

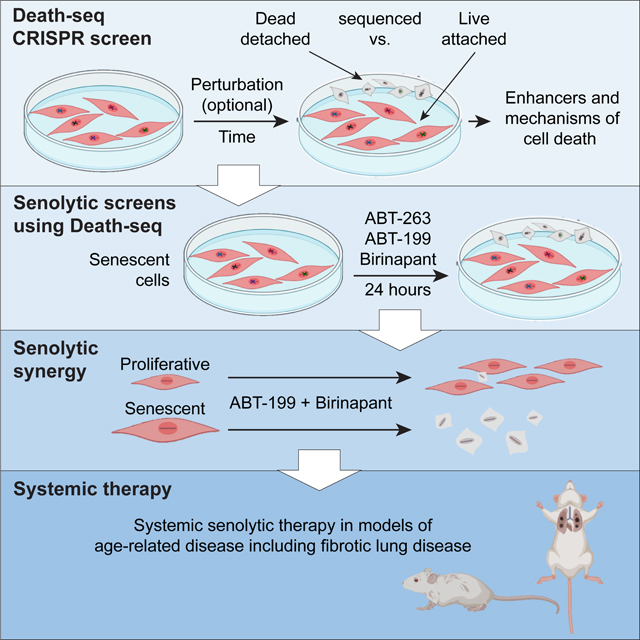
SUMMARY
Selectively ablating damaged cells is an evolving therapeutic approach for age-related disease. Current methods for genome-wide screens to identify genes whose deletion might promote death of damaged or senescent cells are generally underpowered because of the short timescales of cell death as well as the difficulty to scale non-dividing cells. Here, we establish Death-seq, a positive selection CRISPR screen optimized to identify enhancers and mechanisms of cell death. Our screens identified synergistic enhancers of cell death induced by the known senolytic ABT-263. The screen also identified inducers of cell death and senescent cell clearance in models of age-related diseases by a related compound, ABT-199, which alone is not senolytic but exhibits less toxicity than ABT-263. Death-seq enables the systematic screening of cell death pathways to uncover molecular mechanisms of regulated cell death subroutines and identifies drug targets for the treatment of diverse pathological states such as senescence, cancer, and fibrosis.
Keywords: Death screen, Death-seq, CRISPR, positive selection, genome-wide, senescence, senolytics, pulmonary fibrosis, cell death, synthetic lethality
Graphical Abstract
eTOC blurb
Colville et al. present a genetic screening method “Death-seq” to compare positively selected dying cells directly against living cells to improve the ability to systematically identify enhancers and mechanisms of cell death. They then applied this method to identify enhancers of selective cell death in senescent cells in age-related disease.
INTRODUCTION
Cellular senescence is a cell state triggered by various stresses and characterized by prolonged irreversible cell-cycle arrest with secretory features.1,2,3 Elimination of CDKN2A-positive cells, which have been hypothesized to be senescent, extends median lifespan of mice4 and is being developed as a means to treat many age-related diseases5 and to restore tissue homeostasis in aging.6,7 Senolytics, a class of drugs that selectively target senescent cells for death, hold promise for achieving those goals. However, the systemic toxicity and limited potency of current senolytic targets, as well as the lack of definitive characterization of in vivo cellular senescence using selective markers and omics techniques, limit their use to local administration and compromise their clinical application.8
Pooled genetic screens carry potential to identify improved senolytic targets. A recent RNAi approach has been used to identify a new senolytic target,9 but this study failed to identify more than one senolytic target and any hits in classical cell death pathways in part because of the limitations of negative selection growth screens and RNAi interference screens. Negative selection growth screens on cells that rarely divide are even more challenging than on transformed cancer cells as the signal-to-noise ratios are decreased due to the lack of population doubling throughout the screen, which typically last 10–14 days in the case of negative selection growth screens. In contrast, an Annexin V-based enrichment screen outside of the senescence field pioneered the concept of a positive selection screen for dying cells.10 However, performing screens by selection of Annexin V+ cells requires either magnetic columns, which are not suitable for large cells such as senescent cells, or fluorescence-activated cell sorting, which suffers from limited throughput for genome-scale screens with time sensitive readouts like cell death.
To overcome these challenges and to increase the likelihood of uncovering targets in cell death pathways, we developed a method we termed “Death-seq” to positively select dying cells in response to genetic or pharmacologic interventions. We then implemented this approach with a genome-wide CRISPR screen to identify targets to enhance senolytic therapy.
We started with the senolytic therapy ABT-263, an inhibitor of the anti-apoptotic proteins BCL-2, BCL-xL (BCL2L1), and BCL-w (BCL2L2). Senescent cells are resistant to apoptosis which is thought to be mediated in part by the dysregulation of BCL-2 family.2,11 The anti-apoptotic BCL-2 family proteins neutralize BAX and BAK to prevent their oligomerization and inhibit the BH3-only pro-apoptotic proteins that can activate BAX and BAK, thus inhibiting mitochondrial outer membrane permeabilization (MOMP). These interactions that regulate MOMP and apoptosis are dictated by the BCL-2 homology 3 (BH3) domain, and small molecule BH3 mimetics, such as ABT-263, were developed to enhance apoptosis. As a result of senescent cells dysregulation of the BCL-2 family, ABT-263 has been used as a senolytic both in vitro and in vivo.12,13 Second mitochondria-derived activator of caspases (SMAC, also referred to as DIABLO) is released upon MOMP and inactivates IAPs, the inhibitor of apoptosis proteins, to promote apoptosis. The IAP protein family is known to regulate cell survival and three of the eight IAP family members have documented anti-apoptotic roles: X-chromosome-linked IAP (XIAP), cellular IAP 1 and 2 (cIAP1 and cIAP2).14 They interfere with apoptosis signaling via direct and indirect inhibition of caspases 3, 7, and 9. As a natural antagonist of IAPs, SMAC binds to IAPs to prevent their binding to caspases, leading to the activation of the downstream caspase cascade. Like BH3 mimetics, small molecule SMAC mimetics were developed to selectively enhance the death of cancer cells. Our screens systematically identify components of the intrinsic apoptosis pathway, ascertain uncharacterized regulators of senolysis, and uncover a Submitted Manuscript: Confidential synergy between BH3 mimetics and SMAC mimetics in senescent cell elimination with therapeutic potential.
DESIGN
Current genetic screening methods to study viability are not optimized to study phenomena, such as cell death, which happen on short timescales, and are also not optimized to study cells such as postmitotic cells like senescent cells, which are hard to scale. To improve the ability to do so we developed the Death-seq method. Death-seq involves the collection of dying cells through the centrifugation of media containing cells which have detached, or are in the process of detaching, in addition to the usual collection of adherent cells. Subsequently, the method involves sequencing the two populations and comparing the sgRNA populations directly between live vs dying cells. Recent viability screens using primary neurons15 and muscle cells16 highlight a growing opportunity to conduct genome-scale screens in more physiologically relevant cell culture models that are not as amenable to scale-up as immortalized cell lines. Yet applications of genetic screening to these models, such as in neurological diseases, remain scarce due in part to methodology limitations.17 Death-seq amplifies signal and reduces the noise compared to traditional dropout screens by design to further enable screening in more physiologically relevant models. Using Death-seq, we conducted a genome-wide CRISPR knockout (KO) screen in IMR-90 normal human lung fibroblasts and induced them to senescence by doxorubicin (Doxo-SEN) to discover knockouts that would exhibit either increased or decreased sensitivity to senolysis by ABT-263 (also known as navitoclax) (Figure 1A; Figures S1A–S1E). After incubating the cells with ABT-263 for 24 hours and collecting both the floating (dying) and attached (live) cells, the sgRNA distributions of each population were determined by sequencing (“Death-seq”). The casTLE algorithm18 was used to estimate the effect and P value of each gene knockout. Death-seq enables the separate processing of the dying and live cells from the same plates, allowing the direct comparison of sgRNA distributions in each as opposed to comparing to a baseline frequency before the experiment or a DMSO-treated control arm, thereby amplifying signal and cutting tissue culture reagents in half.
Figure 1. A genome-wide CRISPR screen for modifiers of cell death, Death-seq, identifies modifiers of senescent cell death.
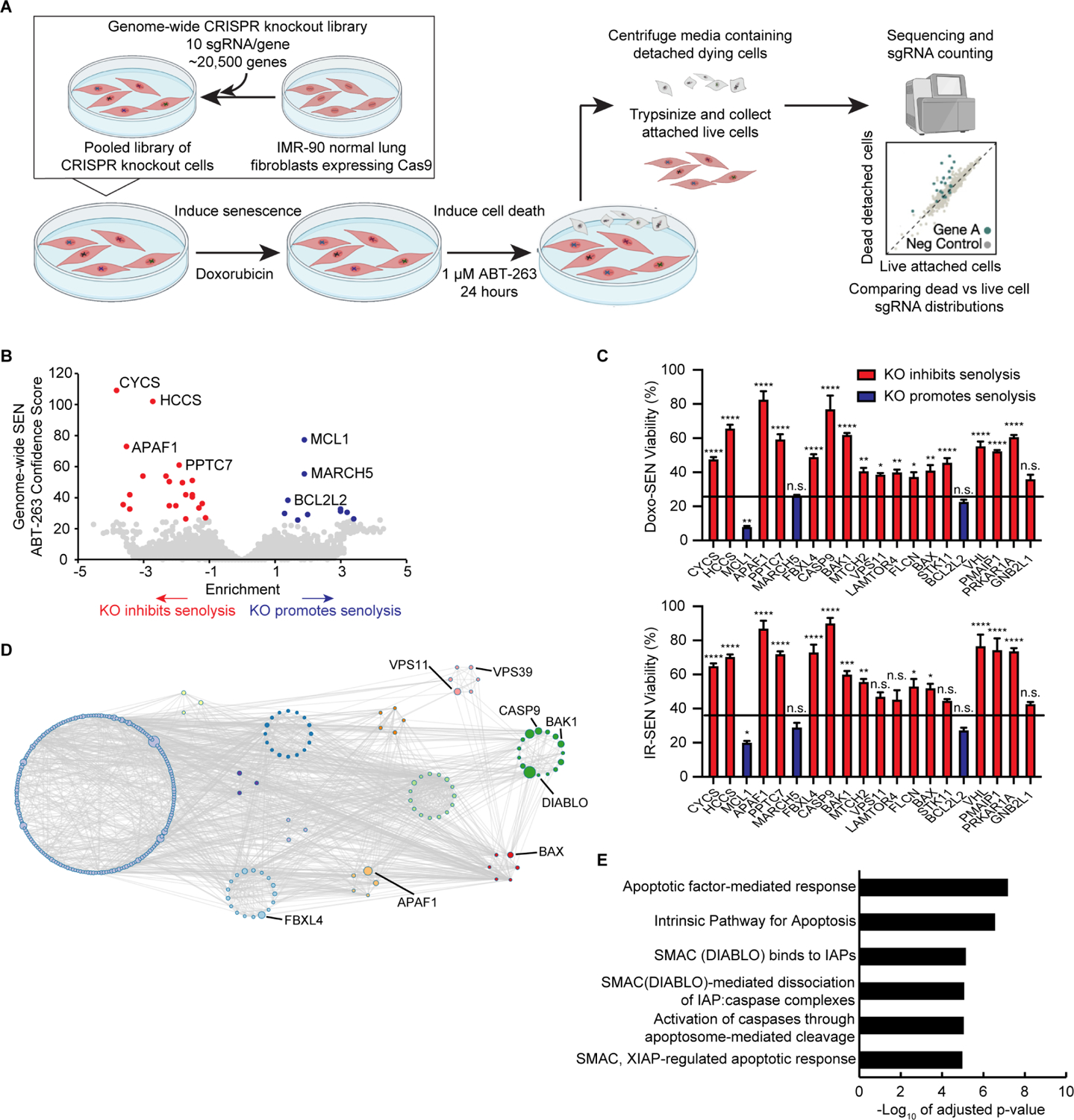
(A) Schematic of Death-seq screening method for genetic modifiers of cell death. The screen 5 was performed in duplicate.
(B) Volcano plot of the effects and confidence scores of all the genes in the genome-wide CRISPR screen in doxorubicin-induced senescent IMR-90s treated with 1 μM ABT-263 for 24 h. Effects and casTLE scores are calculated by casTLE. Labelled are the 31 genes passing 10% FDR for inhibiting (red) or promoting (blue) cell death by ABT-263 when knocked out.
(C) Validation of the top 20 genome-wide screen hits, which in the screen inhibited (red bars) or promoted (blue bars) cell death, using individual-well sgRNA knockouts of the indicated genes compared to control sgRNA viability (represented by the line) after treatment with 1 μM ABT-263 for 3 d in either doxorubicin (Doxo)-induced (top) or irradiation (IR)-induced (bottom) senescent (SEN) IMR-90s. Data are representative of two independent experiments performed in triplicate and are presented as mean ± s.e.m. One-way ANOVA with Dunnett’s post-hoc test relative to control sgRNA-treated cells, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
(D) Visualization of the genetic interaction network of the top 350 enriched genes in the genome-wide ABT-263 screen using Metascape. The size of the nodes corresponds to the confidence score of each gene in the screen. The Molecular Complex Detection (MCODE) algorithm was used to identify densely connected network components identified by circles of colored nodes. Select representative genes are highlighted.
(E) The top Reactome categories enriched in the 64 genes that passed 30% FDR in the genome-wide ABT-263 screen.
See also Figure S1 and the results and raw sequencing counts from screen in Tables S1 and S2 respectively.
RESULTS
Genome-wide senolytic CRISPR screen using Death-seq
Using a 30% false discovery rate (FDR) cutoff, the genome-wide screen identified 64 genetic modifiers which either inhibit or promote the senolytic effect of ABT-263 and 31 hits passing 10% FDR (Figure 1B; Tables S1 and S2). We validated the top 20 hits from the screen using sgRNA KO IMR-90s induced to senescence by doxorubicin or irradiation (IR-SEN) (Figure 1C and S1F). The top hits inhibiting death including CYCS, HCCS, and APAF1 served as expected hits validating the screen given their roles in the intrinsic apoptosis pathway. The fourth hit inhibiting death, PPTC7, a mitochondrial phosphatase localized to the matrix, is poorly characterized19 with no previous connection to apoptosis and was also validated using shRNA knockdowns (Figure S1G). Consistent with recent studies,20,21,22,23 MCL1, another BCL-2 family anti-apoptotic protein which is not targeted by ABT-263, inhibited cell death due to ABT-263. Unlike this recent work, combination treatment of the MCL1 inhibitor S63845 and ABT-263 had a stronger effect on proliferative cells (PROs) compared to senescent cells, as represented by Bliss score (Figures S1H and S1I). VHL, one of the top 20 hits, was further validated as a hit inhibiting cell death given that it had an available small molecule inhibitor (Figure S1J). VHL encodes for the protein Von Hippel–Lindau tumor suppressor which functions as a E3 ubiquitin ligase and has been shown to regulate apoptosis.24 Genetic interaction networks for the top 350 genes and pathway analysis for the 64 hits passing 30% FDR revealed a strong enrichment for pathways related to the intrinsic apoptosis pathway, mTOR signaling, and, unexpectedly, SMAC-IAP-caspase interactions (Figures 1D and 1E; Figure S1K).
