

SUMMARY
The mechanisms by which the early-life microbiota protects against environmental factors that promote childhood obesity remain largely unknown. Using a mouse model in which young mice are simultaneously exposed to antibiotics and a high-fat diet, we show that Lactobacillus species, predominant members of the small intestine microbiota, regulate intestinal epithelial cells (IECs) to limit diet-induced obesity during early-life. A Lactobacillus-derived metabolite, phenyllactic acid (PLA), protects against metabolic dysfunction caused by early-life exposure to antibiotics and a high-fat diet by increasing the abundance of peroxisome proliferator activated receptor γ (PPAR-γ) in small intestine IECs. Therefore, PLA is a microbiota-derived metabolite that activates protective pathways in the small intestinal epithelium to regulate intestinal lipid metabolism and prevent antibiotic-associated obesity during early life.
eTOC blurb
Shelton et al. determine that early-life antibiotics exacerbate diet-induced obesity by disrupting interactions between the gut microbiota and the small intestine epithelium. Their study identifies that a Lactobacillus-derived metabolite, phenyllactic acid, regulates intestinal PPAR-γ to limit fat accumulation, revealing a mechanism by which the early-life microbiota protects against metabolic dysfunction.
Graphical Abstract:
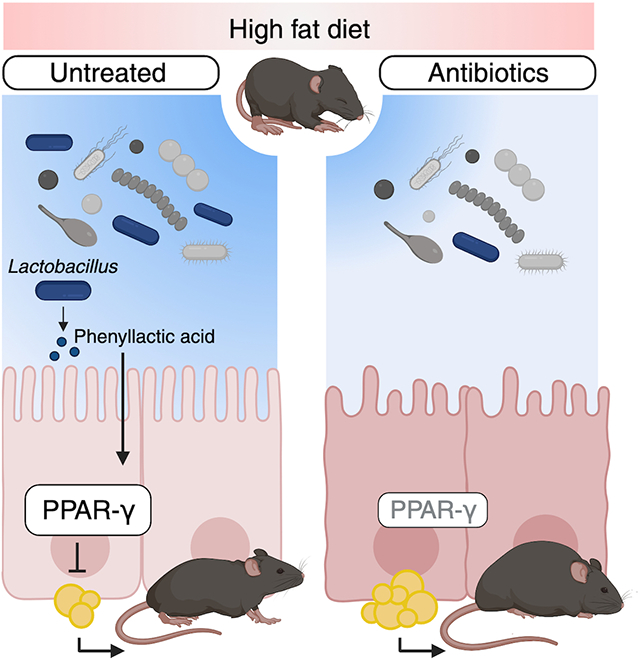
INTRODUCTION
The early-life gut microbiota plays a crucial role in promoting health1,2. In contrast to an adult microbiota, the infant microbiota is characterized by lower bacterial abundance and diversity3–5. As an infant grows, their microbiota becomes more complex and resembles that of an adult by ages 3 – 5 years old4. During this developmental window, the early-life microbiota is uniquely sensitive to perturbations by environmental factors, such as an obesogenic high fat (HF) diet or antibiotic treatment6,7. Disruption to the early-life gut microbiota is hypothesized to have lasting impacts on a child’s health. For instance, early-life antibiotic exposure is correlated with increased weight gain and body-mass-index in children8,9. Previous research using mouse models corroborated human studies and established a causal link between changes in the gut microbiota composition due to early-life antibiotic exposure and increased adiposity10,11. However, the mechanisms by which the gut microbiota exerts its protective effects in the context of childhood obesity are largely unknown.
An unappreciated aspect of the link between early-life antibiotic use and the risk for obesity is the impact of dietary habits on the microbiota. Diet is a key determinant of microbiota composition. For instance, an obesogenic Western-style HF diet perturbs the microbiota and elevates the risk for metabolic diseases12,13. In the United States, children have an elevated intake of saturated fats14 and a high percentage of therapeutic antibiotic usage 15. However, how concurrent exposure to a HF diet and antibiotics alters the risk for childhood obesity remains unknown. Thus, a complete understanding of how early-life antibiotic treatment increases adiposity requires an investigation into the compounding effects of diet and antibiotics on the gut microbiota and host metabolism.
Both the gut microbiota and diet regulate the function of the intestinal epithelium. During consumption of a Western-style obesogenic HF diet, the small intestine epithelium adapts to contend with the increase in dietary fat16. Specifically, the small intestine epithelium increases its’ ability to metabolize fats, protecting the host from features of metabolic dysfunction16–18. Concurrently, the small intestine microbiota changes upon exposure to a HF diet which impacts the function of the small intestine19, as the small intestine microbiota serves as an important regulator of intestinal lipid absorption, metabolism, and secretion19–21. Furthermore, recent work has determined that specific members of the small intestine microbiota can inhibit lipid secretion and thereby limit serum triglycerides during consumption of a HF diet22,23. Therefore, early-life antibiotics may promote obesity through perturbation to microbiota-epithelium interactions that protect against metabolic dysfunction caused by a HF diet.
In this manuscript, we investigate how early-life microbiota disruption promotes HF diet-induced obesity. We simultaneously exposed young mice to antibiotics and a high-fat diet and determined that antibiotic treatment leads to greater adiposity and metabolic dysfunction during consumption of a HF diet. Concurrent exposure to antibiotics and a HF diet depleted Lactobacillus species from the small intestine microbiota and experiments in colonized gnotobiotic identified the loss of Lactobacillus species as the driver of excess fat accumulation in our model. Characterization of the intestinal epithelium in mice given antibiotics and a HF diet revealed dysregulation of genes related to lipid metabolism, leading to increased triglycerides in the small intestine epithelium and serum. We propose that disruption to intestinal lipid metabolism leads to increased adiposity through depletion of peroxisome proliferator-activated receptor γ (PPAR-γ), a regulator of lipid metabolism, in the small intestine epithelium. Untargeted metabolomics revealed that antibiotics and a HF diet reduced the abundance of the Lactobacillus-derived metabolite, phenyllactic acid, in the small intestine lumen. We show that phenyllactic acid upregulated intestinal PPAR-γ and protected against metabolic dysfunction caused by early-life exposure to antibiotics and a high-fat diet. Therefore, PLA is a microbiota-derived metabolite that activates protective pathways in the small intestine epithelium to prevent obesity during early life.
RESULTS
Concurrent exposure to antibiotics and a HF diet during early life promotes long-lasting metabolic dysfunction
To investigate whether early-life concurrent exposure to detrimental environmental factors (e.g., obesogenic HF diet and antibiotics) have a synergistic effect in promoting obesity, we first established a mouse model (Fig. 1A) in which young (3-week-old) mice were exposed to either a low fat (LF) (10%) or high fat (HF) (60%) diet with or without low dose penicillin treatment (LDP, 6.67 mg/L)10. Mice were maintained on their respective diets and antibiotics for 5 weeks until they reached sexual maturity. After 5 weeks, mice exposed to a HF diet and LDP gained significantly more weight (Fig. 1B) and abdominal fat (Fig. 1C) than mice fed a HF diet alone, despite equal food consumption (fig. S1A). Furthermore, no difference in weight gain (Fig. 1B) or abdominal fat (Fig. 1C) was observed between mice fed a LF diet and mice given a LF diet and LDP, indicating that antibiotic exposure alone was insufficient to promote additional adiposity in our model. To assess whether the increased weight gain in mice exposed to HF diet and LDP was due to overall body composition changes (e.g., lean mass or bone mineral density), we performed DEXA scanning in animals exposed to HF diet or HF diet and LDP (fig. S1, B – C). DEXA analysis revealed no differences in lean weight (fig. S1B) or bone mineral density (fig. S1C) between experimental groups, suggesting that a HF diet and LDP specifically increase fat accumulation. Mice treated with LDP and given a HF diet also developed additional features of metabolic dysfunction including increased fasting blood glucose (Fig. 1D) and greater fat accumulation in hepatocytes (Fig. 1, E and F) compared to HF diet fed mice.
Figure 1. Early-life exposure to low dose penicillin (LDP) and a high fat (HF) diet promotes metabolic dysfunction.
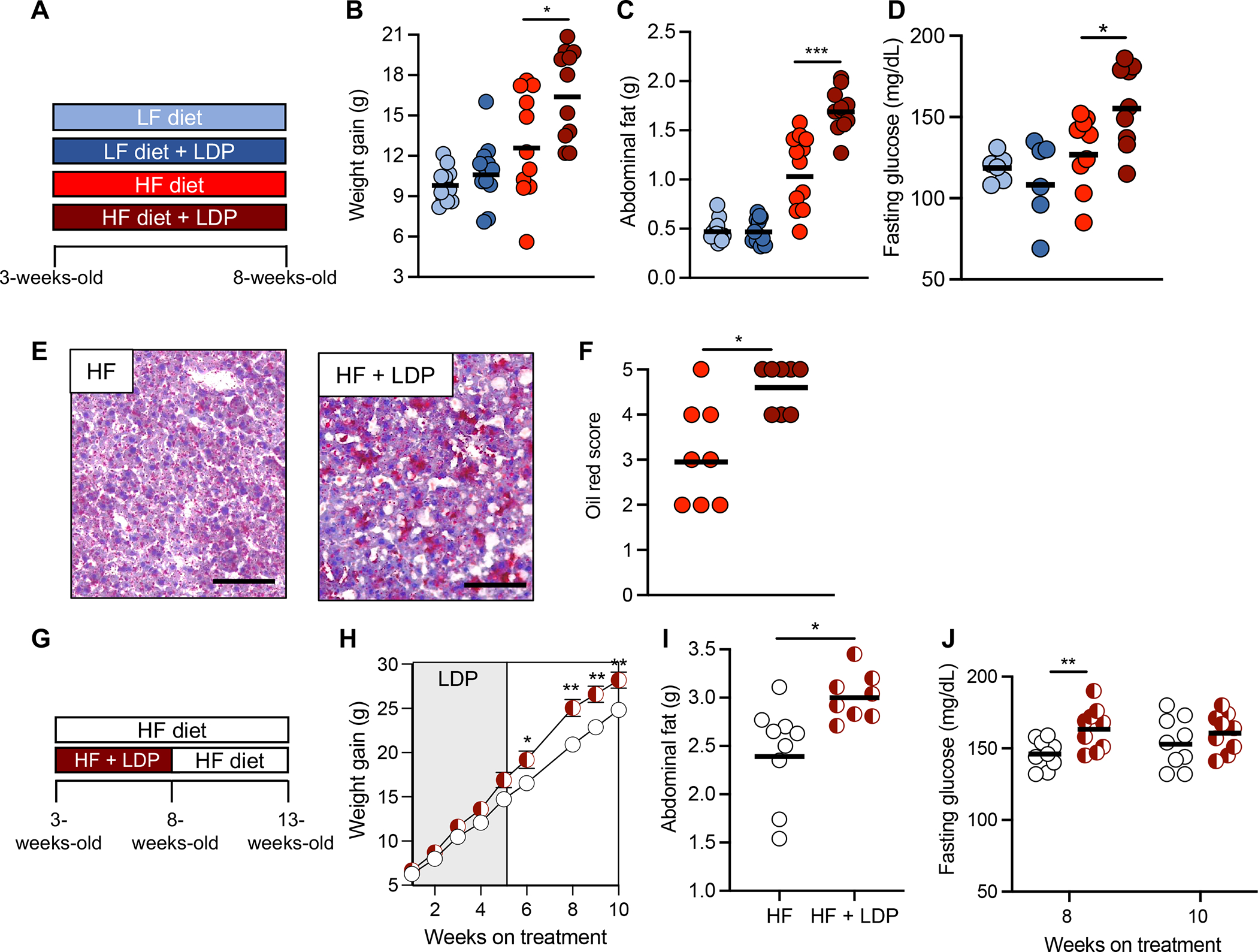
(A, G) Schematics of the experimental models and the groups used. (B) Weight gain, (C) abdominal fat (g), and (D) fasting glucose levels of mice in each group after 5 weeks. (E) Representative images of oil red O-stained sections of liver from HF diet and HF diet + LDP-treated mice. Scale bar represents 100 μm. (F) Combined oil red scores from livers of mice given a HF diet or a HF diet + LDP. (H) Weight gain of mice in each group over the course of the 10-week experiment. (I) Abdominal fat (g) measured after the 10-week diet and antibiotic manipulations. (J) Fasting glucose levels were measured after 8 and 10 weeks on a HF diet. (B – D, F, I, and J) Each dot represents one animal. Bars represent geometric mean. (H) Dots represent mean ± standard error of the mean. (B, C) N = 12 mice/group. Data representative of two independent cohorts (D, F, H – J) N = 9 mice/group. Data representative of one independent cohort. (B-D, I - J) *, p < 0.05; **, p < 0.01; ***, p < 0.005 using an unpaired two-tailed Student’s t test. (F) *, p < 0.05 using Mann-Whitney test. (H) *, p < 0.05; **, p < 0.01 using multiple unpaired two-tailed Student’s t tests.
To determine if the metabolic dysfunction persisted after antibiotic treatment ended, mice exposed to either a HF diet and LDP or a HF diet alone were given only a HF diet for an additional 5 weeks (Fig. 1G). HF diet and LDP-exposed mice gained more weight (Fig. 1H) and abdominal fat (Fig. 1I) than mice only given a HF diet for 10 weeks, despite no longer receiving LDP and consuming equal amounts of food throughout the experiment (fig. S2A). Mice treated with LDP and fed a HF diet also sustained elevated fasting blood glucose 3 weeks after antibiotics were removed (Fig. 1J). Differences between experimental groups were not significant at 10 weeks, when some mice exposed to HF diet alone started to show increased fasting glucose levels24. This data suggests that early life exposure to HF diet and LDP causes increased fasting blood glucose (Fig. 1D), and that glucose levels remain elevated in this experimental group even weeks after antibiotics are removed (Fig. 1J). Consumption of a HF diet was required for LDP to promote lasting adiposity as mice switched onto a LF diet after 5 weeks of exposure to a HF diet and LDP no longer gained additional weight or adiposity (fig. S2, B – D). Moreover, mice treated with LDP and fed a LF diet were also given a LF diet alone for 5 additional weeks and no differences in weight gain or abdominal fat were observed (fig. S2, E – G). Collectively, these data reveal that concurrent early-life exposure to antibiotics and a HF diet leads to early-onset and lasting HF-dependent obesity and metabolic dysfunction through unknown mechanisms.
Depletion of Ligilactobacillus murinus from the small intestine microbiota increases fat accumulation
As disruption of the early-life microbiota by antibiotics was previously shown to promote adiposity11, we hypothesized that simultaneous exposure to antibiotics and a HF diet altered the gut microbiota to accelerate fat deposition. Antibiotic treatment increased metabolic dysfunction after a 5-week treatment only in young mice fed an obesogenic HF diet (Fig. 1, B – H), pointing to a contribution of dietary lipids to our phenotypes. As the small intestine is the primary site for dietary fat absorption and metabolism19, we decided to investigate changes in the small intestine microbiota during concurrent early-life exposure to HF diet and antibiotics. 16S rRNA analysis of the distal small intestine microbiota revealed that diet was the most predominant driver of compositional changes between the 4 experimental groups (fig. S3, A – C) consistent with previously published research25. As antibiotics promoted adiposity only when young mice consumed a HF diet (Fig. 1C), we focused on differences between the small intestine microbiota of mice given a HF diet or a HF diet and LDP and not mice that were fed a LF diet or a LF diet and LDP. Relative to the HF diet only group, exposure to LDP led to significant depletion of bacteria belonging to the Lactobacillaceae family, the most abundant family in their small intestine microbiota (Fig. 2, A and B; fig. S3, C and D). Interestingly, in vitro growth of Lactobacillus species indicated sensitivity to the combination of penicillin and lipids or the combination of penicillin and bile salts (elevated in the intestine during consumption of a high-fat diet26) (fig. S3, F and G), providing an explanation for the depletion of Lactobacillus spp. during exposure to penicillin and a HF diet. While Lactobacillus levels recovered in HF diet and LDP-exposed mice when they were switched onto a HF diet alone, their weight gain still persisted, (Fig 1H; fig. S3, H and I), supporting the hypothesis that short-term microbiota perturbations can promote lasting metabolic changes11.
Figure 2. Ligilactobacillus murinus protects against adiposity during early-life consumption of a HF diet.
