

Summary
The ubiquitin-proteasome system plays critical roles in biology by regulating protein degradation. Despite their importance, precise recognition specificity is known for few of the 600 E3s. Here we establish a two-pronged strategy for identifying and mapping critical residues of internal degrons on a genome scale in HEK-293T cells. We employ Global Protein Stability profiling combined with machine learning to identify 15,800 peptides likely to contain sequence-dependent degrons. We combine this with scanning mutagenesis to define critical residues for over 5,000 predicted degrons. Focusing on Cullin-RING ligase degrons, we generated mutational fingerprints for 219 degrons and developed DegronID, a computational algorithm enabling the clustering of degron peptides with similar motifs. CRISPR analysis enabled the discovery of E3-degron pairs of which we uncover 16 pairs that revealed extensive degron variability and structural determinants. We provide the visualization of this data on the public DegronID Data Browser as a resource for future exploration.
Graphical Abstract
eTOC blurb
Zhang et al. combined a genome-wide screen with machine learning to identify thousands of short linear motifs (degrons) in human proteins that are associated with sequence-dependent protein degradation. They went on to map critical residues, identify interacting ligases, and validate binding for a diverse collection of internal degrons.
Introduction
One of the primary mechanisms through which cells regulate signal transduction is through protein degradation. By selectively modulating protein degradation, cellular signaling pathways can quickly adapt to changing environmental conditions. Selective protein degradation is mainly mediated by proteases, selective autophagy and the Ubiquitin-Proteasome System (UPS), a complex system involving approximately 600 E3 ubiquitin ligases in mammals that selectively interact and ubiquitinate their substrates1. It was estimated that at least 80% of protein degradation is mediated by the UPS2. UPS substrates play crucial roles in nearly all major cellular processes. Dysregulation of protein degradation are frequently associated with many pathological disorders, ranging from cancer, immune pathologies to neurodegeneration3.
In addition to responses to stimuli, constitutive regulation of protein stability is also important for quality control. Protein misfolding is a serious problem for which there are several dedicated pathways involved in quality control to direct misfolded proteins for degradation4. Likewise, cells need mechanisms that can recognize proteins that fail to assemble into proper complexes5,6. Other areas of quality control include addressing failures in the proper localization of proteins to subcellular organelles such as the mitochondria7 or the ER8, or other specific membranes9,10 as seen for BAG6 recognition of mislocalized signal sequences. While some show regulation (e.g. the heat shock response), others act constitutively. Constitutively active E3 ligases may recognize masked degrons when proteins are misfolded or misassembled in complexes, act upon conformational change or post-translational modification of their substrate, or tie the abundance of a protein strictly to it mRNA level regulated in response to some stimuli.
Why are so many different E3s needed for the UPS? Presumably they have diverse modes of regulation and recognize distinct substrates11. However, since the discovery of the first degron in 198612, the number of known degrons remains sparse13. Furthermore, the motifs of many degrons were poorly characterized, making it challenging to predict E3 substrates using these sequences. We previously established the GPS-peptidome technology and applied it to the analysis of C-degrons and N-degrons10,14,15. The simplicity and position of those degrons allowed them to be defined computationally. However, such an approach is unable to define more complex degrons and therefore a systematic attempt to characterize naturally-existing internal degrons (position-independent degrons) is still lacking.
Results
GPS-peptidome screen for internal degrons
To develop a systematic method to identify internal degrons, we followed the strategy depicted in Figure 1. First, we designed 10,14 an oligonucleotide library encoding a peptides tiled across the entire human proteome. The library has ~ 470K peptides of 28 residues, overlapping by 5 residues. The peptidome library was cloned in frame with the C-terminus of the GFP reporter. To avoid the unwanted artificial C-degrons when fused to the C-terminus of GFP, a constant 20 amino acids lacking known C-degrons was positioned at the C-terminus of the library (Step 1 of Figure 1; STAR Methods). For authentic C-teminal degrons, we left in the natural stop codon to serve as previously studied controls14. The cloned GPS-peptidome library was packaged into lentivirus and used to infect HEK293T (ATCC CRL-3216) cells at a low multiplicity of infection (MOI) and selected using hygromycin resistance (Step 1 of Figure 1). The resulting population was sorted into 6 bins by fluorescence-activated cell sorting (FACS) according to the stability of individual GFP-peptide fusion relative to the DsRed protein, followed by DNA sequencing to quantify the relative abundance of each GFP-peptide sequence in each bin. To determine the stability of each GFP-peptide fusion, we employed the Protein Stability Index (PSI), which is calculated as a weighted average score of each peptide in the 6 bins, with PSI=1 being the least stable, and PSI=6 being the most stable10,14 (Figure S1A). We focused our analysis on ~260K peptides (referred to as the 260K peptidome library) with sufficient reads for high-confidence analysis (Table S2; STAR Methods).
Figure 1. Schematic diagram illustrating the workflow of mapping E3-degron pairs.
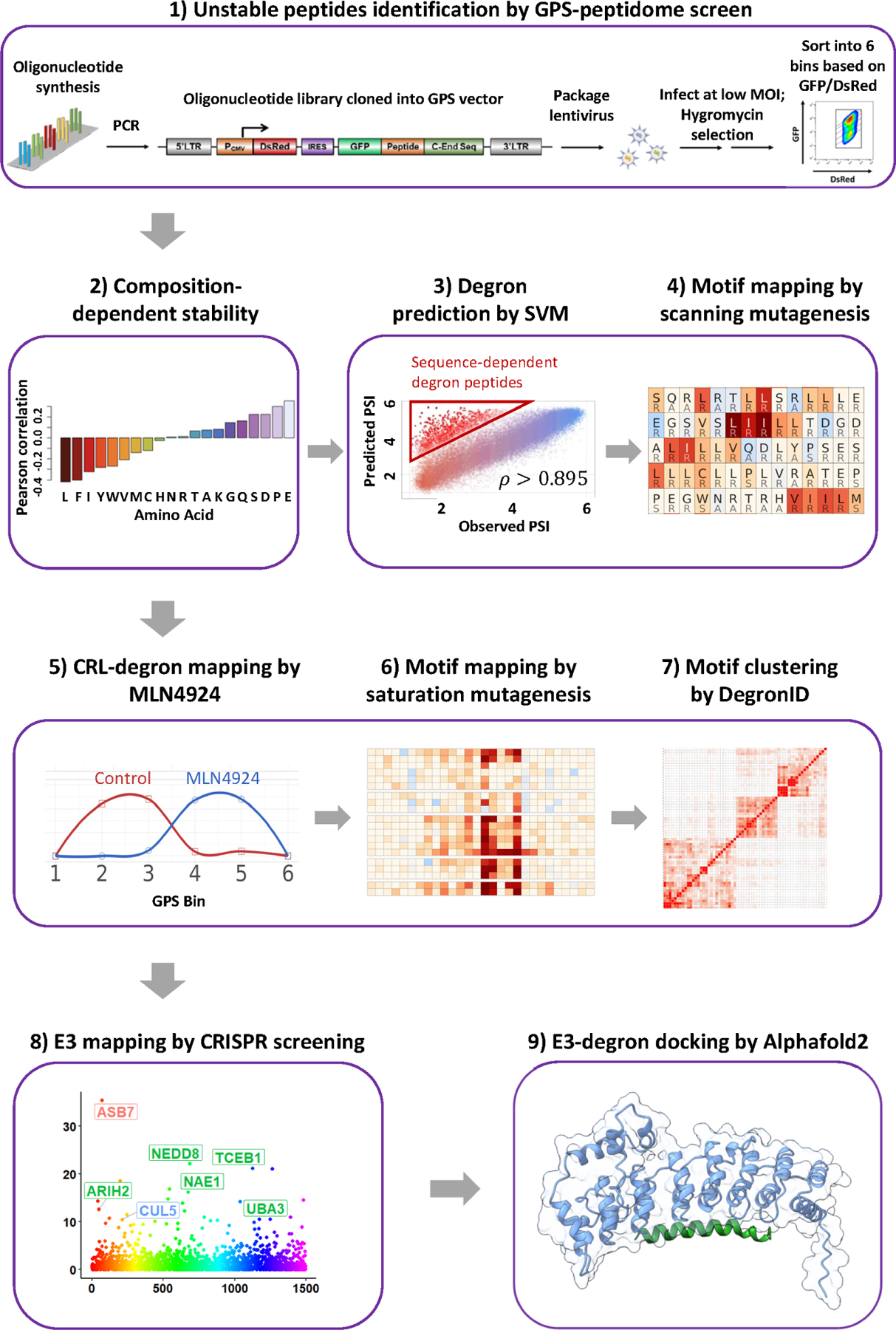
Overview of study workflow.
Correlation between amino acid composition and peptide stability
Peptide instability can be sequence-dependent, i.e. a defined sequential pattern of specific residues, or composition-dependent, i.e. based on the sum of the individual properties of amino acids regardless of order. We are primarily interested in the former class and sought to first identity the class of unstable peptides explained by composition-dependent mechanisms so as to minimize their contribution to our subsequent analysis. We observed a strong correlation between peptide amino acid composition and peptide stability (Step 2 of Figure 1; Figure S1B). The number of amino acids with hydrophobic side chains, except alanine, negatively correlated with peptide stability. Notably, leucine showed a striking Pearson correlation coefficient of −0.42 with peptide PSI. The frequency of amino acids with acidic side chains (glutamic and aspartic acid), and proline, positively correlated with PSI. Glutamic acid demonstrated the most significant correlation with a Pearson correlation coefficient of 0.35 (p-value < 2.2e-16) followed by proline (r = 0.30, p-value < 2.2e-16), aspartic acid (r = 0.23, p-value < 2.2e-16), serine (r = 0.22, p-value < 2.2e-16), glutamine (r = 0.16, p-value < 2.2e-16) and glycine (r = 0.14, p-value < 2.2e-16) (Step 2 of Figure 1). Alanine, threonine, asparagine, arginine, histidine and lysine were relatively neutral while the remaining larger hydrophobics showed a negative correlation with stability. Strong correlations with instability indicate that particular amino acids play central roles in general peptide turnover, consistent previous qualitative observations10,14.
Combining machine learning predictions and genetic analysis to identify sequence-dependent degrons
Next, we sought to use this information for identification of complex degrons. We previously found neural networks could be used to predict sequence-dependent degron motifs in small datasets containing C-terminal degrons of 2 amino acids16 but were unable to easily identify rarer and more complex degrons in larger datasets. We took a two-pronged approach to identify sequence specific degrons. First, we predicted peptides whose stabilities were composition-dependent. Secondly, we focused on those peptides whose stabilities could not be predicted by composition alone and thus were candidates for containing a sequence-dependent degron and carried out genetic analysis to identify sequences within them responsible for instability.
To predict composition-dependent peptides, we trained a support vector machine (SVM) model on 10% of the peptide data, using the counts of each amino acid within the peptide as features to predict PSI (Figure S1B, STAR Methods). We then used this trained model to predict PSI based on composition for the remaining peptides. The predicted PSI and observed PSI showed a Pearson correlation coefficient of approximately 0.9, with more than 92% of peptides exhibiting less than 1 PSI unit of difference between predicted PSI and observed PSI (Step 3 of Figure 1). The difference between the predicted PSI and the experimentally observed PSI is termed as degron index (DI). The larger the DI, the stronger the sequence-specific degron activity of the peptide. For example, C-end peptides with known C-degron motifs served as positive controls and showed significantly larger DIs, as compared with other C-end peptides (Figure S1C). We took the ~15800 peptides with DIs greater than +1 as our pool of peptides that potentially encode a large repertoire of degron peptides.
Many types of E3-regulated peptides should be present in the DI analysis, and we were particularly interested in those regulated by CRLs. Thus, we also measured PSI for cells treated with MLN4924 to block CRL-mediated destruction of CRL-regulated peptides. We calculated the MLN4924 ΔPSI to determine which peptides were likely to be a strong CRL substrates.
Next, we sought to identify the degron motifs within unstable peptides using a genetic approach. We generated a scanning mutagenesis library of containing 283,880 oligonucleotides covering 9817 peptides. This includes 1782 peptides of ΔPSI > 1 for MLN2924 treatment for CRL substrates, and 5790 peptides of DI > 1 for non-CRL substrates, together with other peptides to facilitate even stability distribution of the library and effective sorting (Table S3; STAR Methods). To maximize the chance that each mutation causes a significant change in the amino acid’s chemical properties, we employed an “opposite” scanning mutagenesis scheme where each amino acid was mutated to an amino acid with different chemical properties. “A, G, V, L, I, S, T, C, D and E” were mutated to “R”; “M, W, F and P” were mutated to “S”; “Y, N, Q, K, R and H” were mutated to “A” to generate a library of singly mutated peptides (STAR Methods). This GPS peptide library was screened as described for the original genome-wide library to determine the PSI for each peptide.
As the CRL-degron peptides will be characterized later in greater detail below, we will momentarily focus here on the discussion of putative non-CRL degron peptides: degron peptides with large DIs but no significant stabilization by MLN4924 (MLN4924 ΔPSI < 0.2). Interestingly, we observed a pervasive motif commonly found in many non-CRL degron peptides (Figure 2A), often characterized by a series of hydrophobic residues, sometimes interrupted by one or two hydrophilic residues. For 4 representative degron peptides, we performed CRISPR screens to identify the E3 ligase(s) that target these degron peptides for degradation (Figure 2B, STAR Methods).
Figure 2. SVM machine learning-aided identification of a BAG6 degron motif.
