

Abstract
Objectives:
Recent translational scientific efforts in subglottic stenosis (SGS) support a disease model where epithelial alterations facilitate microbiome displacement, dysregulated immune activation, and localized fibrosis. Yet despite recent advances, the genetic basis of SGS remains poorly understood. We sought to identify candidate risk genes associated with an SGS phenotype, investigate their biological function, and identify the cell types enriched for their expression.
Methods:
The Online Mendelian Inheritance in Man (OMIM) database was queried for single gene variants associated with an SGS phenotype. The functional intersections and molecular roles of the identified genes were explored using pathway enrichment analysis (PEA) computational methods. Cellular localization of the candidate risk genes was measured via transcriptional quantification in an established single cell RNA sequencing (scRNA-seq) atlas of the proximal airway.
Results:
20 genes associated with SGS phenotype were identified. PEA resulted in 24 significantly enriched terms including “cellular response to TGF-ß,” “epithelial-to-mesenchymal transition,” and “adherens junctions.” Mapping the 20 candidate risk genes to the scRNA-seq atlas found 3 (15%) genes were enriched in epithelial cells, 3 (15%) in fibroblasts, and 3 (15%) in endothelial cells. 11 (55%) genes were expressed ubiquitously among tissue types. Interestingly, immune cells were not significantly enriched for candidate risk genes.
Conclusion:
We identify and provide biologic context for 20 genes associated with fibrotic disease of the proximal airway and form the foundation for future detailed genetic study.
Keywords: Idiopathic subglottic stenosis, laryngotracheal stenosis, tracheal stenosis, iSGS, epithelial-to-mesenchymal transition
LAY SUMMARY
Subglottic stenosis (SGS) is a narrowing of the airway that can occur spontaneously or after injury. Patients with SGS experience significant breathing problems that affect their quality of life. We used genetic analysis tools to better understand the biology of SGS.
INTRODUCTION
Subglottic stenosis (SGS) can occur after iatrogenic injury,1 as a manifestation of collagen vascular disease,2 or without an identifiable associated disease process (termed idiopathic SGS: iSGS).3 Given the significant emotional, physical, and financial costs associated with recurrent airway obstruction,4 focused scientific approaches have been employed to identify key elements of SGS disease pathophysiology. Over the last decade, these efforts have made inroads in understanding the biological basis of SGS. The establishment of rare patient cohorts has facilitated linkage of tissue samples with clinical outcomes data.5 The creation of animal models and in vitro systems has also facilitated the delineation of key pathways involved in SGS disease pathogenesis.6 These results support a model where epithelial alterations facilitate microbiome displacement,7,8 dysregulated immune activation,9,10 and localized fibrosis (Figure 1).11–13
Figure 1.
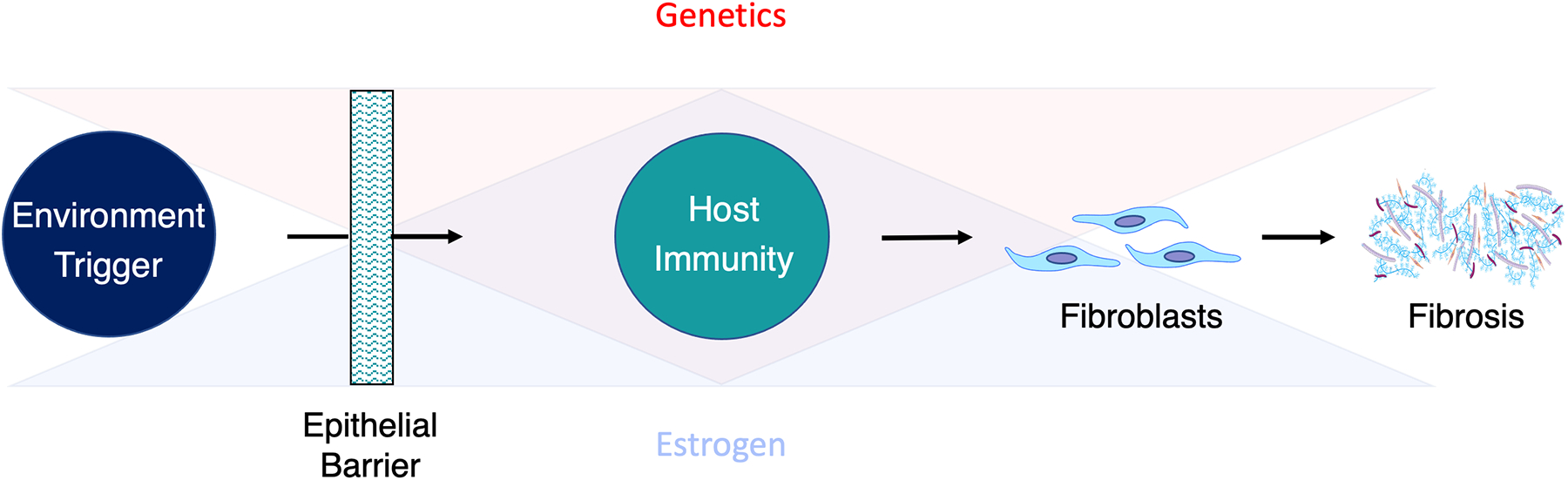
North American Airway Collaborative (NoAAC) model of proximal airway disease pathogenesis. Epithelial dysfunction facilitates displacement of antigenic triggers across the mucosal barrier. Inappropriate host immune responses drive inflammation resulting in pathologic fibroblast activation and subsequent extracellular matrix deposition. Genetics and estrogen may each influence barrier function, immune activation, or fibroblast phenotype in the proximal airway mucosa.
Despite this progress, the genetic basis of SGS still represents a major knowledge gap.14 This gap is particularly apparent for iSGS, where the tight demographics of affected patients would seem to support a genetic basis for disease.3,15 Yet natural history studies in iSGS suggest a rate of familial aggregation of only 2.5%, arguing against strict Mendelian inheritance.16 Similarly, prior genetic analyses of iatrogenic post-intubation stenosis patients (investigating a limited number of candidate genes) did not reveal any clear associations with disease risk.17,18 While published studies have demonstrated a strong association between defined genetic polymorphisms and overall disease risk of granulomatosis with polyangiitis (GPA), the larger GPA population has not been interrogated for genetic variants specifically conferring SGS risk.19,20
Fortunately, the confluence of emerging open-source genetic data repositories, gene set enrichment algorithms, and single cell transcriptional cellular atlases provide an opportunity for rapid acceleration of our knowledge of the genetic basis of proximal airway disease. The Online Mendelian Inheritance in Man (OMIM) database is a free, comprehensive, authoritative compendium of genetic research. Harnessing its well-annotated linkage between phenotype and genotype, we queried OMIM for all genes associated with an SGS phenotype. After identifying 20 candidate genes, we utilized pathway enrichment analysis21 and cellular localization on an established proximal airway single cell RNA sequencing (scRNA-seq) atlas7 to define candidate risk genes for proximal airway stenosis and provide biologic context for the identified genetic risk. The results of this study allow insight into SGS biology and most critically, provide a direction for future genetic discovery.
