of 1.14 and LD score regression intercept of 1.07 indicate modest inflation of the meta-analysis test statistics that is partially driven by polygenicity, consistent with other complex diseases.21
Consistent with previous GWAS of psoriasis, by far the strongest evidence of association was observed within the MHC region on chromosome 6. The association signal peaks at rs12189871 (OR: 3.31, 95% CI 3.21–3.40, P=1.8×10−1524) but genome-wide significant evidence of association (P<5×10−8) was observed from positions chr6:25,622,875 telomeric to and chr6:33,971,609 centromeric to the MHC, reflecting multiple independent associations and complex patterns of linkage disequilibrium (LD)22 (Supplementary Figure 1).
Genome-wide significant psoriasis associations were also observed in the present study at all but two of the previously reported susceptibility loci in Europeans7–9 (Figure 1, Supplementary Table 1). The two loci without genome-wide significant evidence of association (13q14.11 and 21q22.12) were previously reported as psoriasis susceptibility loci in a trans-ethnic meta-analysis23 and fell just short of genome-wide significance in the current study (Supplementary Table 2).
Figure 1 – Manhattan plot summarising genome-wide associations with psoriasis susceptibility.

x-axis, genomic position; y-axis, −log10(P-value) of association; red and green points, regions previously and newly associated, respectively, with psoriasis susceptibility at genome-wide significance (P=5×10−8) in European ancestry populations; solid horizontal line, genome-wide significance threshold; dotted horizontal line, y-axis break at 10−30; chromosomes (labelled 1–22) are alternately shaded for clarity.
We identified associated genetic variants at 109 distinct loci, 50 of which have not been previously implicated in psoriasis susceptibility in European ancestry populations (Figure 1, Supplementary Tables 1 and 3, Supplementary Note). Five of these 50 loci encompass variants previously reported at genome-wide significance in other ancestral populations.22, 24, 25
To resolve the presence of multiple independent association signals at each of these loci we performed conditional and joint analysis26 across 108 susceptibility loci (excluding the MHC). We found evidence of two or more high-confidence independent associations at 27 loci, including evidence of two independent signals each at newly reported loci at 4q27 and 8q12.2. This totals 148 independent non-MHC psoriasis association signals (Supplementary Table 1).
Assuming a prevalence of 1.5%, we estimate that 46.5% (standard error, SE: 4.4%) of variance in the liability to psoriasis is explained by common SNPs outside of the MHC region, increasing to 59.2% (SE: 13.2%) when including the MHC region. The 52 independent association signals at the 50 newly identified susceptibility loci were estimated to contribute 2.9% of the non-MHC common variant liability in addition to the 11.6% accounted for by established loci (Supplementary Table 4).
Fine-mapping of candidate causal variants
Within each susceptibility locus, we sought to identify variants with strong statistical and functional evidence of being the causal variant underlying the psoriasis association signal. We constructed Bayesian 95% credible sets for causal variants at 144 sufficiently well-imputed independent susceptibility signals (Methods; Supplementary Table 1). A single variant had a posterior probability of >0.5 for being causal (PPmax>0.5) at 51 (36%) of the fine-mapped non-MHC signals with resolution to a single putative causal variant (PPmax>0.95) for 22 signals (15%) (Supplementary Table 5).
Our expanded GWAS meta-analysis provides increased power and improved ability to resolve causal variants. Thus, comparison against fine-mapping in the previous meta-analysis7 revealed the same or fewer number of variants in 95% credible sets at 52 (83%) of the 63 established psoriasis susceptibility loci (Figure 2A, Supplementary Figure 2, Supplementary Table 6).
Figure 2 – Statistical and functional fine-mapping.

A. Comparison of 95% Bayesian credible sets to previous GWAS meta-analysis. Each point represents a different association signal established in the previous meta-analysis (Tsoi et al., 2017). Point colour indicates direction of change, blue dashed line indicates equality. B. Prioritisation of protein-altering variants. Points represent protein-altering variants identified in Bayesian credible sets for independent psoriasis signals; x-axis: posterior probability of causality from statistical fine-mapping analysis, y-axis: CADD score estimating deleteriousness of protein altering variant, point colour: whether corresponding susceptibility signal is in a known or newly reported genomic region and whether primary or secondary signal. Note the TRAF3IP2 variant is rs33980500, discussed in the main text. C. Highlighted high-confidence regulatory variants derived from TURF analysis. For each variant, bars show the generic and tissue-specific regulatory probabilities (y-axis) estimated by TURF for all tissues (x-axis). Blood and skin are highlighted in orange and blue, respectively.
To further highlight likely disease-causing variants and identify the biological mechanisms through which they influence pathological processes, we functionally annotated 5,345 variants present in 95% credible sets. For 24 association signals, at least one variant in the credible set was predicted to give rise to altered or truncated protein sequences (Supplementary Table 7). Twenty of these missense and nonsense variants were predicted to be deleterious (CADD score > 15), including well-known alleles influencing psoriasis risk in IL23R, TRAF3IP2, TYK2 and RIGI (formerly known as DDX58).8 Of relevance to RIGI, a deleterious missense variant was also observed in the structurally related gene DHX58 (ENSP00000251642.3:p.Asn461Ser, CADD score 25.0), encoding the nucleic acid receptor LGP2 which, like the protein retinoic acid-inducible gene (RIG-I) encoded by RIGI, contributes to the innate antiviral response. A further six putative causal variants were observed in genes located at novel psoriasis susceptibility loci including high-confidence deleterious variants affecting CPVL (ENSP00000387164.1:p.Tyr168His) and POU2F3 (ENSP00000260264.4:p.His154Arg), and a low-frequency nonsense allele in STAP2 that is protective against psoriasis (ENSP00000468927.1:p.Tyr169Ter, CADD score 41.0, OR = 0.79, 95% CI 0.76–0.82) (Figure 2B).
We also assessed the likelihood that variants within the credible sets have a regulatory function, both across tissues and specifically within blood or skin.27 Consistent with the notion that statistical fine-mapping identifies causal variants, signals that resolve to smaller credible sets are enriched for variants with higher regulatory probability (Pgeneric = 2.0×10−11, Ptissue-specific = 1.8×10−17, Kruskal–Wallis test) (Supplementary Table 8, Supplementary Figure 3). At 14 association signals, a single candidate variant was prioritised with a high generic regulatory probability, both in absolute terms and relative to other variants in the credible set (Supplementary Table 9, Supplementary Figure 4). Notably, this included two independent signals at TRAF3IP2, a gene that encodes a regulator of NF-κB at chromosome 6q21.28–30 We found strong evidence that the well-known psoriasis-associated missense variant rs33980500 (p.Asp10Asn; Figure 2B) itself has a regulatory role, and independently that rs6908585 is a skin-specific regulatory variant (regulatory probability of 0.491; Figure 2C), suggesting that both coding and gene regulation of TRAF3IP2 contribute to psoriasis susceptibility at this locus.
