

Abstract
Cerebral malaria (CM), a potentially fatal encephalopathy caused primarily by infection with Plasmodium falciparum, results in long-term adverse neuro-psychiatric sequelae. Neural cell injury contributes to the neurological deficits observed in CM. Abnormal regulation of tau, an axonal protein pathologically associated with the formation of neurofibrillary lesions in neurodegenerative diseases, has been linked to inflammation and cerebral microvascular compromise and has been reported in human and experimental CM (ECM). Immunotherapy with a monoclonal antibody to pathological tau (PHF-1 mAB) in experimental models of neurodegenerative diseases has been reported to mitigate cognitive decline. We investigated whether immunotherapy with PHF-1 mAB prevented cerebral endotheliopathy, neural cell injury, and neuroinflammation during ECM. Using C57BL/6 mice infected with either Plasmodium berghei ANKA (PbA), which causes ECM, Plasmodium berghei NK65 (PbN), which causes severe malaria, but not ECM, or uninfected mice (Un), we demonstrated that when compared to PbN infection or to uninfected mice, PbA infection resulted in significant memory impairment at 6 days post-infection, in association with abnormal tau phosphorylation at Ser202/Thr205 (pSer202/Thr205) and Ser396–404 (pSer396–404) in mouse brains. ECM also resulted in significantly higher expression of inflammatory markers, in microvascular congestion, and in glial cell activation. Treatment with PHF-1 mAB prevented PbA-induced cognitive impairment and was associated with significantly less vascular congestion, neuroinflammation and neural cell activation in mice with ECM. These findings suggest that abnormal regulation of tau protein contributes to cerebral vasculopathy and is critical in the pathogenesis of neural cell injury during CM. Tau-targeted therapies may ameliorate the neural cell damage and subsequent neurocognitive impairment which occur during disease.
Keywords: Plasmodium berghei ANKA, P. berghei NK65, Memory, Neural cell damage, Glial cell activation, cerebral vascular congestion, Neuroinflammation.
Graphical Abstract
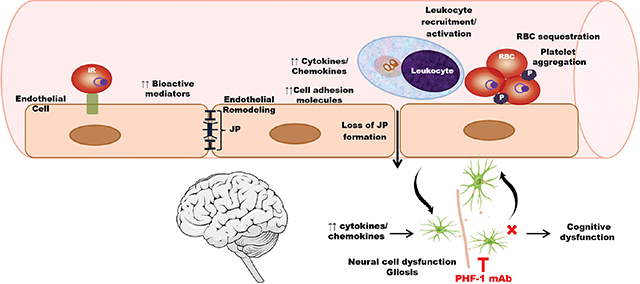
Infection of mice with Plasmodium berghei ANKA, a murine malaria species, induces remodeling of cerebral endothelial cells, a process which primes the endothelium for the recruitment of inflammatory cells and of infected red blood cells to the cerebral microvasculature. Vascular pathology also leads to neuroinflammation, involving the activation of glial cells and abnormal expression of tau proteins in neurons, leading to cognitive dysfunction. Passive immunization to pathological tau with an anti-PHF-1 monoclonal antibody mitigates microglial activation and malaria-induced endotheliopathy, ultimately resulting in preservation of cognitive function.
iR = infected red blood cell; JP = junctional protein; RBC = red blood cell; P = platelet; PHF-1 = paired helical filament 1.
Introduction
Cerebral malaria (CM) remains one of the most severe manifestations of disease caused by infection with Plasmodium species. This neurological syndrome is associated with 20% mortality and is characterized by significant morbidity (Carter et al., 2005; Hochman et al., 2015; John, Bangirana, et al., 2008; Murphy & Breman, 2001; Newton & Krishna, 1998), and an estimated 20 – 25% of CM survivors develop long-term neuropsychiatric complications and learning disabilities despite significant reductions in malaria transmission in the last decade (Boivin et al., 2007; Carter et al., 2006; Carter et al., 2005; Idro, Kakooza-Mwesige, et al., 2010; John, Bangirana, et al., 2008; Senanayake & de Silva, 1994). Vascular congestion and cytokine dysregulation are thought to contribute to the adverse neurological sequelae that occur during CM (Brown, Turner, et al., 1999; Datta et al., 2019; Holmberg et al., 2017; John, Panoskaltsis-Mortari, et al., 2008; Shabani et al., 2017). Several reports have shown that vessel occlusion and thrombosis were associated with axonal damage and demyelination during fatal pediatric CM (Baptista et al., 2010; Dorovini-Zis et al., 2011). Further, sequestration of infected erythrocytes in the brain microvasculature of CM patients has consistently correlated with disease severity (Baptista et al., 2010; Brown, Hien, et al., 1999; Dorovini-Zis et al., 2011; Ponsford et al., 2012). Our laboratory previously demonstrated a significant correlation between decreased cerebral blood flow and axonal damage in a mouse model (Kennan et al., 2005; Medana & Turner, 2006). Thus, it is likely that vascular congestion and reduced cerebral blood flow contribute to neuronal and glial cell damage and subsequently result in behavioral deficits observed in mice and CM patients (Brundel et al., 2012; Dorovini-Zis et al., 2011; Greiner et al., 2015; Ponsford et al., 2012).
We previously reported that tau, a neuronal protein important for microtubule stability, cytoskeletal organization and organellar/protein transport across axons (Lee et al., 2001; Shahani & Brandt, 2002), is abnormally phosphorylated during experimental CM (ECM) (Dai et al., 2012). When abnormally phosphorylated, particularly at Ser199–202–Thr205 and Ser396–404 (Mondragon-Rodriguez et al., 2014; Neddens et al., 2018), tau monomers are incapable of binding axonal microtubules, making them susceptible to misfolding and subsequent aggregation in axonal lumens and other sites within the neurons (Billingsley & Kincaid, 1997; Busciglio et al., 1995; Shahani & Brandt, 2002), or to secretion into the extracellular space (Yamada et al., 2014). Phosphorylation of tau at Ser396–404 significantly contributes to the formation of paired helical filaments (PHFs), the main components of neurofibrillary tangles in neurodegenerative tauopathies (Ihara et al., 1986; Kosik et al., 1986). Although the mechanisms by which tau dysregulation leads to cognitive decline are not fully understood, it is thought that abnormal tau accumulation in both presynaptic and postsynaptic terminals induces neuronal toxicity (Busciglio et al., 1995; Hanger et al., 2014), and that inclusions inside the cells may to lead to damage to neurons involved in memory processes, learning and other cognitive functions (Bendiske et al., 2002; Brunden et al., 2008; Hanger et al., 2014). In addition, when secreted extracellularly, tau interacts with glial cells, promoting inflammation (Leyns & Holtzman, 2017). Activated astrocytes and microglia within amyloid deposits can then further potentiate the neuroinflammatory response (Ahmad et al., 2019; Fakhoury, 2018).
The development of immunotherapy against abnormal tau forms has emerged as a promising approach in preventing neurodegeneration (Chai et al., 2011; Sankaranarayanan et al., 2015). Passive immunization with anti-tau antibodies has been reported to significantly inactivate tau pathology (Dai et al., 2017; Liu et al., 2016) and inflammatory pathways associated with neuronal damage and cognitive impairment during Alzheimer’s Disease (AD) (Medina, 2018). While our laboratory previously reported mitigation of tau pathology and subsequent preservation of cognitive function with lithium treatment (Dai et al., 2012), monoclonal antibodies to tau paired helical filaments (PHF-1 mAB) have never previously been investigated as a potential intervention against neuronal injury during ECM. In this study, we demonstrate for the first time the role of vascular congestion and glial cell activation in the abnormal regulation of tau and subsequently in the development of memory impairment associated with ECM. We herein report that immunotherapy to abnormal tau forms abrogates ECM-induced glial cell activation and vascular congestion, and consequently prevents infection-associated cognitive dysfunction.
Materials and Methods
Ethics Statement.
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The experiments were approved by the Institutional Animal Care and Use Committee at Albert Einstein College of Medicine (Protocol #: 20160609) and Yale School of Medicine (IACUC #: 2018–20196). Mice were housed in standard cages as per our approved protocol, with a maximum of 5 mice per cage. Animals had ad libitum access to food and water. Median weight of mice at baseline was 17 grams. All efforts were made to minimize suffering.
Mouse Infection.
Six-week old female C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME; RRID:MGI:7264769) were divided arbitrarily into three experimental groups: mice infected with Plasmodium berghei ANKA (PbA; Rodriguez Lab, New York University Langone), which developed ECM, mice infected with P. berghei NK65 (PbN; Rodriguez Lab, New York University Langone), which developed severe malaria (Van den Steen et al., 2010), and some degree of cerebral inflammation (Van den Steen et al., 2006), but no parasite accumulation in the brain, and no neurological impairment (Baptista et al., 2010; Van den Steen et al., 2010), and uninfected mice. Based on several experiments, a minimum sample size of 7 mice per group have been sufficient in order for our analyses to detect a mean difference at 0.05 significance. Due to an expected mortality rate of >50% at 7 days post-infection in untreated mice with experimental cerebral malaria, an initial size of 15 mice infected with PbA were used in each experiment been used in order to ensure at least 7 mice would be available for analysis at 7 days post infection. For uninfected mice and mice infected with PbN, an initial size of 7 mice were used for each experiment. The same number of mice was utilized in each replicate of the experiments. Total number of animals per group are listed in the figure legends for each analysis.
Mice were injected intraperitoneally (IP) with either 105 PbA-, PbN- or uninfected- red blood cells (RBCs; Figure S1). RBCs were diluted in 200 μL of phosphate-buffered saline (PBS). Disease progression was monitored every other day by measuring body weight, temperature, locomotor activity and parasitemia via tail blood smears stained with Giemsa (Figure S1; Giemsa Cat. No. SLBQ0140V; Sigma-Aldrich, St. Louis, MO). Fifty percent of the mice in each infection group were euthanized at 5 days post-infection and brains were harvested for analysis. The remaining 50% underwent memory testing at 6 days post infection and were euthanized at 7 days post infection (Figure S1).
For the PbA, PbN, and uninfected experiments, a total number of 27 uninfected, 24 PbA and 24 PbN mice were infected for the Day 5 and Day 7 brain analyses (Figure S1). Brain samples from each infection group were chosen arbitrarily for each analysis, with specific numbers of mice per group noted in each analysis. For the PHF-1 treatment experiments, a total number of 14 uninfected saline, 7 uninfected IgG, 10 uninfected PHF-1, 12 PbA saline, 13 PbA IgG, 13 PbA PHF-1 were used for Day 7 brain analyses. Mice from each infection and treatment group were arbitrarily chosen for each analysis, with the specific number of samples per group noted in each analysis.
As the experiments were designed to allow for comparisons at select time points, all the mice were sacrificed at the chosen timepoints for brain analyses, including moribund mice. A separate survival experiment was conducted for survival analysis and is included in the supplemental information (Figure S4).
PHF-1 mAB experimental design.
PHF-1 mAB, a monoclonal antibody against pathological tau forms was generously provided by Dr. Peter Davies. It recognizes tau phosphorylated on serine amino acids 404 and 396 on C-terminus of tau. The PHF-1 antibody was dissolved in PBS and used. For control, non-specific mouse IgG in PBS (Cat. No. SLM56; Equitech-Bio Inc., Kerrville, TX, USA) was used.
At 3 days post infection (dpi), PbA-infected and uninfected mice were arbitrarily assigned and treated with PHF-1 mAB at a single dose of 12.5 μg/g per mouse IP. For isotype controls, parallel groups of PbA-infected and uninfected mice were treated with a single dose of 12.5 μg/g non-specific IgG (Equitech-Bio) in PBS.
PHF-1 dosing was based on a dose-response experiment in uninfected mice demonstrating no behavior alterations for 7 days after treatment. Saline-treated PbA-infected mice were used as an ECM-positive control in all experiments (Figure S1). These experiments were also used to assess mortality.
Disease progression was monitored every other day by measuring body weight, temperature, locomotor activity and parasitemia via tail blood smears stained with Giemsa (Sigma-Aldrich, St. Louis, MO). All mice underwent memory testing at 6 dpi (Figure S1).
At 7 days post-infection, when ECM mice typically become moribund, all experimental mice were anesthetized with ketamine (150 mg/kg) and xylazine (10 mg/kg) and intracardially perfused with ice-cold saline. Brains were harvested, and for each mouse the brain was divided at mid-line with one hemisphere fixed in 10% buffered formalin and stored at room temperature for future hematoxylin and eosin (H&E) and immunofluorescence analysis, and the other half flash-frozen and stored at –80°C for future protein and RNA analysis.
Quantification of Brain Vessel Congestion.
The degree of brain vessel occlusion with leukocytes and RBCs was quantified on a 0–4 scale by blind assessment of H&E-stained slides as previously published by our laboratory (Dai et al., 2010; Freeman et al., 2016) and adapted from Fauconnier et. al. (Fauconnier et al., 2012). The analysis was performed by two independent investigators by visualization under a microscope. This scoring was performed blindly as slides were marked with a code which did not include infection conditions. Vessels were scored in the following manner: 0 = no congestion and presence of 1–2 endothelial cells; 1 = < 10% congestion and presence of 1–2 leukocytes and/or 1–2 RBCs; 2 = 10–50% congestion; 3 = 50–90% congestion; 4 = > 90% congestion.
Immunostains.
Microglial activity was visualized by immunofluorescence analysis as previously published by our laboratory (Dai et al., 2010; Desruisseaux et al., 2008; Freeman et al., 2016). Briefly, following antigen retrieval, 5μm sagittal slices were probed using rabbit antibody to ionized calcium-binding adaptor molecule (Iba1; Cat. No. 019–91741, Wako Chemicals), a peptide that is selectively expressed in macrophages/microglia, at a dilution of 1:300 at 4°C overnight. Fluorescent labeled goat-anti rabbit (1:1000; Alexa Fluor 594; Cat. No. A32740, Thermo Fisher) secondary antibody was used according to the manufacturer’s protocol. Brain slices were examined with a Zeiss Axio Observer 5 microscope.
