

ABSTRACT
Minority variants of Mycobacterium tuberculosis harboring mutations conferring resistance can become dominant populations during tuberculosis (TB) treatment, leading to treatment failure. Our understanding of drug-resistant within-host subpopulations and the frequency of resistance-conferring mutations in minority variants remains limited. M. tuberculosis sequences recovered from liquid cultures of culture-confirmed TB cases notified between January 2017 and December 2021 in New South Wales, Australia were examined. Potential drug resistance-conferring minority variants were identified using LoFreq, and mixed populations of different M. tuberculosis strains (≥100 SNPs apart) were examined using QuantTB. A total of 1831 routinely sequenced M. tuberculosis strains were included in the analysis. Drug resistance-conferring minority variants were detected in 3.5% (65/1831) of sequenced cultures; 84.6% (55/65) had majority strains that were drug susceptible and 15.4% (10/65) had majority strains that were drug resistant. Minority variants with high-confidence drug resistance-conferring mutations were 1.5 times more common when the majority strains were drug resistant. Mixed M. tuberculosis strain populations were documented in 10.0% (183/1831) of specimens. Minority variants with high-confidence drug resistance-conferring mutations were more frequently detected in mixed M. tuberculosis strain populations (2.7%, 5/183) than in single strain populations (0.6%, 10/1648; P = 0.01). Drug-resistant minority variants require monitoring in settings that implement routine M. tuberculosis sequencing. The frequency with which drug-resistant minority variants are detected is likely influenced by pre-culture requirement. Culture-independent sequencing methods should provide a more accurate reflection of drug-resistant subpopulations.
KEYWORDS: drug resistance, M. tuberculosis, minority variants, mixed strain populations
INTRODUCTION
The emergence and spread of drug-resistant (DR) Mycobacterium tuberculosis strains pose a major global public health challenge (1, 2). The World Health Organization (WHO) estimated that 450,000 new tuberculosis (TB) cases had rifampicin-resistant (RR) or multidrug-resistant (MDR) TB in 2021, although only 166,991 (37.1%) were formally reported (1). Undetected drug-resistant TB (DR-TB) is a major concern since it compromises patient care and facilitates DR-TB transmission (1).
The presence of drug-resistant M. tuberculosis strains as minority subpopulations in diagnostic samples has been documented (3 – 5). These minority subpopulations may represent either “mixed infection” (simultaneous co-infection of the host with more than one strain), or minority variants of the same strain, with heteroresistance defined as concomitant infection with both drug-resistant and drug-susceptible variants (either as mixed strain infections or minority variants). The presence of minority variants with drug resistance-conferring mutations could promote resistance amplification if patients receive a sub-optimal treatment (6), compromising patient outcomes and potentially facilitating the spread of these strains within the community (7 – 9).
The detection of drug-resistant M. tuberculosis can be achieved using either culture-based phenotypic drug-susceptibility testing (pDST) or genotypic drug-resistance prediction (gDST), which relies on rapid molecular tests or whole genome sequencing (WGS) to detect mutations associated with drug resistance (1, 10). Culture-based pDST can potentially detect drug-resistant subpopulations that are present at more than 1% of the cultured population (9, 11). Several rapid gDST methods have been endorsed by the WHO to detect drug resistance directly from clinical specimens, without initial culture. Both Xpert MTB/RIF and MTB/RIF Ultra are reported to be able to detect rifampicin resistance if present at more than 20% of M. tuberculosis population in the sample, while GenoTypeMTBDRplusv2.0 (LPA-Hain) detects isoniazid or rifampicin resistance if present in more than 5% of bacteria at the site of infection (4, 12, 13). All molecular tests are limited by the fact that they can only assess a small number of pre-determined mutations, which is not inclusive of many mutations associated with drug resistance listed in the WHO Catalogue (14).
WGS studies have demonstrated the presence of drug-resistant subpopulations in clinical samples (15, 16), confirming results from earlier molecular strain-typing methods (3, 4). However, WGS analysis of drug-resistance mutations usually focuses on the majority population present within the diagnostic specimen (17, 18), but recent validation of genome-wide variant calling tools for the detection of minority resistance-conferring variants has opened new opportunities to identify mixed strain populations and minority variants (18, 19). Specifically, LoFreq allows detection of minority variants harboring drug resistance-conferring mutations that may not be recognized by standard WGS data analysis pipelines (18, 19). In instances with multi-strain infection, QuantTB (20) quantifies the estimated mixture of constituent strains based on a strain specific single-nucleotide polymorphism (SNP) reference database and the number of SNP differences between strains.
This study leveraged routine prospective sequencing of all culture-confirmed TB cases in New South Wales (NSW), Australia, between 2017 and 2021. It aimed to investigate the incidence with which minority variants and mixed strain infections harbored drug resistance-conferring mutations not detected by standard bioinformatic pipelines. Accurate detection of minority variants with associated drug resistance is important for optimal clinical care and public health surveillance.
MATERIALS AND METHODS
Genomic sequences of M. tuberculosis cultures from TB cases notified between 1 January 2017 and 31 December 2021 in NSW, Australia, were examined together with their respective pDST results (Table S1).
All positive M. tuberculosis cultures were cultured either in BACTEC MGIT 960 (Becton Dickinson) or on Lowenstein Jensen (LJ) slopes. Phenotypic DST was conducted using the modified microdilution method in the BACTEC MGIT 960 system with WHO recommended critical concentrations as previously described (21). All M. tuberculosis isolates were prospectively sequenced in-house. WGS libraries were prepared with DNA extracted from MGIT or LJ cultures using the Nextera XT kit (Illumina, San Diego, California) as previously described (21). Sequencing was performed on an IIlumina NextSeq500 (Illumina, San Diego, California) instrument using 2 × 150bp paired-end chemistry. The quality of short reads was checked using FastQC v0.1 (https://github.com/s-andrews/FastQC). Sequenced reads were trimmed using Trimmomatic v0.36 (https://github.com/usadellab/Trimmomatic).