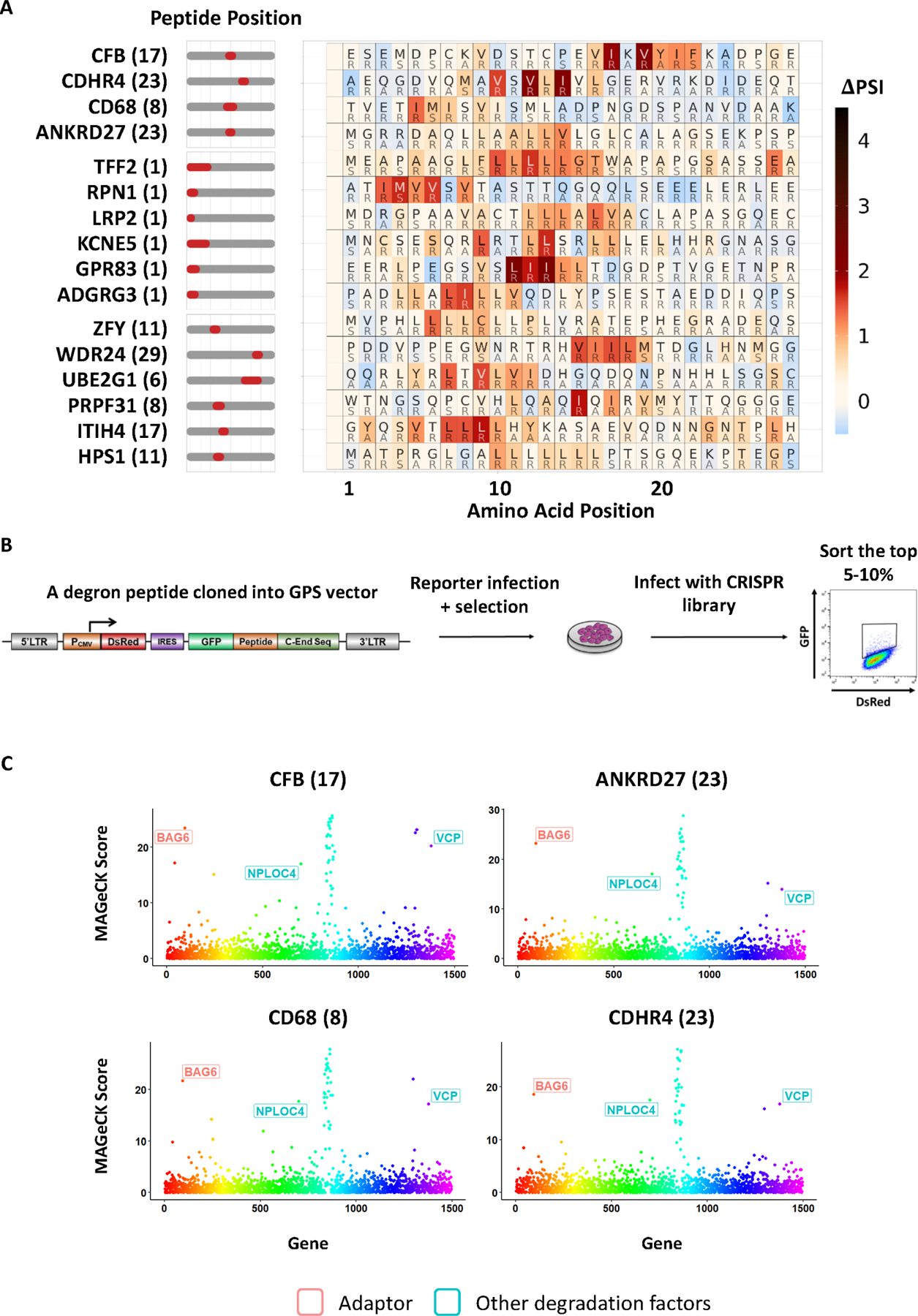
(A) Scanning mutagenesis of non-CRL degron peptides. Peptides in the top group were subjected to CRISPR screens and BAG6 was identified as a destabilizing gene. BAG6-like motifs were found not only in terminal peptides (middle group) deriving from N-terminal signal peptide sequences, but also in internal peptides (bottom group).
(B) Schematic diagram illustrating the workflow of the CRISPR screen designed to identify the genes required for the degradation of each degron peptide.
(C) CRISPR screens of 4 representative degron peptides with motifs described in (A) using a sgRNA lentiviral library of UPS related genes identified BAG6 as a gene required for the degron activity.
Strikingly, all 4 peptides were destabilized by BAG6 (Figure 2C). It had been previously established that BAG6 serves as a cytosolic protein chaperone that works with RNF126 to target proteins with a N-terminal signal peptide that fail to translocate into ER for destruction as a translocation quality control mechanism8,17. RNF126 did not score as a hit because its sgRNAs dropped out during the screening process. It was proposed that BAG6 performs this function through recognizing the exposed signal peptide that should otherwise be buried upon proper translocation. However, the precise degron motif recognized by BAG6 was never resolved at the precise amino acid level. Signal peptides are often 16–30 amino acids in length and possess a tripartite structure with a hydrophobic core region18. Our results suggested that BAG6 recognizes a signal peptide through its hydrophobic core region, as many signal peptides were predicted to encode non-CRL degrons and their scanning mutagenesis revealed BAG6 degron motifs within their hydrophobic regions (Figure 2A). Importantly, the motif recognized by BAG6 can be as short as 3–4 hydrophobic residues as in CD68. As many proteins that do not have a signal peptide also encode a putative BAG6 motif that could be exposed upon misfolding (Figure 2A), our findings suggest that the traditional view that BAG6 specializes in translocation quality control of the secretory pathway might be incomplete4. Instead, BAG6 might work as a general quality control pathway to protect cells from stresses caused by aberrant protein folding and potential aggregation. As hydrophobicity is a major driver for protein folding, most protein misfolding will likely expose a short stretch of hydrophobic sequences that could be recognized by BAG6 for protein quality control through proteasomal degradation.
In addition to the BAG6 degron motif, our scanning mutagenesis also identified degron motif patterns in other putative non-CRL degron peptides (Figure S1D). Though this study primarily focused on characterizing CRL-degron peptides, we anticipate that future research that studies these degron peptides would identify a large set of non-CRL degron pathways.
GPS-peptidome screen with MLN4924 to identify degrons recognized by CRLs
We next focused on characterizing the CRL-regulated degrons. To identify CRL-regulated degron-containing peptides, we performed a GPS-peptidome screen in the presence of MLN4924 (Figure 3A, B, STAR Methods). A total of 4245 peptides showed a MLN4924-ΔPSI score of larger than 0.8. We selected a subset of 180 high-confidence peptides and cloned the DNA encoding them into lentiviral GPS reporters using one of two forms of C-End sequences for downstream analyses (STAR Methods). A total of 101 validated peptides were validated and classified into different Cullin scaffolds using dominant-negative Cullins (Figures 3C, S1E and Table S5). We identified multiple peptides whose stabilities were regulated by five different Cullins. We observed that all peptides stabilized by DN CUL2 were stabilized equally by DN CUL5, but not all CUL5 regulated peptides were stabilized by DN CUL2, indicating a difference in inhibitory function by dominant negative CUL2 and CUL5. Altogether, these confirmed that our approach sampled CRL-degrons without biasing towards a specific degradation pathway.
Figure 3. The GPS-peptidome screen using MLN4924 identified CRL-dependent degron peptides.
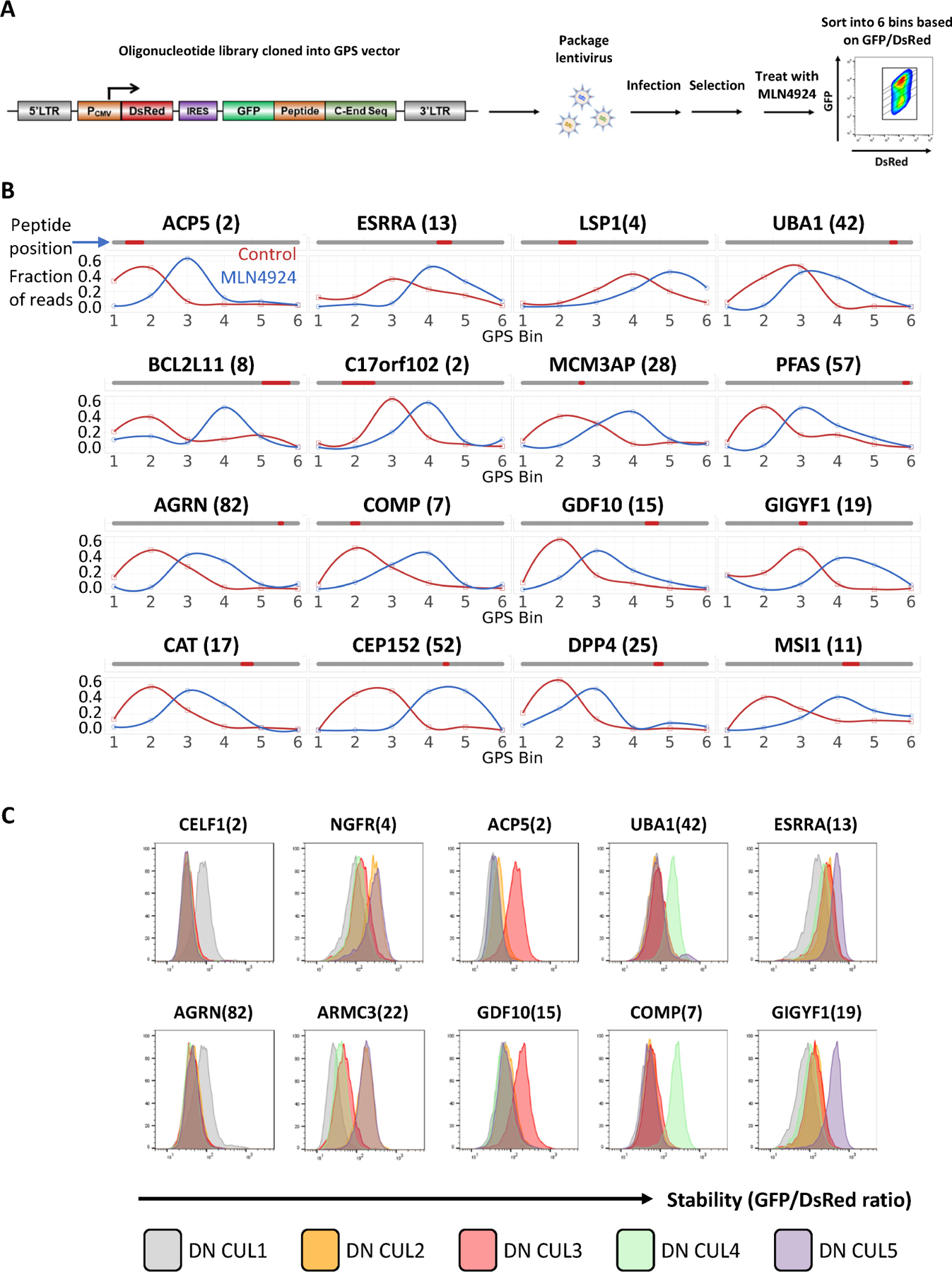
(A) Schematic diagram illustrating the workflow of the GPS-peptidome screen in the presence of MLN4924.
(B) Representative peptides identified in the screen in (A) as responsive to MLN4924 are shown. For each peptide, their relative distribution across the 6 bins in the control condition was compared to that of the MLN4924-treated condition. See also Figure S1.
(C) Representative GPS measurements for peptide stability with dominant-negative (DN) CUL expression. Two peptides selectively stabilized by DN CUL1, DN CUL2/5, DN CUL3, DN CUL4A, DN CUL5 are shown.
DegronID enabled degron peptide clustering based on degron motif similarities
To systematically define degron motifs, we performed a GPS-peptidome screen with a saturation mutagenesis peptidome library containing 133,250 oligonucleotides covering 250 selected peptides stabilized by MLN4924, where every residue of each peptide was mutated to the other 19 amino acids (Table S6). We achieved high resolution mapping of 219 degron footprints. Thirty-one peptides were missed either because of low representation or because they possessed multiple degrons which cannot be easily disambiguated. The saturation mutagenesis revealed distinct degron motifs. However, we also noticed similarities among several degron motifs, raising the possibility that multiple peptides may be recognized by the same cognate E3 ligase.
We then developed a motif clustering algorithm, DegronID, to classify peptides containing similar motifs (Step 7 of Figure 1). Briefly, for a degron footprint with saturation mutagenesis data, DegronID scores the human peptidome library for similarity to the degron footprint. DegronID then computes a similarity score between pairs of footprints and performs hierarchical clustering to group similar motifs (STAR Methods). The peptides from our reference database that were predicted by DegronID to be most similar to our finely mapped degron motifs tend to be less stable than our library as a whole (Figure 4A) and less stable than expected based on our composition-dependent SVM model (Figure S2B). Furthermore, they are more likely to contain more peptides than statistically expected that are stabilized by MLN4924 (Figures 4B). From DegronID’s hierarchical clustering of degron footprints, we limit the dendrogram to 40 groups (DegronID group) for practicality and observe multiple meta-clusters with similarity among multiple adjacent groups (Figure 4C). We additionally annotate alpha helix secondary structures using JPred419 and identified Cullin and E3 ligase adaptors identified from subsequent validation (Figure 4C). As expected, we find multiple peptide substrate with predicted motif similarity share an E3 ligase.As computational validation of DegronID, we find that peptides with shared E3 tend to cluster together and to score as similarly to each other relative to unrelated peptides (Figure 4C, S2D).
Figure 4: Degron ID classified saturation mutagenesis motifs into clusters based on their sequence similarities.