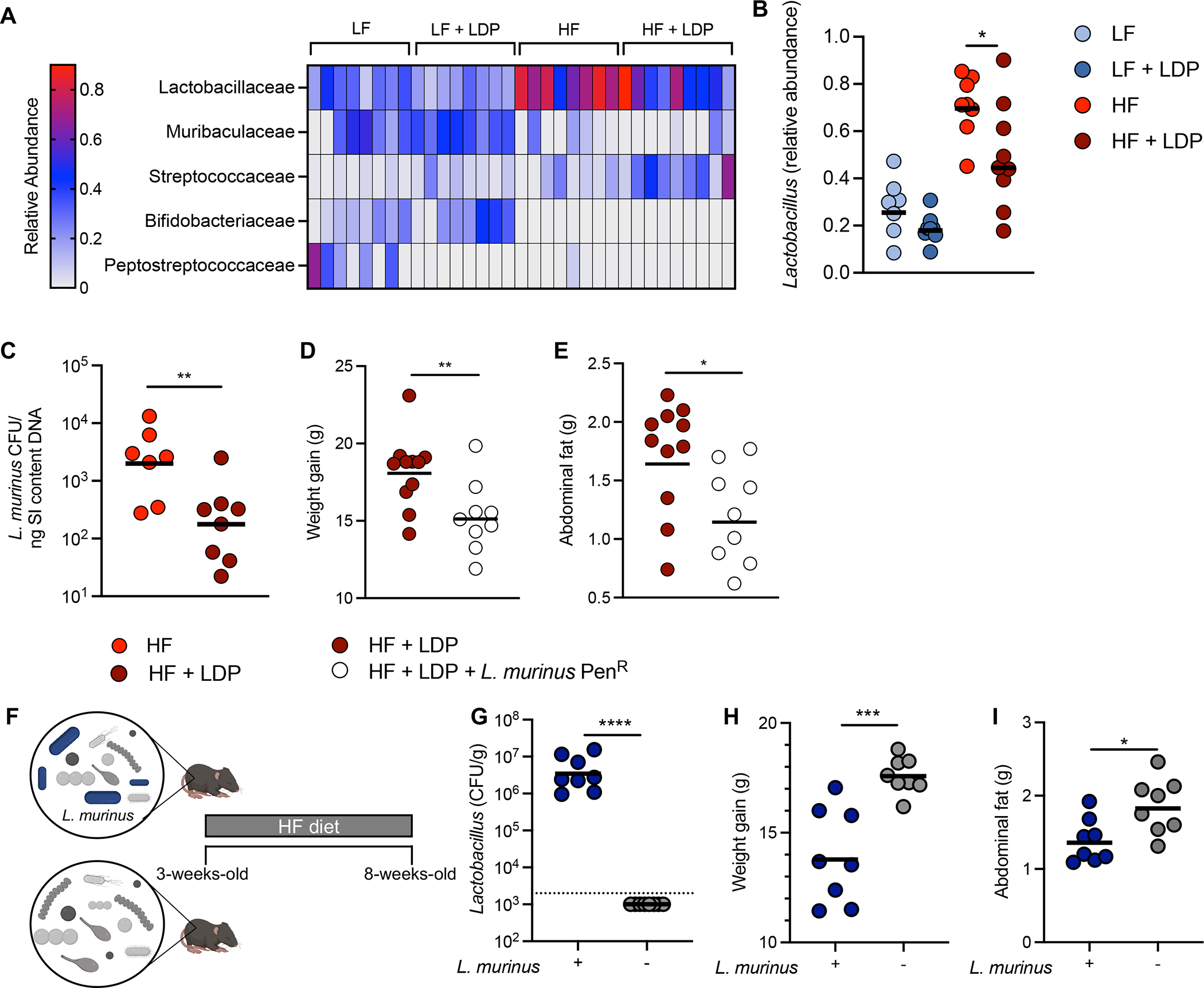
(A) Heat map showing differential abundance of the 5 most abundant families in the small intestine microbiota of mice after 5 weeks of treatment. (B) Relative abundance of the genus Lactobacillus in the small intestine microbiota as determined by 16S rRNA sequencing. (C) The abundance of L. murinus in the small intestine as determined by qPCR. (D) Weight gain and (E) abdominal fat of mice given a HF diet and LDP alone or gavaged with a penicillin resistant strain of L. murinus (L. murinus PenR) after 5 weeks. (F) A schematic representation of the gnotobiotic experiment. (G) Lactobacillus abundance (colony forming unit (CFU) / gram) in small intestine (SI) after 5 weeks on a HF. Dotted line indicates limit of detection for Lactobacillus species in this experiment. (H) Weight gain and (I) abdominal fat (g) of mice after 5 weeks. Each dot represents one animal. Bars represent geometric mean. (A) N = 5 mice/group; (B) N = 9 mice/group; (C) N = 7 – 8 mice/group; (D – E) N = 12 mice/group; (G – H) N = 8 mice/group. Data representative of two independent cohorts. *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001 using an unpaired two-tailed Student’s t test.
We next investigated if the loss of Lactobacillus species promoted metabolic dysfunction in mice exposed to antibiotics and a HF diet. Quantitative PCR revealed that the predominant Lactobacillus species present in the small intestine of untreated mice was Ligilactobacillus murinus (L. murinus) (fig. S3E). Moreover, we determined that L. murinus was significantly depleted from the small intestine microbiota of mice given LDP and a HF diet (Fig. 2C). Therefore, we generated a strain of L. murinus resistant to penicillin (L. murinus PenR) and used this strain to restore the presence of L. murinus in the intestinal lumen by re-colonizing a subset of mice exposed to a HF diet and LDP with L. murinus PenR. Mice given L. murinus PenR gained less weight (Fig. 2D) and abdominal fat (Fig. 2E) than mice exposed to a HF diet and LDP alone. Due to the transient colonization seen with L. murinus PenR (data not shown), we performed an additional experiment using a defined microbiota strategy in gnotobiotic mice. Young (3-week-old) germ-free mice were colonized with either a defined microbiota containing L. murinus or lacking L. murinus (Fig. 2F) and were then maintained on a HF diet for 5 weeks. Mice colonized with a microbiota containing L. murinus (Fig. 2G) gained less weight (Fig. 2H) and accumulated less abdominal fat (Fig. 2I) than mice given a microbiota without L. murinus. Together, these data suggest that L. murinus protects young mice from adiposity during the consumption of a HF diet.
Our model utilizes chronic, low doses of antibiotics27, an exposure that largely results from dietary sources28. However, children can also be treated with therapeutic doses of antibiotics during infections, which our LDP treatment does not replicate. To address if therapeutically relevant concentrations of antibiotics combined with a HF diet led to similar physiological effects as LDP, we exposed young mice to three clinical doses of penicillin (CDP, 0.167 mg/L) (in combination with an LF or HF diet) over 5 weeks (Fig. 3A). Consistent with LDP, concurrent exposure to a HF diet and clinical doses of antibiotics caused marked weight gain (Fig. 3B), as well as increased abdominal fat (Fig. 3C), and fasting blood glucose (Fig. 3D) of mice compared to mice given only a HF diet. As seen in our LDP model, CDP depleted Lactobacillus from the microbiota of young mice (Fig. 3, E and F), supporting the role of Lactobacillus depletion from the small intestine microbiota as a driver of the synergistic effect of clinical and subclinical antibiotic exposure and HF diet in promoting obesity. To further investigate how L. murinus exerts its anti-obesity effects, we chose to utilize our HF diet and LDP model, as mice simultaneously exposed to a HF diet and LDP had sustained Lactobacillus depletion from their small intestinal microbiota (Fig. 1, I and J; fig. S3H).
Figure 3. Repeated treatment with clinical doses of penicillin (CDP) and a high fat diet promote increased adiposity.
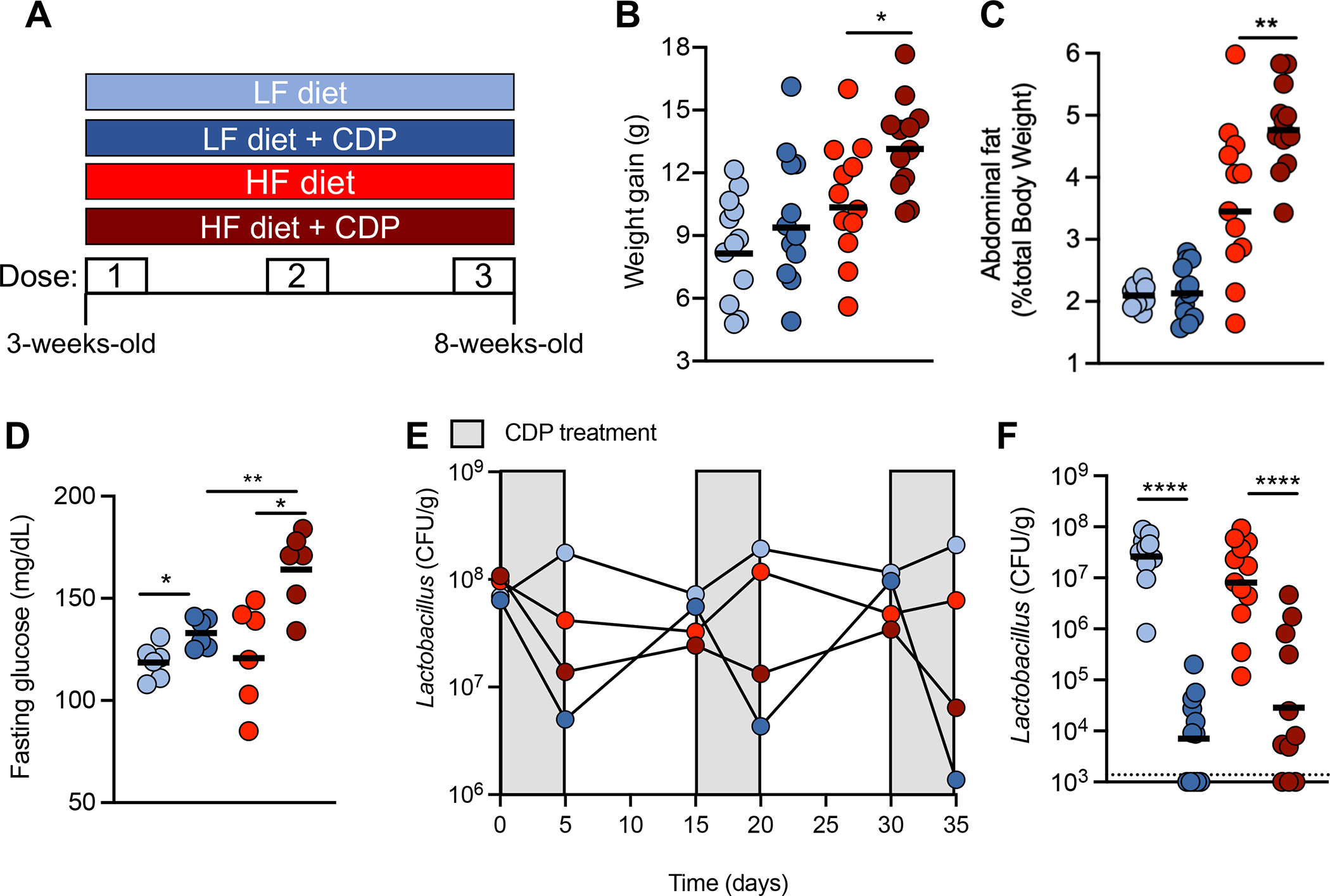
(A) A schematic of the experimental model and the groups used. (B) Weight gain, (C) percent abdominal fat, and (D) fasting glucose levels of mice after 5 weeks. (E) Lactobacillus abundance (Colony forming unit (CFU) / gram in the feces of treated mice over the course of the 5-week experiment. (F) Lactobacillus abundance in the small intestine was determined after 5 weeks. (A – D, F) Each dot represents one animal. Bars represent geometric mean. N = 12 mice/group. (E) Dots indicate geometric mean of abundances from six mice. *, p < 0.05; **, p < 0.01; ****, p < 0.001 using an unpaired two-tailed Student’s t test.
Early-life antibiotics disrupt intestinal lipid metabolism during the consumption of a HF diet
A mechanism by which disruptions to the gut microbiota can lead to metabolic dysfunction is through perturbation of intestinal epithelium function29,30. The intestinal epithelium contributes to a healthy host metabolism through numerous mechanisms, including regulation of lipid metabolism, uptake, and secretion as well as gut hormone production31. Therefore, changes in the early-life gut microbiota due to antibiotic exposure and a HF diet may contribute to childhood obesity by disrupting the activity of intestinal epithelial cells. To determine if the combination of a HF diet and antibiotics affected the intestinal epithelium, we examined sections of the ileum after 5-weeks of exposure to either a LF or HF diet with or without concurrent treatment with LDP (Fig. 4A). Histopathological analysis of sections revealed that the individual impact of either LDP treatment or a HF diet synergized in perturbing the intestinal epithelium in mice exposed to both LDP and a HF diet (Fig. 4B). To gain a deeper understanding of how exposure to a HF diet and LDP altered the function of the intestinal epithelium, we next performed RNA-sequencing of distal small intestine epithelial cells (IECs) in mice given a HF diet or a HF diet and LDP. We determined that concurrent exposure to an obesogenic HF diet and antibiotics altered the expression of approximately 200 genes (compared to mice fed a HF diet alone) belonging to diverse pathways (Fig. 4, C and D). Analysis of gene pathways altered in HF diet and LDP-treated mice revealed perturbed expression of transcripts related to lipid and fatty acid metabolism (Fig. 4D). Quantitative PCR analysis of small intestine IECs confirmed RNA-sequencing results and showed a significant difference in the expression of genes related to lipid digestion (Angptl4) and absorption (Cd36), trafficking (Fabp1 and Fabp2), and secretion (Mttp and ApoB) (Fig. 4E)32–37. Despite increased Cd36 expression, mice that were given a HF diet and LDP did not absorb more fat from the diet compared to mice that were only fed a HF diet (Fig. 4F). However, we measured significantly higher triglycerides in the small intestine IECs of fasted HF diet and LDP treated mice (Fig. 4G). We also determined that mice exposed to a HF diet and LDP had significantly more lipids in their fasted serum than mice given only a HF diet (Fig. 4H). Thus, we reasoned that early-life treatment with LDP perturbed intestinal lipid metabolism, leading to increased lipid transport from the intestine into circulation. To investigate this hypothesis, we injected mice given a HF diet or HF diet and LDP with the lipoprotein lipase inhibitor tyloxapol, and then challenged the mice with a bolus of olive oil. Mice exposed to a HF diet and LDP had significantly more serum triglycerides two hours after the olive oil gavage (Fig. 4I), suggesting that their elevated serum lipids (Fig. 4H) result from greater intestinal lipid secretion.
Figure 4. Exposure to early-life antibiotics alters the response of the intestinal epithelium to a high fat diet.
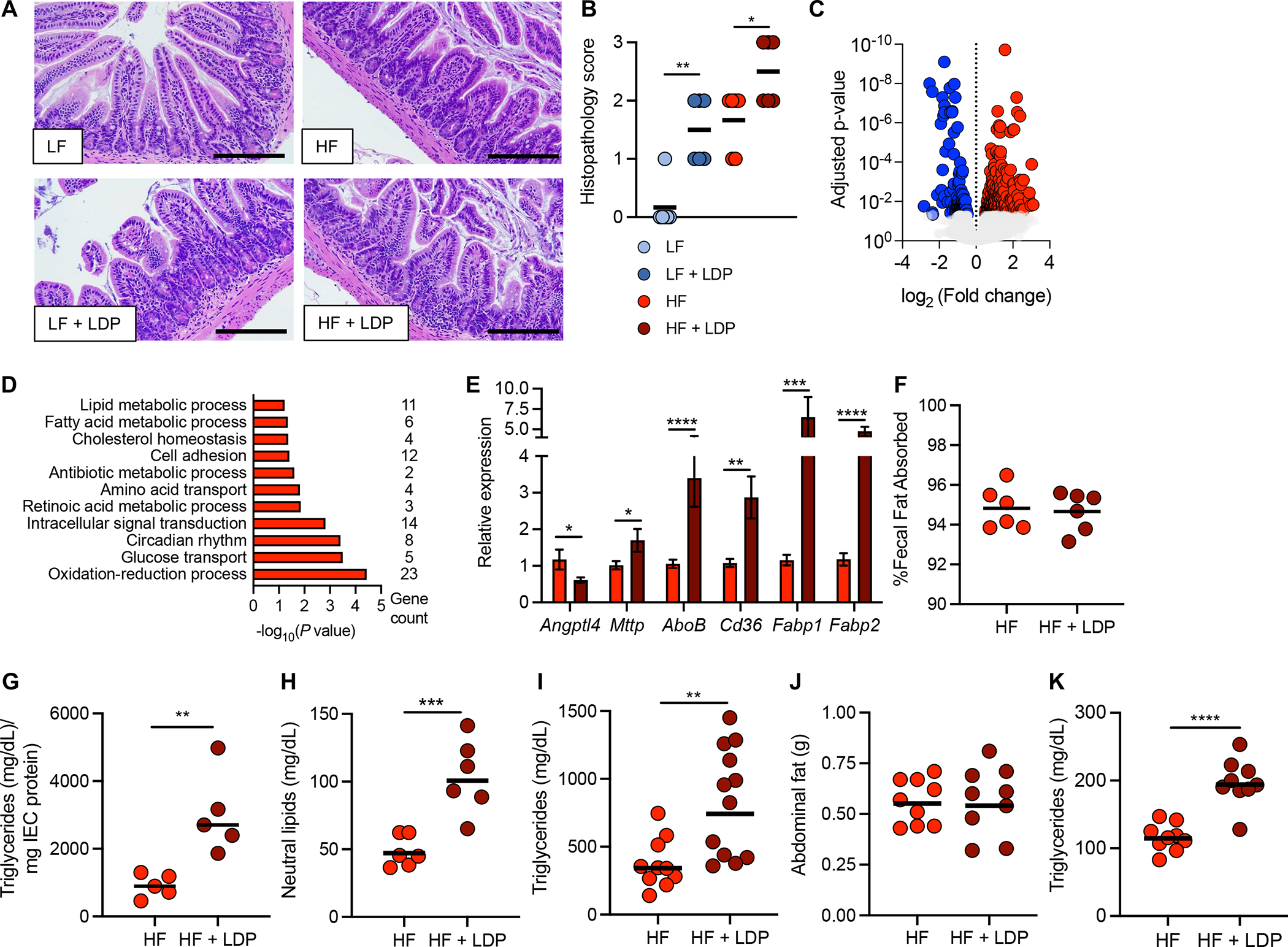
(A) Hematoxylin and eosin-stained sections of the ileum from mice after 5 weeks of exposure to a LF or HF diet ± LDP. (B) Histopathology scores of ileum sections. Scale bar represents 200 μm. (C and D) RNA-sequencing analysis of epithelial cells isolated from the ileum (IECs) of mice after 5 weeks of exposure to a HF diet or a HF diet + LDP. (C) Volcano plot of genes significantly (an adjusted p-value < 0.05) upregulated (red) and downregulated (blue) in mice exposed to a HF diet + LDP compared to mice given only a HF diet. (D) Enriched GO (gene ontology) categories in IECs of mice fed a HF diet + LDP-treatment. (E) Epithelial transcripts of the indicated genes measured by qPCR in mice given a HF diet or a HF diet and LDP. (F) Fecal fat absorption was measured after exposure to a HF diet or HF diet and LDP for 5 weeks. (G - I) Mice exposed to a HF diet or a HF diet and LDP for 5 weeks were fasted and (G) triglyceride abundance in IECs and (H) neutral lipids in the serum were measured. (I) An oral fat tolerance test was performed in mice given a HF diet or a HF diet and LDP for 5 weeks. Serum triglyceride concentration was measured two hours after the bolus of olive oil. (J) Abdominal fat (g) and (K) fasting triglycerides of mice given a HF diet or a HF diet and LDP for 2 weeks. (B and E) Bars represent mean ± standard error of the mean (SEM). (F – K) Bars represent geometric mean. Each dot represents one animal. (B) N = 6 mice/group. Data representative of one independent cohort; (C – D) N = 3 mice/group. (E) N = 14 mice/group. Data representative of 2 independent cohorts. (F – H) N = 5–6 mice/group. Data representative of one independent cohort. (I) N =12 mice/group. Data representative of one independent cohort. (J– K) N = 9 mice/group. Data representative of one independent cohort. (B)*, p < 0.05; **, p < 0.01 using a one-tailed Mann-Whitney test. (E – K) *, p < 0.05; **, p < 0.01; ***, p < 0.005; ****, p < 0.001 using an unpaired two-tailed Student’s t test.