Death-seq head-to-head versus negative selection viability screens
In order to validate the genome-wide screen findings, we constructed a custom lentiviral sgRNA library targeting 384 genes to test the top ~350 genome-wide screen hits as well as select apoptosis and lysosomal related genes (Table S3). We tested Death-seq head-to-head against a traditional negative selection screen design, which used a DMSO-treated control arm, using both an apoptosis sublibrary targeting ~3000 genes as well as the newly constructed custom sgRNA library (Figure 2A). Death-seq identified 3 more hits reaching a 10% FDR threshold in the apoptosis sublibrary (Figure 2B; Tables S1 and S2) and 49 more hits in the custom sublibrary (Figure 2C; Tables S1 and S2) compared to the traditional method while achieving higher correlations between technical replicates. We then plotted the Death-seq combo confidence scores for each gene against those generated with the traditional method to compare the level of signal in the output of each method using both the apoptosis sublibrary (Figure 2D) and the custom sublibrary (Figure 2E). With the new positive selection method comparing live cells directly against dying cells, noise is reduced and signal is amplified enabling higher confidence scores and more significant hit calling.
Figure 2. Head-to-head comparison of Death-seq against traditional negative selection viability screens.
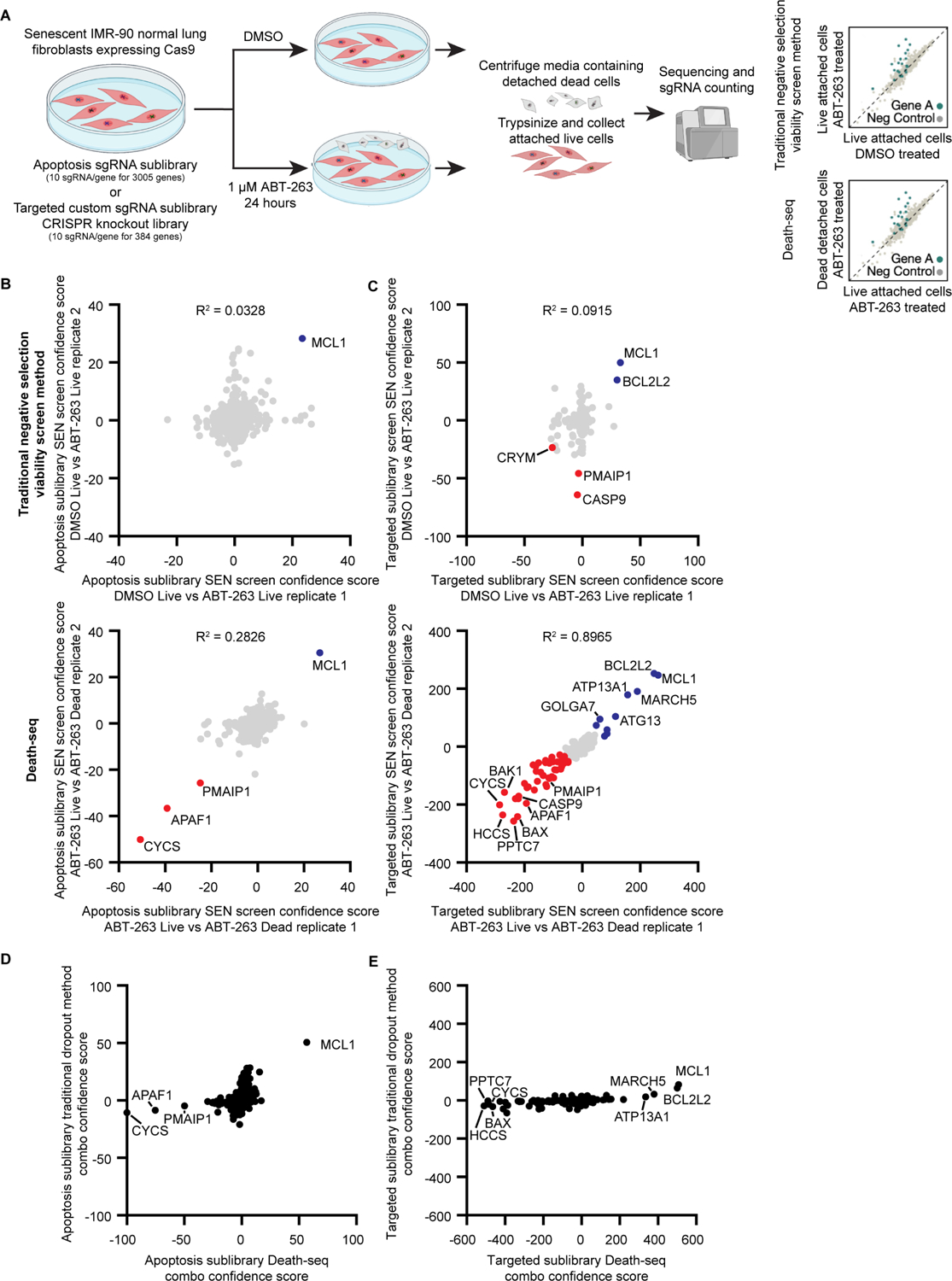
(A) Schematic of head-to-head comparisons of Death-seq screening method against traditional negative selection viability screens for genetic modifiers of cell death induced by ABT-263 treatment. The screens were performed in duplicate.
(B and C) Correlation of casTLE confidence scores for all genes in the apoptosis (ACOC) (B) or targeted custom sublibrary (C) ABT-263 screen technical replicates comparing a traditional negative selection viability screen (top, DMSO-treated live cells vs. ABT-263-treated live cells) against Death-seq (bottom, ABT-263-treated live cells vs. ABT-263-treated dying cells).
Labelled are the genes passing 10% FDR for inhibiting (red) or promoting (blue) cell death by ABT-263 when knocked out. R-squared values are from linear regression models. (D and E) Comparison of combo casTLE confidence scores generated using a traditional negative selection viability screen method (DMSO-treated live cells vs. ABT-263-treated live cells) vs. the Death-seq method (ABT-263-treated live cells vs. ABT-263-treated dying cells) for all genes in the apoptosis (ACOC) (D) or the targeted custom sublibrary (E).
See also the results, raw sequencing counts, and composition of the targeted custom sublibrary from screens in Tables S1, S2, and S3 respectively.
Targeted sublibrary senolytic Death-seq screens highlight importance of SMAC
Given the enrichment of pathways in the genome-wide screen related to the IAP-binding mitochondrial protein SMAC/DIABLO, we validated DIABLO as a hit inhibiting cell death in sgRNA KO senescent IMR-90 cells (Figures 3A and S2A). To further validate the genome-wide ABT-263 screen and in hopes of finding a systemic senolytic therapy, we conducted parallel screens using ABT-263, the SMAC mimetic birinapant, and ABT-199 in Doxo-SENs as well as an additional ABT-263 screen in hydrogen peroxide-induced senescent cells (H2O2-SENs) (Figure 3B). Due to the well-documented BCL-xL on-target toxicity of ABT-263 to platelets limiting its use as a systemic therapy,25 ABT-199, also known as venetoclax, was developed with enhanced BCL-2 specificity and reduced potency to BCL-xL to avoid platelet toxicity.26 Although ABT-199 has been shown to not be senolytic in fibroblasts,11 we hypothesized that a screen may reveal a genetic perturbation that, in combination with ABT-199, acts in a synthetic lethal manner in senescent cells while still avoiding platelet toxicity. The Doxo-SEN ABT-263 custom sublibrary screen identified 80 hits passing a 10% FDR threshold (Figure 3C; Tables S1 and S2), including DIABLO and other members of the intrinsic apoptosis pathway, and the casTLE confidence scores of each gene correlated well with those of the genome-wide screen (Figure S2B). The H2O2-SEN and Doxo-SEN ABT-263 screens correlated strongly, potentially suggesting the two inducers yield similar senescence states (Figure S2C). The birinapant Doxo-SEN screen identified 14 hits which passed 10% FDR, 5 of which also passed 10% FDR in the ABT-263 screen (Figure 3D; Tables S1 and S2). Given their different mechanisms of action (MoAs), the birinapant and ABT-263 screens had a weak correlation. In support of the idea that these hits are combinatorial, BCL2L1 was a hit in the birinapant screen and DIABLO was a hit in the ABT-263 screen. The ABT-199 and ABT-263 screens had a strong correlation given their similar MoAs (Figure 3E; Tables S1 and S2). The ABT-199 screen identified 58 hits which passed 10% FDR including both DIABLO and XIAP. The ABT-199 and ABT-263 screens shared 41 hits at the 10% FDR threshold.
Figure 3. Targeted sublibrary senolytic Death-seq screens highlight importance of SMAC.
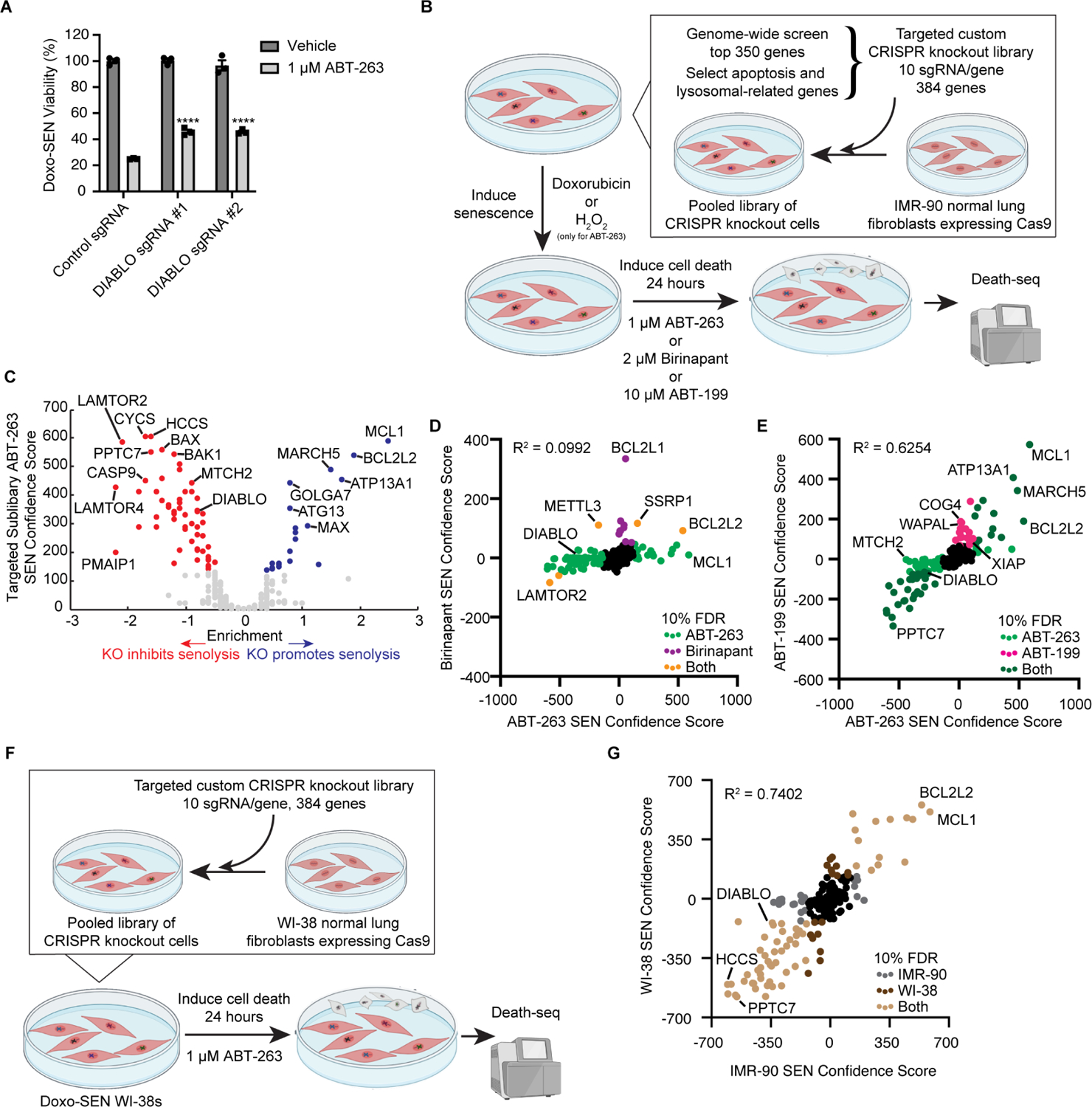
(A) Validation of DIABLO/SMAC sgRNA knockouts compared to control gRNA viability after treatment with vehicle or 1 μM ABT-263 for 3 d in Doxo-induced senescent IMR-90s. Data are representative of two independent experiments performed in triplicate and are presented as mean ± s.e.m. One-way ANOVA with Dunnett’s post-hoc test relative to control sgRNA-treated cells in the corresponding drug treatment, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
(B) Schematic of targeted custom sublibrary screens. The targeted custom library was infected using lentivirus into Cas9-expressing IMR-90 normal lung fibroblasts. The IMR-90s expressing the targeted sublibrary were then induced to senesce with Doxo and treated with 1 μM ABT-263, 2 μM birinapant, or 10 μM ABT-199 for 24 h. A targeted custom sublibrary screen was also performed with H2O2-induced senescent IMR-90s and 1 μM ABT-263 treatment. Like the genome-wide screen, Death-seq was used to compare the dying cell populations directly with the live attached cells. The screens were performed in duplicate.
(C) Volcano plot of the effects and confidence scores of all the genes in the targeted custom CRISPR screen in Doxo-induced senescent IMR-90 cells treated with 1 μM ABT-263 for 24 h. Labelled are the 80 genes passing 10% FDR for inhibiting (red) or promoting (blue) cell death by ABT-263 when knocked out.
(D) Correlation of combo casTLE confidence scores of the ABT-263 and birinapant screens in Doxo-induced senescent IMR-90s for all genes in the targeted custom sublibrary. R-squared value is from a linear regression model. Labelled in light green, purple, and orange are hits passing 10% FDR only in ABT-263 screen, only in birinapant screen, or in both screens, respectively.
(E) Correlation of combo casTLE confidence scores of the ABT-263 and ABT-199 screens in Doxo-induced senescent IMR-90s for all genes in the targeted custom sublibrary. R-squared value is from a linear regression model. Labelled in light green, pink, and dark green are hits passing 10% FDR only in ABT-263 screen, only in ABT-199 screen, or in both screens, respectively.
(F) Schematic of targeted custom sublibrary ABT-263 screen in Doxo-SEN WI-38 normal lung fibroblasts run head-to-head against Doxo-SEN IMR-90s. The screens were performed in duplicate.