MATERIALS AND METHODS
OMIM Database & Literature Search
The OMIM database was queried for the following terms: subglottic stenosis; tracheal stenosis; laryngotracheal stenosis. A resulting list of 28 phenotype-gene relationships was concatenated and cleared of duplicates, yielding a final list of 20 candidate risk genes.22 Each gene was validated by confirming an association with an SGS phenotype via review of the primary literature.
Pathway Enrichment Analysis
We utilized functional pathway enrichment analysis to explore and interpret the interactome of the candidate risk gene set. Broadly, pathway enrichment analysis is a bioinformatics technique to identify shared properties or interactions which may exist among a given set of genes. Pathways themselves are typically defined as models describing the series of mechanistic interactions of genes, proteins, metabolites, and signaling molecules that lead to a certain product or change in a cell or tissue.21
The widely used g:Profiler pipeline was employed for the input list of 20 genes.23 g:Profiler links genes to established biological databases and detects biological pathways implicated in the experimental gene list that are statistically significantly enriched, or over-represented, relative to what is expected by chance. It includes pathways from the Gene Ontology (GO) consortium and its domains, biological process (GO:BP) and molecular function (GO:MF). g:Profiler also includes genetic information from the Kyoto Encyclopedia of Genes and Genomes (KEGG), as well as a standardized vocabulary of human disease phenotypes from Human Phenotype Ontology (HP). Altogether, this bioinformatics approach facilitates useful mechanistic insight into large-scale genomics data. Pathway enrichment analysis was performed with g:Profiler’s g:GOSt tool as previously described.21
Idiopathic Pulmonary Fibrosis Cellular Atlas
In order to validate our computational approach designed to localize candidate risk gene expression to specific cell types, we first analyzed existing data from a published scRNA-seq atlas24 of idiopathic pulmonary fibrosis (IPF). IPF is a fibrotic disease bearing mechanistic similarity to iSGS.25,26 Decades of basic research have strongly implicated epithelial integrity and telomerase mutations in IPF disease pathogenesis, with large-scale genome-wide association studies (GWAS) that have rigorously identified genetic loci associated with IPF disease risk.27 Habermann et al. used the 10x Genomics Chromium scRNA-seq platform and Seurat28,29 R statistical package to generate an IPF cell atlas based on 114,396 single cells from 12 explanted lungs with IPF, 8 fibrotic lungs of other etiologies, and 10 nonfibrotic controls.24
North American Airway Collaborative (NoAAC) Idiopathic Subglottic Stenosis Cell Atlas
Using an analogous approach to the above-mentioned work in IPF, we determined the distribution and phenotype of cellular populations present in iSGS airway scar by analyzing our previously established scRNA-seq dataset7 containing 34,000 cells from 7 iSGS patients and 3 matched unaffected controls. scRNA-seq was performed using the 10x Genomics Chromium platform, with data integration via Harmony,30 and analysis with Seurat.28,29 Unsupervised clustering analysis classified the tissue type of each cluster (immune, epithelial, endothelial, fibroblast/mesenchyme) based on PanglaoDB.31
Mapping and gene localization
We adapted published methods to define the tissue-specificity of SGS candidate risk genes.32–34 The expression levels of candidate risk genes were determined from the average cell expression in the NoAAC scRNA-seq cell atlas. Genes were defined as enriched in a specific cell type if their expression in that cell type exceeded all others by >½ a standard deviation (34.1%). Genes without a cell type fulfilling enrichment criteria were categorized as ubiquitous.
RESULTS
OMIM Database & Literature Search
Twenty genes associated with SGS phenotype were identified (Table 1). Primary literature validation of each genotype-phenotype pair showed virtually all consisting of rare multi-organ system syndromes, with SGS as an observed feature. Phenotypic inheritance patterns were 10 (50%) autosomal dominant, 8 (40%) autosomal recessive, and 2 (10%) X-linked.
Table 1.
Twenty candidate risk genes for stenosis of the proximal airway. Results of Online Mendelian Inheritance in Man (OMIM) database search for following phenotypic terms: “subglottic stenosis,” “tracheal stenosis,” and “laryngotracheal stenosis.”
| Gene | Gene/Locus Name | MIM ID | Location | Phenotype | Inheritance | PMID |
|---|---|---|---|---|---|---|
| AFF4 | AF4/FMR2 family, member 4 | 604417 | 5q31.1 | CHOPS syndrome | AD | 31058441 |
| ELN | Elastin | 130160 | 7q11.23 | Cutis laxa, autosomal dominant | AD | 9873040 |
| FGFR1 | Fibroblast growth factor receptor-1 | 136350 | 8p11.23 | Pfeiffer syndrome | AD | 2208766 |
| FGFR2 | Fibroblast growth factor receptor-2 | 176943 | 10q26.13 | Apert syndrome | AD | 1519659 |
| FLNB | Filamin B | 603381 | 3p14.3 | Larsen syndrome | AD | 9185735 |
| GMNN | Geminin DNA replication inhibitor | 602842 | 6p22.3 | Meier-Gorlin syndrome 6 | AD | 26637980 |
| LTBP3 | Latent TGFB binding protein-3 | 602090 | 11q13.1 | Geleophysic dysplasia 3 | AD | 27068007 |
| MAP3K7 | MAPK kinase 7 | 602614 | 6q15 | Frontometaphyseal dysplasia 2 | AD | 28498505 |
| SMAD4 | SMAD family member 4 | 600993 | 18q21.2 | Myhre syndrome | AD | 31539271 |
| TBX3 | T-box 3 | 601621 | 12q24.21 | Ulnar-mammary syndrome | AD | 3430557 |
| ZEB2 | Zinc finger E box-binding homeobox 2 | 605802 | 2q22.3 | Mowat-Wilson syndrome | AD | 17567886 |
| ADAMTSL2 | ADAMTS-like protein 2 | 612277 | 9q34.2 | Geleophysic dysplasia 1 | AR | 33082559 |
| EFL1 | Elongation factor-like GTPase 1 | 617538 | 15q25.2 | Shwachman-Diamond syndrome 2 | AR | 28331068 |
| HYLS1 | HYLS1 gene | 610693 | 11q24.2 | Hydrolethalus syndrome | AR | 7028327 |
| IDUA | Iduronidase, alpha-L | 252800 | 4p16.3 | Mucopolysaccharidosis Ih | AR | 24767144 |
| PISD | Phosphatidylserine decarboxylase | 612770 | 22q12.2 | Liberfarb syndrome | AR | 30858161 |
| SNIP1 | SMAD nuclear interacting protein 1 | 608241 | 1p34.3 | Craniofacial dysmorphism | AR | 34570759 |
| TONSL | Tonsoku-like DNA repair protein | 604546 | 8q24.3 | Spondyloepimetaphyseal dysplasia | AR | 30773277 |
| EBP | Emopamil-binding protein | 300205 | Xp11.23 | Chondrodysplasia punctata | XLD | 21634086 |
| FLNA | Filamin A, alpha | 300017 | Xq28 | Melnick-Needles syndrome | XLD | 16835913 |
Pathway Enrichment Analysis
Pathway enrichment analysis of the 20 input genes resulted in 24 statistically enriched terms (Figure 2A). The top 3 most significantly enriched GO:BP terms (after collapsing duplicates) were “cellular response to TGF-ß,” “epithelial-to-mesenchymal transition” (EMT), and cardiac “outflow tract morphogenesis.” The single GO:MF term result was “fibroblast growth factor receptor activity.” The top 3 KEGG term results were “signaling pathways regulating pluripotency of stem cells,” “MAPK signaling pathway,” and “adherens junction” (Figure 2B). For a complete list of enriched pathways, see Supplemental Table 1.