To determine if the elevated serum lipids in mice exposed to a HF diet and LDP preceded their increased adiposity, we investigated differences in physiology after only two weeks on a HF diet and LDP. Despite a reduction in Lactobacillus species in the small intestine (fig. S4A), mice given a HF diet and LDP did not gain more weight (fig. S4B) or accumulate more abdominal fat (Fig. 4J) after only two weeks of exposure. However, HF diet and LDP-treated mice had significantly greater triglycerides in their fasted serum (Fig. 4K). To understand if elevated triglycerides in the serum coincided with perturbation of intestinal lipid metabolism after two weeks of exposure to a HF diet and LDP, we measured the expression of the same genes that were altered after 5 weeks in mice given a HF diet and LDP (Fig. 4E). Interestingly, expression of Angptl4 was decreased after exposure to a HF diet and LDP for two weeks, but no significant differences were observed in the expression of the other lipid metabolism genes (fig. S4F). Our data suggests that exposure to a HF diet and LDP dysregulates intestinal lipid metabolism, leading to elevated serum triglycerides, promoting excess weight gain and adiposity.
Activation of intestinal PPAR-γ by L. murinus protects against fat accumulation during consumption of a HF diet
We next investigated how a HF diet and LDP disrupted intestinal lipid metabolism, specifically focusing on mechanisms leading to reduced Angptl4 in small intestine IECs, as this preceded the development of metabolic dysfunction. In the intestine, peroxisome-proliferator activated receptor gamma (PPAR-γ), a nuclear receptor, regulates Angptl4 expression38,39 as well as lipid metabolism in the small intestine40. Importantly, PPAR-γ abundance and activity in colonic IECs decreases during antibiotic treatment38. Therefore, we investigated whether intestinal PPAR-γ was a mechanism connecting microbiota perturbations to dysregulated intestinal lipid metabolism during early-life exposure to a HF diet and LDP. LDP treatment did not impact the gene expression and protein abundance of PPAR-γ in small intestine IECs when mice were fed a LF diet (fig. S5, A–D). However, PPAR-γ gene expression and protein abundance in small intestine IECs was significantly reduced in mice given a HF diet and LDP (relative to mice fed a HF diet alone) (Fig. 5, A – D). No changes in the expression of Pparg or related target genes were observed in the adipose tissue or liver (fig. S5, E and F). Our findings suggest that exposure to a HF diet and antibiotics specifically depleted PPAR-γ from small intestine IECs.
Figure 5. Loss of intestinal PPAR-γ signaling promotes increased adiposity due to exposure to LDP and a HF diet.
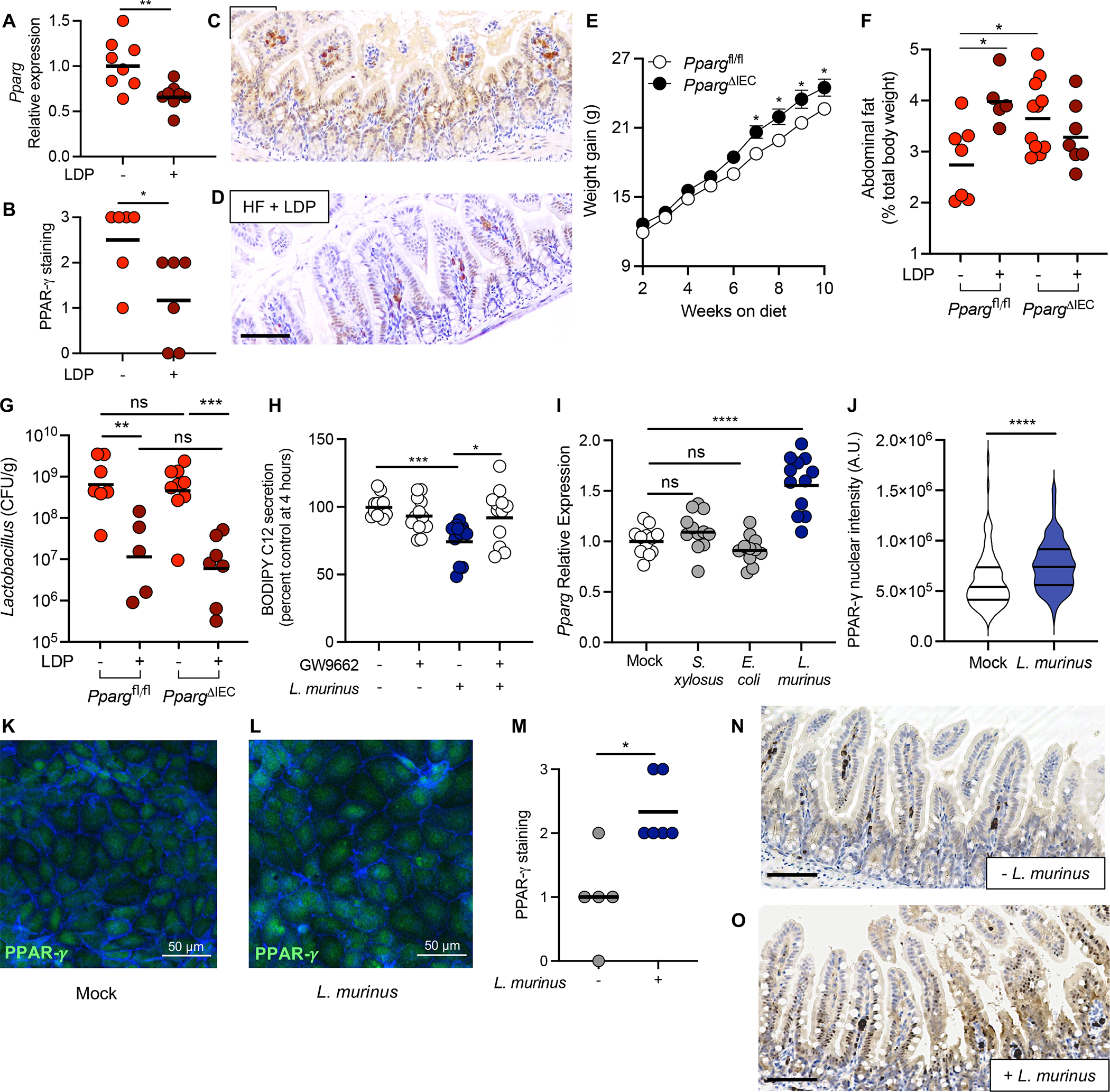
(A) Expression of Pparg in the ileum epithelium of HF diet or HF diet + LDP-treated mice measured by qPCR. (B) PPAR-γ abundance in the epithelium was quantified by scoring stained sections of the ileum. (C – D) Representative images of PPAR-γ staining in HF diet fed mice (C) and HF diet fed + LDP-treated mice (D). Scale bar represents 200 μm. (E) 3-week-old Ppargfl/flVillincre/− mice (PpargΔIEC) and littermate control Ppargfl/flVillin−/− mice (Ppargfl/fl) were fed a HF diet for 10 weeks and weight gain was determined weekly. (F) Abdominal fat (% total body weight) and (G) Lactobacillus abundance (colony forming unit (CFU) / gram) in the small intestine (SI) of PpargΔIEC and Ppargfl/fl measured after the 6-week diet and antibiotic manipulations. (H - L) Intestinal epithelial cells were infected with Ligilactobacillus murinus (L. murinus) at an MOI of 100. (H) Amount of BODIPY C12 secreted by treated Caco-2 cells was measured 4 hours after addition of lipid micelles. (I) Immortalized mouse small intestinal epithelial (MSIE) cells were mock-treated or treated with E. coli Mt1B1, S. xylosus 33-ERD13C, or L. murinus, before expression of Pparg was measured using qPCR. (J - L) MSIE cells were treated with L. murinus (L) or left untreated (K) and incubated for 16 hours. Cells were subsequently stained with anti-PPAR-γ and phalloidin to stain for F-actin. (J) PPAR-γ nuclear intensity was quantified using ImageJ; 15 cells were selected from 2 images from 3 independent experiments. (M - O) Germ-free mice were colonized with a defined microbiota with or without L. murinus and placed on a HF diet for 5 weeks. (M) PPAR-γ abundance in the epithelium was quantified by scoring stained sections of the ileum. Representative images of PPAR-γ staining in mice colonized with (O) or without (N) L. murinus. Scale bar represents 200 μm. (A, F, G) Each dot represents one animal. Bars represent geometric mean. N = 5 – 7 mice/group. (B and M) Each dot represents one animal. Bars represent mean. N = 6 – 7 mice/group. Data representative from one independent cohort. (E) Dots represent mean +/− SEM (N = 8 – 9/ genotype). (H and I) Each dot represents one technical replicate (Data shown from 3 biological replicates). Bars represent geometric mean. (J) Violin plot showing the distribution of PPAR-γ nuclear intensity (n = 90 cells). (A, G, H, I, J) *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 an unpaired two-tailed Student’s t test. (B, M) *, p < 0.05 using a two-tailed Mann-Whitney test. (E) *, p < 0.0332 using a two-way ANOVA with Šídák’s multiple comparisons test. (F) *, p < 0.05 using a one-way ANOVA with Šídák’s multiple comparisons test.
With the reduction of intestinal PPAR-γ observed in HF diet and LDP-treated mice, we next asked if genetic ablation of PPAR-γ specifically in IECs resulted in increased weight gain and metabolic dysfunction. Intestinal epithelial-specific PPAR-γ knockout mice (PpargΔIEC) and littermate control mice (Ppargfl/fl) were fed a LF or HF diet for 10 weeks (Fig. 5E, fig. S5G). PpargΔIEC gained more weight and fat than their littermate controls when fed a HF diet (Fig. 5E, fig. S5H). Consistent with the effects of a HF diet and LDP, PpargΔIEC did not gain more weight or fat when given a LF diet (fig. S5, G and H). To determine if the obesogenic effects of a HF diet and LDP act through depletion of intestinal PPAR-γ, we exposed PpargΔIEC and wildtype Ppargfl/fl to a HF diet or a HF diet and LDP. Wildtype Ppargfl/fl mice developed significantly more abdominal fat when exposed to a HF diet and LDP compared to when only fed a HF diet (Fig. 5F). However, a HF diet and LDP treatment failed to increase the amount of adiposity accumulated by PpargΔIEC mice (Fig. 5F), supporting our hypothesis that LDP promotes fat accumulation during exposure to a HF diet by blunting PPAR-γ signaling in the intestinal epithelium. To confirm that differences in adiposity were not attributable to changes in LDP activity, Lactobacillus abundance in the small intestine of PpargΔIEC and Ppargfl/fl mice was measured. No difference in Lactobacillus abundance was observed between PpargΔIEC and Ppargfl/fl mice fed a HF diet or between PpargΔIEC and Ppargfl/fl mice exposed to a HF diet and LDP (Fig. 5G).
Lactobacillus species can modulate lipid secretion in IECs through activation of the PPARs22,23. As we observed a reduction in Lactobacillus spp. upon exposure to a HF diet and LDP (Fig. 2A), we next investigated whether Lactobacillus spp. could activate PPAR-γ in vitro. As we observed that L. murinus protected young mice from HF diet-induced obesity (Fig. 2I), we utilized this isolate to interrogate the relationship between Lactobacillus spp. and PPAR-γ. In intestinal epithelial (Caco-2) cells, we determined that L. murinus increased the transcriptional activity of PPAR-γ to a similar level as the known PPAR-γ agonist, butyrate38 (fig. S6A). Furthermore, we observed that L. murinus significantly increased the nuclear localization of PPAR-γ (fig. S6, B, C, E). As L. murinus regulated both the abundance and activity of PPAR-γ, we then asked whether L. murinus affected lipid secretion in a PPAR-γ-dependent manner. In agreement with previous findings23, L. murinus decreased secretion of micelles containing a fluorescent fatty acid (BODIPY C12) by Caco-2 cells (Fig. 5H). However, adding a PPAR-γ inhibitor, GW9662, blocked the ability of L. murinus to inhibit lipid secretion (Fig. 5H), suggesting that L. murinus increases PPAR-γ activity in IECs to reduce lipid secretion from the intestine.
To continue our investigation into the relationship between L. murinus and PPAR-γ, we transitioned to the small intestine cell line to recapitulate our mouse model more accurately. Pparg expression was increased in mouse small intestinal epithelial (MSIE) cells treated with L. murinus (Fig. 5I). In contrast, treatment with Escherichia coli Mt1B1 or Staphylococcus xylosus failed to increase expression of Pparg in MSIE cells, suggesting that the increase in Pparg expression is unique to L. murinus and not a result of co-culture with bacteria (Fig. 5I). Moreover, compared to mock-treated cells, L. murinus significantly increased the amount of PPAR-γ in the nucleus in MSIE cells (Fig. 5, J – L) (consistent with our findings in Caco-2 cells; fig. S6). Further evidence of a specific interaction with L. murinus and intestinal PPAR-γ is seen in gnotobiotic mice colonized with a defined microbiota with or without L. murinus (Fig. 2F). In agreement with our in vitro data, mice colonized with a microbiota containing L. murinus have significantly higher levels of PPAR-γ staining in their intestine compared to mice lacking L. murinus (Fig. 5, M – O).
As PPAR-γ is known to be regulated by microbiota-derived metabolites38,41, we next investigated if the cell-free supernatant from L. murinus cultures could increase PPAR-γ nuclear localization. Indeed, the supernatant from L. murinus cultures, but not E. coli Mt1B1, significantly increased the amount of PPAR-γ accumulating in the nucleus (fig. S6, B – I). Together, our in vitro data reveals that Lactobacillus-derived metabolites activate PPAR-γ, leading us to hypothesize that the loss of PPAR-γ activity due to exposure to a HF diet and antibiotics occurred due to the depletion of Lactobacillus-derived metabolites.
Phenyllactic acid is depleted from the small intestine lumen and regulates intestinal PPAR-γ
To identify Lactobacillus metabolites responsible for activating intestinal PPAR-γ, untargeted metabolomics was performed on the small intestine contents from mice given a HF diet or a HF diet and LDP (Fig. 6A). Multiple metabolites known to be produced by Lactobacillus species were significantly depleted in mice exposed to a HF diet and LDP, including phenyllactic acid (PLA) (Fig. 6B). As previous work identified that Lactobacillus-derived PLA upregulates PPARG in adipocytes42, we investigated whether PLA was the L. murinus-derived metabolite in supernatants that regulated PPAR-γ in IECs. First, we determined that intestinal PLA abundance is dependent on the gut microbiota as germ-free mice have significantly less PLA in their small intestine compared to conventional mice when fed a HF diet (Fig. 6C). PLA was then measured in supernatants from cultures of the defined microbiota used to colonize germ-free mice (Fig. 2F). PLA levels were significantly greater in supernatants of the defined microbiota containing L. murinus (Fig. 6D), indicating that L. murinus is the primary producer of PLA compared to other microbes present in our defined small intestine microbiota. Furthermore, we determined that L. murinus, alone, produced PLA in cell culture experiments (Fig. 6E). Importantly, L. murinus PenR (Fig. 2, D – E), another strain of L. murinus (DSM 20452), different Lactobacillus spp., and Bifidobacterium animalis also all produce PLA in vitro (Fig. 6E), highlighting that the ability to produce PLA is conserved between multiple bacterial species present in the gut microbiota. To investigate PLA production by L. murinus in vivo, we measured intestinal PLA levels in mice given L. murinus PenR during exposure to a HF diet and LDP (Fig. 2, D and E). Compared to mice treated with a HF diet and LDP alone, mice colonized with L. murinus PenR had significantly higher PLA levels in their small intestine content (Fig. 6F), supporting our hypothesis that Lactobacillus species are a significant contributor to the intestinal PLA abundance.
Figure 6. Exposure to a HF diet and LDP depletes phenyllactic acid, an activator of intestinal PPAR-γ, from the small intestine.