Real-time PCR analysis (qPCR).
RNA was extracted from frozen half-brains using TRIzol Reagent (Cat. No. 15596026; Invitrogen Life Technologies) then purified using the spin columns from the RNeasy Lipid Tissue Mini Kit (Cat. No. 74804; QIagen, Venlo, The Netherlands) and treated with DNase I (Cat. No. 18068–015; Invitrogen, Carlsbad, CA, USA) as recommended by the manufacturer. Following isolation and purification, RNA was quantified by NanoDrop, and (0.5 ug/sample) 500 ng/sample was reverse transcribed into cDNA using the SuperScript III First-Strand Synthesis System for RT-PCR (Cat. No. 18080–051; Invitrogen Life Technologies) per manufacturer’s protocol.
Transcription levels were assessed by quantitative real-time RT-PCR (qRT-PCR) using Sybr Premix Ex Taq (Cat. No. RR82LR; Takara Bio USA, Inc., Mountain View, CA) and the Bio-Rad C1000 Touch Thermal Cycler Real-Time PCR Detection System. mRNA transcription levels were normalized to β-actin (housekeeping gene; Cat. No. mAB 3700; Cell signaling Technology) and the results are expressed as fold change compared to uninfected controls. Non-template solutions served as controls.
The following primers were used: Mouse Beta-Actin (β-actin) Fw: 5’-GGGAATGGGT CAGAAGGAC-’3 (19 bp) and Rev: 5’-GGTCATCTTTTCACGGTTGG-’3 (20 bp); Mouse Adgre1 (F4/80) Fw: 5’-CTTTGGCTATGGGCTTCCAGTC-’3 (22 pb) and Rev: 5’- GCAAGG AGGACAGAGTTTA-TCGTG-’3 (24 pb); Mouse IL-6: Fw 5’-GCTAAGGACCAAGACCA TCCAAT-3’ (23 bp) and Rev: 5’ GCTTAGGCATAACGCACTAGGTTT-3’ (24 bp); Mouse IL-1β; Fw: 5’-AGA TGAGAGCATCCAGC TTC-’3 (20 bp) and Rev: 5’-CTGTAGTGCAGCTG TCTAATGG-’3 (22 bp); Mouse IL-8 (CXCL-8): Fw: 5’-TTACTGCAACAGAAAGGAAG-3’ (20 bp) and Rev: 5’-GGTATTAACCTGTTAGTAATTG-3’ (22 bp); Mouse IL-1RA: Fw: 5’-AACCAGCTCATT GCTGGGTACTTA-3’ (24 bp) and Rev: 5’-GCCCAAGAACACACT ATGAAGGTC-3’ (24 bp); Mouse TGF-β: Forward: 5’-TTGCTTCAGCTCCACAGAGA-’3 (20 bp) and Rev: 5’-TGGTTGTAGAGGGCAAGGA-’3 (19 bp). qPCR data was calculated and reported as the normalized relative ratio, 2−ΔΔCT, where ΔCT = CT (target) – CT (β-actin), and ΔΔCT = ΔCT (sample) – ΔCT (calibrator) (Livak & Schmittgen, 2001).
Western Blot.
Brain region lysates were prepared by homogenizing frozen extracts in lysis buffer as previously described (Dai et al., 2012) and incubated overnight at 4°C with the following primary antibodies: Anti-Tau pSer396 rabbit monoclonal antibody (Cat. No. ab32057; 1:12000; Abcam), Anti-Tau pSer202 rabbit monoclonal Antibody (Cat. No. ab108387; 1:1000; Abcam), Anti-Tau [TAU-5] mouse monoclonal antibody (Cat. No. ab80579; 1:5000; Abcam), Anti-Glial Fibrillary Acid Protein (GFAP) mouse monoclonal antibody (Cat. No. MAB360; 1:1000; Merck/Millipore), Anti-CD11b rabbit polyclonal antibody (Cat. No. NB110–89474; 1:1000; NovusBio). Following primary antibody incubation, membranes were incubated with Li-COR Donkey anti-rabbit (Cat No 925–68073, LICOR) and/or Goat anti-mouse IgG (Cat. No. 925–68070, LICOR) Secondary antibodies directly labelled with near-infrared dyes (Li-COR, USA). Quantification of immunoblot data was performed using volumetric measurement of fluorescence intensity on a LiCor Odyssey CLx imager.
Memory Testing.
Visual memory by the object recognition test was conducted as previously described (Dai et al., 2010; Desruisseaux et al., 2008; Ennaceur & Meliani, 1992; Freeman et al., 2016). Briefly, experimental mice were subjected to both training and testing periods in a square opaque, plastic arena. During training periods, mice were placed in the arena for 3 min and allowed to explore identical objects. After training, mice were returned to their home cages for 45 min. They were then subjected to testing periods for 3 min where they were once again allowed to explore two objects, one of which was retained from training and the other of which was novel. The expectation was that animals would prefer the novel object to the familiar one during the testing period. Experiments were conducted at multiple stages during the day, in a dark room with red lighting. Each batch of tests included all experimental groups in order to lower the chance of time of observation bias. Data were presented as a % preference (novel object exploration time/total exploration time x 100). A preference of 55% or better was deemed successful and indicated the animal demonstrated functional visual memory. To maintain blinding, original cage cards containing infection and treatment conditions were removed by the experimenter and replaced by a card containing a code (numbers 1, 2, 3 etc). Cages were organized in batches prior to training to ensure that different treatment groups were analyzed within the same time frame during the day. The examiner did not have access to the original cage card information during memory testing.
The primary endpoint of the study was preservation of cognitive function. Secondary endpoints included mitigation of neuroinflammation and of microvascular congestion. histologic changes and protein/ mRNA expression of biomarkers.
Mice were excluded from cognitive testing and analysis if they were moribund; if they had gross delay in locomotor activity; if they did not explore the objects during testing or if less than 2 minutes total were spent exploring the two objects during training. For the PbA, PbN, and uninfected experiments, a total of 16 PbA-infected mice were excluded from memory testing at day 6 based on the above criteria. Multiple experiments of blinded cognitive testing performed at day 6 during the same light/ dark cycles - were pooled to increase the sample size. For the PHF-1 treatment experiments no mice were excluded from testing. Please see Figure S1 legend for the total sample sizes for the memory tests.
Rapid murine coma and behavior score (RMCBS) testing (Carroll et al., 2010) was performed in pooled experiments between control and PbA-infected mice.
Statistical Analysis.
All analyses consisted of comparisons between uninfected and PbA-infected experimental treatment groups or between uninfected, PbA-infected or PbN-infected mice. Group effects were analyzed using one-way ANOVA. Two-way ANOVA was used for analysis of longitudinal data. For one-way and two-way ANOVA, Dunnett’s correction for multiple comparisons was used. For analyses of cytokine and neural biomarker expression, the false discovery rate was used to correct for multiple comparisons. Statistical values of α < 0.05 adjusted for multiple comparisons were deemed significant. Data were not assessed for normality. No test for outliers was conducted, and no data were excluded from analysis, except as specified in cognitive testing above. Analyses were conducted in GraphPad Prism.
Unless otherwise noted, all data are representative of 3 experiments, each performed in triplicates.
Results
ECM results in significant memory impairment
Consistent with our previous findings (Freeman et al., 2016; Martins et al., 2016), PbA infection induced significant impairment of visual working memory at 6 days post-infection (6 dpi; Figure 1) which was not observed in PbN infected mice or uninfected controls. PbA-infected mice did not exhibit memory impairment when compared to uninfected control or PbN-infected mice at 4 dpi (Figure S2).
Figure 1. Visual memory at 6 days post-infection.
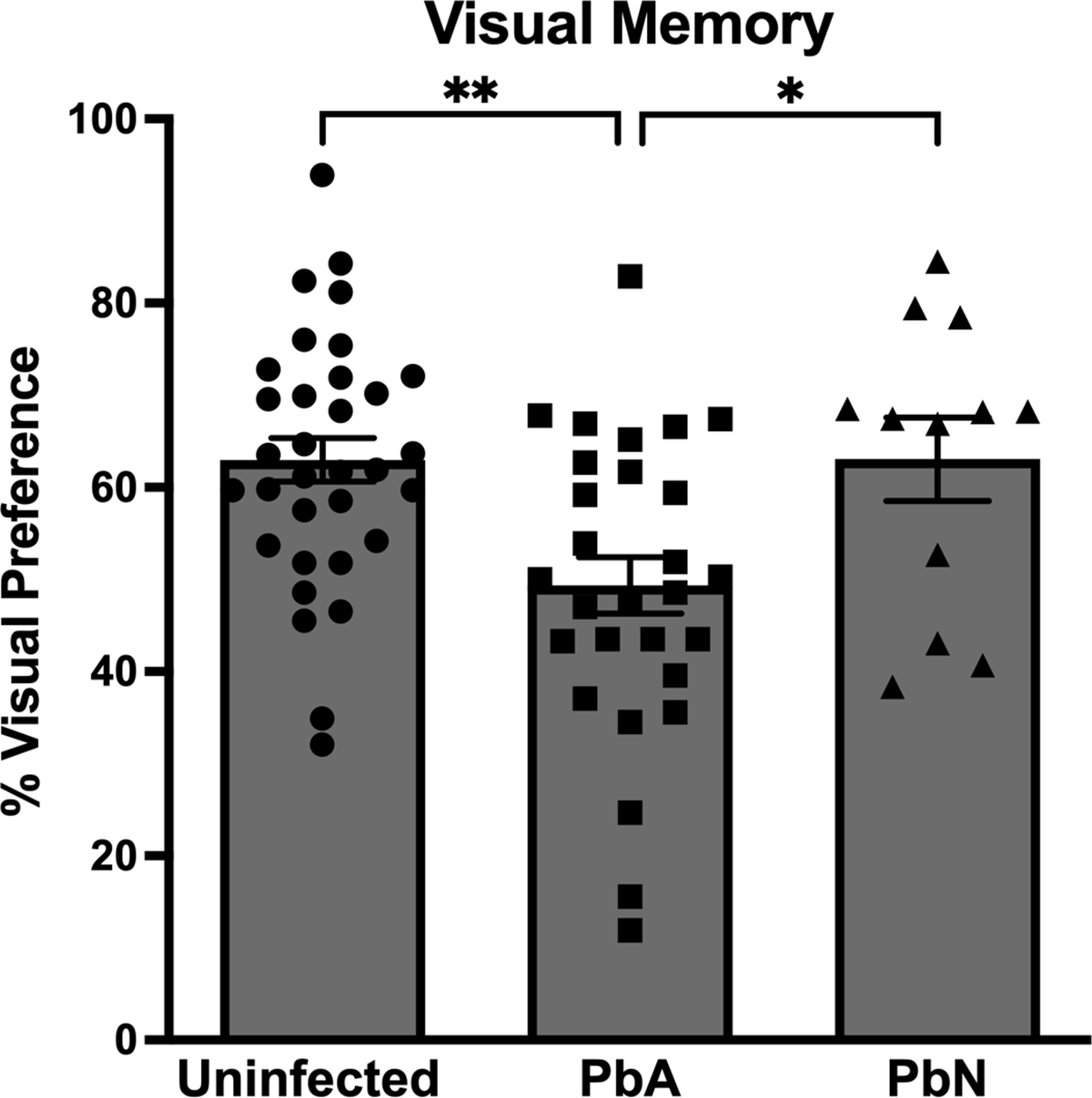
At 6 days post-infection (dpi) PbA-infected mice exhibited severe visual memory impairment during the novel object recognition test compared to PbN-infected and uninfected mice (F(2, 70) = 7.14; p < 0.01). Data were representative of five experiments. PbA = Plasmodium berghei ANKA; PbN = Plasmodium berghei NK65
* = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001; ns = not significant. N = 33 Uninfected control mice/ 28 PbA-infected mice/ 12 PbN-infected mice.
In pooled experiments, there was no correlation between performance on the rapid murine coma and behavior score (Carroll et al., 2010) and memory in healthy or PbA-infected mice (Figure S3) at day 6 post-infection.
PbA infection results in cerebral vascular congestion.
To determine the kinetics of vascular congestion in brain regions associated with memory impairments in our murine model, we quantified vessel occlusion in mouse brain sections. PbA-infected mice exhibited more cerebrovascular congestion at 5 dpi (Figure 2A–C) and at 7 dpi (Figure 2D–F) than was observed in uninfected mice or PbN-infected mice. At 5 dpi, the differences in vascular congestion were observed in the cortex (Figure 2G), hippocampus (Figure 2H), thalamus (Figure 2I), brainstem (Figure 2J) and cerebellum (Figure 2K). The observed vascular occlusion persisted at 7 dpi, with PbA-infected mice showing significantly more microvascular congestion than uninfected mice or PbN-infected mice in the cortex (Figure 2G), hippocampus (Figure 2H), thalamus (Figure 2I) and cerebellum (Figure 2K). PbA-infected mice that survived to 7 dpi exhibited similar vascular congestion to uninfected mice in the brainstem.
Figure 2. Quantification of vessel congestion in the cortex, hippocampus, thalamus, brainstem and cerebellum at days 5 and 7 post-infection.
