

Abstract
β-barrel membrane proteins (βMPs) found in the outer membrane of gram-negative bacteria, mitochondria, and chloroplasts play important roles in membrane anchoring, pore formation and enzyme activities. However, it is often difficult to determine their structures experimentally, and the knowledge of their structures is currently limited. We have developed a method to predict the 3D architectures of βMPs. We can accurately construct transmembrane domains of βMPs by predicting their strand registers, from which full 3D atomic structures are derived. Using 3D Beta-barrel Membrane Protein Predictor (3D-BMPP), we can further accurately model the extended beta barrels and loops in non-TM regions with overall greater structure prediction coverage. 3DBMPP is a general technique that can be applied to protein families with limited sequences as well as proteins with novel folds. Applications of 3DBMPP can be broadly applied to genome-wide βMPs structure prediction.
Keywords: β-barrel membrane proteins, structure prediction, sequence covariation, strand register, computer simulation, loop prediction, sequential Monte Carlo sampling
1. Introduction
β-barrel membrane proteins (βMPs) are medically important as bacterial βMPs provide an important candidate class of molecular targets for development of antimicrobial drugs and vaccines. The advancement in the studies of β-barrel membrane proteins (βMPs) show further promise in bionanotechnology such as bionanopore sensor development. A major hindrance in the studies of βMPs is the limited structural knowledge: As of November 2020, only ∼552 βMP structures, of which∼323 are unique [2] have been deposited in the Protein Data Bank (PDB) that contains over 170,000 structures [3]. This limitation also hinders understanding of structural basis of the function and mechanism of βMPs.
Computational structure prediction can bridge the gap between identified βMP sequences and resolved βMP structures by providing high-resolution and high-accuracy model structures. Here we describe a template-free method for predicting 3D structures of βMPs, which provides significant improvement over previous methods [4]. Our predictor method, named 3D beta-barrel membrane protein predictor (3D-BMPP), is based on a statistical mechanical model [5] that comprises of sequence covariation information and is built upon a parametric structural model of intertwined zigzag coil. In addition, predictions are extended to include non-TM regions, including both extended β-sheets and loops, with significantly enriched coverage of residues. Furthermore, our method can be applied to model structures of βMPs with novel folds, including those from mitochondria of eukaryotes, as corroborated by the accurately modeled structures of VDAC and FimD. Our method is general and can broadly be applied to genome-wide structural prediction of βMPs.
2. Materials
3D-BMPP is a python framework with source code and scripts available from the public git repository: https://github.com/jksr/3dbmpp. In addition, 3DBMPP predictor is dependent on softwares like BBQ algorithm, PSICOV and SCWRL4.
2.1. Equipment:
Linux HPC cluster or workstation equipped with at least 2 GB RAM per computational node.
G++ compiler version ≥4.7 (https://gcc.gnu.org/).
CMake software version ≥4.8 (https://cmake.org/).
Proteins as FASTA files can be obtained from Protein Data Bank [3].
Git software, for obtaining 3D-BMPP and PSICOV source code, version ≥ 1.7.1 (https://git-scm.com)
Python packages, Numpy and Biopython along with JAVA class ultilities are required by our predictor.
2.2. Equipment Setup
We assume access is available to a Linux terminal operating a bash shell. Download and install all listed software. It is recommended to download 3D-BMPP and PSICOV using git command-line.
In a Linux terminal, obtain the 3DBMPP source codes via git-clone command: git clone https://github.com/jksr/3dbmpp-pipe.git
Next, navigate to source folder with cd bin/src followed by make command. cd ../..
Follow analogous procedures to download and install PSICOV and save it in under folder named psicov. In addition, the psicov requires HHBLITS for predicting contacts for a target sequence as alignment tool. Follow the HHBLITS documentation for its installation instructions [6].
To download scwrl, you must apply for a license on their website. We then recommend unpacking it into a folder name scwrl in the top-level folder of this repository.
