

Abstract
The aortic valve consists of valvular interstitial cells (VICs) and endothelial cells (VECs). While these cells are understood to work synergistically to maintain leaflet structure and valvular function, there are few co-culture models of these cell types. In this study, aortic valve co-cultures (AVCCs) were assembled using magnetic levitation and cultured for 3 days. Immunohistochemistry and qRT-PCR were used to assess the maintenance of cellular phenotype and function, and the formation of extracellular matrix. AVCCs stained positive for CD31 and α-smooth muscle actin (αSMA), demonstrating phenotype was maintained. Functional markers endothelial nitric oxide synthase (eNOS), von Willebrand factor (VWF), and prolyl-4-hydroxylase were present. Extracellular matrix components collagen type I, laminin, and fibronectin also stained positive, with reduced gene expression of these proteins in 3D compared to 2D. Genes for collagen type I, lysyl oxidase, and αSMA were expressed less in AVCCs than in 2D cultures, indicating that VICs are quiescent. Co-localization of CD31 and αSMA in the AVCC suggests that endothelial-mesenchymal transdifferentiation (EnMT) is maybe occurring. Differences in VWF and eNOS in VECs cultured in 2D and 3D also suggests that the AVCC possibly has anti-thrombotic potential. Overall, a co-culture model of the aortic valve was designed, and serves as a basis for future experiments to understand heart valve biology.
Keywords: Aortic valve, valvular interstitial cells, valvular endothelial cells, co-culture, 3D cell culture, magnetic levitation
1. Introduction
The aortic valve, located between the left ventricle and the aorta, regulates unidirectional blood flow from the heart to the systemic circulation. The valve consists of three leaflets, each of which is comprised of two cell types: valvular interstitial cells (VIC) that populate the interior of the leaflet, and valvular endothelial cells (VEC) that cover the surface of the leaflet. VICs are a heterogeneous group of cells with fibroblast and smooth muscle cell phenotypes that maintains the extracellular matrix (ECM) of the leaflet [1–3]. VECs regulate the transfer of signals in the bloodstream to the valve interior, mediate inflammatory and hemostatic responses, and have a phenotype similar to vascular endothelial cells, but demonstrate major differences in alignment to flow and mechanobiology [4–7]. Together, both VICs and VECs work to maintain the leaflet structure and valve function.
The interactions between these two cell types are critical for normal valve function. The presence of VECs has been shown to keep VICs in a quiescent state, as defined by the low expression of α-smooth muscle actin (αSMA) [8]. The key role that their interaction plays in valve function is also demonstrated by the finding that dysfunction of both cell types appear in calcific aortic valve disease (CAVD) [9]. Injury to the valvular endothelium leads to thrombosis, inflammation, and lipid accumulation [10–15]. These factors lead to the activation of VICs, or increased expression of αSMA, matrix remodeling, and their progression towards an osteoblastic phenotype, which ultimately result in calcification and stenosis [16–20].
Despite the well-established notion that both cell types and the interaction between the two are critical to valve maintenance, function, and disease, there are few co-culture models of VECs and VICs in literature. The lack of co-culture models of the aortic valve can partly be attributed to the inability to produce such models using traditional 2D cell culture techniques, with which the majority of research on valvular cells is conducted. 2D environments are poor representations of the native 3D environment in which valvular cells reside [21–23]. The inadequacy of 2D cultures for valvular research is highlighted by the varied results of studies on the effect of statins as treatments for calcific aortic valve disease. Statins in 2D in vitro cultures of VICs significantly reduced calcific nodule size and area, as well as αSMA expression [24–28], yet, clinical trials did not show any improvement in outcome for patients with calcific aortic stenosis who took statins [29–33]. Indeed, studies of the effect of statins on VICs in 3D collagen gels showed a smaller, conditional reduction in calcification in comparison to 2D cultures [26]. These results demonstrate the enormous gap in complexity and fidelity between simple 2D cell culture models and the human body, and necessitate the development of cost-effective, clinically relevant, and representative 3D co-culture models of the aortic valve.
To that end, this study used a magnetic levitation method with magnetic nanoparticles to assemble 3D co-cultures of VECs and VICs. In this method, cells are incubated with a nanoparticle assembly consisting of poly-L-lysine, magnetic iron oxide (MIO; Fe3O4, magnetite), and gold nanoparticles that form a gel via electrostatic interactions [34–37]. The uptake of this gel by cells renders them magnetic and allows for their manipulation, specifically by levitating the cells off the surface into the media, where the cells aggregate and interact to form larger 3D structures. This method has previously been used to create 3D cultures of glioblastomas, smooth muscle cells, adipose stem cells, and pulmonary cells [37–41]. Magnetically levitated human glioblastoma cells demonstrated greater proliferation and more in vivo-like protein expression in comparison to 2D cultures [37].
In addition, this method has previously been used to create co-cultures. Layered co-cultures of the lung were sequentially assembled using epithelial cells, smooth muscle cells, fibroblasts, and endothelial cells within 8 hours [41]. After 2 days of culture, ECM (collagen type I, fibronectin, laminin) was formed and organized, and the phenotypes of all four cell types were maintained. After 7 days, epithelial cell function and phenotype were still present [41]. In comparison, previously reported co-culture models of the valve leaflet consisted of collagen-based gels which were embedded and contracted with VICs, and then seeded with VECs on their surfaces in a process that took between 4-8 days to assemble [8,42]. As a result, magnetic levitation is a rapid alternative method for assembling co-culture models.
Based on the success of the magnetically levitated co-culture of the bronchiole, this study used magnetic levitation to create 3D co-cultures of aortic valve cells. VICs and VECs were sequentially assembled into layered co-cultures. Immunohistochemistry (IHC) was used to verify the phenotype and function of both VECs and VICs, and assess ECM formation within the co-culture. Unlike previous studies using magnetic levitation, this study is the first to analyze the gene expression profiles of these cultures using quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR). In addition, the effects of the magnetic nanoparticles and exposure to the magnetic field on cell proliferation were investigated. The expected result of this study was a VIC and VEC co-culture model that maintained cell phenotype and function, and synthesized relevant ECM.
2. Materials and Methods
2.1. Cell Isolation and Culture
Fresh porcine hearts were obtained from a local commercial abattoir (Fisher Ham and Meats, Spring, TX), from which the aortic valves were extracted. Aortic VECs and VICs were harvested as previously described [43,44]. Both cell types were cultured in an incubator (37°C, 5% CO2, 95% humidity) with medium changes every other day.
VECs were isolated from the leaflets via digestion using collagenase II (60 U/mL) and dispase (2U/mL) [43]. VECs were seeded on flasks or glass slides coated with 2.5% gelatin in 1:1 H2O:phosphate buffered saline (PBS, pH~7.4) [45], and cultured in specialized medium (EGM-2, Lonza Biosciences, Walkersville, MD) with 2% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). At the first passage, magnetic cell sorting was used to purify the VECs for CD31+ cells (Mouse monoclonal anti-CD31 antibody TLD-3A12, Millipore, Billerica, MA) [43]. VECs were used at their third passage.
VICs were isolated from the leaflets with a multistep digestion using collagenase II (~450 U/mL), hyaluronidase (~50 U/mL), and collagenase III (~350 U/mL) [44]. VICs were seeded on uncoated flasks or glass slides in DMEM/F12 medium supplemented with 10% bovine growth serum (BGS, Hyclone, Logan, UT), 1% HEPES buffer, and 1% P/S. VICs were used at their third passage.