(G) Correlation of combo casTLE confidence scores of the Doxo-induced senescent IMR-90s and Doxo-induced senescent WI-38s ABT-263 screens for all genes in the targeted custom sublibrary. R-squared value is from a linear regression model. Labelled in grey, dark brown, and light brown are hits passing 10% FDR only in the IMR-90 screen, only in the WI-38 screen, or in both screens, respectively.
See also Figure S2 and the results, raw sequencing counts, and composition of the targeted custom sublibrary from screens in Tables S1, S2, and S3 respectively.
To further visualize the results of the Doxo-SEN ABT-263 custom sublibrary screen, we created a subcellular localization map with selected hits passing the 10% FDR threshold (Figure S2D). Many hits involved mitochondrial biology as previously discussed, serving as a positive control for the ability of the Death-seq method to identify components of cell death subroutine pathways. MARCH5, an E3 ubiquitin ligase known to mediate the degradation of proteins that regulate mitochondrial fission and fusion,27 was one of the mitochondrially located hits which promoted senolysis by ABT-263 and ABT-199 in agreement with recent work in cancer cells.28,29 Recently, additional evidence was published validating our screen findings and revealing the likely mechanism through which our screen hit MTCH2 assists in senolysis by aiding MARCH5 in turnover of the MCL1:NOXA complex.30
As highlighted in the pathway analysis of the genome-wide screen results (Figure S1E), we identified a signal for mTOR signaling with several hits involving lysosomal biology. Our sublibrary screens identified several members of the lysosomal membrane–anchored Rag-Ragulator complex, a regulator of cellular responses to nutrient availability and metabolism. These members include FLCN, all five LAMTOR subunits 1–5, RRAGA, and RRAGC as hits inhibiting senolysis induced by ABT-263. Interestingly, a role of the Rag-Ragulator complex in cell death was not uncovered until a recently published CRISPR screen identified the mechanistic role of the complex in pyroptosis.31 However, this work did not find a role for the complex in apoptosis. Our ABT-263 senescence screens also identified hits involving the lysosomal associated GATOR complex, known for amino acid sensing. These included synergistic hits within the GATOR1 subcomplex, including NPRL2 and NPRL3, and hits inhibiting death in the GATOR2 subcomplex, including WDR24, WDR59, SEH1L, and MIOS. Furthermore, the screens identified hits inhibiting death in the lysosomal associated tethering complexes HOPS and CORVET including VPS16, VPS18, VSP11, VPS41, and VPS39. The breadth and systematic nature of the identified modifiers of senolysis further validate our screening method.
In order to further validate the targeted sublibrary senolytic Death-seq screens and validate the ability of the Death-seq method to work in additional cell types, we conducted additional targeted sublibrary screens in parallel with the previously described Doxo-SEN IMR-90 ABT-263 custom sublibrary screen (Figure 3B). In one such parallel screen, we tested Doxo-SEN WI-38 normal fibroblasts with the same ABT-263 treatment (Figure 3F) and identified 77 hits passing a 10% FDR threshold with a strong correlation to the same screening conditions in IMR-90s (Figure 3G; Tables S1 and S2). Providing further validation, DIABLO and other members of the intrinsic apoptosis pathway were among the hits shared in both screens. In another set of parallel screens, we treated proliferative IMR-90s with the same dose of ABT-263, ABT-199, and birinapant respectively to compare head-to-head the effects of knockouts on proliferative versus senescent cells (Figures S2E–S2G; Tables S1 and S2). These screens revealed MCL1, MARCH5, and ATP13A1 as hits promoting cell death in both senescent and proliferative cells, supporting the strong effect on proliferative cells of an earlier validation experiment with MCL1 inhibition (Figures S1H and S1I). MAX, GOLGA7, ATG13, GTF2F2, TRAPPC1, and COG4 knockouts showed selective effects on senescent cell viability compared to proliferative cells, highlighting the potential of this method to identify specific effects on viability. Importantly, these screens suggest that the combination of DIABLO and BCL2L1 may have a selective effect on senescent cell but not proliferative cell viability as demonstrated by the presence of DIABLO as a SEN-only hit in the ABT-263 and ABT-199 screens while BCL2L1 was a SEN-only hit in the birinapant screen (Figures S2E–G).
BH3 and SMAC mimetics synergize to induce senolysis
We next focused on the intriguing, recurring hit SMAC/DIABLO. Consistent with our genetic screening results, we observed a synergistic inhibition of cell viability in Doxo-SEN IMR-90s using the combination of ABT-263 and SMAC mimetic birinapant as calculated by the Bliss independence model (Figures 4A and 4B) and no synergistic relationship in proliferative cells (Figure S3A). Likewise, the combination of ABT-263 with other SMAC mimetics SM-164 and GDC-0152 also produced this increased senolytic effect (Figures S3B and S3C). We used a slightly lower dose range (up to 0.5 μM which is near the EC50 dose) of ABT-263 than in the screens to be able to identify effects enhancing cell death. We also validated the DIABLO and XIAP hits from the ABT-199 screen with the combination of ABT-199 and birinapant showing an even stronger synergy on senescent cell viability as shown by Bliss score (Figures 4C and 4D) while still sparing proliferative cells (Figure S3D). This result was further validated using ABT-199 in combination with SM-164 or GDC-0152 (Figures S4A and S4B), as well as in IR-SENs with all three SMAC mimetics (Figures S4C–S4E) and we confirmed that inhibition was being achieved at doses which are synergistic (Figure S4F).
Figure 4. BH3 and SMAC mimetics synergize to induce selective death in senescent cells.
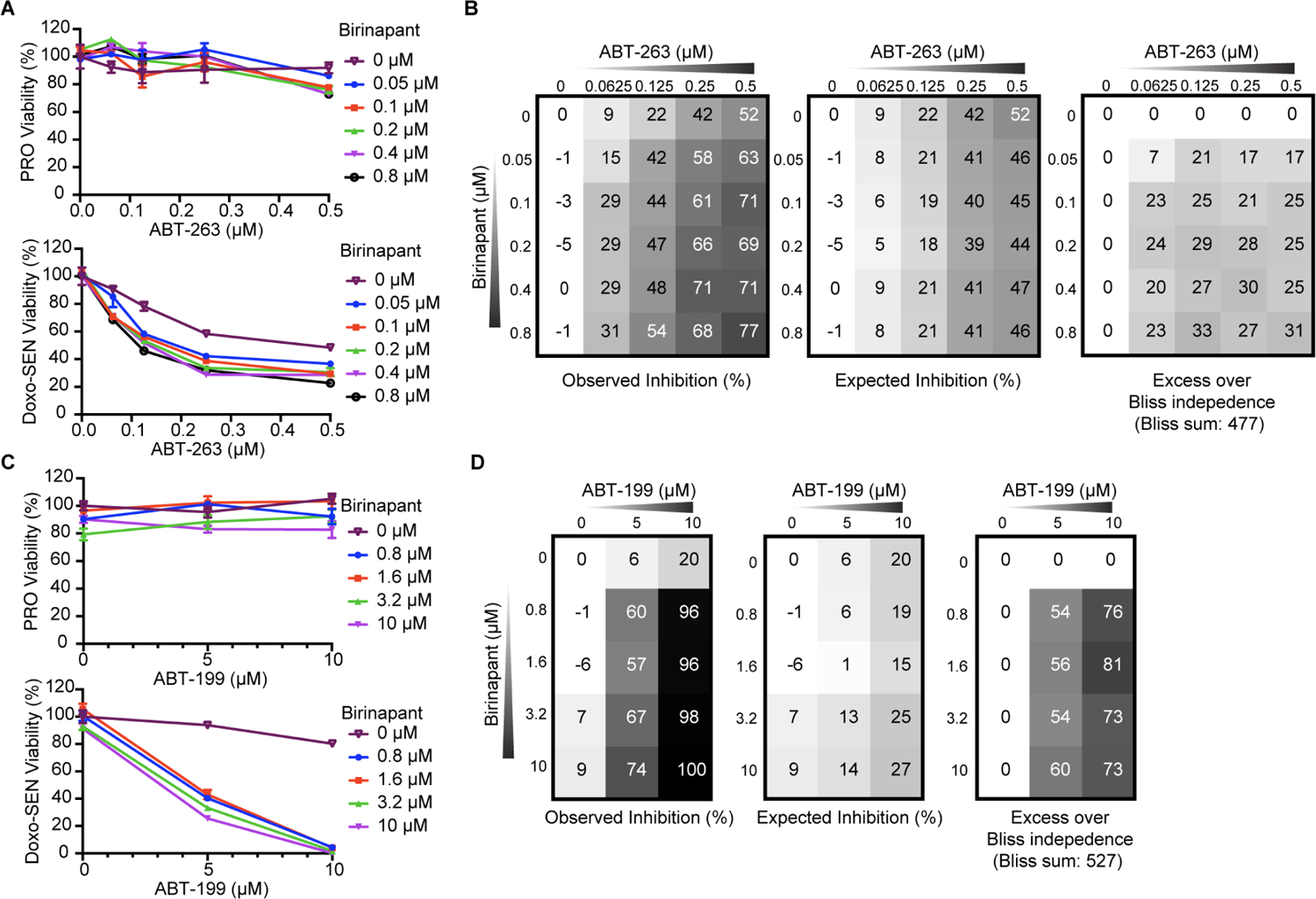
(A) Proliferative (top) and Doxo-induced senescent (bottom) IMR-90s were treated with ABT-263 and birinapant at the indicated concentrations for 3 d before viability was assessed relative to no drug control. Data are representative of two independent experiments performed in triplicate and are presented as mean ± s.e.m.
(B) The percent expected inhibition is subtracted from the percent observed inhibition at each combination of drug doses in Doxo-induced senescent IMR-90s to calculate drug synergy represented by excess over Bliss independence.
(C) Proliferative (top) and Doxo-induced senescent (bottom) IMR-90s were treated with ABT-199 and the SMAC mimetic birinapant at the indicated concentrations for 3 d before viability was assessed relative to no drug control. Data are representative of two independent experiments performed in triplicate and are presented as mean ± s.e.m.
(D) The percent expected inhibition is subtracted from the percent observed inhibition at each combination of drug doses in Doxo-induced senescent IMR-90s to calculate drug synergy represented by excess over Bliss independence.
Having validated the ability of ABT-199 and SMAC mimetics to synergize in senolysis, we next sought to orthogonally confirm cell death, mitochondrial outer membrane permeabilization, and the senolytic ability of the combination in different inducers of senescence and cell types. Using flow cytometry and Annexin V staining, we analyzed the extent of cell death in proliferative and senescent IMR-90s treated with vehicle, ABT-199/SMAC mimetic, or ABT-263, each with or without the pan-caspase inhibitor Q-VD-Oph (QVD). As with ABT-263, the percent of apoptotic cells in the ABT-199/SMAC mimetic group decreased with QVD treatment, suggesting that the combination acts through caspase-mediated apoptosis in senescent cells (Figure 5A). Mitochondrial membrane potential was significantly reduced in senescent cells treated with ABT-199 alone, ABT-199/SMAC mimetic, and ABT-263, but not in proliferative cells (Figure 5B). Caspase 3/7 activity was significantly increased in senescent cells treated with ABT-199/SMAC mimetic and ABT-263, but not in proliferative cells (Figure 5C). The combination of ABT-199 and SMAC mimetic was senolytic across different senescence inducers in IMR-90s including H2O2, bleomycin, palbociclib, and etoposide (Figure S5A). The combination was also senolytic and synergistic in senescent WI-38 normal lung fibroblasts (Figures 5D and S5B), although notably with higher levels of toxicity following treatment with birinapant alone in both proliferative and senescent cells. We further tested the combination in senescent human retinal microvascular endothelial cells (HRMECs), a disease-relevant primary cell population recently utilized in a study which implicated the pathological role of senescent cells in diabetic retinopathy.32 ABT-199/birinapant was also senolytic in HRMECs (Figure S5C).
Figure 5. ABT-199 and SMAC mimetic combination is senolytic through apoptosis across different cell types.
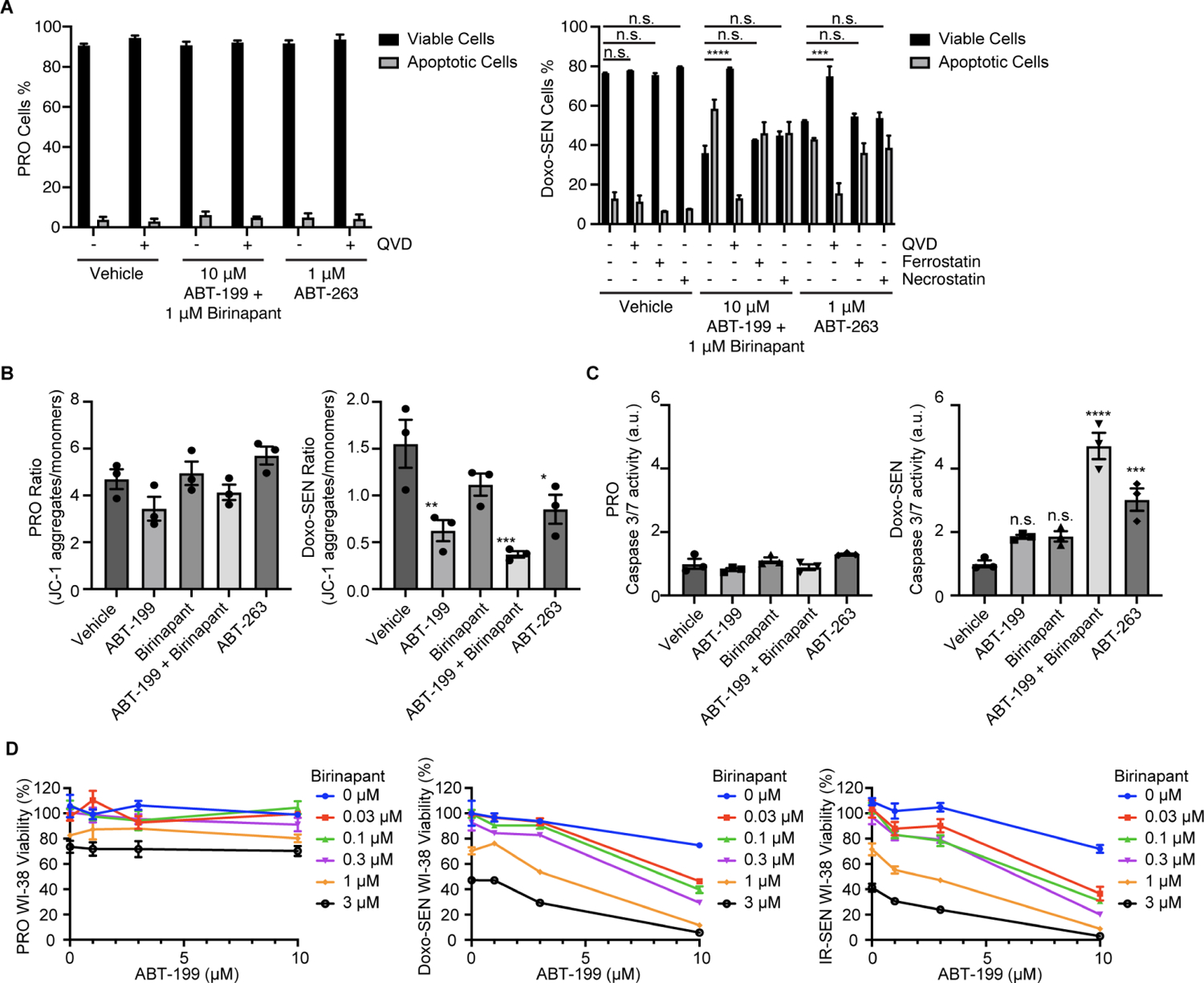
(A) Percentage of viable and apoptotic proliferative (left) and Doxo-induced senescent (right) IMR-90s 24 h after treatment with vehicle, 10 μM ABT-199 and 1 μM birinapant, or 1 μM ABT-263, all with and without 20 μM Q-VD-Oph (QVD), 10 μM ferrostatin, and 10 μM necrostatin.