Figure 2.
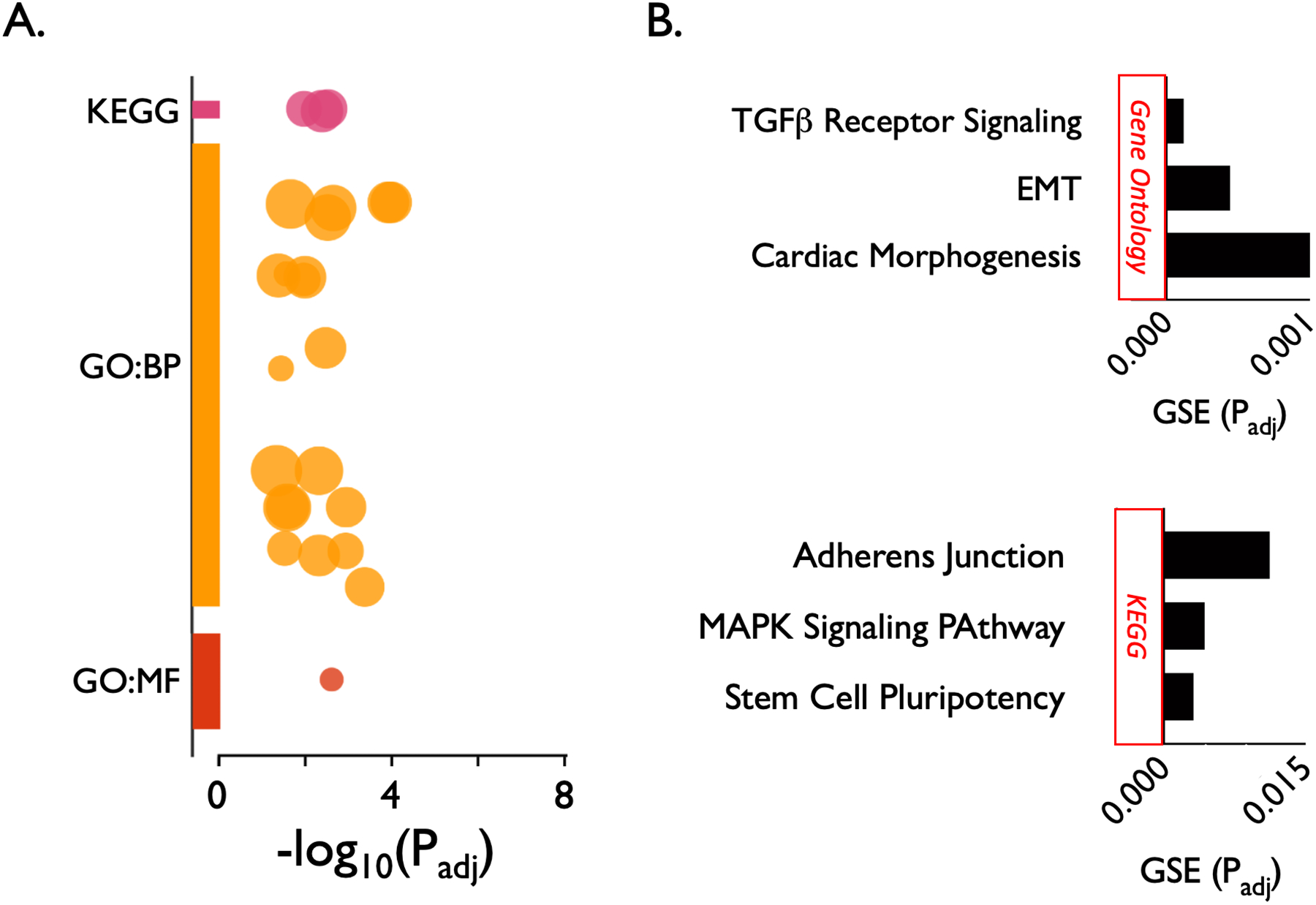
Pathway enrichment analysis of 20 candidate risk genes for subglottic stenosis using g:Profiler. The 24 statistically enriched terms are represented as a Manhattan plot showing each functional term as a circle (A). The top 3 most significantly enriched gene ontology biological process (GO:BP) terms (after collapsing duplicates) were “cellular response to TGF-ß,” “epithelial-to-mesenchymal transition” (EMT), and cardiac “outflow tract morphogenesis.” The single gene ontology molecular function (GO:MF) term was “fibroblast growth factor receptor activity.” The top 3 Kyoto Encyclopedia of Genes and Genomes (KEGG) terms were “signaling pathways regulating pluripotency of stem cells,” “MAPK signaling pathway,” and “adherens junction” (B). X axis represents the adjusted P value of the gene set enrichment (GSE). A complete listing of the 24 significantly enriched biological pathways is listed in Supplemental Table 1.