2.2. Magnetic Levitation
Magnetic levitation using the Bio-Assembler Kit (Nano3D Biosciences, Houston, TX) was employed to create 3D cultures (Fig. 1B) [37]. Confluent flasks of cells were treated with a magnetic nanoparticle assembly (8 μL/cm2 of cell culture surface area or 50 μL/mL medium, NanoShuttle, NS, Nano3D Biosciences) for overnight incubation to allow for cell binding to the nanoparticles. NS was fabricated as previously described, by mixing Au nanoparticles prepared by citrate reduction, poly-L-lysine, and iron oxide [34,35,37]. Treated cells were then detached with trypsin and resuspended in a low attachment 24-well plate with 400 μL of medium. A magnetic driver of 24 neodymium magnets (field strength = 50 G) designed for 24-well plates and a plastic lid insert were placed atop the well plate to levitate the cells to the air-liquid interface.
Figure 1:
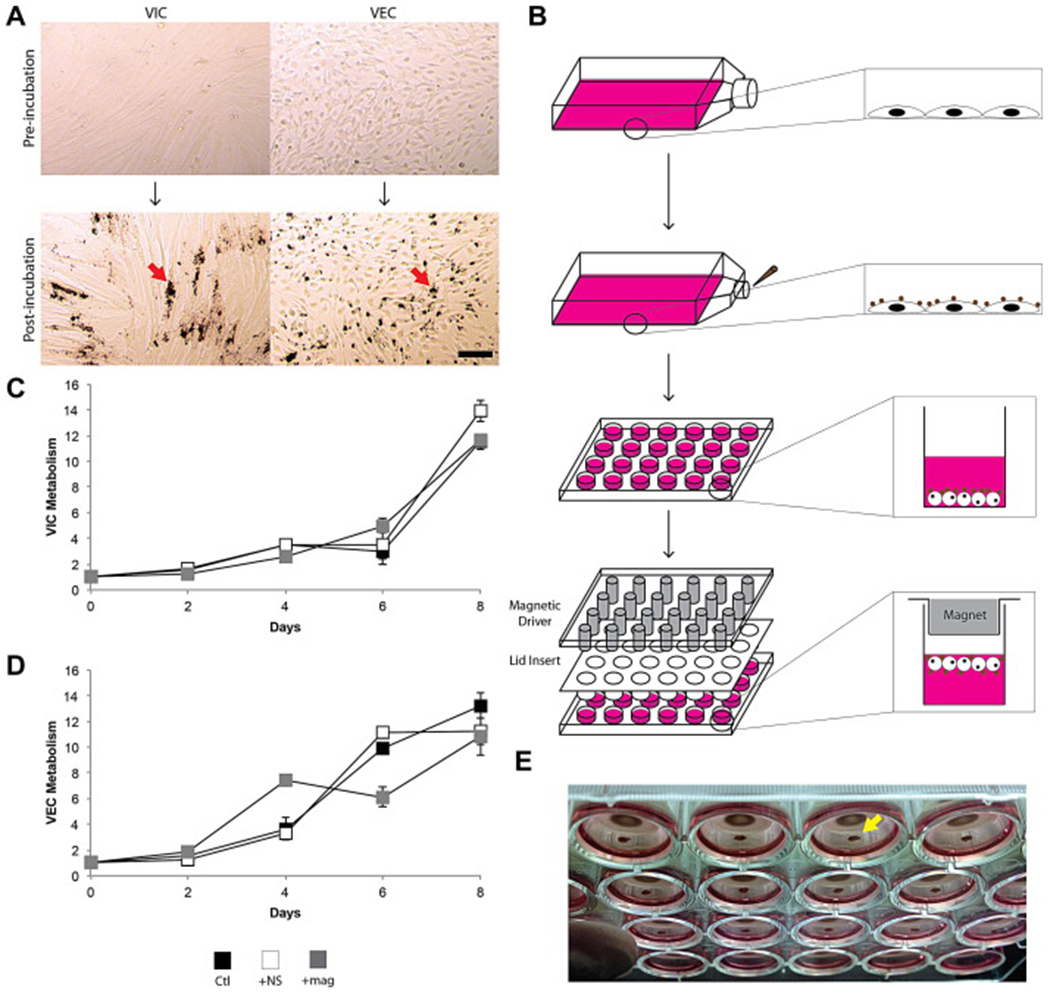
(A) VICs and VECs before and after incubation with NanoShuttle. Note the maintenance of morphology after incubation, and the localization of the magnetic nanoparticles (indicated by red arrow) with the cells. Scale bar = 100 μm. (B) Schematic of magnetic levitation. A confluent flask of cells is incubated with NanoShuttle overnight to allow for cell binding. The next day, the cells are detached from the surface and resuspended in 400 μL of medium in a 24-well plate. A magnetic driver and plastic lid insert is placed atop the well plate to levitate the cell constructs to the air-liquid interface. Metabolic activity of VICs (C) and VECs (D) with incubation with NanoShuttle and exposure to the magnet over 8 days (n=9). There was no significant difference found between these groups for both cell types. Error bars represent standard error of the mean. (E) 3D cultures (indicated by yellow arrow) levitating in a 24-well plate as seen from below the plate.
2.3. Cell Metabolism
The effects of both the NS and magnetic field on cell metabolism over 8 days were measured using an MTT assay. Briefly, MTT reagent (0.5 mg/mL in medium, thiazolyl blue tetrazolium bromide, Sigma-Aldrich, St. Louis, MO) was added to each well. After 3-4 hours of incubation, the medium was aspirated to yield the formazan blue crystals at the bottom of the well. Acidified isopropanol (0.1 N HCl in isopropanol) was added to dissolve the formazan blue crystals, and the absorbance of the resulting solution was read in triplicate on a spectrophotometer (SpectraMax M2, Molecular Devices, Sunnyvale, CA) at 570 nm with background subtraction at 690 nm. All measurements were normalized to day 0 results.
To test the effects of adding NS (+NS) or exposure to a magnetic field (+mag) to VIC and VEC proliferation, 25,000 cells were seeded in 24-well plates. For +NS wells, NS was added to the wells (8 μL/cm2) on day 0 for incubation overnight. For +mag wells, neodymium magnets were placed 1 mm underneath each well (field strength = 300 G). Medium was changed at day 1 and every other day after that for the duration of the study. Cell metabolic activities were measured on days 2, 4, 6, and 8 (n=9).
2.4. Co-Culture Assembly
Magnetic levitation was used to create co-cultures of VICs and VECs (Fig. 2A) [41]. VICs and VECs were incubated overnight with NS, and then levitated into 3D cultures of 500,000 cells each. After 4 hours of levitation, a 0.1875” OD Teflon pen housing a neodymium magnetic rod was used to sequentially pick up a 3D culture of each cell type to assemble the co-culture: first VECs, then VICs. The still attached co-culture was then submerged in 150 μL VEC medium in a 96-well plate for 4 hours. The co-culture was then put back into a 24-well plate by first filling a well with 400 μL of VEC medium, then removing the rod magnet from the Teflon pen, and placing the magnetic driver underneath the well to attract the co-culture into the well. Once detached, the magnetic driver was moved to the top of the well and the co-culture was levitated again. These co-cultures are heretofore referred to as the aortic valve co-culture (AVCC).