Detection of minority variants with drug resistance
The reference genome used for the genomic analysis was M. tuberculosis strain H37Rv (NCBI GenBank accession: NC_000962.3). Minority variants were defined as those with an allelic frequency of 3%–49% detected by LoFreq v2.1.1 (18, 19). Majority variants were defined as nucleobases with an allelic frequency of 50% or more, although 75% is frequently used at the cutoff (21, 22). We focused on the drug resistance-conferring minority variants in rpoB, katG, fabG1, inhA, pncA, panD, embB, embA, gyrA, gyrB, and rrs associated with resistance to first-line (isoniazid, rifampicin, pyrazinamide, and ethambutol) and key second-line TB drugs (fluoroquinolones and the injectables such as amikacin, capreomycin, and kanamycin). Drug resistance-conferring mutations in minority variants in genes Rv1979v, mmpL5, Rv0678, pepQ, mmpS5, fgd1, fbiAB, fbiC, ddn, Rv2983, rrl, and rplC reported in association with resistance to bedaquiline, pretomanid, and linezolid, now recommended for a short-course treatment by WHO (23, 24), were also examined.
Trimmed reads were aligned to the reference genome of M. tuberculosis strain H37Rv using BWA-MEM v0.7.17 (25). Potential drug resistance-conferring minority variants were called using LoFreq v2.1.1 (18, 19) from BAM files produced by BWA-MEM. The indel quality score was inserted into the BAM files and potential minority variants (SNPs and indels) were called. We used the strand bias filter to remove any minority variants with a read depth of less than five reads, an allelic frequency below 3% and a maximum strain variation over 0.01 (18, 19). Potential minority variants were annotated using SnpEFF v4.3 (26).
Drug resistance verification and classification
The potential drug resistance-conferring minority variants called by LoFreq were manually cross-checked with the 2021 WHO Catalogue (23, 24). Mutations that were classified as either in group 1 (Associated w R), group 2 (Associated w R - Interim), or group 3 (Uncertain significance) in the 2021 WHO Catalogue (23, 24) were analyzed. We defined “Associated w R” as high confidence resistance-associated variants, and “Associated w R - Interim” as interim confidence resistance-associated variants. The high confidence drug resistance mutations-associated minority variants were manually reviewed by direct visualization of the read alignments using Geneious Prime 2023.0.4.
Detection of mixed M. tuberculosis strain populations
Mixed population infection was defined as the presence of more than one strain of M. tuberculosis during the same disease episode. We detected mixed populations of M. tuberculosis in WGS data using QuantTB v1.0 (20) with default settings (>100 SNP differences between strains) and defined the population that was present in the highest proportion as the majority population.
Statistical analysis
Prism GraphPad v9.4.1 was used for statistical analyses. Differences in drug resistance between minority variants and mixed populations were assessed using the Fisher’s exact test with a significance level of P < 0.05.
RESULTS
A total of 1,831 M. tuberculosis strains sequenced during routine WGS were included in the analysis, which represent the majority of TB cases identified in NSW during the 5-year study period (Fig. 1; Table S1). All four major M. tuberculosis strain lineages were represented (Lineage 1 n = 571, Lineage 2 n = 544, Lineage 3 n = 285, and Lineage 4 n = 431). In this study, isolates were considered drug resistant if resistance was detected either on routine pDST, or if WHO-defined drug resistance-associated mutations were identified by WGS (if pDST was absent or isolates were reported susceptible by pDST).
Fig 1.
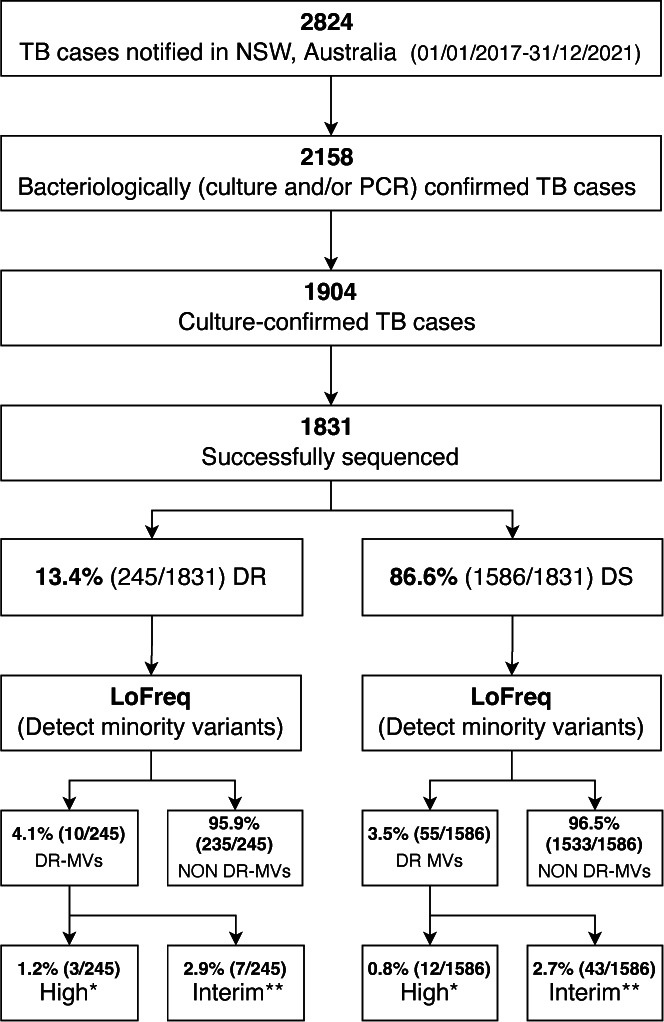
Detection of drug resistance-conferring minority variants in M. tuberculosis isolates using LoFreq. DR: drug resistant (majority variants resistant to any first-line TB drug); DS: drug susceptible (majority variants susceptible to all first-line TB drugs); DR-MVs: drug-resistant minority variants; NSW: New South Wales; PCR: M. tuberculosis-specific polymerase chain reaction; TB: tuberculosis. *Group 1 (“Assoc W R”) drug-resistant variants, as in 2021 WHO Catalogue (23, 24). **Group 2 (“Assoc W R - Interim”) drug-resistant variants, as in 2021 WHO Catalogue (23, 24).
