

SUMMARY
Uveal melanoma (UM) is a rare cancer resulting from the transformation of melanocytes in the uveal tract. Integrative analysis has identified four molecular and clinical subsets of UM. To improve our molecular understanding of UM, we performed extensive multi-omics characterization comparing two aggressive UM patient-derived xenograft models with normal choroidal melanocytes, including DNA optical mapping, specific histone modifications, and DNA topology analysis using Hi-C. Our gene expression and cytogenetic analyses suggest that genomic instability is a hallmark of UM. We also identified a recurrent deletion in the BAP1 promoter resulting in loss of expression and associated with high risk of metastases in UM patients. Hi-C revealed chromatin topology changes associated with the upregulation of PRAME, an independent prognostic biomarker in UM, and a potential therapeutic target. Our findings illustrate how multi-omics approaches can improve our understanding of tumorigenesis and reveal two distinct mechanisms of gene expression dysregulation in UM.
Graphical abstract
In brief
Gentien et al. perform extensive multi-omics of PDX derived from highly aggressive uveal melanomas (UMs) and normal uveal melanocytes to identify genomic, epigenomic, and transcriptomic patterns associated with tumorigenesis. Integrative analyses reveal genomic instability and identify mechanisms of dysregulation for two highly important genes, BAP1 and PRAME, in aggressive UMs.
INTRODUCTION
Uveal melanoma (UM) is a rare cancer (5–7 cases per million per year) that mainly affects adults and represents 5% of all melanomas.1 UM results from the malignant transformation of melanocytes of the uveal tract of the eye, which comprises the iris, the ciliary body, and choroidal membrane.2 UM primary tumors are well controlled by surgery and/or radiotherapy; however, more than 30% of the patients develop metastases, mainly in the liver, with a very poor prognosis. Improvement in the understanding of aggressive UM is essential for identifying efficient new therapeutic approaches.
The vast majority of UMs display activating mutations in GNAQ3 or its paralog GNA11,4 their upstream activator CYSLTR2,5 or downstream effector PLCB4.6 These mutually exclusive Gα/q-related mutations present in 98% of UMs are recognized as a primary event of UM oncogenesis7 and lead to activation of the Gα/q signaling pathway.8,9 Mutations in BAP1, EIF1AX, SF3B1, and SRSF210–13 were identified as secondary mutational events necessary for malignant transformation. Mutations in BAP1, SF3B1, and EIF1AX (so called BSE events) are associated with distinct delays in the appearance of metastasis, with the shortest delay associated with BAP1.14
Over the last two decades, a number of recurrent chromosomal abnormalities have been identified in UM, including monosomy 3 (M3), gain of 6p and 8q, as well as loss of 6q and 8p. These abnormalities are associated with adverse clinical outcome and are currently used for clinical prognosis.15–18 Monosomy 3 and gain of chromosome 8 correlate individually with an intermediate risk of metastasis, and the highest risk of metastasis is associated with combined M3 and gain of 8q.16,18 Integrative analysis including copy number variations, DNA methylation, recurrent protein coding mutations, and gene expression profiles has identified four molecular and clinical subsets in UM.18
To improve our understanding of tumor oncogenesis, we performed extensive multi-omic and FISH characterization of two aggressive UM patient derived xenografts (PDXs) with distinct mutational and chromosomal rearrangement patterns, as well as short-term culture of normal choroidal melanocytes (NMs) for comparison. In addition to investigating somatic DNA alterations and performing RNA sequencing and DNA topology analysis, we performed whole-genome DNA methylation sequencing and chromatin immunoprecipitation (ChIP-seq) of histone marks associated with activating (H3K4me3), repressing (H2A119Ub, H3K27me3), or enhancing (H3K27Ac) gene expression. These complementary analyses improved the characterization of regulated genes and pathways in aggressive UM.
RESULTS
Samples studied
We sorted UM cells from two aggressive PDX models, MP41 and MP46,19–21 to obtain both a pure tumor population and a sufficient number of cells for molecular profiling.22 The MP41 model was generated from enucleation of a UM occurring in a 50-year-old female patient who had a metastasis 31 months after the initial diagnosis and who died 43 months after the diagnosis of multiple metastases (including bone, lung, and subcutaneous lesions). The MP46 model was established from enucleation of a tumor occurring in a 69-year-old male patient. This patient developed a liver metastasis 6 months after diagnosis of the primary tumor and died 7 months from initial diagnosis (Figure 1A). These two aggressive models harbor canonical activating mutations in GNAQ/11 and share 8q and 6p gains. MP46 displayed isodisomy of chromosome 3 and was deficient in BAP1 by immunohistochemistry (IHC) (Figure 1B), even though no BAP1 mutations were identified by Sanger sequencing. MP41 is BAP1 proficient by IHC (Figure 1B), and no mutations were identified in BAP1, SF3B1, or EIF1AX by Sanger sequencing.