Methodological Validation: Idiopathic Pulmonary Fibrosis Cell Atlas & Localization of Candidate Risk Gene Expression
Using canonical lineage-defining markers to annotate clusters, Habermann et al. defined 31 cell types/states in the idiopathic pulmonary fibrosis (IPF) lung.24 These were further clustered into 4 basic tissue types for visualization: immune, epithelial, endothelial, and mesenchymal (Figure 3A).24
Figure 3.
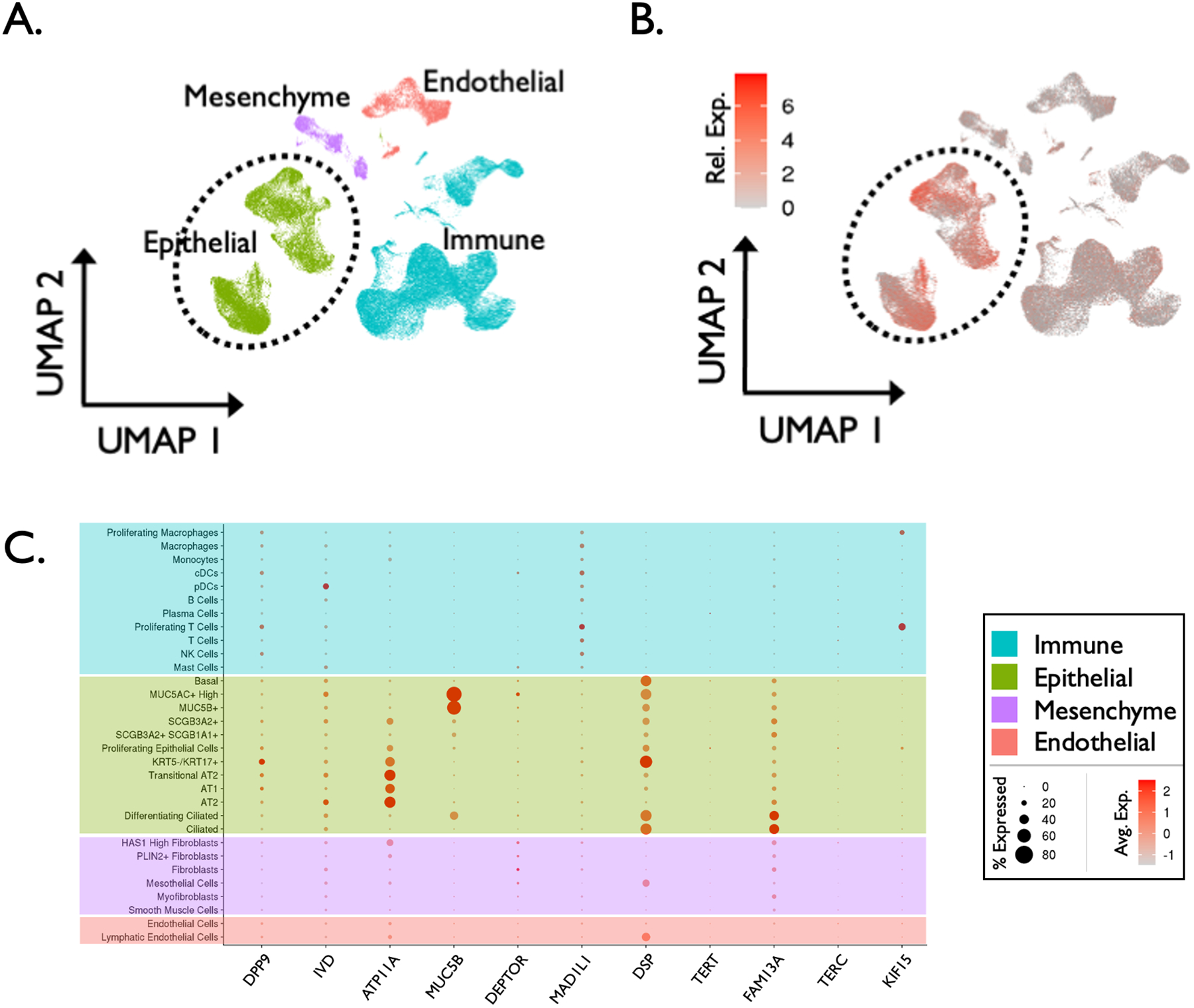
Method validation: cell localization of idiopathic pulmonary fibrosis (IPF) candidate risk genes. (A) Single cell RNA sequencing (scRNA-seq) atlas comprised of 114,396 cells from 12 explanted IPF lung samples, clustered into 4 tissue types and visualized via UMAP projection. (B) Differential expression of 11 established IPF risk genes (RNA expression denoted by color gradient) among the 4 broad cell types demonstrating enriched expression of disease risk genes in epithelial cells in the IPF lung. (C) Additional dot plot depicting expression of 11 established IPF risk genes within 31 distinct cell types/states in the IPF lung (RNA expression denoted by color gradient, percentage of expressing cells within a cell type denoted by dot size).
Fourteen genes have been rigorously associated with IPF risk via GWAS.27 Of these 14 genes, 11 were expressed in the scRNA-seq IPF atlas. We then investigated which cell types expressed the risk genes. Of the 11 genes expressed, the majority significantly localized to epithelial cells (Figure 3B). Specifically, 9 epithelial cell subtypes demonstrated the majority of expression of the 11 IPF risk genes (Figure 3C). Beyond epithelial cells, a small minority of immune, endothelial, or mesenchymal cell populations expressed any of the established 11 IPF risk genes.
These findings are consistent with the currently accepted model of IPF as primarily a disease of aberrant epithelial repair. Together, these results 1) validate our bioinformatics methods in a similar disease model and 2) reinforce the concept that localizing candidate risk genes to specific cell populations can indeed yield meaningful insight into disease biology.
Idiopathic Subglottic Stenosis Cell Atlas & Localization of Candidate Risk Gene Expression
scRNA-seq data from iSGS airway scar were embedded in a uniform manifold approximation and projection (UMAP) and clustered into 22 cell types/states within 4 broad tissue classes: immune, epithelial, endothelial, and mesenchymal (Figure 4A). Eleven (55%) of the 20 candidate risk genes identified via OMIM search were found to be expressed ubiquitously among all tissue types (Figure 4B). Three (15%) genes (EBP, FGFR2, GMNN) were defined as enriched in epithelial cells, 3 (15%) (ELN, FGFR1, TBX3) in fibroblasts, and 3 (15%) (ADAMTSL2, FLNB, TONSL) in endothelial cells (Figure 4C). Interestingly, we did not observe differential expression of candidate risk genes among immune cells. For a complete list of tissue-specific expression categories, see Supplemental Table 2.