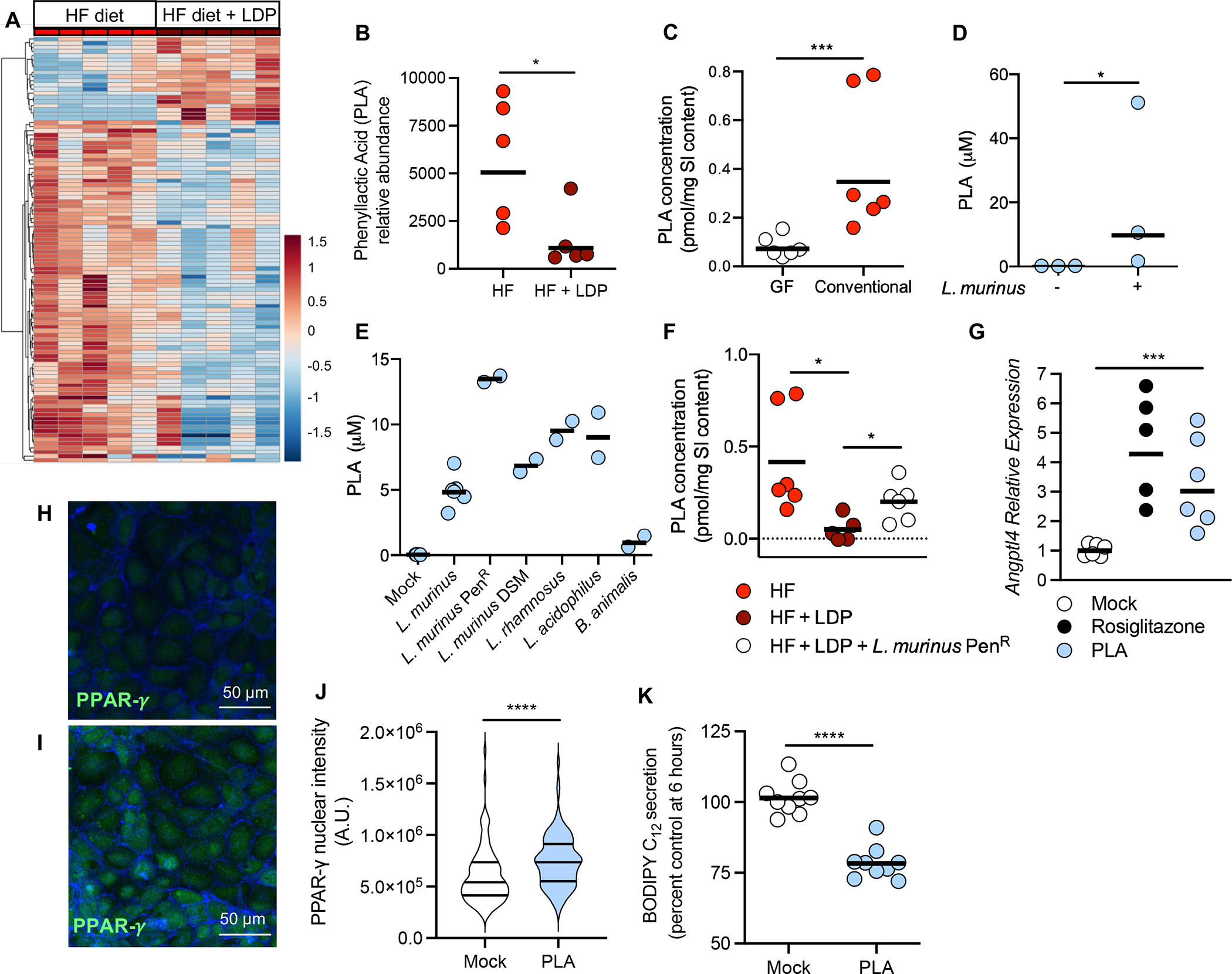
(A and B) Untargeted metabolomics was performed on ileum content from mice given a HF diet or a HF diet + LDP (N = 5/phenotype). (A) Heat map clustering of the experimental sample groups and metabolites. Samples (columns) are clustered by group and relative abundance (rows) across different groups, ordered from low (blue) to high (red) abundance. (B) Normalized relative abundance, as determined by untargeted metabolomics, of phenyllactic acid (PLA) in the small intestine content of mice fed a HF diet or a HF diet + LDP-treated. (C) Concentration (pmol/mg of intestinal content) of PLA in the small intestine of germ-free (GF) and conventional mice fed a HF diet for 5 weeks. (D) Concentration (μM) of PLA in supernatants from defined microbiota cultures lacking or containing L. murinus. (E) Concentration (μM) of PLA in the supernatant of lactic acid bacteria grown in cell culture media. (F) Concentration (pmol/mg of intestinal content) of PLA in the small intestine of mice. Dotted line indicates limit of detection = 0.025 pmol/mg. (G) Mouse enteroids were treated with either 10 μM Rosiglitazone or 5 mM PLA or left untreated. (H and I) Immortalized mouse small intestinal cells were treated with 5 mM PLA (F) or left untreated (E). Cells were subsequently stained with anti-PPAR-γ and phalloidin to identify F-actin. (J) PPAR-γ nuclear intensity was quantified using ImageJ; 15 cells were selected from 2 images from 3 independent experiments. (K) Amount of BODIPY C12 secreted by treated Caco-2 cells was measured 6 hours after addition of lipid micelles. (B, C,) Each dot represents one animal. Lines represent geometric mean. (F) Each dot represents one animal. Lines represent mean. (B, C, and F) Data represents one independent cohort. N = 5 or 6 mice/group. (D- E) Each dot represents a biological replicate. Lines show geometric mean. (G) Each dot represents one technical replicate. Data shown from three biological replicates. Lines show geometric mean. (J) Violin plot showing the distribution of PPAR-γ nuclear intensity (n = 90 cells). (K) Each dot represents one technical replicate. Data shown from three biological replicates. Lines show geometric mean. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 using an unpaired two-tailed Student’s t test.
To determine if PLA activated PPAR-γ in IECs, we treated mouse enteroids with PLA and observed increased expression of Angptl4 in mouse enteroids to a similar level as the PPAR-γ agonist, Rosiglitazone (Fig. 6G). Furthermore, PLA treatment significantly increased PPAR-γ accumulation in the nucleus of IECs compared to mock treated cells (Fig. 6, H – J). PLA also reduced lipid micelle secretion by Caco-2 cells (Fig. 6K), suggesting that this Lactobacillus-derived metabolite may play a role in protecting the host against metabolic dysfunction during consumption of a HF diet by regulating lipid metabolism in IECs through PPAR-γ.
Phenyllactic acid protects young mice from HF-diet induced metabolic dysfunction
Based upon our in vitro data, we asked whether supplementation with PLA increased PPAR-γ abundance in the small intestine of mice exposed to a HF diet and LDP. In agreement with the untargeted metabolomics data (Fig. 6A), mice exposed to a HF diet and LDP had significantly less PLA in their small intestine compared to mice given only a HF diet, and supplementation with PLA restored PLA abundance in HF diet and LDP-treated mice to a similar level as mice given a HF diet alone (Fig. 7A). Elevated intestinal PLA (Fig. 7A) significantly increased the abundance of PPAR-γ in the intestinal epithelium when mice were exposed to a HF diet and LDP compared to mice given only a HF diet (Fig. 7, B and C). Consistent with an increase in intestinal PPAR-γ, HF diet and LDP exposed mice treated with PLA developed less adiposity and gained significantly less weight (Fig. 7, D and E, fig. S7A) than mice exposed to a HF diet and antibiotics. PLA supplementation also significantly inhibited LDP-induced fat accumulation in hepatocytes (Fig. 7, F and G) and reduced fasting blood glucose (relative to mice exposed to a HF diet and LDP) (fig. S7B). Changes in physiology due to PLA supplementation occurred despite mice treated with PLA consuming more food and having similar reductions in Lactobacillus abundance in the small intestine (fig. S7, C and D). We determined that PLA, a metabolite produced by the small intestine resident gut microbiota, protects against early-life metabolic dysfunction during exposure to a HF diet by regulating PPAR-γ function in IECs.
Figure 7. Phenyllactic acid upregulates intestinal PPAR- γ and inhibits HF + LDP induced metabolic dysfunction.
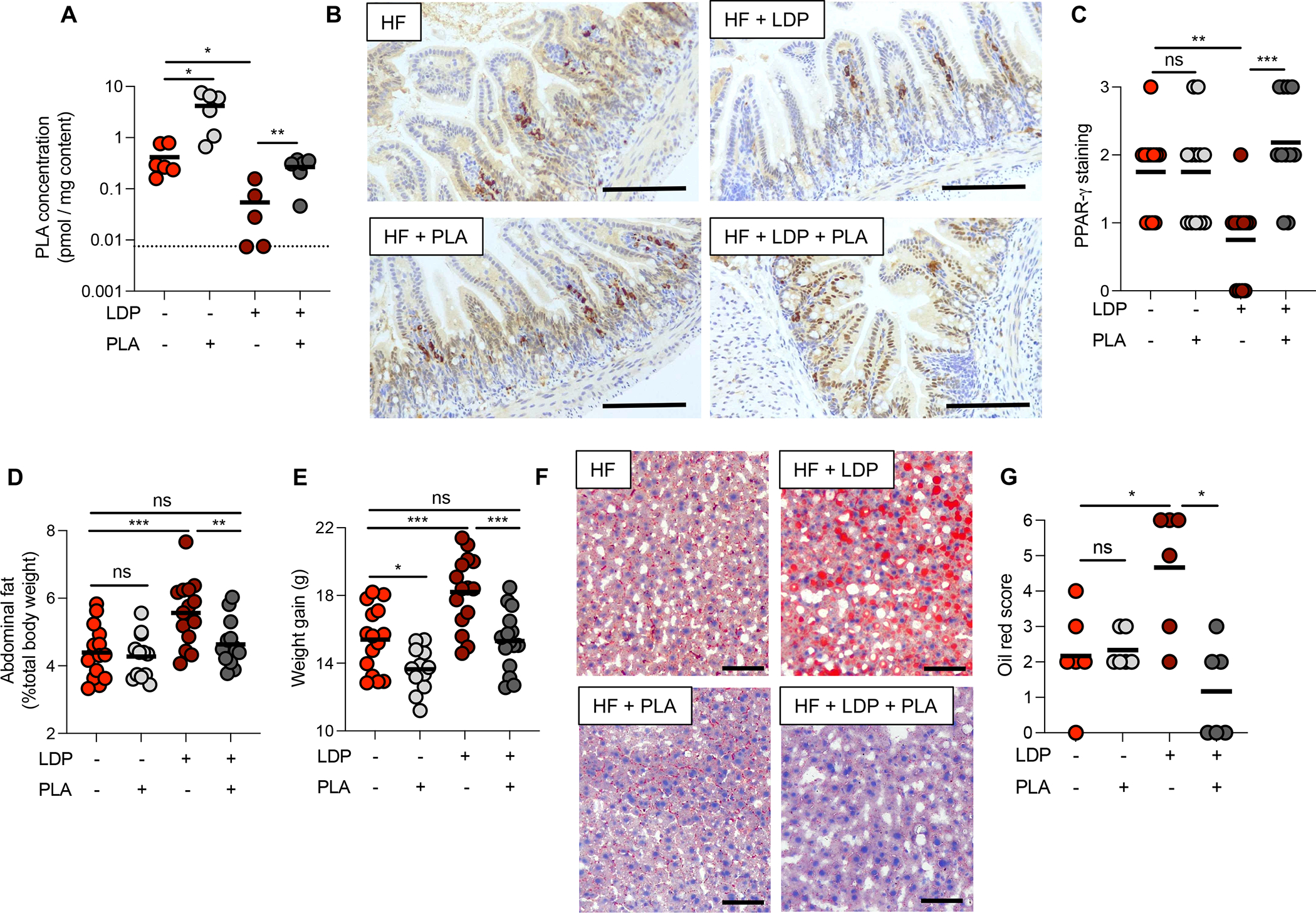
(A) Concentration (pmol/mg of intestinal content) of PLA in the small intestine of mice. Dotted line indicates limit of detection = 0.025 pmol/mg. (B) Sections from the ileum of HF diet mice given 10 mM PLA in their drinking water and/or LDP for 5 weeks were stained for PPAR-γ. Scale bar represents 200 μm. (C) PPAR-γ abundance in the epithelium was quantified by scoring blinded sections of the ileum. (D) Abdominal fat (% total body weight) and (E) weight gain of mice measured after 5-week on a HF diet ± PLA ± LDP. (F) Representative images of oil red O-stained sections of the liver. Scale bar represents 100 μm. (G) Combined oil red scores from livers of mice after 5 weeks of treatment. (A, G) N = 6 mice/group. (C – E) N =12 mice/group. (A, C – E, G) Each dot represents one animal. Lines represent geometric mean. Data representative of two independent cohorts. (A, D, and E) *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 using an unpaired two-tailed Student’s t test. (C and G) *, p < 0.05; **, p < 0.01 using Mann-Whitney test.
DISCUSSION
The worldwide prevalence of childhood obesity has risen dramatically over the last thirty years 43. In the United States, approximately 20% of school age children are considered obese, resulting in a significant public health concern44. The etiology of childhood obesity is complex and both genetic and environmental factors contribute to the development of the disease45. Recently, early-life disruption to the gut microbiota by antibiotic treatment has been proposed as a risk factor for childhood obesity10,11. However, epidemiological surveys have produced conflicting results regarding whether early-life antibiotics correlate with a higher body-mass-index, prompting further research into how the early-life microbiota could be protective against obesity8,46. Importantly, these epidemiological surveys did not consider the children’s diet and, therefore may be missing relevant synergistic effects between antibiotics and other environmental factors8,46. To answer this gap in knowledge, we developed a mouse model in which young mice were concurrently exposed to antibiotics and an obesogenic HF diet. Specifically, we investigated the effects of penicillin, which belongs to the most commonly prescribed class of antibiotics in the United States47. In this paper, we determined that early-life penicillin exposure accelerates the development of high fat diet-induced obesity by disrupting interactions between the small intestine microbiota and IECs. We further demonstrated that depletion of Lactobacillus species in the small intestine of mice given antibiotics and a HF diet led to excess adiposity. Specifically, a HF diet and antibiotics reduced the abundance of the Lactobacillus-derived metabolite, PLA, which regulates PPAR-γ in IECs and protects against HF diet-induced obesity.
We determined that early-life concurrent exposure to antibiotics and a HF diet perturbs lipid metabolism in IECs and provides further evidence for the small intestine’s significant role in regulating whole-body lipid homeostasis48. In obese patients, small intestine lipid metabolism is significantly altered, suggesting that dysregulation of intestinal lipid metabolism contributes to metabolic dysfunction49. Interestingly, proteins increased in the small intestine of obese patients49 (FABP1, FABP2, MTP) corresponded to genes upregulated in the small intestine of mice exposed to a HF diet and antibiotics. We determined that mice given a HF diet and antibiotics accumulate significantly more triglycerides in their IECs and serum but did not absorb more fat from their diet. Together, this data supports the hypothesis that concurrent exposure to a HF diet and antibiotics perturbs intestinal lipid metabolism and leads to greater lipoprotein secretion, consistent with what is observed in obese patients49. We propose that these changes in intestinal lipid metabolism, at least in part, are due to loss of PPAR-γ signaling. Previous research has demonstrated that disruption to the gut microbiota decreases PPAR-γ activity in the intestine38 consistent with our observation that a HF diet and LDP treatment reduces PPAR-γ in the small intestine. The role of PPAR-γ in lipid metabolism is cell-type specific. For instance, in the adipose tissue, PPAR-γ promotes lipid storage and lipogenesis50 whereas in macrophages PPAR-γ reduces free fatty acid and triglyceride accumulation51. In the intestine, deletion of PPAR-γ resulted in elevated serum triglycerides40, agreeing with our model in which depletion of intestinal PPAR-γ coincides with significantly greater triglycerides in fasted serum. Therefore, intestinal PPAR-γ presents a promising target to regulate lipid metabolism and reduce metabolic dysfunction during exposure to a HF diet. Indeed, treatment with the PPAR-γ agonist, Rosiglitazone, represses the expression of intestinal apoB and reduces triglyceride secretion in a hamster model52. Collectively, these findings highlight the role of intestinal lipid metabolism, specifically PPAR-γ, in protecting against metabolic dysfunction.
The depletion of Lactobacillus species from the small intestine in mice exposed to a HF diet and antibiotics poses a significant threat to the early-life microbiota-host interaction. Lactobacillus is one of the earliest colonizers of the gut and is a predominant member of the infant microbiota4,53. Despite a reduction in the relative abundance of Lactobacillus as a child matures4, Lactobacillus species remain key constituents of the gut microbiota54. Importantly, antibiotic treatment, specifically with penicillin, leads to long-lasting reductions in Lactobacillus in children9. Therefore, investigations into how loss of Lactobacillus impacts host health and development remain clinically relevant. Previous research has determined that specific Lactobacillus species, L. paracasei F19 and L. reuteri ATCC 4659, limit the effects of an obesogenic HF diet in adult mice55,56, but the mechanism by which these strains exert their protection remains relatively unknown. Furthermore, the ability of Lactobacillus to prevent metabolic dysfunction is species-specific as some species seem to offer no protection or exacerbate the effect of a HF diet55,57. Even more so, probiotic intervention studies have shown that Lactobacillus species largely fail to alter body composition in children58,59, highlighting the critical need to understand how Lactobacillus species protect against HF-diet induced obesity. In our paper, we determine that L. murinus, a predominant Lactobacillus species in the human gut54, inhibits lipid secretion from IECs by upregulating intestinal PPAR-γ.
This paper did not explore the impact of other small intestine microbiota members on host adiposity during early-life exposure to a HF diet. However, in our 16s rRNA sequencing of the small intestine microbiota of mice exposed to a HF diet and LDP, we determined that the relative abundance of the Streptococcaceae family was increased. As Streptococcus species are a dominant group in the human small intestine microbiota60, this increase in our model warrants further investigation. Streptococcus species are highly abundant in atherosclerotic plaques and correlate positively with HDL cholesterol level61, suggesting that elevated Streptococcaceae in HF diet and LDP treated mice could be adding to their metabolic dysfunction. Interestingly, commensal Streptococcus species have been shown to downregulate PPAR-γ activity62 which may contribute to the inhibition of intestinal PPAR-γ in mice given a HF diet and LDP. Therefore, future work will investigate how interactions between multiple small intestine microbiota members influence host metabolism through intestinal PPAR-γ.
Due to the transient colonization of probiotics63, direct administration of the beneficial Lactobacillus-derived metabolite may have better success in preventing metabolic dysfunction. Therefore, we identified a Lactobacillus-derived metabolite, phenyllactic acid (PLA), that activates intestinal PPAR-γ and protects young mice from metabolic dysfunction due to concurrent exposure to a HF diet and antibiotics. Multiple bacterial species produce phenyllactic acid, including species belonging to the Bifidobacteriaceae and Peptostreptococcaceae families64–66. Interestingly, we see a depletion of Bifidobacteriaceae and Peptostreptococcaceae in mice fed a HF diet and mice exposed to a HF diet and antibiotics (Fig. 2A), suggesting that multiple bacterial species may contribute to PLA production in the intestine. Significantly, PLA is present in the feces of infants and correlates with the abundance of Bifidobacterium64, indicating that this metabolite is a component of the early-life gut metabolome in humans. Previous work revealed that PLA upregulated an adipose tissue-specific isoform of PPAR-γ42, and we have now determined that PLA also increases intestinal PPAR-γ activity and abundance. We then extended these findings by showing that PLA inhibits lipid secretion in intestinal epithelial cells and prevents antibiotic-induced excess adiposity in young mice during consumption of an obesogenic HF diet. Collectively, we identified a previously unknown mechanism by which the early-life microbiota communicates with the intestinal epithelium to decrease HF-induced obesity, providing new evidence linking early-life antibiotic exposure to childhood obesity.
STAR METHODS
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mariana Byndloss (mariana.x.byndloss@vumc.org).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact without restriction.
Data availability
16S rRNA sequencing data has been deposited in the European Nucleotide Archive under the accession number: PRJEB52997. RNA sequencing data has been deposited in the European Nucleotide Archive under the accession number: PRJEB53157. The mass spectrometry data are available in the Metabolomics Workbench public repository, with the data set identifier as ST002186. All data sets are publicly available. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL DETAILS
Animal Experiments
All experiments in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Vanderbilt University Medical Center.