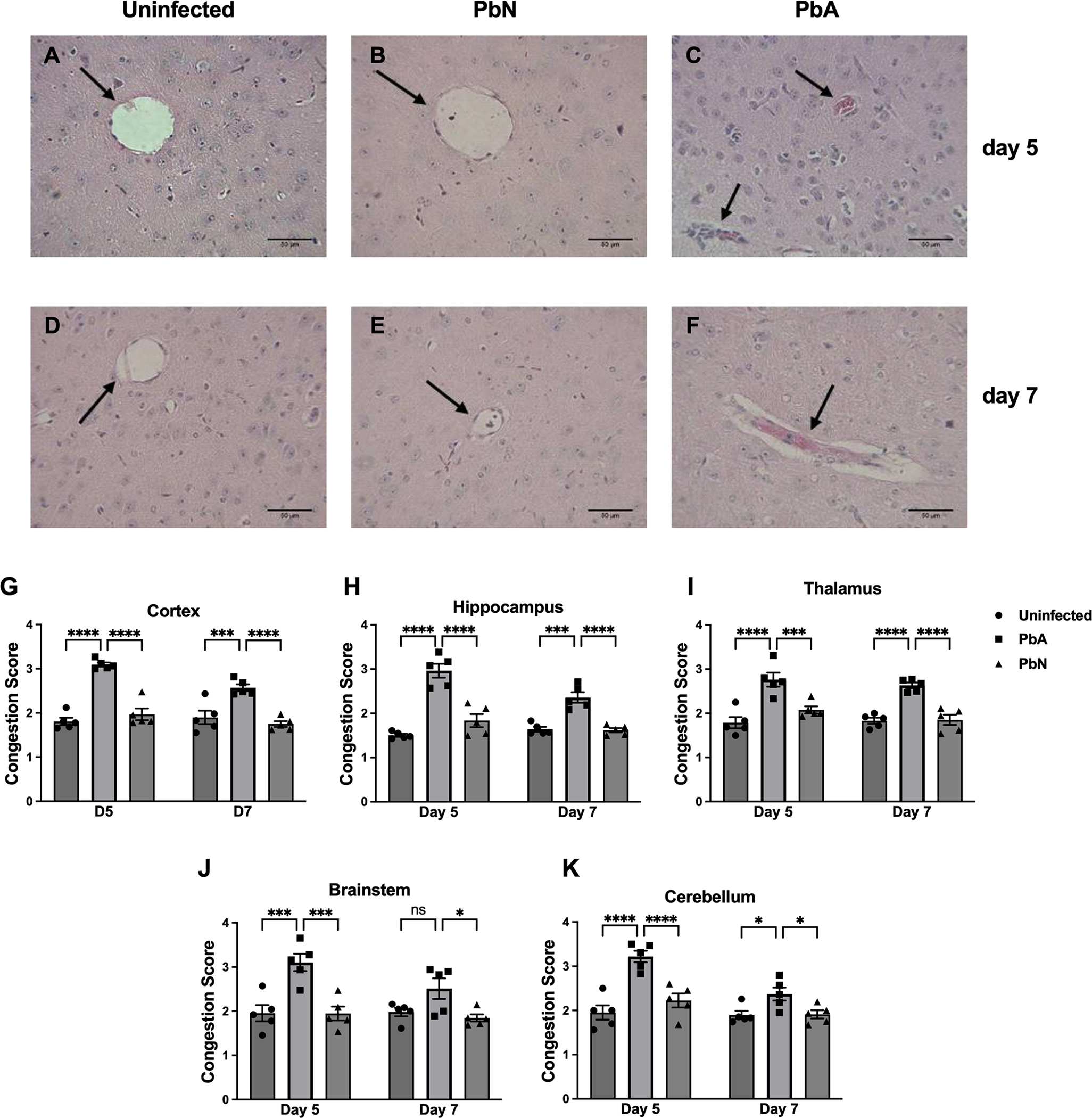
Representative H&E-stained brain sections demonstrating vessel congestion in the thalamus (A-F): Uninfected (A), PbN (B) and PbA (C) at 5 dpi and Uninfected (D), PbN (E) and PbA (F) at 7 dpi. Black arrows point to vessels. Note the non-occluded vessels in uninfected (A, D) and PbN-infected (B, E) mice in contrast to heavily congested vessels with dark-stained monocytes, hemozoin and pink-colored RBCs in PbA-infected mice (C, F). Brain vessel congestion in the Cortex (G), Hippocampus (H), Thalamus (I), Brainstem (J) and Cerebellum (K) of PbA- and PbN-infected mice sacrificed at days 5 and 7 post-infection. Results showed sustained vessel congestion in PbA infected mice compared to PbN and uninfected mice at day 5 and 7 post-infection. Congestion was quantified on a 0–4 scale by blind assessment of leukocyte and red blood cell accumulation in a specific vessel (0 = no congestion; 1 = < 10% congestion; 2 = 10–50% congestion; 3 = 50–90% congestion; 4 = > 90% congestion). Scale bar 50 μm. Magnification = 40X. PbA = Plasmodium berghei ANKA; PbN = Plasmodium berghei NK65.
Cortex: Infection factor F(2, 24) = 64.4, p < 0.0001; Time factor F(1, 24) = 7.2, p = 0.01; Interaction F(2, 24) = 4.8, p < 0.05.
Hippocampus: Infection factor F(2, 24) = 61.9, p < 0.0001; Time factor F(1, 24) = 7, p = 0.01; Interaction F(2, 24) = 6.1, p < 0.01.
Thalamus: Infection factor F(2, 24) = 38.9, p < 0.0001; Time factor F(1, 24) = 1.4, p = ns; Interaction F(2, 24) = 0.8, p = ns.
Brainstem: Infection factor F(2, 24) = 18.5, p < 0.0001; Time factor F(1, 24) = 2.7, p = ns; Interaction F(2, 24) = 1.9, p = ns.
Cerebellum: Infection factor F(2, 24) = 24.4, p < 0.0001; Time factor F(1, 24) = 13.82, p < 0.01; Interaction F(2, 24) = 4.6, p < 0.05.
* = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001; ns = not significant. N = 5 mice/group.
PbA infection results in increased inflammation in the brain
Several pro- and anti-inflammatory cytokines and chemokines, important in immune cell recruitment and adhesion, are modulated during CM and are associated with enhanced disease severity (Idro, Marsh, et al., 2010; John, Panoskaltsis-Mortari, et al., 2008; John, Park, et al., 2008; Lyke et al., 2004; Oakley et al., 2018). We quantified the mRNA expression of inflammatory mediators in whole brains of PbA-infected, PbN-infected and uninfected mice during the course of ECM, at intermediate (5 dpi) and terminal stages (7 dpi) of disease. At 5 dpi, PbA infection resulted in significantly higher mRNA expression of IL-6 (Figure 3A), IL-8 (Figure 3B), and IL-1RA (Figure 3C) compared to uninfected or to PbN-infected mice. While PbA infection induced higher expression of TGF-β, this was similar to TGF-β expression in PbN-infected mice (Figure 3D). No differences were observed in mRNA expression of IL-1β (Figure 3E) at 5 dpi between the three groups. At 7 dpi, PbA-infected mice sustained higher mRNA expression of IL-6 (Figure 3A), IL-8 (Figure 3B), IL-1RA (Figure 3C), TGF-β (Figure 3D), and IL-1β (Figure 3E) when compared to uninfected and to PbN-infected mice.
Figure 3. mRNA expression of select cytokines and chemokines in PbA-infected, PbN-infected and uninfected mice brains at days 5 and 7 post-infection.

Fold relative expression of markers associated with proinflammatory response was assessed by q-PCR in brain of PbA-infected, PbN-infected and uninfected mice sacrificed at days 5 and 7 post-infection. All genes were normalized to beta-actin (β-actin). Increased mRNA expression levels in TGF-β (A), IL-8 (B) and IL-6 (C) at day 5 post-infection. Sustained increase in mRNA expression levels in TGF-β (A), IL-8 (B), IL-6 (C), IL-1RA (D) and IL-1β at day 7 post-infection. PbA = Plasmodium berghei ANKA; PbN = Plasmodium berghei NK65. IL = Interleukin; TGF = transforming growth factor. IL-6: Infection factor F(2, 66) = 43.6, p < 0.0001; Time factor F(1, 66) = 37, p < 0.0001; Interaction F(2, 66) = 7.6, p < 0.01.
IL-8: Infection factor F(2, 66) = 36.8, p < 0.0001; Time factor F(1, 66) = 34.8, p < 0.0001; Interaction F(2, 66) = 8.2, p < 0.001.
IL1-RA: Infection factor F(2, 66) = 87.7, p < 0.0001; Time factor F(1, 66) = 63.9, p < 0.0001; Interaction F(2, 66) = 16.8, p < 0.0001.
TGF-β: Infection factor F(2, 72) = 21.8, p < 0.0001; Time factor F(1, 72) = 0.3, p = ns; Interaction F(2, 72) = 1.4, p = ns.
IL-1β: Infection factor F(2, 68) = 23, p < 0.0001; Time factor F(1, 68) = 35.7, p < 0.0001; Interaction F(2, 68) = 9.2, p < 0.001.
* = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001; ns = not significant. N = 13 uninfected mice/ 12 PbA-infected mice/ 12 PbN-infected mice.
ECM-induced memory impairment is associated with glial and neuronal damage
While glial cells play an essential role in maintaining normal brain function during physiologic conditions (Farfara et al., 2008), they are capable of inducing the secretion of several inflammatory mediators that provoke pathological changes in the CNS when activated (Benarroch, 2013; Forman et al., 2005; Higuchi et al., 2002; Higuchi et al., 2005; Schwartz et al., 2013). To determine the contribution of glial activation to the increased production of inflammatory cytokines in the brain during ECM, we measured the kinetics of microglial activation and of astrogliosis in brain homogenates by quantifying the mRNA expression of the adhesion G protein coupled receptor E1 (Adgre1), a gene which encodes for the F4/80 protein found on the surface of murine microglia and monocytes (Oakley et al., 2018) and by quantification of GFAP expression, the major intermediate filament protein and component of the cytoskeleton in mature astrocytes (Ishiki et al., 2016; Middeldorp & Hol, 2011), at intermediate and late stages of disease. PbA infection induced significantly higher expression of Adgre1 at 5 dpi (Figure 4A), and at 7 dpi (Figure 4A) when compared to uninfected or to PbN-infected mice. There were no differences in GFAP expression at 5 dpi (Figure 4B); however, PbA infection induced higher levels of GFAP in the brains of ECM mice at 7 dpi (Figure 4B) when compared to uninfected, but at levels similar to PbN-infected mice (Figure 4B).
Figure 4. Expression of Adgre1 (F4/80) and GFAP in brain lysates of PbA-infected, PbN-infected, and uninfected mice brains at days 5 and 7 post-infection.

(A). Fold relative expression of marker associated with proinflammatory response was assessed by q-PCR in brain of PbA-infected, PbN-infected and uninfected mice sacrificed at days 5 and 7 post-infection. Adgre1 gene was normalized to beta-actin (β-actin). Increased mRNA expression levels in Adgre1 at day 5 post-infection and sustained increase in mRNA expression levels in Adgre1 at day 7 post-infection in PbA-infected mice. N = 12 mice/group.
(B) Representative western blot images and protein levels illustrating GFAP and a GDI loading control in PbA-infected, PbN-infected and uninfected mice at day 5 and 7 post-infection. PbA = Plasmodium berghei ANKA; PbN = Plasmodium berghei NK65. Adgre1 = Adhesion G Protein-Coupled Receptor E1; GFAP = Glial fibrillary acidic protein. GDI = Guanosine nucleotide dissociation inhibitor (housekeeping protein). Western blots for figures 4 and 5 were stripped and re-probed.
Adgre1: Infection factor F(2, 66) = 30.9, p < 0.0001; Time factor F(1, 66) = 14.4, p < 0.001; Interaction F(2, 66) = 4.1, p < 0.05.
GFAP: Infection factor F(2, 69) = 4.3, p < 0.05; Time factor F(1, 69) = 0.7, p = 0.4; Interaction F(2, 69) = 1.2, p = ns.
* = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001; ns = not significant. N = 13–14 uninfected mice/ 12 PbA-infected mice/ 12 PbN-infected mice.
PbA infection results in abnormal tau phosphorylation at terminal stages of disease
Several reports demonstrate that extracellular tau is internalized by microglia (Bolos et al., 2017; Maphis et al., 2015; Sanchez-Mejias et al., 2016; Vogels et al., 2019). This results in microglial activation and eventual degeneration as well as impairment of microglial function (Bolos et al., 2017; Sanchez-Mejias et al., 2016), it also contributes abnormal tau propagation in the brain (Bolos et al., 2017; Maphis et al., 2015; Perea et al., 2018; Vogels et al., 2019). Using immunoblots of whole brain lysates, we herein demonstrate that PbA infection induced significantly more hyperphosphorylation of tau at Ser202/Thr205 (Figure 5A, C) and at Ser396–404 (Figure 5B, D) at 7 dpi following microglial activation. Infection also induced greater expression of total tau overall at late stages of disease (Figure 5 A–B, E–F). There were no significant differences observed in tau phosphorylation or in total tau expression between uninfected, PbA- and PbN-infected mice at 5 dpi.
Figure 5. Tau modifications in brain lysates of PbA-infected, PbN-infected, and uninfected mice at day 7 post-infection.

(A) Representative western blot images and protein quantification levels of phosphorylated Ser202 tau (pSer202 tau), total tau and a GDI loading control in PbA-, PbN- and uninfected mice at day 5 and 7 post-infection. (B) Representative western blot images and protein quantification levels of phosphorylated Ser396−404 tau (pSer396−404 tau) in PbA- and PbN-infected mice sacrificed at day 5 and 7 post-infection. (C-D) Quantification of immunoblot data was performed using volumetric measurement of fluorescence intensity on a LiCor Odyssey CLx imager. Values shown reflect change relative to uninfected mice. PbA = Plasmodium berghei ANKA; PbN = Plasmodium berghei NK65. Western blots for figures 4 and 5 were stripped and re-probed.