Figure 4.
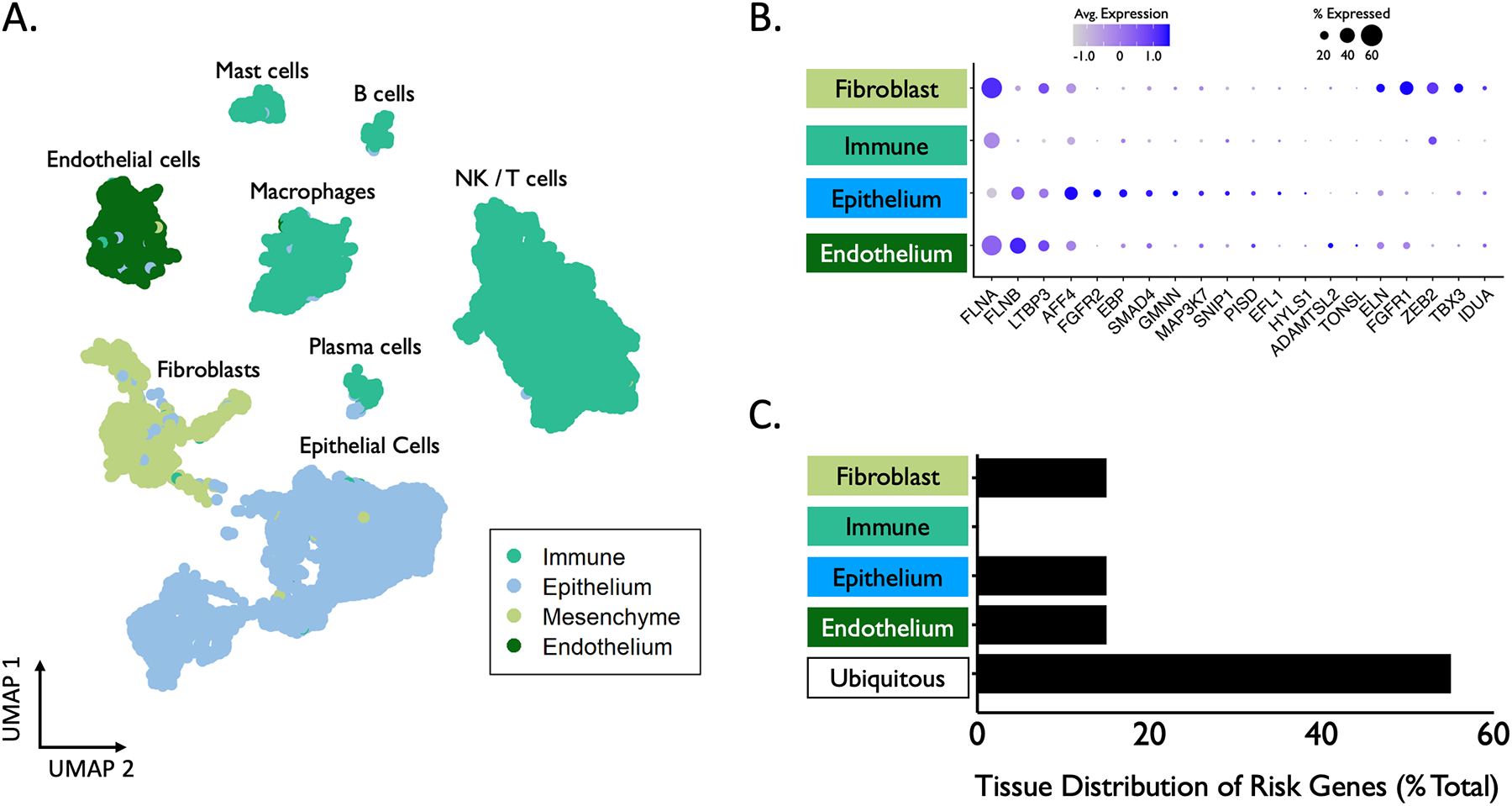
North American Airway Collaborative (NoAAC) cell atlas of idiopathic subglottic stenosis (iSGS). (A) Single cell RNA sequencing (scRNAseq) results of 7 iSGS airway scar samples consisting of 34,000 cells with unsupervised clustering analysis for cell subtype. (B) Tissue-specific expression of 20 candidate risk genes associated with proximal airway stenosis (average quantitative RNA expression denoted by color gradient, percentage of cells showing expression within a tissue type denoted by size of dot). (C) Percentage of genes significantly enriched in each tissue type. Genes were defined as enriched in a specific cell type if their expression in that cell type exceeded all others by >½ a standard deviation (34.1%). Genes without a cell type fulfilling enrichment criteria were categorized as “Ubiquitous”.
Summary of Enriched Genes Among Tissue Types
Among genes enriched in epithelial cells, EBP encodes an integral membrane protein of the endoplasmic reticulum and causes abnormal cartilage and bone development when mutated. FGFR2 is a member of the fibroblast growth factor receptor family related to skin development and osteogenesis. GMNN encodes a nuclear regulatory protein that inhibits the cell cycle.
Among genes enriched in fibroblasts, ELN encodes elastin, with mutations associated with aortic stenosis. FGFR1 is a member of the fibroblast growth factor receptor family and is involved in limb induction. TBX3 encodes a transcriptional repressor involved in the regulation of pluripotent stem cells.
Among genes enriched in endothelial cells, ADAMTSL2 encodes a secreted glycoprotein that interacts with latent transforming growth factor beta (TGF-ß) binding protein 1. FLNB encodes a cytoskeletal product that plays an important role in chondrogenesis and ossification. TONSL is believed to be a negative regulator of NF-kappa-B, with a canonical role in the host inflammatory response and maintenance of immune homeostasis.
DISCUSSION
Summary of Key Findings
This study sought to explore the genetic architecture of SGS susceptibility and utilize the findings to drive meaningful biological insight into disease pathogenesis. The OMIM database provided unbiased identification of SGS phenotypes associated with 20 defined genetic mutations. These patients would not be classified as having idiopathic SGS; rather, they demonstrate an SGS phenotype in the setting of a genetic syndrome, composed of a constellation of other clinical findings. However, the OMIM database reveals that several unique genotypes can independently produce a common phenotype, i.e., SGS. Given the identification of 20 such genotypes, investigation of how these unique genes work together allows new insights into common pathways of subglottic remodeling across diverse diseases.