Early-life antibiotic treatment using clinical doses has been linked to increased childhood body mass index in males67. Additionally, the effects of diet-induced obesity are significantly greater in young male mice compared to females68. As we focused our study on the synergistic effects of antibiotics and a high fat (HF) diet, male mice were used for this work. Male C57BL/6N mice (#027), 3-weeks-old, were obtained from Charles River Laboratory. Prior to experiments, fecal samples were collected from mice and initial Lactobacillus levels determined by homogenizing fecal content in 1 mL of sterile PBS, followed by serial dilutions of the samples which were plated on De Man, Rogosa, and Sharpe (MRS) agar (BD Difco #288210). After overnight incubation under anaerobic conditions, colony forming units (CFUs) were calculated to obtain Lactobacillus colonization levels. The initial weight of mice was also collected. Mice were then assigned to groups so that no significant differences in weight or Lactobacillus abundance between groups occurred before beginning treatments.
Female and male C57BL/6J Ppargfl/flVillincre/− and littermate Ppargfl/flVillin−/− (control) mice were generated at Vanderbilt University Medical Center by mating Ppargfl/fl mice (The Jackson Laboratory, #004584) with Ppargfl/flVillincre/− mice38. Genotype of offspring determined by PCR using protocol primers described by The Jackson Laboratory. Mice were weaned at 3 weeks of age and assigned to one of the treatment groups described below.
Animals were fed either a 60% fat diet (HF) (Research Diets Inc., #D12492) or a 10% fat control diet (LF) (Research Diets Inc., #D12450J) for 5 weeks. Groups of LF or HF diet mice were also given low doses of penicillin (Sigma Aldrich #P1382)(LDP) (6.67 mg/L,10) in their drinking water throughout the experiment or clinical doses of penicillin (0.167 g/L,69) in their drinking water from days 0 – 5, 15 – 20, and 30 – 35. For long-term experiments, mice were fed either a LF or HF diet or a LF or HF diet with LDP for 5 weeks before being switched to a HF or LF diet alone for an additional 5 weeks. Ppargfl/flVillincre/− and littermate Ppargfl/flVillin−/− mice were maintained on a HF diet or a HF diet and LDP treatment for 6 weeks as they required additional time to reach a similar weight as C57BL/6N mice on a HF diet (data not shown). In some experiments, drinking water was supplemented with 10 mM D-(+)-3-phenyllactic acid (Sigma-Aldrich #376906) for 5 weeks.
C57Bl/6N germ-free mice (Taconic, B6NTac – GF) were bred in-house and moved into a positive pressure cage system at 3-weeks-old.
Over the course of the experiments, mice were weighed weekly. Food consumption of groups of mice was measured twice per week and normalized to the group or cage weight. In addition, fecal samples were collected weekly or bi-weekly, depending on the experiment, and plated onto MRS agar to measure Lactobacillus abundance. After 5, 8, or 10 weeks, mice were fasted for approximately 6 hours before fasting blood glucose was measured using a glucometer.
At the end of the experiment (5-weeks or 10-weeks after starting diet and antibiotic treatments), mice were humanely euthanized by CO2 administration. Afterwards, serum, ileum (distal small intestine) content, ileum tissue (for epithelial isolation as described below), and samples for histopathology, IHC, and oil-red staining were collected. Liver and adipose tissue was flash frozen for RNA extraction. In addition, the epididymal (abdominal) fat from mice was removed and weighed.
Bacterial culture conditions
The bacterial strains used in this study are listed in Table S1. Lactobacillus strains were routinely grown anaerobically at 37 °C in MRS broth (BD Difco # 288130) or on MRS agar plates. E. coli Mt1B1 was grown aerobically in LB broth (BD Difco #244620) at 37 °C. Muribaculum intestinale was grown anaerobically at 37 °C in Anaerobic Akkermanisa Medium70. Clostridium clostridioforme and Clostridium sporogenes were grown anaerobically at 37 °C on Brain Heart Infusion (BHI) agar (BD Difco #211059) supplemented with 5% (v/v) defibrinated sheep blood (Thermo Scientific #R54016) and single colonies used to inoculate Supplemented Brain Heart Infusion (BHIS) medium 71. Streptococcus daniellae ERD01G was routinely grown anaerobically at 37 °C in Tryptic Soy Broth (TSB) (BD Difco #211825). Staphylococcus xylosus 33-ERD13C was routinely grown aerobically at 37 °C in LB broth (BD Difco #244620).
Mouse small intestinal epithelial cells
MSIE cells were generously provided by Dr. Fan Yang and were previously generated from the small intestine of 6- to 8-week-old C57BL/6 mice harboring a thermolabile mutation (tsA58) under the control of an IFN-γ-inducible H-2Kb promoter and a temperature-sensitive simian virus 40 (SV40) large T antigen72. Expression of the SV40 large T antigen was induced by culturing cells at 33°C with IFN-γ which allowed for cell proliferation. MSIE cells were routinely cultured in RPMI 1640 medium (Gibco #11875093) supplemented with 5% Fetal Bovine Serum (Gibco #16140071), 5 U/ml of mouse IFN-γ (R&D Systems #485-MI), 100 U/ml penicillin and streptomycin (Gibco #15140122), 5 μg/ml insulin, 5 μg/ml transferrin, and 5 ng/ml selenous acid (Sigma-Aldrich #I3146) at 33°C (permissive temperature) under 5% CO2. Cells were passaged every 2 to 3 days. MSIE cells were seeded into 12-well plates (1 × 105 cells/ well) or 6-well plates (6 × 105 cells/well), incubated until confluent (usually 2 to 3 days), and then treated.
Caco-2 cell culture
Caco-2 cells (ATCC #HTB-37) were grown in Caco-2 cell culture media (Minimal Essential Media (MEM) (Gibco #11090081), 10% Fetal Bovine Serum (Gibco #16140071), 1% Glutamax (Gibco #35030061), 1% MEM Nonessential Amino Acids (Corning #11140050), and 1% Sodium Pyruvate (Gibco #11360070)) at 37°C and 5% CO2. Cells were passaged every 3–4 days in T75 flasks according to ATCC protocols. For lipid secretion assays, 70 – 80% confluent T75 flasks were diluted, and approximately 1 × 105 cells seeded onto 12-well transwell plates (Corning Costar #3493) containing collagen coated 0.4 μm pore size inserts. Cells were differentiated for 14 days on the transwell inserts with the media being replaced every two days. For immunofluorescence experiments, 70% - 80% confluent T75 flasks were diluted, and then approximately 6 × 105 cells seeded onto glass coverslips in 6-well plates.
Mouse enteroids
Enteroids were generated from 10–12-week-old female C57BL/6J mice. Mice were humanely euthanized, and the ileum removed, rinsed with ice-cold PBS, and then opened. Tissue was incubated in 0.5% bleach in PBS for 5 minutes before rinsing with 0.1 mg/mL Primocin (InvivoGen #ant-pm-05) in PBS (PBS/P). Crypts were then isolated from the tissue by gently rocking the ileum in chelation buffer (1M dithiothreitol and 20 mM EDTA/EGTA in PBS) at 4°C for 30 minutes. The supernatant was then discarded, and additional PBS/P was added before shaking the tissue vigorously. The supernatant was filtered through a 70 μm filter and then spun at 500 x g for 5 minutes. The cell pellet was resuspended in PBS/P and Matrigel (Corning #356234) (final concentration of Matrigel was approximately 80%). 30 μL of the mixture was aliquoted into a 6-well non-tissue culture treated plate (6 domes/ well) and incubated at 37°C until solidified. Media (Intesticult (Stemcell Technologies #06005), 10 μM Y-27632 (bio-techne | TOCRIS #1254), and Primocin) was then added and enteroids incubated for 2 days at 37°C, 5% CO2. Y-27632 was removed after two days. Media was replaced every 2–3 days and enteroids were passaged every 7–10 days. Enteroids were passaged by dissociating organoids from Matrigel and adding TrypLE Express (Gibco #12604013).
Differentiated enteroids (10 – 14 days after passaging) were treated with either 10 μM Rosiglitazone (Sigma-Aldrich #R2408) or 5 mM PLA and incubated for 24 hours. Media was replenished during the incubation at 8 hours. Organoids were then dissociated out of the Matrigel, and RNA extracted from the cell pellets (Norgen BioTek Corp. #17200).
Method details
Body composition
Mice were fed a HF diet or exposed to a HF diet and LDP for 5 weeks. Then, dual-energy X-ray absorptiometry (DEXA) was performed before their sacrifice on anesthetized mice by using a Faxitron UltraFocus (Hologic). The instrument was calibrated for offset images, dark images, and flat-field images before the measurement by a method provided by the manufacturer.
Oil Red O Staining
Portions of liver were frozen in optimal cutting temperature (O.C.T.) compound (Fisher Scientific #23730571) and 5 μm thick sections were cut and mounted to slides. Slides were stored at −80 °C until staining. To stain lipids, slides were brought to room temperature and then placed in 10% neutral-buffered formalin solution for 10 minutes. The Newcomer Supply Tech Oil Red O, Propylene Glycol staining kit (Newcomer Supply #12772B) was used for visualization. Slides were then coverslipped with aqueous mounting medium. Scoring of blinded tissue sections were performed by a veterinary pathologist based on the criteria listed in Table S4. Representative images were taken using a Leica DM750 microscope and a Leica ICC50W camera.
16s rRNA sequencing and data analysis
Content from the distal small intestine was collected at the end of the 5-week experiment and stored on ice. DNA was then extracted from the samples using the DNeasy PowerSoil Kit (Qiagen #47016) according to the manufacturer’s instructions. Sequencing libraries were prepared by the University of California San Diego (UCSD) Microbiome Core using protocols and primers published on the Earth Microbiome Project website and sequencing performed on Illumina NovaSeq 6000 according to the manufacturer’s instructions. Other samples were sequenced by Seqmatic on Illumina MiSeq. Single end sequences were imported into QIIME2 (version 2022.2). Reads were trimmed to exclude regions that fell below a quality score of 25. Features tables for each data set were constructed and then merged following denoising. Data was rarefied based on minimum read quantity. Phylogenetic trees were then constructed, and taxonomy was assigned based on the Silva genes classifier. Pseudocounts added and ANCOM was performed to identify alterations in gut microbiota composition based on diet or administration of low-dose penicillin. Feature tables constructed for taxonomic levels 5(genera), 6(family), and 2(phylum). Pseudocounts added to each of the collapsed feature tables and ANCOMs performed at for each experimental variable as previously performed. Distance matrices (jaccard, bray-curtis, weighted, and unweighted) were exported from QIIME2 for further processing in R-studio. Alpha diversity metrics (Shannon, Faith_pd, and Observed features) were extracted from QIIME2 for downstream applications. PERMANOVAs performed for each distance matrix, as well as the creation of non-metric multidimensional scaling (nmds) plots based on each distance matrix.
L. murinus-specific qPCR
To measure the abundance of L. murinus in the small intestine, a protocol was adapted from Yildez et al.73. Briefly, a DNA (ng) and CFU correlation was generated using L. murinus (isolate collected from male C57BL/6N mice (#027), 3-weeks-old; species identified by 16S rRNA sequencing of the colony by Genewiz). L. murinus was cultured anaerobically in MRS broth overnight and then 1 mL of culture was serially diluted and plated on MRS agar for determination of CFU. A second 1 mL aliquot of L. murinus culture was lysed and genomic DNA extracted using the PureLink Genomic DNA Mini Kit (Invitrogen #K1820–01). CFU/ng of L. murinus was calculated in triplicate using the results from above.
DNA from content in the distal small intestine was extracted as described above using the DNeasy PowerSoil Kit. Primers specific for either the 16S rRNA gene of Lactobacillus species74 or L. murinus (Table S2) were combined with iQ SYBR Green Supermix (Bio-Rad #1708882) and 20 ng of DNA from either the small intestine content or a standard curve of L. murinus gDNA. Quantitative PCR (qPCR) was performed using the following thermal cycling program: initial denaturation step at 95°C for 5 min, followed by 40 cycles of denaturation at 95°C for 30 s and annealing/extension at 60 °C for 60 s. Cq values obtained from the qPCR reactions were compared to a L. murinus gDNA standard curve to determine the amount of L. murinus gDNA present in each sample. Then, CFU/ng of small intestine content DNA was calculated using the correlation calculated as described above.
Colonization of HF and LDP-treated mice with L. murinus PenR
To create a strain of L. murinus resistant to low doses of penicillin used in the mouse experiments described above, a single colony of L. murinus was inoculated into MRS broth containing 50 μg/L penicillin (Sigma Aldrich #P1382). After 24 hours, the culture was diluted 1:100 into MRS broth containing 100 μg/L penicillin. The culture was passaged as described into increasing concentrations of penicillin until L. murinus grew in MRS broth containing 6.67 mg/L penicillin. To isolate single colonies, passaged L. murinus was plated on MRS agar containing 6.67 mg/L penicillin. Single colonies were inoculated into fresh MRS broth and cultures used to make glycerol stocks.
To colonize mice with a L. murinus PenR (strain generation described below), male C57BL/6N mice (#027), 3-weeks-old, from Charles River Laboratory were inoculated by intragastric gavage (1 × 109 CFU in 0.1 mL MRS broth) with L. murinus PenR. At 48 hours post colonization, mice were switched onto a HF diet and given LDP in their drinking water. Mice were given L. murinus PenR by intragastric gavage (1 × 109 in 0.1 mL MRS broth) weekly for the 5 weeks of exposure to a HF diet and LDP.
Bacterial culture with bile salts and penicillin
For measuring the growth of bacteria upon exposure to penicillin, bile salts, lipids or a combination, bacterial species were cultured as follows: L. murinus was streaked onto MRS agar and single colonies used to start cultures in MRS broth. Overnight cultures were harvested by centrifugation at 4,000 × g for 5 minutes and then pellets were resuspended in 1 mL fresh media. Optical density at 600 nm (OD600) was measured and cultures adjusted to an OD600 = 0.1. Cultures were then diluted again to an OD600 = 0.001. Stocks of penicillin (Sigma Aldrich #P1382) were prepared at a concentration of 250 mg/L in dH2O and then batches at 10x the concentration tested in the assay were made in MRS broth (pH = 6.4). Bile salts (Sigma Aldrich #B8756) were dissolved in dH2O at a concentration of 250 mM and then diluted in MRS broth (pH = 6.4) to a concentration of 0.9375 mM. Lipid mixture (Sigma Aldrich #L0288) was diluted to a concentration of 12.5% (v/v) in MRS broth. For measuring the growth of untreated bacterial species, 10 μl of adjusted bacterial culture was added to 90 μl of media in a 96 well plate. To measure the impact of penicillin on bacterial growth, 10 μl of 10x penicillin stocks were added to wells containing 10 μl of adjusted bacterial culture and 80 μl of media. The effect of bile salts on bacterial growth was determined by adding 80 μl of media containing 0.9375 mM bile salts to 10 μl of adjusted culture and 10 μl of plain media. The impact of lipids on bacterial growth was determined by adding 80 μl of media containing 12.5% lipids to 10 μl of adjusted culture and 10 μl of plain media. To measure the effect of the combination of penicillin and bile salts or penicillin and lipids on bacterial growth, 10 μl of adjusted culture, 10 μl of 10x penicillin stocks, and 80 μl of media containing 0.9375 mM bile salts, or 12.5% bile salts were combined. Plates were incubated anaerobically at 37 °C. OD600 was measured every hour for 24 hours using the Epoch 2 plate reader (BioTek). In vitro growth assays were performed in triplicate with different colonies.
Gnotobiotic mouse experiments
C57Bl/6N germ-free mice (from our in-house colony) were colonized with a defined microbiota community representative of a small intestine microbiota at 3 weeks of age. The defined microbiota included 1 × 109 CFU of the following bacteria: Muribaculum intestinale, Clostridium clostridioforme, Clostridium sporogenes, and Streptococcus daniellae ERD01G. Ligilactobacillus murinus (isolation and characterization described below) was added to the defined microbiota (1 × 1010 CFU) for a subset of mice. Germ-free mice were intragastrically inoculated with 0.1 mL of the defined microbiota and maintained on a chow diet for 5 days. Colonized mice were then given 1 × 108 CFU of Escherichia coli Mt1b1 and 48-hours later were switch onto a HF diet (Research Diets, Inc., D12492i). Mice were maintained on a HF diet for 5 weeks. Groups of mice colonized with L. murinus continued to be gavaged weekly with 1 × 1010 CFU of the bacterium and L. murinus was also supplied in their drinking water (1 × 108 CFU/mL).
Histopathology Analysis
Tissue from the distal small intestine was fixed in 10% neutral buffered formalin, paraffin-embedded and 5 μm thick sections were cut and mounted to slides and stained with hematoxylin and eosin. Scoring of blinded tissue sections were performed by a veterinary pathologist based on the criteria listed in Table S3. Representative images were taken using a Leica DM750 microscope and a Leica ICC50W camera.
Enterocyte isolation
The ileum (distal small intestine) was removed from mice and opened lengthwise before placing in cold phosphate buffered saline (PBS) (Gibco #14190144). Tubes were gently inverted four times. Tissue was then placed in a 15 mL conical centrifuge tube containing dissociation reagent #1 (30 mM EDTA, 1.5 mM DTT, pH 8 in PBS) and incubated for 20 minutes buried in ice. Then, tissue was transferred to a new 15 mL conical centrifuge tube containing pre-warmed (37°C) dissociation reagent #2 (30 mM EDTA, pH 8 in PBS) and samples were incubated at 37°C for 10 min. After incubation, tubes containing the samples were shaken vigorously for 30 seconds to release epithelium from the basement membrane. Cell suspension was transferred to a new 15 mL conical centrifuge tube, leaving any remnant tissue, and cells were pelleted by centrifugation at 800 × g for 5 minutes at 4°C. Supernatant was removed and the cell pellet was flash frozen in liquid nitrogen for subsequent RNA extraction or triglyceride measurements.
RNA extraction and quantitative real-time PCR
Liver tissue and enterocyte cell pellets were homogenized using a FastPrep-24 and RNA isolated by the TRIzol method (Invitrogen #15596018) following the manufacturer’s protocol. Adipose tissue was homogenized using a Fastprep-24 and RNA isolated using QIAzol Lysis Reagent (Qiagen #79306) according to the manufacturer’s protocol. RNA was reverse transcribed using iScript gDNA Clear cDNA Synthesis Kit (Bio-Rad #1725035). Quantitative real-time PCR was performed using iQ SYBR Green Supermix (Bio-Rad #1708882) and the appropriate primer sets (Table S2). Relative gene expression was calculated using Act2b as the housekeeping gene.