Figure 2:
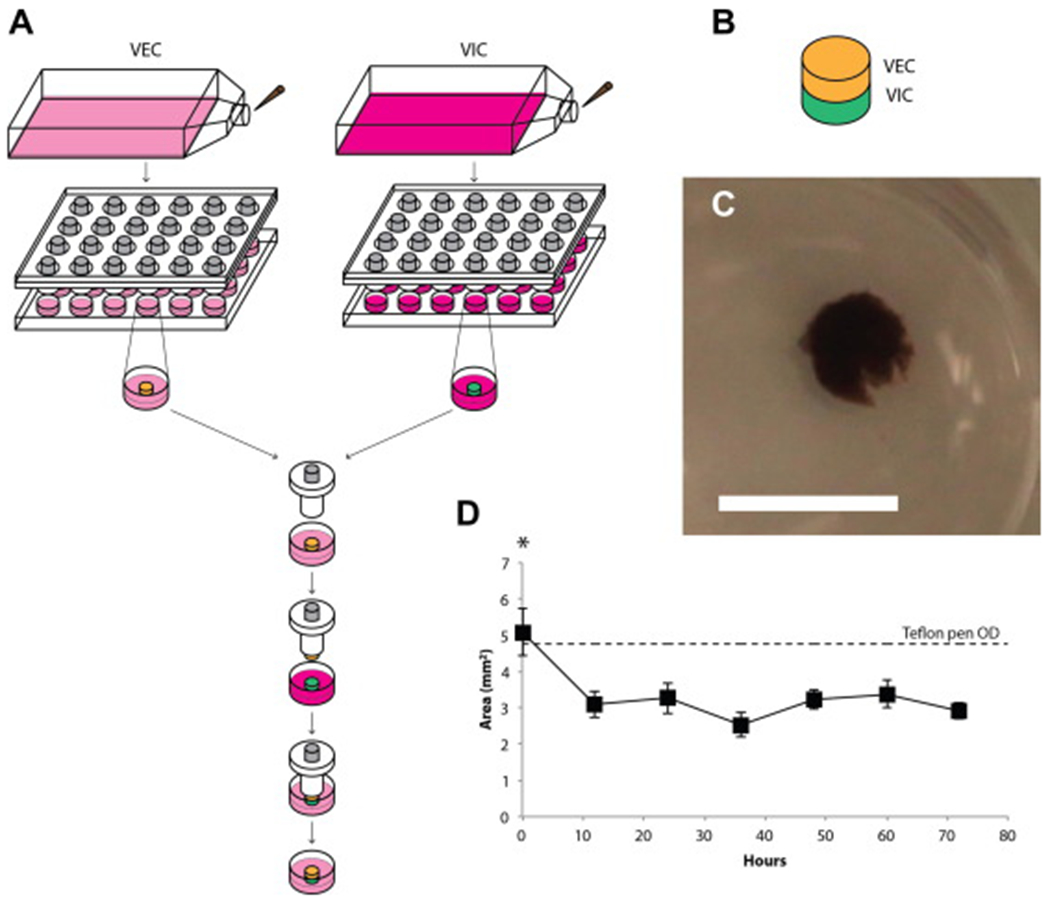
(A) Schematic of co-culture assembly using magnetic levitation. 3D cultures of VECs (yellow) and VICs (green) were separately levitated for 4 hours. A Teflon pen was used to then sequentially assemble the co-culture together, VECs then VICs. The co-culture is submerged in medium, still attached to the Teflon pen, for 4 hours. The co-culture is then detached off the Teflon and levitated for 3 days in VEC medium. (B) Schematic of the co-culture, within which the VEC layer sits atop the VIC layer. (C) A resulting VIC-VEC culture after 3 days of levitation as seen from above the plate. Scale bar = 5 mm. (D) The planar size of the co-culture over 3 days (n=9). The co-culture starts close to the size of the Teflon pen used to assemble the co-culture. By 12 hours, the co-culture significantly shrinks in size and maintains that size over the next 60 hours. *: p<0.05 versus the rest.
2.5. Immunohistochemistry
IHC was used to verify the maintenance of phenotype and function, and the formation of ECM. AVCCs were fixed in 4% paraformaldehyde for at least 5 hours, then dehydrated, embedded in paraffin, and sectioned according to standard procedures. To preserve the structure of the AVCC, the Teflon pen used for co-culture assembly was used to hold the co-culture in place and maintain its structure during processing. For 2D immunocytochemistry (ICC), VICs and VECs were seeded on glass chamber slides at 50,000 cells/well. The next day, cells were fixed with 4% paraformaldehyde.
Sections to be stained were rehydrated and underwent antigen retrieval using a citrate buffer solution (Antigen Decloaker, Biocare Medical, Concord, CA) at 80°C for 30 min. For intracellular antigens, sections were then permeabilized using 0.2% Triton X-100 for 15 min. All sections were then washed and blocked using 1% donkey serum (GeneTex, Irvine, CA) in PBS for 1 hour at room temperature. Experimental sections were then incubated overnight at 4°C with the primary antibody of interest at the manufacturer’s recommended dilution in PBS with 1% bovine serum albumin. Negative controls were left incubating with donkey serum. The next day, all sections were washed and incubated with a fluorescent secondary antibody (AlexaFluor 488/633, Invitrogen, Carlsbad, CA) for 1 hour at room temperature and counterstained with DAPI (KPL, Gaithersburg, MD) for 15 min. All slides were then washed, mounted, and imaged. 2D ICC slides were stained similarly to 3D cultures after permeabilization with Triton X-100. Images were captured on a confocal microscope (LSM 510 META NLO, Zeiss).
The antigens stained for in this study included: αSMA (Abcam, Cambridge, MA) for VIC phenotype; CD31 (Abcam) for VEC phenotype; collagen type I (Col I, Abcam), laminin (Lam, Abcam), and fibronectin (FN, Abcam) for ECM; prolyl 4-hydroxylase (P4H, Bioss, Woburn, MA) for collagen synthesis; endothelial nitric oxide synthase (eNOS, Santa Cruz Biotechnology, Santa Cruz, CA) and von Willebrand factor (VWF, Abcam) for endothelial function; and VE-cadherin (VE-cad, Cell Signaling Technology, Danvers, MA) and N-cadherin (N-cad, Invitrogen) for cell-cell interactions. For each antigen, the AVCC, 3D and 2D cultures were stained at the same time.
2.6. Quantitative reverse transcriptase polymerase chain reaction
Sample mRNA was extracted using Trizol Reagent (Invitrogen) mediated lysis and a series of ethanol washes and centrifugations. The mRNA was reverse-transcribed into cDNA using Primescript 1st strand cDNA Synthesis Kit (Takara Bio, Otsu, Japan). The cDNA samples were stored at −20°C until use. Using the 2X QuantiTect SYBR Green PCR Master Mix (Clontech Laboratories, Mountain View, CA), qRT-PCR on the cDNA was performed (Mastercycler ep realplex, Eppendorf, Hamburg, Germany) to assess differences in gene expression levels between the AVCC, 3D VEC and VIC cultures, and 2D VEC and VIC cultures. For quantification, the GAPDH gene (Integrated DNA Technologies, Coralville, IA) was used as the housekeeping gene, and sample group gene expression levels were normalized to the corresponding expression levels of the AVCC. The AVCC was used as the standard for the purposes of ease of analysis and comparison, given that some markers are specific to either VICs or VECs.
qRT-PCR was performed (n=3-5) to measure the positive gene expression of: αSMA for VIC phenotype; CD31 for VEC phenotype; COL1A1, FN, Lam-β1 for ECM; lysyl oxidase (LOX) for collagen fibrillogenesis; and eNOS and VWF for endothelial function (Integrated DNA Technologies) (Table 1).