To rigorously assign functional biological context to the 20 identified candidate risk genes, we first performed pathway enrichment analysis using open-source bioinformatics software. Consistent with prior reports, our results highlight the importance of TGF-ß signaling and the phenomenon of epithelial-to-mesenchymal transition (EMT) in SGS disease pathogenesis.7,35
Next, we employed a new technique to understand which cell types might contribute to subglottic mucosal remodeling. Given the novelty of this approach, we first validated the computational technique in a more well-studied disease (IPF). The approach uses the insight that not all cell types express the same RNA transcripts despite sharing the same DNA (i.e., certain genes are only expressed in specific cell types). We mapped the cellular location of each identified risk gene to the tissue types present in SGS scar using a scRNA-seq atlas. On its own, identifying syndromic candidate SGS risk genes via OMIM does not answer the question of which gene(s) are causative in non-syndromic etiologies of SGS (idiopathic, GPA-related, post-intubation, etc.). However, our data does suggest what pathways and cell types (epithelial, fibroblasts, etc.) are sufficient to drive subglottic tissue remodeling.
Surprisingly, many of the OMIM risk genes are expressed in fibroblasts and epithelial cells, with minimal expression in immune cells. We interpret this finding to suggest that epithelial barrier dysfunction and wound healing are central to the process of subglottic remodeling, while primary immune dysfunction is not. This appears to be in contrast with other mucosal diseases, such as inflammatory bowel disease, which has several associated genetic polymorphisms present in immune pathways.
Role of Fibroblasts
While the pathogenesis of SGS is undoubtedly multifactorial, the role of fibroblasts in generating clinically relevant fibrosis and scar contracture appears certain. Prior work in iSGS has demonstrated that local inflammatory mediators IL-17A and TGF-ß synergistically activate fibroblasts and promote fibrotic extracellular matrix remodeling.12 TGF-ß is considered a fibrosis “master switch,” inducing fibroblast collagen deposition within multiple organ systems.36–38 Histopathologic evidence shows TGF-ß at increased concentrations within the subglottic submucosa of iSGS patients.39 Blockade of fibroblast TGF-ß receptor function affords protection from mechanical epithelial injury40 and bleomycin-induced fibrosis41 in animal models of airway injury.
While fibroblasts in the proximal airway can derive from local cell activation or an influx of circulating fibrocytes, epithelial-to-mesenchymal transition (EMT) may represent another potential source of pathologic fibroblasts. Driven in large part by TGF-ß signaling,42 EMT is a molecular program in which epithelial cells lose their polarity and transform towards a mesenchymal phenotype, leading to excessive deposition of collagen-rich extracellular matrix (ECM) proteins.43 Loss of E-cadherin and disruption of cell-cell junctions are other key features.43 EMT was the second-most significant result in our pathway enrichment analysis, indicating that the candidate risk genes may interact to promote EMT-related cellular changes in the airway epithelium. The IPF literature has implicated EMT in the formation of contractile myofibroblasts during pulmonary fibrogenesis.44,45 The extent to which EMT contributes to SGS pathogenesis remains to be determined and merits further research.
Endothelial Cells
SGS occurs in 10–20% of GPA patients.2 This has raised the possibility for a role for aberrant proximal airway small vessel physiology in SGS disease pathogenesis. Interestingly, structural and functional changes in the airway endothelium have been observed in asthma, COPD, and IPF.46,47 In pulmonary fibrosis, endothelial changes stimulated by TGF-ß and vascular endothelial growth factor (VEGF) impair endothelial barrier function and promote paracrine secretion of inflammatory mediators by endothelial cells, resulting in fibroblast activation. The role of endothelium in SGS pathogenesis remains unresolved. However, given the clinical parallels between idiopathic SGS and proximal airway scar arising in GPA, the role of endothelial cell signaling in SGS pathogenesis may be substantial and merits continued investigation.
Epithelial Barrier Function
The ciliated pseudostratified columnar epithelium of the subglottis and trachea plays a crucial role in barrier defense. Epithelial barrier dysfunction is implicated in multiple airway diseases including IPF,25,26 COPD,48 and asthma.49 Data from our group demonstrates displacement of the native luminal microbiota into the submucosal tissues in iSGS samples, while animal data suggests that bacteria are essential to the inflammatory remodeling that occurs after subglottic epithelial injury.7 In the present study, the localization of 15% of SGS candidate risk genes to the airway epithelium supports the hypothesis that epithelial barrier dysfunction plays a major role in disease pathogenesis.
Controversies Raised by This Study: The Role of the Host Immune Response
Sustained inflammation of the airway mucosa is a well-known histologic characteristic of SGS airway scar.9,38 Murine data demonstrate that aberrant mucosal immune activation is a key feature of fibrotic remodeling in the proximal airway.7,10 Human specimens show increased numbers of resident memory CD8+ T cells in iSGS,50 and increased CD4+ T-lymphocytes in iatrogenic subglottic fibrosis,10 supporting a mechanistic role of adaptive immunity. Yet surprisingly, in the present study, we did not observe enrichment of candidate risk genes in immune cells. This suggests that iSGS may resemble IPF, where repeated injury to vulnerable alveolar epithelium leads to a dysregulated tissue repair process.51 Several well-powered GWAS in IPF have identified genetic variants associated with increased disease risk.27 These genes appear to primarily impact epithelial function and surfactant production. Despite this, the presence of lymphoid aggregates in the lung tissue, together with autoantibodies in the serum, suggests that the immune system is likely to play a role in either initiation or progression of IPF. In addition to unique circulating autoantibody levels, CD4+ T cells from IPF patients possess augmented effector function, demonstrate oligoclonal expansion, and proliferate in response to antigen present in diseased lung tissues.52 Similarly in iSGS, a host immune system free of pathogenic genetic mutation may be directed inappropriately against a novel antigen (either self or pathogen) to promote disease progression after an initial epithelial injury.
Strengths and Limitations
This study employed an innovative, unbiased bioinformatics approach to interrogate genetic risk in SGS. However, there are several key limitations. First, familial iSGS comprises only 2.5% of the iSGS population,16 and in contrast to IPF, may be largely driven by non-Mendelian genetics. Relying on the OMIM database to identify candidate pathways and cell types may prove limited due to its inability to capture candidate risk genes for diseases with non-Mendelian inheritance patterns. Second, hile the current scRNA-seq cell atlas provides unique insights, increased refinement of the atlas with a greater number of cells from more patients will continue to improve our understanding of cellular interactions in complex pathways. Additionally, the scRNA-seq cell atlas used as reference data is derived from iSGS patients; future work should integrate transcriptome data from non-idiopathic SGS patients, i.e. those arising after intubation injury and in GPA. Third, the sequencing depth in scRNA-seq is inferior to bulk tissue sequencing, making future work confirming the localization of candidate risk genes to the different cell types in subglottic scar important.