Tau pSer202: Infection factor F(2, 49) = 6.5, p < 0.01; Time factor F(1, 49) = 2, p = ns; Interaction F(2, 49) = 1.1, p = ns.
Total tau: Infection factor F(2, 56) = 4.5, p < 0.05; Time factor F(1, 56) = 6.8, p < 0.05; Interaction F(2, 56) = 1.3, p = ns.
Tau pSer396−404: Infection factor F(2, 51) = 6.5, p < 0.01; Time factor F(1, 51) = 0.08, p = ns; Interaction F(2, 51) = 2.1, p = ns.
Total tau: Infection factor F(2, 54) = 6, p < 0.01; Time factor F(1, 54) = 4.7, p < 0.05; Interaction F(2, 54) = 0.3, p = ns.
* = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001; ns = not significant. N = 8–12 uninfected mice/ 8–9 PbA-infected mice/ 8–10 PbN-infected mice.
Administration of monoclonal antibody to paired helical filaments prevents ECM-induced memory impairment.
We previously reported that lithium treatment, through its inhibition of glycogen synthase kinase 3β (Noble et al., 2005), a major kinase in the regulation of tau function, mitigated tau pathology and prevented cognitive dysfunction during ECM (Dai et al., 2012). However, as lithium does not directly modulate tau properties, we herein tested the ability of immunotherapy against pathological tau to mitigate the CNS manifestations of ECM. Treatment of PbA-infected mice PHF-1 mAB, resulted in preservation of visual working memory. PbA-infected mice treated with PHF-1 mAB had significantly higher preference scores during the novel object recognition test in comparison to untreated PbA-infected mice at 6 dpi (Figure 6A).
Figure 6. PHF-1 mAB administration improves visual memory during ECM.

(A) At 6 days post-infection, preference scores were significantly higher in PbA-infected mice treated with PHF-1 mAB than in untreated infected mice or mice infected mice treated with non-specific IgG (F(5, 68) = 2.7; p < 0.05). PbA = Plasmodium berghei ANKA; PbN = Plasmodium berghei NK65. PHF = paired helical filament.
* = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001; ns = not significant. N = 24 uninfected + saline/ 20 PbA-infected + saline/ 7 uninfected + IgG/ 8 PbA-infected + IgG/ 7 uninfected + PHF-1 mAB/ 8 PbA + PHF-1 mAB.
Notably, PHF-1 mAB effects on cognition occurred in the absence of antiparasitic effects. Parasitemia was similar in both PHF-1 mAB treated PbA-infected and in untreated PbA-infected mice (Figure S4A). In addition, PHF-1 mAb had no effect on mortality in PbA-infected mice (Figure S4B) consistent with our previous reports using lithium treatment (Dai et al., 2012).
PHF-1 mAB prevents the cerebral vascular congestion that occurs during ECM
Tau pathology has been linked to vascular activation and remodeling (Merlini et al., 2016; Vogels et al., 2019). Thus, to test whether targeting abnormal tau with PHF-1 mAB prevented microvascular congestion during ECM, we examined H&E sagittal sections of perfused and fixed brain tissue from uninfected and PbA-infected mice with and without treatment with PHF-1 mAB. Treatment of PbA-infected mice with PHF-1 mAB mitigated PbA-induced cerebrovascular congestion in mice at 7 dpi (Figure 7A–F). PHF-1 mAB treatment prevented vessel occlusion in the cortex (Figure 7G, Figure S5A), hippocampus (Figure 7H; Figure S5B), thalamus (Figure 7I), and brainstem (Figure 7J; Figure S5C). Although congestion in the cerebellum was lower in PHF-1 mAB treated infected mice, this difference did not reach significance (Figure 7K; Figure S5D; adjusted p = 0.09).
Figure 7. PHF-1 mAB treatment inhibits vascular congestion in the brains of PbA-infected mice.

(A-F) Representative H&E-stained brain image demonstrating microvessels congestion in the brainstem of uninfected mice and PbA-infected mice treated with saline, non-specifc IgG, or PHF-1 mAB: Un-Saline (A), PbA-Saline (D), Un-IgG (B), PbA-IgG (E), Un-PHF-1 mAB (C) and PbA-PHF-1 mAB (F) at 7 dpi. Black arrows point to vessels. Note the non-occluded vessels in Un-Saline (A), the vessels heavily congested with dark-stained monocytes, hemozoin and pink-colored RBCs in PbA-Saline (D) and PbA-IgG (E) and less occlusion in PbA-PHF-1 mAB (F) mice. (G-K) Brain microvascular congestion was quantified on a 0–4 scale by blind assessment of leukocyte and red blood cell accumulation in a specific vessel (0 = no congestion; 1 = < 10% congestion; 2 = 10–50% congestion; 3 = 50–90% congestion; 4 = > 90% congestion) in the cortex (G; F(5, 57) = 11.4; p < 0.0001), hippocampus (H; F(5, 40) = 7.8; p < 0.0001), thalamus (I; F(5, 40) = 12.2; p < 0.0001), brainstem (J; F(5, 35) = 4.8; p < 0.01), and cerebellum (K; F(5, 43) = 8; p < 0.0001) of PHF-1 mAB treated PbA-infected compared to untreated PbA-infected mice at terminal stage of disease. PbA = Plasmodium berghei ANKA; PbN = Plasmodium berghei NK65; Un = uninfected. PHF = paired helical filament.
* = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001; ns = not significant. N = 6–30 mice/group.
Both PHF-1 mAB and non-immune IgG mitigated neuroinflammation in PbA-infected mice
Previous studies in mice and humans have reported that increased secretion of pro-inflammatory mediators promotes pathogenesis of CM (de Kossodo & Grau, 1993; de Vries et al., 1996; Jakobsen et al., 1994; John, Panoskaltsis-Mortari, et al., 2008). The mRNA expression of TGF-β, IL-8, IL-6, IL-1RA, and IL-1β were quantified in whole brains of untreated PbA-infected, PHF-1 mAB treated PbA-infected and treated and untreated uninfected control mice at 7 dpi using quantitative real-time polymerase chain reaction (q-PCR). PHF-1 mAB treated infected mice significantly lower transcription of TGF-β (Figure 8A), IL-8 (Figure 8B), and IL-6 (Figure 8C). Notably, infected mice treated with non-immune IgG control also expressed significantly lower expression of IL-8 (Figure 8B) and IL-6 (Figure 8C). PHF-1 mAB treatment had no effect on the expression of IL-1RA (Figure 8D) or IL-1β (Figure 8E).
Figure 8. Inflammatory responses to PHF-1 mAB in the brains of uninfected and PbA-infected mice.

At 7 dpi, PbA-infected mice had higher TGF-β (A; p < 0.05; q < 0.05), IL-8 (B; p < 0.05; q < 0.05), IL-6 (C; p < 0.05; q < 0.05), IL-1RA (D; p < 0.05; q < 0.05) and IL-1β (E; p < 0.05; q < 0.05) mRNA expression compared to uninfected mice. PHF-1 mAB treatment resulted in lower expression of TGF-β in PHF-1 mAB treated infected mice (A). Both non-specific IgG and PHF-1 mAB treatment resulted in lower levels of IL-8 (B) and IL-6 (C) in PbA-infected mice. PHF-1 mAB had no effect on IL-1RA (D) or IL-1β (E) expression. Multiple comparisons were corrected using the false discovery rate (q). An α < 0.05 was considered significant for false discovery. PbA = Plasmodium berghei ANKA; PbN = Plasmodium berghei NK65; Un = uninfected; IL = interleukin; IL-1RA = interleukin 1 receptor antagonist; TGF = transforming growth factor.
* = false discovery rate (q) < 0.05; ns = false discovery rate (q) not significant. N = 7–8 mice/group.
PHF-1 mAB prevents PbA-induced microglial activation, but not astrogliosis
Glial cells are potent amplifiers of inflammation within the central nervous system (CNS) during CM infection (Medana et al., 1997; Shrivastava et al., 2017; Wilson et al., 2018). To test PHF-1 mAB effects on glial cell protein and mRNA expression of glial cell markers were quantified. Consistent with our previous reports (Dai et al., 2010; Desruisseaux et al., 2008), PbA infection resulted in morphological changes in Iba1 positive cells, and in their distribution to perivascular spaces in the cerebral cortex (Figure 9A), the hippocampus (Figure S6A), the brainstem (Figure S6B), and the cerebellum (Figure S6C). PHF-1 mAB prevented these changes in the cortex (Figure 9A), hippocampus (Figure S6A), and the cerebellum (Figure S6C), but not in the brainstem (Figure S6B). IgG treated infected mice had similar microglial morphology and distribution as seen in saline-treated infected mice in all regions. Quantification of microglial activity revealed higher protein expression of CD11b, an integrin family member expressed on the surface of myeloid cells, including microglia, monocytes, and macrophages (Howe et al., 2012; Rentsendorj et al., 2018) in PbA-infected mice and in IgG-treated mice than observed in mice treated with PHF-1 mAb (Figure 9B). Both PHF-1 mAb and non-specific IgG treatment resulted in significantly lower expression of Adgre1 in PbA-infected mice than untreated mice (Figure 9C). Both non-immune IgG and PHF-1 mAB similarly resulted in lower expression of GFAP in the brains of PbA infected mice (Figure 9C), suggesting that PHF-1 mAB had no effects on the transcription of Adgre1 nor provided protective effects on astrocytosis beyond those of non-specific IgG.
Figure 9. PHF-1 mAB administration prevented microglial and monocyte cell (myeloid cells) but not astrocyte activation during PbA infection.
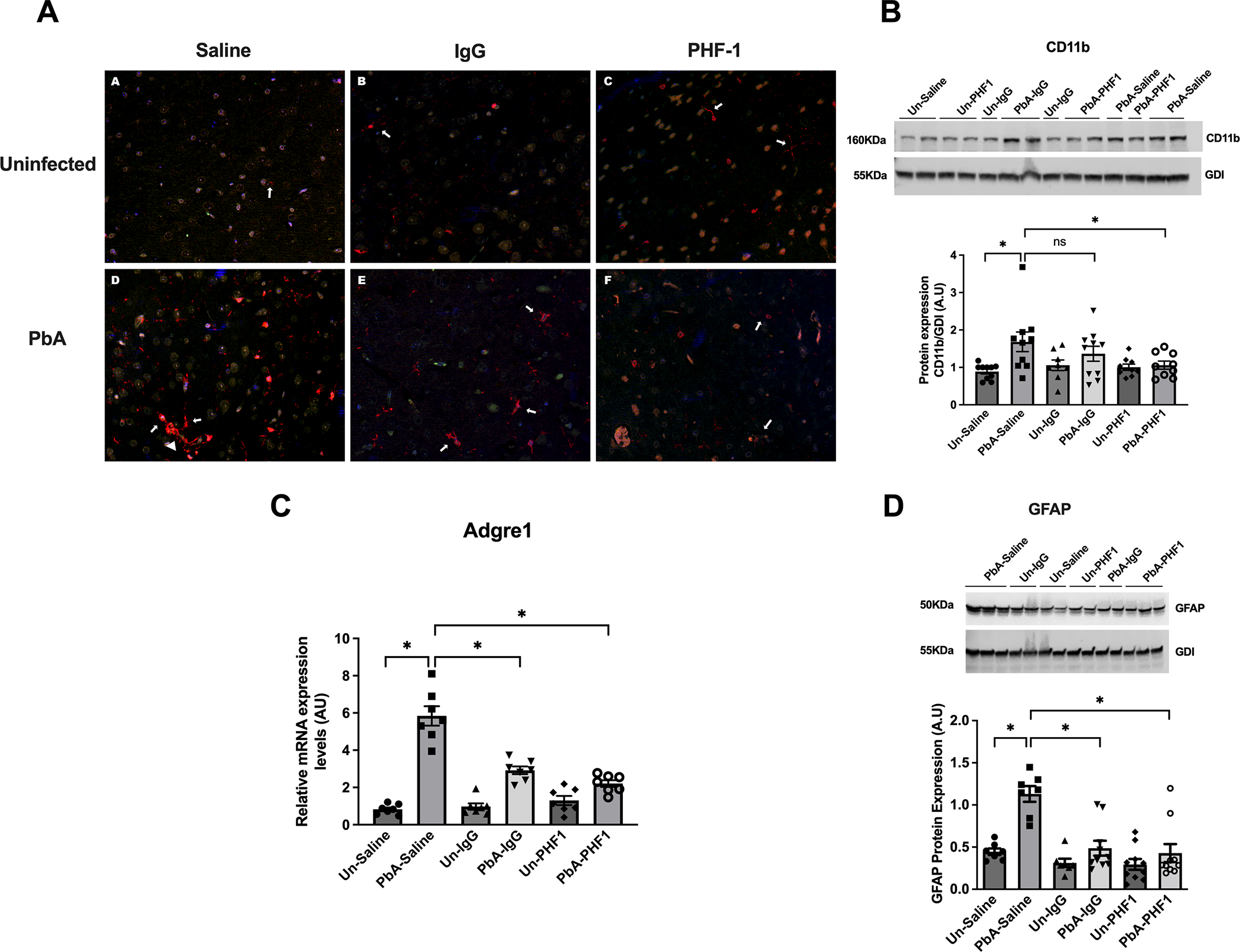
(A) Illustrative immunofluorescent stains for ionized calcium-binding adaptor molecule (Iba1) positive cells demonstrated rhomboid microglia in saline-treated and non-specific IgG treated PbA-infected mice (arrows), some polarized to the perivascular region, surrounding congested vessels (Saline PbA arrowhead). Microglia from PHF-1 treated PbA-infected mice expressed a more ramified morphology, similar to their uninfected counterparts (arrows). (B) PHF-1 mAB treatment resulted in significantly lower levels of CD11b in whole brain lysates of infected mice (p < 0.05; q < 0.05) while non-specific IgG had no effect (p = ns; q = ns) on the expression of CD11b in PbA-infected mice 7 days post-infection when compared to saline-treated mice. (C-D) Both PHF-1 mAB and non-specific IgG treatment resulted in lower levels of Adgre1 mRNA (C) GFAP protein (D) expression in PbA-infected mice compared to untreated mice (p < 0.05; q < 0.05). Multiple comparisons were corrected using the false discovery rate (q). An α < 0.05 was considered significant for false discovery. PbA = Plasmodium berghei ANKA; PbN = Plasmodium berghei NK65; Un = uninfected; Adgre1 = Adhesion G Protein-Coupled Receptor E1; GFAP = Glial fibrillary acidic protein. GDI = Guanosine nucleotide dissociation inhibitor (housekeeping protein). Blots were stripped and re-probed with respective antibodies. * = false discovery rate (q) < 0.05; ns = false discovery rate (q) not significant. N = 7 mice/group.