Detection of drug resistance-conferring minority variants
Drug resistance-conferring minority variants were detected in 3.5% (65/1,831) of sequenced strains (Fig. 1); 70.8% of the 1,831 sequenced strains were cultured from respiratory specimens with roughly similar proportions of resistance-conferring minority variants identified in respiratory and non-respiratory specimens (Fig. S1a). Of the minority variants with drug-resistance mutations, 15/65 (23.1%) had high confidence mutations (Table 1) and 50/65 (76.9%) had interim confidence mutations (Table S2). In addition, we identified mutations in minority variants associated with resistance to BPaLM drugs, as well as those classified as group 3 (uncertain significance) in the 2021 WHO Catalogue (Table S3). All drug-resistance associated minority variants identified were supported by at least four overlapping reads with at least one forward and one reverse read; strand bias was less than 0.001. The allelic frequency of minority variants associated with drug resistance varied from 3.3% to 23.0% (Fig. 2). Minority variants with drug-resistance mutations were found in all major lineages (Fig. S1b), but more commonly associated with Lineage 1 compared to other lineages; however, this was not statistically significant (P = 0.08).
TABLE 1.
Overview of “high-confidence drug resistance-conferring mutations” a identified in M. tuberculosis minority variants, including mixed strain infections
| Strain ID | Source | Smear microscopy | Lineage | gDST profile | pDST profile | Mixed strain population b (Y/N) |
|---|---|---|---|---|---|---|
| 18-1773-0435 | Pus mediastinal | Negative | Lineage 1 | S | INH-R | Y |
| 17-3391-0281 | Lavage | Negative | Lineage 1 | S | S | Y |
| 17-3391-0008 | Fluid - neck | Negative | Lineage 1 | S | S | Y |
| 17-3391-0083 | Sputum | Negative | Lineage 2 | fabG1 -15 c > t | INH-R | Y |
| 18-1773-0007 | Sputum | Negative | Lineage 3 | S | S | Y |
| 20-002-0186 | Sputum | Negative | Lineage 1 | fabG1 -15 c > t | INH-R | N |
| 20-002-0197D1 | Sputum | Positive | Lineage 1 | S | S | N |
| 21-002-0305 | Pleura | Negative | Lineage 1 | S | S | N |
| 21-002-0237 | Tissue - lung | Negative | Lineage 1 | S | S | N |
| 17-3391-0013 | Lymph node | Negative | Lineage 2 | S | S | N |
| 17-3391-0018 | Sputum | Negative | Lineage 2 | S | S | N |
| 21-002-0252D1 | Sputum | Negative | Lineage 2 | S | S | N |
| 18-1773-0451 | Uncertain | Negative | Lineage 4 | S | S | N |
| 20-002-0411 | Washing | Negative | Lineage 4 | S | S | N |
| 21-002-0328 | Pleura | Negative | Lineage 4 | S | S | N |
Fig 2.

Allelic frequency of drug resistance-conferring mutations detected in identified minority M. tuberculosis variants. DR: drug resistant (majority variants resistant to any first-line TB drugs); DS: drug susceptible (majority variants susceptible to all first-line TB drugs); DR-MVs: drug-resistant minority variants; LPA: line probe assay; pDST: phenotypic drug-susceptibility testing; WGS: whole genome sequencing. Horizontal red line (at 3%) and dashed lines indicate the limit of detection of minority variants for each method.
The majority of the 65 specimens with drug-resistant minority variants (84.6%, 55/65) had majority variants that were phenotypically and genomically drug susceptible. The remaining 10 (15.4%, 10/65) had majority variants with drug resistance along with minority variants with additional resistance (Fig. 1 and 2). High-confidence drug resistance-conferring mutations in minority variants were 1.5 times more common when majority variants were drug resistant (1.2%, 3/245) compared to drug susceptible (0.8%, 12/1,586; P = 0.44).
Identification of mixed M. tuberculosis strain populations
We identified mixed M. tuberculosis strain populations in 183 (10.0%, 183/1,831) specimens (Fig. 3); 177 specimens were determined to be mixed with two distinct populations (96.7%, 177/183), while six specimens showed signatures of three distinct populations (3.3%, 6/183) (Table S1). Specimens with mixed M. tuberculosis strain populations were distributed across all four major lineages, but significantly more common in Lineage 3 compared to other lineages (Fig. S2a) (P = 0.0002). The majority of specimens with mixed populations were cultured from respiratory specimens (124/183, 67.8%); 32.2% of non-respiratory specimens had mixed populations (Fig. S2b).
Fig 3.