(B) Proliferative (left) and Doxo-induced senescent (right) IMR-90s were treated for 24 h with vehicle, 10 μM ABT-199, 1 μM birinapant, 10 μM ABT-199 and 1 μM birinapant, or 1 μM ABT-263. The treated cells were then stained and measured by flow cytometry to determine the ratio of JC-1 aggregates to JC-1 monomers. Data are representative of two independent experiments performed in triplicate and are presented as mean ± s.e.m. One-way ANOVA with Dunnett’s post-hoc test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
(C) Normalized caspase 3/7 activity in proliferative (left) and Doxo-induced senescent (right) IMR-90s that were treated for 24 h with vehicle, 10 μM ABT-199, 1 μM birinapant, 10 μM ABT-199 and 1 μM birinapant, or 1 μM ABT-263. Data are representative of two independent experiments performed in triplicate and are presented as mean ± s.e.m. One-way ANOVA with Dunnett’s post-hoc test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
(D) Proliferative (left), Doxo-induced senescent (middle), and IR-induced senescent (right) WI-38s were treated with ABT-199 and birinapant at the indicated concentrations for 3 d before viability was assessed relative to no drug control.
See also Figure S5.
ABT-199 and SMAC mimetic combination spares human platelets and reduces levels of senescent cell markers in vivo
Despite that BCL-2 is not required for platelet survival,33 the effect of simultaneous inhibition of BCL-2 and IAPs on platelets is still unclear. To determine if the combination of ABT-199 and birinapant is toxic to platelets, we cultured human platelets in either ABT-263, ABT-199, or the ABT-199/birinapant combination before assessing viability. ABT-263 significantly depleted platelets starting at 300 nM while the combination did not affect human platelet viability (Figures 6A, S6A, and S6B). To further test for toxicity to platelets, we treated mice with either ABT-263 or the ABT-199/birinapant combination and assessed platelet counts (Figure S6C). We found that ABT-263 led to profound thrombocytopenia, whereas the ABT-199/birinapant combination exhibited minimal platelet toxicity after treatment and no evidence of toxicity after four treatments (Figures 6B and S6D).
Figure 6. ABT-199 and SMAC mimetic combination spares human platelets and reduces levels of senescent cell markers in vivo.
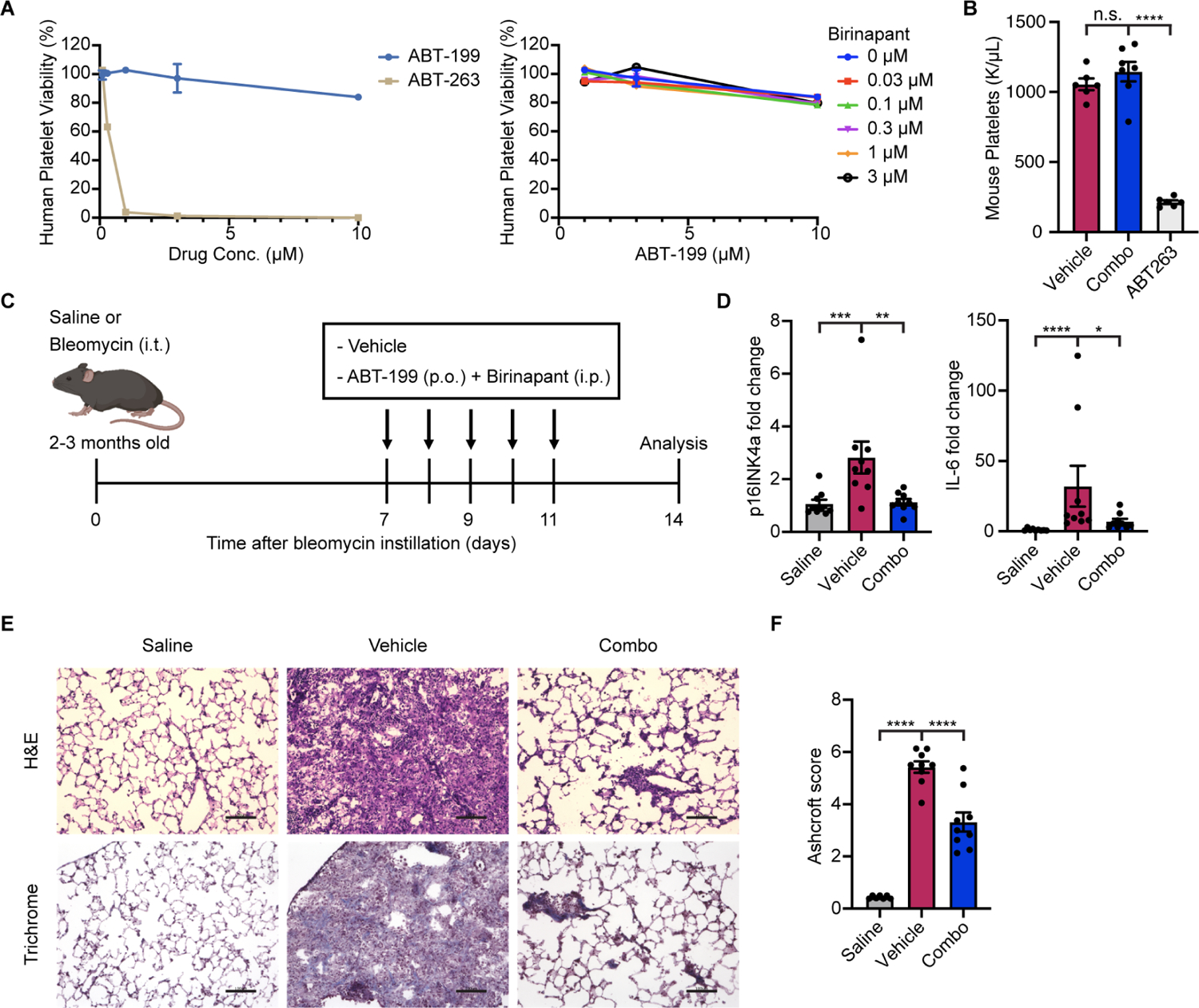
(A) Human platelets were treated with ABT-199 or ABT-263 (left) or ABT-199 in combination with birinapant at the indicated concentrations (right) for 3 d before viability was assessed relative to no drug control. Data are representative of two independent experiments performed in triplicate and are presented as mean ± s.e.m.
(B) Platelet count from mice treated with vehicle (n = 6), ABT-199 in combination with birinapant (n = 7), or ABT-263 (n = 6), eight days after the initiation of treatment.
(C) Schematic of bleomycin-induced IPF mouse model experiment.
(D) mRNA expression of p16INK4a and Il6 in murine lungs were quantified by qRT-PCR and normalized by Tbp levels. Fold-increase was calculated with respect to the mRNA levels in saline-treated control mice (n = 9 in each group). Data are representative of two independent experiments performed in triplicate and are presented as mean ± s.e.m. One-way ANOVA with Dunnett’s post-hoc test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
(E) Representative images of hematoxylin and eosin (top) and Masson’s trichrome (bottom) stained lung sections from saline control (left), vehicle-treated (middle), and ABT-199/birinapant-treated (right) mice. Scale bar: 100 μm.
(F) Pulmonary fibrosis was quantified with modified Ashcroft scores (n = 7 control group, n = 9 vehicle and combo groups). Data are representative of two independent experiments performed in triplicate and are presented as mean ± s.e.m. One-way ANOVA with Dunnett’s post-hoc test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
See also Figure S6.
Given the potency and selectivity of the ABT-199/birinapant combination against senescence in vitro and the absence of toxicity to human and mouse platelets, we wondered whether the combination could be used therapeutically in a systemic fashion. Recent evidence has suggested a causal role of senescent cells in the pathogenesis of idiopathic pulmonary fibrosis (IPF).34,35,36,37 IPF is a deadly chronic lung disease characterized by progressive lung scarring with a median survival of less than three years and aging as a major risk factor.38,39,40 Consistently, studies have demonstrated an accumulation of senescent cells in the lungs of patients with IPF.34,39,41,42 Moreover, the importance of human genetic variants in telomerase in both sporadic and familial disease suggests the pathological contribution of cellular senescence.43,44,45 Removal of senescent cells in experimental lung fibrosis models alleviated pulmonary fibrosis and improved lung function.34,46,47 We therefore tested the ability of the systemic combination to clear senescent cells in the bleomycin-injury IPF model, in which bleomycin induces senescence in various lung cells including alveolar epithelial cells and myofibroblasts.34,48,49 Mice were treated with a single dose of bleomycin through intratracheal administration and subsequently treated with vehicle or the ABT-199/birinapant combination (Figure 6C). The levels of senescent cell markers increased 14 days after bleomycin injury and, while treatment was unable to significantly restore some markers like p21 and Col1a1 (Figures S6E and S6F), several others including p16INK4a, Il6, Col1a2, and senescence-associated β-galactosidase (SA-βgal), were significantly decreased by the combination treatment (Figures 6D; Figures S6F and S6G). Strikingly, treatment with the combination significantly reduced the severity of pulmonary fibrosis assessed by histological staining and Ashcroft histopathology scoring (Figures 6E and 6F).
Next, we tested the combination in an independent disease model. Senescence is associated with metabolic disease and obesity-induced disorders including nonalcoholic steatohepatitis (NASH).50,51 Targeting senescent cells has been shown to mitigate liver damage in mice receiving high-fat diet.9,52,53,54 In a mouse model of NASH induced by feeding of a choline-deficient, high-fat diet (CDA-HFD), mice were treated with vehicle, the ABT-199/birinapant combination, or ABT-263 after four weeks on a CDA-HFD (Figure S6H). Supporting the induction of senescence in the NASH model, levels of liver p16INK4a mRNA, serum alanine aminotransferase (ALT), and collagen content were significantly increased by the diet in the vehicle CDA-HFD group compared to normal chow controls (Figures S6I, S6J, and S6K). We noted a reduction in liver p16INK4a levels by the combination (Figure S6I), as well as a lower level of serum ALT, suggesting that liver function was improved (Figure S6J). Overall, this systemic senolytic combination treatment resulted in the reduction of senescent cell markers consistent with their clearance and partially ameliorated some aspects of age-related pathologies.
Death-seq identifies enhancers of senescent cell death in the absence of any senolytic drug
Given the ability of the Death-seq screening methodology to identify genetic modifiers of cell death in response to small molecule perturbations, we next tested if this methodology could be used to identify genetic modifiers of senescent cell death in the absence of pharmacologic induction. Toward this end, we conducted a Death-seq genome-wide CRISPR KO screen in Doxo-SEN IMR-90s, collecting dying cells at days 7 and 9 after senescence induction (Figure 7A). Using a 30% FDR cutoff, this genome-wide screen identified 13 gene KOs which increased senescent cell death (Figure 7B; Tables S1 and S2). Only 1 of these 13 genes, MARCH5, was also a hit in the ABT-263 genome-wide screen. BCL2L1 was one of the top 13 hits, providing validation since this was an expected hit. Several hits, including the second most significant hit, TRAPPC13, have not been previously implicated to play a role in senescence or as a target to increase cell death.
Figure 7. Death-seq identifies modifiers of cell death in the absence of drugs as well as in nonapoptotic cell death.
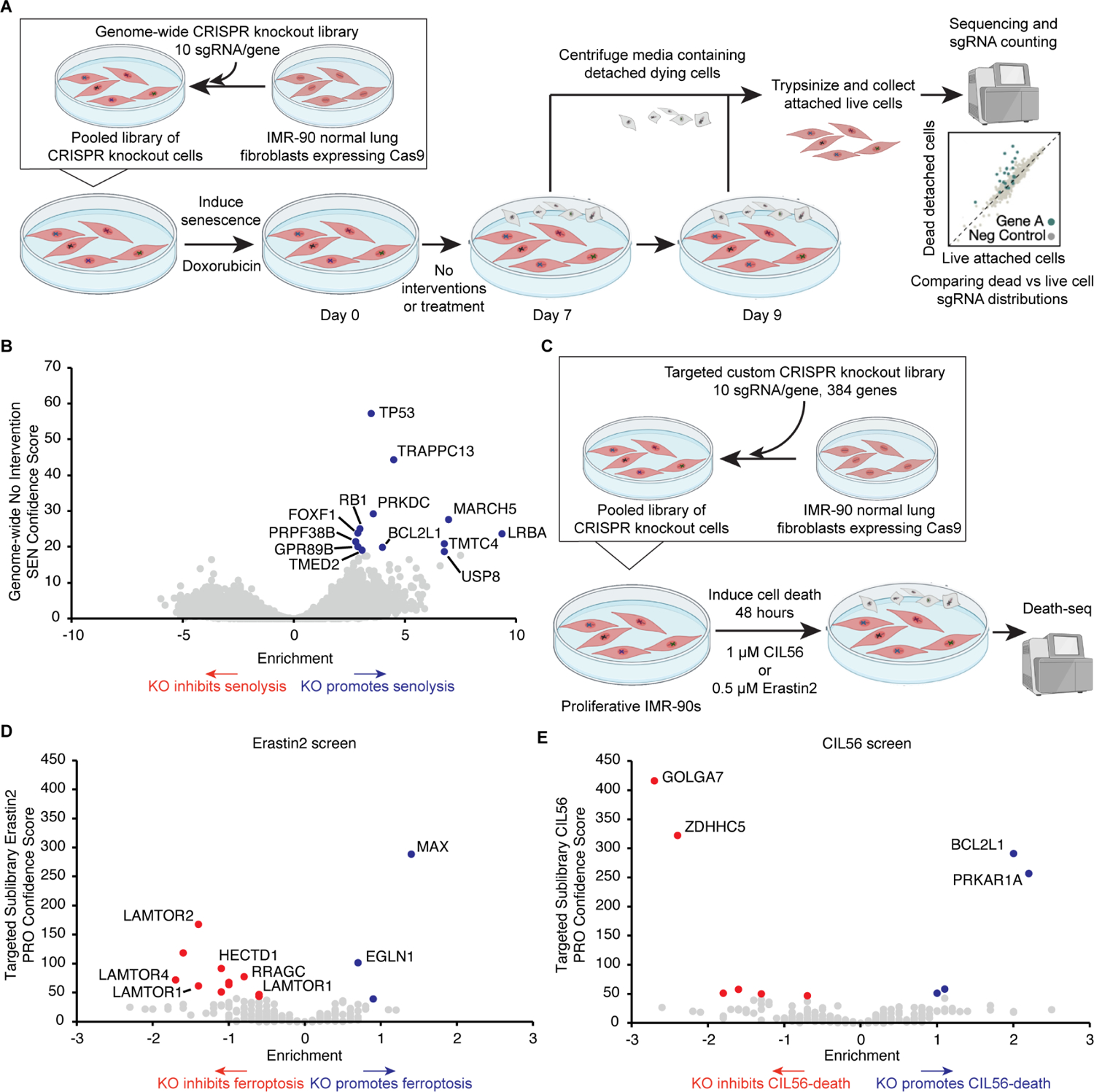
(A) Schematic of Death-seq genome-wide CRISPR KO screen for modifiers of Doxo-SEN cell death in the absence of small molecules or perturbation. The screen was performed in duplicate.
