

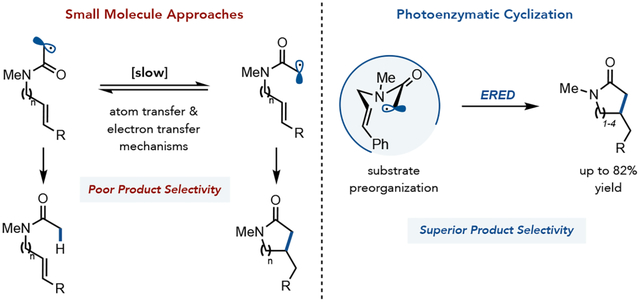
Abstract
Reductive radical cyclizations are ubiquitous in organic synthesis and have been applied to the synthesis of structurally complex molecules. N-heterocyclic motifs can be prepared through the cyclization of α-haloamides; however, slow rotation around the amide C–N bond results in preferential formation of an acyclic hydrodehalogenated product. Here, we compare four different methods for preparing γ, δ, ε, and ζ-lactams via radical cyclization. We found that a photoenzymatic method using flavin-dependent ‘ene’-reductases affords the highest level of product selectivity. We suggest that through selective binding of the cis amide isomer, the enzyme preorganizes the substrate for cyclization, helping to avoid premature radical termination.
Keywords: Biocatalysis, Lactam synthesis, Radical Chemistry
Graphical Abstract
Reductive radical cyclizations are classic transformations for organic synthesis.1 Due to their broad functional group tolerance and ability to form bonds with unactivated coupling partners, this family of reactions has been deployed to prepare various natural products and pharmaceuticals.2–5 However, the termination of radical intermediates prior to C–C bond formation is a significant challenge for some molecules. Consequently, substrates are often selected which favor reactive conformations or have low energetic barriers to bond rotation.6
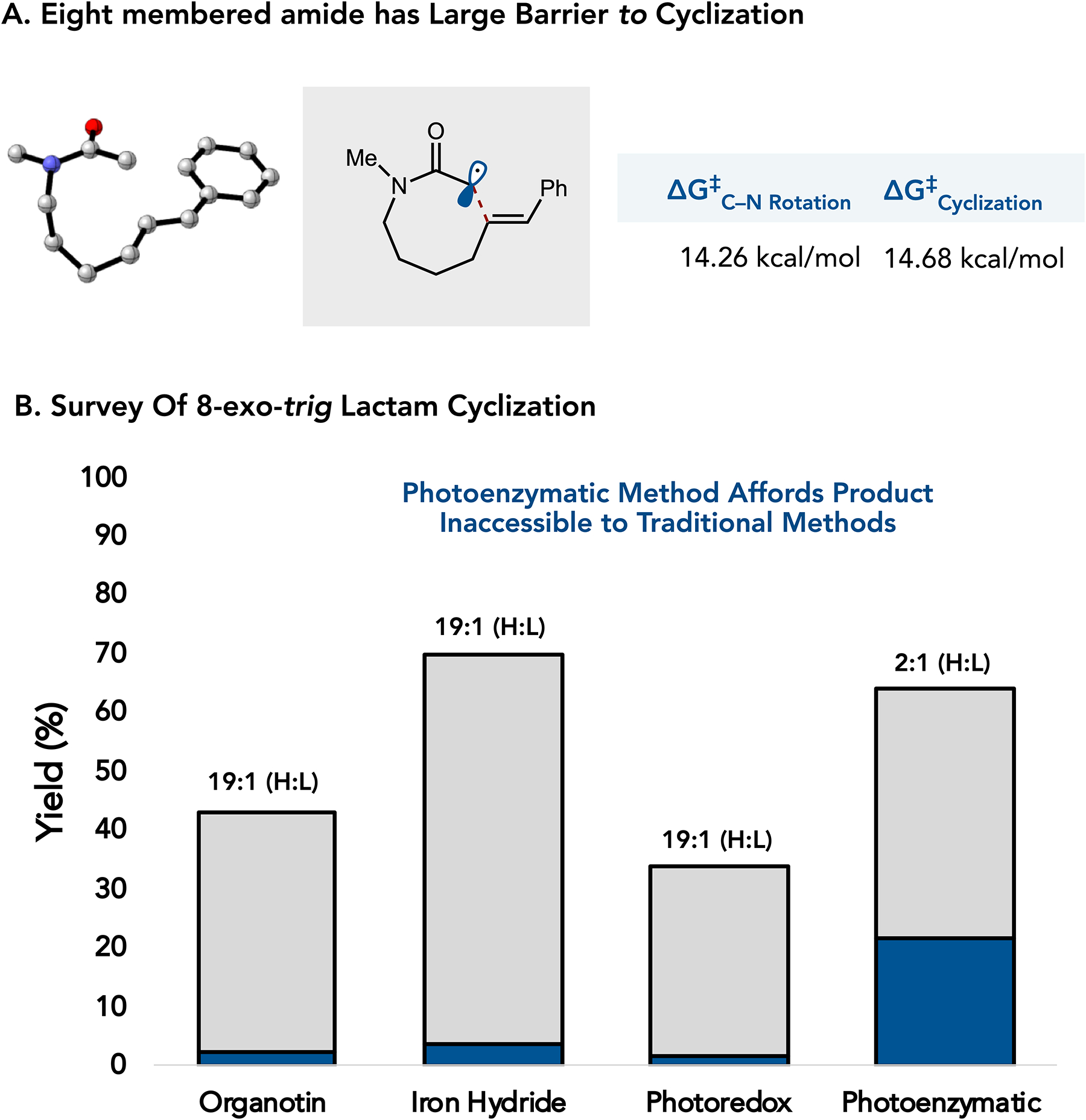
Cyclizations involving α-halo esters and amides are attractive for preparing lactones and lactams, respectively. However, these substrates have significant barriers to rotation around the C–O and C–N bonds of the ester and amide. These substrates often favor the trans-isomer, leading to hydrodehalogenation of the starting material when radical termination is faster than C–O or C–N bond rotation (Figure 1a).7 Stork and Ueno demonstrated the use of acetals instead of esters as a function of fast C–O bond rotation (Figure 1b).8 Alternatively, Curran found that atom transfer radical cyclization can be used with amides which reversibly terminates the α-acyl radical until the thermodynamically lactam is formed (Figure 1b).9 We questioned whether modern methods for reductive radical cyclization could overcome the limitations of the traditional nBu3SnH/AIBN reductive cyclization conditions. Herein, we survey four distinct strategies for the reductive radical cyclization of α-chloroamides to access γ, δ, ε, and ζ lactams, i) a traditional atom transfer radical cyclization using nBu3SnH/AIBN, ii) an electron transfer mediated reaction involving in situ generation of LnFeH, iii) a photoredox method involving reductive dehalogenation using an Iridium photocatalyst and nBu3N as a hydride source, and iv) a photenzymatic method involving electron transfer from a flavin cofactor (Figure 1c).