Identification of candidate genes
Given the demonstrated importance of putative regulatory variants at many of the established and newly identified psoriasis susceptibility loci, we sought to identify genes whose expression is associated with psoriasis susceptibility variants in blood and skin (both sun exposed and unexposed) by means of a transcriptome-wide association study (TWAS) derived from Genotype-Tissue Expression (GTEx) project data.31 As expected given the large effects of psoriasis MHC associations and the extensive LD characteristic of the region, and as characterised in detail previously,22 there are multiple genes within the MHC with predicted expression differences (Supplementary Figure 5).
Outside of the MHC, transcriptome-wide significant gene expression differences (P<2.18×10−6; Methods) were observed at 47 susceptibility loci (12 newly reported) and a further six loci within 1Mb of association signals with suggestive evidence of association with psoriasis (Pmeta<10−5) (Supplementary Table 10). Interestingly, the gene predicted to be differentially expressed was the closest gene to the lead variant of the association signal at only 19 (40%) of these 47 loci (Supplementary Figure 6, Supplementary Table 11). Thirty-two of the non-MHC TWAS genes have been previously highlighted as candidate psoriasis genes.10 Amongst genes at new loci, IRF5, which encodes an interferon (IFN) regulatory factor that activates type I IFN responses, has been previously implicated in immune-mediated inflammatory disease through its association with systemic lupus erythematosus.32, 33
To identify groups of genes across psoriasis susceptibility loci with related biological function we employed DEPICT.34 This highlighted a series of immune pathways and gene sets whose membership is over-represented at psoriasis risk loci (1,632 gene sets at false discovery rate <5%; Supplementary Figure 7, Supplementary Table 12A); this includes protein-protein interaction subnetworks for the genes CBL (proto-oncogene), EGFR (epidermal growth factor receptor; EGFR signalling being regulated via CBL35), ESX1 and TEC (Tec Protein Tyrosine Kinase) (Supplementary Table 12B) that are implicated for the first time in this study.
Cellular and functional contexts of psoriasis associations
To identify the cellular contexts of genes whose transcription is mediated by psoriasis susceptibility variation we investigated expression patterns in single-cell transcriptomes derived from lesional and non-lesional skin of up to 14 chronic plaque psoriasis patients. 140 genes that were detectably expressed in scRNA-seq and identified in the TWAS analysis formed five clusters based on average expression patterns across twelve cell types (Figure 3). The clusters exhibited prominent upregulation of genes in: (1) lymphatic endothelial and eccrine cells, (2) keratinocytes, (3) T cells and dendritic cells, (4) melanocytes, and (5) fibroblasts, lymphatic endothelial cells and pericytes, respectively. As expected, the genes located in the epidermal differentiation complex at chr1q21.3 were preferentially expressed in epidermal keratinocytes. Genes involved in the antiviral response were identified as being enriched in the T cell/dendritic cell, melanocyte and keratinocyte clusters (Supplementary Figure 8).
Figure 3 – Relative expression of TWAS genes in single-cell skin transcriptomes of psoriasis patients.
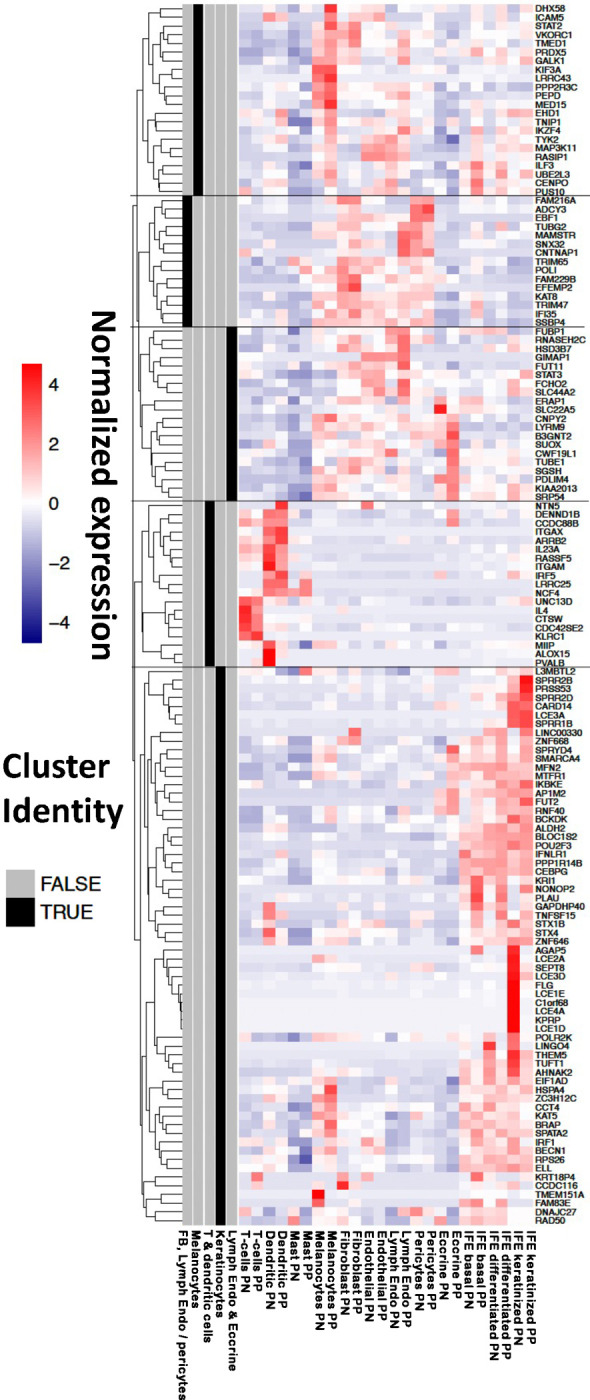
Expression level (cell colour; purple-red scale) represents mean value among cells in the corresponding cell type/condition (x-axis). For each gene (y-axis), the values were standardised and expression patterns were used for clustering (dendrogram, left hand side). Five clusters were identified (grey/black bars, left hand side) and labelled according to the cell types that exhibit highest expression for the genes in the cluster. PP, psoriasis lesions; PN, non-lesional skin; Lymph Endo, lymphatic endothelial cells; IFE, interfollicular epidermis. Clusters are identified based on enrichment for: (1) lymphatic endothelial and eccrine cells; (2) keratinocytes; (3) T cells and dendritic cells; (4) melanocytes; (5) fibroblasts, lymphatic endothelial cells and pericytes.