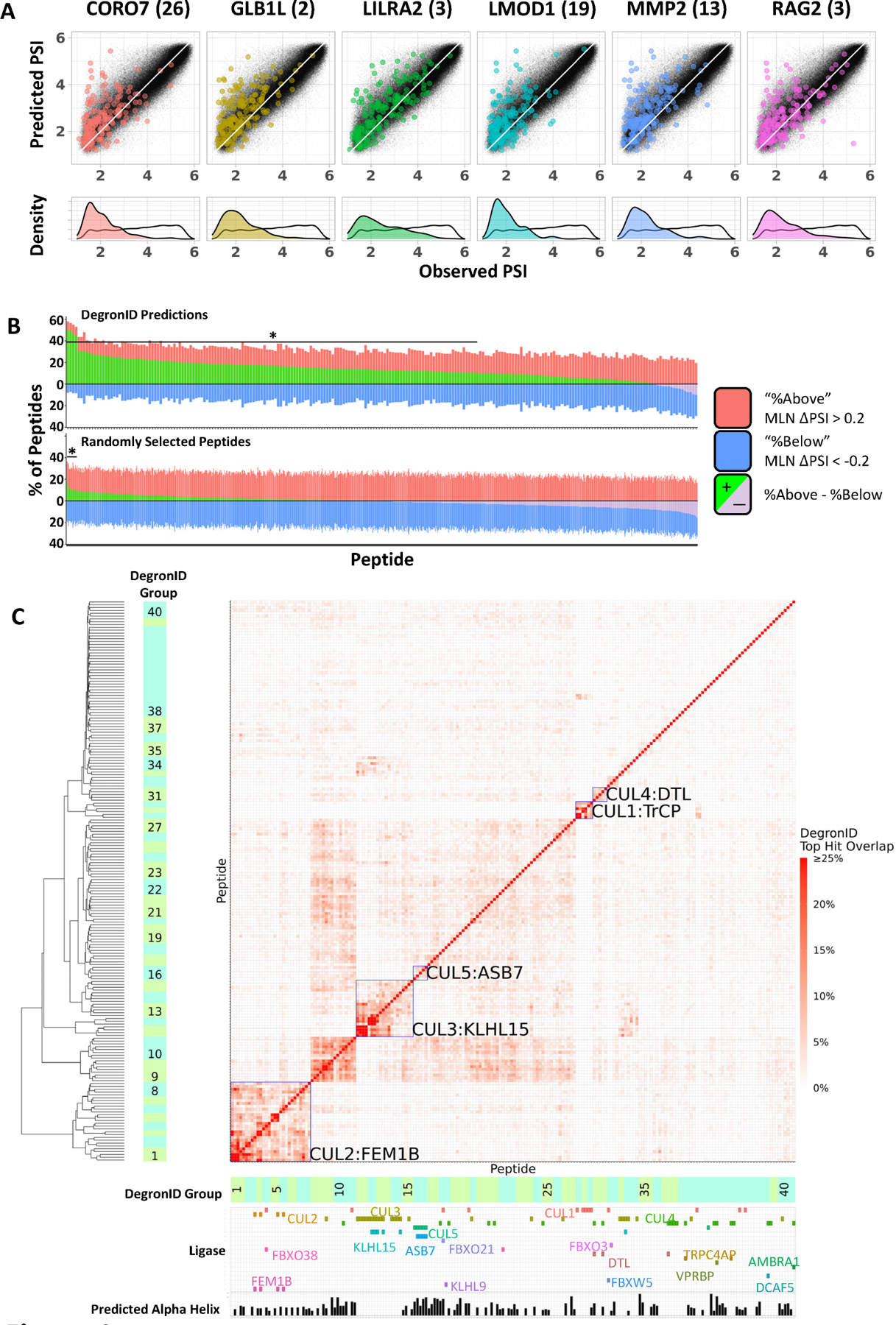
(A) Distribution of peptides in terms of PSI observed in GPS screen (x) and composition-based PSI prediction (y) from the 260k library (black) and the top 200 scoring hits by DegronID (color).
(B) Summary of top 200 hits by DegronID by MLN ΔPSI for (top) DegronID predictions for 198 CRL peptides; (bottom) iterations of random selections of 200 peptides from our 260k library. The bracket and asterisk indicate the instances for which the value of the green bar is greater than or equal to that which would be expected with an FDR of 0.1.
(C) Hierarchical clustering of saturation mutagenesis degron footprints. Alpha helical structure predictions and paired ligases from validation experiments are indicated below the clustergram. Meta-clusters that correspond to groups of degrons with multiple members sharing the same CRL are boxed and labelled (see Figure S3). See also Figure S2.
We also validated that the DegronID scoring algorithm chooses unstable peptides by examining sequences that most closely resemble other degrons that have been previously characterized. D-box motif degrons from the APC/C degron repository20 and C-terminal degron motifs containing either a GG* or RG* terminus from our previous study14 also predict unstable peptides (Figure S2A, STAR Methods). Furthermore, we checked that DegronID can properly predict known degrons among the top hits for a particular ligase. For example, for the BTRCP recognized degron motif, 2 known substrates, CDC25A and CDC25B, rank in the top 0.02% of prediction ranking 40th and 41st, respectively. In addition, known substrates degraded by KLHL15 score among the top 0.2% for similarity to a strong FRY domain from GLB1L(2) that we characterized by saturation mutagenesis (Figure S2C, top left). Additionally, of the 52 proteins identified as interacting with KLHL15 in BioPlex 3.0, DegronID identifies 16 proteins (31%) containing an [FL]R[FY] motif at the sequence that most closely matches the GLB1L(2) FRYV motif, with 13 additional proteins containing a weaker version of the [FL]R[FY] degron. By contrast, only 8.8% of the proteins in our library contained the [FL]R[FY] motif (Figure S2C). By Fisher’s exact test, the enrichment of [FL]R[FY] in KLHL15 interacting proteins compared to all human proteins in our library has a significance of p < 6.7e-06. This suggests that DegonID may be useful to prune high-throughput immunoprecipitation data for substrates that may be degraded by a particular E3.
To examine the possibility of using DegronID to characterize endogenous substrates of a particular E3, we chose six KLHL15 interacting proteins from BioPlex 3.0 that contained a FRY-like motif and which exhibited sensitivity to MLN4924 in our peptidome screen. The full-length ORFs for these proteins were cloned into the GPS 6.0 DEST vector and then stably expressed in either WT or KLHL15 KO HEK293T cells. ZNF511 was strongly stabilized in KLHL15 KO cells, while GPS-ORF constructs containing other KLHL15 interacting proteins exhibited a more subtle increase in stability in KLHL15 KO cells, while control GPS-ORF constructs containing randomly selected proteins that do not contain a FRY-like motif and are not known to interact with KLHL15 show no difference in stability in WT or KLHL15 KO cells (Figure S5D).
CRISPR screen identified the cognate E3 ligases of CRL-degron peptides
DegronID identifies clusters of motifs that look similar by saturation mutagenesis (Figure S3). To identify the cognate E3 ligases responsible for the degron activity of peptides with apparently distinct motifs, we performed CRISPR screens for degron peptides in different clusters. Fifteen CRL-adaptors were identified to be responsible for the degron activity of the peptides: including CUL1FBXO21, CUL1FBXO3, CUL1FBXO38, CUL1FBXW5, CUL1b-TrCP, CUL2FEM1B, CUL3KLHL9, CUL3KLHL15, CUL4DTL, CUL4DCAF5, CUL4TRPC4AP, CUL4AMBRA1, CUL4VPRBP, CUL5ASB7, CUL5ASB3 (Figures 6, S4; STAR Methods). One peptide was mapped to CUL1 but with no scoring CUL1 adaptor, raising the possibility that the sgRNAs targeting the corresponding adaptor dropped out during the screening process or was redundant. Subsequent examination of the degron motif showed that it matches a canonical b-TrCP degron motif21,22 of which there are two paralogs, b-TrCP1 and b-TrCP2. While the canonical b-TrCP degron motifs require one or two phosphoserines, our degrons have acidic residues that act as phosphomimetics as seen in CDC25B. We also performed the CRISPR screen for peptide DLST(7), one of the 19 peptides for which we did not achieve a high-resolution mapping of its footprint, and mapped it to CUL5ASB3. In addition to the substrate receptors, each of these screens identified commonly known components of CRL pathways including ARIH1, ARIH2, NEDD8, NAE1 and other core UPS components including VCP and proteosome subunits.
Figure 6. CRISPR screens identified the cognate E3 ligases for CRL-degron peptides.
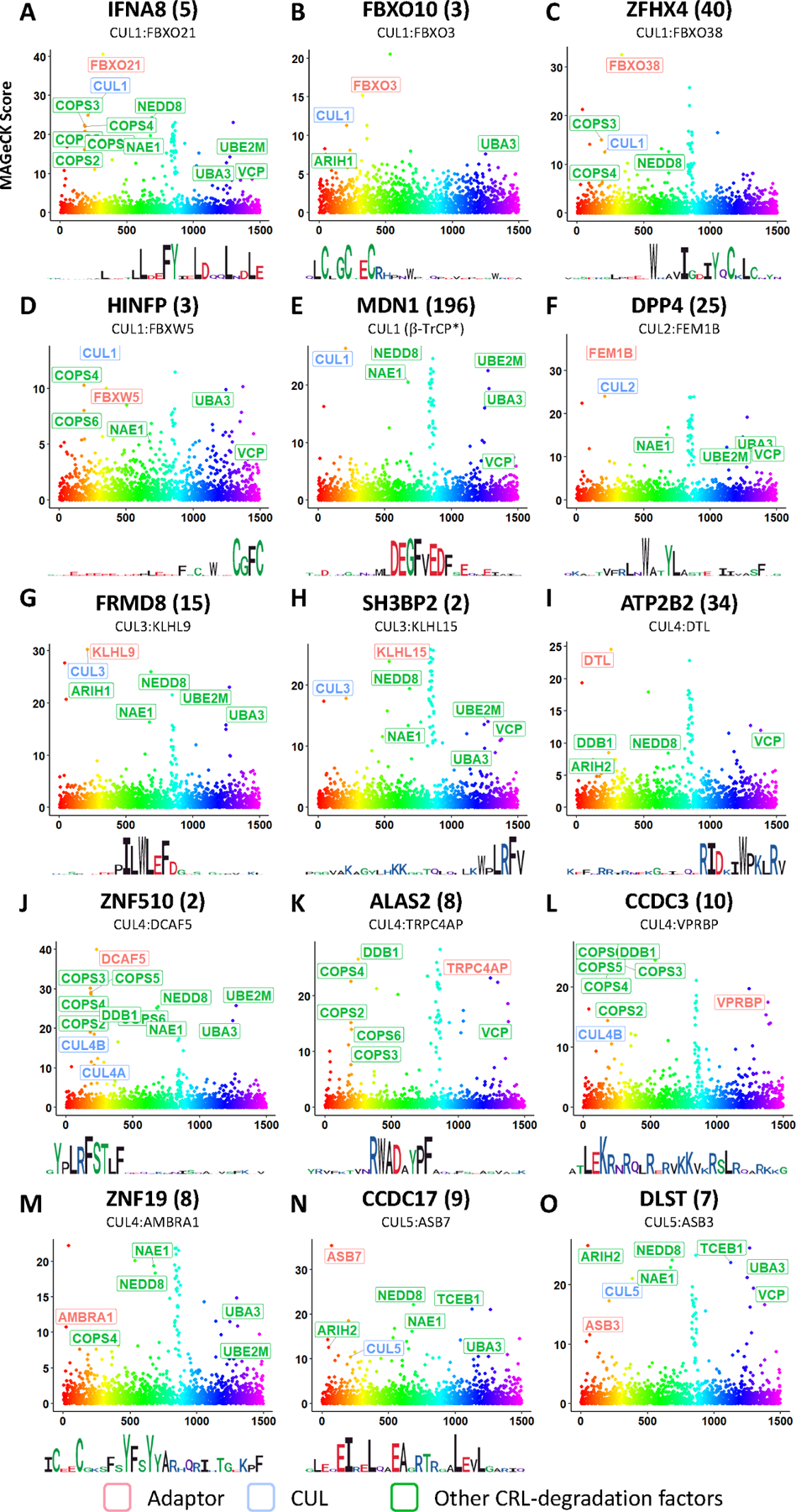
MAGeCK scores from CRISPR screens to identify cognate E3 ligase of each shown peptide. (STAR Methods). Logo plots derived from the each peptide’s mutagenesis footprint are also shown. See also Table S7, Figure S4 and Figure S5.
Among the 15 CRL adaptors, CUL1b-TrCP and CUL4DTL have been studied extensively and their cognate degron motifs have been accurately defined21,23. For CUL2FEM1B, CUL3KLHL15 and CUL4TRPC4AP, although instances of their degron motifs have been identified, our results here include many new degrons that are distinct variants of the previously known degron motifs. Some new motif variants are related to previously established degron motifs, as for the case of CUL3KLHL15, or could be completely unrelated sequences that bind to the same E3 ligase via a different binding site, as predicted to be the case of CUL2FEM1B (see below). To our knowledge, for the other 10 E3 ligases that we identified, including CUL1FBXO21, CUL1FBXO3, CUL1FBXO38, CUL1FBXW5, CUL3KLHL9, CUL4DCAF5, CUL4AMBRA1, CUL4VPRBP, CUL5ASB7, CUL5ASB3, no instances of precisely defined degron motifs have been identified. Degrons detected by FBXO21 and VPRBP were studied previously but were not precisely defined24–26.
FEM1B degron
The largest degron meta-cluster (clusters 1–8), was characterized by a degron motif resembling WxxYL and more generally W[VAC]x[YRT][ILT] (Figure S3B). A CRISPR screen identified CUL2FEM1B as a destabilizing factor of DPP4(25), a representative peptide of this meta-cluster (Figure 5F, 6F). Several other members of meta-cluster sharing similar motifs were also tested and confirmed to be FEM1B substrates by CRISPR mediated FEM1B KO (Figure 4C, S5C). This connection was unexpected because previous results from our group identified a R* C-end degron recognized by FEM1B. The presence of 2 structurally distinct classes of degrons recognized by the same E3 ligase suggests that the same E3 ligase could possess different degron binding domains27,28.
Figure 5. Saturation mutagenesis identified degron motifs in peptides stabilized by MLN4924.