Discussion
The pathogenesis of neuropsychiatric impairment with cerebral malaria is not completely understood. Reports in Ugandan children suggest an association between cognitive dysfunction and small vessel ischemic injury (Idro et al., 2016) and reveal a positive correlation with increased secretion of pro-inflammatory cytokines and chemokines in the cerebral spinal fluid (CSF; (John, Panoskaltsis-Mortari, et al., 2008)). Studies in both pediatric CM and ECM have reported that congestion of the brain and retinal vasculature (Brown, Hien, et al., 1999; Dorovini-Zis et al., 2011; Freeman et al., 2016; Martins et al., 2016), associated with endothelial damage, breakdown of the blood-brain barrier (BBB), leukocyte infiltration, and ultimately resulting in myelin and axonal damage (Brown, Hien, et al., 1999; Dorovini-Zis et al., 2011). The axonal protein, tau had previously been linked to CM in children and adults (Medana et al., 2007; Medana et al., 2005). Abnormal tau in the CSF and in the plasma have been linked to more severe disease and with neuropsychiatric dysfunction and coma in Ugandan children with CM (Datta et al., 2021; Datta et al., 2019). However, the link between vascular damage and tau pathology leading to neuropsychiatric dysfunction during CM has not been defined.
We previously demonstrated that changes to the cerebral microvasculature during ECM included vascular remodeling, recruitment of inflammatory cells and breach of the BBB (Dai et al., 2012; Freeman et al., 2016; Martins et al., 2016). These changes were the earliest changes observed in mice with ECM and preceded the development of memory impairment (Dai et al., 2010; Desruisseaux et al., 2008; Freeman et al., 2016). Our laboratory and others also previously reported that ECM induces astrocyte and microglia activation, and neuronal degeneration close to areas or sites of monocyte vascular adhesion (Ampawong et al., 2014; Desruisseaux et al., 2008). This has also been observed in fatal human disease (Medana et al., 2001). However, the implications of this topographical relationship have not been investigated. In this experiment, we demonstrate that PbA-induced endothelial activation resulted in the release of inflammatory mediators at intermediate stages of disease which worsened, in association with microglial activation, as disease progressed. In conjunction with the increase in the expression of inflammatory mediators, PbA-infected mice also exhibited astrogliosis during later stages of disease. Microglia are resident phagocytic cells of the CNS and key innate immune cells of the brain (Ampawong et al., 2014; Medana et al., 1997). Activated microglia have been shown to cause astrocyte activation and impair astrocytic function and coupling (Faustmann et al., 2003; Hwang et al., 2006; Kirkley et al., 2017). Stable microglial-astrocyte-neuronal structure and function are critical for normal neurocognitive function (Bano et al., 2011; Brundel et al., 2012; Crews & Masliah, 2010; Jellinger & Stadelmann, 2001; Okouchi et al., 2007). The data observed in our experiments suggest that the remodeling of endothelial cells during ECM primes the neurovascular unit for abluminal release of inflammatory mediators which induce microglial activation, and this in turn potentiates the neuroinflammatory response (Yenari et al., 2010).
Microglia are crucial in mediating the neuronal response to inflammatory insults (Pascual et al., 2012); thus, when activated, can both augment the neuroinflammatory response (Hwang et al., 2006) and provoke pathological changes in the CNS (Bartels et al., 2009; Benarroch, 2013; Bloom, 2014; Capuccini et al., 2016; Forman et al., 2005; Higuchi et al., 2002; Higuchi et al., 2005; Schwartz et al., 2013; Tunon-Ortiz & Lamb, 2019; Wilson et al., 2018; Zlokovic, 2008), including inducing neurodegeneration and facilitating the propagation of pathological tau protein in the brain (Bolos et al., 2017; Kempuraj et al., 2016; Maphis et al., 2015; Perea et al., 2018; Vogels et al., 2019).
In normal healthy brains, tau protein is expressed mostly in neurons, where it localizes to axons to promote axonal transport and neuronal integrity (Binder et al., 1985). Phosphorylation of tau at specific residues, such as Ser202/Thr205 and Ser396–404, occurs during tau inclusion formation (Mondragon-Rodriguez et al., 2014; Morishima-Kawashima, Hasegawa, Takio, Suzuki, Yoshida, Titani, et al., 1995; Morishima-Kawashima, Hasegawa, Takio, Suzuki, Yoshida, Watanabe, et al., 1995; Strang et al., 2017) and are strongly linked to neuronal injury neurodegeneration, and ultimately to deficits in cognitive function (Ballatore et al., 2007; Billingsley & Kincaid, 1997; Buee et al., 2000; Feany, 1996; Flament et al., 1990; Goedert et al., 1995; Iqbal et al., 2005; Kuret et al., 2005; Shahani & Brandt, 2002; Sorrentino & Bonavita, 2007; Trojanowski & Lee, 1994a, 1994b, 1995). Notably, several stimuli have been reported to result in an increase in pathological and extracellular tau, including ischemia (Pluta et al., 2018) and loss of essential nutrients (Mohamed et al., 2014). Cerebral endotheliopathy has long been associated with neurological impairment during CM and ECM (Baptista et al., 2010; Carter et al., 2005; Dorovini-Zis et al., 2011; Freeman et al., 2016; Greiner et al., 2015; Hochman et al., 2015; Idro, Marsh, et al., 2010; Kennan et al., 2005; Martins et al., 2016; Ponsford et al., 2012; Shikani et al., 2012; Thumwood et al., 1988; Wilson et al., 2018).
Conversely, neurodegenerative diseases are associated significant disruption of the cerebral vascular endothelium, even prior to the onset of symptoms (Govindpani et al., 2019). Tau protein is reported to play a vital role in the regulation of the neurovascular unit during physiological conditions (Michalicova et al., 2020). However, when abnormally regulated, pathological tau has been linked to vascular activation and remodeling (Merlini et al., 2016; Pluta et al., 2018; Vogels et al., 2019). Tau-mediated destabilization of the cerebral endothelium is thought to be induced by the activation of glial cells which results in deformities in the endothelial basement membrane, alteration of tight junctional expression and distribution, and an increase in the expression of cell adhesion molecules on the surface of endothelial cells (Kovac et al., 2011; Majerova et al., 2019), creating a neurotoxic feedback loop. This process likely plays an important role in the recruitment of leukocytes to the endothelium observed in our experiments. Our data presented herein suggest that blocking pathological tau with PHF-1 mAb and subsequent stabilization of glial cells mitigate tau-mediated action on the cerebral endothelium, including the recruitment of leukocytes.
Our laboratory was the first to report that during ECM, tau protein was abnormally hyperphosphorylated at pSer202 and Ser396–404, and that this was associated with long-term memory impairment (Dai et al., 2012). We reported higher levels of phosphorylated tau and misfolded tau in the brains of mice with ECM compared to uninfected mice (Dai et al., 2012). We also demonstrated that ECM resulted in abnormal activation of tyrosine kinases, including GSK3 which is known to phosphorylate tau (Dai et al., 2012). Activation of other kinases involved in tau phosphorylation, including MAP kinases, have also been reported with ECM (Anand et al., 2013; Patel et al., 2007). Inhibition of GSK3 with Lithium resulted in similar tau expression and phosphorylation as in uninfected mice and in preservation of memory (Dai et al., 2012). In corroboration with those data, we again demonstrated pathologically phosphorylated tau during advanced ECM in association with cognitive impairment. This was specific to ECM as it was not observed in mice infected with PbN, which does not induce ECM symptoms (Freeman et al., 2016; Martins et al., 2016). Likewise, abnormally elevated tau protein in the CSF of children with CM correlate with disease severity and with neuropsychiatric impairment (Datta et al., 2019). Preventing tau pathology has been proposed as a potential therapeutic strategy for AD-like diseases (Boutajangout et al., 2011; Gu et al., 2013; Otvos et al., 1994).
We previously reported that lithium treatment mitigated tau pathology and prevented cognitive dysfunction during ECM (Dai et al., 2012) via inhibition of glycogen synthase kinase 3β, a major kinase in the regulation of tau function (Noble et al., 2005). However the molecular effects of lithium are pleiotropic, targeting multiple cell survival and transcriptional pathways (Abdul et al., 2018; Akkouh et al., 2019; Malhi et al., 2013); thus, its therapeutic outcomes are broad and potentially harmful (Abdul et al., 2018; Malhi et al., 2013). In addition, reports show that lithium induces dose-dependent antiparasitic effects in rodent malaria models (Nurul Aiezzah et al., 2010). To avoid confounding off-target effects of lithium on tau-mediated cognitive dysfunction during ECM, we tested the ability of a monoclonal antibody against pathological tau, PHF-1 mAB, to modulate the course of ECM.
PHF-1 mAB is a monoclonal antibody which specifically targets pathological tau aggregates, phosphorylated at Ser396−404, and is capable of recruiting inert tau, causing their misfolding, and inducing neurotoxicity (DeVos et al., 2018; Gu et al., 2013; Otvos et al., 1994). Anti-tau antibodies, including PHF-1 mAB, target the proliferation of pathological tau facilitated by microglia in the brain (Andersson et al., 2019; Luo et al., 2015; Vogels et al., 2019). They have been shown to work by restoring normal microglial function and enhancing clearance of abnormal tau (Andersson et al., 2019; Luo et al., 2015; Vogels et al., 2019). Interestingly in our experiment PHF-1 mAb treatment on microglia was region-specific. Immunotherapy had no effects on infection induced morphological changes and temporal distribution of microglia in the brainstem, which contains the respiratory center, providing a potential pathophysiological explanation for the lack of effect of PHF-1 mAb on mortality in infected mice.
Anti-PHF-1 agents have no antimalarial properties to our knowledge, and this was seen in our model as tau immunization had no effect on parasitemia or mortality. Rather, in our experiments, the antibody is used to target pathological tau and to abrogate the neural cell injury that occurs with abnormal tau phosphorylation. Specifically targeting pathological tau by administration of PHF-1 mAB to PbA-infected mice prevented memory impairment due to ECM. The preservation of cognitive function was associated with concurrent mitigation of intravascular congestion in the brain and of abrogation of microglial activation resulting in a reduced neuroinflammatory response with PHF-1 mAB treatment of PbA-infected mice. Furthermore, treatment with PHF-1 mAB mitigated malaria-induced cerebral endotheliopathy, resulting in preservation of cognitive function. These data suggest that tau-mediated microglial dysfunction and vascular congestion during ECM are important drivers of neural cell toxicity and ensuing neurological deficits observed with disease.
In our experiments, PbA-induced astrocytosis occurred at later stages of disease, following the establishment of cognitive impairment. Aditionally, PHF-1 mAB treatment mediated mitigation of astrocytosis which were due to non-specific IgG effects. Thus, our data suggest, for the first time, that although astrocyte may be important in clearing parasite-derived vesicles during ECM during terminal disease (Shrivastava et al., 2017), these cells likely do not contribute to the ensuing cognitive impairment that occurs during ECM. Likewise, aside from TGF-β, PHF-1 mAB treatment had no effect on the expression of inflammatory cytokines and chemokines in the brain beyond those of non-specific IgG, and in fact had no effect at all on the expression of IL-1β or IL-1RA in the brains of infected mice. Interestingly, John et. al. previously reported that although admission levels of several cytokines and chemokines were elevated in the CSF of children with cerebral malaria, only TNF-α correlated with impaired cognitive function at 6-month follow-up (John, Panoskaltsis-Mortari, et al., 2008). This observation is supported by data presented herein. The lack of effect of immunotherapy on the expression of IL-1β and of IL-1RA suggests that these cytokines were not critical to the development of neurological pathology during ECM, consistent with previous reports in Ghanaian children showing that IL-1β and IL-1RA did not contribute to the development of severe malaria (Gyan et al., 2002).
The mechanisms that underlie neurocognitive dysfunction during CM are not entirely understood. Treatment with antimalarials eliminates the parasite burden but does not improve neurocognition (Bitta et al., 2017; Dai et al., 2010; Varo et al., 2018), prompting a need for the development of adjunct therapeutics that can mitigate or prevent the neuropsychiatric effects of CM. Tau has been linked to CM in children and adults (Datta et al., 2021; Datta et al., 2019; Medana et al., 2007; Medana et al., 2005), and in the data presented herein, is associated with abnormal glial profile and neuroinflammation which occur concurrently with cerebral endotheliopathy and vascular congestion, and ensuing in memory impairment. The precise mechanism for the development and progression of abnormal tau regulation during CM has not been well investigated. In these experiments, we report that PbA infection results in activation of the cerebral microvasculature and of microglia at intermediate stages of illness and this likely contributes to the propagation of abnormal tau and the associated neurologic deficits in PbA-infected mice. Tau antibody-mediated prevention or reduction of tau pathology in tauopathy models has been studied in AD mouse models (Boutajangout et al., 2011; Dai et al., 2018; Ittner et al., 2015; Ittner et al., 2010). The current study provides the first evidence that treatment with PHF-1 mAB prevented the endothelial and microglial activation associated with ECM, and that this led to mitigation memory impairment in PbA-infected mice. Further studies are needed to elucidate the precise effects of vascular insult on microglial clearance of abnormal tau during ECM as these interactions could provide useful molecular targets for potential neuroprotective therapy in human CM.