RNA-sequencing
RNA was extracted from enterocyte cell pellets as described above and samples sequenced by the Vanderbilt Technologies for Advanced Genomics (VANTAGE). VANTAGE performed an mRNA enrichment and then prepared a cDNA library using the stranded mRNA (polyA-selected) library preparation kit (New England BioLabs #E7760). Sequencing was then performed at paired-end 150 bp on the Illumina NovaSeq 6000 targeting an average of 25 M reads per sample. Results were then analyzed by Vanderbilt Technologies for Advanced Genomics Analysis and Research Design (VANGARD) as follows. Adapters were trimmed by Cutadapt (v2.10). After trimming, reads were mapped to the mouse reference genome GRCm38.p6 using STAR (v2.7.3a) 75 and quantified by featureCounts (v2.0.0) 76. DESeq2 (v.1.24.0) 77 was used to detect differential expression between two groups. WebGestaltR (v0.4.4) was used to perform functional enrichment analysis against Gene Ontology and KEGG. Gene Ontology analysis was performed using the online tool DAVID Bioinformatics Resources (https://david.ncifcrf.gov/).
Lipid Measurements
After 5 weeks of exposure to a HF diet or a HF diet and LDP, mice were fasted overnight and then humanely euthanized by CO2. Plasma was collected and then the amount of neutral lipids (Cell Biolabs STA-617), triglycerides (Cell Biolabs STA-396), free fatty acids (Cell Biolabs STA-618), or cholesterol (Cell Biolabs STA-390) were measured using the appropriate assay kit. Enterocytes from fasted mice were collected as described above and triglycerides measured in the cell lysate. The concentration of triglycerides was normalized to protein abundance in the cell lysate as determined by a standard Bradford assay (Bio-Rad #5000006).
Non-invasive Measurement of Intestinal Fat Absorption
Measurements of dietary fat absorption were based off the protocol by Jandacek et al.78. C57BL/6N were given a HF diet or a HF diet and LDP for 5 weeks as described above. After the 5 weeks of treatment, mice were placed in a clean cage and switched to a diet containing sucrose polybehenate (Research Diets, Inc., #D12492 with 5% of the fat replaced with sucrose polybehenate). The mice were moved again to a clean cage after 24 hours and maintained on the diet containing sucrose polybehenate. On the third day of feeding with sucrose polybehenate, fecal pellets were collected from the mice before placing animals in a clean cage. After 4 days on the diet, fecal pellets were collected again.
Fecal samples were saponified with methanolic sodium hydroxide to obtain free fatty acids which were then methylated using 14% BF3 methanol to produce fatty acid methyl esters. Hexane was then used to extract the fatty acid methyl esters. The fatty acids were detected using gas chromatography and compared to a set of standards and an internal standard. The percentage of fat absorption was determined based on the ratio of total fatty acids to behenic acid in the diet and in the feces.
Oral fat tolerance test
Mice were given a HF diet or a HF diet and LDP for 5 weeks before being fasted for 6 hours. Then, mice were injected intraperitoneally with 0.5 g/kg tyloxapol (Sigma Aldrich #1704003; dissolved in sterile PBS) 30 minutes before they were given 200 μl of olive oil via intragastric gavage. Blood samples were collected from the tail 2 hours after olive oil gavage. Samples were then spun at 1000 × g for 20 minutes at 4 °C and the supernatant collected and stored at −80 °C. Triglycerides were measured in the serum as described below.
PPAR-γ Immunostaining
Pieces of the distal small intestine were fixed in 10% neutral-buffered formalin, paraffin-embedded and 5 μm thick sections were cut and mounted to slides. Slides were placed on the Leica Bond-Rx IHC Stainer. All steps besides dehydration, clearing and coverslipping were performed on the Bond-Rx. Slides were deparaffinized. Heat induced antigen retrieval was performed on the Bond Max using their Epitope Retrieval 2 solution (Leica Biosystems #AR9640) for 20 minutes. Slides were placed in a protein block (Agilent DAKO # x0909) for 10 minutes. Slides were then placed in a PowerVision Block (Leica Biosystems #PV6122) for 60 minutes. The sections were incubated with anti-PPAR-γ (Santa Cruz Biotechnology #sc-7273), diluted 1:100, for 30 minutes. The Bond Refine Polymer detection system (Leica Biosystems #DS9800) was used for visualization. Slides were the dehydrated, cleared and coverslipped. Scoring of blinded tissue sections were performed by a veterinary pathologist based on the criteria listed in Table S5. Representative images were taken using a Leica DM750 microscope and a Leica ICC50W camera.
Lipid secretion assay in Caco-2 cells
Differentiated Caco-2 wells were treated with either 3 μM GW9662 (Sigma-Aldrich #M6191), live L. murinus (prepared as described above), both 3 μM GW9662 (Sigma-Aldrich #M6191) and live L. murinus, or 5 mM D-(+)-3-phenyllactic acid (Sigma-Aldrich #376906) for 16 hours. Lipid micelles were prepared as previously described22,23,79 in serum-free Caco-2 cell culture media. Micelles, containing 0.6 mM oleic acid (Sigma-Aldrich #O1008), 2 mM sodium taurocholate (Sigma-Aldrich #86339), 0.2 mM 2-mono-oleoylglycerol (Sigma-Aldrich #M7765), 0.05 mM cholesterol (Sigma-Aldrich #C8667), 0.2 mM L-α-lysophosphatidylcholine (Sigma-Aldrich #L4129) and 0.02 mM BODIPY C12 fatty acid (Invitrogen #D3835), were added to the apical compartment of cells. After a 10-minute incubation, media containing lipid micelles was removed and replaced with serum-free Caco-2 cell culture media. Four and six hours after addition of lipid micelles, 150 μL from the basal compartment was removed and 50 μL added in triplicate to wells of black 96-well plate. Fluorescence was measured (excitation at 548 nm and emission at 578 nm) (BioTek).
PPAR-γ fluorescent staining
MSIE or Caco-2 cells were seeded onto coverslips as described above and incubated for 48 hours. Then, cells were mock-treated or treated with live L. murinus (prepared as described above), L. murinus or E. coli Mt1B1 supernatant (prepared as described above), or 5 mM D-(+)-3-phenyllactic acid (Sigma-Aldrich #376906) for 16 hours. All conditions were buffered with 20 mM HEPES (Gibco #15630080). After treatment, media was removed, and cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences #15710-S) for 15 minutes at 37 °C. Cells were then washed 3 times for 5 minutes in PBS (Gibco #14190144). Subsequently, cells were treated with 0.1% Triton X-100 (Acros Organics #AC327371000) for 15 minutes and then washed again 3 times for 5 minutes with PBS (Gibco #14190144). Cells were blocked in 5% BSA (Alfa Aesar #J64655) for one hour before being incubated with anti-PPAR-γ (Santa Cruz Biotechnology #sc-7273) (Diluted 1:50) for one hour at room temperature. After incubation with primary antibody, cells were washed 3 times for 5 minutes in PBS (Gibco #14190144) and then goat anti-mouse IgG Alexa Fluor 488 (Invitrogen #A-11001) (diluted 1:1000) was added for 1 hour at room temperature. Coverslips were washed in PBS (Gibco #14190144) and then Alexa Fluor 546-Phalloidin (Thermo Fisher #A22283) or Alexa Fluor 647-Phalloidin (Thermo Fisher #A22287) was added for 20 minutes. Afterwards, coverslips were washed in PBS (Gibco #14190144) and then mounted on glass slides in ProLong Diamond Antifade Mountant with DAPI (Molecular Probes #P36962). Imaging was performed on a Zeiss LSM880 Airyscan Confocal Microscope. Representative images shown in paper were collected using a Nikon A1 Microscope equipped with 405, 488, 561, and 645 nm LASERs and a Plan Fluor 40X/ 1.30 NA immersion objective. For imaging in all microscope modalities, imaging acquisition parameters were matched between samples during image acquisition. Images were contrast enhanced and cropped using FIJI/ImageJ software (NIH). Abundance of nuclear PPAR-γ was calculated using FIJI/ImageJ measurement tools and dimensions of DAPI stained nuclei.
PPAR Reporter Assay
Eighty to ninety percent confluent Caco-2 cells were seeded into 96 well plates at a concentration of approximately 1 × 104 cells/well. Forty-eight hours post seeding, wells containing Caco-2 cells were transfected with 150 ng of PPRE x3-TK-Luc80 using Lipofectamine 3000 (Thermo Fisher L3000008) according to manufacturer’s instructions. After overnight incubation, transfected cells were mock-treated or treated with 8 mM sodium butyrate (Acros Organics #263191000), or live L. murinus for 5 hours. Media was then removed, and cells washed with 100 μl PBS. Cells were then lysed and luciferase assay substrate (Promega #E1500) added according to manufacturer’s instructions. Luciferase was then measured using plate reader (BioTek).
Co-culture of bacterial strains and intestinal epithelial cells
For in vitro experiments, strains were grown overnight as follows: L. murinus was grown anaerobically in MRS broth and E. coli Mt1B1 and S. xylosus 33-ERD13C were grown aerobically in LB broth. Overnight cultures were harvested at 4000 × g for 5 minutes. L. murinus was then adjusted to an OD = 1 in Caco-2 cell culture media, E. coli Mt1B1 was adjusted to an OD = 0.025, and S. xylosus 33-ERD13C was adjusted to an OD = 0.05. Strains were adjusted to different starting concentrations due to the ability of E. coli and S. xylosus 33-ERD13C to grow in the cell culture media. Bacterial strains were added to Caco-2 or MSIE cells for co-culture experiments. Similar bacterial burdens between strains were observed after 16 hours of co-culture. To prepare Lactobacillus and E. coli Mt1B1 supernatants, Caco-2 media was inoculated with a single colony of L. murinus and grown aerobically (37 °C, 5% CO2) for 24 hours. Culture was then harvested at 4000 × g for 5 minutes and the supernatant filter sterilized (0.2 μm pore size). The volume of the supernatant used was normalized by the OD of the bacterial culture when two different strains were used. Both live cultures and supernatants were buffered using 20 mM HEPES. MSIE cells were incubated with live bacterial cultures for 16 hours and then either RNA was extracted from the cells (Norgen BioTek Corp. #17200), or the cells were stained for PPAR-γ (protocol described below). Caco-2 cells were incubated with either live bacteria or their supernatant for 16 hours before cells were stained for PPAR-γ (protocol described below).
Untargeted metabolomics
Optima grade LC-MS solvents for the mass spectrometry analyses were purchased from Thermo Fisher Scientific, USA (Thermo Fisher Scientific, Waltham, MA). Individual isotopically labeled standards were purchased from Cambridge Isotope Laboratories (Tewksbury, MA) and CDN Isotopes (Pointe-Claire, Quebec, Canada).
Metabolomics sample preparation:
Frozen mouse intestinal content samples (n=20, 5 biological replicates for each sample group) were lysed in 500 μl ice-cold lysis buffer (1:1:2, v:v:v, acetonitrile: methanol: ammonium bicarbonate 0.1M - pH 8.0) and sonicated individually using a probe tip sonicator at 50% power (10 pulses). The lysis buffer contained isotopically labeled standards (n=2) to determine sample process variability (see Table S6). Homogenized samples were normalized by weight to the smallest amount of tissue sample such that each sample contained an equal amount of tissue. Proteins were precipitated from individual samples by addition of 800 μL of ice-cold methanol followed by overnight incubation at −80°C. Precipitated proteins were pelleted by centrifugation (15,000 rpm, 15 min) and metabolite extracts were dried down in vacuo and stored at −80°C. Individual samples were reconstituted in 100 μL of reverse phase liquid chromatography reconstitution buffer (acetonitrile/water with 0.1% formic acid, 3:97, v/v) containing isotopically labeled standards (n=2) to assess instrument variability (see Table S6). A pooled quality control (QC) sample was prepared by pooling equal volumes (10 μL) from each individual sample following reconstitution.
Metabolomics based reverse phase liquid chromatography high resolution tandem mass spectrometry analysis:
Prepared samples were analyzed by RPLC-HRMS/MS in the Vanderbilt Center for Innovative Technology (CIT) using a modified version of a reversed phase chromatography negative ionization method81. Metabolites were separated on a Thermo Fisher Scientific (Waltham, MA) Hypersil Gold C18 column (100 × 2.1 mm, 1.9 μm particle size) using water/acetonitrile gradient with formic acid (0.1%) added to both mobile phases. Samples were analyzed on a high-resolution Thermo Fisher Scientific Q-Exactive HF hybrid quadrupole-Orbitrap mass spectrometer (Bremen, Germany) equipped with a Thermo Fisher Scientific Vanquish UHPLC binary system and autosampler controlled by Xcalibur 4.4 software (Waltham, MA). Liquid chromatography was performed at 250 μL min−1 using solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in 80:20 acetonitrile:water) with a gradient length of 30 min as previously described in81. Solvents and column were held at 45°C.
Mass spectrometry (full MS) analyses (5 μL injection volume) were acquired in negative ion mode at maximum resolution of 120,000 and scanned from m/z 70 to m/z 1050 with a scan rate of 3.5 Hz, automatic gain control (AGC) target of 1 × 106, and maximum ion injection time of 100 ms. MS/MS spectra were collected at 15,000 resolution, AGC target of 2 × 105 ions, and maximum ion injection time of 100 ms. Pooled QC samples were used for column conditioning (10x pooled QC sample prior to individual sample analysis and 2x pooled QC sample was injected every five samples), retention time alignment and to assess mass spectrometry instrument reproducibility (intra-batch) throughout the sample set. A total of 20 samples (randomized injection 1x) were analyzed by RPLC-HRMS/MS analysis after conditioning the analytical platform with pooled QC samples. Quality assurance (QA) practices were applied to assess the analytical method performance. Further, a system suitability sample (LCMS QC reference material, Waters Corporation, Milford, MA) was used to verify the stability of retention times, peak shapes and areas before and after sample batch analysis.
Metabolomics data processing and statistical analysis:
Spectral features (retention time, m/z pairs) were extracted from the RPLC-HRMS/MS data using Progensis QI v. 3.0 (Non-linear Dynamics, Waters Corporation, Milford, MA). Briefly, the data from five (n=5) biological replicates from each sample type were imported, processed, normalized (integration) and reviewed. All MS and MS/MS sample runs were aligned against a pooled QC sample reference run. Spectral features (retention time, m/z pairs) were further de-adducted and de-isotoped to generated compounds or metabolites. Data was further curated by applying QA practices to the data. Specifically, compounds or metabolites with spectral features with >25% coefficient of variation (CV) in the pooled QC samples were removed (total: 2270 metabolites). Data were normalized to all compounds using Progenesis QI.
Principal component Analysis (PCA) was used to identify and remove outliers in the data set (see fig 8A). Heatmap analysis was performed in Metaboanalyst 5.0, data was pareto scaled and log transformed using Pearson distance and average clustering (see fig. 8B) to illustrate differences based on metabolite abundances across the experimental groups. Sample process and instrument variability were also assessed (see Table S6) to determine sample and batch acceptance. Briefly, QA metrics for sample process variability and instrument variability are ≤ 25% CV and ≤ 10% CV, respectively. In these studies, no samples were identified as outliers. Statistical analyses were performed in Progensis QI using variance stabilized measurements achieved through log normalization to calculate p-values by one-way analysis of variance (ANOVA) test. Significantly changed metabolites were chosen with the criteria p-value <0.05 and |fold change| > 2.
Tentative (Level 3, L3) and putative annotations (Level 2, L2) were determined within Progenesis QI software using accurate mass measurements (<5 ppm error), isotope distribution similarity, retention time matching and fragmentation spectrum matching using database searches against Human Metabolome Database (HMDB) 82, METLIN 83, the National Institute of Standards and Technologydatabase 84 and an in-house library. Annotations (Level 1–3) 85 were determined for significant metabolites by searching the above libraries or databases. The metabolite annotations of significantly different compounds (pairwise comparisons between HF diet and HF diet + LDP-treated mice) are represented in Table S7. The MS data are available in the Metabolomics Workbench public repository, with the data set identifier as ST002186.
Preparation of defined microbiota supernatants
To measure PLA in supernatants from defined microbiota cultures, fecal samples were collected from gnotobiotic mice colonized with the defined microbiota described above (including or lacking L. murinus) prior to colonization with E. coli Mt1B1. Fecal samples were resuspended at a concentration of 1 g feces to 25 mL PBS and combined in a 1:1 ratio with 50% sterile glycerol. Samples were then stored at −80. 1 mL of the defined microbiota fecal sample (either with or without L. murinus) was used to inoculate 10 mL of Anaerobic Akkermanisa Medium. Cultures were grown anaerobically for 48 hours. After 48 hours, cultures were harvested at 4000 × g for 10 minutes. The resulting pellets were resuspended in MRS broth and separated into 3 5 mL aliquots of Caco-2 media (previously incubated anaerobically for 48 hours). Cultures were then incubated for 16 hours before being harvested at 4000 × g for 10 minutes. The supernatant was filter sterilized and PLA measured as described below.
Phenyllactic Acid Measurements
Extraction and normalization.
Intestinal contents were weighed, diluted to a final density of 40 mg/mL in water/acetonitrile (3:2) buffered with 250 mM sodium phosphate pH 2.5 and homogenized/extracted using ceramic beads (2.8 mm dia.) and a Bead Mill 4 homogenizer (Thermo Fisher; Waltham, MA). Insoluble debris was removed by centrifugation (10,000 × g, 30 min, 5 °C); the supernatants were transferred to clean Eppendorf tubes and stored at −20 °C until the day of analysis.
LC-HRMS Analysis.