(B) Volcano plot of the effects and confidence scores of all the genes in the genome-wide CRISPR screen in the absence of small molecules. Effects and casTLE scores are calculated by casTLE. Labelled are the 13 genes passing 30% FDR for promoting (blue) senescent cell death when knocked out.
(C) Schematic of Death-seq sublibrary CRISPR KO screens in proliferative IMR-90s for modifiers of ferroptosis induced by treatment with 0.5 μM erastin2 for 48 h and cell death induced by treatment with 1 μM CIL56 for 48 h. The screens were performed in duplicate.
(D) Volcano plots of the effects and confidence scores of all the genes in the sublibrary CRISPR screens for modifiers of erastin2-induced ferroptosis (left) and CIL56-induced cell death (right) in proliferative IMR-90s. Effects and casTLE scores are calculated by casTLE. Labelled are the genes passing 30% FDR for inhibiting (red) or promoting (blue) cell death when knocked out. See also the results and raw sequencing counts from screens in Tables S1 and S2 respectively.
Death-seq does not bias towards apoptosis and identifies modifiers of nonapoptotic forms of regulated cell death
In order to examine Death-seq in different cell types and to further test if this method is biased towards revealing genes in the apoptosis pathway, we conducted a CRISPR KO screen with the custom targeted sublibrary in human iPSC-derived cardiomyocytes to test for modifiers of cardiotoxicity induced by the MCL1 inhibitor S63845 (Figure S7A). In agreement with recent evidence55,56 and helping to confirm that the method does not bias to apoptosis, the screen did not reveal apoptosis machinery as a mechanism of MCL1 inhibitor induced cardiotoxicity (Figure S7B; Tables S1 and S2). A genome-wide screen, instead of one using a library biased towards apoptosis pathway members, is likely necessary to reveal the non-apoptotic mechanism behind MCL1 inhibitor induced cardiotoxicity.
To further test if Death-seq did not bias towards apoptosis and could also identify modifiers of nonapoptotic forms of regulated cell death, we conducted CRISPR KO screens in proliferative IMR-90s treated with erastin2 to induce ferroptosis and the small molecule CIL56 to induce an unconventional form of nonapoptotic cell death (Figure 7C).57 We used non-senescent, proliferating cells for these screens as ferroptosis and CIL56-induced cell death are phenomena mostly characterized in proliferative cells. These screens were conducted using the previously described custom targeted sublibrary enriched with hits from the ABT263 Doxo-SEN screen. As such, this library is enriched with modifiers of apoptosis and does not contain major regulators of ferroptosis. Using a 10% FDR cutoff, the erastin2 and CIL56 Death-seq screens identified 14 and 6 gene KOs, respectively, which either inhibit or promote death by each drug (Figure 7D and 7E; Tables S1 and S2). Notably, none of the apoptosis pathway members were included in these hits despite many being hits in the ABT-263 screens. The erastin2 screen, like the ABT-263 screen, featured a number of hits inhibiting cell death which were members of the Rag-Ragulator complex, such as LAMTOR1–4 and RRAGC. KOs of MAX and the prolyl hydroxylase domain (PHD) enzyme EGLN1 were the top two hits enhancing cell death. The CIL56 screen revealed PRKAR1A as a hit demonstrating that its KO enhanced cell death which was interesting as it was a hit with the opposite direction of effect, its KO inhibited cell death, in the ABT-263 Doxo-SEN IMR-90 screen suggesting a very different role of the gene in CIL56-induced cell death compared to ABT-263-induced senolysis. The top two hits from the CIL56 screen inhibiting death were GOLGA7 and ZDHHC5, which form a plasma membrane protein acyltransferase complex required for CIL56-induced cell death,57 thus providing validation for the screen and the ability of Death-seq to identify regulators of nonapoptotic cell death.
DISCUSSION
Here, we present a screening method to systematically assess modifiers of cell death. We discovered a number of robust modifiers of ABT-263 senolytic therapy and demonstrated the ability to identify genetic modifiers of death, even with drugs that do not alone induce significant amounts of cell death such as ABT-199 and birinapant. Using the new screening method, we were able to generate a high-quality catalog of genes that served as modifiers of senescent cell death. These data sets of modifiers of senolysis will serve as an important resource for clinicians and experts in the fields of cell death, BH3 mimetics, SMAC mimetics, mitochondrial biology, and cellular senescence. The BCL-2 family of proteins has many intra-family interactions and a high degree of complexity that makes it difficult to dissect and predict cell fate with standard molecular biology techniques.58 Our results will help provide a resource and technique to further the molecular understanding of BCL-2 inhibitors, especially in the context of cellular senescence where there is a dearth of work relative to the cancer field. The data sets not only contain a number of well characterized members of the apoptosis pathway, but also less well characterized genes in apoptosis pathways such as HCCS which encodes for the enzyme holocytochrome c-type synthase. Many hits intriguingly have no previous connection to apoptosis, cell death, or senescence. An example of one of these hits is PPTC7, a strong hit inhibiting death from both our ABT-263 and ABT-199 senolysis screens. PPTC7 encodes for a poorly characterized PP2C phosphatase localized to the mitochondrial matrix. Published work has revealed evidence for the role of PPTC7 in mitochondrial metabolism.19 However, to our knowledge, no evidence exists to suggest a role of PPTC7 in cell death. Two screen hits which promoted senolysis, GOLGA7 and ATP13A1, are two more examples of surprising hits, given the lack of documented connection to senolysis or apoptosis, with GOLGA7 recently found to be necessary for an unconventional, nonapoptotic form of cell death.57 Our ABT-263 and ABT-199 screen resistance hits also pointed to the role of the Rag-Ragulator complex, the GATOR complex, the HOPS complex, and the CORVET complex in cell death. Future work is required to help tease apart the mysteries revealed by Death-seq, including the role of PPTC7, GOLGA7, ATP13A1, and the lysosomal associated complexes in senolysis by BH3 mimetics, as well as the many other hits not currently associated with cell death or senescence.
We validated the unexpected finding from our screens that the BH3 mimetics ABT-263 and ABT-199 act in a synergistic fashion with SMAC mimetics to induce senolysis. Preliminary in vitro studies in the cancer field hint at the potential of a synergy between BCL-2 inhibition and SMAC mimetics to improve efficacy and reduce toxicity.59,60 This work suggested that the dysregulation of the IAP proteins in cancer cells may hinder the death of cancer cells. Just like cancer cells, senescent cells are regarded as resistant to cell death due to their general dysregulation of anti-apoptotic proteins.2 However, the exact mechanisms of this anti-apoptotic phenotype remain to be investigated. Many questions remain to be answered regarding the molecular mechanisms of the synergy between ABT-199 and SMAC mimetics in senolysis and whether this relates to the general phenomenon of senescent cells resistance to death.
Selectively ablating damaged cells, including through the use of senolytics, has shown promise as a strategy to address age-related disease, but because of toxicities, most senolytics have been limited to local rather than systemic administration.8 Likewise, the lack of definitive characterization of cellular senescence in vivo during physiological aging using selective markers and omics techniques has bottlenecked progress to translating senolytics to target aging.61,62 Although not included in this manuscript, we analyzed single-cell RNA-seq datasets of physiological aging in mice for markers of senescence, such as Cdkn2a and markers of the senescence-associated secretory phenotype, to identify better target cell types to screen with Death-seq in vitro and to see if aged mice could be treated with the senolytic combination. The analyzed datasets included the Tabula Muris Senis transcriptomic atlas looking across 23 different organs and tissues,63 as well as a brain-specific atlas.64 Despite an increase in both the percent of cells expressing Cdkn2a and expression levels of Cdkn2a increasing when pooling cells across 23 organs,63 we were unable to find cells with multiple markers of senescence in physiologically aged mice in either atlas, in accordance with another recent independent cell atlas dataset.65
Given the world’s dramatically increasing population of persons over the age of 65, it is important to develop strategies to extend and restore healthspan.66,67,68 The COVID-19 pandemic has demonstrated the enhanced vulnerability of persons over 65 and those with age-related disease to hospitalization, death, and other adverse events as a result of infections, in this case with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Senescent cells have been highlighted as a potential reason for this enhanced vulnerability of persons over 65,69 with a recent study demonstrating that senescent cells have an amplified inflammatory response to SARS-CoV-2 and that in old mice infected with SARS-CoV-2 related virus, senolytics reduce mortality and increase antiviral antibodies.70 A systemic senolytic treatment with minimal side effects could be used in the future to protect persons over 65 and those with age-related disease from SARS-CoV-2 and future pandemics. Likewise, a tolerable systemic senolytic treatment will be critical for clinical trials in other diseases that cannot be treated with local administration such as IPF, COPD, atherosclerosis, and neurodegeneration.8 With this goal in mind, we find that the combination of ABT-199 and birinapant is not toxic to platelets and reduces levels of senescent cell markers in mouse models of IPF and NASH, establishing therapeutic potential. ABT-199 is FDA approved and birinapant has completed several Phase 2 clinical trials which could accelerate the clinical use of this combination. At least eight SMAC mimetics have shown to be well tolerated with reasonable safety profiles in human trials.14 The drug class has failed so far to advance to approval due to limited clinical activity as a single-agent in oncology.
Death-seq, through its comparison of guide composition in dying cells directly to live cells, allows more efficient and effective genome-wide screens and unlocks the potential for new findings in the senescence field and in a wide variety of fields outside of senescence. Due to the limitations of conventional negative selection screens, CRISPR screens have yet to be used to identify senolytic targets. Considering the pleiotropic phenotypes of senescent cells, other groups have stated the need to shift from traditional reductionist scientific methods to new systematic high-throughput methods to study senescence.3 Similar to the cancer field, systematic efforts are needed to untangle the anti-apoptotic defense mechanisms of senescent cells. In addition to enabling the study of senescent cells, Death-seq can be of benefit to many other areas of biological and pathological study. The simplicity of this screening method and its ability to increase signal and decrease noise will enable the identification of enhancers and mechanisms of cell death subroutines in other fields where it was previously not possible with currently available techniques. For instance, although not explored in this work, this method is well suited to identify genetic modifiers of cell death in neurodegeneration and cardiovascular disease as well as mechanisms of drug resistance and synthetic lethality in cancer. Additionally, Death-seq is amenable to performing screens in more physiologically relevant culture systems such as 3D organoid systems,22 which are more difficult to scale-up and more resource-intensive compared to the use of immortalized cell lines.
Limitations of the study
Death-seq with the readout included in this manuscript is an assay for cell detachment accompanying cell death. Thus, any knockout which interferes with cell attachment may show up as a hit in the screens. For the same reason, the assay could be useful for studying the biological phenomenon of adhesion or anoikis, a form of regulated cell death occurring upon cell detachment. In order to use Death-seq in contexts where the readout of cell detachment is not possible, such as for suspension cells or in vivo, the readout of Death-seq would need to be adapted. For example, for suspension cells using a readout of density-gradient centrifugation to separate less dense dying cells from living cells or for in vivo screens using a FACS-based readout with genetic mouse models with fluorescent reporters for cell death based, for example, on caspase activation or Annexin V expression. For senescence screens, we generated knockouts while the cells were still proliferative over the span of at least two weeks. We then induced senescence and conducted the treatment. As a result, knockouts of genes which are essential for cell proliferation were significantly reduced or eliminated before the screen was conducted. This was useful for the senescence screens as we were targeting knockouts of genes which were preferentially essential to senescent cells. Also, it was possible that knockouts of genes which promote senescence or potentially even different senescent-like states occurred before treatment.
Although we provide evidence that combinations of BH3 and SMAC mimetics are senolytic across a variety of cell types and inducers of senescence, senescence is a heterogeneous state and as such it is possible that the results from our screen could be specific to the cell states used in screening. As understanding of which damaged cell types, including senescent cell types, play pathogenic versus beneficial or negligible roles in age-related disease and aging continues to improve, we expect that Death-seq will be utilized in additional cell types based on their diseasespecific pathogenic relevance. Just like the Cancer Dependency Project has profiled hundreds of cancer cell models to identify genetic vulnerabilities for therapeutic development,70 we anticipate that Death-seq will enable a similar project in the field of “senescent cells” across different models and inducers of senescence. Additionally, given the nascent and non-selective tools which exist to study in vivo senescence,71 the mouse models of “senescence” used in this study are limited by the number of robust in vivo biomarkers available to confirm senolytic efficacy.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Thomas Rando (trando@mednet.ucla.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data S1 refers to unprocessed data underlying the display items in the manuscript, related to all Figures 1–7 and S1–S7. Tables S1–S4 contain the screening data produced from this work as well as sgRNA/shRNA sequences.
This paper does not report original code. All screening data was analyzed with the casTLE scripts version 1.0 that are available at https://github.com/elifesciences-publications/dmorgens-castle. Flow cytometry data was analyzed using FlowJo 10 software. Statistics were calculated using GraphPad Prism 9 software.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell culture
Media was changed every two to four days and cells were cultured in tissue culture plates in a humidified 37°C incubator set at 5% CO2 and 5% O2. IMR-90s (sex = female) and WI-38s (sex = female) were cultured in Dulbecco’s Modified Eagle Medium (Corning, 10013CV) supplemented with 10% heat-inactivated FBS (Omega Scientific FB-02) and 1% pen-strep (Omega Scientific PS-20). HRMECs were cultured in Endothelial Cell Growth Medium MV2 kit (PromoCell, C-22022). Cells were periodically tested to ensure absence of mycoplasma. To induce cellular senescence by doxorubicin, cells were treated with doxorubicin at 250 nM for the first 24 hours then 83.3 nM for the subsequent 48 hours before washing out drug and culturing for another 4 days before experimentation. To induce cellular senescence by irradiation, cells were exposed to 10 Gy X-rays in an X-RAD 320 before changing the media and waiting 10 days before experimentation. To induce cellular senescence by H2O2, cells were treated with 200 μM H2O2 for 2 hours every other day for three total treatments and experimentation began 7 days after the first treatment. To induce cellular senescence with bleomycin, cells were treated with 40 μg/mL bleomycin for 2 hours and experimentation began 7 days after initial treatment. To induce cellular senescence with palbociclib, cells were treated with 10 μM palbociclib for 7 days before experimentation. To induce cellular senescence with etoposide, cells were treated with 50 μM etoposide for 48 hours and experimentation began 7 days after initial treatment.