Figure 1.
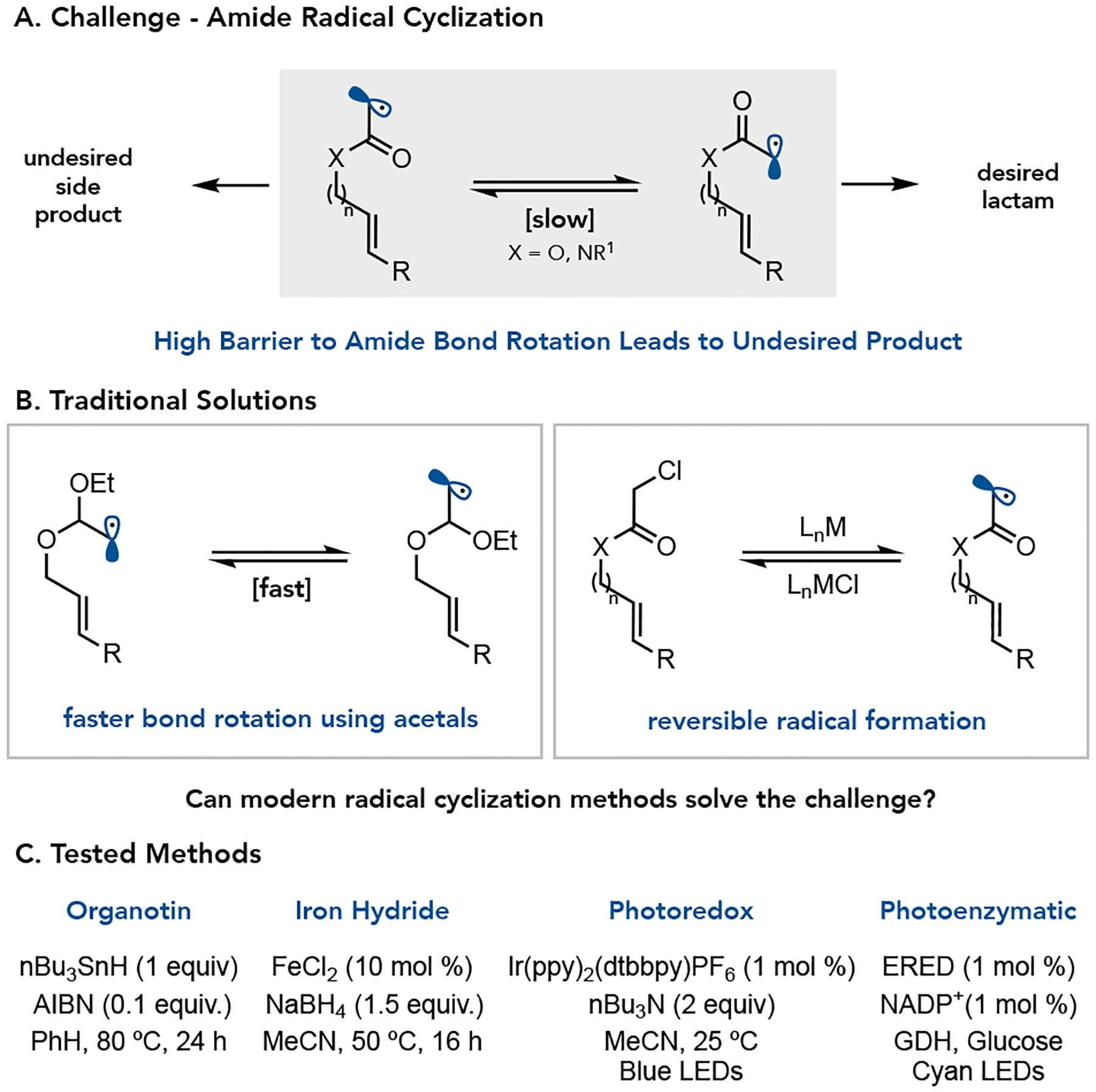
A. Challenges in Amide Cyclization B. Traditional Solutions C. Tested Methods.
We began by exploring a 5-exo-trig cyclization to afford a γ-lactam. Density Functional Theory (DFT) calculations to determine the barrier to rotation around the amide to be 14.83 kcal/mol, with the activation barrier to cyclization being 8.16 kcal/mol. These calculations indicate that cyclization is faster than rotation around the amide.10,11,12 (Figure 2a). Using nBu3SnH and catalytic AIBN as a radical initiator, the reaction occurred in 34% yield with a 2.8:1 ratio of hydrodehalogenated and cyclized product, consistent with previous reports (Figure 2b).13,14,15 Yield is defined as the isolated mixture of HDH and Lactam. Reported product ratios are determined from crude NMR.