Table 1:
Summary of the DNA primer sequences used to assess sample gene expression using qRT-PCR.
| Gene | 5’← DNA Sequence → 3’ | Product Size | Accession Number | |
|---|---|---|---|---|
| ACTA2 (αSMA) | Forward | AATAGAACACGGCATCATC | 77 | FJ547477 |
| Reverse | CACGAAGCTCATTGTAGAA | |||
| PECAM1 (CD31) | Forward | ACTGCTAACAACCAGAATT | 80 | X98505 |
| Reverse | GCTTGACAGGAGAATAATATAAC | |||
| COL1A1 | Forward | AGTTGTCTTATGGCTATGATGAG | 78 | XM_003483014 |
| Reverse | GACCACGAGGACCAGAAG | |||
| FN1 | Forward | CTACTATTACTGGTCTGGAA | 75 | AY839862 |
| Reverse | CACTCTTCTGATTGTTCTT | |||
| LAMB1 | Forward | CACCACGGATTCCAACAG | 75 | AF329358 |
| Reverse | TGCTCCAACATCAAGTCT | |||
| LOX | Forward | CAGTGGATTGATATTACAG | 99 | NM_001206403 |
| Reverse | ATTGTTGGAATAGTCTGA | |||
| VWF | Forward | CGAACCCAAGAAGAGAAT | 108 | S78431 |
| Reverse | ATCACTTCCTCCACAAAC | |||
| NOS3 (eNOS) | Forward | AGAGAATGGAGAGAGTTT | 104 | AY266137 |
| Reverse | TATTGAAGCGGATTTTGT | |||
| GAPDH | Forward | CATTGACCTCCACTACAT | 119 | AF017079 |
| Reverse | AGATGGTGATGGGATTTC |
2.7. Statistical Analysis
One-way ANOVAs were performed on the data (JMP, SAS, Cary, NC), with significance defined as p<0.05. If a significant effect was observed, post-hoc Tukey’s testing was used to observe pairwise comparisons. Data is presented as mean ± standard error of the mean.
3. Results
3.1. Magnetic Levitation
Both VICs and VECs were successfully levitated into 3D cultures. Both VICs and VECs were able to bind with the NanoShuttle without grossly affecting morphology (Fig. 1A). Neither incubation with NanoShuttle nor exposure to the magnetic field significantly affected the metabolism of VICs and VECs (Fig. 1C-D).
3.2. Co-Culture Assembly
AVCCs were successfully assembled and maintained for 3 days. At day 3, the AVCC still maintained its competent structure (Fig. 2B). After 12 hours of levitation, the planar size of these co-cultures decreased significantly, but did not change for the remaining 60 hours (Fig. 2C). The AVCC was approximately 500 μm thick after culture. Hematoxylin and eosin staining showed two color stains, demonstrating the presence of two distinct cell types (see Supplemental Figure S3).
3.3. Phenotypic Markers
Immunohistochemistry demonstrated the presence of VICs and VECs within the AVCC by positive staining for αSMA and CD31 (Fig. 3A, for negative controls see Supplemental Figure S4). Both 3D and 2D VIC cultures stained positively for αSMA. Similarly, both 3D and 2D VEC cultures stained positively for CD31, and both 3D monotype cultures stained positively for αSMA. qRT-PCR confirmed IHC results, as AVCCs expressed both αSMA and CD31 mRNA (Fig. 3B). There was no difference in αSMA gene expression between the AVCC and 3D monotype cultures, but these cultures expressed significantly less αSMA mRNA than 2D VIC cultures (652.078X v. AVCC, p<0.05). CD31 gene expression in the AVCC was statistically similar to that in 3D monotype cultures and 2D VEC cultures. There was virtually no CD31 expression by 3D VIC cultures (0.007X v. AVCC), and CD31 expression by 3D VEC cultures was significantly higher than that in 3D VIC cultures (471.990X v. 3D VIC, p<0.05), whereas CD31 expression by 2D VEC cultures trended higher than 3D VIC cultures (427.439X v. 3D VIC, p=0.055).
Figure 3:
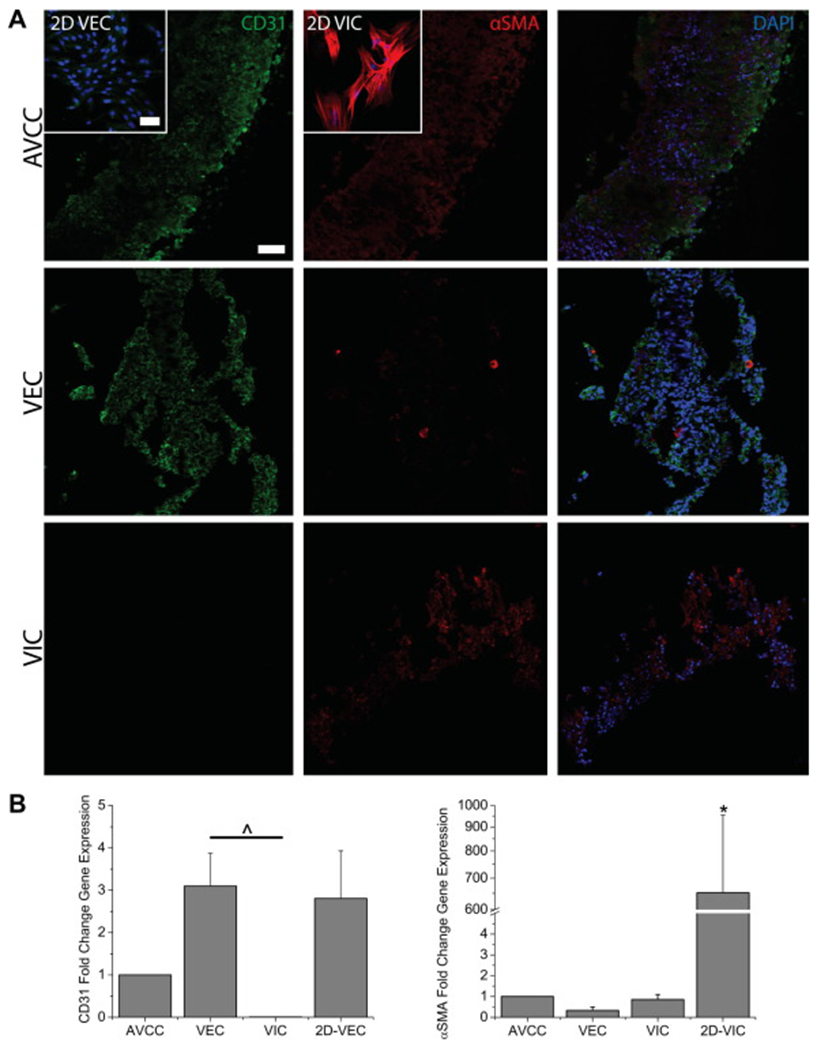
(A) IHC stains for the phenotypic markers αSMA (red) and CD31 (green) in the AVCC, 3D VIC and VEC cultures, and 2D VIC and VEC cultures (insets). Nuclei are counterstained using DAPI (blue). The AVCC stained positively for both phenotypic markers, indicating the presence of both VICs and VECs. CD31 was limited to the outer edges of the AVCC, while αSMA was distributed throughout. Scale bar = 50 μm. (B) qRT-PCR results for the phenotypic markers αSMA and CD31 (n=3-5). αSMA gene expression for the AVCC and 3D monotype cultures was significantly lower than 2D VIC cultures. CD31 gene expression is non-existent in 3D VIC cultures. *: p<0.05 v. other groups. ^: p<0.05 within bracket. Error bars represent standard error of the mean.
3.4. Functional Markers
All functional markers of interest stained positively in the AVCC (Fig. 4A, for negative controls see Supplemental Figure S5). VE-cad was limited to the outer edges of the AVCC and co-localized with CD31. Similarly, VWF was located towards the edge of the AVCC. N-cad, P4H, and eNOS all stained positively throughout the AVCC. qRT-PCR demonstrated that LOX gene expression was significantly less in the AVCC than in both 2D VIC (14.963X v. AVCC, p<0.005) and VEC (11.310X v. AVCC, p<0.05) cultures, but similar to both 3D VIC and VEC cultures (Fig. 4B). VWF gene expression in 2D VEC cultures was the highest, and significantly higher than 3D monotype cultures and the AVCC (48.571X v. AVCC, p<0.05). Similarly, eNOS gene expression was highest in 3D VEC cultures, where it was significantly higher than 3D VIC cultures, 2D VEC cultures, and the AVCC (5.606X v. AVCC, p<0.005).