Human platelet isolation and viability assay
Human platelets were isolated and cultured as described.73 In brief, human platelet-rich plasma (PRP) was purchased within 3 days after harvest. The PRP was transferred into a 50 mL conical tube with 5 mL acid citrate buffer (Santa Cruz Biotechnology, sc-214744). This mixture was centrifuged at 250g for 20 minutes at room temperature without the centrifuge brake. PRP was collected into a 15 mL polypropylene conical tube and prostaglandin E1 (PGE1) (Santa Cruz Biotechnology, sc-201223A) and apyrase (Sigma-Aldrich, A6237) were added to final concentrations of 1 μM and 0.2 units/mL, respectively, to prevent clotting. After gently mixing the solution, platelets were pelleted by centrifugation at 1200g for 10 minutes without the centrifuge brake and the pelleted platelets were gently washed without disrupting the pellet in 2 mL HEPES Tyrode’s buffer (Boston BioProducts, PY-921WB) containing 1 μM PGE1 and 0.2 units/mL apyrase. After washing, pellets were slowly resuspended in 10 mL HEPES Tyrode’s buffer containing 1 μM PGE1, 0.2 units/mL apyrase, and 10% FBS. For the viability assays, platelet number was adjusted to 2 × 108/mL in HEPES Tyrode’s buffer containing 1 μM PGE1, 0.2 units/mL apyrase, and 10% FBS. Each treatment was conducted in 2 mL of platelet suspension in 15 mL polypropylene conical tubes on a rotating platform at room temperature and the viability of platelets was measured after treatment for indicated time points using the XTT assay with the Cell Proliferation Kit II according to the manufacturer’s protocol. Experimental values were normalized using a vehicle-treated and an ABT-263-treated (50 μM) condition to set the maximal and minimal viability values, respectively. Extra precautions such as room temperature storage/incubation, centrifuging without a deceleration brake, gently pipetting and 20 mixing, and rotating platelets during incubation were taken to avoid platelet activation, aggregation, and spontaneous apoptosis.
Animals
Animal procedures were approved by the Administrative Panel on Laboratory Animal Care of the VA Palo Alto Health Care System. C57BL6 male mice were housed in specific pathogen-free conditions in barrier protected rooms under a twelve-hour light-dark cycle and were fed ad libitum.
In vivo treatment
ABT-263 and ABT-199 were prepared in 10% ethanol, 30% polyethylene glycol 400, and 60% Phosal 50 PG, followed by sonication at room temperature for 2 hours. Birinapant was prepared in 12.5% Captisol adjusted to pH 4. ABT-263 and ABT-199 were administered by oral gavage (50 mg/kg). Birinapant was administrated by intraperitoneal injection (15 mg/kg).
Bleomycin-induced pulmonary fibrosis model
8- to 10-week-old C57BL/6 male mice were anesthetized and intratracheally instilled with 50 μL of sterile saline (control group) or USP grade bleomycin (1.8U/kg). Body weights were monitored throughout the study. Bleomycin-treated animals with less than 10% weight loss over the first 7 days were excluded from the study. Mice were treated with 50 mg/kg of ABT-199 and 15 mg/kg of birinapant, or vehicle control for 5 days starting 7 days after bleomycin instillation. Animals were euthanized at 14 days after bleomycin injury, and lungs were harvested for qRT-PCR and histological analysis as described below.
Non-alcoholic steatohepatitis (NASH) model
6-week-old C57BL/6 male mice were fed with a choline-deficient, L-amino acid defined, high-fat diet (CDA-HFD: A06071302i, Research Diets) for 7 weeks. After 4 weeks of feeding, mice were treated with 50 mg/kg of ABT-199 and 15 mg/kg of birinapant, 50 mg/kg of ABT-263 or vehicle control seven times in 3 weeks. After 7 weeks of high-fat diet, livers were harvested for qRT-PCR analysis and hydroxyproline assay as described below, and blood was collected and analyzed by Stanford Animal Diagnostic Laboratory. Hydroxyproline assay was performed using the Hydroxyproline Assay Kit (Sigma-Aldrich, MAK008–1KT) according to manufacturer’s instructions.
Mouse platelet assay
8- to 10-week-old C57BL/6 male mice were treated with 50 mg/kg of ABT-199 and 15 mg/kg of birinapant, 50 mg/kg of ABT-263 or vehicle control on days 0, 2, 4, and 7. Blood was collected on days 1 and 8 and a complete blood count was performed by Stanford Animal Diagnostic Laboratory.
METHOD DETAILS
Genome-wide Death-seq positive selection cell death CRISPR screening
For the genome-wide ABT-263 CRISPR–Cas9 screen, the CRISPR/Cas9 deletion library with ten sgRNAs per gene was synthesized, cloned, and infected into Cas9-expressing IMR-90s as previously described.74 This library includes 5644 non-targeting negative control sgRNAs and 6750 safe-targeting negative control sgRNAs. ~500 million IMR-90s stably expressing EF1α-Cas9-BLAST were infected with the genome-wide sgRNA library at an MOI < 1. Infected cells underwent puromycin selection (1.5 μg/ml) for 4 days, after which puromycin was washed out and replaced with normal growth medium. Deep sequencing after selection confirmed sufficient sgRNA library representation. Cells were split into two technical replicates and maintained for ~2 weeks before inducing senescence by adding doxorubicin at 250 nM for the first 24 hours then 83.3 nM for the subsequent 48 hours before washing out doxorubicin for 4 days in normal growth medium. 7 days after doxorubicin addition, the ~350 million senescent cells in each technical replicate at ~1,500x coverage (~1,500 cells containing each sgRNA) were treated with 1 μM ABT-263 for 24 hours. To harvest DNA from the dying cells, the medium was collected from above the adherent cells to collect detached cells. The tissue culture plates were washed two times in PBS and both washes were also collected and combined with the media. The plates were washed one more time in PBS and then treated with DNase I (Worthington Biochemical Corporation) for 10 minutes at 37°C to remove any remaining genomic DNA from dying cells as described15 and then washed three more times in PBS to remove residual DNase I and dying cells. The collected media containing dying cells was centrifuged for 5 minutes at 550g while the remaining attached living cells on the plates were trypsinized and centrifuged for 5 minutes at 300g before genomic DNA was harvested from all screen populations separately by with a DNeasy Blood and Tissue Kit (Qiagen) including proteinase K digestion to inactivate residual DNase I. Deep sequencing on an Illumina Nextseq platform with a NextSeq 500/550 High Output kit was used to monitor library composition which was then directly compared between the dying and live cell populations (“Death-seq”) using casTLE (https://github.com/elifesciences-publications/dmorgens-castle). In brief, the casTLE algorithm compares each set of 10 gene-targeting guides to the non-targeting and “safe-targeting” control sgRNAs. The enrichment of individual guides was calculated as the log ratio between dying and living cell populations, and gene-level effects were calculated from the ten guides targeting each gene. A confidence score (casTLE score) was then derived as a log-likelihood ratio describing the significance of the gene-level effect. P values were then estimated by permutating the targeting guides as previously described,18 and hits were called using FDR thresholds calculated via the Benjamini-Hochberg procedure. See Table S1 and Table S2 for the complete results for all CRISPR screens. The top 350 genes ranked by casTLE score in the genome-wide ABT-263 CRISPR screen were used to produce a protein-protein interaction enrichment analysis using the Metascape tool75 and the interaction network was visualized using Cytoscape.76 Reactome enrichment analysis was performed by inputting all genes passing a 30% FDR in the ABT-263 genome-wide screen into the Reactome tool (https://reactome.org/PathwayBrowser)77 and sorting the results on adjusted P value. Reactome IDs for Reactome categories reported in Figure 1 are: R-HSA-111471, R-HSA-109606, R-HSA-111463, R-HSA-111464, R-HSA-111459, R-HSA-111469. For the genome-wide CRISPR–Cas9 screen for modifiers of Doxo-SEN cell death in the absence of small molecules or perturbation, the same protocol as the genome-wide ABT-263 screen was used with the exceptions that no drug treatment occurred and that dying cells were extracted from collected media at two time points, day 7 and day 9, post-doxorubicin addition and pooled together for sequencing.
Sublibrary Death-seq positive selection cell death CRISPR screening
The targeted custom sublibrary targets a total of 384 genes (10 sgRNA per gene): the top ~350 genes as ranked by casTLE score from the genome-wide screen as well as select apoptosis and lysosomal-related genes. It also contains 374 control “safe-targeting” sgRNAs. See Table S3 for the complete sgRNA composition of the targeted custom sublibrary. Agilent Technologies synthesized the sublibrary oligonucleotides which were cloned into pMCB320 using BstXI/BlpI overhangs after PCR amplification. To conduct head-to-head comparisons of Death-seq versus traditional negative selection screening, both our targeted custom sublibrary and the 10 sgRNA/gene apoptosis and cancer sublibrary were used in screens following the genome-wide screen protocol described above with the exception of the addition of the DMSO-treated control arm, where half the plates of cells in each technical replicate were treated with DMSO instead of ABT-263. The head-to-head screens also had a different screen coverage (in each technical replicate, ~8,000x coverage for the targeted custom sublibrary head-to-head screens and ~1,500x coverage for the apoptosis and cancer sublibrary screens). To conduct parallel screens testing Death-seq with different small molecules, a different inducer of senescence (H2O2), proliferative IMR-90s, and doxorubicin-induced senescent WI-38s the genome-wide screen protocol described above was followed except with the targeted custom sublibrary and with a higher coverage of ~20,000x in each technical replicate. The eight treatment groups included one in H2O2-induced senescent IMR-90s with 24 hours of 1 μM ABT-263 treatment, three in doxorubicin-induced senescent IMR-90s with 24 hours of treatment with 1 μM ABT-263, 10 μM ABT-199, or 2 μM birinapant in each respective treatment arm, three in proliferative IMR-90s with the same respective treatment arms, and one in doxorubicin-induced senescent WI-38s with 24 hours of 1 μM ABT-263 treatment. Subcellular localizations of proteins encoded by screen hits were taken from UniProtKB78 and MitoCarta3.0,79 one of the 80 hits was left off, RP11–45M22.4, as it is an annotated region of the genome not a gene. MitoCarta3.0’s biological pathway classifications were used to determine the apoptosis pathway members. To conduct a screen testing Death-seq in the context of cardiotoxicity induced by the MCL1 inhibitor S63845 in human iPSC-derived cardiomyocytes, the genome-wide screen protocol described above was followed with the following exceptions. The targeted custom sublibrary was used with a coverage of ~10,000x. Cardiomyocytes were derived from human iPSCs as described80 and cultured in T75 flasks. hiPSC derived cardiomyocytes were treated with 10 μM MCL1 inhibitor S63845 for 48 hours before takedown. To conduct a screen testing Death-seq in the context of ferroptosis and non-apoptotic cell death induced by the small molecule CIL56, the genome-wide screen protocol described above was followed with the following exceptions. The targeted custom sublibrary was used with a coverage of ~20,000x in each technical replicate. Proliferative IMR-90s were treated in parallel with either 0.5 μM erastin2 or 1 μM CIL56 for 48 hours before cells were collected to end the screens.
Lentivirus production and infection
HEK293T cells were transfected with third-generation packaging plasmids and individual sgRNA vectors or sgRNA libraries using PEI max (Polysciences, 26008–5). Lentivirus was harvested after 48 hours and 72 hours and filtered through a 0.45-μm polyethersulfone (PES) filter (Corning 431096). 4 μg/mL of polybrene was used to deliver sgRNA vectors and libraries into IMR-90s. To validate results of genome-wide and sublibrary screens, we infected Cas9-expressing IMR-90 cells with plasmids expressing a single specific sgRNA and puromycin resistance. Two days after infection, the infected cells were puromycin-selected for 4 days and allowed to recover for at least 2 days before experimentation. To additionally validate the PPTC7 screen hit, doxorubicin-induced senescent IMR-90s were infected with plasmids expressing a 10 single specific shRNA and puromycin resistance (see Table S4 for sgRNA/shRNA sequences).
Cell viability and drug synergy
The cells were plated in triplicate in 96-well plates (Corning, 3603) (typically 70,000 senescent and 20,000 proliferative cells per mL of media when plating to attempt to have similar numbers of senescent and proliferative cells at the timepoint of viability assay). Unless otherwise indicated, cells were plated at least one day before drug treatment began and cell viability was assessed after 3 days of drug treatment, using the XTT assay with the Cell Proliferation Kit II according to the manufacturer’s protocol. Experimental values were normalized using a vehicle-treated and a puromycin-treated (10 μg/ml) condition to set the maximal and minimal viability values, respectively. Drug synergy was assessed as previously described.81 In brief, drug synergy was assessed using the Bliss independence model.82 Using the equation FA +FB − (FA × FB), where FA and FB are the fractional viability inhibitions of the individual drugs A and B at a given dose, the drug combination’s predicted fractional viability inhibition is calculated. The Bliss excess is the difference between the expected viability inhibition and the observed inhibition from the drug combination. Bliss sum is the sum of individual Bliss scores in each matrix of drug doses. Bliss scores greater than zero, close to zero, and less than zero denote synergy, additivity, and antagonism, respectively.
Apoptosis assays
To assess apoptosis, Annexin V assays and caspase 3/7 activation assays were used. For the Annexin V assays, APC PI Annexin V solutions were utilized according to the manufacturer’s protocol. In brief, after treatment of the cells with the indicated doses of drugs including some conditions with 20 μM Q-VD-OPH, 10 μM ferrostatin, or 10 μM necrostatin, cells were washed and stained with 1:20 APC Annexin V and 1:10 Propidium Iodide Solution for 15 minutes at room temperature. The fluorescence of the cells was measured on a BD-FACS Aria III flow cytometer. In the case of adherent cells, detached dying cells in the media following drug treatment were centrifuged down and combined with the remaining adherent cells, which were trypsinized. For the caspase 3/7 activation assay, the EarlyTox Caspase-3/7-D NucView 488 assay kit was utilized according to the manufacturer’s protocol. IMR-90s were treated with the indicated doses of vehicle or drugs and 10 μM caspase substrate before reading out fluorescence on a plate reader.
Mitochondria membrane potential
Mitochondrial membrane potential was measured using JC-1 staining solution according to the manufacturer’s protocol. IMR-90s were treated with small molecules then stained with JC-1 solution for 5 minutes and the fluorescence of the cells was measured on a BD-FACS Aria III flow cytometer.