3-Phenyllactic acid (PLA) and a deuterium-labeled internal standard (PLA-d3) were derivatized with the reagent dansyl hydrazine and the carboxyl activating agent 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) to their corresponding dansyl hydrazone derivatives. Due to the limited stability of EDC under acidic conditions, stock solutions of EDC were made up fresh in water and used immediately. Briefly, 25 μL aliquots of thawed extracts were spiked with 2 nmol PLA-d3, diluted 1:3 with homogenization buffer, and derivatized at room temperature with dansyl hydrazine (25 μL × 50 mg/mL) and EDC (25 μL × 150 mg/mL). After one hour at room temperature, 25 μL methanol/water (1:1) containing 500 mM glucose was added to quench the excess reagents. Following centrifugation (10,000 × g, 30 min, 5 °C), quenched reaction mixtures were transferred to 2-mL autosampler vials equipped with low-volume polypropylene inserts and Teflon-lined rubber septa. The sample injection volume was 10 μL. PLA calibration standards were prepared in water and derivatized in the same manner. LC-HRMS analysis was performed using a Thermo Q Exactive HF hybrid quadrupole/orbitrap high resolution mass spectrometer interfaced to a Vanquish Horizon HPLC system (Thermo Fisher). The mass spectrometer was operated in positive ion mode, with targeted selected ion monitoring (t-SIM) detection of specified precursor ions at a resolving power of 30,000, an isolation window of 2.0 m/z, and the following HESI source parameters: spray voltage 4 kV; capillary temperature 300°C; HESI temperature 100 °C; S-lens RF level 50; N2 sheath gas 40; N2 auxiliary gas 10; sweep gas 2.0; in-source CID off. Extracted ion chromatograms were constructed for PLA and PLA-d3 with the following exact masses and a mass tolerance of +/− 5 ppm: PLA: [M+H]+ 414.1482; PLA-d3: [M+H]+ 417.1670. Data acquisition and quantitative spectral analysis were done using Thermo Xcalibur version 4.1.31.9 and Thermo LCQuan version 2.7, respectively. Calibration curves were constructed by plotting peak areas against analyte concentrations for a series of eleven calibration standards, ranging from 0.010 to 1000 μM. A weighting factor of 1/C2 was applied in the linear least-squares regression analysis to maintain homogeneity of variance across the concentration range. An Acquity BEH C18 reverse phase analytical column (2.1 × 100 mm, 1.7 μm, Waters, Milford, MA) was used for all chromatographic separations. Mobile phases were made up of 0.2 % HCOOH in (A) water and in (B) CH3CN. Gradient conditions were as follows: 0–1.0 min, B = 5 %; 1–6 min, B = 5–95 %; 6–8 min, B = 95 %; 8–8.5 min, B = 95–5 %; 8.5–12 min, B = 5 %. The flow rate was maintained at 300 μL/min, and the total chromatographic run time was 12 min. A software-controlled divert valve was used to transfer the LC eluent from 0 to 4.5 min and from 6.5 to 12 min of each chromatographic cycle to waste.
QUANTIFICATION AND STATISTICAL ANALYSIS
Fold changes of ratios (mRNA relative expression, PPRE activity, BODIPY C12 secretion) as well as percent abdominal fat, fasting glucose, PLA concentration (relative abundance and absolute concentration), and bacterial abundance were transformed logarithmically prior to statistical analysis. For quantification of PPAR-γ nuclear intensity, the integrated density of PPAR-γ fluorescent signal within the DAPI stained nucleus was measured. The integrated density was then transformed logarithmically prior to statistical analysis. An unpaired Student’s t test was used on the transformed data to determine whether differences between groups were statistically significant (p < 0.05). Comparisons between more than 3 groups were determined using a one-way ANOVA followed by Tukey’s or Šídák’s multiple comparison test. When comparisons between multiple groups and containing two independent variables were assessed, statistically significant differences between groups were determined by two-way ANOVA followed by Tukey’s or Šídák’s multiple comparison test. Significance of differences in histopathology, PPAR-γ immunohistochemistry, and oil red O staining were determined by a two-tailed non-parametric test (Mann-Whitney).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-PPAR-γ | Santa Cruz Biotechnology | Cat #: sc-7273 |
| Goat anti-mouse IgG Alexa Fluor 488 | Thermo Fisher Scientific | Cat #: A-11001 |
| Bacterial and virus strains | ||
| Ligilactobacillus murinus | This study | CS010 |
| Muribaculum intestinale | DSMZ | YL27 |
| Clostridium clostridioforme | DSMZ | YL32 |
| Clostridium sporogenes | DSMZ | B-CC-163–3B |
| Streptococcus daniellae ERD01G | DSMZ | ERD01G |
| Staphylococcus xylosus 33-ERD13C | DSMZ | 33-ERD13C |
| Escherichia coli Mt1B1 | DSMZ | Mt1B1 |
| Ligilactobacillus murinus PenR | This study | CS052 |
| Ligilactobacillus murinus | DSMZ | M-6244–3B |
| Lacticaseibacillus rhamnosus | ATCC | Cat #: 53103 |
| Lactobacillus acidophilus | ATCC | Scav; Cat #: 4356 |
| Bifidobacterium longum subsp. animalis | DSMZ | YL2 |
| Chemicals, peptides, and recombinant proteins | ||
| Difco Lactobacilli MRS agar | Becton Dickinson | Cat #: 288210 |
| Difco Lactobacilli MRS broth | Becton Dickinson | Cat #: 288130 |
| Penicillin V potassium salt | Sigma | Cat #: P1382 |
| D-(+)-3-phenyllactic acid | Sigma | Cat #: 376906 |
| Difco LB broth | Becton Dickinson | Cat #: 244620 |
| Difco Brain Heart Infustion agar | Becton Dickinson | Cat #: 241830 |
| Bacto Yeast Extract | Becton Dickinson | Cat #: 212750 |
| Bacto Tryptic soy broth | Becton Dickinson | Cat #: 211825 |
| Dipotassium hydrogen phosphate | Fisher Chemicals | Cat #: P288 |
| Hemin | Sigma | Cat #: 51280 |
| D(+)-Glucose | Acros Organics | Cat #: 41095 |
| Sodium carbonate | Fisher Chemicals | Cat #: 5263 |
| (L) – cysteine | Sigma | Cat #: 168149 |
| Menadione | Sigma | Cat #: 47775 |
| Heat-Inactivated Fetal Bovine Serum | Gibco | Cat #: 16140–071 |
| RPMI 1640 Medium | Gibco | Cat #: 11875093 |
| Recombinant Mouse IFN-gamma Protein | R&D Systems | Cat #: 485-MI-100 |
| Penicillin-Streptomycin | Gibco | Cat #: 15140122 |
| ITS Liquid Media Supplement | Sigma | Cat #: I3146 |
| Minimal Essential Media | Gibco | Cat #: 11095–080 |
| Glutamax | Gibco | Cat #: 35050–061 |
| Nonessential amino acids | Gibco | Cat #: 11140–050 |
| Sodium pyruvate | Gibco | Cat #: 11360070 |
| Trypsin-EDTA (0.05%) | Gibco | Cat #: 25300054 |
| Primocin | InvivoGen | Cat #: ant-pm-1 |
| Dithiothreitol | Acros Organics | Cat #: 16568 |
| EDTA | Sigma | Cat #: E5134 |
| EGTA | Sigma | Cat #: E3889 |
| Matrigel Membrane matrix | Corning | Cat #: 354234 |
| Intesticult Organoid Growth Medium (Mouse) | STEMCELL Technologies | Cat #: 06005 |
| Y-27632 dihydrochloride | TOCRIS | Cat #: 1254 |
| Rosiglitazone | Sigma | Cat #: R2408 |
| O.C.T. Compound | Fisher Chemicals | Cat #: 4585 |
| iQ SYBR Green Supermix | BioRad | Cat #: 1708880 |
| Bile salts | Sigma | Cat #: B8756 |
| Lipid mixture | Sigma | Cat #: L0288 |
| TRIzol Reagent | Invitrogen | Cat #: 15596026 |
| QIAzol Lysis Reagent | Qiagen | Cat #: 79306 |
| BioRad Protein Reagent | BioRad | Cat #: 5000006 |
| Tyloxapol | Sigma | Cat #: 1704003 |
| GW9662 | Sigma | Cat #: M6191 |
| Oleic acid | Sigma | Cat #: O1008 |
| Sodium taurocholate | Sigma | Cat #: T4009 |
| 2-mono-oleoylglycerol | Sigma | Cat #: M7765 |
| Cholesterol | Sigma | Cat #: C8667 |
| L-a-lysophosphatidylcholine | Sigma | Cat #: L4129 |
| BODIPY 558/568 C12 | Invitrogen | Cat #: D3835 |
| HEPES | Gibco | Cat #: 15630–080 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat #: 15710–5 |
| Triton X-100 | Acros Organics | Cat #: 327372500 |
| Alexa Fluor 647 Phalloidin | Invitrogen | Cat #: A22287 |
| ProLong Diamond Antifade Mountant with DAPI | Invitrogen | Cat #: P36971 |
| Butyric acid, sodium salt | Acros Organics | Cat #: 263191000 |
| DL-3-phenyllactic Acid-d3 | Toronto Research Chemicals | Cat #: P335567 |
| DL-3-phenyllactic acid | Toronto Research Chemicals | Cat #: H953713 |
| Critical commercial assays | ||
| Total RNA Purification Kits | Norgen Biotek Corp. | Cat #: 37500 |
| Oil Red O, Propylene Glycol staining kit | Newcomer Supply | Cat #: 12772 |
| DNeasy PowerSoil Kit | Qiagen | Cat #: 47014 |
| PureLink Genomic DNA Mini Kit | Invitrogen | Cat #: K182001 |
| iScript gDNA Clear cDNA synthesis kit | BioRad | Cat #: 1725034 |
| NEBNext® Ultra™ II Directional RNA Library Prep Kit for Illumina | New England BioLabs Inc. | Cat #: E7760 |
| Lipid Quantification Kit | Cell BioLabs, Inc. | Cat #: STA-617 |
| Serum Triglyceride Quantification kit | Cell BioLabs, Inc. | Cat #: STA-396 |
| Free Fatty Acid Assay Kit | Cell BioLabs, Inc. | Cat #: STA-618 |
| Total Cholesterol Assay Kit | Cell BioLabs, Inc. | Cat #: STA-384 |
| Bond Refine Polymer detection system | Leica Biosystems | Cat #: DS9800 |
| Luciferase Assay System | Promega | Cat #: E1500 |
| Deposited data | ||
| 16S rRNA sequencing of distal small intestine contents | European Nucleotide Archive | ENA: PRJEB52997 |
| RNA sequencing of distal small intestine epithelial cells | European Nucleotide Archive | ENA: PRJEB53157 |
| Untargeted metabolomics of distal small intestine contents | Metabolomics Workbench | ST002186 |
| Experimental models: Cell lines | ||
| Mouse: small intestinal cells from H-2Kb-tsA58 transgenic mice | Whitehead et al., 1993) | MSIE |
| Human: Caco-2 cells | ATCC | HTB-37 |
| Experimental models: Organisms/strains | ||
| Mouse: B6.Cg-Tg(Vil1-cre)997Gum/J | The Jackson Laboratory | JAX: 004586 |
| Mouse: B6.129-Ppargtm2Rev/J | The Jackson Laboratory | JAX: 004584 |
| Mouse: Ppargfl/flVillincre/- | Byndloss et al., 2017 | N/A |
| Mouse: C57BL/6NCrl |
Charles River Laboratory | CRL: 027 |
| Mouse: Germ-free C57Bl/6N | In-house colony | N/A |
| Oligonucleotides | ||
| Information regarding oligonucleotides used in this study is listed in Table S2. | ||
| Recombinant DNA | ||
| PPRE x3-TK-Luc | Addgene | Plasmid #: 1015 |
| Software and algorithms | ||
| Excel | Microsoft | N/A |
| Prism 9 (Version 9.2.0) | Graphpad | https://www.graphpad.com/features |
| QIIME2 (version 2022.2) | N/A | https://qiime2.org/ |
| STAR (v2.7.3a) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| featureCounts (v2.0.0) | (Liao et al., 2014) | https://subread.sourceforge.net/ |
| DESeq2 (v.1.24.0) | (Love et al., 2014) | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| WebGestaltR (v0.4.4) | N/A | https://www.webgestalt.org/ |
| Progensis QI v. 3.0 | Non-linear Dynamics, Water Corporation | https://www.nonlinear.com/progenesis/qi/v3.0/download/ |
| Metaboanalyst 5.0 | N/A | https://www.metaboanalyst.ca/MetaboAnalyst/home.xhtml |
Highlights.
Early-life exposure to antibiotics and a high-fat (HF) diet exacerbates obesity.
Loss of small intestinal Lactobacillaceae leads to increased adiposity.
Antibiotics and a HF diet exacerbate adiposity via depletion of intestinal PPAR-γ.
Lactobacillus-derived phenyllactic acid protects against antibiotic-induced obesity.
ACKNOWLEDGMENTS
We would like to acknowledge the Translational Pathology Shared Resource at Vanderbilt University Medical Center which is supported by NCI/NIH Cancer Center Support Grant P30CA068485 and the Shared Instrumentation Grant S10 OD023475-01A1 for the Leica Bond RX. Additionally, we would like to thank the University of California San Diego (UCSD) Microbiome Core as this publication includes data generated at the UC San Diego IGM Genomics Center utilizing an Illumina NovaSeq 6000 that was purchased with funding from a National Institutes of Health SIG grant (#S10 OD026929). This work was supported in part using the resources of the Center for Innovative Technology (CIT) at Vanderbilt University. The authors also wish to acknowledge the expert technical support of the Vanderbilt Technologies for Advanced Genomics Analysis and Research Design (VANGARD) core facilities. Immunofluorescent images were collected through the use of the Vanderbilt Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126) using a Zeiss LSM880 Airyscan ConfocalMicroscope (supported by NIH grant 1 S10 OD021630 1).
C.D.S. was supported by the NIH (T32AI112541 and 1F31AI161882-01A1), 2021–2022 Danone North America Gut Microbiome, Yogurt and Probiotics Fellowship, and VICTR (VR55193 and VR54476). N.G.S. was supported by NIH T32 Training Grant T32ES007028-46 and GT15104 from the Howard Hughes Medical Institute through the James H. Gilliam Fellowships for Advanced Study program. N.J.F. was supported by NIH T32 Training Grant T32DK007673. Work in N.O.M.’s lab was funded by Department of Veterans Affairs (BX005699). Worked in S.H.P.’s lab was funded by the Department of Veterans Affairs (1IK2BX005401) and funds from the Department of Medicine at Vanderbilt University Medical Center. M.X.B. is an HHMI Freeman Hrabowski Scholar. Work in M.X.B.’s lab was funded by The V Foundation for Cancer Research V Scholar grant (V2020–013), the NIH (R01DK131104-01 and 1R01AI168302-01A1), The Pew Charitable Trusts (2022-A-19568), and the Vanderbilt Institute for Clinical and Translational Research (VICTR) (VR53102).
Footnotes
DECLARATION OF INTEREST
Authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Information
Tables S1 – S6 and Figures S1 – S8 included in separate supplementary materials file.