Figure 4:
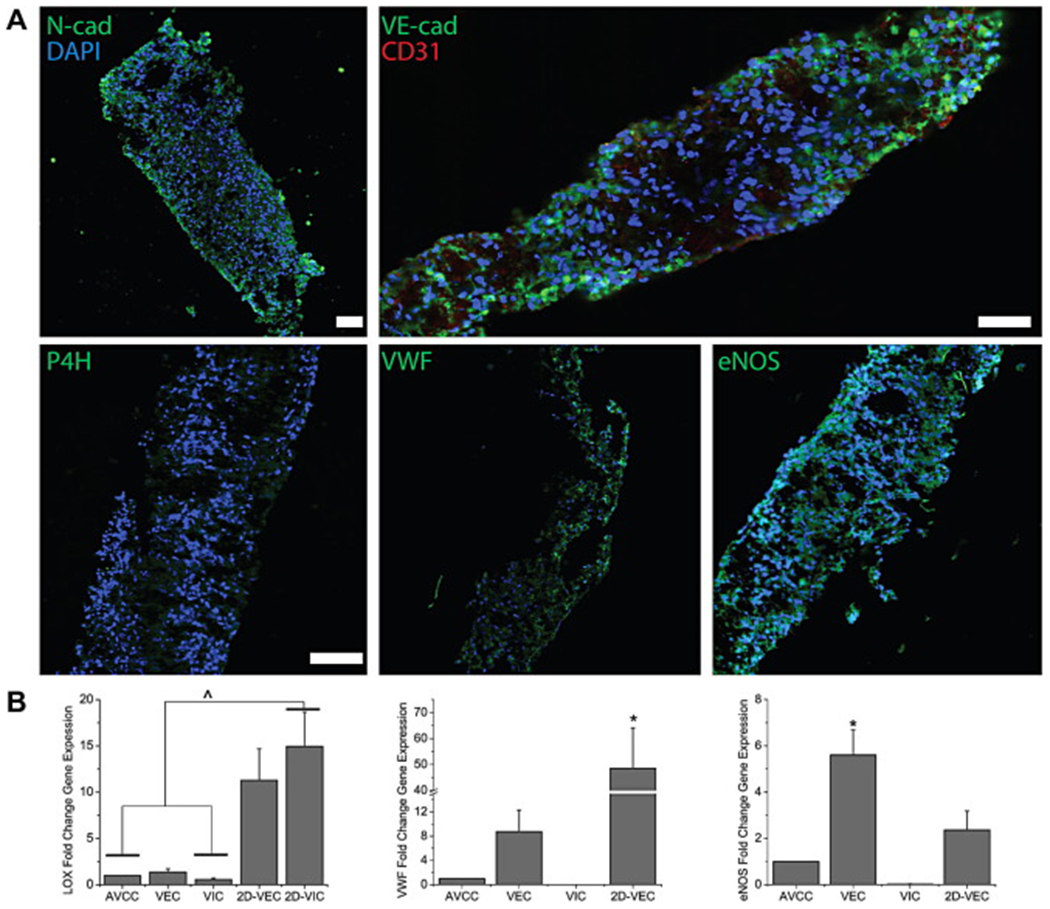
(A) IHC stains (green) for the functional markers N-cad, P4H, VWF, eNOS, and VE-cad (double-stained with CD31 in red) in the AVCC. Nuclei are counterstained using DAPI (blue). The AVCC stained positively for all these functional markers. VE-cad and VWF were localized to the outer edges of the AVCC, while N-cad, P4H, and eNOS were distributed evenly throughout. Scale bar = 100 μm. (B) qRT-PCR results for the functional markers LOX, VWF, and eNOS (n=3-5). Gene expression for LOX and VWF was significantly lower in the AVCC in comparison to 2D VIC and VEC cultures, respectively, while for eNOS, expression in the AVCC was significantly lower than expression in 3D VEC cultures. *: p<0.05 v. other groups. ^: p<0.05 within bracket. Error bars represent standard error of the mean.
3.5. Extracellular Matrix
With regards to the ECM components of interest, Col I, FN, and Lam stained positively, and evenly distributed within IHC stains of the AVCC (Fig. 5A, for negative controls see Supplemental Figure S6). In 2D cultures, Col I and FN stained more intensely and prominently in 2D VIC cultures than 2D VEC cultures, while Lam stain intensity was higher in 2D VEC cultures. Gene expression for COL1A1 was the highest in 2D VIC cultures, where it was also significantly higher than the other groups (98.135X v. AVCC, p<0.0001) (Fig. 5B). There was a significant difference in FN mRNA expression between 2D cultures and all 3D cultures (94.967-113.285X v. AVCC, p<0.01). 2D VEC cultures expressed significantly higher amounts of Lam-β1 mRNA than 3D monotype cultures (2.822-5.735X v. 3D cultures, p<0.05), but statistically similar amounts to 2D VIC cultures and the AVCC (2.247X v. AVCC, p=0.095).
Figure 5:
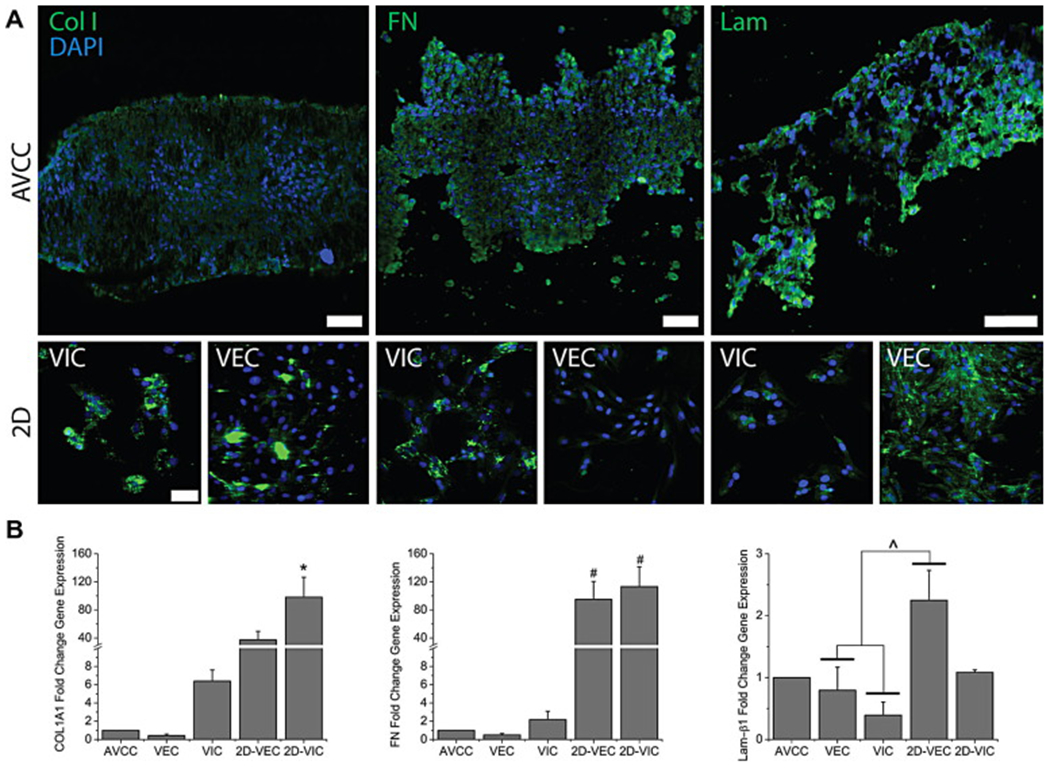
(A) IHC stains (green) for the ECM components Col I, FN, and Lam in the AVCC and 2D VIC and VEC cultures. Nuclei are counterstained using DAPI (blue). These ECM components are present and distributed throughout. In 2D cultures, Col I and FN are more prominent with VICs, while Lam is more prominent with VECs. Scale bar = 50 μm. (B) qRT-PCR results for the ECM markers COL1A1, FN, and Lam-β1. AVCC expressions of COL1A1 and FN were significantly less than 2D VIC cultures. Lam-β1 for all groups was found to be statistically similar with expression in the AVCC, but was significantly larger in 2D VEC cultures, than 3D monotype cultures. *: p<0.05 v. other groups. ^: p<0.05 within bracket. #: p<0.05 v. 3D monotype cultures. Error bars represent standard error of the mean.