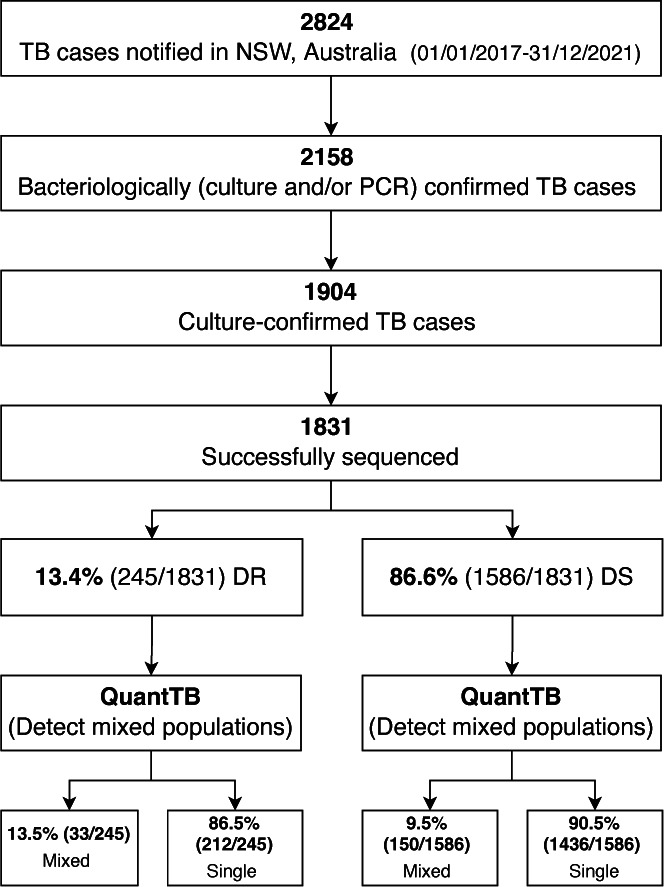
Identification of mixed strain populations in M. tuberculosis isolates using QuantTB. DR: drug resistant (majority variants resistant to any first-line TB drugs); DS: drug susceptible (majority variants susceptible to all first-line TB drugs); Mixed: mixed strain population; NSW: New South Wales; PCR: M. tuberculosis-specific polymerase chain reaction; Single: single strain population; TB: tuberculosis.
Mixed population infections were 42.1% more common in strains where the majority population was drug resistant (13.5%, 33/245) compared to drug susceptible (9.5%, 150/1,586; P = 0.07). The combined results of LoFreq and QuantTB, to determine the proportion of strains with drug-resistant minority variants that represent mixed populations, are represented in Fig. 4. Drug-resistant minority variants were documented in 6.0% (11/183) of specimens with mixed populations, regardless of whether the dominant strain was considered drug resistant or susceptible (Fig. 4 and 5). Specifically, in 0.8% (2/245) where the majority strain was considered drug resistant and in 0.6% (9/1,586) where the majority strain was considered drug susceptible, while 3.3% (54/1,648) of specimens with a single strain population harbored drug-resistant minority variants.
Fig 4.

Combined assessment of drug-resistant minority variants and mixed M. tuberculosis strain populations detected in specimens with majority drug-resistant or drug-susceptible strains. DR: drug resistant (majority variants resistant to any first-line TB drugs); DS: drug susceptible (majority variants susceptible to all first-line TB drugs); DR-MVs: drug-resistant minority variants; MDR: multidrug resistant; Mixed: mixed strain population; NSW: New South Wales; PCR: M. tuberculosis-specific polymerase chain reaction; Single: single strain population; TB: tuberculosis. *Group 1 (“Assoc W R”) drug-resistant variants, as in 2021 WHO Catalogue (23, 24). **Group 2 (“Assoc W R - Interim”) drug-resistant variants, as in 2021 WHO Catalogue (23, 24).
Fig 5.

Overview of M. tuberculosis drug-resistant minority variants and mixed strain populations detected. DR: drug resistant (majority variants resistant to any first-line TB drugs); DS: drug susceptible (majority variants susceptible to all first-line TB drugs); DR-MVs: drug-resistant minority variants; Mixed: mixed strain population with/without drug-resistance mutations; Single: single strain population. Numbers (N) in the brackets next to legend indicate the total number of isolates per legend.
Assignment of drug resistance-conferring minority variants
The assignment of high-confidence drug-resistance mutations detected in minority variants in single strain population specimens is summarized in Table 2 (interim confidence mutations in Table S2). One specimen considered phenotypically and genotypically isoniazid mono-resistant (majority variant fabG1 -15 c > t) also had a rpoB_H445N mutation detected as a minority variant at 11.4% allelic frequency, indicating potential for selection of this MDR strain with inappropriate treatment (Table 2). Minority variants with high-confidence drug resistance-conferring mutations were more frequently detected in specimens with mixed strain populations (2.7%, 5/183) than in those with a single strain population (0.6%, 10/1,648) (P = 0.01). Due to limitations of the bioinformatics software and short-read sequencing technology used, minority drug-resistance mutations were unable to be assigned to either the minority or majority population within specimens with mixed population (Table 3).
TABLE 2.
“High-confidence drug resistance-conferring mutations” a identified in minority variants from specimens with a single M. tuberculosis strain (N = 1,648)
| Strain ID | pDST profile | Majority drug-resistant variant | Minority drug-resistant variant | Predicted MDR c (Y/N) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mutation | POS | Nucleotide change | Read depth | AF (%) | Predicted DR b | ||||
| 20-002-0186 | INH-R, RIF-S | fabG1 -15 c > t | rpoB_H445N | 761139 | C to A | 70 | 11.4 | RR | Y |
| 18-1773-0451 | S | S | rpoB_H445N | 761139 | C to A | 83 | 12 | RR | N |
| 17-3391-0013 | S | S | rpoB_H445N | 761139 | C to A | 97 | 6.2 | RR | N |
| 20-002-0411 | S | S | rpoB_H445N | 761139 | C to A | 73 | 5.5 | RR | N |
| 20-002-0197D1 | S | S | katG_S315T | 2155168 | C to G | 57 | 8.8 | INH-R | N |
| 21-002-0305 | S | S | katG_W328L | 2155129 | C to A | 256 | 3.5 | INH-R | N |
| 21-002-0328 | S | S | katG_W328L | 2155129 | C to A | 135 | 3.7 | INH-R | N |
| 21-002-0252D1 | S | S | katG_W328L | 2155129 | C to A | 168 | 4.2 | INH-R | N |
| 21-002-0252D1 | S | S | pncA_G97C | 2288953 | C to A | 175 | 3.4 | PZA-R | N |
| 17-3391-0018 | S | S | pncA_V180G | 2288703 | A to C | 91 | 11 | PZA-R | N |
| 21-002-0237 | S | S | pncA_A146T | 2288806 | C to T | 255 | 6.7 | PZA-R | N |
TABLE 3.