Human subjects: Involves human subjects:
If yes: Informed consent & ethics approval achieved:
if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
Supplementary Material
Acknowledgments:
The authors acknowledge Dr. Louis M. Weiss and the late Dr. Herbert Tanowitz for their invaluable advice and comments on the manuscript; Mr. Dazhi Zhao and Ms. Fengying Chen for considerable assistance with the maintenance, infection and treatment of the mice.
Funding Statement:
This work was supported by the National Institutes of Health: T32 NS007098–31 (HJS); F31 NS084746 (HJS); T32 AI070117 (BDF); R01 NS069577 (MSD). The Burroughs-Wellcome Fund Career Awards for Medical Scientists (MSD). Yale University Dean’s Diversity Initiative Faculty Excellence award (MSD).
List of abbreviations
- AD
Alzheimer’s Disease
- Adgre1
Adhesion G protein coupled receptor E1
- ANOVA
Analysis of variance
- BBB
Blood-brain barrier
- CM
Cerebral malaria
- CSF
Cerebral spinal fluid
- DPI
Days post infection
- ECM
Experimental cerebral malaria
- GFAP
Glial Fibrillary Acid Protein
- H&E
Hematoxylin and eosin
- IL
Interleukin
- IP
Intraperitoneally
- mAB
Monoclonal antibody
- PbA
Plasmodium berghei ANKA
- PbN
Plasmodium berghei NK65
- PBS
Phosphate-buffered saline
- PHF
Paired helical filament
- RBC
Red blood cell
- RRID
Research Resource Identifier (see scicrunch.org)
- TGF
Transforming growth factor
- Un
Uninfected
Footnotes
The authors declare no competing financial interests.
Data Availability Statement:
The authors confirm that the data supporting the findings of this study are available within the article. Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
References
- Abdul A, De Silva B, & Gary RK (2018). The GSK3 kinase inhibitor lithium produces unexpected hyperphosphorylation of beta-catenin, a GSK3 substrate, in human glioblastoma cells. Biol Open, 7(1). 10.1242/bio.030874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad MH, Fatima M, & Mondal AC (2019). Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer’s disease: Rational insights for the therapeutic approaches. J Clin Neurosci, 59, 6–11. 10.1016/j.jocn.2018.10.034 [DOI] [PubMed] [Google Scholar]
- Akkouh IA, Skrede S, Holmgren A, Ersland KM, Hansson L, Bahrami S, Andreassen OA, Steen VM, Djurovic S, & Hughes T (2019). Exploring lithium’s transcriptional mechanisms of action in bipolar disorder: a multi-step study. Neuropsychopharmacology. 10.1038/s41386-019-0556-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ampawong S, Chaisri U, Viriyavejakul P, Nontprasert A, Grau GE, & Pongponratn E (2014). Electron microscopic features of brain edema in rodent cerebral malaria in relation to glial fibrillary acidic protein expression. Int J Clin Exp Pathol, 7(5), 2056–2067. https://www.ncbi.nlm.nih.gov/pubmed/24966914 [PMC free article] [PubMed] [Google Scholar]
- Anand SS, Maruthi M, & Babu PP (2013). The specific, reversible JNK inhibitor SP600125 improves survivability and attenuates neuronal cell death in experimental cerebral malaria (ECM). Parasitol Res, 112(5), 1959–1966. 10.1007/s00436-013-3352-0 [DOI] [PubMed] [Google Scholar]
- Andersson CR, Falsig J, Stavenhagen JB, Christensen S, Kartberg F, Rosenqvist N, Finsen B, & Pedersen JT (2019). Antibody-mediated clearance of tau in primary mouse microglial cultures requires Fcgamma-receptor binding and functional lysosomes. Sci Rep, 9(1), 4658. 10.1038/s41598-019-41105-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, & Trojanowski JQ (2007). Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci, 8(9), 663–672. 10.1038/nrn2194 [DOI] [PubMed] [Google Scholar]
- Bano D, Agostini M, Melino G, & Nicotera P (2011). Ageing, neuronal connectivity and brain disorders: an unsolved ripple effect. Mol Neurobiol, 43(2), 124–130. 10.1007/s12035-011-8164-6 [DOI] [PubMed] [Google Scholar]
- Baptista FG, Pamplona A, Pena AC, Mota MM, Pied S, & Vigario AM (2010). Accumulation of Plasmodium berghei-infected red blood cells in the brain is crucial for the development of cerebral malaria in mice. Infect Immun, 78(9), 4033–4039. 10.1128/IAI.00079-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels AL, Kortekaas R, Bart J, Willemsen AT, de Klerk OL, de Vries JJ, van Oostrom JC, & Leenders KL (2009). Blood-brain barrier P-glycoprotein function decreases in specific brain regions with aging: a possible role in progressive neurodegeneration. Neurobiol Aging, 30(11), 1818–1824. 10.1016/j.neurobiolaging.2008.02.002 [DOI] [PubMed] [Google Scholar]
- Benarroch EE (2013). Microglia: Multiple roles in surveillance, circuit shaping, and response to injury. Neurology, 81(12), 1079–1088. 10.1212/WNL.0b013e3182a4a577 [DOI] [PubMed] [Google Scholar]
- Bendiske J, Caba E, Brown QB, & Bahr BA (2002). Intracellular deposition, microtubule destabilization, and transport failure: an “early” pathogenic cascade leading to synaptic decline. J Neuropathol Exp Neurol, 61(7), 640–650. 10.1093/jnen/61.7.640 [DOI] [PubMed] [Google Scholar]
- Billingsley ML, & Kincaid RL (1997). Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J, 323 (Pt 3), 577–591. https://www.ncbi.nlm.nih.gov/pubmed/9169588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, & Rebhun LI (1985). The distribution of tau in the mammalian central nervous system. J Cell Biol, 101(4), 1371–1378. 10.1083/jcb.101.4.1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitta MA, Kariuki SM, Mwita C, Gwer S, Mwai L, & Newton C (2017). Antimalarial drugs and the prevalence of mental and neurological manifestations: A systematic review and meta-analysis. Wellcome Open Res, 2, 13. 10.12688/wellcomeopenres.10658.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom GS (2014). Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol, 71(4), 505–508. 10.1001/jamaneurol.2013.5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin MJ, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM, & John CC (2007). Cognitive impairment after cerebral malaria in children: a prospective study. Pediatrics, 119(2), e360–366. 10.1542/peds.2006-2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolos M, Llorens-Martin M, Perea JR, Jurado-Arjona J, Rabano A, Hernandez F, & Avila J (2017). Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol Neurodegener, 12(1), 59. 10.1186/s13024-017-0200-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutajangout A, Ingadottir J, Davies P, & Sigurdsson EM (2011). Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem, 118(4), 658–667. 10.1111/j.1471-4159.2011.07337.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown H, Hien TT, Day N, Mai NT, Chuong LV, Chau TT, Loc PP, Phu NH, Bethell D, Farrar J, Gatter K, White N, & Turner G (1999). Evidence of blood-brain barrier dysfunction in human cerebral malaria. Neuropathol Appl Neurobiol, 25(4), 331–340. https://www.ncbi.nlm.nih.gov/pubmed/10476050 [DOI] [PubMed] [Google Scholar]
- Brown H, Turner G, Rogerson S, Tembo M, Mwenechanya J, Molyneux M, & Taylor T (1999). Cytokine expression in the brain in human cerebral malaria. J Infect Dis, 180(5), 1742–1746. 10.1086/315078 [DOI] [PubMed] [Google Scholar]
- Brundel M, de Bresser J, van Dillen JJ, Kappelle LJ, & Biessels GJ (2012). Cerebral microinfarcts: a systematic review of neuropathological studies. J Cereb Blood Flow Metab, 32(3), 425–436. 10.1038/jcbfm.2011.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden KR, Trojanowski JQ, & Lee VM (2008). Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J Alzheimers Dis, 14(4), 393–399. 10.3233/jad-2008-14406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, & Hof PR (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev, 33(1), 95–130. https://www.ncbi.nlm.nih.gov/pubmed/10967355 [DOI] [PubMed] [Google Scholar]
- Busciglio J, Lorenzo A, Yeh J, & Yankner BA (1995). beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron, 14(4), 879–888. https://www.ncbi.nlm.nih.gov/pubmed/7718249 [DOI] [PubMed] [Google Scholar]
- Capuccini B, Lin J, Talavera-Lopez C, Khan SM, Sodenkamp J, Spaccapelo R, & Langhorne J (2016). Transcriptomic profiling of microglia reveals signatures of cell activation and immune response, during experimental cerebral malaria. Sci Rep, 6, 39258. 10.1038/srep39258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RW, Wainwright MS, Kim K-Y, Kidambi T, Gómez ND, Taylor T, & Haldar K (2010). A Rapid Murine Coma and Behavior Scale for Quantitative Assessment of Murine Cerebral Malaria. PLoS One, 5(10), e13124. 10.1371/journal.pone.0013124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter JA, Lees JA, Gona JK, Murira G, Rimba K, Neville BG, & Newton CR (2006). Severe falciparum malaria and acquired childhood language disorder. Dev Med Child Neurol, 48(1), 51–57. 10.1017/S0012162206000107 [DOI] [PubMed] [Google Scholar]
- Carter JA, Mung’ala-Odera V, Neville BG, Murira G, Mturi N, Musumba C, & Newton CR (2005). Persistent neurocognitive impairments associated with severe falciparum malaria in Kenyan children. J Neurol Neurosurg Psychiatry, 76(4), 476–481. 10.1136/jnnp.2004.043893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, Buckner N, Hanmer J, Davies P, O’Neill MJ, Hutton ML, & Citron M (2011). Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem, 286(39), 34457–34467. 10.1074/jbc.M111.229633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L, & Masliah E (2010). Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet, 19(R1), R12–20. 10.1093/hmg/ddq160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai CL, Hu W, Tung YC, Liu F, Gong CX, & Iqbal K (2018). Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated Tau-induced pathology in 3 x Tg-AD mice. Alzheimers Res Ther, 10(1), 13. 10.1186/s13195-018-0341-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai CL, Tung YC, Liu F, Gong CX, & Iqbal K (2017). Tau passive immunization inhibits not only tau but also Abeta pathology. Alzheimers Res Ther, 9(1), 1. 10.1186/s13195-016-0227-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Freeman B, Shikani HJ, Bruno FP, Collado JE, Macias R, Reznik SE, Davies P, Spray DC, Tanowitz HB, Weiss LM, & Desruisseaux MS (2012). Altered regulation of Akt signaling with murine cerebral malaria, effects on long-term neuro-cognitive function, restoration with lithium treatment [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. PLoS One, 7(10), e44117. 10.1371/journal.pone.0044117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M, Reznik SE, Spray DC, Weiss LM, Tanowitz HB, Gulinello M, & Desruisseaux MS (2010). Persistent cognitive and motor deficits after successful antimalarial treatment in murine cerebral malaria. Microbes Infect, 12(14–15), 1198–1207. 10.1016/j.micinf.2010.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta D, Bangirana P, Opoka RO, Conroy AL, Co K, Bond C, Zhao Y, Kawata K, Saykin AJ, & John CC (2021). Association of Plasma Tau With Mortality and Long-term Neurocognitive Impairment in Survivors of Pediatric Cerebral Malaria and Severe Malarial Anemia. JAMA Netw Open, 4(12), e2138515. 10.1001/jamanetworkopen.2021.38515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta D, Conroy AL, Castelluccio PF, Ssenkusu JM, Park GS, Opoka RO, Bangirana P, Idro R, Saykin AJ, & John CC (2019). Elevated cerebrospinal fluid tau protein concentrations on admission are associated with long-term neurologic and cognitive impairment in Ugandan children with cerebral malaria. Clin Infect Dis. 10.1093/cid/ciz325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kossodo S, & Grau GE (1993). Role of cytokines and adhesion molecules in malaria immunopathology. Stem Cells, 11(1), 41–48. 10.1002/stem.5530110108 [DOI] [PubMed] [Google Scholar]
- de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, Breimer DD, & Kuiper J (1996). The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol, 64(1), 37–43. https://www.ncbi.nlm.nih.gov/pubmed/8598388 [DOI] [PubMed] [Google Scholar]
- Desruisseaux MS, Gulinello M, Smith DN, Lee SC, Tsuji M, Weiss LM, Spray DC, & Tanowitz HB (2008). Cognitive dysfunction in mice infected with Plasmodium berghei strain ANKA [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. J Infect Dis, 197(11), 1621–1627. 10.1086/587908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Corjuc BT, Oakley DH, Nobuhara CK, Bannon RN, Chase A, Commins C, Gonzalez JA, Dooley PM, Frosch MP, & Hyman BT (2018). Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain [Original Research]. Frontiers in Neuroscience, 12(267). 10.3389/fnins.2018.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorovini-Zis K, Schmidt K, Huynh H, Fu W, Whitten RO, Milner D, Kamiza S, Molyneux M, & Taylor TE (2011). The neuropathology of fatal cerebral malaria in malawian children [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Am J Pathol, 178(5), 2146–2158. 10.1016/j.ajpath.2011.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennaceur A, & Meliani K (1992). A new one-trial test for neurobiological studies of memory in rats. III. Spatial vs. non-spatial working memory. Behav Brain Res, 51(1), 83–92. http://www.ncbi.nlm.nih.gov/pubmed/1482548 [DOI] [PubMed] [Google Scholar]