4. Discussion
This study used magnetic levitation to construct a layered co-culture of the aortic valve, the AVCC. This is the first study to both magnetically levitate aortic valve cells, and then use this technique to assemble such co-cultures. AVCCs were assembled and cultured for 3 days in VEC medium. The size of the AVCC decreased by 12 hours of levitation, likely due to cell contraction, then was held constant for the remaining 60 hours. Immunohistochemistry was used to stain for markers of cellular phenotype, function, and ECM. qRT-PCR was used to quantitatively measure the gene expression for these same markers in these 3D cultures, the first time it has been used on magnetically levitated cultures. Positive stains and gene expression for αSMA and CD31 verified VIC and VEC phenotype. The presence of P4H, LOX, VWF, and eNOS indicate the preservation of function by both cell types. ECM formation in the form of Col I, FN, and Lam was demonstrated. The positive stain for N-cad affirmed the cell-cell interactions within the AVCC, while the positive stain for VE-cad showed that VECs in the AVCCs form tight junctions. This study also affirms the use of magnetic levitation as a simple method to create 3D co-cultures.
The AVCC and 3D VIC cultures were found to have significantly reduced gene expression of αSMA, as well as COL1A1, FN, and P4H, than 2D VIC cultures, suggesting that magnetically levitated VICs in the AVCC are in a quiescent state [1,46,47]. This quiescence could be a result of the difference in substrate stiffness between 2D cultures on glass, and 3D self-aggregated cultures. Higher stiffnesses have previously been shown to increase αSMA expression and the formation of stress fibers [48]. In addition, these cells reverted to a quiescent state from an activated state when levitated from 2D to 3D, confirming previous results that VIC activation is both plastic and reversible [47,49]. The presence of VECs could also have reduced VIC activation, as VICs in a collagen-gel based co-culture with VECs demonstrated reduced αSMA expression [8]. This role of VECs is supported by the fact that the gene expressions of COL1A1 and FN were reduced by more than half in AVCCs compared to 3D VIC cultures, even though VICs made up half of the AVCC. As an activated VIC state is commonly associated with matrix remodeling and the onset of CAVD [16,17,50], the quiescent state of VICs suggests that the AVCC can be used to study the activation of VICs under various environmental conditions. Further research is required to understand VIC quiescence and activation within the AVCC.
Interestingly, while VIC phenotype was found to be quiescent and ECM gene expression was significantly less in the AVCC compared to 2D cultures, the AVCC stained positively for ECM components, such as Col I, FN, and Lam. The formation and organization of ECM in the AVCC within 3 days would presumably require VIC activation for matrix remodeling [18]. IHC and qRT-PCR were conducted after 3 days of culture to find a quiescent state, but activation and matrix remodeling could have occurred earlier, yielding the ECM that was observed. Col I was found to have formed in the AVCC as early as 2 days of levitation (data not shown), while magnetically levitated pulmonary fibroblasts and tracheal smooth muscle cells have been demonstrated to produce and extrude Lam into the extracellular space within 6 hours [41]. As with other studies in heart valve biology, αSMA was used alone as a marker of VIC activation [48,51] and to distinguish VICs from VECs within the AVCC, but other markers for VIC activation, including non-muscle myosin heavy chain (NMM) and embryonic smooth-muscle myosin heavy chain (SMemb) [46,52], should be explored in future studies. Future studies using this model will also require time-based studies to further characterize VIC activation and ECM formation within the AVCC.
The positive stains for CD31, VWF, eNOS, Lam, and VE-cad verify that VEC phenotype and function is maintained within the AVCC and competent endothelium is being formed. Unexpectedly, on the edges of AVCCs and throughout 3D VEC cultures, CD31 was co-localized with αSMA. In addition, gene expression for eNOS, which catalyzes nitric oxide in endothelial cells and has previously been used as a functional marker for VECs for its important role in vasoactivity and resistance to oxidative stress [53–55], in the AVCC was less than half that of 3D VEC cultures, while consisting of half VECs. Gene expression for CD31, VWF, and Lam in the AVCC also trended similarly to eNOS. Together, these results suggest the possibility of endothelial to mesenchymal transdifferentiation (EnMT) occurring within the AVCC. VECs commonly differentiate into myofibroblast phenotypes in 2D in vitro culture [43], but the fact that the AVCC was cultured in VEC media and without a stiff substrate suggest that this result is unique. VECs have been shown to undergo EnMT in vitro in a manner stimulated by TGF-β1 and inhibited by vascular endothelial growth factor (VEGF) [56,57]. EnMT has been implicated in valvular disease and mechanically regulated [58], but the finding that the AVCC was positive for VE-cad, and its VICs were in a quiescent state, would suggest that EnMT is not occurring as a response to injury or endothelial dysfunction within the AVCC. Rather, VECs could possibly be undergoing EnMT to replenish or supplement VICs, a role previously suggested and investigated as a possible source for progenitor cells in engineered heart valves [7,59]. Further research using the AVCC will require a deeper look into EnMT within the model, and could provide further understanding of EnMT and its utility.
An interesting result of this study was that 3D VEC cultures showed significantly reduced expression of VWF, but increased expression of eNOS, relative to 2D VEC cultures. As VWF and eNOS are both VEC-mediated proteins essential in facilitating endothelial pro- [60–62] and anti-thrombotic [63,64] responses, respectively, these results suggest the possibility that 3D VEC cultures are in a more stable, anti-thrombotic state as opposed to 2D cultures. The expression of these proteins is highly influenced by the surrounding environment. Previous studies have shown that when subjected to physiological shear flow conditions, vascular endothelial cells produce less pro-thrombogenic components such as VWF [65,66]. Thus, the changes in VWF and eNOS gene expression between 2D and 3D are possibly a result of the lower stiffness and higher cell density of the 3D environment compared to 2D. Therefore, culturing valvular cells in 3D culture environments may not only maintain cell phenotype and function, but possibly enhance overall homeostatic cell function relative to 2D cultures. Further studies of hemostatic factors within the AVCC are necessary to understand its anti-thrombotic potential.
A major concern for this study was the choice of medium for co-culture, which was the medium used to culture VECs. In our other study using magnetic levitation to assemble co-cultures of the bronchiole, epithelial medium was used to maintain the co-cultures [41]. The rationale behind that choice was that epithelial cells were the definitive cell type of the bronchiole, and the most sensitive to serum due to fears of epithelial-mesenchymal transdifferentiation (EMT). As such, in that study, those co-cultures were cultured in epithelial medium with and without 1% FBS, and both cases showed the maintenance of epithelial phenotype and cell survival after 7 days [41]. Similarly, in this study VEC medium, which contained 2% FBS, was chosen over VIC medium, which contained 10% BGS, in order to preserve VEC phenotype against the possibility of EnMT. The results showed VEC phenotype was ultimately maintained, as confirmed by positive stains and gene expression for CD31, VWF, and eNOS, although co-localization with αSMA was present. Further research using this model should look at further optimization of the medium used for co-culture.
5. Conclusions
Magnetic levitation was successfully used to assemble co-cultures using VICs and VECs. Immunostaining and gene expression analysis were used to verify that the phenotype and functions of both cell types were maintained, and relevant ECM was formed. The result is a model that maintains phenotypes and induces ECM formation with a faster assembly time compared to other co-cultures. The AVCC could in the future be used for a wide variety of experiments, such as those involving mechanobiology, or the progression of CAVD. This study also affirms the versatility and utility of magnetic levitation to create representative 3D cell culture models.