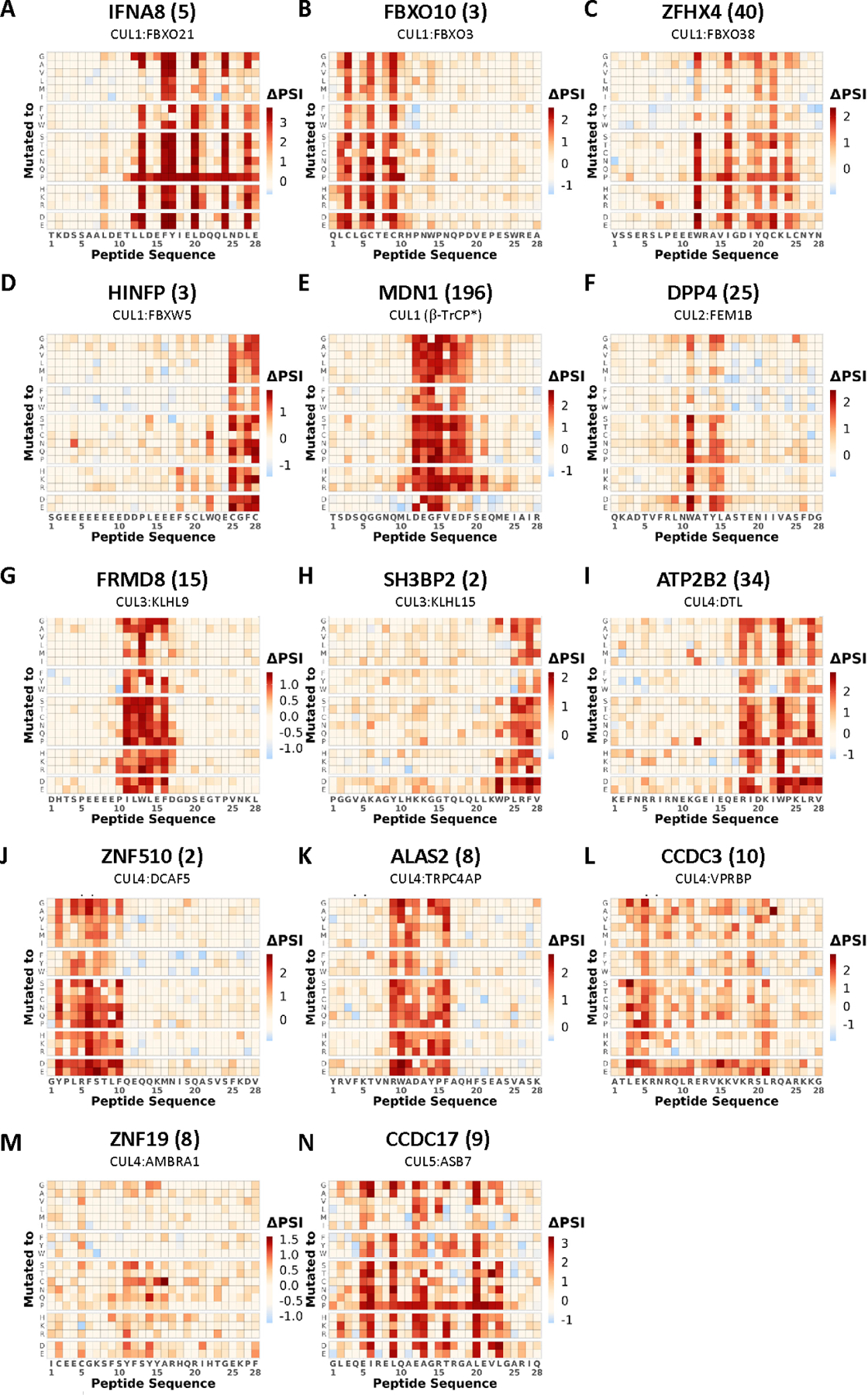
Saturation mutagenesis degron footprints for selected CRL degron peptides. The cognate E3 identified from subsequent CRISPR screening is also indicated. See also Figure 6 and Figure S3.
KLHL15 degron
The second largest degron meta-cluster (clusters 11–15), was characterized by a degron motif resembling LRF (Figure S3C). CRISPR screen identified CUL3KLHL15, as a destabilizing factor of the peptide SH3BP2(2), a representative member of this cluster (Figure 5H, 6H). Several other members of cluster sharing similar motifs were also tested and confirmed to be KLHL15 substrates by CRISPR mediated KLHL15 KO (Figure S5B). It was previously established that KLHL15 recognizes a tripeptide FRY degron, with 3 identified substrates containing the FRY degron to date29–31. However, our results here indicate that the actual motif is much more nuanced, as KLHL15 not only recognizes the motif, but also other variants, notably FRF, LRF, LRY (Figure S3C). Moreover, there are also residues adjacent to the core tripeptide motif that appear to be required in some contexts pointing to flexibility in degron E3 recognition. These underscored how a precise of understanding of degron motifs could enable more specific motif prediction while at the same time expanding the potential substrate repertoire recognized by the E3 ligase.
FBXO21 degron
CRISPR screening identified CUL1FBXO21 as a regulator of IFNA8(5) stability. Interestingly, our saturation mutagenesis revealed that the degron containing IFNA8(5) peptide whose stability is regulated by FBXO21 has discontinuous stretches of motif residues (Figures 5A, 6A). The spacing was reminiscent of an amphipathic alpha-helix and analysis by PROTEUS232 determined it was highly likely to form an alpha-helical structure (Figure S6B). Additionally, while most substitutions were tolerated in non-essential residues within the motif region of IFNA8(5), mutating any residue within this region to proline, which is known to disrupt alpha helices, completely abolished the degron activity throughout the stretch of amino acids supporting the need of an alpha helix for degron activity (Figure 5A).
ASB7 degron
Peptides of cluster 16 are among several clusters that showed regularly spaced critical residues in their degrons (Figure S3E, 5N). CRISPR screening identified CUL5ASB7 as a regulator of CCDC17(9) stability, a representative member of this cluster (Figure 5). Several additional peptides of this cluster sharing similar motifs, GIGYF1(19), LZTS1(16), and CEP152(52), were also tested and confirmed to be regulated by ASB7 (Figure S5A). These peptides are predicted to form an alpha-helical structure whose degron activity is clearly disrupted by prolines at any position within the predicted helical region (Figure S6). Interestingly, the 4 peptide substrates of CUL5ASB7 motifs show substantial flexibility over what are clearly related extended motifs (Figure S3E). The typical ASB7-regulated degron is spread out over 16–19 amino acids with 6 or 7 critical amino acids all along the same surface of an alpha helix. At each position it appears that one or two amino acids can be important although there are certain positions where pairs of amino acids seem to be dominant. Going from left to right, at position 1 (residue +1) there is an acidic residue, usually E, often followed by a hydrophobic residue such as L or I. At position 2, (residue +5), is frequently an L residue but can tolerate an I or M. Position 3 (residue +8) is another acidic acid residue, usually E, which can sometimes tolerate a T or a hydrophobic. Position 4 (residue +12) is often an L but is often flanked with charged residues, Positions 5 (residue +16) and 6 (residue +19) are often leucine that can tolerate I or M substitutions. As the surface interaction is extensive, suboptimal amino acids in one position might be compensated for by stronger interactions at one of the other positions to generate sufficient binding efficiency to interact with the E3 ligase. This makes prediction of this degron complex.
AlphaFold2-assisted global docking
E3 ubiquitin ligases typically recognize key residues in the degron of their substrate, as has been shown by several crystal structures28,33–35. Given recent advances in structural prediction algorithms, we sought to explore predicted complexes between our degrons and their cognate E3 ligases. Thus, we leveraged the AlphaFold2-multimer algorithm36,37 (STAR Methods). We found that docking for FEM1B, FBXO21, and ASB7 showed predicted interaction interfaces that were consistent with our degron saturation mutagenesis results.
The ASB7 adaptor is an ankyrin repeat protein that encodes seven ankyrin repeats with each repeat forming a helix-turn-helix structure38. Multiple ankyrin repeats thus have the potential to recognize dispersed interacting residues on target proteins and could possibly recognize the distributed residues in the alpha-helix degron. Strikingly, docking of CCDC17(9) and LZTS1(16) onto ASB7 predicted an interaction surface that mapped directly onto the critical residues we previously determined by saturation mutagenesis with each point of interaction with the critical residues on the degron peptide being recognized by a distinct ankyrin repeat (Figure 7A). The docking of these peptide critical residues to ASB7 was stable across the top three ranked AlphaFold structure prediction models. The notion that dispersed residues may be able to align to E3 substrate adaptors that are composed of multiple repeats, as many CRL adaptors are, may be a general property that allows evolution of many distinct substrate specificities.
Figure 7. E3-degron docking by AlphaFold2 identified critical degron residues consistent with that revealed by saturation mutagenesis, co-immunoprecipitation, and GPS.
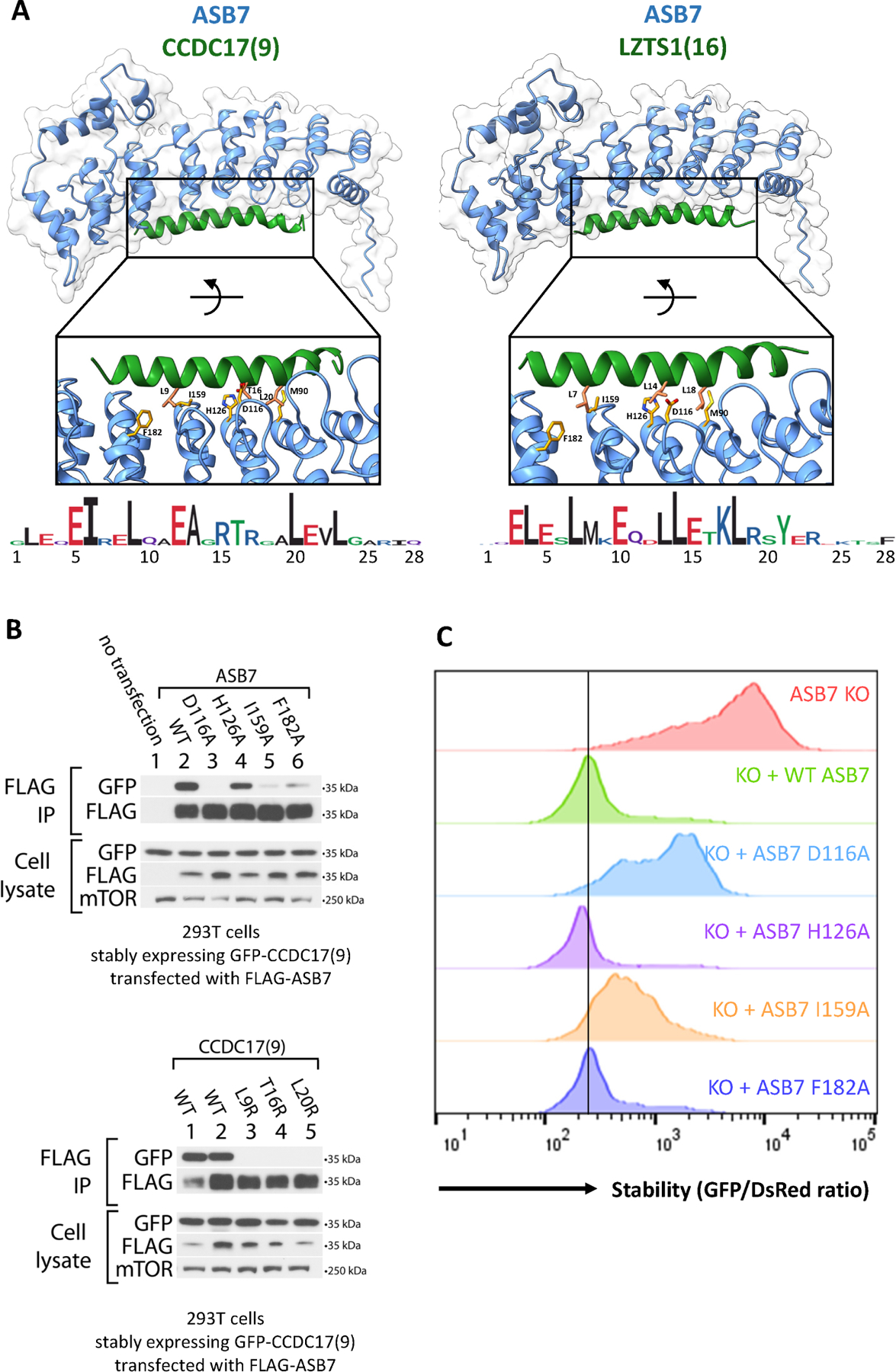
(A) Alphafold2 multimer docking of ASB7 degron peptides onto ASB7. Selected residues at the interaction interface are shown and labelled.
(B) Immunoblots for ASB7:CCDC17(9) binding in 293T ASB7 KO cells. (top) Co-immunoprecipitation of FLAG-ASB7 (WT or the indicated mutant) in cells stably expressing GFP-CCDC17(9) (WT). (bottom) Co-immunoprecipitation of FLAG-ASB7 (WT) in cells stably expressing GFP-CCDC17(9) (WT or the indicated mutant). Cells from lanes 1, 3, 4, and 5 stably express the specified GFP-degrons fusions flanked with C end sequence A (QGRARPNQEVQIGEMENQLS); while cells from lane 2 stably express GFP-CCDC17(9) flanked with C end sequence B (QGRARPNQEVQIGEMENQLD).
(C) Flow stability data for CCDC17(9) GPS reporter peptide with KO, stably expressed WT, or stably expressed mutant ASB7. See also Figure S6 and Figure S7.
While many F-box protein adaptors contain repeats such as WD40 and LRR repeats, a substantial fraction lack such repeats and belong to the FBXO class. One of the successful docking predictions was for FBXO21. Motif residues found by saturation mutagenesis in peptide IFNA8(5) were predicted as interacting residues in the docking structures (Figure S6C).
We also applied AlphaFold2 to predict degron docking for FEM1B substrates and found that the prediction successfully identified the critical W residue in the FEM1B motif to be crucial for FEM1B binding (Figure S7A). The structure of FEM1B binding to a R* degron peptide was solved earlier. However, docking of internal FEM1B degron peptides onto FEM1B showed that they bind to the FEM1B through a binding region distinct to that of R*. We postulate that the presence of 2 degron binding sites on the same E3 ligase could enable selective degradation regulation through binding distinct surfaces, thereby allowing this E3 ligase to very flexible in substrate recognition27,28.
Characterization of E3-degron binding
Based on AlphaFold2 docking of E3-degron multimers, we sought to test whether mutagenesis of putative critical residues would affect E3-degron binding. For ASB7, FEM1B, and FBXO21, we performed co-immunoprecipitation experiments to measure binding in HEK293T cells stably expressing GFP-peptide degrons and transiently transfected with N-FLAG-tagged E3 ligase. We tested WT E3 and also E3 and degron mutants as shown (Figures 7B, S6D, S7B). We observe effective immunoprecipitation of wild-type E3 with paired degron peptide and that mutagenesis of certain E3 and degron mutants that we identified as potentially critical for binding based on AlphaFold2 docking structures disrupted the ability of the E3 to co-IP the degron peptide with the exception of ASB7 H126A. We also tested the effect of E3 mutants on degron stability by GPS, and we find that the E3 mutants that disrupt binding by co-IP also disrupt the efficacy of the ligase to destabilize the degron peptide by flow (Figures 7C, S7C).
Measurement of degron peptide half life
We also tested the effect of ASB7, FEM1B, and FBXO21 mutants on the stability of their cognate degron peptides by cycloheximide chase (Figure S6A). In all three cases, we find that presence of the E3 ligase results in shorter half-life of the corresponding GPS reporter peptide.