Despite the T cell centric regulation highlighted by previous studies, our results show that psoriasis-associated variants also govern gene regulation in stromal cells and keratinocytes. We therefore investigated whether specific cytokines regulate subsets of genes whose expression is influenced by psoriasis susceptibility variants, by examining the transcript abundance of non-MHC TWAS genes in transcriptome profiles from keratinocytes following a series of cytokine challenges (Supplementary Figure 9, Supplementary Table 13).36 The transcriptomic shift most strongly enriched for TWAS genes was induced by a combination of IL17A and TNF (FC=14.2; FDR=2.3×10−5), followed by TNF alone (FC=11.8; FDR=1.82×10−4). These results suggest potential context-specific biological effects in keratinocytes for the implicated genes and highlight the roles of psoriasis loci in regulating inflammatory response in keratinocytes.
Genome-wide correlations and causal inference
A series of recent studies have investigated the shared genetic architecture of psoriasis with other disease and health-related traits and have identified putative causal relationships with smoking, obesity and lifetime risk of cardiovascular disease.37–40 To further investigate the shared genetic liability and potential causal relationships, we assessed genetic correlation between psoriasis and 345 disease and health-related traits and evaluated asymmetry in the correlation structure (specifically, the mixed fourth moments between effect sizes41) that are consistent with causal relationships.
Positive genetic correlations were observed between psoriasis and 36 diseases and health-related traits (Supplementary Table 14) including evidence of a substantial shared genetic architecture with colitis, generalised pain, angina and pulmonary disorders (rg≥0.30, P<6.8×10−6 in all cases; Supplementary Figure 10). Significant genetic correlations were also observed with 42 physical and functional measures (Supplementary Figure 11), and 11 traits related to lifestyle and quality of life (Supplementary Figure 12).
Among traits for which we found evidence of a genetic correlation with psoriasis, 12 have evidence of asymmetry in the correlation structure that is consistent with a causal relationship (FDR<0.05) (Supplementary Table 15, Supplementary Figure 13). Susceptibility to stroke, triglyceride levels and multiple measures of adiposity have genetic support for a putative causal role in psoriasis (all genetic causal proportion (GCP)<−0.67, P≤4.6×10−3), the latter consistent with previous reports using Mendelian randomisation.37, 38 Notably, we also observed evidence indicating a causal role of psoriasis genetic risk in the development of other traits including back pain and generalised pain, fracture risk, diabetes and periodontitis (all GCP>0.63, P<3.2×10−3).
Discussion
This work represents the largest genetic study of psoriasis susceptibility undertaken to date. Relative to previous efforts7, the increased statistical power of our larger meta-analysis sample size has resulted in the identification of 45 new psoriasis susceptibility loci with genome-wide significant evidence of association. A further five risk loci have been observed for the first time at genome-wide significance in populations of European ancestry.
A range of recent and emerging targeted therapies have proven highly effective in psoriasis.1 Many of these exemplify the concordance between a drug’s efficacy and the presence of disease susceptibility variants that disrupt the targeted pathway,17 including biologics targeting IL-23 (encoded by IL23A and IL12B at the 12q13.3 and 5q33.3 psoriasis susceptibility loci, respectively) and deucravacitinib42 a small-molecule TYK2 inhibitor whose development was directly informed by the presence of loss-of-function alleles of TYK2 that confer protection to psoriasis.8 Furthermore, the efficacy of TNF inhibitors has been associated with genetic variants at the 6q23.3 susceptibility locus, where TNFAIP3 encodes A20, an inhibitor of TNF signalling.43 The current study establishes two new psoriasis susceptibility loci harbouring genes encoding therapeutic targets (Supplementary Table 16). We found novel psoriasis susceptibility variants centred on the 5’ untranslated region of IL17RA at chromosome 22q11.1 (lead variant rs917864, OR: 1.08, P=3.9×10−9). The interleukin-17 receptor A subunit encoded by this gene44 is bound with high affinity by brodalumab, a biologic therapy demonstrated to confer effective long-term control of psoriasis.45, 46 Similarly, the aryl hydrocarbon receptor is encoded by AHR, the closest protein-coding gene to the novel 7p21.1 association signal (lead variant rs78233367, OR: 1.06, P=1.3×10−8). Activation of this receptor by the recently approved topical AHR-modulating agent Tapinarof effectively controls psoriasis symptoms.47–49
In addition to AHR, a series of other transcription factors were implicated as putative causal genes by our functional fine mapping approach. We observed examples of newly reported association signals mapping to fundamental processes of cell differentiation, proliferation and trafficking that contribute to established psoriasis pathomechanisms including interferon response, T cell regulation and keratinocyte hyperproliferation. The elongation factor gene ELL, which maps to the newly identified 19p13.11 locus, is predicted by TWAS to be upregulated in blood in the presence of psoriasis-associated alleles. Moreover, by stabilizing RNA polymerase II within the super elongation complex, ELL has been demonstrated to help sustain the expression of the epidermal proliferation genes that are greatly upregulated in psoriasis.50 A deleterious missense variant in POU2F3 (rs7110845, p.His154Arg, P=2.3×10−13), which encodes an epidermal transcription factor critical to keratinocyte differentiation,51 is reported here for the first time. CCAAT enhancer binding transcription factors, implicated in myeloid lineage commitment and regulation of inflammatory cytokines,52–54 are encoded by CEBPB at the known 20q13.13 locus, and CEBPA and CEBPG at the newly reported 19q13.11 locus. The latter gene is predicted by TWAS to be downregulated in sun-exposed and unexposed skin in the presence of psoriasis risk variants and would therefore be consistent with previous reports that C/EBPγ suppresses proinflammatory cytokine activity.55
Transcriptional regulation of interferon responses is further coordinated by genes at a series of psoriasis susceptibility loci. The interferon regulatory factor genes IRF1 and IRF5 are predicted by TWAS to be upregulated in sun-exposed skin and across tissues, respectively, in the presence of psoriasis risk variants at the known 5q31.1 (IRF1) and newly reported 7q32.1 (IRF5) loci. We also identified a susceptibility locus at 20q13.2 that contains NFATC2, encoding a transcription factor that interacts with interferon regulatory factor 4 (encoded by IRF4 at the 6p25.3 locus) to regulate T cell development via enhanced IL-4 expression. Epigenetic regulation of interferon signalling is implicated through the histone acetyltransferase genes KAT5 (11q13.1 locus) and KAT8 (16p11.2), which regulate either directly or indirectly the transcriptional activity of IRF356, 57, and are predicted to be upregulated in skin.