Supplementary Material
6. Acknowledgements
This study was funded in part by a predoctoral fellowship from the American Heart Association (to H.T.); a National Science Foundation (NSF) Small Business Innovation Research (SBIR) Award Phase I (0945954) and Phase II (1127551) from NSF IIP Division of Industrial Innovation and Partnerships; and the State of Texas Emerging Technology Fund (ETF).
Footnotes
Author Disclosure Statement
The University of Texas M. D. Anderson Cancer Center (UTMDACC) and Rice University, along with their researchers, have filed patents on the technology and intellectual property reported here. If licensing or commercialization occurs, the researchers are entitled to standard royalties. Glauco R. Souza, Robert M. Raphael, T. C. Killian have equity in Nano3D Biosciences, Inc. UTMDACC and Rice University manage the terms of these arrangements in accordance to their established institutional conflict-of-interest policies.
8. References
- [1].Taylor PM, Batten P, Brand NJ, Thomas PS, Yacoub MH. The cardiac valve interstitial cell. Int J Biochem Cell Biol 2003;35:113–8. [DOI] [PubMed] [Google Scholar]
- [2].Chester AH, Taylor PM. Molecular and functional characteristics of heart-valve interstitial cells. Philos Trans R Soc Lond B Biol Sci 2007;362:1437–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mulholland DL, Gotlieb AI. Cell biology of valvular interstitial cells. Can J Cardiol 1996;12:231–6. [PubMed] [Google Scholar]
- [4].Butcher JT, Tressel S, Johnson TL, Turner D, Sorescu G, Jo H, Nerem RM. Transcriptional profiles of valvular and vascular endothelial cells reveal phenotypic differences: influence of shear stress. Arterioscler Thromb Vasc Biol 2006;26:69–77. [DOI] [PubMed] [Google Scholar]
- [5].Butcher JT, Penrod AM, García AJ, Nerem RM. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arterioscler Thromb Vasc Biol 2004;24:1429–34. [DOI] [PubMed] [Google Scholar]
- [6].Gould ST, Srigunapalan S, Simmons CA, Anseth KS. Hemodynamic and cellular response feedback in calcific aortic valve disease. Circ Res 2013;113:186–97. [DOI] [PubMed] [Google Scholar]
- [7].Bischoff J, Aikawa E. Progenitor cells confer plasticity to cardiac valve endothelium. J Cardiovasc Transl Res 2011;4:710–9. [DOI] [PubMed] [Google Scholar]
- [8].Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Eng 2006;12:905–15. [DOI] [PubMed] [Google Scholar]
- [9].Li C, Xu S, Gotlieb AI. The progression of calcific aortic valve disease through injury, cell dysfunction, and disruptive biologic and physical force feedback loops. Cardiovasc Pathol 2012. [DOI] [PubMed] [Google Scholar]
- [10].Poggianti E, Venneri L, Chubuchny V, Jambrik Z, Baroncini LA, Picano E. Aortic valve sclerosis is associated with systemic endothelial dysfunction. J Am Coll Cardiol 2003;41:136–41. [DOI] [PubMed] [Google Scholar]
- [11].Olsson M, Rosenqvist M, Nilsson J. Expression of HLA-DR antigen and smooth muscle cell differentiation markers by valvular fibroblasts in degenerative aortic stenosis. J Am Coll Cardiol 1994;24:1664–71. [DOI] [PubMed] [Google Scholar]
- [12].Olsson M, Dalsgaard CJ, Haegerstrand A, Rosenqvist M, Rydén L, Nilsson J. Accumulation of T lymphocytes and expression of interleukin-2 receptors in nonrheumatic stenotic aortic valves. J Am Coll Cardiol 1994;23:1162–70. [DOI] [PubMed] [Google Scholar]
- [13].Müller AM, Cronen C, Kupferwasser LI, Oelert H, Müller K-M, Kirkpatrick CJ. Expression of endothelial cell adhesion molecules on heart valves: up-regulation in degeneration as well as acute endocarditis. J Pathol 2000;191:54–60. [DOI] [PubMed] [Google Scholar]
- [14].Olsson M, Thyberg J, Nilsson J. Presence of oxidized low density lipoprotein in nonrheumatic stenotic aortic valves. Arterioscler Thromb Vasc Biol 1999;19:1218–22. [DOI] [PubMed] [Google Scholar]
- [15].Mahler GJ, Butcher JT. Inflammatory regulation of valvular remodeling: the good(?), the bad, and the ugly. Int J Inflam 2011;2011:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol 2007;171:1407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-β: Implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res 2004;95:253–60. [DOI] [PubMed] [Google Scholar]
- [18].Hinton RB, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res 2006;98:1431–8. [DOI] [PubMed] [Google Scholar]
- [19].Rajamannan NM. Calcific aortic valve disease: cellular origins of valve calcification. Arterioscler Thromb Vasc Biol 2011;31:2777–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Monzack EL, Masters KS. Can valvular interstitial cells become true osteoblasts? A side-by-side comparison. J Heart Valve Dis 2011;20:449–63. [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang S Beyond the Petri dish. Nat Biotechnol 2004;22:151–2. [DOI] [PubMed] [Google Scholar]
- [22].Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol 2006;7:211–24. [DOI] [PubMed] [Google Scholar]
- [23].Abbott A Cell culture: biology’s new dimension. Nature 2003;424:870–2. [DOI] [PubMed] [Google Scholar]
- [24].Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G, Baumgartner H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation 2004;110:1291–5. [DOI] [PubMed] [Google Scholar]
- [25].Monzack EL, Masters KS. A time-course investigation of the statin paradox among valvular interstitial cell phenotypes. Am J Physiol Heart Circ Physiol 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Monzack EL, Gu X, Masters KS. Efficacy of simvastatin treatment of valvular interstitial cells varies with the extracellular environment. Arterioscler Thromb Vasc Biol 2009;29:246–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Benton JA, Kern HB, Leinwand LA, Mariner PD, Anseth KS. Statins block calcific nodule formation of valvular interstitial cells by inhibiting alpha-smooth muscle actin expression. Arterioscler Thromb Vasc Biol 2009;29:1950–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Osman L, Yacoub MH, Latif N, Amrani M, Chester AH. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation 2006;114:I547–52. [DOI] [PubMed] [Google Scholar]
- [29].Rossebø AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Bärwolf C, Holme I, Kesäniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med 2008;359:1343–56. [DOI] [PubMed] [Google Scholar]
- [30].Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med 2005;352:2389–97. [DOI] [PubMed] [Google Scholar]
- [31].Moura LM, Ramos SF, Zamorano JL, Barros IM, Azevedo LF, Rocha-Gonçalves F, Rajamannan NM. Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J Am Coll Cardiol 2007;49:554–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chan KL, Teo K, Dumesnil JG, Ni A, Tam J. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation 2010;121:306–14. [DOI] [PubMed] [Google Scholar]
- [33].Peltonen T, Ohtonen P, Näpänkangas J, Ohukainen P, Ruskoaho H, Taskinen P. Statin treatment and gene expression of anti-atherogenic factor C-type natriuretic peptide system in stenotic aortic valves. J Heart Valve Dis 2011;20:545–51. [PubMed] [Google Scholar]