Figure 2.
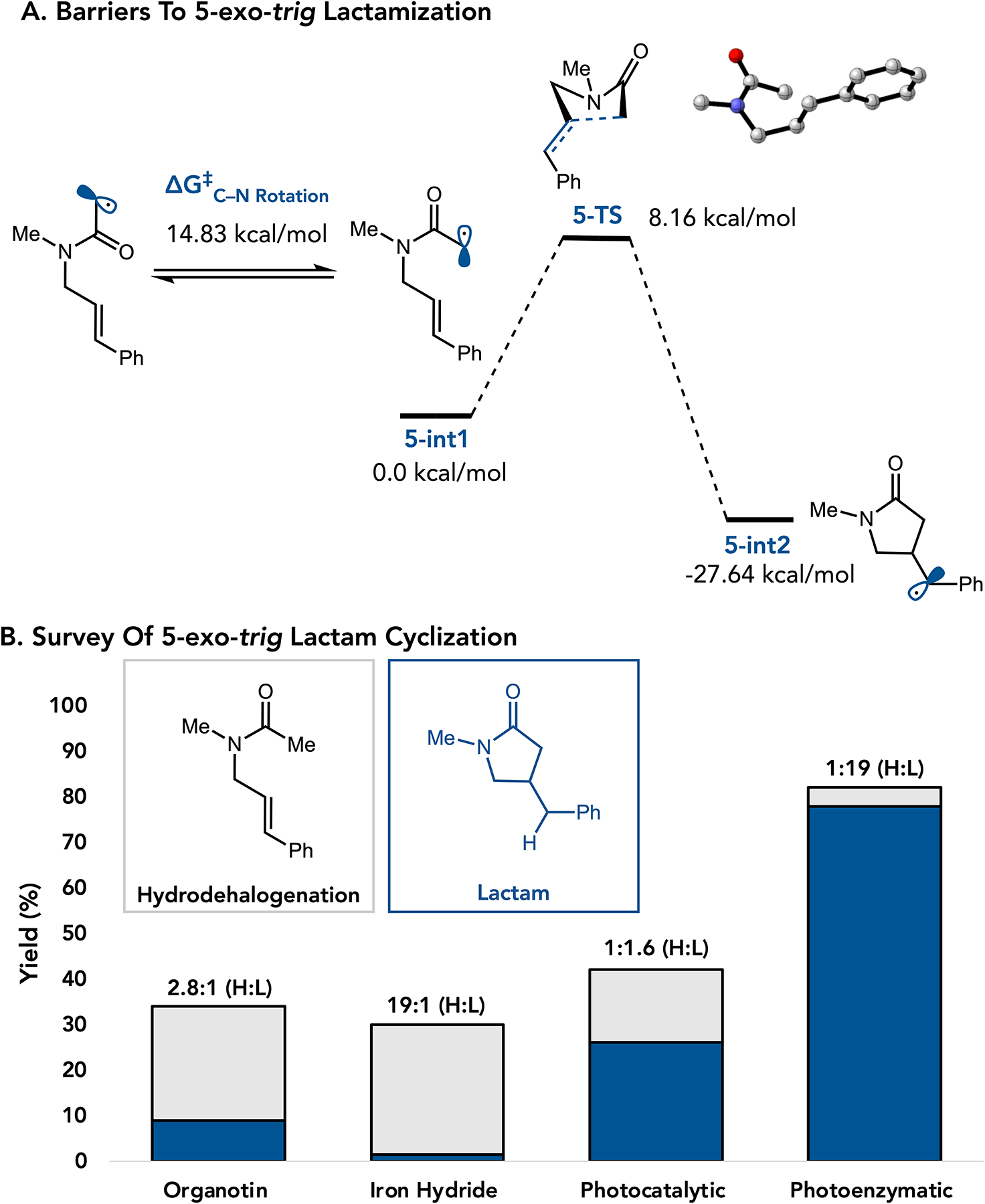
Survey of the 5-exo-trig Cyclization A. DFT Calculations B. Method Yields and Product Selectivity.
Fensterbank and coworkers described a reductive cyclization using FeCl2 and NaBH4.16,17 Under these conditions, FeCl2 is reduced to generate a metal hydride which functions as a radical initiator with NaBH4 hypothesized to serve as a hydrogen atom source. This method, however, proved ineffective, providing a >95:5 ratio of undesired product to cyclization at a modest 30% yield (Figure 2b).18
Next, we considered a photoredox method where radical initiation occurs via reductive cleavage of the C–Cl bond. Reuping and coworkers demonstrated that iridium photoredox catalysts could catalyze a 5-endo-trig cyclization using α-chloroamides as substrates and tributylamine as a terminal reductant.19 Under these conditions led to 42% conversion of starting material to a 1.6:1 ratio of lactam to hydrodehalogenated product (Figure 2b).20 We hypothesize that the slight preference for the lactam product is due to slow radical termination. The change in rate can be attributed to the strength of the C–H bonds of tributylamine by comparison to the strength of Sn–H or B–H bonds.19b Alternatively, reductive dehalogenation may occur preferentially from the cis-amide isomer, reorganizing the radical for cyclization.