3. Methods
To predict structures of βMPs, we proceed in three steps: predicting strand registers (interstrand hydrogen bond contacts), predicting 3D coordinates of TM residues, and modeling non-TM residues (Figure 1). Detailed information of the methods can be found in [1] (in its SI Appendix, sections 2–5).
Figure 1:
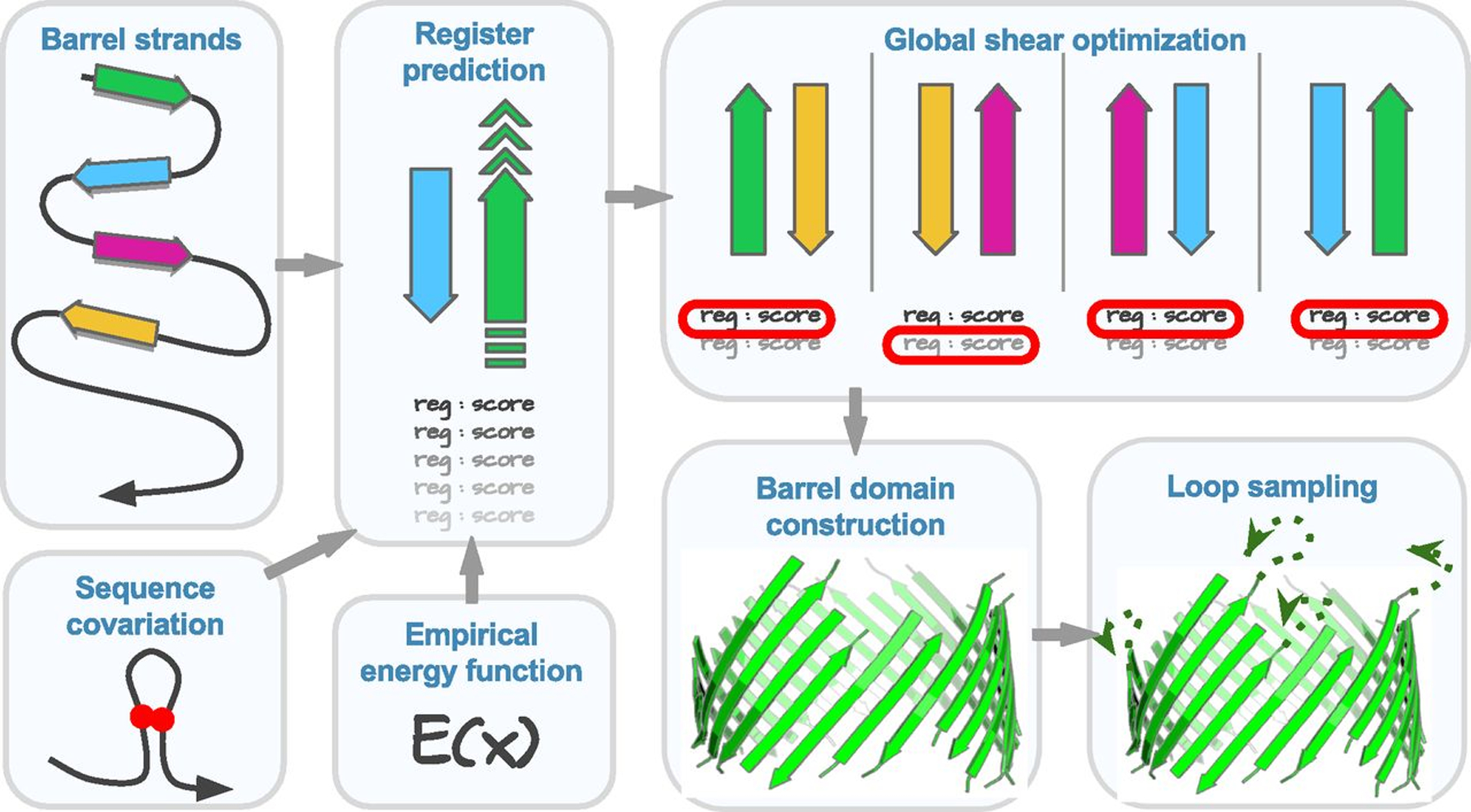
The flowchart of βMP structure prediction method 3D-BMPP [1].