We shed further light on the cellular context of the interferon response in psoriasis through our single-cell expression data. Consistent with its role in in epidermal proliferation50, we found that the ELL gene described above is a TWAS candidate predominantly expressed in keratinocytes (Figure 3). Another cluster of TWAS target genes, including many innate immune genes (e.g., DHX58, STAT2, TYK2) are highly expressed in melanocytes from psoriasis lesions. In addition to producing melanin for photoprotection, melanocytes assume an activated phenotype in psoriasis lesions58 and express functional toll-like receptors TLR2, 3, 4, 7 and 959, enabling innate antibacterial and antiviral immune responses. Melanocytes have also been recently proposed as target cells of the HLA-C*06:02-restricted autoimmune response in psoriasis via the melanocyte autoantigen ADAMTSL5, which could provide a skin-specific target for autoimmune attack.60, 61 More generally, our single-cell expression analysis suggests that psoriasis genetic risk is mediated by a range of cell types beyond the established triad of T cells, dendritic cells and keratinocytes.
We found predicted deleterious protective variants in two genes encoding members of the RIG-I like receptor (RLR) family, linking recognition of viral RNAs to interferon signalling62, 63: DHX58 at chromosome 17q21.2 and RIGI (formerly DDX58) at 9p21.1 (Supplementary Table 7). Prior to this study, STAT3 had been considered the leading functional candidate in 17q21.2 due to its prominent role in IL-23 signal transduction.10 Previous reports have implicated deleterious coding variants in IFIH1, the third member of the RLR family, as conferring protection against psoriasis8, 64 and psoriatic arthritis65; the protective IFIH1 variants rs35667974 and rs1990760 are both highly significant in the current meta-analysis (P=5.0×10−23 and 7.0×10−50, respectively) but not prioritised in credible sets due to insufficient effective sample size and the presence of a more strongly associated intronic variant (rs2111485), respectively. While the RIGI and DHX58 coding variants both show a low posterior probability of being causal (the intronic variant rs11795343 being strongly preferred in RIGI, with posterior probability 0.922), the fact that potentially deleterious coding variants are found to be associated with reduced psoriasis risk in all three known human RLR family members is intriguing. These findings are consistent with reports of rare gain-of-function mutations in RIGI and IFIH1 underlying Singleton-Merton syndrome, in which psoriasis is a clinical feature.66, 67 Much remains to be learned about how the role of RLR family members in driving interferon signalling is affected by psoriasis-associated genetic variation.
This study represents a major advance in our understanding of the genetic basis of psoriasis. The number of documented psoriasis susceptibility signals has approximately doubled, with better refinement of known loci to highlight plausible biological mechanisms through which they influence psoriasis risk. We propose novel disease mechanisms, including a role for the disruption of basic cellular machinery such as transcription and epigenetic modulation in regulating the inflammatory process in psoriasis. For the first time, genetic susceptibility signals are contextualised to specific skin cell types and cytokine signalling pathways. Our data point to the participation in psoriasis pathogenesis of previously underappreciated cell types such as melanocytes. This work will underpin the next era of molecular studies in psoriasis and psoriasis therapeutics.
Methods
Contributing GWAS studies
Genotype data for chronic plaque psoriasis cases and unaffected or population-based controls were compiled for 18 contributing studies, each of which underwent stringent QC at one of eight contributing analysis centres (Supplementary Table 17). Full details are provided in Supplementary Methods. Prior to association testing, inter-dataset duplicated and first- or second-degree related participants were identified using KING (version 2.0)68 by sharing subsets of between 2,502 and 6,864 genotyped markers outside known psoriasis-associated regions. After removing duplicated and related samples, the final sample size was 36,466 cases and 458,078 controls. The cumulative effective sample size was 103,614 (Supplementary Table 17). All participants were of European ancestry. The contributing analysis centres were responsible for genome-wide imputation and association testing (further details in Supplementary Methods).
Meta-analysis
Summary statistics from individual studies were aligned based on GRCh37 positions and alleles; reference alleles were checked for consistency across studies. In many datasets, more than one imputation reference panel was used to maximise imputation quality;9 summary statistics for each variant were preferentially taken from the version with highest imputation quality (INFO or R2) score. Standard error-weighted fixed effects meta-analysis was performed using METAL v2020–05-05.69
Genomic inflation was calculated based on a set of 170,786 LD-independent variants outside of previously established psoriasis susceptibility loci having minor allele frequency > 0.05 (Supplementary Methods).20 LD score regression intercept was calculated using LDSC software with default settings and precomputed LD scores derived from 1000 Genomes data.21
Associated regions and independent signal identification
Associated genomic regions were identified using Genome-wide Complex Trait Analysis conditional and joint analysis (GCTA-COJO).26 To facilitate this, the autosomes were first partitioned into distinct LD blocks (Supplementary Methods). A region (LD block) was considered associated with psoriasis susceptibility if at least one variant tested in three or more contributing studies and with Neff > 10,000 (11,808,957 eligible variants in total) achieved genome-wide significance (P<5×10−8). The LD block that includes the MHC region (chr6, 24.0–36.3Mb) extends beyond the established boundaries of the extended MHC (chr6, 25.7–33.4Mb70); we observe an association outside of the established boundaries that could plausibly be driven by weak LD with the strongly associated MHC variants (Supplementary Figure 1).
For the identification of additional independent association signals within associated regions we used a more stringent subset of variants having Neff > 93,252 (90% of maximum possible), with five non-MHC regions omitted due to lack of variants meeting this sample size threshold. We employed GCTA-COJO with a custom reference panel to determine independently associated lead variants using a stepwise model selection procedure, and to estimate conditional association statistics for each signal in LD blocks with multiple independent signals (Supplementary Methods). We were unable to estimate additional independent signals within the MHC block (Supplementary Methods).
To annotate our genomic regions against psoriasis susceptibility loci previously established at genome-wide significance in European populations or other ancestries, we reviewed recent GWAS meta-analyses7–9, 22–25, 71 and assessed all psoriasis associations in GWAS Catalog (accessed 31 October 2022).72
We used LDSC21 to estimate the total common SNP heritability for psoriasis on the liability scale, assuming a population prevalence of 1.5%. We note that the heritability estimated by LDSC when including the MHC region is to be interpreted with caution due to its complex genetic architecture.21, 73 Variance explained by our genome-wide significant associations was estimated using the Mangrove package in R,74 based on the jointly estimated effect sizes from COJO and with allele frequencies estimated from 1000 Genomes data. Since independent signals were not estimated in the LD block containing the MHC region, only the marginal effect size of the lead SNP was included, which is likely to underestimate the heritability attributable to this region.