Our group recently reported a biocatalytic reductive radical cyclization using flavin-dependent ‘ene’-reductases (EREDs). While the hallmark of this reactivity is high enantioselectivity, we recognized that preferential formation of the lactam product would be synthetically valuable.21–28 We attribute the high level of product selectivity to the enzyme selectively binding the cis-amide isomer, preorganizing the substrate for cyclization.21 We found that a small collection of ERED homologs can facilitate different amide radical cyclization.21 With the goal of identifying a single catalyst that would be effective for a variety of cyclization modes, we screened a small selection of mutants of ERED from Gluconobacter. (GluER). We found that GluER-T36A-W66A can react with many kinds of substrates to primarily afford the desired lactam product.29 When GluER-T36A-W66A is used for the model 5-exo-trig cyclization, the desired product is formed in 82% conversion with a >19:1 ratio of products favoring the desired cycloadduct.30
Next, we expanded our study to investigate the formation of six (δ) and seven (ε) member lactams. We postulated that the larger ring size would increase the kinetic barrier to cyclization, resulting in more hydrodehalogenated product.31 The barriers to rotation abound the amide C–N bond was calculated to be 13.97 and 13.36 kcal/mol for the substrate that would form the six and 7-membered rings, respectively, similar to the value calculated for the 5-membered ring substrate. The barrier to cyclization for the 6-membered ring is 7.72 kcal/mol, slightly decreased by comparison to the 5-membered ring formation. Cyclization to form the 7-membered ring δ-lactam has a barrier of 9.07 kcal/mol.10,11,12 When these substrates were tested using the organotin method, both afforded the hydrodehalogenated product primarily.15 These results are consistent with relative rates of cyclization by comparison to amide bond rotation being responsible for product outcome. The metal hydride method was again ineffective, affording a >20:1 of hydrodehalogenated product by comparison to lactam.18,32,33 The photoredox method showed an increase in hydrodehalogenated product over the lactams for both 6 and 7-exo-trig cyclizations.20,32,33 Finally, the photoenzymatic reaction using GluER-T36A-W66A afforded product at >20:1 ratio of lactam to hydrodehalogenation amide, indicating superior product selectivity across all three ring sizes.30,32,33
Finally, we investigated the synthesis of ζ-lactams via an 8-exo-trig ζ-cyclization. This substrate is unique as it has a higher activation barrier for cyclization (calculated by DFT to be 14.68 kcal/mol) than the other substrates tested.10,11,12 Surveying the traditional methods, we observe very little lactam product from organotin, metal hydride, and photoredox methods, consistent with cyclization being significantly slower than radical termination.15,18,20,34 Interestingly, GluER-T36A-W66A formed a 2:1 ratio of the hydrodehalogenated product to lactam.30,34 While this enzyme would require further protein engineering to achieve better product ratios, it highlights the opportunity for an enzyme to facilitate a reaction that would be challenging using small molecule methods.
In conclusion, we surveyed four strategies for amide radical cyclization and found the photoenzymatic method to provide the highest yields of the desired product. This study highlights the opportunity of enzymes to address challenges in chemical synthesis beyond enantioselectivity. We hope this study can be of value to practitioners interested in utilizing radical cyclizations for chemical synthesis.
Supplementary Material
Figure 3.
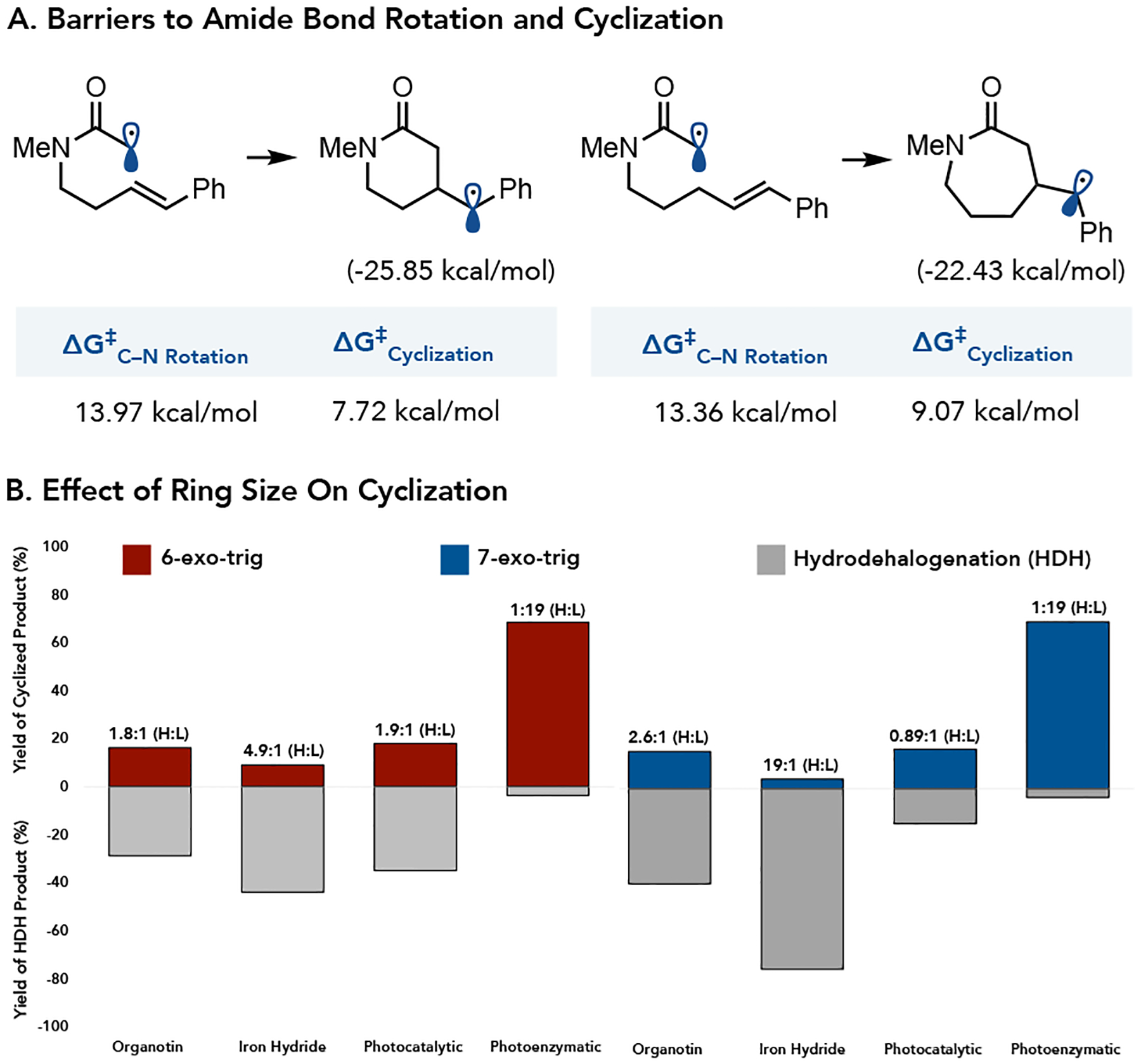
Survey of the 6 and 7 exo-trig cyclization’s
Figure 4.
Survey of the eight exo-trig cyclization
Acknowledgment
We thank Prof. David Collum (Cornell) for computational resources.