DegronID Data Browser
To allow easy access to this large mutagenesis dataset, we created a web-hosted application to share data visualizations. The website provides visualizations of the effect of Cullin inhibitor MLN4924 on stability measured by GPS (Figure 3B) and degron footprints from scanning and saturation mutagenesis (Figure 2A, 5). Additionally, the website incorporates the degron clustering result of DegronID and enables the user to easily explore groups of related degron footprints. The application is freely accessible at https://brandonsie.shinyapps.io/DegronID/
Discussion
Systematic elucidation of the cellular degradome is critical to understanding the protein stability landscape and its role in shaping both homeostatic and regulatory biology. Here we identify peptides with the potential to be degrons in proteins, dependent on their accessibility within a protein, which can be affected by allosteric actions, complex formation or misfolding. The vast majority of the peptides in this library are likely recognized by general quality control mechanisms responsive to disordered sequences with simple amino acid compositions as their stabilities can be predicted by summing the contributions due to composition alone. The amino acid correlations to stability were similar to those we previously observed with N- and C-end degrons such as hydrophobicity correlating with instability and acidic residues correlating with stability, however, a few important differences stand out. Most prominently we found that proline, glutamine, glycine and serine correlated with stability and threonine trended in that direction. Unexpectedly, all amino acid constituents of the so called “PEST” sequences39 correlated with stability. PEST sequences have long been considered to be regions that confer instability on proteins39,40. Our findings suggest that, statistically speaking, these amino acids alone are very unlikely to cause instability. Either the PEST hypothesis is incorrect, or it is a particular arrangement of those residues or phosphorylation that generate the degrons within these low complexity regions. How these sequences confer stability on proteins remains to be determined.
From our scanning mutagenesis screen, we found that the most frequent class of non-CRL degrons were regulated by BAG6, which is an E3-associated chaperone that was known to recognize hydrophobic signal sequences on mis-localized membrane proteins. We found that BAG6 recognizes short hydrophobic degrons of 5–7 residues in length of a defined composition that is not relegated to stretches of hydrophobic residues but can accommodate internal neutral or even charged residues. There are clearly many additional sequence-specific degrons in the non-CRL class that are not BAG6 regulated as determined by our scanning mutagenesis patterns that should expand this degron class as well.
Mapping degron peptides to their cognate E3 ligases revealed 15 distinct sets of cognate CRLs, including CUL1FBXO21, CUL1FBXO3, CUL1FBXO38, CUL1FBXW5, CUL1b-TrCP, CUL2FEM1B, CUL3KLHL9, CUL3KLHL15, CUL4DTL, CUL4DCAF5, CUL4TRPC4AP, CUL4AMBRA1, CUL4VPRBP, CUL5ASB7, CUL5ASB3. Notably, except for CUL1b-TrCP & CUL4DTL, our study identified previously undefined sets of precisely defined degron motifs recognized by the other 13 CRLs and based on the motif separation by DegronID, many additional degrons remain unexplored and we expect many more orphan E3-degron pairs to emerge from future analysis of these degron motifs. The same strategy developed in this study could be applied to study the non-CRL degrome. Furthermore, cellular degrome under different cellular conditions or in different cell types with differential E3 ligase activities could also be surveyed using this strategy that we built here.
We found that the degron motifs grouped together by DegronID tend to have visually noticeable similarities in their degron footprints. Degrons that cluster together by DegronID do not necessarily have the same E3. Multiple E3 ligases may recognize similar degron recognition sequences. However, by exploring degron motif clusters generated by DegronID, we did find sets of degron motifs that cluster together and do share the same E3 ligase, which has provided deeper insights into degron and E3 plasticity. A case in point is CUL3KLHL15, an E3 ligase known to recognize the FRY degron. However, our saturation mutagenesis detected a much more nuanced degron that allowed many substitutions and expanded the number of additional residues involved in recognition from 3 to between 4 and 7 residues with both required and avoided residues at each position. Presumably degrons that deviate from the central FRY motif can take advantage of distal residues to improve their degrative potentials by gaining additional interactions. For example, many FRY degrons have a hydrophobic residue, often V, at the +4 position. Those having L at +1 position, e.g. CILP (51) and SYT2 (11), often have a hydrophobic at −2 and occasionally at −3 or − 4.
Secondly, we identified a distinct FEM1B degron motif lacking the R* motif , W[VC]xxL, which makes use of a different degron binding pocket present on FEM1B as predicted by AlphaFold2 modelling. In the case of R* degron, the C-terminal R binds to the D131 on the ANK3 repeat28. However, for the W[VC]xxL motif, it was predicted that the W of the degron coordinates with W367 located near the TPR domain. In the case of WxxYL, the Y of the degron coordinates with F501 on FEM1B (Figure S7A). We have validated that FEM1B W367 and F501 are critical to binding between FEM1B and a WVTYL-containing peptide by co-immunoprecipitation and critical to destabilization of a WVTYL-containing peptide by GPS (Figure S7B, C). By contrast, FEM1B mutations H345A and Y84A, which are not predicted to contact critical residues of the WVTYL degron (Figure S7A), do not impair FEM1B:WVTYL binding by co-immunoprecipitation.
Third, while many degrons reside in unstructured regions of proteins13, we identified peptides that stood out as clearly involving stable secondary structures, notably the CCDC17(9) peptide regulated by CUL5ASB7 and the IFNA8(5) peptide regulated by CUL1FBXO21 (Figure 7A, S6C). Both peptides were predicted to be alpha helical, and the hydrophobic residues required for degradation were primarily located on one face of the helix. Consistent with this structural prediction, placement of the alpha helix-breaking proline and, to a lesser extent, glycine at any position along the degron region caused stabilization (Figure 5A, N), supporting a role for the helical structure in degron function. Importantly, docking these degron peptides with their E3 showed that the residues on one helical face that were predicted to be critical for degron activity were precisely the same residues that made contact with the E3. In the case of the ankyrin repeat protein ASB7, loops at the ends of the ankyrin repeats were spaced with the same pitch as the alpha helix to identify equivalently spaced critical residues on the helical face. We have validated that mutations D116A or I159A on ASB7 perturb the ability of ASB7 to bind or destabilize the CCDC17(9) alpha helix (Figure 7B, C). We also find for both ASB7:CCDC17(9) and FBXO21:IFNA8(5) that mutation of the putative critical residues along one helical face of the degron peptide impairs E3:degron binding (Figure 7B, S6D).
The large number of degrons mapped in this study have the potential to expand the Proteolysis Targeting Chimeras (PROTACs) toolbox. PROTACs are an increasingly important concept in therapeutics41. Despite the profound excitement generated by PROTACs in the past decade, its complete potential has not been fully realized. This is in part limited by the relatively few E3 ligases with precise definition of their cognate degrons42. PROTACS often mimic endogenous substrates43. It was demonstrated that a chemical mimicking a known FEM1B degron enabled the discovery of a new PROTACs therapeutic44. This ligand binds to FEM1B by targeting a previously identified degron binding pocket on FEM1B involving ANK3-ANK627. This illustrates the feasibility and importance of developing new PROTACs therapeutics through understanding natural degron motifs.
Through the 40-year history of degron analysis, most degrons were discovered one per study and the reported motifs were often incomplete. We began developing a high-throughput system for characterizing degrons but were previously only able to easily identify patterns and the N- and C-termini of peptides which were simple and gained specificity from being positioned at protein ends. However, the vast majority of degrons are internal. Here, we extended our earlier work on N/C-degrons and apply GPS to systematically study internal degrons. To gauge the degree of our progress relative to previous progress in the field on internal degrons over the last 40 years, the Eukaryotic Linear Motif (ELM) database currently presented a collection of internal mammalian degrons recognized by 14 ubiquitin ligases45. Our proof-of-concept work involved mutagenic fingerprinting of a large set of internal degrons recognized by 16 ubiquitin ligases and highlighted many additional peptides with potential degron activity. Of these 16, only two E3s overlapped with the ELM collection, b-TrCP and CDT2 (DTL). Consequently, anticipate that many more degrons await to be uncovered in this dataset and their knowledge will fuel discovery of portions of full-length proteins regulated by cognate E3s and possibly, coupled with structural prediction programs like AlphaFold2. Predictions of full-length substrates may become possible and may be facilitated by filtering of high-throughput immunoprecipitation data by motif search similar to DegronID. Future studies would be aided by methods for high throughput screening of E3:degron pairs, which have recently been developed46. We foresee that future research into these degrons will help unravel the complex but yet under-explored cellular degradome and their physiological roles in biological systems.
Limitations of the study
We chose a 28-mer tile peptidome approach to represent human internal degrons, and thus we are likely to inherently miss some structural or conformational motifs. However, our peptidome screen was able to effectively capture many alpha helical degron motifs. The E3-degron pairs in this study were characterized genetically and, therefore, in some cases the destabilization of the degron peptide by the E3 could in theory be due to indirect effects. However, the results from our substrate binding assays for FBXO21, ASB7, and FEM1B demonstrate that these effects are direct as do the impacts of the stabilizing mutations in the peptide that disrupt E3 binding. This concurs with analysis from our previous studies on N- and C-terminal degrons in which the effects have all been direct when tested by ourselves and others10,14,28,33–35.
Some peptides with an E3-interacting motif may not score as a degron peptide for several reasons: (1) Activation of degron activity requires a spatially accessible lysine for ubiquitination. While there are 18 surface-accessible lysine residues in GFP, we cannot rule out the chance that in some cases the E3s recruited by degrons cannot access a lysine for ubiquitination. (2) The cognate E3s for some degrons may not be expressed in HEK293T cells. (3) The CRL-degrons require activation by post-translation modifications such as phosphorylation or acetylation. This peptide system likely lacks most protein modifications and will miss many of those degrons. However, although CUL1b-TrCP required phosphorylation of its degrons, we identified 6 degrons that lack phosphosites but contain clusters of acidic residues that are likely to act as phosphomimetics (Figure S5A). Thus, we may be able to detect E3 ligases that require phosphates on some degrons.
Although we were able to use the AlphaFold2 prediction models to identify critical E3 residues for degron binding that disrupted Co-IPs when mutagenized, confirmed structures would certainly aid in identification of such residues.
Despite these limitations, these systematic studies represent a substantial contribution to the available knowledge in this field and should provide the basis of the discovery of many additional degron-E3 pairs in the future.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen J. Elledge (selledge@genetics.med.harvard.edu).
Materials Availability
Any reagents that are unique to this study will be made available upon request.
Data and code availability
Original GPS peptidome screen data and raw western blot images were deposited to Mendeley Data and Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. The DOIs are listed in the key resources table under “Deposited Data” as “GPS peptidome processed screen data and raw images” (Mendeley Data) and “GPS peptidome raw and processed screen data” (GEO). Detailed GPS peptidome screen data, mutagenesis screen data, CRISPR screen data, and DN CUL analyses are available in Tables S1, S2, S3, S4, S5, S6, S7.
All original code has been deposited to Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table under “Software and algorithms” as “DegronID”.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-mTOR | Cell Signaling Technology | 2983S |
| Rabbit monoclonal anti-FLAG | Cell Signaling Technology | 14793S |
| Rabbit monoclonal anti-GFP | Cell Signaling Technology | 2956S |
| Goat anti-Rabbit secondary antibody, HRP conjugate | Cell Signaling Technology | 7074S |
| Chemicals, peptides, and recombinant proteins | ||
| MLN4924 | Active Biochem | A-1139 |
| Polybrene | Santa Cruz | sc-134220 |
| Critical commercial assays | ||
| QIAEX II Gel Extraction Kit | QIAGEN | 20051 |
| PCR purification kit | QIAGEN | 28106 |
| Gentra Puregene Cell Kit | QIAGEN | 158767 |
| Deposited data | ||
| GPS peptidome processed screen data and raw images | This paper | http://dx.doi.org/10.17632/32h94v2yv2.2 (Mendeley Data) |
| GPS peptidome raw and processed screen data | This paper |
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE240610 (Gene Expression Omnibus) |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | ATCC CRL-3216 |
| Oligonucleotides | ||
| sg-FEM1B: GTGACATAGCCAAGCAGATAG | Integrated DNA Technologies | N/A |
| sg-KLHL15: GATTTCGGCGTAAACATCGA | Integrated DNA Technologies | N/A |
| sg-ASB7: GCCAACATCGACATTCAGAA | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pHAGE-GPS6.0-DEST | This paper | N/A |
| pHAGE-GPS6.0-peptide libraries | This paper | N/A |
| lentiCRISPR v2 | Addgene | 52961 |
| Ultimate ORF Collection | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/life-science/cloning/clone-collections/ultimate-orf-clone-collection.html |
| Software and algorithms | ||
| Bowtie 2 | Langmead and Salzberg, 2012; Langmead et al., 200950,51 | http://bowtie-bio.sourceforge.net/index.shtml |
| Cutadapt | Martin, 201149 | http://cutadapt.readthedocs.io/en/stable/index.html |
| MAGeCK | Li et al., 201452 | https://sourceforge.net/projects/mageck/ |
| Flowjo | Flowjo | https://www.flowjo.com |
| ColabFold | Mirdita et al., 202237 | https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb |
| ChimeraX | Pettersen et al., 202155 | https://www.rbvi.ucsf.edu/chimerax/ |
| Proteus2 | Montgomerie et al., 200832 | http://www.proteus2.ca/proteus2/ |
| JPred4 | Drozdetskiy et al., 201519 | https://www.compbio.dundee.ac.uk/jpred/ |
| DegronID | This paper |
https://doi.org/10.5281/zenodo.8103915 (Zenodo) |
| R 4.1.2 | R Core Team | https://www.r-project.org/ |
| Caret | Kuhn, 200854 | https://doi.org/10.18637/jss.v028.i05 |
| Tidyverse | Wickham et al., 201957 | http://dx.doi.org/10.21105/joss.01686 |
| Biostrings | Bioconductor | https://doi.org/10.18129/B9.bioc.Biostrings |
| aplot | CRAN | https://cran.r-project.org/web/packages/aplot |
| ggdendro | CRAN | https://cran.r-project.org/web/packages/ggdendro |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell Lines
We cultured HEK293T (ATCC CRL-3216) cells at 37°C and 5% CO2 in DMEM, high glucose, GlutaMAX™ Supplement, pyruvate (ThermoFisher Scientific) added with 10% fetal bovine serum (HyClone), 1% of 10,000 U/mL Penicillin-Streptomycin (ThermoFisher Scientific). For MLN4924 experiment, we incubated cells overnight with 1 mM MLN4924 and then analyzed the GFP/DsRed ratio by flow cytometry. For DN CULs experiment, we infected cells with the DN CULs lentivirus and analyzed the GFP/DsRed ratio by flow cytometry 2 days post-infection.