Complex diseases emerge from interactions between genetic polymorphisms and environmental exposures. While this study investigated genetic risk in SGS, environmental exposures in genetically susceptible hosts may also be an important contributor to SGS development and remains a question for future study.
Future Research Directions
Ongoing work from our group7 and others within the NoAAC consortium8,35,53 support a central role for airway epithelial dysfunction in SGS pathogenesis (termed the NoAAC proximal airway model). Molecular and histologic evidence of epithelial dysfunction in SGS is substantiated by the observed displacement of bacteria beneath the subglottic lamina propria. Animal models confirm that bacteria are necessary for pathologic proximal airway fibrosis after epithelial injury via activation of host adaptive immunity.
These new genetic results support the NoAAC proximal airway model with localization of several candidate risk genes to airway epithelial cells. Additionally, the expression patterns of fibroblasts and endothelial cells provide new insight into fibrotic proximal airway disease risk. Future studies incorporating whole-exome and whole-genome sequencing may uncover additional alleles contributing to disease risk, which can be localized to specific cell types using identical methodology. Identifying the particular cell populations manifesting pathologic transcriptional regulation may expose new precision therapeutic targets. Additional research into the genetic variants associated with SGS in GPA2 and post-intubation injury54 may reveal common pathways in the development of proximal airway scar, as well as offer new avenues for treatment. The genetic study of familial iSGS patients also holds great potential to illuminate the genetic architecture of subglottic remodeling and is an active area of research within the NoAAC consortia.
Conclusion
We offer a functional characterization of the genetic architecture underlying SGS. Our study integrates existing knowledge on genotype-phenotype associations, biological pathway interactions, and localization of candidate risk genes to individual cell types within SGS airway scar. Our findings reinforce the mechanistic role of TGF-ß signaling pathways and EMT in SGS pathogenesis. They also suggest that genetic variation may specifically impact epithelium, fibroblasts, and endothelium to impart an elevated risk of fibrotic disease in the proximal airway.
Supplementary Material
Supplemental Table 1. Complete results of g:Profiler pathway enrichment analysis of 20 candidate risk genes.
Supplemental Table 2. Complete cell localization results for 20 candidate risk genes.
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health (NIH) / National Heart, Lung, and Blood Institute (NHLBI) award number R01HL146401 (A.G.); Patient-Centered Outcomes Research Institute (PCORI) award number 1409-22214 (A.G.). We thank the members of the North American Airway Collaborative for their contributions to this research.
Footnotes
Disclosures: The authors report no conflicts of interest.
This work appeared as a poster presentation at the Triological Society 2023 Combined Sections Meeting, January 26–28, 2023, Coronado, California.
LEVEL OF EVIDENCE
N/A
REFERENCES
- 1.Gelbard A, Francis DO, Sandulache VC, Simmons JC, Donovan DT, Ongkasuwan J. Causes and consequences of adult laryngotracheal stenosis. The Laryngoscope 2015; 125:1137–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Quinn KA, Gelbard A, Sibley C, et al. Subglottic stenosis and endobronchial disease in granulomatosis with polyangiitis. Rheumatology 2019; 58:2203–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gelbard A, Anderson C, Berry LD, et al. Comparative Treatment Outcomes for Patients With Idiopathic Subglottic Stenosis. JAMA otolaryngology-- head & neck surgery 2020; 146:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gnagi SH, Howard BE, Anderson C, Lott DG. Idiopathic Subglottic and Tracheal Stenosis: A Survey of the Patient Experience. Ann Otol Rhinol Laryngol 2015; 124:734–739. [DOI] [PubMed] [Google Scholar]
- 5.Verma SP, Goshtasbi K, Berry LD, Anderson C, Francis DO, Gelbard A. Utilization and Influence of Online Support Communities in Idiopathic Subglottic Stenosis Patients. Laryngoscope 2021; 131:1821–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hillel AT, Namba D, Ding D, Pandian V, Elisseeff JH, Horton MR. An In Situ, In Vivo Murine Model for the Study of Laryngotracheal Stenosis. JAMA Otolaryngology–Head & Neck Surgery 2014; 140:961. [DOI] [PubMed] [Google Scholar]
- 7.Gelbard A, Shilts MH, Strickland B, et al. Idiopathic subglottic stenosis arises at the interface of host and pathogen. medRxiv [Preprint.] February 04, 2022. Available at: 10.1101/2022.02.02.22270308 [DOI] [Google Scholar]
- 8.Lina IA, Tsai H-W, Berges AJ, et al. Phenotypic Epithelial Changes in Laryngotracheal Stenosis. The Laryngoscope 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gelbard A, Katsantonis N-G, Mizuta M, et al. Idiopathic subglottic stenosis is associated with activation of the inflammatory IL-17A/IL-23 axis. The Laryngoscope 2016; 126:E356–E361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hillel AT, Ding D, Samad I, Murphy MK, Motz K. T-Helper 2 Lymphocyte Immunophenotype Is Associated With Iatrogenic Laryngotracheal Stenosis. The Laryngoscope 2019; 129:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lina IA, Berges A, Ospino R, et al. Identifying Phenotypically Distinct Fibroblast Subsets in Type 2 Diabetes-Associated Iatrogenic Laryngotracheal Stenosis. Otolaryngology--Head and Neck Surgery: Official Journal of American Academy of Otolaryngology-Head and Neck Surgery 2022; 166:712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrison RJ, Katsantonis N-G, Motz KM, et al. Pathologic Fibroblasts in Idiopathic Subglottic Stenosis Amplify Local Inflammatory Signals. Otolaryngology--Head and Neck Surgery: Official Journal of American Academy of Otolaryngology-Head and Neck Surgery 2019; 160:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motz K, Lina I, Murphy MK, et al. M2 Macrophages Promote Collagen Expression and Synthesis in Laryngotracheal Stenosis Fibroblasts. The Laryngoscope 2021; 131:E346–E353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pizzuto M, Donaldson D, Brodsky L. A role for genetic predisposition in subglottic stenosis. International Journal of Pediatric Otorhinolaryngology 1998; 44:279–284. [DOI] [PubMed] [Google Scholar]
- 15.Gelbard A, Donovan DT, Ongkasuwan J, et al. Disease homogeneity and treatment heterogeneity in idiopathic subglottic stenosis. The Laryngoscope 2016; 126:1390–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drake VE, Gelbard A, Sobriera N, et al. Familial Aggregation in Idiopathic Subglottic Stenosis. Otolaryngology--Head and Neck Surgery: Official Journal of American Academy of Otolaryngology-Head and Neck Surgery 2020; 163:1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anis MM, Zhao Z, Khurana J, Krynetskiy E, Soliman AMS. Translational genomics of acquired laryngotracheal stenosis. The Laryngoscope 2014; 124:E175–179. [DOI] [PubMed] [Google Scholar]