Statistical fine-mapping
For each independent association signal, we used the method suggested by Wen and Stephens,75 and implemented in previous work,22 to calculate posterior probabilities (PP) for each variant being causal. Within each LD block, PPs were calculated for variants with Neff > 90% of maximum, using the joint meta-analysis association p-values estimated by GCTA-COJO to account for the presence of multiple association signals. Bayesian 95% credible sets were subsequently constructed by incorporating variants in decreasing PP order until the cumulative PP exceeded 0.95. Since independent signals could not be established for the LD block containing the MHC region, the 95% credible set for the lead signal was based on unconditional association statistics. We did not estimate credible sets for five signals where no variants had Neff > 90% of maximum.
We also assessed improvement in statistical fine-mapping relative to the GWAS datasets available in the previous (2017) psoriasis GWAS meta-analysis.7 To this end, we selected 63 psoriasis susceptibility loci that were genome-wide statistically significant in either the 2017 meta-analysis and/or other published studies of white European ancestry populations and, for comparability, re-computed associations for these loci in the 2017 meta-analysis without the PAGE dataset (which was typed using the Immunochip and therefore lacks full genome-wide coverage). For the comparison, 95% credible sets were calculated in both the current and (re-computed) 2017 meta-analysis within windows of 200kb around the 2017 lead markers, based on unconditional association statistics; credible set construction was restricted to variants well-imputed for a Neff > 90% of the maximum possible meta-analysis sample size.
Prioritisation of causal variants
For variants identified in 95% credible sets (5,344 distinct variants), we assessed the potential contribution to disease risk via altered protein-coding or regulation. Variants with predicted protein-altering consequence were identified using the Variant Effect Predictor,76 along with associated Combined Annotation Dependent Depletion (CADD) scores.77 To identify regulatory variants likely to influence psoriasis risk we calculated generic and tissue-specific regulatory prediction scores with the RegulomeDB-based method TURF.27 The TURF analyses used default settings and reference data, and excluded indels (5,102 SNPs in total). For each independent association signal, we checked for prioritised regulatory variants, which we defined as variants with: (i) generic regulatory probability >0.7, (ii) at least 50% share of generic regulatory probability among variants within their credible set, and (iii) no other credible set variant having generic regulatory probability >0.5. We assessed blood- and skin-specific regulatory probabilities for prioritised variants and compared these to 49 other tissues with tissue-specific regulatory probabilities.
Transcriptome-wide association study
Predictions of differential gene expression in the presence of psoriasis-associated genetic variation were generated using the S-PrediXcan method78 implemented in the Complex Trait Genetics Virtual Lab (CTG-VL).79 We predicted expression differences in whole blood (n = 6,275 genes with prediction models available), sun-exposed skin (n = 9,085) and sun-unexposed skin (n = 7,617) using GTEx v7 reference data.80 We employed a Bonferroni-adjusted transcriptome-wide significance threshold of 2.18×10−6 (22,977 tests performed) to assess differential expression associated with psoriasis.
To establish the physical location of psoriasis-associated TWAS genes relative to susceptibility signals, we considered all protein-coding and long non-coding RNA genes from Ensembl (GRCh37 version 109).81 Genes were allocated to psoriasis-associated genomic regions based on having transcription start site (TSS) within the corresponding LD block, or within 1Mb of the lead variant. Gene distances in each region were compared based on the distance from TSS to the lead psoriasis-associated variant. Significant TWAS genes outside of psoriasis-associated genomic regions were mapped based on distance to the variant within 1Mb having lowest meta-analysis p-value (all psoriasis-suggestive variants, Pmeta<10−5).
Single-cell functional analyses
To delineate the specific skin cell types underlying psoriasis-associated TWAS genes, we studied their expression profiles using scRNA-seq data that we recently reported.82 For each TWAS gene, we computed the average expression in each of 12 cell types derived from non-lesional or lesional skin of psoriasis cases (24 biopsy type/cell type combinations in total; nnon-lesional=11, nlesional=14), and subsequently calculated the standardized expression across cell types for each gene. We grouped genes with consistent expression profiles and identified five clusters using hierarchical clustering.
We also investigated the overlap between psoriasis-associated TWAS genes and transcriptomic changes induced by psoriasis-linked cytokines. Specifically, gene expression profiles were generated for keratinocytes from 50 healthy adult donors before and after stimulation with a range of cytokines (IL-4, IL-13, IFN-α, IFN-γ, TNF-α, and IL-17A), as described previously.36, 83 We compared and tested for statistical enrichment (hypergeometric test) our list of non-MHC TWAS genes against the top 90 most statistically significant upregulated genes in each keratinocyte cytokine challenge.
Biological pathway analysis
We used DEPICT v1.1 to identify sets of genes with coordinated function in which genes from psoriasis susceptibility loci are highly expressed.34 As input we used a list of 214 independent and genome-wide significant variants derived from the full meta-analysis summary statistics via distance- and LD-based clumping (r2>0.05 within 2Mb windows) in PLINK84 based on 1000 Genomes European samples,85 except for the extended HLA region where only the lead SNP was included. For comparison purposes, we repeated the analysis using a subset of 158 variants within 1Mb of previously established psoriasis susceptibility loci. In each case, functional gene sets were considered significantly enriched at a false discovery rate (FDR) of <0.05 based on 14,462 gene sets tested.
Correlation and causation analyses
Genetic correlations86 were estimated in CTG-VL using summary statistics for an initial set of 1,376 traits (CTG-VL default list), a majority of which were derived from UK Biobank by the Neale lab (http://www.nealelab.is/uk-biobank). We excluded traits that were too general or non-specific, represented follow-up questions for specific groups (e.g., details of presentation for severe mental illnesses) or detailed lifestyle questions (e.g., specifics of diet or occupation), psoriasis and duplicated or otherwise difficult-to-interpret traits, resulting in 592 traits categorised into disease and health (n=345), physical and functional measures (n=210) or lifestyle and quality of life (n=37) (Supplementary Table 14). Significant genetic correlation was identified among these traits based on a Bonferroni-adjusted p-value threshold of 8.45×10−5 (592 tests performed), and we checked that all significant traits had heritability z-score >4.
Partial genetic causality between psoriasis and a range of other traits was estimated using a latent causal variable (LCV) model41 implemented in CTG-VL. Of the 592 traits for which genetic correlation was assessed (above), LCV results were available for 585. We focused on 108 traits with significant genetic correlation at FDR<0.05. Traits with evidence for a causal relationship were subsequently identified based on a significant genetic causality proportion (FDR<0.05 among 108 traits).