METHOD DETAILS
Transfection and lentivirus production
We generated lentivirus by transfection of HEK293T cells using lentiviral vector together DNA with plasmids encoding Gag-Pol, Rev, Tat and VSV-G using PolyJet In Vitro DNA Transfection Reagent (SignaGen Laboratories) based on the recommendation from the manufacturer. We collected the supernatants containing lentivirus after 2 days and used them to infect target cells. We added polybrene at 8 mg/ml for lentiviral infections when performing screens where there is need to increase infectivity.
Plasmids
Dominant-negative Cullin constructs were kindly provided as a gift from W. Harper. For the cloning of selected degron substrates into the GPS6.0 DEST vector for downstream analyses (DN CULs analysis, CRISPR screens, E3-degron pair validation using individual sgRNAs), the degron peptides were cloned into GPS6.0 DEST vector in different batches using either C-End sequence A (QGRARPNQEVQIGEMENQLS, same linker as the one in the peptidome libraries), or C-End sequence B (QGRARPNQEVQIGEMENQLD) (see Table S5 for details). Both C-End sequences have no known C-degrons and differ only by the C-terminal residue. For individual gene-disruption experiments by CRISPR/Cas9, sgRNAs were cloned into lentiCRISPR v2 (Addgene #52961, deposited by Feng Zhang) as described47. The sgRNA sequences involved in this study were listed in the Key Resources Table. For co-immunoprecipitation, E3 ligase ORFs were cloned into the pHAGE-CMV-2xFlag-N DEST vector. For flow-based stability assays, E3 ligase ORFs were cloned into pHAGE EF1𝛼 BFP.
Flow cytometry
HEK293T cells were detached from plates using trypsin and then analyzed on a CytoFLEX (Beckman Coulter). We collected flow cytometry data using CytoFLEX (Beckman Coulter) and performed analyses using FlowJo. We performed the cell sorting via the MoFlo Astrios (Beckman Coulter) instrument.
Design and generation of GPS libraries
GPS-peptidome library
The human proteome sequences were truncated into 28 residues with 5 residues overlaps between neighboring peptides and with 15 bp flanking primers, encoding GIRSG-28-mer-QGRAR. Oligonucleotides were synthesized by Agilent and amplified by PCR. The amplicons were then cloned into the lentiviral GPS vector pHAGE-GPS6.0-DEST via NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs) at 6x coverage. The 28-mer peptides were followed by a 20 amino acid C-End sequence (QGRARPNQEVQIGEMENQLS).
Mutagenesis libraries
The scanning mutagenesis libraries included a total of 9817 peptides, including 5790 peptides of degron index greater than 1, 1782 selected peptides of MLN4924-ΔPSI greater than 1, together with 3023 other peptides for the purpose balancing the GFP/DsRed distribution of the entire library to assist sorting. For scanning mutagenesis oligo libraries, we mutated each residue from each selected peptide was in succession to an amino acid of different side-chain chemical property: “A, G, V, L, I, S, T, C, D, E” were mutated to “R”; “M, W, F, P” were mutated to “S”; “Y, N, Q, K, R, H” were mutated to “A”. For saturation mutagenesis, we mutated each residue from each selected peptide was in succession to all of the other 19 possible amino acids. For scanning mutagenesis of C-terminal peptides, mutations to “R” at the third position from C-terminus were substituted with mutations to “H”, and mutations to “A” at the first and second positions from C-terminus were substituted with mutations to “S”, for the purpose of avoiding the generation of known C-degrons by the mutagenesis. We added two constant 15 bp flanking sequences at the two ends for amplification purposes. Oligonucleotides were synthesized by Agilent and amplified by PCR. The amplicons were then cloned into the lentiviral GPS vector pHAGE-GPS6.0-DEST by NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs) at >100x coverage. The 28-mer peptides were followed by a 20 amino acid C-End sequence (QGRARPNQEVQIGEMENQLS).
GPS screens
We packaged GPS-peptidome lentiviral libraries and transduced HEK293T at low multiplicity of infection (MOI). Two days after transduction, hygromycin was added to eliminate untransduced cells and 7–9 days after transduction, cells were sorted into 6 bins in approximately 1:1 ratio according to the GFP/DsRed ratio. Sorted cells were immediately subjected to genomic DNA extraction or were expanded by cell culture for 2–3 days before downstream genomic DNA extraction.
For each bin, we performed genomic DNA extraction (QIAGEN Gentra Puregene Cell Kit). We amplified the peptide sequences cloned into GFP by PCR with Q5 Hot Start Polymerase (NEB) using PCR primers annealing to constant sequences shared across the library. We then purified the amplicons (QIAGEN PCR purification kit). For each bin, we added >100 ng purified amplicons as template for another PCR reaction that adds an Illumina P5 sequence and nucleotides of 1–7 bp at the 5’ end for the purpose of staggering, together with an Illumina index and a P7 sequence at the 3’ end48. We then quantified samples by agarose gel electrophoresis and pooled them together. We then purified the resulting sample by agarose gel electrophoresis followed by gel extraction (QiaQuick Gel Extraction kit). The purified sample was then sequenced by an Illumina NextSeq instrument. We aimed to maintain >50-fold representation throughout the screening process.
CRISPR screens
We packaged the sgRNA lentiviral libraries targeting ~1500 genes related to ubiquitination and transduced HEK293T expressing specific GFP-peptide fusions at a low MOI. Two days after transduction, puromycin was added to eliminate untransduced cells. 7–9 d after transduction, the top 5–10% cells with the largest GFP/DsRed ratio were isolated by FACS. For sgRNA lentiviral libraries prepared from the same batch of CRISPR library DNA, one group of unsorted population was collected as a control. sgRNA sequences in sorted samples and unsorted controls were then amplified by 2-steps PCR as described for the GPS screens and quantified by Illumina sequencing. We aimed to maintain >100 fold representation throughout the screening process. However, for some samples, coverages were compromised at the genomic DNA extraction step.
Measurement of protein half-life.
HEK293T cells were separately mutated for ASB7, FBXO21, or FEM1B using lentiCRISPR v2 as mentioned earlier in Methods. E3 mutant and wild-type 293T cells were infected with lentivirus to induce stable expression of a GPS peptide construct that we identified from the GPS peptidome screen as regulated by that E3. For ASB7, we used CCDC17(9). For FBXO21, we used IFNA8(5). For FEM1B, we used UNC13D(26). The 28-mer peptides were each followed by C-end sequence A (QGRARPNQEVQIGEMENQLS). E3 knockout and WT cells stably expressing GPS peptide reporter constructs were seeded onto 6-well plates at 0.4M cells/well and incubated in a CO2 incubator at 37°C. After 48 h of incubation, the media was replaced with media containing 100 ug/mL cycloheximide (Millipore Sigma 239765) for inhibition of protein synthesis, or DMSO as a control. Cells were exposed to cycloheximide for 0, 0.5, 1, 2, 4, or 6 hours. Then, cells were rinsed once with PBS and lysed using RIPA buffer supplemented with 1x protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific 78441). Cell lysates were centrifuged at 21,000xg for 10 min at 4°C. Protein concentration in the supernatant was normalized across samples using BCA assay (Thermo Fisher Scientific 23225) prior to immunoblotting.
Immunoblotting
Expression levels of GFP-peptide fusions were measured by western blot with anti-GFP antibody. mTOR levels were also measured by western blot as a control. Samples for immunoblotting were diluted in Tris-Glycine SDS sample buffer and loaded into 4–12% Tris-Glycine 15-well pre-cast gels (Thermo Fisher Scientific XP04125BOX) with Tris-Glycine SDS running buffer (Thermo Fisher Scientific LC2675–4). A 10–250 kDa prestained protein ladder was run alongside samples (Thermo Fisher Scientific 26619). Eletrophoresis was run at 165V until the ladder reached the bottom of the gel. The protein was then loaded into a Trans-Blot Turbo Transfer System (BioRad) and transferred from the gel to a 0.2 𝜇m nitrocellulose membrane (BioRad 1704158). Membranes were blocked in 1x TBST (CST 9997S) with 5% milk for 30 min at room temperature. Then rabbit anti-mTOR (CST 2983S), rabbit anti-FLAG (CST 14793S), or rabbit anti-GFP (CST 2956S) primary antibodies were added to the blocking solution at 1:1000. Membranes were then incubated at 4°C overnight with gentle rocking. Membranes were then rinsed repeatedly with 1x TBST, and HRP-linked anti-rabbit secondary antibody (CST 7074S) were added at 1:2000 in 1x TBST with 5% milk. Blots were incubated with secondary antibody for 1 h at room temperature with gentle rocking, then washed again with 1x TBST. Blots were then exposed to Pierce ECL western blotting substrate (Thermo Fisher Scientific 32106) and exposed to high sensitivity autoradiography film (Denville Scientific E3218).
Co-Immunoprecipitation
HEK 293T cells that had been previously mutated for ASB7, FBXO21, or FEM1B by lentiCRISPR v2 were made to stably express corresponding GPS reporter peptides by lentiviral infection (ASB7:CCDC17(9), FBXO21:IFNA8(5), FEM1B:WVTYL_28mer). The 28-mer peptides were each followed by C-end sequence A (QGRARPNQEVQIGEMENQLS). Cells were seeded onto 100 mm plates at 1.1M cells/plate. 48 h later, cells were transfected with 3 ug DNA/plate of WT or mutant E3 ligase in the pHAGE-CMV-2xFlag-N DEST vector. Approximately 24 h later, media was aspirated and replaced with media containing 1 𝜇M MLN4924. After 16 h of incubation with MLN4924, cells were rinsed with ice-cold PBS and collected by scraping in 0.7 mL of lysis buffer containing 0.5% CHAPS, 40 mM HEPES pH 7.4, 100 mM NaCl, 4 mM EDTA, and 1x protease and phosphatase inhibitor cocktail. Cell lysates were incubated at 4°C with end-over-end rotation for 10 min, then centrifuged at 21,000xg for 15 min at 4°C. A 50 uL aliquot of the supernatant was collected as input, and the rest of the supernatant was transferred to a clean Eppendorf tube for immunoprecipitation. Anti-FLAG magnetic beads (Sigma M8823) were rinsed three times in lysis buffer, and the equivalent of 15 𝜇L of beads was added to each sample. Sample and bead mixtures were incubated at 4°C with end-over-end rotation for 90 min, then washed three times with lysis buffer and resuspended in Tris-Glycine SDS sample buffer containing 10% 2-mercaptoethanol. Samples were then heated to 95°C for 3 min to elute proteins for subsequent immunoblotting.
QUANTIFICATION AND STATISTICAL ANALYSIS
CRISPR screens
For each Illumina read, constant sequences of lentiCRISPRv2 vector were trimmed away via Cutadapt49. The number of reads perfectly mapped to each sgRNA sequences was counted via Bowtie250,51. Top performing genes targeted by multiple sgRNAs enriched in the sorted population were ranked using the MAGeCK method52. For some peptide samples belonging to certain batches of CRISPR screens, bench-top contamination of pre-cloned sgRNAs were observed. We did not annotate the MAGeCK hits considered to be due to contamination in the CRISPR screen MAGeCK plots. The full MAGeCK results for each screen are shown in Table S4 and Table S7.
GPS-peptidome screens
For each Illumina read, constant sequences of the GPS6.0 vector were trimmed away via Cutadapt49. For each bin, the number of reads perfectly mapped to each peptide was counted via Bowtie250. After normalizing the differences in sequencing depth for each bin, the PSI of each peptide was calculated as a weighted average of its distribution across the 6 bins, leading to a PSI score in between 1 (most unstable) and 6 (most stable):
where i denotes the bin (from 1 to 6) and Ri denotes the fraction of peptide reads in that particular bin.
Two replicates were performed for the control GPS-peptidome screen, and one was performed for the MLN4924 treated GPS-peptidome screen. We focused our analysis on ~260K peptides with >25 reads across all three samples (Table S2). Non-filtered data are available in Table S1.
An MLN4924-ΔPSI score was calculated by subtracting the peptide PSI in the absence of MLN4924 from the peptide PSI in the presence of MLN4924.
GPS-peptidome mutagenesis screens
Perfectly mapped peptide reads were used for the calculation of the PSI of each peptide. ΔPSI of each peptide mutant was generated by subtracting the PSI of the WT from the PSI of the mutant. Heatmaps were generated using ggplot253 for R and showed the ΔPSI of each mutant. The darker the color, the more stabilized the mutant. The mutagenesis data details were included in Table S3 for scanning mutagenesis and Table S6 for saturation mutagenesis.
Prediction of peptide PSI from composition
Ten percent of the ~260K peptidome data was used as the training set. For each peptide, the number of occurrences of each amino acid was counted as a feature. The training set, which included 20 features (count of 20 amino acids) and one output variable PSI, was then fed into the SVM-based machine learning algorithm for regression learning using the Caret package in R54. The learned algorithm was then used to predict the PSI of the entire ~260K peptidome data as the testing set. Degron index of each peptide was then calculated by subtracting observed PSI from predicted PSI.
Protein-peptide docking using AlphaFold2
For each protein-peptide docking, the full sequence of the cognate CRL adaptor and the 28-mer degron peptide were used as input for AlphaFold2 prediction. AlphaFold2-multimer prediction was performed on ColabFold v1.5.2 to generate 5 predicted unrelaxed docking structures with default parameters37. For each docking prediction, the highest scoring predicted structure is shown and illustrated using ChimeraX55. We examined all 5 predicted structure models and emphasize residues for which the intermolecular interactions are stable across models as described in the text.
DegronID: scoring peptide:degron similarity
We performed saturation mutagenesis on 250 28 amino acid sequences from the human peptidome to characterize their degron motifs. Data for 19 motifs were ruled insufficient due to lack of data for multiple point mutations. Data for an additional 12 motifs were ruled insufficient due to poor degron mapping – for example, no individual mutation produced a substantial ΔPSI stabilization above 1. We proceeded to process data for the 219 remaining motifs. We then made further use of the mutagenesis ΔPSI values determined from these mutagenesis experiments to predict degron motif similarity to other sequences in the human peptidome. We use a custom implementation of a paradigm for scoring position specific scoring matrices56. More specifically, we use the position specific scoring matrices from our saturation matrices to generate a numerical prediction of the potential of any other sequence of interest to contain a closely related degron. Under the assumption that each amino acid position contributes independently to the degron, the basic structure of the predicted degron score for a given sequence is termed the ΔPSI summation score (S) and is calculated as
Where indicates one of the 20 standard amino acids, indicates position along the mutagenized 28-mer, is the experimentally determined stability measure for that amino acid substitution at that position, is equal to 1 if the queried amino acid sequence has amino acid at position and is equal to 0 otherwise, and is an additional weight described below.