Figure 1. Principal characteristics of MP41 and MP46 PDXs established from aggressive uveal melanomas.
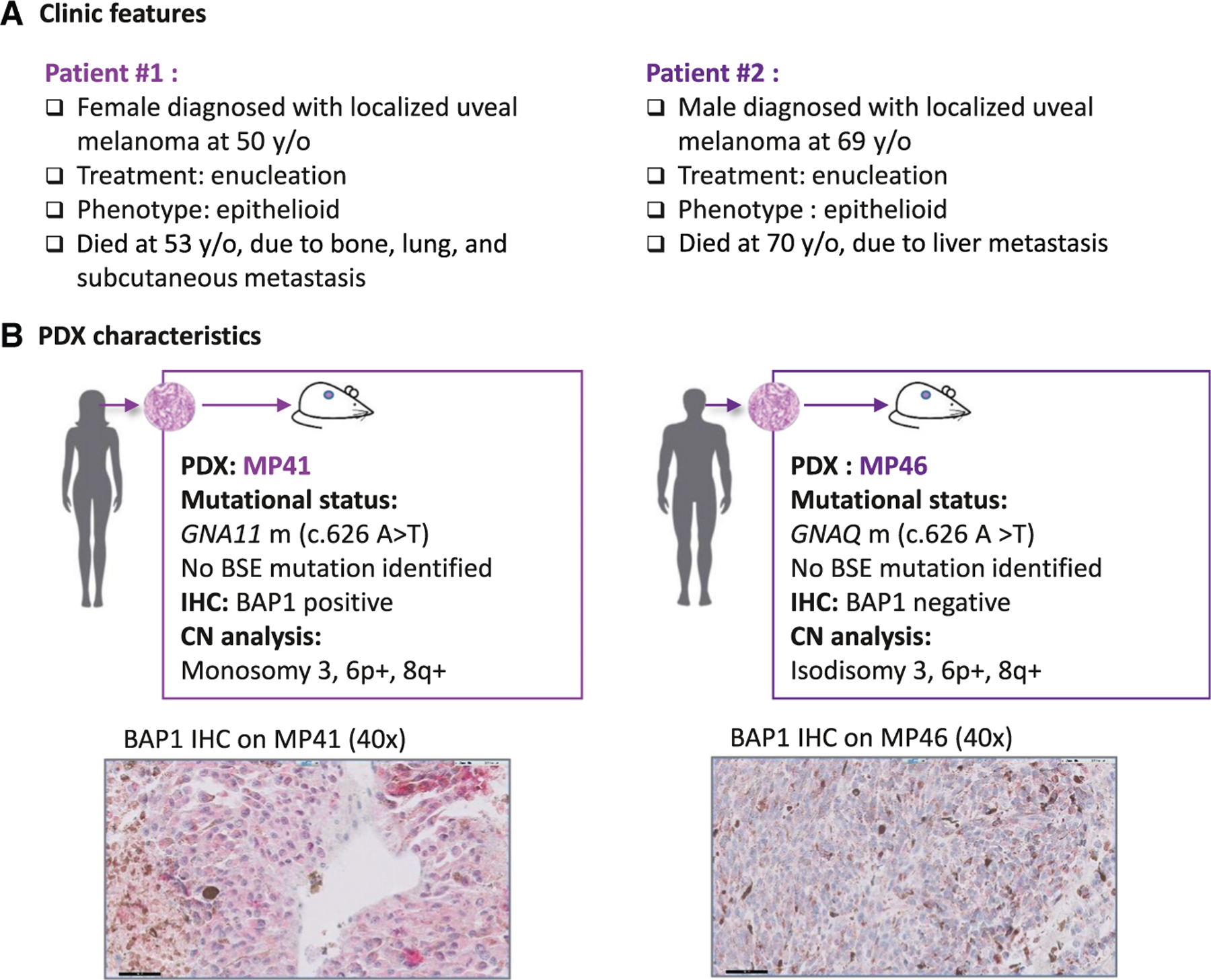
(A) Clinical characteristics of UM cases.
(B) Main molecular characteristics of corresponding patient-derived xenograft models established and characterized previously.21 Mutational status was assessed with Sanger sequencing (GNAQ/GNA11, BAP1, SF3B1, EIF1AX), with Cytoscan HD microarrays for copy number analysis and BAP1 immunohistochemistry from formalin-fixed paraffin-embedded (FFPE) tissue section.