“High-confidence drug resistance-conferring mutations” a identified in minority variants from specimens with more than one M. tuberculosis strain (N = 183)
| Strain ID | pDST profile | Majority drug-resistant variant | Mixed strain population ratio | Minority drug-resistant variant | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mutation | POS | Nucleotide change | Read depth | AF (%) | |||||
| 17-3391-0083 | INH-R, RIF-S | fabG1 -15 c > t | 63%:37% | rpoB_H445N | 761139 | C to A | 222 | 4.1 | |
| 18-1773-0435 | INH-R | S b | 81.9%:18.1% | katG_S315T | 2155168 | C to G | 62 | 11.3 | |
| 18-1773-0007 | S | S | 80.4%:19.6% | katG_S315T | 2155168 | C to G | 89 | 20.2 | |
| 17-3391-0281 | S | S | 57.9%:42.1% | pncA_V128G | 2288859 | A to C | 244 | 6.6 | |
| 17-3391-0008 | S | S | 51.7%:48.3% | embB_D354A | 4247574 | A to C | 140 | 8.6 | |
DISCUSSION
This study is the first to quantify the frequency with which minority variants that harbor mutations conferring drug resistance are observed in routinely sequenced isolates from a low TB incidence setting. It is also the first to differentiate same strain minority variants from those representing mixed M. tuberculosis strain populations. From a programmatic perspective, the assessment of drug-resistant minority variants improves clinical risk management and should provide patient benefits, especially in settings where disease relapse is a concern.
Drug resistance-conferring minority variants are not routinely interrogated during WGS-based gDST, but may be detected if pDST is performed, leading to discrepancies between WGS and phenotypic data. Our analysis of WGS data from culture-confirmed clinical M. tuberculosis strains confirmed previous findings that minority strains carrying high-confidence drug-resistance mutations (such as katG_S315T at 11.3% allelic frequency) can account for occasional discrepancies between pDST and gDST (8, 27). Routinely assessing for minority variants with drug resistance-conferring mutations in clinical M. tuberculosis cultured specimens would enhance drug-resistance surveillance and have particular clinical relevance to prevent drug-resistance amplification and disease relapse.
Minority variants with drug resistance-conferring mutations, especially in strains demonstrating phenotypic drug resistance, may potentially develop additional drug resistance if exposed to sub-optimal treatment. Therefore, the presence of these minority variants provides an early risk indication for treatment failure (7, 8, 17, 28). The presence of minority variants with additional resistance to rifampicin or isoniazid is particularly important when majority variants have mono-resistance to either of these first-line drugs, as these strains have the potential to become MDR. In our analysis, we identified the presence of minority variants with additional rifampicin resistance in 5/245 (2.0%) of TB cases with isoniazid monoresistance. However, genomically and phenotypically undetectable MDR minority variants may be more common in high-incidence settings. The early detection of drug resistant minority variants should guide optimal treatment to reduce the risk of failure or relapse.
Our findings confirmed previous observations that minority M. tuberculosis variants with drug resistance-conferring mutations can be detected by WGS, even when this is not apparent on pDST (4). Unrecognized minority variants with drug resistance could be selected for during sub-optimal treatment, potentially leading to treatment failure and clinical relapse (29). The sensitivity of pDST has been reported to be as high as 1% of the strain population (9, 11); however, our gDST data reflected instances where 6.8% of strains had high-confidence drug-resistance mutations but still tested susceptible on pDST. In total, 3.5% (55/1,586) of strains that tested drug susceptible on pDST had minority variants above the 3% level with drug resistance-associated mutations. Instances of sub-optimal accuracy and sensitivity of pDST are well recognized (1, 23), especially when the drug-resistant subpopulations represent only a small proportion of strains. These discrepant results warrant further interrogation as undetected drug resistance has important laboratory reporting and clinical management implications.
Our observations indicate that minority variants with high-confidence drug resistance-conferring mutations were more frequently detected in cases of mixed infections. Infection with more than one strain (mixed-strain infection) will increase the risk of drug resistance-conferring mutations being present purely by chance. Infection with more than one strain is likely to occur in a high-transmission setting, where the risk of drug-resistant strains being present is elevated (30). The presence of minority variants from the same strain with drug-resistance mutations may be a marker of previous sub-optimal treatment exposure, which may have facilitated their selection. Although only a small number of patients had documented retreatment, exposure to over-the-counter TB drugs is not uncommon in South-East Asian countries from which many of the cases detected in Australia originate (31).
Important limitations of our analysis need to be acknowledged. In our analysis, the detection of minority variants with drug-resistance mutations was based on the WHO drug-resistant TB mutations catalogue released in 2021, which has well-recognized limitations. Use of the new WHO drug-resistant TB mutations catalogue, to be released in 2023, which includes additional mutations, should improve the accuracy of drug-resistance detection. Ongoing curation and improvement of TB drug-resistance databases should further enhance subpopulation analyses in future studies. Laboratory subcultures of M. tuberculosis can reduce clonal diversity, with culture selection leading to a loss of the information on resistant-minority populations in clinical samples (32, 33). The drug-resistant minority variants observed from culture-derived WGS data may not capture all relevant M. tuberculosis population dynamics (7). Culture-independent sequencing, without any pre-selection bias from culture, may be more reflective of the bacterial population in the clinical sample. Considering this, the incorporation of metagenomic sequencing or targeted next-generation sequencing (tNGS) directly from respiratory samples offers promising alternative approaches (34, 35). It would be valuable to have more data demonstrating the feasibility and accuracy of direct metagenomic sequencing or tNGS as alternatives to culture-based methods. Investigating and optimizing culture-independent WGS and the application of tNGS to detect drug-resistant minority variants may enhance patient management and surveillance of drug-resistant variants.