Funding Information
Financial support provided by the NIH (R01 GM127703). This work made use of the Cornell University NMR Facility, which is supported, in part, by the NSF through MRI award CHE-1531632.
Footnotes
Supporting Information
Is there Supporting Information to be published? Click here to indicate YES or NO (text and links will be updated prior to publication).
Primary Data
Is there Primary Data to be associated with this manuscript? Click here to indicate YES or NO (text and links will be updated prior to publication).
References and Notes
- (1).Jasperse CP; Curran DP; Fevig TL Chem. Rev 1991, 91, 6, 1237–1286 [Google Scholar]
- (2).Liao J; Yang X; Ouyang L; Lai Y; Huang J; Luo R Organic Chemistry Frontiers, 2021, 8, 1345–1363. [Google Scholar]
- (3).Corsello MA, Kim J; Garg NK Nature Chemistry 2017, 9, 944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hung K;Hu X; & Maimone TJ Natural Product Reports 2018, 35, 174–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Fantinati A; Zanirato V; Marchetti P; Trapella C ChemistryOpen, 2020, 9, 100–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Song L; Fang X; Wang Z; Liu K; Li C The Journal of Organic Chemistry, 2016, 81, 2442–2450. [DOI] [PubMed] [Google Scholar]; (b) Brill ZG; Grover HK; Maimone TJ Science, 2016, 352, 1078–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Clark AJ; Curran DP; Fox DJ; Ghelfi F; Guy CS; Hay B; James N; Phillips JM; Roncaglia F; Sellars PB; Wilson P; Zhang H The Journal of Organic Chemistry, 2016, 81, 5547–5565. [DOI] [PubMed] [Google Scholar]
- (8).(a) Stork G; Mook R Jr.; Biller SA; Rychnovsky SD Journal of the American Chemical Society, 1983, 105, 3741–3742. [Google Scholar]; (b) Ueno Y; Moriya O; Chino K; Watanbe M; Okawara M J. Chem. Soc., Perkin Trans. 1 1986,1351–1356. [Google Scholar]; (c) Ueno Y; Chino K; Watanabe M; Moriya O; Okawara M J. Am. Chem. Soc 1982,104, 5564–5566. [Google Scholar]
- (9).Curran DP; Tamine J The Journal of Organic Chemistry, 1991, 56, 2746–2750. [Google Scholar]
- (10).Frisch MJ et al. Gaussian, Inc, Wallingford CT, 2009. [Google Scholar]
- (11).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; et al. Gaussian, Inc: Wallingford, CT: 2013. [Google Scholar]
- (12).Marenich AV; Cramer CJ; Truhlar DG J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- (13).Sato T; Wada Y; Nishimoto M; Ishibashi H; Ikeda M; J. Chem. Soc, 1989, 879–886. [Google Scholar]
- (14).Sato T; Chono N; Ishibashi H; Ikeda M. J. Chem. Soc, 1, 1995, 1115. [Google Scholar]
- (15). General Procedure for Organotin Method. Adapted from Sato et al. and detailed below. The chloroamide starting material (0.224 mmol) was dissolved in dry benzene (4 ml). To a solution of Bu3SnH (1 equiv.) and AIBN (9 mol%) in dry benzene (6 ml) was added via a syringe during 40 min under reflux and the mixture was further refluxed for 12 h. After cooling, the solvent was evaporated off and the residue was chromatographed on silica gel. The crude residue is purified using automated silica gel chromatography. Fractions containing product are combined and concentrated and weighed for isolated yield determination. (5-exo-trig : 34% Yield 2.8:1 Hydrodehalogenation (HDH):Lactam). Yield Determined as a Ratio of Products. Product Ratio Determined Using Crude NMR. 5exo-trig-substrate.21 1H-NMR (500 MHz, CDCl3) δ 7.38 – 7.28 (m, 5H), 6.51 (t, J = 13 Hz, 1H), 6.10–6.18 (m, 1H), 4.17–4.14 (m, 2H), 4.11 (s, 2H), 3.04 (d, J = 30 Hz, 3H).13C-NMR (126 MHz, CDCl3) δ 166.73, 166.39, 152.98, 136.35, 135.83, 133.50, 132.63, 128.64, 127.90, 126.50, 123.45, 123.30, 52.20, 50.10, 41.43, 41.03, 35.02, 34.05. 5exo-trig-HDH21: 1H-NMR (500 MHz, CDCl3) δ 7.38 – 7.28 (m, 5H), 6.51 (t, J = 13 Hz, 1H), 6.10–6.18 (m, 1H), 4.17–4.14 (m, 2H), 4.11 (s, 2H), 3.04 (d, J = 30 Hz, 3H).13C-NMR (126 MHz, CDCl3) δ 166.73, 166.39, 152.98, 136.35, 135.83, 133.50, 132.63, 128.64, 127.90, 126.50, 123.45, 123.30, 52.20, 50.10, 41.43, 41.03, 35.02, 34.05. 5exo-trig-Lactam21: 1H-NMR 500 MHz, CDCl3) δ 7.28 (t, J=7 Hz, 2H), 7.20 (t, J=7 Hz, 1H), 7.13 (d, 2H), 3.27 – 3.23 (m, 2H), 2.92 (s, 3H), 2.62 (dd, J = 13, 6 Hz, 1H), 2.59 (dd, J = 13, 6 Hz, 1H), 2.46 (m, 1H), 2.06 (m, 2H), 1.86 (m, 1H), 1.48 (m, 1H).13C-NMR (126 MHz, CDCl3) δ 169.55, 139.18, 128.89, 128.53, 126.30, 49.10, 42.02, 38.47, 35.24, 34.41, 28.56.