Whole-genome sequencing and copy number analysis confirmed that MP41 and MP46 were high-risk UM
First, MP41 and MP46 were subjected to whole-genome sequencing (WGS) to perform single nucleotide variant (SNV) and copy number analyses. To facilitate the identification of somatic alterations, WGS was performed on matched healthy tissue adjacent to original primary tumors.
Somatic point mutation analysis revealed less than one somatic mutation per Mb (0.42 and 0.37 SNV/Mb in MP41 and MP46 respectively) as observed previously in UM.18 Based on Cancer Genome Interpreter23 and VarSome24 classification, a unique known driver mutation associated to a pathogenic role was identified in both MP41 (GNA11 c.626A>T, allele frequency [AF]:68%) and MP46 (GNAQ c.626A>T, AF: 43%). Such mutations in GNAQ/11 paralogs on the most frequent hotspot known in UM are in agreement with previous characterization of PDX and cells.21 Tables S1 and S2 list all the SNVs annotated as passenger mutations, having a moderate to high impact on amino acid sequence, or affecting ncRNAs for MP41 and MP46, respectively. Additional mutations identified in MP41 included a premature stop codon in KMT2C (KMT2C:[p.Tyr987*]), a known driver mutation, and 13 predicted passenger mutations (based on OncoDrive MUT algorithm). In MP46, we identified 21 putative passenger mutations in 19 genes, of which eight were predicted to be pathogenic. Furthermore, no SNVs were detected in BAP1, SF3B1, SFRSF2, or EIF1AX genes even at deeper coverage sequencing (Cas9-targeted Oxford Nanopore Technologies, Figure S1A; Table S3).
The TCGA UM study distinguished four copy number subtypes that had diverse aneuploid events and divided disomy 3 (D3)-UM and M3-UM into two subgroups, based on somatic copy number alterations.18 Somatic copy number alterations as losses (L), gains (G), and monosomy (M) identified from WGS of MP41 and MP46 models include notably for MP41: M3, G6p, L6q, L8p, G8q, and for MP46: isodisomy 3, G6p, L8p, G8q21 (Figures 2A and 2B; Table S4; Figure S1B). Based on the TCGA copy number subtypes of UMs, MP41 and MP46 were classified into group 2 and group 4 respectively.18 The classification of MP46 is consistent with the enrichment of BAP1-deficient tumors in group 4. Although TCGA group 2 is enriched in SF3B1-mutated UM, no SF3B1 or SRSF2 mutations and no SF3B1 splicing patterns have been observed in MP41.13
Figure 2. Genome and gene expression global overview.
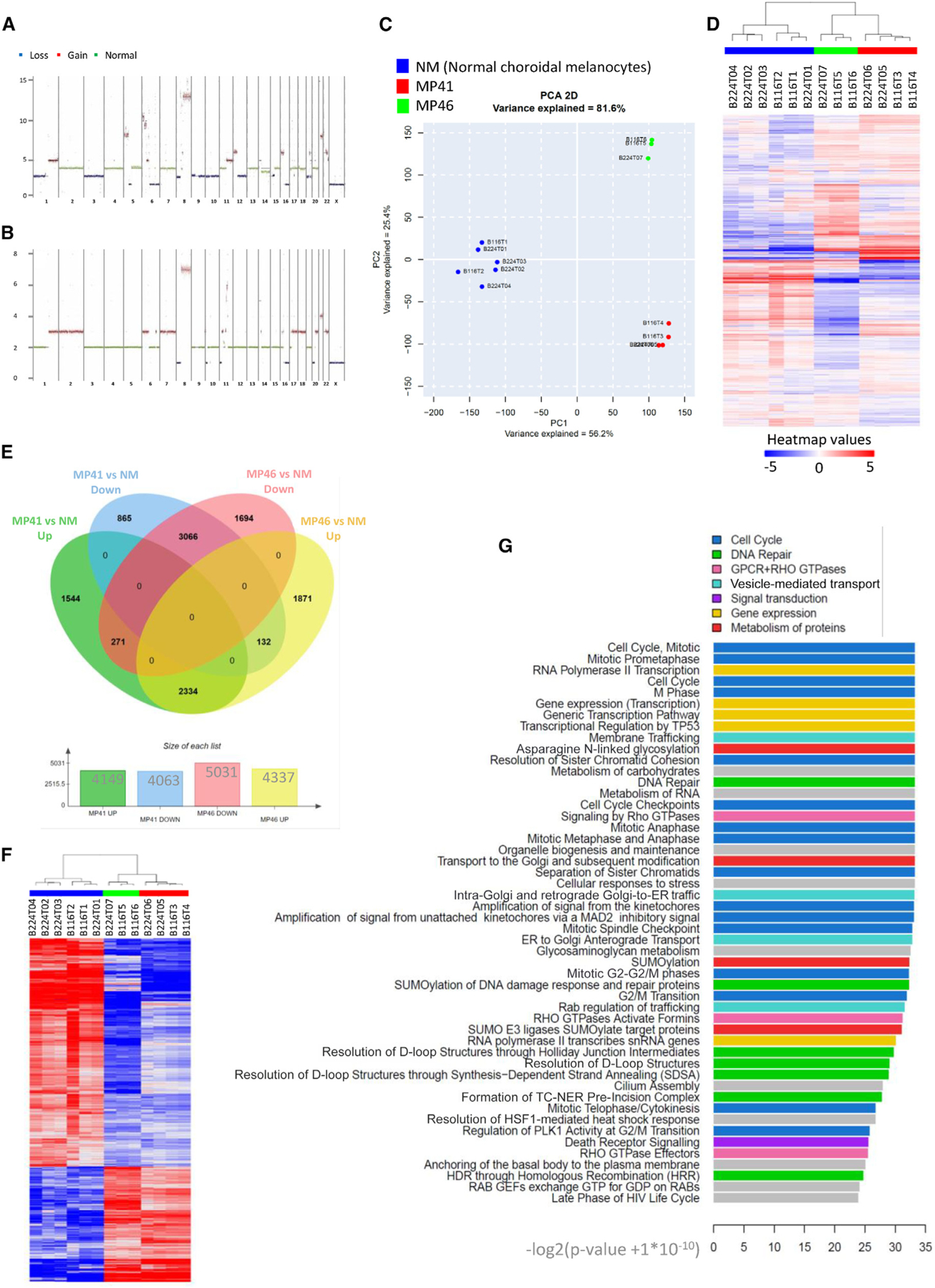
(A and B) MP41 (A) and MP46 (B) copy number profile established from whole-genome sequencing. Losses, gains, and normal regions are colored respectively in blue, red, and green.