- Fakhoury M (2018). Microglia and Astrocytes in Alzheimer’s Disease: Implications for Therapy. Curr Neuropharmacol, 16(5), 508–518. 10.2174/1570159X15666170720095240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfara D, Lifshitz V, & Frenkel D (2008). Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer’s disease. J Cell Mol Med, 12(3), 762–780. 10.1111/j.1582-4934.2008.00314.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauconnier M, Palomo J, Bourigault ML, Meme S, Szeremeta F, Beloeil JC, Danneels A, Charron S, Rihet P, Ryffel B, & Quesniaux VF (2012). IL-12Rbeta2 is essential for the development of experimental cerebral malaria. J Immunol, 188(4), 1905–1914. 10.4049/jimmunol.1101978 [DOI] [PubMed] [Google Scholar]
- Faustmann PM, Haase CG, Romberg S, Hinkerohe D, Szlachta D, Smikalla D, Krause D, & Dermietzel R (2003). Microglia activation influences dye coupling and Cx43 expression of the astrocytic network. Glia, 42(2), 101–108. 10.1002/glia.10141 [DOI] [PubMed] [Google Scholar]
- Feany MB (1996). Neuropeptide modulation of learning and memory processes. Rev Neurosci, 7(2), 151–164. 10.1515/revneuro.1996.7.2.151 [DOI] [PubMed] [Google Scholar]
- Flament S, Delacourte A, & Mann DM (1990). Phosphorylation of Tau proteins: a major event during the process of neurofibrillary degeneration. A comparative study between Alzheimer’s disease and Down’s syndrome. Brain Res, 516(1), 15–19. https://www.ncbi.nlm.nih.gov/pubmed/2142011 [DOI] [PubMed] [Google Scholar]
- Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VM, & Trojanowski JQ (2005). Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosci, 25(14), 3539–3550. 10.1523/JNEUROSCI.0081-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman BD, Martins YC, Akide-Ndunge OB, Bruno FP, Wang H, Tanowitz HB, Spray DC, & Desruisseaux MS (2016). Endothelin-1 Mediates Brain Microvascular Dysfunction Leading to Long-Term Cognitive Impairment in a Model of Experimental Cerebral Malaria. PLoS Pathog, 12(3), e1005477. 10.1371/journal.ppat.1005477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Crowther RA, Vanmechelen E, Probst A, Gotz J, Burki K, & Cohen P (1995). Molecular dissection of the paired helical filament. Neurobiol Aging, 16(3), 325–334. https://www.ncbi.nlm.nih.gov/pubmed/7566342 [DOI] [PubMed] [Google Scholar]
- Govindpani K, McNamara LG, Smith NR, Vinnakota C, Waldvogel HJ, Faull RL, & Kwakowsky A (2019). Vascular Dysfunction in Alzheimer’s Disease: A Prelude to the Pathological Process or a Consequence of It? J Clin Med, 8(5). 10.3390/jcm8050651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner J, Dorovini-Zis K, Taylor TE, Molyneux ME, Beare NA, Kamiza S, & White VA (2015). Correlation of hemorrhage, axonal damage, and blood-tissue barrier disruption in brain and retina of Malawian children with fatal cerebral malaria. Front Cell Infect Microbiol, 5, 18. 10.3389/fcimb.2015.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Congdon EE, & Sigurdsson EM (2013). Two novel Tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce Tau protein pathology. J Biol Chem, 288(46), 33081–33095. 10.1074/jbc.M113.494922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyan B, Goka B, Cvetkovic JT, Perlmann H, Lefvert AK, Akanmori B, & Troye-Blomberg M (2002). Polymorphisms in interleukin-1beta and interleukin-1 receptor antagonist genes and malaria in Ghanaian children. Scand J Immunol, 56(6), 619–622. 10.1046/j.1365-3083.2002.01161.x [DOI] [PubMed] [Google Scholar]
- Hanger DP, Lau DH, Phillips EC, Bondulich MK, Guo T, Woodward BW, Pooler AM, & Noble W (2014). Intracellular and extracellular roles for tau in neurodegenerative disease. J Alzheimers Dis, 40 Suppl 1, S37–45. 10.3233/JAD-132054 [DOI] [PubMed] [Google Scholar]
- Higuchi M, Ishihara T, Zhang B, Hong M, Andreadis A, Trojanowski J, & Lee VM (2002). Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron, 35(3), 433–446. 10.1016/s0896-6273(02)00789-4 [DOI] [PubMed] [Google Scholar]
- Higuchi M, Zhang B, Forman MS, Yoshiyama Y, Trojanowski JQ, & Lee VM (2005). Axonal degeneration induced by targeted expression of mutant human tau in oligodendrocytes of transgenic mice that model glial tauopathies. J Neurosci, 25(41), 9434–9443. 10.1523/JNEUROSCI.2691-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochman SE, Madaline TF, Wassmer SC, Mbale E, Choi N, Seydel KB, Whitten RO, Varughese J, Grau GE, Kamiza S, Molyneux ME, Taylor TE, Lee S, Milner DA Jr., & Kim K (2015). Fatal Pediatric Cerebral Malaria Is Associated with Intravascular Monocytes and Platelets That Are Increased with HIV Coinfection. MBio, 6(5), e01390–01315. 10.1128/mBio.01390-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg D, Franzen-Rohl E, Idro R, Opoka RO, Bangirana P, Sellgren CM, Wickstrom R, Farnert A, Schwieler L, Engberg G, & John CC (2017). Cerebrospinal fluid kynurenine and kynurenic acid concentrations are associated with coma duration and long-term neurocognitive impairment in Ugandan children with cerebral malaria. Malar J, 16(1), 303. 10.1186/s12936-017-1954-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe CL, Lafrance-Corey RG, Sundsbak RS, & Lafrance SJ (2012). Inflammatory monocytes damage the hippocampus during acute picornavirus infection of the brain. J Neuroinflammation, 9, 50. 10.1186/1742-2094-9-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SY, Jung JS, Kim TH, Lim SJ, Oh ES, Kim JY, Ji KA, Joe EH, Cho KH, & Han IO (2006). Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol Dis, 21(3), 457–467. 10.1016/j.nbd.2005.08.006 [DOI] [PubMed] [Google Scholar]
- Idro R, Kakooza-Mwesige A, Asea B, Ssebyala K, Bangirana P, Opoka RO, Lubowa SK, Semrud-Clikeman M, John CC, & Nalugya J (2016). Cerebral malaria is associated with long-term mental health disorders: a cross sectional survey of a long-term cohort. Malar J, 15(1), 184. 10.1186/s12936-016-1233-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idro R, Kakooza-Mwesige A, Balyejjussa S, Mirembe G, Mugasha C, Tugumisirize J, & Byarugaba J (2010). Severe neurological sequelae and behaviour problems after cerebral malaria in Ugandan children. BMC Res Notes, 3, 104. 10.1186/1756-0500-3-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idro R, Marsh K, John CC, & Newton CR (2010). Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review]. Pediatr Res, 68(4), 267–274. https://doi.org/10.1203/00006450-201011001-00524 10.1203/PDR.0b013e3181eee738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihara Y, Nukina N, Miura R, & Ogawara M (1986). Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J Biochem, 99(6), 1807–1810. 10.1093/oxfordjournals.jbchem.a135662 [DOI] [PubMed] [Google Scholar]
- Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, & Grundke-Iqbal I (2005). Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta, 1739(2–3), 198–210. 10.1016/j.bbadis.2004.09.008 [DOI] [PubMed] [Google Scholar]
- Ishiki A, Kamada M, Kawamura Y, Terao C, Shimoda F, Tomita N, Arai H, & Furukawa K (2016). Glial fibrillar acidic protein in the cerebrospinal fluid of Alzheimer’s disease, dementia with Lewy bodies, and frontotemporal lobar degeneration. J Neurochem, 136(2), 258–261. 10.1111/jnc.13399 [DOI] [PubMed] [Google Scholar]
- Ittner A, Bertz J, Suh LS, Stevens CH, Gotz J, & Ittner LM (2015). Tau-targeting passive immunization modulates aspects of pathology in tau transgenic mice. J Neurochem, 132(1), 135–145. 10.1111/jnc.12821 [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, & Gotz J (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell, 142(3), 387–397. 10.1016/j.cell.2010.06.036 [DOI] [PubMed] [Google Scholar]
- Jakobsen PH, McKay V, Morris-Jones SD, McGuire W, van Hensbroek MB, Meisner S, Bendtzen K, Schousboe I, Bygbjerg IC, & Greenwood BM (1994). Increased concentrations of interleukin-6 and interleukin-1 receptor antagonist and decreased concentrations of beta-2-glycoprotein I in Gambian children with cerebral malaria. Infect Immun, 62(10), 4374–4379. https://www.ncbi.nlm.nih.gov/pubmed/7927698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, & Stadelmann C (2001). Problems of cell death in neurodegeneration and Alzheimer’s Disease. J Alzheimers Dis, 3(1), 31–40. https://www.ncbi.nlm.nih.gov/pubmed/12214070 [DOI] [PubMed] [Google Scholar]
- John CC, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM, Wu B, & Boivin MJ (2008). Cerebral malaria in children is associated with long-term cognitive impairment. Pediatrics, 122(1), e92–99. 10.1542/peds.2007-3709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John CC, Panoskaltsis-Mortari A, Opoka RO, Park GS, Orchard PJ, Jurek AM, Idro R, Byarugaba J, & Boivin MJ (2008). Cerebrospinal fluid cytokine levels and cognitive impairment in cerebral malaria. Am J Trop Med Hyg, 78(2), 198–205. http://www.ncbi.nlm.nih.gov/pubmed/18256412 [PMC free article] [PubMed] [Google Scholar]
- John CC, Park GS, Sam-Agudu N, Opoka RO, & Boivin MJ (2008). Elevated serum levels of IL-1ra in children with Plasmodium falciparum malaria are associated with increased severity of disease. Cytokine, 41(3), 204–208. 10.1016/j.cyto.2007.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, Zaheer S, Iyer SS, & Zaheer A (2016). Neuroinflammation Induces Neurodegeneration. J Neurol Neurosurg Spine, 1(1). https://www.ncbi.nlm.nih.gov/pubmed/28127589 [PMC free article] [PubMed] [Google Scholar]
- Kennan RP, Machado FS, Lee SC, Desruisseaux MS, Wittner M, Tsuji M, & Tanowitz HB (2005). Reduced cerebral blood flow and N-acetyl aspartate in a murine model of cerebral malaria. Parasitol Res, 96(5), 302–307. 10.1007/s00436-005-1349-z [DOI] [PubMed] [Google Scholar]
- Kirkley KS, Popichak KA, Afzali MF, Legare ME, & Tjalkens RB (2017). Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J Neuroinflammation, 14(1), 99. 10.1186/s12974-017-0871-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, & Selkoe DJ (1986). Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A, 83(11), 4044–4048. 10.1073/pnas.83.11.4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac A, Zilka N, Kazmerova Z, Cente M, Zilkova M, & Novak M (2011). Misfolded truncated protein tau induces innate immune response via MAPK pathway. J Immunol, 187(5), 2732–2739. 10.4049/jimmunol.1100216 [DOI] [PubMed] [Google Scholar]
- Kuret J, Congdon EE, Li G, Yin H, Yu X, & Zhong Q (2005). Evaluating triggers and enhancers of tau fibrillization. Microsc Res Tech, 67(3–4), 141–155. 10.1002/jemt.20187 [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, & Trojanowski JQ (2001). Neurodegenerative tauopathies. Annu Rev Neurosci, 24, 1121–1159. 10.1146/annurev.neuro.24.1.1121 [DOI] [PubMed] [Google Scholar]
- Leyns CEG, & Holtzman DM (2017). Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener, 12(1), 50. 10.1186/s13024-017-0192-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Zhao L, Blackman B, Parmar M, Wong MY, Woo T, Yu F, Chiuchiolo MJ, Sondhi D, Kaminsky SM, Crystal RG, & Paul SM (2016). Vectored Intracerebral Immunization with the Anti-Tau Monoclonal Antibody PHF1 Markedly Reduces Tau Pathology in Mutant Tau Transgenic Mice. J Neurosci, 36(49), 12425–12435. 10.1523/JNEUROSCI.2016-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, & Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25(4), 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Luo W, Liu W, Hu X, Hanna M, Caravaca A, & Paul SM (2015). Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep, 5, 11161. 10.1038/srep11161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyke KE, Burges R, Cissoko Y, Sangare L, Dao M, Diarra I, Kone A, Harley R, Plowe CV, Doumbo OK, & Sztein MB (2004). Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect Immun, 72(10), 5630–5637. 10.1128/IAI.72.10.5630-5637.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerova P, Michalicova A, Cente M, Hanes J, Vegh J, Kittel A, Kosikova N, Cigankova V, Mihaljevic S, Jadhav S, & Kovac A (2019). Trafficking of immune cells across the blood-brain barrier is modulated by neurofibrillary pathology in tauopathies. PLoS One, 14(5), e0217216. 10.1371/journal.pone.0217216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi GS, Tanious M, Das P, Coulston CM, & Berk M (2013). Potential mechanisms of action of lithium in bipolar disorder. Current understanding. CNS Drugs, 27(2), 135–153. 10.1007/s40263-013-0039-0 [DOI] [PubMed] [Google Scholar]
- Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, & Bhaskar K (2015). Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain, 138(Pt 6), 1738–1755. 10.1093/brain/awv081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins YC, Freeman BD, Akide Ndunge OB, Weiss LM, Tanowitz HB, & Desruisseaux MS (2016). Endothelin-1 Treatment Induces an Experimental Cerebral Malaria-Like Syndrome in C57BL/6 Mice Infected with Plasmodium berghei NK65. Am J Pathol, 186(11), 2957–2969. 10.1016/j.ajpath.2016.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medana IM, Chaudhri G, Chan-Ling T, & Hunt NH (2001). Central nervous system in cerebral malaria: ‘Innocent bystander’ or active participant in the induction of immunopathology? Immunol Cell Biol, 79(2), 101–120. 10.1046/j.1440-1711.2001.00995.x [DOI] [PubMed] [Google Scholar]
- Medana IM, Hunt NH, & Chan-Ling T (1997). Early activation of microglia in the pathogenesis of fatal murine cerebral malaria. Glia, 19(2), 91–103. https://www.ncbi.nlm.nih.gov/pubmed/9034826 [DOI] [PubMed] [Google Scholar]
- Medana IM, Idro R, & Newton CR (2007). Axonal and astrocyte injury markers in the cerebrospinal fluid of Kenyan children with severe malaria [Research Support, Non-U.S. Gov’t]. J Neurol Sci, 258(1–2), 93–98. 10.1016/j.jns.2007.03.005 [DOI] [PubMed] [Google Scholar]
- Medana IM, Lindert RB, Wurster U, Hien TT, Day NP, Phu NH, Mai NT, Chuong LV, Chau TT, Turner GD, Farrar JJ, & White NJ (2005). Cerebrospinal fluid levels of markers of brain parenchymal damage in Vietnamese adults with severe malaria. Trans R Soc Trop Med Hyg, 99(8), 610–617. 10.1016/j.trstmh.2004.11.017 [DOI] [PubMed] [Google Scholar]
- Medana IM, & Turner GD (2006). Human cerebral malaria and the blood-brain barrier. Int J Parasitol, 36(5), 555–568. 10.1016/j.ijpara.2006.02.004 [DOI] [PubMed] [Google Scholar]
- Medina M (2018). An Overview on the Clinical Development of Tau-Based Therapeutics. Int J Mol Sci, 19(4). 10.3390/ijms19041160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlini M, Wanner D, & Nitsch RM (2016). Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer’s disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol, 131(5), 737–752. 10.1007/s00401-016-1560-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalicova A, Majerova P, & Kovac A (2020). Tau Protein and Its Role in Blood-Brain Barrier Dysfunction. Front Mol Neurosci, 13, 570045. 10.3389/fnmol.2020.570045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middeldorp J, & Hol EM (2011). GFAP in health and disease. Prog Neurobiol, 93(3), 421–443. 10.1016/j.pneurobio.2011.01.005 [DOI] [PubMed] [Google Scholar]
- Mohamed NV, Plouffe V, Remillard-Labrosse G, Planel E, & Leclerc N (2014). Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci Rep, 4, 5715. 10.1038/srep05715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragon-Rodriguez S, Perry G, Luna-Munoz J, Acevedo-Aquino MC, & Williams S (2014). Phosphorylation of tau protein at sites Ser(396–404) is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol Appl Neurobiol, 40(2), 121–135. 10.1111/nan.12084 [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, & Ihara Y (1995). Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem, 270(2), 823–829. 10.1074/jbc.270.2.823 [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Watanabe A, Titani K, & Ihara Y (1995). Hyperphosphorylation of tau in PHF. Neurobiol Aging, 16(3), 365–371; discussion 371–380. 10.1016/0197-4580(95)00027-c [DOI] [PubMed] [Google Scholar]
- Murphy SC, & Breman JG (2001). Gaps in the childhood malaria burden in Africa: cerebral malaria, neurological sequelae, anemia, respiratory distress, hypoglycemia, and complications of pregnancy. Am J Trop Med Hyg, 64(1–2 Suppl), 57–67. 10.4269/ajtmh.2001.64.57 [DOI] [PubMed] [Google Scholar]
- Neddens J, Temmel M, Flunkert S, Kerschbaumer B, Hoeller C, Loeffler T, Niederkofler V, Daum G, Attems J, & Hutter-Paier B (2018). Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol Commun, 6(1), 52. 10.1186/s40478-018-0557-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton CR, & Krishna S (1998). Severe falciparum malaria in children: current understanding of pathophysiology and supportive treatment. Pharmacol Ther, 79(1), 1–53. http://www.ncbi.nlm.nih.gov/pubmed/9719344 [DOI] [PubMed] [Google Scholar]
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, Gaynor K, Wang L, LaFrancois J, Feinstein B, Burns M, Krishnamurthy P, Wen Y, Bhat R, Lewis J, Dickson D, & Duff K (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A, 102(19), 6990–6995. https://doi.org/0500466102 [pii] 10.1073/pnas.0500466102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurul Aiezzah Z, Noor E, & Hasidah MS (2010). Suppression of Plasmodium berghei parasitemia by LiCl in an animal infection model. Trop Biomed, 27(3), 624–631. https://www.ncbi.nlm.nih.gov/pubmed/21399604 [PubMed] [Google Scholar]
- Oakley MS, Chorazeczewski JK, Aleshnick M, Anantharaman V, Majam V, Chawla B, Myers TG, Su Q, Okoth WA, Takeda K, Akue A, KuKuruga M, Aravind L, & Kumar S (2018). TCRbeta-expressing macrophages induced by a pathogenic murine malaria correlate with parasite burden and enhanced phagocytic activity. PLoS One, 13(7), e0201043. 10.1371/journal.pone.0201043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okouchi M, Ekshyyan O, Maracine M, & Aw TY (2007). Neuronal apoptosis in neurodegeneration. Antioxid Redox Signal, 9(8), 1059–1096. 10.1089/ars.2007.1511 [DOI] [PubMed] [Google Scholar]
- Otvos L Jr., Feiner L, Lang E, Szendrei GI, Goedert M, & Lee VM (1994). Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res, 39(6), 669–673. 10.1002/jnr.490390607 [DOI] [PubMed] [Google Scholar]
- Pascual O, Ben Achour S, Rostaing P, Triller A, & Bessis A (2012). Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A, 109(4), E197–205. 10.1073/pnas.1111098109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SN, Lu Z, Ayi K, Serghides L, Gowda DC, & Kain KC (2007). Disruption of CD36 impairs cytokine response to Plasmodium falciparum glycosylphosphatidylinositol and confers susceptibility to severe and fatal malaria in vivo. J Immunol, 178(6), 3954–3961. 10.4049/jimmunol.178.6.3954 [DOI] [PubMed] [Google Scholar]
- Perea JR, Llorens-Martin M, Avila J, & Bolos M (2018). The Role of Microglia in the Spread of Tau: Relevance for Tauopathies. Front Cell Neurosci, 12, 172. 10.3389/fncel.2018.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta R, Ulamek-Koziol M, Januszewski S, & Czuczwar SJ (2018). Tau Protein Dysfunction after Brain Ischemia. J Alzheimers Dis, 66(2), 429–437. 10.3233/JAD-180772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsford MJ, Medana IM, Prapansilp P, Hien TT, Lee SJ, Dondorp AM, Esiri MM, Day NP, White NJ, & Turner GD (2012). Sequestration and microvascular congestion are associated with coma in human cerebral malaria. J Infect Dis, 205(4), 663–671. 10.1093/infdis/jir812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentsendorj A, Sheyn J, Fuchs DT, Daley D, Salumbides BC, Schubloom HE, Hart NJ, Li S, Hayden EY, Teplow DB, Black KL, Koronyo Y, & Koronyo-Hamaoui M (2018). A novel role for osteopontin in macrophage-mediated amyloid-beta clearance in Alzheimer’s models. Brain Behav Immun, 67, 163–180. 10.1016/j.bbi.2017.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Mejias E, Navarro V, Jimenez S, Sanchez-Mico M, Sanchez-Varo R, Nunez-Diaz C, Trujillo-Estrada L, Davila JC, Vizuete M, Gutierrez A, & Vitorica J (2016). Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol, 132(6), 897–916. 10.1007/s00401-016-1630-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Barten DM, Vana L, Devidze N, Yang L, Cadelina G, Hoque N, DeCarr L, Keenan S, Lin A, Cao Y, Snyder B, Zhang B, Nitla M, Hirschfeld G, Barrezueta N, Polson C, Wes P, Rangan VS, … Ahlijanian M (2015). Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS One, 10(5), e0125614. 10.1371/journal.pone.0125614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Kipnis J, Rivest S, & Prat A (2013). How do immune cells support and shape the brain in health, disease, and aging? J Neurosci, 33(45), 17587–17596. 10.1523/JNEUROSCI.3241-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senanayake N, & de Silva HJ (1994). Delayed cerebellar ataxia complicating falciparum malaria: a clinical study of 74 patients. J Neurol, 241(7), 456–459. https://www.ncbi.nlm.nih.gov/pubmed/7931448 [DOI] [PubMed] [Google Scholar]
- Shabani E, Ouma BJ, Idro R, Bangirana P, Opoka RO, Park GS, Conroy AL, & John CC (2017). Elevated cerebrospinal fluid tumour necrosis factor is associated with acute and long-term neurocognitive impairment in cerebral malaria. Parasite Immunol, 39(7). 10.1111/pim.12438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahani N, & Brandt R (2002). Functions and malfunctions of the tau proteins. Cell Mol Life Sci, 59(10), 1668–1680. https://www.ncbi.nlm.nih.gov/pubmed/12475178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikani HJ, Freeman BD, Lisanti MP, Weiss LM, Tanowitz HB, & Desruisseaux MS (2012). Cerebral malaria: we have come a long way. Am J Pathol, 181(5), 1484–1492. 10.1016/j.ajpath.2012.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava SK, Dalko E, Delcroix-Genete D, Herbert F, Cazenave PA, & Pied S (2017). Uptake of parasite-derived vesicles by astrocytes and microglial phagocytosis of infected erythrocytes may drive neuroinflammation in cerebral malaria. Glia, 65(1), 75–92. 10.1002/glia.23075 [DOI] [PubMed] [Google Scholar]
- Sorrentino G, & Bonavita V (2007). Neurodegeneration and Alzheimer’s disease: the lesson from tauopathies. Neurol Sci, 28(2), 63–71. 10.1007/s10072-007-0789-x [DOI] [PubMed] [Google Scholar]
- Strang KH, Goodwin MS, Riffe C, Moore BD, Chakrabarty P, Levites Y, Golde TE, & Giasson BI (2017). Generation and characterization of new monoclonal antibodies targeting the PHF1 and AT8 epitopes on human tau. Acta Neuropathol Commun, 5(1), 58. 10.1186/s40478-017-0458-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thumwood CM, Hunt NH, Clark IA, & Cowden WB (1988). Breakdown of the blood-brain barrier in murine cerebral malaria. Parasitology, 96 (Pt 3), 579–589. 10.1017/s0031182000080203 [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, & Lee VM (1994a). Paired helical filament tau in Alzheimer’s disease. The kinase connection. Am J Pathol, 144(3), 449–453. https://www.ncbi.nlm.nih.gov/pubmed/8129030 [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ, & Lee VM (1994b). Phosphorylation of neuronal cytoskeletal proteins in Alzheimer’s disease and Lewy body dementias. Ann N Y Acad Sci, 747, 92–109. 10.1111/j.1749-6632.1994.tb44403.x [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, & Lee VM (1995). Phosphorylation of paired helical filament tau in Alzheimer’s disease neurofibrillary lesions: focusing on phosphatases. FASEB J, 9(15), 1570–1576. https://www.ncbi.nlm.nih.gov/pubmed/8529836 [DOI] [PubMed] [Google Scholar]
- Tunon-Ortiz A, & Lamb TJ (2019). Blood brain barrier disruption in cerebral malaria: Beyond endothelial cell activation. PLoS Pathog, 15(6), e1007786. 10.1371/journal.ppat.1007786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Steen PE, Geurts N, Deroost K, Van Aelst I, Verhenne S, Heremans H, Van Damme J, & Opdenakker G (2010). Immunopathology and dexamethasone therapy in a new model for malaria-associated acute respiratory distress syndrome. Am J Respir Crit Care Med, 181(9), 957–968. 10.1164/rccm.200905-0786OC [DOI] [PubMed] [Google Scholar]
- Van den Steen PE, Van Aelst I, Starckx S, Maskos K, Opdenakker G, & Pagenstecher A (2006). Matrix metalloproteinases, tissue inhibitors of MMPs and TACE in experimental cerebral malaria [Research Support, Non-U.S. Gov’t]. Lab Invest, 86(9), 873–888. 10.1038/labinvest.3700454 [DOI] [PubMed] [Google Scholar]
- Varo R, Crowley VM, Sitoe A, Madrid L, Serghides L, Kain KC, & Bassat Q (2018). Adjunctive therapy for severe malaria: a review and critical appraisal. Malar J, 17(1), 47. 10.1186/s12936-018-2195-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogels T, Murgoci AN, & Hromadka T (2019). Intersection of pathological tau and microglia at the synapse. Acta Neuropathol Commun, 7(1), 109. 10.1186/s40478-019-0754-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KD, Ochoa LF, Solomon OD, Pal R, Cardona SM, Carpio VH, Keiser PH, Cardona AE, Vargas G, & Stephens R (2018). Elimination of intravascular thrombi prevents early mortality and reduces gliosis in hyper-inflammatory experimental cerebral malaria. J Neuroinflammation, 15(1), 173. 10.1186/s12974-018-1207-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, Cirrito JR, Patel TK, Hochgrafe K, Mandelkow EM, & Holtzman DM (2014). Neuronal activity regulates extracellular tau in vivo. J Exp Med, 211(3), 387–393. 10.1084/jem.20131685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenari MA, Kauppinen TM, & Swanson RA (2010). Microglial activation in stroke: therapeutic targets. Neurotherapeutics, 7(4), 378–391. 10.1016/j.nurt.2010.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron, 57(2), 178–201. 10.1016/j.neuron.2008.01.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article. Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.