- (16).Kyne SH; Lévêque C; Zheng S; Fensterbank L; Jutand A; Olivier C Tetrahedron, 2016, 72, 7727–7737 [Google Scholar]
- (17).Ekomié A; Lefèvre G; Fensterbank L; Lacôte E; Malacria M; Ollivier C; Jutand A Angewandte Chemie International Edition, 2012, 51, 6942–6946. [DOI] [PubMed] [Google Scholar]
- (18).General Procedure for Iron Hydride Method. Adapted from Kyne et al. and detailed here. The FeCl2 (10 mol%) and NaBH4 (2 equiv) were added to a screw cap tube in a glovebox. Acetonitrile (0.375 mL) was added under argon, and the mixture stirred for 15 min at room temperature. A solution of chloroamide (0.224 mmol) in acetonitrile (0.125 mL) was added under argon. The reaction was sealed, removed from the glovebox, heated to 50°C and allowed to procced overnight. The reaction was cooled to room temperature, quenched with water and the aqueous phase extracted with DCM. The combined organic phase was washed with brine, dried with sodium sulfate, and the solvent removed in vacuo. The crude residue is purified using automated silica gel chromatography. Fractions containing product are combined and concentrated and weighed for isolated yield determination. (5 exo-trig 30%, 95:5 HDH:Lactam)
- (19).(a) Fava E; Nakajima M; Tabak MB; Rueping M Green Chemistry, 2016, 18, 4531–4535 [Google Scholar]; (b) deB B. Darwent, National Bureau of Standards, 1970, 31, [Google Scholar]
- (20).General Procedure for Photoredox Method. Photocatlytic Method. Adapted from Fava et al and detailed below. An 8 dram vial was charged with chloroamide (0.25 mmol 1 equiv.), Ir(ppy)2(dtb-bpy)PF6 (PC, 1 mol%) and NBu3 (2 equiv.) under nitrogen in a glovebox. Degassed acetonitrile (12.5 ml, 0.02M) was added and the reaction sealed. The reaction was then removed from glovebox and irradiated with a 450 nm Kessil Lamp for 48 hrs. After this period, the mixture was diluted with Et2O and the organic phase was extracted three times with brine, dried over MgSO4, filtered, and evaporated under reduce pressure. The crude residue is purified using automated silica gel chromatography. Fractions containing product are combined and concentrated and weighed for isolated yield determination. (5 exo-trig 42%, 1:1.6 HDH:Lactam)
- (21).Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK Science, 2019, 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ye Y; Fu H; Hyster TK Activation Modes in Biocatalytic Radical Cyclization Reactions. Journal of Industrial Microbiology and Biotechnology, 2021, 48. 10.1093/jimb/kuab021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Page CG; Cooper SJ; DeHovitz JS; Oblinsky DG; Biegasiewicz KF; Antropow AH; Armbrust KW; Ellis JM; Hamann LG; Horn EJ; Oberg KM; Scholes GD; Hyster TK. Journal of the American Chemical Society, 2020, 143, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gao X; Turek-Herman J; Choi YJ; Cohen R; Hyster T J. Am. Chem. Soc 2021, 10.1021/jacs.1c09828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Fu H; Lam H; Emmanuel MA; Kim JH; Sandoval BA; Hyster TK Journal of the American Chemical Society, 2021, 143, 9622–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Grosheva D; Hyster TK Flavin-Based Catalysis, 2021, 291–313. [Google Scholar]
- (27).Sandoval BA; Clayman PD; Oblinsky DG; Oh S; Nakano Y; Bird M; Scholes GD; Hyster TK Journal of the American Chemical Society, 2020, 143, 1735–1739. [DOI] [PubMed] [Google Scholar]