Supplementary Material
Acknowledgements
The BSTOP steering committee comprises: Professor David Burden (Chair), Professor Catherine Smith, Professor Jonathan Barker, Professor Sara Brown, Dr Nick Dand, Dr Satveer Mahil, Helen McAteer, Dr Julia Schofield and Professor Stefan Siebert. The Estonian Biobank research team comprises: Tõnu Esko, Andres Metspalu, Lili Milani, Reedik Mägi and Mari Nelis.
This research has been conducted using the UK Biobank Resource (approved project 15147). It uses data provided by patients and collected by the NHS as part of their care and support.
We gratefully acknowledge the contributions of patients and family members, other research participants and clinical staff involved in recruitment.
Funding
ND is funded by Health Data Research UK (MR/S003126/1). Support for the study was received from the Department of Health through the National Institute for Health and Care Research (NIHR) Bio-Resource Clinical Research Facility and comprehensive Biomedical Research Centre awards to Guy’s and St Thomas’ National Health Service Foundation Trust in partnership with King’s College London and King’s College Hospital National Health Service Foundation Trust (BRC_1215_20006). We acknowledge support from the Psoriasis Association in relation to Biomarkers of Systemic Treatment Outcomes in Psoriasis (RG2/10, RG2/10) and an award to PDM (ST1/19) and FC (ST3/20). This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement number 821511 (Biomarkers in Atopic Dermatitis and Psoriasis). The JU receives support from the European Union’s Horizon 2020 research and innovation programme and the European Federation of Pharmaceutical Industries and Associations (EFPIA). This publication reflects only the authors’ views and the JU is not responsible for any use that may be made of the information it contains. SKM is funded by a NIHR Advanced Fellowship (NIHR302258). This study presents independent research supported by NIHR BioResource Centre Maudsley, NIHR Maudsley Biomedical Research Centre (BRC) at South London and Maudsley NHS Foundation Trust and Institute of Psychiatry, Psychology and Neuroscience (IoPPN), King’s College London. The BioResource gratefully acknowledge capital equipment funding from the Maudsley Charity (Grant Ref. 980) and Guy’s and St Thomas’s Charity (STR130505). PDM reports a Translational Research Grant (814364) from the National Psoriasis Foundation. LCT received funding from the National Institutes of Health (NIH) (K01 AR072129, P30 AR075043, UC2 AR081033). This work was supported by awards from NIH (R01AR042742, R01AR050511, R01AR054966, R01AR063611, R01AR065183), the National Psoriasis Foundation, and the Babcock Memorial Trust. JTE is supported by the Ann Arbor Veterans Hospital. MTP is supported by a Research Career Development Award from the Dermatology Foundation. XW is supported by NIH (R01ES033634, R35GM138121). MKS reports a Translational Research Grant from the National Psoriasis Foundation. ACB was supported by the University of Michigan A. Alfred Taubman Medical Research Institute via Taubman Emerging Scholar funds and the NIH (K08 AR078251, P30 AR075043). JEG is supported by NIH (P30AR075043) and the Taubman Medical Research Institute. Part of this study was funded by a grant to AR and UH from the Bundesministerium für Bildung und Forschung (BMBF Metarthros 01EC1407A), by a grant to UH from the German Research Foundation (CRC1181–2, project A05) and by a grant to HB and FB from the Bundesministerium für Bildung und Forschung (BMBF ArthroMark 01EC1401C). The HNR study (Erlangen cohort) is supported by the Heinz Nixdorf Foundation (Germany). Additionally, the study is funded by the German Ministry of Education and Science and the German Research Council (DFG; Project SI 236/8–1, SI236/9–1, ER 155/6–1). The genotyping of the Illumina HumanOmni-1 Quad BeadChips of the HNR subjects was financed by the German Centre for Neurodegenerative Disorders (DZNE), Bonn. T.Traks and KK report support from the Estonian Research Council (PUT1465, PRG1189). SK is supported by MSWA, The Michael J. Fox Foundation, Shake It Up Australia, and Perron Institute for Neurological and Translational Science. MT-L was supported by the European Union through the European Regional Development Fund (Project No. 2014–2020.4.01.15–0012). RM was supported by Estonian Research Council grant PRG1911. TE was supported by Estonian Research Council grant PRG1291. The Trøndelag Health Study (The HUNT Study) is a collaboration between HUNT Research Center (Faculty of Medicine and Health Sciences, NTNU - Norwegian University of Science and Technology), Trøndelag County Council, Central Norway Regional Health Authority, and the Norwegian Institute of Public Health. The genotyping in HUNT was financed by the National Institutes of Health; University of Michigan; the Research Council of Norway; the Liaison Committee for Education, Research, and Innovation in Central Norway; and the Joint Research Committee between St Olavs hospital and the Faculty of Medicine and Health Sciences, NTNU. LT, KH and ML work in a research unit funded by Stiftelsen Kristian Gerhard Jebsen; Faculty of Medicine and Health Sciences, NTNU - Norwegian University of Science and Technology; The Liaison Committee for Education, Research and Innovation in Central Norway; the Joint Research Committee between St. Olavs Hospital (Trondheim, Norway) and the Faculty of Medicine and Health Sciences, NTNU - Norwegian University of Science and Technology. ML is supported by grants from the Liaison Committee for Education, Research and Innovation in Central Norway and the Joint Research Committee between St Olav’s hospital and the Faculty of Medicine and Health Sciences, NTNU. The study received support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Cluster of Excellence 2167 “Precision Medicine in Chronic Inflammation (PMI) (EXC 2167–390884018). This work was supported by a grant from Versus Arthritis (21754). This research was co-funded by the NIHR Manchester Biomedical Research Centre (NIHR203308). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. DJ acknowledges that his research was supported by Cambridge Arthritis Research Endeavour (CARE) and the NIHR Cambridge Biomedical Research Centre (BRC-1215–20014). WL acknowledges funding from NIH (R01AR065174, U01AI119125). The UCSF-MS DNA biorepository is supported by RG-1611–26299 from the National Multiple Sclerosis Society to JRO. VC is supported by a Pfizer Chair Research Award, Rheumatology, University of Toronto. The Schroeder Arthritis Institute Psoriatic Disease Program is supported by the Krembil Foundation.