We employed two tools; LoFreq to determine minority variants associated with drug resistance and QuantTB to detect mixed M. tuberculosis strain infections. However, neither software was able to assign minority variants associated with drug resistance to majority or minority populations within the isolate. It is possible that the drug-resistant minority variant belongs to the minority population due to the fact that the vast majority of the strains tested were phenotypically and genotypically consistent. However, it is also possible that the minority drug-resistant variants arose from the majority strain population but were not detected using pDST. For instance, if the minority variant, rpoB_H445N, was present in the majority population of our sample with a majority variant fabG1 -15 c>t, there would be the potential risk of developing MDR-TB. In addition to the limitations with bioinformatic pipelines, the use of the short-read sequencing technology can also impact the assignment of drug resistance. It is unlikely to capture both drug-resistant minority variants and strain population differences in a single 150bp read. This highlights the need for enhanced bioinformatic pipelines with the ability to detect drug resistance-conferring mutations in both majority and minority populations and to assign these correctly, ideally together with M. tuberculosis strain/lineage identification. The false-positive detection of mixed strain co-infection by QuantTB remains a possibility but recent incorporation of additional validation steps and an updated SNP database should have increased its reliability. Determination of the (sub)lineages responsible for the mixed strain infection was not possible but might be beneficial to include in future software tools.
The allelic frequency of minority variants with resistance detected in our analysis ranged from 3.3% to 23.0%. Previous studies suggested that the presence of drug-resistant minority is associated with poor treatment outcomes (7, 36), especially if these minority variants have an allelic frequency of more than 5% (17, 36). However, some studies indicate that even when present at very low (<1%) allelic frequencies, these minority variants may be experiencing amplification during sub-optimal treatment, leading to establishment of resistance and a subsequent enhancement in fitness (15, 37). This study did not have the opportunity to access and integrate individual-level patient outcome data. We acknowledge this as a major limitation and hope to incorporate such data in future analyses to assess the impact of these minority variants on treatment outcomes.
In conclusion, our findings demonstrate that minority variants of M. tuberculosis with drug resistance-conferring mutations can be detected from routine WGS data using enhanced bioinformatic pathways. High-resolution interrogation of drug-resistant minority variants could improve clinical risk assessment and strengthen surveillance of drug resistant strains.
The findings of this study are equally applicable to other low incidence settings. The use of similar approaches also requires consideration in high TB incidence settings to assess its relevance and optimal implementation, in order to promote equitable access to the benefits associated with advanced pathogen genomics. However, low incidence settings do have an important role to explore and refine new applications to push the technological boundaries, while acknowledging that the appropriate use of different approaches and their relative value-add might be setting-specific.
ACKNOWLEDGMENTS
The authors thank the Sydney Informatics Hub and the University of Sydney’s high-performance computing cluster, Artemis.
X.Z. is funded by NHMRC Centre for Research Excellence in Tuberculosis. The funders of this study had no role in the study design, data collection, data analysis and interpretation, or writing of the report. The corresponding author had full access to study data and final responsibility for the decision to submit for publication.
B.J.M. and V.S. designed the study and guided the data analysis. Data collection was done by T.C., E.M., and C.L. E.S. and X.Z. performed bioinformatic analysis and wrote the first manuscript draft with editing from all authors. The final manuscript was approved by all authors.
The authors declare no competing interests.
Contributor Information
Xiaomei Zhang, Email: xiaomei.zhang@sydney.edu.au.
Christine Y. Turenne, University of Manitoba, Winnipeg, Manitoba, Canada
DATA AVAILABILITY
Raw de-identified pathogen WGS data was deposited in the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA) under BioProject number PRJNA899911.
ETHICS APPROVAL
The study was approved by the Western Sydney Local Health District (WSLHD) Human Research Ethics Committee (approval no. 2019/PID14240).
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jcm.00485-23.
M. tuberculosis strains with drug resistance-conferring minority variants; (a) the percentage of respiratory and non-respiratory specimens (b) the number and percentage of strains with minority variants detected in all four major lineages.
M. tuberculosis strains with mixed populations; (a) the number and percentage of mixed strain populations detected in different M. tuberculosis lineages, (b) the percentage in respiratory and non-respiratory specimen sources.
Figure S1: M. tuberculosis strains with drug resistance-conferring minority variants; (a) the percentage of respiratory and non-respiratory specimens (b) the number and percentage of strains with minority variants detected in all four major lineages; Figure S2: M. tuberculosis strains with mixed populations; (a) the number and percentage of mixed strain populations detected in different M. tuberculosis lineages (b) the percentage in respiratory and non-respiratory specimen sources.