Position weighting. Although the mutagenesis was performed over 28-mer peptide tiles, the sequence interval corresponding to a particular degron may by much shorter, often between three and ten amino acids in length. To quantify this, we devise a “position importance score” to represent how important each amino acid position is to the integrity of the degron. Each column of the mutagenesis scoring matrix has scores 𝑠 for each amino acid . “Position importance score” is then calculated for each column of the mutagenesis matrix as , which is then normalized to the maximum position importance score for any column of that matrix. A degron-relevant interval is determined (purple bracket) by the shortest continuous interval containing all amino acid positions with at least 0.5 conservation score, with two additional amino acids flanking on each side when possible.
Amino acid weighting. In addition to position-based weighting, we give weight to particularly disruptive amino acids even if substitution occurs at an otherwise lowly conserved position. For each matrix, a per-amino acid “amino acid importance score” is calculated for each row analogously to “position importance score” for each column above. Prior to calculating amino acid weighting, the 28-column saturation mutagenesis matrix is cropped to the degron-relevant interval described in the paragraph above.
A composite weight considering both position conservation and amino acid disruption is used in the final scoring and is defined as the maximum value of the separate position and amino acid weights. That is,
We use this scoring paradigm to query the human peptidome for sequences most similar to the degrons represented in our 250 saturation mutagenesis experiments. For the analysis reported here, we focus on sequences represented in our 260k peptide library, but it is possible to query much larger databases without much added computational burden.
DegronID: hierarchical clustering
Degron prediction scores were generated for all amino acid sequences of the appropriate length from the human peptidome. We mapped our 260k library to matching k-mer sequences from the human peptidome and assigned each 28mer the score of the best scoring k-mer contained within. For 198 high-confidence degron footprints, 200 top predicted hits were chosen as the 200 28mer peptides with the lowest ΔPSI summation score.
Hierarchical clustering of degron footprints from saturation mutagenesis was performed using ward agglomeration and Euclidean distance to group degrons based on the overlap in the top 200 DegronID hits between pairs of degron footprints. The dendrogram was cut into 40 small clusters, then manually reassembled into larger meta-clusters. DegronID analyses were performed in R version 4.1.2 with help of the O2 High Performance Compute Cluster, supported by the Research Computing Group, at Harvard Medical School. Analyses were conducted using the tidyverse57 collection of R packages, along with Biostrings, aplot, and ggdendro (see key resources table).
DegronID: benchmarking validation
Amino acid frequency at each position of the D box motif was calculated from aligned D box sequences from the APC/C degron repository.20 This amino acid frequency matrix was used in place of a saturation mutagenesis matrix for input to DegronID. The 200 peptides from the human peptidome library that DegronID scores as most similar to the D-box motif are identified and their stability is compared to the library as a whole. For C-terminal motifs, we performed DegronID using our previously published saturation mutagenesis motifs as input.14
Secondary structure predictions
For the saturation mutagenesis motifs, we use JPred419 to predict the secondary structure of each peptide. For each of the 28-mer sequences, we concatenate the 25 N-terminal and 20 C-terminal flanking residues from the GPS construct, then run JPred4 predictions in bulk using the web server (https://www.compbio.dundee.ac.uk/jpred/). The consensus predictions were extracted, then cropped to the degron-relevant interval for each peptide as defined above. For each degron, we extract the length of degron sequence that is predicted to be an alpha helix or beta sheet by JPred4.
ADDITIONAL RESOURCES
Visualizations of degron footprints, peptide stability information, and DegronID clustering are publicly available via the DegronID Data Browser: https://brandonsie.shinyapps.io/DegronID/.
Supplementary Material
Sheet “I28 Control Replicate 1” contains Protein Stability Index (PSI) data for the first replicate of the control GPS peptidome screen. Sheet “I28 Control Replicate 2” contains PSI Data for the second replicate of the control GPS peptidome screen. Sheet “I28 MLN4924” contains PSI data for the GPS peptidome screen with pan-Cullin inhibitor MLN4924. The first column contains a peptide ID which is unique for each peptide in the library and consists of a UniProt accession ID, a gene name, and a numeric peptide ID to indicate where the peptide falls within the overall protein sequence. The second column indicates the sum of peptide reads across the six GPS bins. The third column indicates the protein stability index value, which is calculated as described in Methods.
Stability data for peptides from the “260K” peptidome library for which main analysis was performed. The first column contains peptide ID with the same format as in Table S1. The second column contains amino acid sequence of each peptide. The third column contains the change in PSI in the MLN4924 condition compared to control. The fourth column contains the measured PSI for the control GPS peptidome screen. The fifth column contains the PSI predicted by our composition-dependent SVM machine learning model. The sixth column contains the difference between the composition-based prediction of expected PSI from SVM and the experimentally observed PSI from the control GPS peptidome screen.
Data related to scanning mutagenesis GPS peptidome screen. The first column contains the peptide ID of the unmutated (wild-type) peptide. Each residue of the wild-type amino acid sequence was mutated to a chemically different amino acid. Column 2 contains the amino acid position that was mutated (value of 0 indicates wild-type sequence). Column 3 contains the amino acid sequence, including any mutations. Column 4 contains the protein stability index from GPS screening of the peptide. Column 5 contains the difference between mutant PSI and the wild-type PSI. Column 6 contains the identity of the mutant amino acid.
MAGeCK data for CRISPR screens of BAG6 peptides. Document contains tabs for screens of four peptides: CFB(17), ANKRD27(23), CD68(8), and CDHR4(23). Column 1 contains the ID of the screened gene. Columns correspond to the columns of gene_summary_txt as described in the MAGeCK wiki: https://sourceforge.net/p/mageck/wiki/output/.
Column 1 contains gene and peptide ID using the same format as Table S1. Column 2 contains the identity of the DN CUL that stabilized the peptide. Column 3 contains the version of the 20aa C-end flanking sequence for each peptide. Columns 4 and 5 contain the identity of the experimentally identified E3 ligase, either by CRISPR screen or by individual testing.
Data from saturation mutagenesis GPS peptidome screen. The first column contains peptide ID, starting with the same format as Table S1, then appended with mutation information in the form _mutNX, where N is the numeric position of the residue within the 28-mer, and X is the amino acid residue at that position. The second column contains the amino acid sequence for each peptide. The third column contains the normalized sum of reads from the six GPS bins. The fourth column contains protein stability index (PSI) score for each peptide.
Document contains tabs for 26 peptides and are named correspondingly. Column 1 contains the ID of the screened gene. Columns correspond to the columns of gene_summary_txt as described in the MAGeCK wiki: https://sourceforge.net/p/mageck/wiki/output/.
Highlights.
Genome-wide Global Protein Stability (GPS) assay identifies 15,800 degron peptides.
Critical degron residues were mapped by scanning and saturation mutagenesis.
CRISPR screening reveals cognate E3 ligases for 16 distinct degrons.
E3-degron pairs were docked by Alphafold2 and validated by co-immunoprecipitation.
Acknowledgments
We thank members of the Elledge lab for helpful suggestions and reagents. We thank the Harvard University Medical School Department of Immunology Flow Cytometry Core Facility for their invaluable help in cell sorting. The alpha helix icon used in the graphical abstract was designed by DBCLS https://togotv.dbcls.jp/en/pics.html and is licensed under CC-BY 4.0 Unported https://creativecommons.org/licenses/by/4.0/. Z.Z. is a Croucher Ph.D. Scholar. R.T.T. is a Sir Henry Wellcome Postdoctoral Fellow (201387/Z/16/Z) and a Pemberton-Trinity Fellow. This work was supported by the National Institutes of Health Aging grant AG11085 (S.J.E.) S.J.E. is a member of the Ludwig Center at Harvard and an Investigator with the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
S.J.E. is a founder of TSCAN Therapeutics, MAZE Therapeutics, ImmuneID and Mirimus, serves on the scientific advisory boards of Homology Medicines, ImmuneID, MAZE Therapeutics and TSCAN Therapeutics, is an advisor for MPM Capital, and is on the Editorial Board for Molecular Cell; none of which affect this work.
REFERENCES
- 1.Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, and Joazeiro CAP (2008). Genome-Wide and Functional Annotation of Human E3 Ubiquitin Ligases Identifies MULAN, a Mitochondrial E3 that Regulates the Organelle’s Dynamics and Signaling. PLoS One 3, e1487. 10.1371/JOURNAL.PONE.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins GA, and Goldberg AL (2017). The Logic of the 26S Proteasome. Cell 169, 792–806. 10.1016/j.cell.2017.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popovic D, Vucic D, and Dikic I (2014). Ubiquitination in disease pathogenesis and treatment. Nat Med 20, 1242–1253. 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- 4.Juszkiewicz S, and Hegde RS (2018). Quality Control of Orphaned Proteins. Mol Cell 71, 443–457. 10.1016/J.MOLCEL.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yanagitani K, Juszkiewicz S, and Hegde RS (2017). UBE2O is a quality control factor for orphans of multiprotein complexes. Science (1979) 357, 472–475. 10.1126/science.aan0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mena EL, Kjolby RAS, Saxton RA, Werner A, Lew BG, Boyle JM, Harland R, and Rape M (2018). Dimerization quality control ensures neuronal development and survival. Science (1979) 362, eaap8236. 10.1126/science.aap8236. [DOI] [PubMed] [Google Scholar]
- 7.Mårtensson CU, Priesnitz C, Song J, Ellenrieder L, Doan KN, Boos F, Floerchinger A, Zufall N, Oeljeklaus S, Warscheid B, et al. (2019). Mitochondrial protein translocation-associated degradation. Nature 569, 679–683. 10.1038/s41586-019-1227-y. [DOI] [PubMed] [Google Scholar]
- 8.Hessa T, Sharma A, Mariappan M, Eshleman HD, Gutierrez E, and Hegde RS (2011). Protein targeting and degradation are coupled for elimination of mislocalized proteins. Nature 475, 394–397. 10.1038/nature10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khmelinskii A, Blaszczak E, Pantazopoulou M, Fischer B, Omnus DJ, Le Dez G, Brossard A, Gunnarsson A, Barry JD, Meurer M, et al. (2014). Protein quality control at the inner nuclear membrane. Nature 516, 410–413. 10.1038/nature14096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Timms RT, Zhang Z, Rhee DY, Harper JW, Koren I, and Elledge SJ (2019). A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science (1979) 365, eaaw4912. 10.1126/science.aaw4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lucas X, and Ciulli A (2017). Recognition of substrate degrons by E3 ubiquitin ligases and modulation by small-molecule mimicry strategies. Curr Opin Struct Biol 44, 101–110. 10.1016/j.sbi.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 12.Bachmair A, Finley D, and Varshavsky A (1986). In Vivo Half-Life of a Protein Is a Function of Its Amino-Terminal Residue. Science (1979) 234, 179–186. 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 13.Mészáros B, Kumar M, Gibson TJ, Uyar B, and Dosztányi Z (2017). Degrons in cancer. Sci Signal 10, eaak9982. 10.1126/scisignal.aak9982. [DOI] [PubMed] [Google Scholar]
- 14.Koren I, Timms RT, Kula T, Xu Q, Li MZ, and Elledge SJ (2018). The Eukaryotic Proteome Is Shaped by E3 Ubiquitin Ligases Targeting C-Terminal Degrons. Cell 173, 1622–1635.e14. 10.1016/j.cell.2018.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherpa D, Chrustowicz J, and Schulman BA (2022). How the ends signal the end: Regulation by E3 ubiquitin ligases recognizing protein termini. Mol Cell 82, 1424–1438. 10.1016/j.molcel.2022.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tokheim C, Wang X, Timms RT, Zhang B, Mena EL, Wang B, Chen C, Ge J, Chu J, Zhang W, et al. (2021). Systematic characterization of mutations altering protein degradation in human cancers. Mol Cell 81, 1292–1308.e11. 10.1016/j.molcel.2021.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodrigo-Brenni MC, Gutierrez E, and Hegde RS (2014). Cytosolic Quality Control of Mislocalized Proteins Requires RNF126 Recruitment to Bag6. Mol Cell 55, 227–237. 10.1016/j.molcel.2014.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarjanazi H, Savas S, Pabalan N, Dennis JW, and Ozcelik H (2007). Biological implications of SNPs in signal peptide domains of human proteins. Proteins: Structure, Function, and Bioinformatics 70, 394–403. 10.1002/prot.21548. [DOI] [PubMed] [Google Scholar]
- 19.Drozdetskiy A, Cole C, Procter J, and Barton GJ (2015). JPred4: a protein secondary structure prediction server. Nucleic Acids Res 43, W389–W394. 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davey NE, and Morgan DO (2016). Building a Regulatory Network with Short Linear Sequence Motifs: Lessons from the Degrons of the Anaphase-Promoting Complex. Mol Cell 64, 12–23. 10.1016/j.molcel.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Low TY, Peng M, Magliozzi R, Mohammed S, Guardavaccaro D, and Heck R, A.J. (2014). A systems-wide screen identifies substrates of the SCFβTrCP ubiquitin ligase. Sci Signal 7, rs8. 10.1126/scisignal.2005882. [DOI] [PubMed] [Google Scholar]