- (28).Clayman PD; Hyster TK Journal of the American Chemical Society, 2020, 142, 15673–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nicholls B; Oblinsky D; Kurtoic S; Grosheva D; Ye Y; Scholes G; Hyster,. Angewandte Chemie International Edition 2021; Just Accepted. 10.1002/anie.202113842 [DOI] [PubMed] [Google Scholar]
- (30).General Procedure for Photoenzymatic Method. Adapted from Biegasiewicz et al. and detailed here. All reactions are run with 0.224 mmol of chloroamide starting material. Solid (D)-glucose (6 equiv.) and GDH-105 lyophilized lysate (0.2 mg lysate/mg of starting material) are weighed out into a 25 mL round bottom flask equipped with a magnetic stir bar. This, along with thoroughly degassed reaction buffer (100 mM KPi, pH = 8, 10% v:v glycerol) and the weighed out starting material are taken into a Coy® anaerobic chamber. Reaction buffer, NADP+ (made as a 5 mg/mL solution in reaction buffer, 1 mol%), and purified GluER T36A W66A solution (1 mol%) are added such that the final liquid volume added (12.5 mL) creates a reaction mixture with a starting material concentration of 17.92 mM. Starting material is dissolved in degassed THF cosolvent (2 μL/mg of starting material). This solution is taken up and pipetted directly into the reaction flask. The reaction flask is capped and sealed with a rubber septum and taken out of the anaerobic chamber where it is placed to stir at 400rpm with fan cooling the reaction setup under nitrogen atmosphere irradiated with cyan light (50 W Chanzon high power LED chip, λmax= 490 nm, measured photon flux = 12,000 mM/m2 s) for 36 h. Workup is performed as follows: the contents of the reaction flask are poured into a 125 mL Erlenmeyer flask containing 50 mL of 1 M aqueous hydrochloric acid and 50 mL of dichloromethane. This is stirred vigorously for 45 minutes, after which time the biphasic mixture is filtered through a thick pad of Celite® to remove precipitated material. The filtrate is poured into a separatory funnel and the dichloromethane layer is collected. The aqueous layer is extracted with dichloromethane (2 × 50 mL) and the combined organic layers are dried with anhydrous sodium sulfate and concentrated. Fractions containing product are combined and concentrated and weighed for isolated yield determination. (5 exo-trig 82%, 5:95 HDH:Lactam).
- (31).Smith TW; Butler GB The Journal of Organic Chemistry, 1978, 43, 6–13 [Google Scholar]
- (32). 6-exo-trig results. Yield Determined as a Ratio of Products. Product Ratio Determined Using Crude NMR. Organotin (45%, 64:36 HD: Lactam), Iron Hydride (53%, 83:17), Photoredox (66:34), Photoenzymatic (72%, 5:95). 6-exo-subtrate21: 1H-NMR 500 MHz, CDCl3) δ 7.34 – 7.27 (m, 4H), 7.24 – 7.18 (m, 1H), 6.40 (t, J = 15 Hz 1H), 6.14 (m, 1H), 4.07 (d, J = 11 Hz, 2H), 3.51 (m, 2H), 3.05 (d, J = 35 Hz, 3H), 2.45 (m, 2H).13C-NMR (126 MHz, CDCl3) δ 166.41, 137.28, 136.75, 133.30, 132.32, 128.63, 128.55, 127.66, 127.26, 126.53, 126.11, 125.00, 50.28, 48.25, 41.46, 40.94, 36.16, 33.84, 32.06, 30.90. 6exo-HDH : 1H-NMR (400 MHz, CDCl3) δ 7.28 (m, 4H), 7.12 (m,1H), 6.38 (dd, J= 15, 11, 1H), 6.09 (m, 1H), 3.40 (dt, J = 33, 9 Hz, 2H), 2.90 (d, J = 19 Hz, 3H), 2.40 (m, 2H), 2.00 (d, J=16 Hz, 3H). 13C-NMR (126 MHz, CDCl3) δ 171.14, 137.43, 136.96, 132.78, 131.85, 128.62, 128.53, 127.49, 127.15, 126.06, 125.63, 50.73, 47.42, 36.56, 33.40, 32.77, 32.06, 31.25, 21.88, 21.39. IR: (cm−1) 3024, 2931, 1621, 1492, 1400, 1359, 1260, 1198, 1030, 966, 743, 589 HR-MS[M+1]: calculated 204.1382, found 204.138. 6exoLactam21: 1H-NMR 500 MHz, CDCl3) δ 7.28 (t, J=7 Hz, 2H), 7.20 (t, J=7 Hz, 1H), 7.13 (d, 2H), 3.27 – 3.23 (m, 2H), 2.92 (s, 3H), 2.62 (dd, J = 13, 6 Hz, 1H), 2.59 (dd, J = 13, 6 Hz, 1H), 2.46 (m, 1H), 2.06 (m, 2H), 1.86 (m, 1H), 1.48 (m, 1H).13C-NMR (126 MHz, CDCl3) δ 169.55, 139.18, 128.89, 128.53, 126.30, 49.10, 42.02, 38.47, 35.24, 34.41, 28.56.