-
Step 1:
First, a folder with protein PDB name needs to be created for the required structure to store all the input files and results.
-
Step 2:
Put the corresponding fasta file into the created folder.
-
Step 3:
Put a file with rough information of the starting and ending points of beta strands into the folder. Please refer to example/1bxw.strands.
The sequence ID, seqid of the starting and ending points should be consistent with the fasta file. The start and end points of beta strands can be approximately determined via any third party software for secondary structure prediction listed in http://www.ompdb.org/links.php. We recommend using consensus secondary structure prediction from PRED-TMBB2 [7], BOCTOPUS2 [8], and BetAware [9]. Information from the PSICOV sequence covariation analysis [10] may aid the structure prediction.
This is not a mandatory input. An empty file can be created with filename ending with .psicov in the folder to skip this step. However, this might affect the accuracy of the prediction.
-
Step 4:
In this method, the transmembrane beta barrel proteins are classified into five groups depending on the number of beta strands. The examples of the five groups are shown in the following Table 1:
-
Step 5:
Run the following command from the base 3bmpp repository folder to predict the 3D structure of the TMB proteins:
Table1:
Five groups of TMBs based on the number of their strands
| Groups | Description | Example PDB ids |
|---|---|---|
| 1 | Small TMBs (strand#<16) w/o inplugs or outclamps | 1bxw, 1qj8, 1p4t, 2f1t, 1thq, 2erv, 3dzm, 1qd6, 2f1c, 1k24,1i78, 2wjr, 4pr7 |
| 2 | Small TMBs (strand#<16) w/ inplugs or outclamps | 1t16, 1uyn, 1tlt, 3aeh, 3bs0, 3dwo, 3fid, 3kvn, 4e1s |
| 3 | Medium oligomeric TMBs (16≤strand#<20) | 2mpr, 1a0s, 2omf, 2por, 1prn, 1e54, 2o4v, 3vzt, 4n75 |
| 4 | Medium monomeric TMBs (16≤strand#<20) | 2qdz, 2ynk, 3emn, 3rbh, 3syb, 3szv, 4c00,4gey |
| 5 | Large TMBs (strand#≥20) | 1fep, 2fcp, 1kmo, 1nqe, 1xkw, 2vqi, 3csl, 3rfz, 3v8x, 4q35 |
python 3dbmpp.py --group *group_id* --folder *input_folder_name* --scwrlpath “*relative_path_to_scwrl4_executable*”
For example:
python 3dbmpp.py --group 1 --folder example -–scwrlpath “*relative_path_to_scwrl4_executable*”
Result:
The result is a pdb file that is stored in the current working folder and can be inspected using PyMOL.
Using 3D-BMPP, we can also predict the structure of novel beta barrel protein. Figure 3 shows the structure predicted for TonB-dependent transporter YncD (PDB ID: 6v81) found in E.Coli, which contains 24 beta strands, whose structure was recently determined experimentally.
Figure 3:

Structure prediction of the TM region of a novel protein TonB-dependent transporter YncD (PDB ID: 6v81) found in E. Coli,. Predicted structure of the TM regions (green) is superimposed on experimentally determined structure (cyan).
Information for this protein was not used in deriving the empirical potential function or training of various weight parameters of our model. The predicted structure has an RMSD of 2.51 Å (213 residues) when superimposing the predicted structure of the TM region (green) with experimentally determined structure (cyan).
Conclusion:
Due to the difficulty in determining the structures of membrane protein experimentally, there are currently a limited number of nonhomologous βMPs structures. Computational modeling can provide working 3D models based on βMPs sequences, facilitating applications in nanopore engineering, drug discovery and delivery, as well as aiding in the understanding of structural basis of the function and mechanism of these βMPs. The 3D-BMPP predictor incorporates statistical mechanical model, sequence covariation information, and global register optimization with a parametric structural model of intertwined zigzag coils. Results showed increased accuracy of structure prediction, with broadened scope of extended β-sheets and loops. Overall, our method opens the possibility of structural studies of many βMPs, including those in eukaryotic mitochondria and chloroplasts.