- 22.Loveless TB, Topacio BR, Vashisht AA, Galaang S, Ulrich KM, Young BD, Wohlschlegel JA, and Toczyski DP (2015). DNA Damage Regulates Translation through β-TRCP Targeting of CReP. PLoS Genet 11, e1005292. 10.1371/journal.pgen.1005292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Havens CG, and Walter JC (2009). Docking of a Specialized PIP Box onto Chromatin-Bound PCNA Creates a Degron for the Ubiquitin Ligase CRL4Cdt2. Mol Cell 35, 93–104. 10.1016/j.molcel.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JM, Lee JS, Kim H, Kim K, Park H, Kim J-Y, Lee SH, Kim IS, Kim J, Lee M, et al. (2012). EZH2 Generates a Methyl Degron that Is Recognized by the DCAF1/DDB1/CUL4 E3 Ubiquitin Ligase Complex. Mol Cell 48, 572–586. 10.1016/j.molcel.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Arceci A, Bird K, Mills CA, Choudhury R, Kernan JL, Zhou C, Bae-Jump V, Bowers A, and Emanuele MJ (2017). VprBP/DCAF1 Regulates the Degradation and Nonproteolytic Activation of the Cell Cycle Transcription Factor FoxM1. Mol Cell Biol 37, e00609–16. 10.1128/MCB.00609-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C, Li X, Adelmant G, Dobbins J, Geisen C, Oser MG, Wucherpfenning KW, Marto JA, and Kaelin WG (2015). Peptidic degron in EID1 is recognized by an SCF E3 ligase complex containing the orphan F-box protein FBXO21. Proceedings of the National Academy of Sciences 112, 15372–15377. 10.1073/pnas.1522006112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manford AG, Mena EL, Shih KY, Gee CL, McMinimy R, Martínez-González B, Sherriff R, Lew B, Zoltek M, Rodríguez-Pérez F, et al. (2021). Structural basis and regulation of the reductive stress response. Cell 184, 5375–5390.e16. 10.1016/j.cell.2021.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Liao S, Makaros Y, Guo Q, Zhu Z, Krizelman R, Dahan K, Tu X, Yao X, Koren I, et al. (2021). Molecular basis for arginine C-terminal degron recognition by Cul2FEM1 E3 ligase. Nat Chem Biol 17, 254–262. 10.1038/s41589-020-00704-3. [DOI] [PubMed] [Google Scholar]
- 29.Ferretti LP, Himmels S-F, Trenner A, Walker C, von Aesch C, Eggenschwiler A, Murina O, Enchev RI, Peter M, Freire R, et al. (2016). Cullin3-KLHL15 ubiquitin ligase mediates CtIP protein turnover to fine-tune DNA-end resection. Nat Commun 7, 12628. 10.1038/ncomms12628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song J, Merrill RA, Usachev AY, and Strack S (2021). The X-linked intellectual disability gene product and E3 ubiquitin ligase KLHL15 degrades doublecortin proteins to constrain neuronal dendritogenesis. Journal of Biological Chemistry 296, 100082. 10.1074/jbc.RA120.016210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oberg EA, Nifoussi SK, Gingras A-C, and Strack S (2012). Selective Proteasomal Degradation of the B′β Subunit of Protein Phosphatase 2A by the E3 Ubiquitin Ligase Adaptor Kelch-like 15. Journal of Biological Chemistry 287, 43378–43389. 10.1074/jbc.M112.420281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montgomerie S, Cruz JA, Shrivastava S, Arndt D, Berjanskii M, and Wishart DS (2008). PROTEUS2: a web server for comprehensive protein structure prediction and structure-based annotation. Nucleic Acids Res 36, W202–W209. 10.1093/nar/gkn255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan X, Li Y, Wang G, Zhou Z, Song G, Feng Q, Zhao Y, Mi W, Ma Z, and Dong C (2021). Molecular basis for recognition of Gly/N-degrons by CRL2ZYG11B and CRL2ZER1. Mol Cell 81, 3262–3274.e3. 10.1016/j.molcel.2021.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Yan X, Wang X, Li Y, Zhou M, Li Y, Song L, Mi W, Min J, and Dong C (2021). Molecular basis for ubiquitin ligase CRL2FEM1C-mediated recognition of C-degron. Nat Chem Biol 17, 263–271. 10.1038/s41589-020-00703-4. [DOI] [PubMed] [Google Scholar]
- 35.Rusnac D-V, Lin H-C, Canzani D, Tien KX, Hinds TR, Tsue AF, Bush MF, Yen H-CS, and Zheng N (2018). Recognition of the Diglycine C-End Degron by CRL2KLHDC2 Ubiquitin Ligase. Mol Cell 72, 813–822.e4. 10.1016/j.molcel.2018.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsaban T, Varga JK, Avraham O, Ben-Aharon Z, Khramushin A, and Schueler-Furman O (2022). Harnessing protein folding neural networks for peptide–protein docking. Nat Commun 13, 176. 10.1038/s41467-021-27838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, and Steinegger M (2022). ColabFold: making protein folding accessible to all. Nat Methods 19, 679–682. 10.1038/s41592-022-01488-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Mahajan A, and Tsai M-D (2006). Ankyrin Repeat: A Unique Motif Mediating Protein−Protein Interactions. Biochemistry 45, 15168–15178. 10.1021/bi062188q. [DOI] [PubMed] [Google Scholar]
- 39.Rogers S, Wells R, and Rechsteiner M (1986). Amino Acid Sequences Common to Rapidly Degraded Proteins: The PEST Hypothesis. Science (1979) 234, 364–368. 10.1126/science.2876518. [DOI] [PubMed] [Google Scholar]
- 40.Rechsteiner M, and Rogers SW (1996). PEST sequences and regulation by proteolysis. Trends Biochem Sci 21, 267–271. 10.1016/S0968-0004(96)10031-1. [DOI] [PubMed] [Google Scholar]
- 41.Burslem GM, and Crews CM (2020). Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 181, 102–114. 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paiva S-L, and Crews CM (2019). Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol 50, 111–119. 10.1016/j.cbpa.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ichikawa S, Flaxman HA, Xu W, Vallavoju N, Lloyd HC, Wang B, Shen D, Pratt MR, and Woo CM (2022). The E3 ligase adapter cereblon targets the C-terminal cyclic imide degron. Nature 610, 775–782. 10.1038/s41586-022-05333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henning NJ, Manford AG, Spradlin JN, Brittain SM, Zhang E, McKenna JM, Tallarico JA, Schirle M, Rape M, and Nomura DK (2022). Discovery of a Covalent FEM1B Recruiter for Targeted Protein Degradation Applications. J Am Chem Soc 144, 701–708. 10.1021/jacs.1c03980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar M, Michael S, Alvarado-Valverde J, Mészáros B, Sámano-Sánchez H, Zeke A, Dobson L, Lazar T, Örd M, Nagpal A, et al. (2022). The Eukaryotic Linear Motif resource: 2022 release. Nucleic Acids Res 50, D497–D508. 10.1093/nar/gkab975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Timms RT, Mena EL, Leng Y, Li MZ, Tchasovnikarova IA, Koren I, and Elledge SJ (2023). Defining E3 ligase-substrate relationships through multiplex CRISPR screening. Nature Cell Biology, in press. [DOI] [PMC free article] [PubMed]
- 47.Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meyer M, and Kircher M (2010). Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harb Protoc 2010, pdb.prot5448. 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 49.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 10–12. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 50.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ginestet C (2011). ggplot2: Elegant Graphics for Data Analysis. J R Stat Soc Ser A Stat Soc 174, 245–246. 10.1111/j.1467-985X.2010.00676_9.x. [DOI] [Google Scholar]
- 54.Kuhn M (2008). Building Predictive Models in R Using the caret Package. J Stat Softw 28, 1–26. 10.18637/jss.v028.i05.27774042 [DOI] [Google Scholar]
- 55.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, and Ferrin TE (2021). UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Science 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao Y, Gran B, Pinilla C, Markovic-Plese S, Hemmer B, Tzou A, Whitney LW, Biddison WE, Martin R, and Simon R (2001). Combinatorial Peptide Libraries and Biometric Score Matrices Permit the Quantitative Analysis of Specific and Degenerate Interactions Between Clonotypic TCR and MHC Peptide Ligands. The Journal of Immunology 167, 2130–2141. 10.4049/jimmunol.167.4.2130. [DOI] [PubMed] [Google Scholar]
- 57.Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, et al. (2019). Welcome to the Tidyverse. J Open Source Softw 4, 1686. 10.21105/joss.01686. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sheet “I28 Control Replicate 1” contains Protein Stability Index (PSI) data for the first replicate of the control GPS peptidome screen. Sheet “I28 Control Replicate 2” contains PSI Data for the second replicate of the control GPS peptidome screen. Sheet “I28 MLN4924” contains PSI data for the GPS peptidome screen with pan-Cullin inhibitor MLN4924. The first column contains a peptide ID which is unique for each peptide in the library and consists of a UniProt accession ID, a gene name, and a numeric peptide ID to indicate where the peptide falls within the overall protein sequence. The second column indicates the sum of peptide reads across the six GPS bins. The third column indicates the protein stability index value, which is calculated as described in Methods.
Stability data for peptides from the “260K” peptidome library for which main analysis was performed. The first column contains peptide ID with the same format as in Table S1. The second column contains amino acid sequence of each peptide. The third column contains the change in PSI in the MLN4924 condition compared to control. The fourth column contains the measured PSI for the control GPS peptidome screen. The fifth column contains the PSI predicted by our composition-dependent SVM machine learning model. The sixth column contains the difference between the composition-based prediction of expected PSI from SVM and the experimentally observed PSI from the control GPS peptidome screen.
Data related to scanning mutagenesis GPS peptidome screen. The first column contains the peptide ID of the unmutated (wild-type) peptide. Each residue of the wild-type amino acid sequence was mutated to a chemically different amino acid. Column 2 contains the amino acid position that was mutated (value of 0 indicates wild-type sequence). Column 3 contains the amino acid sequence, including any mutations. Column 4 contains the protein stability index from GPS screening of the peptide. Column 5 contains the difference between mutant PSI and the wild-type PSI. Column 6 contains the identity of the mutant amino acid.
MAGeCK data for CRISPR screens of BAG6 peptides. Document contains tabs for screens of four peptides: CFB(17), ANKRD27(23), CD68(8), and CDHR4(23). Column 1 contains the ID of the screened gene. Columns correspond to the columns of gene_summary_txt as described in the MAGeCK wiki: https://sourceforge.net/p/mageck/wiki/output/.
Column 1 contains gene and peptide ID using the same format as Table S1. Column 2 contains the identity of the DN CUL that stabilized the peptide. Column 3 contains the version of the 20aa C-end flanking sequence for each peptide. Columns 4 and 5 contain the identity of the experimentally identified E3 ligase, either by CRISPR screen or by individual testing.
Data from saturation mutagenesis GPS peptidome screen. The first column contains peptide ID, starting with the same format as Table S1, then appended with mutation information in the form _mutNX, where N is the numeric position of the residue within the 28-mer, and X is the amino acid residue at that position. The second column contains the amino acid sequence for each peptide. The third column contains the normalized sum of reads from the six GPS bins. The fourth column contains protein stability index (PSI) score for each peptide.
Document contains tabs for 26 peptides and are named correspondingly. Column 1 contains the ID of the screened gene. Columns correspond to the columns of gene_summary_txt as described in the MAGeCK wiki: https://sourceforge.net/p/mageck/wiki/output/.
Data Availability Statement
Original GPS peptidome screen data and raw western blot images were deposited to Mendeley Data and Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. The DOIs are listed in the key resources table under “Deposited Data” as “GPS peptidome processed screen data and raw images” (Mendeley Data) and “GPS peptidome raw and processed screen data” (GEO). Detailed GPS peptidome screen data, mutagenesis screen data, CRISPR screen data, and DN CUL analyses are available in Tables S1, S2, S3, S4, S5, S6, S7.
All original code has been deposited to Zenodo and is publicly available as of the date of publication. The DOI is listed in the key resources table under “Software and algorithms” as “DegronID”.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-mTOR | Cell Signaling Technology | 2983S |
| Rabbit monoclonal anti-FLAG | Cell Signaling Technology | 14793S |
| Rabbit monoclonal anti-GFP | Cell Signaling Technology | 2956S |
| Goat anti-Rabbit secondary antibody, HRP conjugate | Cell Signaling Technology | 7074S |
| Chemicals, peptides, and recombinant proteins | ||
| MLN4924 | Active Biochem | A-1139 |
| Polybrene | Santa Cruz | sc-134220 |
| Critical commercial assays | ||
| QIAEX II Gel Extraction Kit | QIAGEN | 20051 |
| PCR purification kit | QIAGEN | 28106 |
| Gentra Puregene Cell Kit | QIAGEN | 158767 |
| Deposited data | ||
| GPS peptidome processed screen data and raw images | This paper | http://dx.doi.org/10.17632/32h94v2yv2.2 (Mendeley Data) |
| GPS peptidome raw and processed screen data | This paper |
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE240610 (Gene Expression Omnibus) |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | ATCC CRL-3216 |
| Oligonucleotides | ||
| sg-FEM1B: GTGACATAGCCAAGCAGATAG | Integrated DNA Technologies | N/A |
| sg-KLHL15: GATTTCGGCGTAAACATCGA | Integrated DNA Technologies | N/A |
| sg-ASB7: GCCAACATCGACATTCAGAA | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| pHAGE-GPS6.0-DEST | This paper | N/A |
| pHAGE-GPS6.0-peptide libraries | This paper | N/A |
| lentiCRISPR v2 | Addgene | 52961 |
| Ultimate ORF Collection | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/life-science/cloning/clone-collections/ultimate-orf-clone-collection.html |
| Software and algorithms | ||
| Bowtie 2 | Langmead and Salzberg, 2012; Langmead et al., 200950,51 | http://bowtie-bio.sourceforge.net/index.shtml |
| Cutadapt | Martin, 201149 | http://cutadapt.readthedocs.io/en/stable/index.html |
| MAGeCK | Li et al., 201452 | https://sourceforge.net/projects/mageck/ |
| Flowjo | Flowjo | https://www.flowjo.com |
| ColabFold | Mirdita et al., 202237 | https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb |
| ChimeraX | Pettersen et al., 202155 | https://www.rbvi.ucsf.edu/chimerax/ |
| Proteus2 | Montgomerie et al., 200832 | http://www.proteus2.ca/proteus2/ |
| JPred4 | Drozdetskiy et al., 201519 | https://www.compbio.dundee.ac.uk/jpred/ |
| DegronID | This paper |
https://doi.org/10.5281/zenodo.8103915 (Zenodo) |
| R 4.1.2 | R Core Team | https://www.r-project.org/ |
| Caret | Kuhn, 200854 | https://doi.org/10.18637/jss.v028.i05 |
| Tidyverse | Wickham et al., 201957 | http://dx.doi.org/10.21105/joss.01686 |
| Biostrings | Bioconductor | https://doi.org/10.18129/B9.bioc.Biostrings |
| aplot | CRAN | https://cran.r-project.org/web/packages/aplot |
| ggdendro | CRAN | https://cran.r-project.org/web/packages/ggdendro |