- (33). 7-exo-trig results. Yield Determined as a Ratio of Products. Product Ratio Determined Using Crude NMR. Organotin (55%, 72:28 HD: Lactam), Iron Hydride (79%, 95:5), Photoredox (31%, 47:53), Photoenzymatic (73%, 5:95). 7-exo-subtrate211H-NMR 500 MHz, CDCl3) δ 7.36–7.36 (m, 4H), 7.25 – 7.17 (m, 1H), 6.39 (t, J = 14 Hz, 1H), 6.19 (m, 1H), 4.07 (d, J = 6.2 Hz, 2H), 3.41 (dt, J= 24, 6 Hz, 2H), 3.03 (d, J = 53.3 Hz, 3H), 2.26 (m, 2H), 1.87 – 1.66 (m, 2H).13C-NMR (126 MHz, CDCl3) δ 166.44, 137.55, 137.17, 131.37, 130.58, 129.52, 128.52, 127.32, 127.03, 125.99, 49.80, 48.04, 41.49, 40.93, 35.72, 33.72, 27.94, 26.59.7exo-HDH 1H- NMR (400 MHz, CDCl3) δ 7.30(m 4H), 7.21(m 1H), 6.40 (m, 1H), 6.20 (m, 1H), 3.36 (dt J= 8 and 40 Hz, 2H), 2.97 (d, J = 24, 3H), 2.23 (p, J = 7 Hz, 2H), 2.08 (d, J = 7 Hz, 3H), 1.73 (m, 2H).13C-NMR (126 MHz, CDCl3) 170.50, 137.66, 137.29, 131.12, 130.34, 129.86, 128.89, 128.60, 128.50, 127.24, 126.95, 125.99, 50.27, 47.21, 36.20, 33.23, 30.36, 30.00, 27.90, 27.00, 21.99, 21.30. IR: (cm−1) 2928, 1637, 1490, 1433, 1397, 1012, 964, 743, 692, 601HR-MS[M+1]: calculated 218.1539, found 218.1537. 7-exo Lactam21: 1H-NMR 500 MHz, CDCl3) δ7.27 (t, J=7 Hz, 2H), 7.19 (t, J= 7 Hz, 1H), 7.15 (d, J= 7 Hz, 2H), 3.46 (dd, J = 14, 11 Hz, 1H), 3.20 (dd, J = 15, 6 Hz, 1H), 2.97 (s, 3H), 2.72 (dd, J = 13, 5 Hz, 1H), 2.56 – 2.44 (m, 3H), 1.93 (m, 1H), 1.79 (m, 2H), 1.46 (m, 1H), 1.28 (m, 1H).13C-NMR (126 MHz, CDCl3) δ 174.38, 139.91, 129.41, 128.29, 125.86, 51.24, 43.12, 36.19, 35.25, 26.93.
- (34).8-exo-trig results. Yield Determined as a Ratio of Products. Product Ratio Determined Using Crude NMR. Organotin (43%, 95:5 HD: Lactam), Iron Hydride (70%, 95:5), Photoredox (34%, 95:5), Photoenzymatic (64%, 66:34).8-exo-substrate: 1H-NMR 400 MHz, CDCl3) δ 7.1 (m, 4H), 7.20 (m, 1H), 6.38 (m 1H), 6.19 (m, 1H), 4.06 (s 2H), 3.37 (dt, J = 8 and 25 Hz, 2H), 3.01 (dd, J = 9 and 43 Hz, 3H), 2.26 (p, J = 7 Hz, 2H), 1.57 (m, 4H). 13C-NMR (126 MHz, CDCl3) δ 169.48, 166.71, 137.43, 131.40, 130.77, 130.32, 129.61, 128.51, 127.04, 126.93, 125.71, 50.35, 48.23, 41.35, 40.77, 35.65, 33.81, 32.56, 27.89, 26.34.IR: (cm−1) 2931, 1742, 1648, 1617, 1446, 1405, 965, 744, 693. HR-MS[M+1]: calculated 266.1306, found 266.1299. 8-Exo-HDH: 1H-NMR (400 MHz, CDCl3) δ 7.33 (m, 4H), 7.19 (m, 1H), 6.38 (dd, J = 6 and 16 Hz, 1H), 6.20 (m, 1H), 3.34 (dt, J = 8 and 40 Hz, 2H), 2.93 (dd, J = 8 and 24 Hz, 3H), 2.25 (p, J = 7 Hz, 2H), 2.09 (d, J = 6 Hz, 3H), 1.49 (m, 4H).13C- NMR (126 MHz, CDCl3) δ 170.40, 137.66, 137.29, 131.12, 130.34, 128.50, 126.95, 125.98, 50.26, 47.21, 36.20, 33.23, 30.36, 30.00, 27.90, 26.93, 21.99, 21.29.IR: (cm-1) : 3023, 2829, 2856, 1637, 1491, 1433, 1397, 1184, 964, 743, 602, 468 HR-MS [M+1]: calculated 232.1695, found 232.1689. 8-exo-lactam. 1H-NMR (400 MHz, CDCl3) δ 7.29 (m 2H), 7.22 (m, 3H), 3.68 (m, 1H), 3.29 (dt, J = 4, 48 Hz, 1H), 2.94 (s, 3H), 2.75 (dd, J = 7 and 13 Hz, 1H), 2.50 (m, 3H), 2.16 (m, 1H), 1.75 (m, 3H), 1.51 (m, 1H), 1.18 (m, 2H).13C-NMR (126 MHz, CDCl3) δ 174.09, 161.27, 140.32, 129.33, 128.29, 126.03, 49.17, 43.14, 41.36, 38.92, 33.28, 28.41, 21.88.IR: (cm−1) 2922, 1634, 1453, 1423, 1396, 1236, 1137, 764, 527, 432. HR-MS[M+1]: calculated 232.1695, found 232.1692
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.