Figure 2:
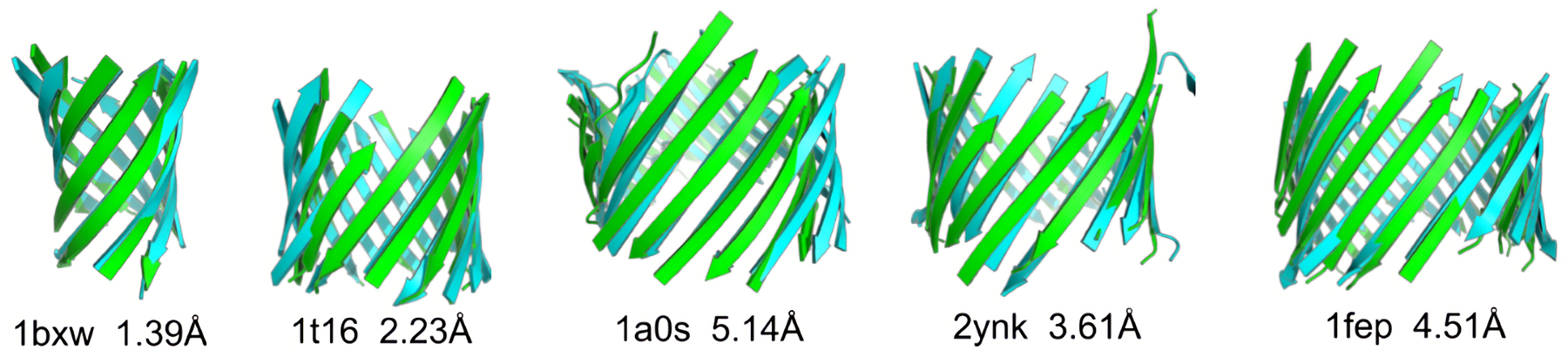
Structure prediction of TM regions. Representatives of predicted structures of the TM regions (green) from each group mentioned in Table 1, which are superimposed on experimentally determined structures (cyan) [1].
Acknowledgements:
This work is support by NIH grant R35 GM127084.
Footnotes
The method was described in [1]. Among 59 non-homologous βMPs (resolution 1.45 Å – 3.2 Å) with less than 30% pairwise sequence identity, of which predictions were made for 51 proteins after excluding multichain βMPs to restrict over-estimation of repetitive interactions. For 3D structure construction, complete dssp strands are used [11].
We take the canonical model of TM strands based on the physical interactions between strands described in [5] and [12]. We have developed a model incorporating both the empirical potential scores of physical interactions between strands from our previous study [5] along with the sequence covariation PSICOV [10] scores of each residue pairs in TM regions, that can identify medium-to-large range residue contacts based on the concept that spatially close residues might coevolve.
The energetic contributions comprise of the adjacent strands’ interactions, interstrand loop entropy, a penalty for left-handedness, and sequence covariation.
Information about odds files can be found in the 3dbmpp/odds folder. Also, to understand the strand register prediction you may check 3dbmpp/bin/src.
For each pair of adjacent strands, we enumerate all possible registers in a reduced conformational space and predict the registers followed by the global shear optimization. The inclusion of global register optimization increases the accuracy of the predicted structures by 0.24 Å on average, suggesting that the global hydrogen bond network cannot be approximated accurately using local strand register alone [1].
We use intertwined zigzag coils, a parametric structural model method from previous study to calculate the positions of Cα atoms [4]. Our parametric model of intertwined zigzag coils captures the zigzag nature of a polypeptide and the varied distance between atoms of two adjacent strands, depending on whether the corresponding residues share a main-chain hydrogen bond. This results in significant improvement in rmsd for all atoms in general and particularly side-chain atoms.