Footnotes
Conflicts of Interest
FC reports grants and consultancy fees from Boehringer Ingelheim. SKM reports departmental income from Abbvie, Almirall, Eli Lilly, Janssen, Leo Pharma, Novartis, Pfizer, Sanofi and UCB, outside the submitted work. MJN has received consultancy fees and/or research funding from Abbvie, Amgen, Celgene, Eli Lilly, Janssen, Pfizer, Novartis and UCB. T.Tejasvi is a member of an advisory board for L’Oreal Teledermatology. VC has received research grants from AbbVie, Amgen, and Eli Lilly and has received honoraria for advisory board member roles from AbbVie, Amgen, BMS, Eli Lilly, Janssen, Novartis, Pfizer, and UCB. His spouse is an employee of AstraZeneca. JEG received research support from Eli Lilly, Kyowa Kirin, Janssen, Almirall, Celgene/BMS, Prometheus, Novartis, Galderma and AnaptysBio, and is a member of an advisory board for Novartis, AbbVie, Eli Lilly, Almirall, Galderma, Boehringer Ingelehim, Celgene/BMS, Sanofi, Janssen and AnaptysBio. SK is a founder of Prion OÜ, Geneto OÜ, Sportsgene OÜ and Genomic Therapeutics Pty Ltd. WL has received research grant funding from Abbvie, Amgen, Janssen, Leo, Novartis, Pfizer, Regeneron, and TRex Bio. PDM reports consultancy fees from Unilever and speaker’s fees from Sanofi and BMS. LCT reports support from Janssen, Galderma, and Novartis. The remaining authors report no conflicts of interest.
References
- 1.Griffiths C. E. M., Armstrong A. W., Gudjonsson J. E. & Barker J. N. Psoriasis W. N.. Lancet 397, 1301–1315 (2021). [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Global report on psoriasis. (World Health Organization, 2016). https://apps.who.int/iris/handle/10665/204417. [Google Scholar]
- 3.Parisi R. et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ 369, m1590 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villacorta R. et al. A multinational assessment of work-related productivity loss and indirect costs from a survey of patients with psoriasis. Br. J. Dermatol. 183, 548–558 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brezinski E. A., Dhillon J. S. & Armstrong A. W. Economic Burden of Psoriasis in the United States: A Systematic Review. JAMA Dermatol. 151, 651–658 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Grjibovski A. M., Olsen A. O., Magnus P. & Harris J. R. Psoriasis in Norwegian twins: contribution of genetic and environmental effects. J. Eur. Acad. Dermatol. Venereol. 21, 1337–1343 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Tsoi L. C. et al. Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat. Commun. 8, 15382 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dand N. et al. Exome-wide association study reveals novel psoriasis susceptibility locus at TNFSF15 and rare protective alleles in genes contributing to type I IFN signalling. Hum. Mol. Genet. 26, 4301–4313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patrick M. T. et al. Genetic signature to provide robust risk assessment of psoriatic arthritis development in psoriasis patients. Nat. Commun. 9, 4178–6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stuart P. E., Tsoi L. C., Hambro C. A. & Elder J. T. Genetics of psoriasis. In Oxford Textbook of Psoriatic Arthritis (eds FitzGerald O. & Gladman D.) (Oxford University Press, 2018). [Google Scholar]
- 11.Nair R. P. et al. Psoriasis bench to bedside: genetics meets immunology. Arch. Dermatol. 145, 462–464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahil S. K., Capon F. & Barker J. N. Genetics of psoriasis. Dermatol. Clin. 33, 1–11 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Gudjonsson J. E. & Elder J. T. Psoriasis. In Fitzpatrick’s Dermatology, 9e (ed Kang S. et al.) (McGraw-Hill Education, New York, NY, 2019). [Google Scholar]
- 14.Zhou W. et al. Global Biobank Meta-analysis Initiative: Powering genetic discovery across human disease. Cell. Genom. 2, 100192 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Doval I. et al. Risk of serious adverse events associated with biologic and nonbiologic psoriasis systemic therapy: patients ineligible vs eligible for randomized controlled trials. Arch. Dermatol. 148, 463–470 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Yiu Z. Z. N. et al. Drug Survival Associated With Effectiveness and Safety of Treatment With Guselkumab, Ixekizumab, Secukinumab, Ustekinumab, and Adalimumab in Patients With Psoriasis. JAMA Dermatol. 158, 1131–1141 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson M. R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Roseno N. A. L., Lorup E. H., Richardson C., Alarcon I. & Egeberg A. Exploring disease comorbidities and temporal disease progression of psoriasis: an observational, retrospective, multi-database, cohort study. Br. J. Dermatol. 188, 372–379 (2023). [DOI] [PubMed] [Google Scholar]
- 19.Armstrong A. W. & Read C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. JAMA 323, 1945–1960 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Devlin B., Roeder K. & Wasserman L. Genomic control, a new approach to genetic-based association studies. Theor. Popul. Biol. 60, 155–166 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Bulik-Sullivan B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stuart P. E. et al. Transethnic analysis of psoriasis susceptibility in South Asians and Europeans enhances fine-mapping in the MHC and genomewide. HGG Adv. 3, 10.1016/j.xhgg.2021.100069. Epub 2021 Nov 6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin X. et al. Genome-wide meta-analysis identifies multiple novel associations and ethnic heterogeneity of psoriasis susceptibility. Nat. Commun. 6, 6916 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuo X. et al. Whole-exome SNP array identifies 15 new susceptibility loci for psoriasis. Nat. Commun. 6, 6793 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang C. et al. Exome-Wide Rare Loss-of-Function Variant Enrichment Study of 21,347 Han Chinese Individuals Identifies Four Susceptibility Genes for Psoriasis. J. Invest. Dermatol. 140, 799–805.e1 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Yang J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong S. & Boyle A. P. Prioritization of regulatory variants with tissue-specific function in the non-coding regions of human genome. Nucleic Acids Res. 50, e6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellinghaus E. et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 42, 991–995 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert S., Swindell W. R., Tsoi L. C., Stoll S. W. & Elder J. T. Dual Role of Act1 in Keratinocyte Differentiation and Host Defense: TRAF3IP2 Silencing Alters Keratinocyte Differentiation and Inhibits IL-17 Responses. J. Invest. Dermatol. 137, 1501–1511 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lambert S. et al. Neutrophil Extracellular Traps Induce Human Th17 Cells: Effect of Psoriasis-Associated TRAF3IP2 Genotype. J. Invest. Dermatol. 139, 1245–1253 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B. & Ritchie M. D. From GWAS to Gene: Transcriptome-Wide Association Studies and Other Methods to Functionally Understand GWAS Discoveries. Front. Genet. 12, 713230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Graham R. R. et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc. Natl. Acad. Sci. U. S. A. 104, 6758–6763 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niewold T. B. et al. IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic lupus erythematosus. Ann. Rheum. Dis. 71, 463–468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pers T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun. 6, 5890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang R., Langdon W. Y. & Zhang J. Negative regulation of receptor tyrosine kinases by ubiquitination: Key roles of the Cbl family of E3 ubiquitin ligases. Front. Endocrinol. (Lausanne) 13, 971162 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsoi L. C. et al. Cytokine responses in nonlesional psoriatic skin as clinical predictor to anti-TNF agents. J. Allergy Clin. Immunol. 149, 640–649.e5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogawa K. et al. A Transethnic Mendelian Randomization Study Identifies Causality of Obesity on Risk of Psoriasis. J. Invest. Dermatol. 139, 1397–1400 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Budu-Aggrey A. et al. Evidence of a causal relationship between body mass index and psoriasis: A mendelian randomization study. PLoS Med. 16, e1002739 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patrick M. T. et al. Shared genetic risk factors and causal association between psoriasis and coronary artery disease. Nat. Commun. 13, 6565–4 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei J. et al. Alcohol consumption and smoking in relation to psoriasis: a Mendelian randomization study. Br. J. Dermatol. 187, 684–691 (2022). [DOI] [PubMed] [Google Scholar]