REFERENCES
- 1.Gensollen T, Iyer SS, Kasper DL, and Blumberg RS (2016). How colonization by microbiota in early life shapes the immune system. Science 352, 539–544. 10.1126/science.aad9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maynard CL, Elson CO, Hatton RD, and Weaver CT (2012). Reciprocal interactions of the intestinal microbiota and immune system. Nature 489, 231–241. 10.1038/nature11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Derrien M, Alvarez AS, and de Vos WM (2019). The Gut Microbiota in the First Decade of Life. Trends Microbiol 27, 997–1010. 10.1016/j.tim.2019.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Roswall J, Olsson LM, Kovatcheva-Datchary P, Nilsson S, Tremaroli V, Simon MC, Kiilerich P, Akrami R, Kramer M, Uhlen M, et al. (2021). Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe 29, 765–776 e763. 10.1016/j.chom.2021.02.021. [DOI] [PubMed] [Google Scholar]
- 5.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, A, D.L., Wu, F., Perez-Perez, G.I., Chen, Y., et al. (2016). Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med 8, 343ra382. 10.1126/scitranslmed.aad7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNamara MP, Singleton JM, Cadney MD, Ruegger PM, Borneman J, and Garland T (2021). Early-life effects of juvenile Western diet and exercise on adult gut microbiome composition in mice. J Exp Biol 224. 10.1242/jeb.239699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chelimo C, Camargo CA Jr., Morton SMB, and Grant CC (2020). Association of Repeated Antibiotic Exposure Up to Age 4 Years With Body Mass at Age 4.5 Years. JAMA Netw Open 3, e1917577. 10.1001/jamanetworkopen.2019.17577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korpela K, Salonen A, Virta LJ, Kekkonen RA, Forslund K, Bork P, and de Vos WM (2016). Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat Commun 7, 10410. 10.1038/ncomms10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho I, Yamanishi S, Cox L, Methe BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I, et al. (2012). Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488, 621–626. 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D, et al. (2014). Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158, 705–721. 10.1016/j.cell.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoo W, Zieba JK, Foegeding NJ, Torres TP, Shelton CD, Shealy NG, Byndloss AJ, Cevallos SA, Gertz E, Tiffany CR, et al. (2021). High-fat diet-induced colonocyte dysfunction escalates microbiota-derived trimethylamine N-oxide. Science 373, 813–818. 10.1126/science.aba3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turnbaugh PJ, Backhed F, Fulton L, and Gordon JI (2008). Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3, 213–223. 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Guglielmo D, and Welsh JA (2018). Consumption of sugars, saturated fat, and sodium among US children from infancy through preschool age, NHANES 2009–2014. Am J Clin Nutr 108, 868–877. 10.1093/ajcn/nqy168. [DOI] [PubMed] [Google Scholar]
- 15.Zetts RM, Stoesz A, Smith BA, and Hyun DY (2018). Outpatient Antibiotic Use and the Need for Increased Antibiotic Stewardship Efforts. Pediatrics 141. 10.1542/peds.2017-4124. [DOI] [PubMed] [Google Scholar]
- 16.Clara R, Schumacher M, Ramachandran D, Fedele S, Krieger JP, Langhans W, and Mansouri A (2017). Metabolic Adaptation of the Small Intestine to Short- and Medium-Term High-Fat Diet Exposure. J Cell Physiol 232, 167–175. 10.1002/jcp.25402. [DOI] [PubMed] [Google Scholar]
- 17.Zembroski AS, D’Aquila T, and Buhman KK (2021). Characterization of cytoplasmic lipid droplets in each region of the small intestine of lean and diet-induced obese mice in response to dietary fat. Am J Physiol Gastrointest Liver Physiol 321, G75–G86. 10.1152/ajpgi.00084.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondo H, Minegishi Y, Komine Y, Mori T, Matsumoto I, Abe K, Tokimitsu I, Hase T, and Murase T (2006). Differential regulation of intestinal lipid metabolism-related genes in obesity-resistant A/J vs. obesity-prone C57BL/6J mice. Am J Physiol Endocrinol Metab 291, E1092–1099. 10.1152/ajpendo.00583.2005. [DOI] [PubMed] [Google Scholar]
- 19.Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, Pierre JF, Miyoshi J, Sontag TJ, Cham CM, et al. (2018). Small Intestine Microbiota Regulate Host Digestive and Absorptive Adaptive Responses to Dietary Lipids. Cell Host Microbe 23, 458–469 e455. 10.1016/j.chom.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semova I, Carten JD, Stombaugh J, Mackey LC, Knight R, Farber SA, and Rawls JF (2012). Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 12, 277–288. 10.1016/j.chom.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El Aidy S, Merrifield CA, Derrien M, van Baarlen P, Hooiveld G, Levenez F, Dore J, Dekker J, Holmes E, Claus SP, Reijngoud DJ, and Kleerebezem M (2013). The gut microbiota elicits a profound metabolic reorientation in the mouse jejunal mucosa during conventionalisation. Gut 62, 1306–1314. 10.1136/gutjnl-2011-301955. [DOI] [PubMed] [Google Scholar]
- 22.Tazi A, Araujo JR, Mulet C, Arena ET, Nigro G, Pedron T, and Sansonetti PJ (2018). Disentangling Host-Microbiota Regulation of Lipid Secretion by Enterocytes: Insights from Commensals Lactobacillus paracasei and Escherichia coli. mBio 9. 10.1128/mBio.01493-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Araujo JR, Tazi A, Burlen-Defranoux O, Vichier-Guerre S, Nigro G, Licandro H, Demignot S, and Sansonetti PJ (2020). Fermentation Products of Commensal Bacteria Alter Enterocyte Lipid Metabolism. Cell Host Microbe 27, 358–375 e357. 10.1016/j.chom.2020.01.028. [DOI] [PubMed] [Google Scholar]
- 24.Kappe C, Zhang Q, Nystrom T, and Sjoholm A (2014). Effects of high-fat diet and the anti-diabetic drug metformin on circulating GLP-1 and the relative number of intestinal L-cells. Diabetol Metab Syndr 6, 70. 10.1186/1758-5996-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poteres E, Hubert N, Poludasu S, Brigando G, Moore J, Keeler K, Isabelli A, Ibay ICV, Alt L, Pytynia M, Ciancio M, and Martinez-Guryn K (2020). Selective Regional Alteration of the Gut Microbiota by Diet and Antibiotics. Front Physiol 11, 797. 10.3389/fphys.2020.00797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murakami Y, Tanabe S, and Suzuki T (2016). High-fat Diet-induced Intestinal Hyperpermeability is Associated with Increased Bile Acids in the Large Intestine of Mice. J Food Sci 81, H216–222. 10.1111/1750-3841.13166. [DOI] [PubMed] [Google Scholar]
- 27.Andersson DI, and Hughes D (2014). Microbiological effects of sublethal levels of antibiotics. Nat Rev Microbiol 12, 465–478. 10.1038/nrmicro3270. [DOI] [PubMed] [Google Scholar]
- 28.Chen J, Ying GG, and Deng WJ (2019). Antibiotic Residues in Food: Extraction, Analysis, and Human Health Concerns. J Agric Food Chem 67, 7569–7586. 10.1021/acs.jafc.9b01334. [DOI] [PubMed] [Google Scholar]
- 29.Everard A, Geurts L, Caesar R, Van Hul M, Matamoros S, Duparc T, Denis RG, Cochez P, Pierard F, Castel J, et al. (2014). Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat Commun 5, 5648. 10.1038/ncomms6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Kuang Z, Yu X, Ruhn KA, Kubo M, and Hooper LV (2017). The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science 357, 912–916. 10.1126/science.aan0677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ko CW, Qu J, Black DD, and Tso P (2020). Regulation of intestinal lipid metabolism: current concepts and relevance to disease. Nat Rev Gastroenterol Hepatol 17, 169–183. 10.1038/s41575-019-0250-7. [DOI] [PubMed] [Google Scholar]
- 32.Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, Song Y, Cohen P, Friedman JM, and Attie AD (2002). Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc Natl Acad Sci U S A 99, 11482–11486. 10.1073/pnas.132384699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hussain MM, Kancha RK, Zhou Z, Luchoomun J, Zu H, and Bakillah A (1996). Chylomicron assembly and catabolism: role of apolipoproteins and receptors. Biochim Biophys Acta 1300, 151–170. 10.1016/0005-2760(96)00041-0. [DOI] [PubMed] [Google Scholar]
- 34.Mattijssen F, Alex S, Swarts HJ, Groen AK, van Schothorst EM, and Kersten S (2014). Angptl4 serves as an endogenous inhibitor of intestinal lipid digestion. Mol Metab 3, 135–144. 10.1016/j.molmet.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lynes M, Narisawa S, Millan JL, and Widmaier EP (2011). Interactions between CD36 and global intestinal alkaline phosphatase in mouse small intestine and effects of high-fat diet. Am J Physiol Regul Integr Comp Physiol 301, R1738–1747. 10.1152/ajpregu.00235.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lagakos WS, Gajda AM, Agellon L, Binas B, Choi V, Mandap B, Russnak T, Zhou YX, and Storch J (2011). Different functions of intestinal and liver-type fatty acid-binding proteins in intestine and in whole body energy homeostasis. Am J Physiol Gastrointest Liver Physiol 300, G803–814. 10.1152/ajpgi.00229.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng H, Chiu TY, Liang YJ, Lee CJ, Liu CS, Suen CS, Yen JJ, Chen HT, Hwang MJ, Hussain MM, Yang HC, and Yang-Yen HF (2021). PRAP1 is a novel lipid-binding protein that promotes lipid absorption by facilitating MTTP-mediated lipid transport. J Biol Chem 296, 100052. 10.1074/jbc.RA120.015002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byndloss MX, Olsan EE, Rivera-Chavez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y, et al. (2017). Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357, 570–575. 10.1126/science.aam9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alex S, Lange K, Amolo T, Grinstead JS, Haakonsson AK, Szalowska E, Koppen A, Mudde K, Haenen D, Al-Lahham S, et al. (2013). Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol Cell Biol 33, 1303–1316. 10.1128/MCB.00858-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duszka K, Oresic M, Le May C, Konig J, and Wahli W (2017). PPARgamma Modulates Long Chain Fatty Acid Processing in the Intestinal Epithelium. Int J Mol Sci 18. 10.3390/ijms18122559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nepelska M, de Wouters T, Jacouton E, Beguet-Crespel F, Lapaque N, Dore J, Arulampalam V, and Blottiere HM (2017). Commensal gut bacteria modulate phosphorylation-dependent PPARgamma transcriptional activity in human intestinal epithelial cells. Sci Rep 7, 43199. 10.1038/srep43199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ilavenil S, Kim DH, Valan Arasu M, Srigopalram S, Sivanesan R, and Choi KC (2015). Phenyllactic Acid from Lactobacillus plantarum PromotesAdipogenic Activity in 3T3-L1 Adipocyte via Up-Regulationof PPAR-gamma2. Molecules 20, 15359–15373. 10.3390/molecules200815359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di Cesare M, Soric M, Bovet P, Miranda JJ, Bhutta Z, Stevens GA, Laxmaiah A, Kengne AP, and Bentham J (2019). The epidemiological burden of obesity in childhood: a worldwide epidemic requiring urgent action. BMC Med 17, 212. 10.1186/s12916-019-1449-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.National Health and Nutrition Examination Survey 2017–March 2020 Prepandemic Data Files Development of Files and Prevalence Estimates for Selected Health Outcomes. (2021). In National Center for Health S, ed. National Health Statistics Reports. 10.15620/cdc:106273. [DOI] [Google Scholar]
- 45.Kumar S, and Kelly AS (2017). Review of Childhood Obesity: From Epidemiology, Etiology, and Comorbidities to Clinical Assessment and Treatment. Mayo Clin Proc 92, 251–265. 10.1016/j.mayocp.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 46.Leong KSW, McLay J, Derraik JGB, Gibb S, Shackleton N, Taylor RW, Glover M, Audas R, Taylor B, Milne BJ, and Cutfield WS (2020). Associations of Prenatal and Childhood Antibiotic Exposure With Obesity at Age 4 Years. JAMA Netw Open 3, e1919681. 10.1001/jamanetworkopen.2019.19681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Outpatient antibiotic prescriptions — United States , 2021. (2021). Centers for Disease Control and Prevention. [Google Scholar]
- 48.Duca FA, Waise TMZ, Peppler WT, and Lam TKT (2021). The metabolic impact of small intestinal nutrient sensing. Nat Commun 12, 903. 10.1038/s41467-021-21235-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veilleux A, Grenier E, Marceau P, Carpentier AC, Richard D, and Levy E (2014). Intestinal lipid handling: evidence and implication of insulin signaling abnormalities in human obese subjects. Arterioscler Thromb Vasc Biol 34, 644–653. 10.1161/ATVBAHA.113.302993. [DOI] [PubMed] [Google Scholar]
- 50.He W (2013). Adipose tissue-specific PPARgamma gene targeting. Methods Mol Biol 952, 117–135. 10.1007/978-1-62703-155-4_8. [DOI] [PubMed] [Google Scholar]
- 51.Ye G, Gao H, Wang Z, Lin Y, Liao X, Zhang H, Chi Y, Zhu H, and Dong S (2019). PPARalpha and PPARgamma activation attenuates total free fatty acid and triglyceride accumulation in macrophages via the inhibition of Fatp1 expression. Cell Death Dis 10, 39. 10.1038/s41419-018-1135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lewis GF, Uffelman K, Naples M, Szeto L, Haidari M, and Adeli K (2005). Intestinal lipoprotein overproduction, a newly recognized component of insulin resistance, is ameliorated by the insulin sensitizer rosiglitazone: studies in the fructose-fed Syrian golden hamster. Endocrinology 146, 247–255. 10.1210/en.2004-1143. [DOI] [PubMed] [Google Scholar]
- 53.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, and Knight R (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 107, 11971–11975. 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rossi M, Martinez-Martinez D, Amaretti A, Ulrici A, Raimondi S, and Moya A (2016). Mining metagenomic whole genome sequences revealed subdominant but constant Lactobacillus population in the human gut microbiota. Environ Microbiol Rep 8, 399–406. 10.1111/1758-2229.12405. [DOI] [PubMed] [Google Scholar]
- 55.Fak F, and Backhed F (2012). Lactobacillus reuteri prevents diet-induced obesity, but not atherosclerosis, in a strain dependent fashion in Apoe−/− mice. PLoS One 7, e46837. 10.1371/journal.pone.0046837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aronsson L, Huang Y, Parini P, Korach-Andre M, Hakansson J, Gustafsson JA, Pettersson S, Arulampalam V, and Rafter J (2010). Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin-like 4 protein (ANGPTL4). PLoS One 5. 10.1371/journal.pone.0013087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong W, Wang H, Yang Y, Zhang Y, Lai H, Cheng Y, Yu H, Feng N, Huang R, Liu S, et al. (2022). High-protein diet prevents fat mass increase after dieting by counteracting Lactobacillus-enhanced lipid absorption. Nat Metab 4, 1713–1731. 10.1038/s42255-022-00687-6. [DOI] [PubMed] [Google Scholar]
- 58.Karlsson Videhult F, Ohlund I, Stenlund H, Hernell O, and West CE (2015). Probiotics during weaning: a follow-up study on effects on body composition and metabolic markers at school age. Eur J Nutr 54, 355–363. 10.1007/s00394-014-0715-y. [DOI] [PubMed] [Google Scholar]
- 59.Luoto R, Kalliomaki M, Laitinen K, and Isolauri E (2010). The impact of perinatal probiotic intervention on the development of overweight and obesity: follow-up study from birth to 10 years. Int J Obes (Lond) 34, 1531–1537. 10.1038/ijo.2010.50. [DOI] [PubMed] [Google Scholar]
- 60.Zoetendal EG, Raes J, van den Bogert B, Arumugam M, Booijink CC, Troost FJ, Bork P, Wels M, de Vos WM, and Kleerebezem M (2012). The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J 6, 1415–1426. 10.1038/ismej.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koren O, Spor A, Felin J, Fak F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, and Backhed F (2011). Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A 108 Suppl 1, 4592–4598. 10.1073/pnas.1011383107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Couvigny B, de Wouters T, Kaci G, Jacouton E, Delorme C, Dore J, Renault P, Blottiere HM, Guedon E, and Lapaque N (2015). Commensal Streptococcus salivarius Modulates PPARgamma Transcriptional Activity in Human Intestinal Epithelial Cells. PLoS One 10, e0125371. 10.1371/journal.pone.0125371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maldonado-Gomez MX, Martinez I, Bottacini F, O’Callaghan A, Ventura M, van Sinderen D, Hillmann B, Vangay P, Knights D, Hutkins RW, and Walter J (2016). Stable Engraftment of Bifidobacterium longum AH1206 in the Human Gut Depends on Individualized Features of the Resident Microbiome. Cell Host Microbe 20, 515–526. 10.1016/j.chom.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 64.Laursen MF, Sakanaka M, von Burg N, Morbe U, Andersen D, Moll JM, Pekmez CT, Rivollier A, Michaelsen KF, Molgaard C, et al. (2021). Bifidobacterium species associated with breastfeeding produce aromatic lactic acids in the infant gut. Nat Microbiol 6, 1367–1382. 10.1038/s41564-021-00970-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Valerio F, Lavermicocca P, Pascale M, and Visconti A (2004). Production of phenyllactic acid by lactic acid bacteria: an approach to the selection of strains contributing to food quality and preservation. FEMS Microbiol Lett 233, 289–295. 10.1016/j.femsle.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 66.Yu S, Zhou C, Zhang T, Jiang B, and Mu W (2015). Short communication: 3-phenyllactic acid production in milk by Pediococcus pentosaceus SK25 during laboratory fermentation process. J Dairy Sci 98, 813–817. 10.3168/jds.2014-8645. [DOI] [PubMed] [Google Scholar]
- 67.Murphy R, Stewart AW, Braithwaite I, Beasley R, Hancox RJ, Mitchell EA, and Group IPTS (2014). Antibiotic treatment during infancy and increased body mass index in boys: an international cross-sectional study. Int J Obes (Lond) 38, 1115–1119. 10.1038/ijo.2013.218. [DOI] [PubMed] [Google Scholar]
- 68.Salinero AE, Anderson BM, and Zuloaga KL (2018). Sex differences in the metabolic effects of diet-induced obesity vary by age of onset. Int J Obes (Lond) 42, 1088–1091. 10.1038/s41366-018-0023-3. [DOI] [PubMed] [Google Scholar]
- 69.Nobel YR, Cox LM, Kirigin FF, Bokulich NA, Yamanishi S, Teitler I, Chung J, Sohn J, Barber CM, Goldfarb DS, et al. (2015). Metabolic and metagenomic outcomes from early-life pulsed antibiotic treatment. Nat Commun 6, 7486. 10.1038/ncomms8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eberl C, Ring D, Munch PC, Beutler M, Basic M, Slack EC, Schwarzer M, Srutkova D, Lange A, Frick JS, Bleich A, and Stecher B (2019). Reproducible Colonization of Germ-Free Mice With the Oligo-Mouse-Microbiota in Different Animal Facilities. Front Microbiol 10, 2999. 10.3389/fmicb.2019.02999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bacic MK, and Smith CJ (2008). Laboratory maintenance and cultivation of bacteroides species. Curr Protoc Microbiol Chapter 13, Unit 13C 11. 10.1002/9780471729259.mc13c01s9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, and Jat PS (1993). Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci U S A 90, 587–591. 10.1073/pnas.90.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yildiz S, Pereira Bonifacio Lopes JP, Berge M, Gonzalez-Ruiz V, Baud D, Kloehn J, Boal-Carvalho I, Schaeren OP, Schotsaert M, Hathaway LJ, et al. (2020). Respiratory tissue-associated commensal bacteria offer therapeutic potential against pneumococcal colonization. Elife 9. 10.7554/eLife.53581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barman M, Unold D, Shifley K, Amir E, Hung K, Bos N, and Salzman N (2008). Enteric salmonellosis disrupts the microbial ecology of the murine gastrointestinal tract. Infect Immun 76, 907–915. 10.1128/IAI.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 77.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jandacek RJ, Heubi JE, and Tso P (2004). A novel, noninvasive method for the measurement of intestinal fat absorption. Gastroenterology 127, 139–144. 10.1053/j.gastro.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 79.Pauquai T, Bouchoux J, Chateau D, Vidal R, Rousset M, Chambaz J, and Demignot S (2006). Adaptation of enterocytic Caco-2 cells to glucose modulates triacylglycerol-rich lipoprotein secretion through triacylglycerol targeting into the endoplasmic reticulum lumen. Biochem J 395, 393–403. 10.1042/BJ20051359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim JB, Wright HM, Wright M, and Spiegelman BM (1998). ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proceedings of the National Academy of Sciences 95, 4333–4337. doi: 10.1073/pnas.95.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Popay TM, Wang J, Adams CM, Howard GC, Codreanu SG, Sherrod SD, McLean JA, Thomas LR, Lorey SL, Machida YJ, et al. (2021). MYC regulates ribosome biogenesis and mitochondrial gene expression programs through its interaction with host cell factor-1. Elife 10. 10.7554/eLife.60191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, et al. (2013). HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res 41, D801–807. 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith CA, O’Maille G, Want EJ, Qin C, Trauger SA, Brandon TR, Custodio DE, Abagyan R, and Siuzdak G (2005). METLIN: a metabolite mass spectral database. Ther Drug Monit 27, 747–751. 10.1097/01.ftd.0000179845.53213.39. [DOI] [PubMed] [Google Scholar]
- 84.Jablonski F, Salvat F, Powell CJ & Lee AY (2016). NIST Standard Reference Database Number 64. [Google Scholar]
- 85.Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, and McLean JA (2016). Untargeted Metabolomics Strategies-Challenges and Emerging Directions. J Am Soc Mass Spectrom 27, 1897–1905. 10.1007/s13361-016-1469-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
16S rRNA sequencing data has been deposited in the European Nucleotide Archive under the accession number: PRJEB52997. RNA sequencing data has been deposited in the European Nucleotide Archive under the accession number: PRJEB53157. The mass spectrometry data are available in the Metabolomics Workbench public repository, with the data set identifier as ST002186. All data sets are publicly available. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.