Main-chain atoms and side chains are added using Gront et al.’s [13] and SCWRL algorithm [14]. The BBQ utilities are in java class 3dbmpp/bin/BBQ. We then use an improved version of the m-DiSGro algorithm [15] to sample loop ensembles.
Readers may wish to check out https://github.com/nerrull/BetaBarrelRefactor for a refactored version of 3DBMPP.
Due to the computation complexity, this package only provides the structure prediction for the barrel domains of the TMBs. For the loop sampling mentioned in Tian et. al, please refer to https://github.com/uic-lianglab/ompg-public.
Currently this ideal cylindrical model does not capture ellipticity, twist, and curvature of local surface of the deformed barrel domains such as those observed in PapC and LptD.
References:
- 1.Tian W, Lin M, Tang K, Liang J, Naveed H (2018) High-resolution structure prediction of β-barrel membrane proteins. Proceedings of the National Academy of Sciences, 115 (7):1511–1516. This article contains supporting information online at 10.1073/pnas.1716817115/-/DCSupplemental. Accessed: December 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40 (Database issue): D370-D376 . Accessed: December 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berman H, et al. (2000) The Protein Data Bank. Nucleic Acids Res 28:235–242. Accessed: December 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naveed H, Xu Y, Jackups R Jr, Liang J (2012) Predicting three-dimensional structures of transmembrane domains of beta-barrel membrane proteins. J Am Chem Soc,134(3):1775–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackups R Jr, Liang J (2005) Interstrand pairing patterns in beta-barrel membrane proteins: the positive-outside rule, aromatic rescue, and strand registration prediction. J Mol Biol, 354(4):979–93. [DOI] [PubMed] [Google Scholar]
- 6.Remmert M, Biegert A, Hauser A, Söding J (2012) HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nature Methods, 9(2):173–175. [DOI] [PubMed] [Google Scholar]
- 7.Tsirigos KD, Elofsson A, Bagos PG (2016) PRED-TMBB2: improved topology prediction and detection of beta-barrel outer membrane proteins. Bioinformatics, 32(17):i665–i671. [DOI] [PubMed] [Google Scholar]
- 8.Hayat S, Peters C, Shu N, Tsirigos KD, Elofsson A (2016) Inclusion of dyad-repeat pattern improves topology prediction of transmembrane β-barrel proteins. Bioinformatics, 32(10):1571–3. [DOI] [PubMed] [Google Scholar]
- 9.Savojardo C, Fariselli P, Casadio R (2013) BETAWARE: a machine-learning tool to detect and predict transmembrane β-barrel proteins in Prokaryotes, Bioinformatics, 29(4): 504–505. [DOI] [PubMed] [Google Scholar]
- 10.Jones D, Buchan D, Cozzetto D, Pontil M (2012) PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics, 28(2):184–190. [DOI] [PubMed] [Google Scholar]
- 11.Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22(12):2577–2637. [DOI] [PubMed] [Google Scholar]
- 12.Ho B, Curmi P (2002) Twist and shear in beta-sheets and beta-ribbons. J Mol Biol, 317:291–308. [DOI] [PubMed] [Google Scholar]
- 13.Gront D, Kmiecik S, Kolinski A (2007) Backbone building from quadrilaterals: A fast and accurate algorithm for protein backbone reconstruction from alpha carbon coordinates. J Comput Chem 28:1593–1597. [DOI] [PubMed] [Google Scholar]
- 14.Krivov G, Shapovalov M, Dunbrack R Jr (2009) Improved prediction of protein side-chain conformations with SCWRL4. Proteins, 77(4):778–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang K, Wong S, Liu J, Zhang J, Liang J (2015) Conformational sampling and structure prediction of multiple interacting loops in soluble and beta-barrel membrane proteins using multi-loop distance-guided chain-growth Monte Carlo method. Bioinformatics 31:2646–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]