- [34].Souza GR, Yonel-Gumruk E, Fan D, Easley J, Rangel R, Guzman-Rojas L, Miller JH, Arap W, Pasqualini R. Bottom-up assembly of hydrogels from bacteriophage and Au nanoparticles: the effect of cis- and trans-acting factors. PLoS One 2008;3:e2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Souza GR, Christianson DR, Staquicini FI, Ozawa MG, Snyder EY, Sidman RL, Miller JH, Arap W, Pasqualini R. Networks of gold nanoparticles and bacteriophage as biological sensors and cell-targeting agents. Proc Natl Acad Sci U S A 2006;103:1215–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hajitou A, Trepel M, Lilley CE, Soghomonyan S, Alauddin MM, Marini FC, Restel BH, Ozawa MG, Moya CA, Rangel R, Sun Y, Zaoui K, Schmidt M, von Kalle C, Weitzman MD, Gelovani JG, Pasqualini R, Arap W. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell 2006;125:385–98. [DOI] [PubMed] [Google Scholar]
- [37].Souza GR, Molina JR, Raphael RM, Ozawa MG, Stark DJ, Levin CS, Bronk LF, Ananta JS, Mandelin J, Georgescu M-M, Bankson JA, Gelovani JG, Killian TC, Arap W, Pasqualini R. Three-dimensional tissue culture based on magnetic cell levitation. Nat Nanotechnol 2010;5:291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Molina JR, Hayashi Y, Stephens C, Georgescu M-M. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia 2010;12:453–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee JS, Morrisett JD, Tung C-H. Detection of hydroxyapatite in calcified cardiovascular tissues. Atherosclerosis 2012;224:340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Daquinag AC, Souza GR, Kolonin MG. Adipose tissue engineering in three-dimensional levitation tissue culture system based on magnetic nanoparticles. Tissue Eng Part C Methods 2013;19:336–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tseng H, Gage JA, Raphael RM, Moore RH, Killian TC, Grande-Allen KJ, Souza GR. Assembly of a three-dimensional multitype bronchiole coculture model using magnetic levitation. Tissue Eng Part C Methods 2013;19:665–75. [DOI] [PubMed] [Google Scholar]
- [42].Flanagan TC, Wilkins B, Black A, Jockenhövel S, Smith TJ, Pandit AS. A collagen-glycosaminoglycan co-culture model for heart valve tissue engineering applications. Biomaterials 2006;27:2233–46. [DOI] [PubMed] [Google Scholar]
- [43].Cheung W-Y, Young EWK, Simmons CA. Techniques for isolating and purifying porcine aortic valve endothelial cells. J Heart Valve Dis 2008;17:674–81. [PubMed] [Google Scholar]
- [44].Stephens EH, Carroll JL, Grande-Allen KJ. The use of collagenase III for the isolation of porcine aortic valvular interstitial cells: rationale and optimization. J Heart Valve Dis 2007;16:175–83. [PubMed] [Google Scholar]
- [45].Kumar TRS, Krishnan LK. A stable matrix for generation of tissue-engineered nonthrombogenic vascular grafts. Tissue Eng 2002;8:763–70. [DOI] [PubMed] [Google Scholar]
- [46].Della Rocca F, Sartore S, Guidolin D, Bertipaglia B, Gerosa G, Casarotto D, Pauletto P. Cell composition of the human pulmonary valve: a comparative study with the aortic valve--the VESALIO Project. Vitalitate Exornatum Succedaneum Aorticum labore Ingegnoso Obtinebitur. Ann Thorac Surg 2000;70:1594–600. [DOI] [PubMed] [Google Scholar]
- [47].Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis 2004;13:841–7. [PubMed] [Google Scholar]
- [48].Quinlan AMT, Billiar KL. Investigating the role of substrate stiffness in the persistence of valvular interstitial cell activation. J Biomed Mater Res A 2012;100:2474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang H, Haeger SM, Kloxin AM, Leinwand LA, Anseth KS. Redirecting valvular myofibroblasts into dormant fibroblasts through light-mediated reduction in substrate modulus. PLoS One 2012;7:e39969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jian B, Narula N, Li Q, Mohler ER III, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg 2003;75:457–65. [DOI] [PubMed] [Google Scholar]
- [51].Cushing MC, Liao J-T, Anseth KS. Activation of valvular interstitial cells is mediated by transforming growth factor-beta1 interactions with matrix molecules. Matrix Biol 2005;24:428–37. [DOI] [PubMed] [Google Scholar]
- [52].Bertipaglia B, Ortolani F, Petrelli L, Gerosa G, Spina M, Pauletto P, Casarotto D, Marchini M, Sartore S. Cell characterization of porcine aortic valve and decellularized leaflets repopulated with aortic valve interstitial cells: the VESALIO Project (Vitalitate Exornatum Succedaneum Aorticum Labore Ingenioso Obtenibitur). Ann Thorac Surg 2003;75:1274–82. [DOI] [PubMed] [Google Scholar]
- [53].Butcher JT, Simmons CA, Warnock JN. Mechanobiology of the aortic heart valve. J Heart Valve Dis 2008;17:62–73. [PubMed] [Google Scholar]
- [54].Butcher JT, Nerem RM. Valvular endothelial cells and the mechanoregulation of valvular pathology. Philos Trans R Soc Lond B Biol Sci 2007;362:1445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Simmons CA, Grant GR, Manduchi E, Davies PF. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res 2005;96:792–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Paranya G, Vineberg S, Dvorin EL, Kaushal S, Roth SJ, Rabkin E, Schoen FJ, Bischoff J. Aortic valve endothelial cells undergo transforming growth factor-beta-mediated and non-transforming growth factor-beta-mediated transdifferentiation in vitro. Am J Pathol 2001;159:1335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yang J-H, Wylie-Sears J, Bischoff J. Opposing actions of Notch1 and VEGF in post-natal cardiac valve endothelial cells. Biochem Biophys Res Commun 2008;374:512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Balachandran K, Alford PW, Wylie-Sears J, Goss JA, Grosberg A, Bischoff J, Aikawa E, Levine RA, Parker KK. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci U S A 2011;108:19943–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sales VL, Mettler BA, Engelmayr GC Jr., Aikawa E, Bischoff J, Martin DP, Exarhopoulos A, Moses MA, Schoen FJ, Sacks MS, Mayer JE Jr.. Endothelial progenitor cells as a sole source for ex vivo seeding of tissue-engineered heart valves. Tissue Eng Part A 2010;16:257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Badimon L, Badimon JJ, Turitto VT, Vallabhajosula S, Fuster V. Platelet thrombus formation on collagen type I. A model of deep vessel injury. Influence of blood rheology, von Willebrand factor, and blood coagulation. Circulation 1988;78:1431–42. [DOI] [PubMed] [Google Scholar]
- [61].Ruggeri ZM, Zimmerman TS. von Willebrand factor and von Willebrand disease. Blood 1987;70:895–904. [PubMed] [Google Scholar]
- [62].Houdijk WPM, de Groot PG, Nievelstein PFEM, Sakariassen KS, Sixma JJ. Subendothelial proteins and platelet adhesion. von Willebrand factor and fibronectin, not thrombospondin, are involved in platelet adhesion to extracellular matrix of human vascular endothelial cells. Arteriosclerosis 1986;6:24–33. [DOI] [PubMed] [Google Scholar]
- [63].Furchgott RF, Vanhoutte PM. Endothelium-derived relaxing and contracting factors. FASEB J 1989;3:2007–18. [PubMed] [Google Scholar]
- [64].Sase K, Michel T. Expression and regulation of endothelial nitric oxide synthase. Trends Cardiovasc Med 1997;7:28–37. [DOI] [PubMed] [Google Scholar]
- [65].Moake JL, Turner NA, Stathopoulos NA, Nolasco LH, Hellums JD. Shear-induced platelet aggregation can be mediated by vWF released from platelets, as well as by exogenous large or unusually large vWF multimers, requires adenosine diphosphate, and is resistant to aspirin. Blood 1988;71:1366–74. [PubMed] [Google Scholar]
- [66].Moake JL, Turner NA, Stathopoulos NA, Nolasco LH, Hellums JD. Involvement of large plasma von Willebrand factor (vWF) multimers and unusually large vWF forms derived endothelial cells in shear stress-induced platelet aggregation. J Clin Invest 1986;78:1456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.