- 41.O’Connor L. J. & Price A. L. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat. Genet. 50, 1728–1734 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papp K. et al. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 379, 1313–1321 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Tejasvi T. et al. TNFAIP3 gene polymorphisms are associated with response to TNF blockade in psoriasis. J. Invest. Dermatol. 132, 593–600 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chung S., Ye X. & Iwakura Y. Interleukin-17 family members in health and disease. Int. Immunol. 33, 723–729 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Papp K. A. et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 366, 1181–1189 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Papp K. et al. Long-term efficacy and safety of brodalumab in psoriasis through 120 weeks and after withdrawal and retreatment: subgroup analysis of a randomized phase III trial (AMAGINE-1). Br. J. Dermatol. 183, 1037–1048 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Meglio P. et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity 40, 989–1001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith S. H. et al. Tapinarof Is a Natural AhR Agonist that Resolves Skin Inflammation in Mice and Humans. J. Invest. Dermatol. 137, 2110–2119 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Lebwohl M. G. et al. Phase 3 Trials of Tapinarof Cream for Plaque Psoriasis. N. Engl. J. Med. 385, 2219–2229 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Li J., Bansal V., Tiwari M., Chen Y. & Sen G. L. ELL Facilitates RNA Polymerase II-Mediated Transcription of Human Epidermal Proliferation Genes. J. Invest. Dermatol. 141, 1352–1356.e3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neumann C., Bigliardi-Qi M., Widmann C. & Bigliardi P. L. The delta-opioid receptor affects epidermal homeostasis via ERK-dependent inhibition of transcription factor POU2F3. J. Invest. Dermatol. 135, 471–480 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pundhir S. et al. Enhancer and Transcription Factor Dynamics during Myeloid Differentiation Reveal an Early Differentiation Block in Cebpa null Progenitors. Cell. Rep. 23, 2744–2757 (2018). [DOI] [PubMed] [Google Scholar]
- 53.Nerlov C. The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 17, 318–324 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Roy S. K., Wachira S. J., Weihua X., Hu J. & Kalvakolanu D. V. CCAAT/enhancer-binding protein-beta regulates interferon-induced transcription through a novel element. J. Biol. Chem. 275, 12626–12632 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Huggins C. J. et al. C/EBPgamma suppresses senescence and inflammatory gene expression by heterodimerizing with C/EBPbeta. Mol. Cell. Biol. 33, 3242–3258 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma G. et al. Histone acetyl transferase TIP60 inhibits the replication of influenza a virus by activation the TBK1-IRF3 pathway. Virol. J. 15, 172–3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huai W. et al. KAT8 selectively inhibits antiviral immunity by acetylating IRF3. J. Exp. Med. 216, 772–785 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abdel-Naser M. B. et al. Increased Activity and Number of Epidermal Melanocytes in Lesional Psoriatic Skin. Dermatology 232, 425–430 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Yu N. et al. Cultured human melanocytes express functional toll-like receptors 2–4, 7 and 9. J. Dermatol. Sci. 56, 113–120 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Arakawa A. et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J. Exp. Med. 212, 2203–2212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prinz J. C. Melanocytes: Target Cells of an HLA-C*06:02-Restricted Autoimmune Response in Psoriasis. J. Invest. Dermatol. 137, 2053–2058 (2017). [DOI] [PubMed] [Google Scholar]
- 62.Rehwinkel J. & Gack M. U. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat. Rev. Immunol. 20, 537–551 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Onomoto K., Onoguchi K. & Yoneyama M. Regulation of RIG-I-like receptor-mediated signaling: interaction between host and viral factors. Cell. Mol. Immunol. 18, 539–555 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li Y. et al. Carriers of rare missense variants in IFIH1 are protected from psoriasis. J. Invest. Dermatol. 130, 2768–2772 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Budu-Aggrey A. et al. A rare coding allele in IFIH1 is protective for psoriatic arthritis. Ann. Rheum. Dis. 76, 1321–1324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jang M. et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet. 96, 266–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rutsch F. et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am. J. Hum. Genet. 96, 275–282 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Manichaikul A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willer C. J., Li Y. & Abecasis G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horton R. et al. Gene map of the extended human MHC. Nat. Rev. Genet. 5, 889–899 (2004). [DOI] [PubMed] [Google Scholar]
- 71.Tsoi L. C. et al. Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci. Nat. Commun. 6, 7001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Buniello A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Finucane H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jostins L., Levine A. P. & Barrett J. C. Using genetic prediction from known complex disease Loci to guide the design of next-generation sequencing experiments. PLoS One 8, e76328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wen X. & Stephens M. Bayesian Methods for Genetic Association Analysis with Heterogeneous Subgroups: from Meta-Analyses to Gene-Environment Interactions. Ann. Appl. Stat. 8, 176–203 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McLaren W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122–4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kircher M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barbeira A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825–1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gabriel Cuellar-Partida et al. Complex-Traits Genetics Virtual Lab: A community-driven web platform for post-GWAS analyses. bioRxiv(Cold Spring Harbor Laboratory Press, 2019). https://search.proquest.com/docview/2178837824. [Google Scholar]
- 80.Consortium GTEx et al. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cunningham F. et al. Ensembl 2022. Nucleic Acids Res. 50, D988–D995 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma F. et al. Single cell and spatial sequencing define processes by which keratinocytes and fibroblasts amplify inflammatory responses in psoriasis. Nat. Commun. 14, 3455–4 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsoi L. C. et al. Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J. Invest. Dermatol. 139, 1480–1489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chang C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7–8. eCollection 2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bulik-Sullivan B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.