Supplementary Tables.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. World Health Organization. Global tuberculosis report 2022. [Google Scholar]
- 2. McBryde ES, Meehan MT, Doan TN, Ragonnet R, Marais BJ, Guernier V, Trauer JM. 2017. The risk of global epidemic replacement with drug-resistant Mycobacterium tuberculosis strains. Int J Infect Dis 56:14–20. doi: 10.1016/j.ijid.2017.01.031 [DOI] [PubMed] [Google Scholar]
- 3. Zhang X, Zhao B, Liu L, Zhu Y, Zhao Y, Jin Q. 2012. Subpopulation analysis of heteroresistance to fluoroquinolone in Mycobacterium tuberculosis isolates from Beijing, China. J Clin Microbiol 50:1471–1474. doi: 10.1128/JCM.05793-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tolani MP, D’souza DTB, Mistry NF. 2012. Drug resistance mutations and heteroresistance detected using the genotype MTBDRplus assay and their implication for treatment outcomes in patients from Mumbai, India. BMC Infect Dis 12:9. doi: 10.1186/1471-2334-12-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cohen T, van Helden PD, Wilson D, Colijn C, McLaughlin MM, Abubakar I, Warren RM. 2012. Mixed-strain Mycobacterium tuberculosis infections and the implications for tuberculosis treatment and control. Clin Microbiol Rev 25:708–719. doi: 10.1128/CMR.00021-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rinder H, Mieskes KT, Löscher T. 2001. Heteroresistance in Mycobacterium tuberculosis. Int J Tuberc Lung Dis 5:339–345. [PubMed] [Google Scholar]
- 7. Ley SD, de Vos M, Van Rie A, Warren RM. 2019. Deciphering within-host microevolution of Mycobacterium tuberculosis through whole-genome sequencing: the phenotypic impact and way forward. Microbiol Mol Biol Rev 83:e00062-18. doi: 10.1128/MMBR.00062-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Metcalfe JZ, Streicher E, Theron G, Colman RE, Allender C, Lemmer D, Warren R, Engelthaler DM. 2017. Cryptic microheteroresistance explains Mycobacterium tuberculosis phenotypic resistance. Am J Respir Crit Care Med 196:1191–1201. doi: 10.1164/rccm.201703-0556OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Folkvardsen DB, Thomsen VØ, Rigouts L, Rasmussen EM, Bang D, Bernaerts G, Werngren J, Toro JC, Hoffner S, Hillemann D, Svensson E. 2013. Rifampin heteroresistance in Mycobacterium tuberculosis cultures as detected by phenotypic and genotypic drug susceptibility test methods. J Clin Microbiol 51:4220–4222. doi: 10.1128/JCM.01602-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lozano N, Lanza VF, Suárez-González J, Herranz M, Sola-Campoy PJ, Rodríguez-Grande C, Buenestado-Serrano S, Ruiz-Serrano MJ, Tudó G, Alcaide F, Muñoz P, García de Viedma D, Pérez-Lago L. 2021. Detection of minority variants and mixed infections in Mycobacterium tuberculosis by direct whole-genome sequencing on noncultured specimens using a specific-DNA capture strategy. mSphere 6:e0074421. doi: 10.1128/mSphere.00744-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Canetti G, Fox W, Khomenko A, Mahler HT, Menon NK, Mitchison DA, Rist N, Smelev NA. 1969. Advances in techniques of testing mycobacterial drug sensitivity, and the use of sensitivity tests in tuberculosis control programmes. Bull World Health Organ 41:21–43. [PMC free article] [PubMed] [Google Scholar]
- 12. Blakemore R, Story E, Helb D, Kop J, Banada P, Owens MR, Chakravorty S, Jones M, Alland D. 2010. Evaluation of the analytical performance of the Xpert MTB/RIF assay. J Clin Microbiol 48:2495–2501. doi: 10.1128/JCM.00128-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ng KCS, Supply P, Cobelens FGJ, Gaudin C, Gonzalez-Martin J, de Jong BC, Rigouts L. 2019. How well do routine molecular diagnostics detect rifampin heteroresistance in Mycobacterium tuberculosis? J Clin Microbiol 57:e00717-19. doi: 10.1128/JCM.00717-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tiberi S, Utjesanovic N, Galvin J, Centis R, D’Ambrosio L, van den Boom M, Zumla A, Migliori GB. 2022. Drug resistant TB - latest developments in epidemiology, diagnostics and management. Int J Infect Dis 124 Suppl 1:S20–S25. doi: 10.1016/j.ijid.2022.03.026 [DOI] [PubMed] [Google Scholar]
- 15. Sun G, Luo T, Yang C, Dong X, Li J, Zhu Y, Zheng H, Tian W, Wang S, Barry CE III, Mei J, Gao Q. 2012. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis 206:1724–1733. doi: 10.1093/infdis/jis601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Black PA, de Vos M, Louw GE, van der Merwe RG, Dippenaar A, Streicher EM, Abdallah AM, Sampson SL, Victor TC, Dolby T, Simpson JA, van Helden PD, Warren RM, Pain A. 2015. Whole genome sequencing reveals genomic heterogeneity and antibiotic purification in Mycobacterium tuberculosis isolates. BMC Genomics 16:857. doi: 10.1186/s12864-015-2067-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vargas R, Freschi L, Marin M, Epperson LE, Smith M, Oussenko I, Durbin D, Strong M, Salfinger M, Farhat MR. 2021. In-host population dynamics of Mycobacterium tuberculosis complex during active disease. Elife 10:e61805. doi: 10.7554/eLife.61805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goossens SN, Heupink TH, De Vos E, Dippenaar A, De Vos M, Warren R, Van Rie A. 2022. Detection of minor variants in Mycobacterium tuberculosis whole genome sequencing data. Brief Bioinform 23:bbab541. doi: 10.1093/bib/bbab541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilm A, Aw PPK, Bertrand D, Yeo GHT, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. 2012. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res 40:11189–11201. doi: 10.1093/nar/gks918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anyansi C, Keo A, Walker BJ, Straub TJ, Manson AL, Earl AM, Abeel T. 2020. QuantTB - a method to classify mixed Mycobacterium tuberculosis infections within whole genome sequencing data. BMC Genomics 21:80. doi: 10.1186/s12864-020-6486-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lam C, Martinez E, Crighton T, Furlong C, Donnan E, Marais BJ, Sintchenko V. 2021. Value of routine whole genome sequencing for Mycobacterium tuberculosis drug resistance detection. Int J Infect Dis 113 Suppl 1:S48–S54. doi: 10.1016/j.ijid.2021.03.033 [DOI] [PubMed] [Google Scholar]