- 18.Anis MM, Krynetskaia N, Zhao Z, Krynetskiy E, Soliman AMS. Determining Candidate Single Nucleotide Polymorphisms in Acquired Laryngotracheal Stenosis. The Laryngoscope 2018; 128:E111–E116. [DOI] [PubMed] [Google Scholar]
- 19.Xie G, Roshandel D, Sherva R, et al. Association of granulomatosis with polyangiitis (Wegener’s) with HLA-DPB1*04 and SEMA6A gene variants: evidence from genome-wide analysis. Arthritis Rheum 2013; 65:2457–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med 2012; 367:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reimand J, Isserlin R, Voisin V, et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nature Protocols 2019; 14:482–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amberger JS, Hamosh A. Searching Online Mendelian Inheritance in Man (OMIM): A Knowledgebase of Human Genes and Genetic Phenotypes. Curr Protoc Bioinformatics 2017; 58:1.2.1–1.2.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raudvere U, Kolberg L, Kuzmin I, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Research 2019; 47:W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Habermann AC, Gutierrez AJ, Bui LT, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv 2020; 6:eaba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011; 364:1503–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selman M, Pardo A. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med 2014; 189:1161–1172. [DOI] [PubMed] [Google Scholar]
- 27.Allen RJ, Guillen-Guio B, Oldham JM, et al. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med 2020; 201:564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell 2019; 177:1888–1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 2019; 20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 2019; 16:1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Franzén O, Gan LM, Björkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford) 2019; 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadota K, Ye J, Nakai Y, Terada T, Shimizu K. ROKU: a novel method for identification of tissue-specific genes. BMC Bioinformatics 2006; 7:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang S, Li Y, Be X, Howes S, Liu W. Detecting and profiling tissue-selective genes. Physiological Genomics 2006; 26:158–162. [DOI] [PubMed] [Google Scholar]
- 34.Schug J, Schuller W-P, Kappen C, Salbaum JM, Bucan M, Stoeckert CJ. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biology 2005; 6:R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang C, Wang S, Moura MC, et al. RNA Sequencing of Idiopathic Subglottic Stenosis Tissues Uncovers Putative Profibrotic Mechanisms and Identifies a Prognostic Biomarker. Am J Pathol 2022. [DOI] [PubMed] [Google Scholar]
- 36.Sime PJ, O’Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol 2001; 99:308–319. [DOI] [PubMed] [Google Scholar]
- 37.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008; 214:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Motz KM, Gelbard A. The role of inflammatory cytokines in the development of idiopathic subglottic stenosis. Translational Cancer Research 2020; 9:2102–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scioscia KA, Miller F, April MM, Gruber BL. Growth factors in subglottic stenosis. Ann Otol Rhinol Laryngol 1996; 105:936–943. [DOI] [PubMed] [Google Scholar]
- 40.Simpson CB, White S, McGuff HS. Anti-transforming growth factor beta as a treatment for laryngotracheal stenosis in a canine model. Laryngoscope 2008; 118:546–551. [DOI] [PubMed] [Google Scholar]
- 41.Zhao J, Shi W, Wang YL, et al. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 2002; 282:L585–593. [DOI] [PubMed] [Google Scholar]
- 42.Mahmood MQ, Reid D, Ward C, et al. Transforming growth factor (TGF) β(1) and Smad signalling pathways: A likely key to EMT-associated COPD pathogenesis. Respirology 2017; 22:133–140. [DOI] [PubMed] [Google Scholar]
- 43.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119:1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim KK, Kugler MC, Wolters PJ, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A 2006; 103:13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ward C, Forrest IA, Murphy DM, et al. Phenotype of airway epithelial cells suggests epithelial to mesenchymal cell transition in clinically stable lung transplant recipients. Thorax 2005; 60:865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olivieri D, Chetta A. Therapeutic perspectives in vascular remodeling in asthma and chronic obstructive pulmonary disease. Chem Immunol Allergy 2014; 99:216–225. [DOI] [PubMed] [Google Scholar]
- 47.Gaikwad AV, Eapen MS, McAlinden KD, et al. Endothelial to mesenchymal transition (EndMT) and vascular remodeling in pulmonary hypertension and idiopathic pulmonary fibrosis. Expert Rev Respir Med 2020; 14:1027–1043. [DOI] [PubMed] [Google Scholar]
- 48.Polosukhin VV, Richmond BW, Du RH, et al. Secretory IgA Deficiency in Individual Small Airways Is Associated with Persistent Inflammation and Remodeling. Am J Respir Crit Care Med 2017; 195:1010–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schleimer RP, Berdnikovs S. Etiology of epithelial barrier dysfunction in patients with type 2 inflammatory diseases. J Allergy Clin Immunol 2017; 139:1752–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gelbard A, Wanjalla C, Wootten CT, et al. The Proximal Airway Is a Reservoir for Adaptive Immunologic Memory in Idiopathic Subglottic Stenosis. The Laryngoscope 2021; 131:610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kropski JA, Blackwell TS. Progress in Understanding and Treating Idiopathic Pulmonary Fibrosis. Annu Rev Med 2019; 70:211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wissinger EL, Stevens WW, Varga SM, Braciale TJ. Proliferative expansion and acquisition of effector activity by memory CD4+ T cells in the lungs following pulmonary virus infection. J Immunol 2008; 180:2957–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Treviño-Villarreal JH, Reynolds JS, Langston PK, Thompson A, Mitchell JR, Franco RA Jr. Down-Regulation of a Profibrotic Transforming Growth Factor-β1/Cellular Communication Network Factor 2/Matrix Metalloprotease 9 Axis by Triamcinolone Improves Idiopathic Subglottic Stenosis. Am J Pathol 2021; 191:1412–1430. [DOI] [PubMed] [Google Scholar]
- 54.Johnson RF, Bradshaw S, Jaffal H, Chorney SR. Estimations of Laryngotracheal Stenosis After Mechanical Ventilation: A Cross-Sectional Analysis. Laryngoscope 2022; 132:1723–1728. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Complete results of g:Profiler pathway enrichment analysis of 20 candidate risk genes.
Supplemental Table 2. Complete cell localization results for 20 candidate risk genes.