Quantitative RT-PCR
RNA was extracted from murine lungs and livers using the RNeasy Plus Mini Kit, and cDNA was prepared using High-Capacity cDNA Reverse Transcription Kit according to the manufacturer’s instructions. qPCR was carried out on an ABI 7900HT thermocyler using TaqMan Fast Advanced Master Mix (Applied Biosystems, 4444557) with following Taqman gene expression assays (Applied Biosystems): Col1a1 (Mm00801666_g1), Col1a2 (Mm00483888_m1), Il6 (Mm00446190_m1), Hprt (Mm01545399_m1) and Tbp (Mm00446973_m1). The primers and probe used for the detection of p16INK4a were as follows: p16-F 5′-CGGTCGTACCCCGATTCAG-3′; p16-R 5′-GCACCGTAGTTGAGCAGAAGAG-3′; Probe 5′- [FAM] AACGTTGCCCATCATCA [MGB]-3′.
Immunoblotting
Equal numbers of cells were lysed in RIPA Buffer, followed by boiling and centrifugation. SDS-PAGE was performed with 40 μg of total protein as determined by BCA assay. Proteins were separated in 4–20% gradient polyacrylamide gels (Bio-Rad) and immobilized onto PVDF membranes. The membranes were probed with antibodies to detect the following proteins: p16 (1:1000, Proteintech 10883-1-AP), p21 (1:500, BD 556430), cIAP1 (1:500, Cell Signaling Technology 7065T), and β-actin (1:25000, Sigma A3854). Bands were detected with WesternBright ECL reagent (Advansta) and imaged and quantified with a ChemiDoc™ Imaging System (Bio-Rad).
Immunofluorescence / Fluorescent Microscopy
The incorporation of EdU into cells was detected with the Click-iT EdU Imaging Kit (ThermoFisher) according to manufacturer’s instructions. Immunofluorescence (IF) imaging was performed with an AxioObserver Z1 epifluorescence microscope (Carl Zeiss) equipped with an Orca-R2 CCD camera (Hamamatsu Photonics). The percentage of EdU+ cells was quantified automatically using the Volocity software. Detection of p21 was performed in lung transverse 10 μm cryosections. Sections were fixed with 2% formaldehyde for ten minutes and blocked in 2% BSA in 0.3% Tween/PBS overnight at 4°C. The lungs were then stained for p21 using a rabbit anti-p21 antibody (1:300, Abcam 188224) for 2 hrs at room temperature. After washing out primary antibodies in 0.3% Tween/PBS, specimens were incubated with secondary antibodies produced in donkey and conjugated with the proper fluorochrome for one hour at room temperature. DNA was stained with DAPI. Specimens were next washed with 0.3% Tween/PBS and mounted with FluoroSave (Calbiochem). IF imaging was performed with BZ-X710 inverted fluorescence phase contrast microscope (Keyence). The percentage of p21+ cells was quantified automatically using the ImageJ software.
AlamarBlue cell proliferation assay
Cell viability was determined using the alamarBlue reagent (Invitrogen). For growth curve experiments, proliferative, Doxo-SEN, and IR-SEN IMR90 cells were seeded onto 48-well plates at a density of 5×103 cells/cm2 and cultured for seven days. After one day, this time point was denoted as day 1. Absorbance at 570 nm using 600 nm as reference wavelength was measured every 2 days using a Spark Microplate Reader (Tecan). Before determination, cells were incubated in normal medium containing 10% alamarBlue reagent for 4 h. Data are expressed as relative values (absorbance of each day divided by absorbance at day 1).
Histopathological evaluation of pulmonary fibrosis
The right lung was inflated and embedded in OCT compound, and 10-μm frozen sections were prepared and stained using hematoxylin and eosin and Masson’s trichrome stain kit according to the manufacturer’s instructions. The severity of pulmonary fibrosis was scored blindly according to the modified Ashcroft scale83 using Masson’s trichrome stained slides.
Senescence-associated β-galactosidase
Senescence-associated β-galactosidase (SA-βgal) assay was performed using the Senescence β-Galactosidase Staining Kit #9860 from Cell Signaling Technology according to manufacturer instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Flow cytometry data was analyzed using FlowJo software. Unless otherwise stated, P values were calculated using ANOVA followed by Dunnett’s multiple comparison test to the vehicle group using GraphPad Prism 9 software. Specific data representation details and statistical procedures are also indicated in the figure legends. All data are presented as means ± s.e.m., n.s. = P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Supplementary Material
Table S1. Results from all CRISPR KO screens, related to Figures 1, 2, 3, 7, S1, S2, and S7.
Table S2. Sequencing counts from all CRISPR KO screens, related to Figures 1, 2, 3, 7, S1, S2, and S7.
Table S4. Sequences of sgRNAs and shRNAs used in this study, related to Figures 1, 3, S1, and S2.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| MultiShot FlexPlate Stbl3 Competent Cells | Thermo Fisher Scientific | Cat#C7381201 |
| Biological samples | ||
| Human platelet-rich plasma (PRP) | ZenBio | Cat#SER-PRP-SDS |
| Chemicals, peptides, and recombinant proteins | ||
| Doxorubicin | Cayman Chemical | 15007; CAS: 25316-40-9 |
| Bleomycin | Cayman Chemical | 13877; CAS: 9041-93-4 |
| USP grade Bleomycin | Meitheal Pharmaceuticals | 71288-106-10 |
| Hydrogen peroxide (H2O2) | Sigma-Aldrich | H1009; CAS: 7722-84-1 |
| Palbociclib | LC Laboratories | P-7788; CAS: 827022-33-3 |
| Etoposide | Sigma-Aldrich | E1383; CAS: 33419-42-0 |
| Q-VD-OPH | Cayman Chemical | 15260; CAS: 1135695-98-5 |
| Ferrostatin-1 | Cayman Chemical | 17729; CAS: 347174-05-4 |
| Necrostatin-1 | Cayman Chemical | 11658; CAS: 4311-88-0 |
| ABT-263 | Cayman Chemical | 11500; CAS: 923564-51-6 |
| ABT-199 | Cayman Chemical | 16233; CAS: 1257044-40-8 |
| Birinapant | Cayman Chemical | 19699; CAS: 1260251-31-7 |
| GDC-0152 | Cayman Chemical | 17810; CAS: 873652-48-3 |
| SM-164 | Cayman Chemical | 28632; CAS: 957135-43-2 |
| VH298 | Cayman Chemical | 21133; CAS: 2097381-85-4 |
| S63845 | Cayman Chemical | 21131; CAS: 1799633-27-4 |
| Erastin2 | Cayman Chemical | 27087; CAS: 1695533-44-8 |
| CIL56 | Acme Bioscience | N/A |
| Phosal 50 PG | MedChemExpress | HY-Y1903; CAS: 774594-96-6 |
| Captisol | CyDex Pharmaceuticals | RC-0C7; CAS: 182410-00-0 |
| Critical commercial assays | ||
| RNeasy Plus Mini kit | QIAGEN | Cat#74134 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368813 |
| NextSeq 500/550 High Output kit v2.5 (75 Cycles) | Illumina | Cat#20024906 |
| Masson’s trichrome stain | Polysciences | Cat#25088-100 |
| Hydroxyproline Assay Kit | Sigma-Aldrich | Cat#MAK008-1KT |
| Cell Proliferation Kit II | Roche | Cat#11465015001 |
| alamarBlue Cell Viability Reagent | Thermo Fisher Scientific | Cat#DAL1100 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling Technology | Cat#9860 |
| APC Annexin V Apoptosis Detection Kit with PI | BioLegend | Cat#640932 |
| JC-1 Mitochondrial Membrane Potential Flow Cytometry Assay Kit | Cayman Chemical | Cat#701560 |
| EarlyTox Caspase-3/7-D NucView 488 assay Kit | Molecular Devices | Cat#R8348 |
| Experimental models: Cell lines | ||
| IMR-90 normal human lung fibroblasts | ATCC | CCL-186 |
| WI-38 normal human lung fibroblasts | ATCC | CCL-75 |
| Human induced pluripotent stem cell line | Stanford Cardiovascular Institute biobank | N/A |
| Human retinal microvascular endothelial cells (HRMECs) | Neuromics | HEC09 |
| Deposited data | ||
| Data S1 - Source Data | This paper | Data S1 - Source Data |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL6 | The Jackson Laboratory | JAX: 000664 |
| Oligonucleotides | ||
| sgRNAs and shRNAs used for in vitro validation, see Table S4 | This paper | N/A |
| Primer: p16-Forward: CGGTCGTACCCCGATTCAG | This paper | N/A |
| Primer: p16-Reverse GCACCGTAGTTGAGCAGAAGAG | This paper | N/A |
| Primer: p21-Forward: CCTCATCCCGTGTTCTCCTTT | This paper | N/A |
| Primer: p21-Reverse GTACCACCCAGCGGACAAGT | This paper | N/A |
| p16 probe 5′-[FAM] AACGTTGCCCATCATCA [MGB]-3′ | This paper | N/A |
| Col1a1 (Mm00801666_g1) | Life Technologies | N/A |
| Col1a2 (Mm00483888_m1) | Life Technologies | N/A |
| Il6 (Mm00446190_m1) | Life Technologies | N/A |
| Hprt (Mm01545399_m1) | Life Technologies | N/A |
| Tbp (Mm00446973_m1) | Life Technologies | N/A |
| Recombinant DNA | ||
| pMCB320 | Addgene | Cat#89359 |
| Mouse CRISPR Deletion Library - Apoptosis and cancer | Addgene | Cat#1000000121 |
| Targeted custom sublibrary, see Table S3 | This paper | N/A |
| Software and algorithms | ||
| UniProtKB | The UniProt Consortium, 2021 | https://www.uniprot.org/ |
| GraphPad Prism 9.1.0 | GraphPad Software, Inc. | https://www.graphpad.com/ |
| MitoCarta3.0 | Rath et al., 2021 | https://www.broadinstitute.org/mitocarta |
| FlowJo 10 | FlowJo, LLC | https://www.flowjo.com/ |
| Metascape 3.5 | Zhou et al., 2019 | https://metascape.org/ |
| Cytoscape_v3.8.2 | Altaf-Ul-Amin et al., 2006 | https://cytoscape.org |
| Reactome | Jassal et al., 2020 | https://reactome.org/PathwayBrowser |
| casTLE scripts version 1.0 | Morgens et al., 2016 | https://github.com/elifesciences-publications/dmorgens-castle |
| Other | ||
| Results from all CRISPR KO screens, see Table S1 | This paper | N/A |
| Sequencing counts from all CRISPR KO screens, see Table S2 | This paper | N/A |
Highlights.
Death-seq is a screening method that enables the systematic study of cell death
Death-seq enables the identification of enhancers and inhibitors of cell death
CRISPR screens with Death-seq identify senolytics for age-related disease
ACKNOWLEDGMENTS
We thank M. Buckley for assisting in the analysis of single-cell RNA-seq datasets from aged animals to search for markers of senescent cells. We thank C. Cain, B. Carter, and the Palo Alto VA Flow Cytometry Core for assistance with flow cytometry experiments. A.C. thanks J. Pluvinage, R. Kamber, M. Schultz, Y. Lu, and M. Bonkowski (deceased) for scientific mentorship. We thank Y. Yu and the entire Rando and Bassik labs for laboratory assistance and discussion. Some illustrations were created with BioRender.com. We acknowledge our funding sources: the National Science Foundation Graduate Research Fellowship DGE-1656518 (A.C.), the Glenn Foundation for Medical Research (T.A.R.), the National Institutes of Health grant P01AG036695 (T.A.R.), the National Institutes of Health grant R01AR073248 (T.A.R.), and the Departments of Veterans Affairs BLR&D Merit Review I01BX002324 (T.A.R.).
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
A.C. and T.A.R. have filed a patent application related to the subject matter of this paper. A.C. was formerly a paid consultant during this work for Maze Therapeutics and Rubedo Life Sciences.
SUPPLEMENTARY INFORMATION
Supplementary information is available for this paper.