- 22. Kohl TA, Utpatel C, Schleusener V, De Filippo MR, Beckert P, Cirillo DM, Niemann S. 2018. MTBseq: a comprehensive pipeline for whole genome sequence analysis of Mycobacterium tuberculosis complex isolates. PeerJ 6:e5895. doi: 10.7717/peerj.5895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walker TM, Miotto P, Köser CU, Fowler PW, Knaggs J, Iqbal Z, Hunt M, Chindelevitch L, Farhat M, Cirillo DM, Comas I, Posey J, Omar SV, Peto TE, Suresh A, Uplekar S, Laurent S, Colman RE, Nathanson C-M, Zignol M, Walker AS, CRyPTIC Consortium, Seq&Treat Consortium, Crook DW, Ismail N, Rodwell TC. 2022. The 2021 WHO catalogue of Mycobacterium tuberculosis complex mutations associated with drug resistance: a genotypic analysis. Lancet Microbe 3:e265–e273. doi: 10.1016/S2666-5247(21)00301-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. World Health Organization. 2021.. Catalogue of mutations in Mycobacterium tuberculosis complex and their association with drug resistance. [Google Scholar]
- 25. Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. doi: 10.48550/arXiv.1303.3997 [DOI] [Google Scholar]
- 26. Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. doi: 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang D, Gomez JE, Chien JY, Haseley N, Desjardins CA, Earl AM, Hsueh PR, Hung DT. 2016. Genomic analysis of the evolution of fluoroquinolone resistance in Mycobacterium tuberculosis prior to tuberculosis diagnosis. Antimicrob Agents Chemother 60:6600–6608. doi: 10.1128/AAC.00664-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crofton J. 1960. Drug treatment of tuberculosis. I. standard chemotherapy. Br Med J 2:370–373. doi: 10.1136/bmj.2.5195.370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shin SS, Modongo C, Zetola NM. 2016. The impact of mixed infections on the interpretation of molecular epidemiology studies of tuberculosis. Int J Tuberc Lung Dis 20:423–424. doi: 10.5588/ijtld.15.1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Atre SR, Jagtap JD, Faqih MI, Dumbare YK, Sawant TU, Ambike SL, Bhawalkar JS, Bharaswadkar SK, Jogewar PK, Adkekar RS, Hodgar BP, Jadhav V, Mokashi ND, Golub JE, Dixit A, Farhat MR. 2022. Tuberculosis pathways to care and transmission of multidrug resistance in India. Am J Respir Crit Care Med 205:233–241. doi: 10.1164/rccm.202012-4333OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. NSW Tuberculosis Program CDB. 2023. Tuberculosis in NSW – Surveillance Report 2021. Sydney: Health Protection NSW. [Google Scholar]
- 32. Nimmo C, Shaw LP, Doyle R, Williams R, Brien K, Burgess C, Breuer J, Balloux F, Pym AS. 2019. Whole genome sequencing Mycobacterium tuberculosis directly from sputum identifies more genetic diversity than sequencing from culture. BMC Genomics 20:433. doi: 10.1186/s12864-019-5841-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shockey AC, Dabney J, Pepperell CS. 2019. Effects of host, sample, and in vitro culture on genomic diversity of pathogenic mycobacteria. Front Genet 10:477. doi: 10.3389/fgene.2019.00477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu SH, Xiao YX, Hsiao HC, Jou R. 2022. Development and assessment of a novel whole-gene-based targeted next-generation sequencing assay for detecting the susceptibility of Mycobacterium tuberculosis to 14 drugs. Microbiol Spectr 10:e0260522. doi: 10.1128/spectrum.02605-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jouet A, Gaudin C, Badalato N, Allix-Béguec C, Duthoy S, Ferré A, Diels M, Laurent Y, Contreras S, Feuerriegel S, Niemann S, André E, Kaswa MK, Tagliani E, Cabibbe A, Mathys V, Cirillo D, de Jong BC, Rigouts L, Supply P. 2021. Deep amplicon sequencing for culture-free prediction of susceptibility or resistance to 13 anti-tuberculous drugs. Eur Respir J 57:2002338. doi: 10.1183/13993003.02338-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nimmo C, Brien K, Millard J, Grant AD, Padayatchi N, Pym AS, O’Donnell M, Goldstein R, Breuer J, Balloux F. 2020. Dynamics of within-host Mycobacterium tuberculosis diversity and heteroresistance during treatment. EBioMedicine 55:102747. doi: 10.1016/j.ebiom.2020.102747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Trauner A, Liu Q, Via LE, Liu X, Ruan X, Liang L, Shi H, Chen Y, Wang Z, Liang R, Zhang W, Wei W, Gao J, Sun G, Brites D, England K, Zhang G, Gagneux S, Barry CE III, Gao Q. 2017. The within-host population dynamics of Mycobacterium tuberculosis vary with treatment efficacy. Genome Biol 18:71. doi: 10.1186/s13059-017-1196-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
M. tuberculosis strains with drug resistance-conferring minority variants; (a) the percentage of respiratory and non-respiratory specimens (b) the number and percentage of strains with minority variants detected in all four major lineages.
M. tuberculosis strains with mixed populations; (a) the number and percentage of mixed strain populations detected in different M. tuberculosis lineages, (b) the percentage in respiratory and non-respiratory specimen sources.
Figure S1: M. tuberculosis strains with drug resistance-conferring minority variants; (a) the percentage of respiratory and non-respiratory specimens (b) the number and percentage of strains with minority variants detected in all four major lineages; Figure S2: M. tuberculosis strains with mixed populations; (a) the number and percentage of mixed strain populations detected in different M. tuberculosis lineages (b) the percentage in respiratory and non-respiratory specimen sources.
Supplementary Tables.
Data Availability Statement
Raw de-identified pathogen WGS data was deposited in the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA) under BioProject number PRJNA899911.