REFERENCES
- 1.Collado M, Blasco MA, and Serrano M (2007). Cellular senescence in cancer and aging. Cell 130, 223–233. 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 2.Di Micco R, Krizhanovsky V, Baker D, and di Fagagna FDA (2021). Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol 22, 75–95. 10.1038/s41580-020-00314-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, et al. (2019). Cellular senescence: defining a path forward. Cell 179, 813–827. 10.1016/j.cell.2019.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. (2016). Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189. 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He S, and Sharpless NE (2017). Senescence in health and disease. Cell 169, 10001011. 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, et al. (2018). Senolytics improve physical function and increase lifespan in old age. Nat. Med 24, 1246–1256. 10.1038/s41591-018-0092-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, et al. (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169, 132–147. 10.1016/j.cell.2017.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Deursen JM (2019). Senolytic therapies for healthy longevity. Science 364, 636–637. 10.1126/science.aaw1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johmura Y, Yamanaka T, Omori S, Wang T, Sugiura Y, Matsumoto M, Suzuki N, Kumamoto S, Yamaguchi K, Hatakeyama S, et al. (2021). Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 371, 265–270. 10.1126/science.abb5916. [DOI] [PubMed] [Google Scholar]
- 10.Arroyo JD, Jourdain AA, Calvo SE, Ballarano CA, Doench JG, Root DE, and Mootha VK (2016). A genome-wide CRISPR death screen identifies genes essential for oxidative phosphorylation. Cell Metab. 24, 875–885. 10.1016/j.cmet.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, et al. (2016). Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun 7, 11190. 10.1038/ncomms11190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu YI, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, et al. (2016). Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435. 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al. (2016). Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med 22, 78–83. 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrish E, Brumatti G, and Silke J (2020). Future Therapeutic Directions for SmacMimetics. Cells 9, 406. 10.3390/cells9020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kramer NJ, Haney MS, Morgens DW, Jovičić A, Couthouis J, Li A, Ousey J, Ma R, Bieri G, Tsui CK, et al. (2018). CRISPR–Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet 50, 603–612. 10.1038/s41588-018-0070-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lek A, Zhang Y, Woodman KG, Huang S, DeSimone AM, Cohen J, Ho V, Conner J, Mead L, Kodani A, et al. (2020). Applying genome-wide CRISPR-Cas9 screens for therapeutic discovery in facioscapulohumeral muscular dystrophy. Sci. Trans. Med 12, eaay0271. 10.1126/scitranslmed.aay0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.So RWL, Chung SW, Lau HHC, Watts JJ, Gaudette E, Al-Azzawi ZAM, Bishay J, Lin LT-W, Joung J, Wanget X, et al. (2019). Application of CRISPR genetic screens to investigate neurological diseases. Mol. Neurodegener 14, 41. 10.1186/s13024-019-0343-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morgens DW, Deans RM, Li A, and Bassik MC (2016). Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol 34, 634–636. 10.1038/nbt.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niemi NM, Wilson GM, Overmyer KA, Vögtle F-N, Myketin L, Lohman DC, Schueler KL, Attie AD, Meisinger C, Coon JJ, et al. (2019) Pptc7 is an essential phosphatase for promoting mammalian mitochondrial metabolism and biogenesis. Nat. Commun 10, 3197. 10.1038/s41467-019-11047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li F, Huangyang P, Burrows M, Guo K, Riscal R, Godfrey J, Lee KE, Lin N, Lee P, Blair IA, et al. (2020). FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat. Cell Biol 22, 728–739. 10.1038/s41556-020-0511-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shahbandi A, Rao SG, Anderson AY, Frey WD, Olayiwola JO, Ungerleider NA, and Jackson JG (2020). BH3 mimetics selectively eliminate chemotherapy-induced senescent cells and improve response in TP53 wild-type breast cancer. Cell Death Differ. 27, 3097–3116. 10.1038/s41418-020-0564-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han K, Pierce SE, Li A, Spees K, Anderson GR, Seoane JA, Lo Y-H, Dubreuil M, Olivas M, Kamber RA, et al. (2020). Nature 580, 136–141. 10.1038/s41586-020-2099-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohli J, Ge C, Fitsiou E, Doepner M, Brandenburg SM, Faller WJ, Ridky TW, and Demaria M (2022). Targeting anti-apoptotic pathways eliminates senescent melanocytes and leads to nevi regression. Nat. Commun 13, 7923. 10.1038/s41467-022-35657-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M and Kim WY (2011). Two sides to every story: the HIF-dependent and HIF-independent functions of pVHL. J. Cell. Mol. Med 15, 187–195. 10.1111/j.1582-4934.2010.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, Josefsson EC, Alwis I, Ono A, Willcox A, et al. (2011). Bcl-xL–inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood 118, 1663–1674. 10.1182/blood-2011-04-347849. [DOI] [PubMed] [Google Scholar]
- 26.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. (2013). ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med 19, 202–208. 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 27.Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura-Hoshino M, Sada K, Hotta H, Yamamura H, et al. (2006). A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO 25, 3618–3626. 10.1038/sj.emboj.7601249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arai S, Varkaris A, Nouri M, Chen S, Xie L, and Balk SP (2020). MARCH5 mediates NOXA-dependent MCL1 degradation driven by kinase inhibitors and integrated stress response activation. eLife 9, e54954. 10.7554/eLife.54954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subramanian A, Andronache A, Li Y-C, and Wade M (2016). Inhibition of MARCH5 ubiquitin ligase abrogates MCL1-dependent resistance to BH3 mimetics via NOXA. Oncotarget 7, 15986–16002. 10.18632/oncotarget.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Djajawi TM, Liu L, Gong J-N, Huang AS, Luo M-J, Xu Z, Okamoto T, Call MJ, Huang DCS, and Delft MF (2020). MARCH5 requires MTCH2 to coordinate proteasomal turnover of the MCL1:NOXA complex. Cell Death Differ. 27, 2484–2499. 10.1038/s41418-020-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Z, Deng W, Bai Y, Miao R, Mei S, Zhang Z, Pan Y, Wang Y, Min R, Deng F, et al. , (2021). The lysosomal Rag-Ragulator complex licenses RIPK1– and caspase-8–mediated pyroptosis by Yersinia. Science 372, eabg0269. 10.1126/science.abg0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crespo-Garcia S, Tsuruda PR, Dejda A, Ryan RD, Fournier F, Chaney SY, Pilon F, Dogan T, Cagnone G, Patel P, et al. (2021). Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell 33, 818–332. 10.1016/j.cmet.2021.01.011. [DOI] [PubMed] [Google Scholar]
- 33.Debrincat MA, Pleines I, Lebois M, Lane RM, Holmes ML, Corbin J, Vandenberg CJ, Alexander WS, Ng AP, Strasser A, et al. (2015). BCL-2 is dispensable for thrombopoiesis and platelet survival. Cell Death Dis. 6, e1721. 10.1038/cddis.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, et al. (2017). Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun 8, 14532. 10.1038/ncomms14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, Mulay A, Soukiasian HJ, David G, Weigt SS, et al. (2020). Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 203, 707–717. 10.1164/rccm.202004-1274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merkt W, Bueno M, Mora AL, and Lagares D (2020). Senotherapeutics: Targeting senescence in idiopathic pulmonary fibrosis. Semin. Cell Dev. Biol 101, 104–110. 10.1016/j.semcdb.2019.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu R-M, and Liu G (2020). Cell senescence and fibrotic lung diseases. Exp. Gerontol 132, 110836. 10.1016/j.exger.2020.110836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider JL, Rowe JH, Garcia-de-Alba C, Kim CF, Sharpe AH, and Haigis MC (2021). The aging lung: Physiology, disease, and immunity. Cell 184, 1990–2019. 10.1016/j.cell.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, and Wells AU (2017). Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 3, 17074. 10.1038/nrdp.2017.74. [DOI] [PubMed] [Google Scholar]
- 40.Barnes T (2013). Perspective of a Daughter Turned Advocate. Ann. Am. Thorac. Soc 10, 403–405. 10.1513/AnnalsATS.201305-108OT. [DOI] [PubMed] [Google Scholar]
- 41.Álvarez D, Cárdenes N, Sellarés J, Bueno M, Corey C, Hanumanthu VS, Peng Y, D’Cunha H, Sembrat J, Nouraie M, et al. (2017). IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Lung Cell. Mol. Physiol 313, L1164–L1173. 10.1152/ajplung.00220.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, et al. (2020). Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv 6, eaba1983. 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore C, Blumhagen RZ, Yang IV, Walts A, Powers J, Walker T, Bishop M, Russell P, Vestal B, Cardwell J, et al. (2019). Resequencing Study Confirms That Host Defense and Cell Senescence Gene Variants Contribute to the Risk of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med 200, 199–208. 10.1164/rccm.201810-1891OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armanios MY, Chen JJL, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips III JA, et al. (2007). Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med 356, 1317–1326. 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 45.Povedano JM, Martinez P, Flores JM, Mulero F, and Blasco MA (2015). Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 12, 286–299. 10.1016/j.celrep.2015.06.028. [DOI] [PubMed] [Google Scholar]
- 46.Wiley CD, Brumwell AN, Davis SS, Jackson JR, Valdovinos A, Calhoun C, Alimirah F, Castellanos CA, Ruan R, Wei Y, et al. (2019). Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight, 4(24). 10.1172/jci.insight.130056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, Lin S, Huang L, Chung EJ, Citrin DE, et al. (2017). Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. International Journal of Radiation Oncology* Biology* Physics, 99, 353–361. 10.1016/j.ijrobp.2017.02.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aoshiba K, Tsuji T, and Nagai A (2003). Bleomycin induces cellular senescence in alveolar epithelial cells. Eur. Respir. J 22, 436–443. 10.1183/09031936.03.00011903. [DOI] [PubMed] [Google Scholar]
- 49.Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, Meldrum E, Sanders YY, and Thannickal VJ (2014). Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med 6, 231ra47. 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, Prata LG, van Dijk TH, Verkade E, Casaclang-Verzosa G, et al. (2019). Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18, e12950. 10.1111/acel.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papatheodoridi AM, Chrysavgis L, Koutsilieris M, and Chatzigeorgiou A (2020). The role of senescence in the development of nonalcoholic fatty liver disease and progression to nonalcoholic steatohepatitis. Hepatology 71, 363–374. 10.1002/hep.30834. [DOI] [PubMed] [Google Scholar]
- 52.Omori S, Wang TW, Johmura Y, Kanai T, Nakano Y, Kido T, Susaki EA, Nakajima T, Shichino S, Ueha S, et al. (2020). Generation of a p16 reporter mouse and its use to characterize and target p16high Cells In Vivo. Cell Metab. 32, 814–828. 10.1016/j.cmet.2020.09.006. [DOI] [PubMed] [Google Scholar]
- 53.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, et al. (2017). Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun 8, 15691. 10.1038/ncomms15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amor C, Feucht J, Leibold J, Ho Y, Zhu C, Alonso-Curbelo D, Mansilla-Soto J, Boyer JA, Li X, Giavridis T, et al. (2020). Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132. 10.1038/s41586-020-2403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rasmussen ML, Taneja N, Neininger AC, Wang L, Robertson GL, Riffle SN, Shi L, Knollmann BC, Burnette DT, Gama V (2020). MCL-1 Inhibition by Selective BH3 Mimetics Disrupts Mitochondrial Dynamics Causing Loss of Viability and Functionality of Human Cardiomyocytes. iScience. 23, 101015. 10.1016/j.isci.2020.101015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, and Opferman JT (2013). Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes & Dev. 27, 1351–1364. 10.1101/gad.215855.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ko P-J, Woodrow C, Dubreuil MM, Martin BR, Skouta R, Bassik MC, and Dixon SJ (2019). A ZDHHC5-GOLGA7 Protein Acyltransferase Complex Promotes Nonapoptotic Cell Death. Cell Chemical Biology 26, 1716–1724. 10.1016/j.chembiol.2019.09.014. [DOI] [PubMed] [Google Scholar]
- 58.Montero J, and Letai Anthony. (2018). Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ 25, 56–64. 10.1038/cdd.2017.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perimenis P, Galaris A, Voulgari A, Prassa M, and Pintzas A (2016). IAP antagonists Birinapant and AT-406 efficiently synergise with either TRAIL, BRAF, or BCL-2 inhibitors to sensitise BRAFV600E colorectal tumour cells to apoptosis. BMC Cancer 16, 624. 10.1186/s12885-016-2606-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen K-F, Lin JP, Shiau C-W, Tai W-T, Liu CY, Yu H-C, Chen P-J, and Cheng A-L (2012). Inhibition of Bcl-2 improves effect of LCL161, a SMAC mimetic, in hepatocellular carcinoma cells. Biochem Pharmacol. 84, 268–277. 10.1016/j.bcp.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 61.Cohn RL, Gasek NS, Kuchel GA, and Xu M (2022). The heterogeneity of cellular senescence: insights at the single-cell level. Trends Cell Biol. 33, 9–17. 10.1016/j.tcb.2022.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gurkar AU, Gerencser AA, Mora AL, Nelson AC, Zhang AR, Lagnado AB, Enninful A, Benz C, Furman D, Beaulieu D, et al. (2003). Spatial mapping of cellular senescence: emerging challenges and opportunities. Nat Aging 3, 776–790. 10.1038/s43587-023-00446-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.The Tabula Muris Consortium. (2020). A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595. 10.1038/s41586-020-2496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buckley MT, Sun ED, George BM, Liu L, Schaum N, Xu L, Reyes JM, Goodell MA, Weissman IL, Wyss-Coray T, et al. (2022). Cell-type-specific aging clocks to quantify aging and rejuvenation in neurogenic regions of the brain. Nature Aging. 10.1038/s43587-022-00335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kimmel JC, Penland L, Rubinstein ND, Hendrickson DG, Kelley DR, and Rosenthal AZ (2019). Murine single-cell RNA-seq reveals cell-identity- and tissue-specific trajectories of aging. Genome Res. 29, 2088–2103. 10.1101/gr.253880.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, Franceschi C, Lithgow GJ, Morimoto RI, Pessin JE, et al. (2014). Geroscience: Linking Aging to Chronic Disease. Cell 159, 709–713. 10.1016/j.cell.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olshansky J (2018). From lifespan to healthspan. JAMA 320, 1323–1324. 10.1001/jama.2018.12621. [DOI] [PubMed] [Google Scholar]
- 68.Scott AJ, Ellison M, and Sinclair D (2021). The economic value of targeting aging. Nature Aging 1, 616–623. 10.1038/s43587-021-00080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nehme J, Borghesan M, Mackedenski S, Bird TG, and Demaria M (2020). Cellular senescence as a potential mediator of COVID-19 severity in the elderly. Aging Cell 19, e13237. 10.1111/acel.13237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Camell CD, Yousefzadeh MJ, Zhu Y, Prata LGPL, Huggins MA, Pierson M, Zhang L, O’Kelly RD, Pirtskhalava T, Xun P, et al. (2021). Senolytics reduce coronavirus-related mortality in old mice. Science 373, eabe4832. 10.1126/science.abe4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576. 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roy AL, Sierra F, Howcroft K, Singer DS, Sharpless N, Hodes RJ, Wilder EL, Anderson JM, et al. (2020). A Blueprint for Characterizing Senescence. Cell 5, 1143–1146. 10.1016/j.cell.2020.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He Y, Zhang X, Chang J, Kim H, Zhang P, Wang Y, Khan S, Liu X, Zhang X, Lv D, et al. (2020). Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat. Commun 11, 1996. 10.1038/s41467-020-15838-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morgens DW, Wainberg M, Boyle EA, Ursu O, Araya CL, Tsui CK, Haney MS, Hess GT, Han K, Jeng EE, et al. (2017). Genome-scale measurement of offtarget activity using Cas9 toxicity in high-throughput screens. Nat. Commun 8, 15178. 10.1038/ncomms15178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Altaf-Ul-Amin M, Shinbo Y, Mihara K, Kurokawa K, and Kanaya S (2006). Development and implementation of an algorithm for detection of protein complexes in large interaction networks. BMC Bioinformatics 7, 207. 10.1186/1471-2105-7-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, et al. (2020). The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503. 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.The UniProt Consortium. (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW, et al. (2021). MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547. 10.1093/nar/gkaa1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rhee J-W, Yi H, Thomas D, Lam CK, Belbachir N, Tian L, Qin X, Malisa J, Lau E, Paik DT, et al. (2020). Modeling Secondary Iron Overload Cardiomyopathy with Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Cell Reports 32, 107886. 10.1016/j.celrep.2020.107886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han K, Jeng EE, Hess GT, Morgens DW, Li A, and Bassik MC (2017). Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol 35, 463–474. 10.1038/nbt.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bliss CI (1939). The toxicity of poisons applied jointly. Ann. Appl. Biol 26, 585–615. 10.1111/j.1744-7348.1939.tb06990.x. [DOI] [Google Scholar]
- 83.Hübner R-H, Gitter W, El Mokhtari NE, Mathiak M, Both M, Bolte H, Freitag-Wolf S, and Bewig B (2008). Standardized quantification of pulmonary fibrosis in histological samples. Biotechniques 44, 507–517. 10.2144/000112729. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Results from all CRISPR KO screens, related to Figures 1, 2, 3, 7, S1, S2, and S7.
Table S2. Sequencing counts from all CRISPR KO screens, related to Figures 1, 2, 3, 7, S1, S2, and S7.
Table S4. Sequences of sgRNAs and shRNAs used in this study, related to Figures 1, 3, S1, and S2.
Data Availability Statement
Data S1 refers to unprocessed data underlying the display items in the manuscript, related to all Figures 1–7 and S1–S7. Tables S1–S4 contain the screening data produced from this work as well as sgRNA/shRNA sequences.
This paper does not report original code. All screening data was analyzed with the casTLE scripts version 1.0 that are available at https://github.com/elifesciences-publications/dmorgens-castle. Flow cytometry data was analyzed using FlowJo 10 software. Statistics were calculated using GraphPad Prism 9 software.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.