(C) Principal component analysis of RNA-seq of six normal choroidal melanocyte samples (blue), four preparations of MP41 UM cells (red). and three preparations of MP46 UM cells (green).
(D) Hierarchical clustering of the same profiles.
(E) Differential gene expression analysis of MP41 vs. NM and MP46 vs. NM to identify genes with a Log2 fold change greater than 1.5 and a p value lower than 0.05.
(F) Heatmap of commonly regulated genes in MP41 vs. NM and MP46 vs. NM.
(G) 50 most highly regulated pathways by reactome analysis of commonly regulated genes listed according to the significance (−log2 [p value + 1E-10]).
Overall, WGS analysis of MP41 and MP46 models confirmed the presence of a unique oncogenic driver mutation in the Gαq pathway, the presence of M3 and G8q, and revealed an association of TCGA copy number group 2 for MP41 and group 4 for MP46.
Gene expression analysis reveals upregulation of well-known genes and highlights DNA repair pathways
We performed a gene expression analysis to compare the transcriptome of UM models with those of NMs. The RNA-seq dataset was composed of five normal melanocytes cell lines (NM) including one technical replicate, four MP41 biological replicates, and three MP46 biological replicates. Using unsupervised principal components analysis and hierarchical clustering of all differentially expressed genes compared to NM controls (log2 fold change [FC] > 1.5, p value ≤ 0.05) we found a high reproducibility of the replicates and clear separation between UM models and NM (Figures 2C and 2D). To control for effects of growth condition on gene expression, we also generated cell lines from each PDX and performed RNA-seq. Unsupervised analyses show clear clustering of each cell line with their matched PDX, consistent with cell culture having a minimal effect on expression profiles in this context (Figure S1C).
To identify consistently differentially expressed genes (DEGs) in aggressive UM, we compared each PDX to the NM, and then we compared the resulting gene lists (Figure 2E; Table S5). For MP41, 8,212 DEG were identified (4,149 upregulated and 4,063 downregulated). Among the 9,368 genes identified in MP46, 4,337 were overexpressed and 5,031 were underexpressed. The overlapping 3,066 downregulated genes and 2,334 upregulated genes between MP41 and MP46 (Figure 2F) were subjected to further analyses.
Cancer testis antigens were significantly enriched among the consistently overexpressed genes in MP41 and MP46, with PRAME25 being the highest cancer testis antigen expressed in both MP41 and MP46 (log2 FC: ~12). PLCB4 and RASGRP3, two key genes in UM oncogenesis, were among the top 50 upregulated genes. A small percentage of patients with UM display activating mutations in the PKC regulator PLCB4, which are mutually exclusive to GNAQ/GNA11/CYSLTR2 mutations.6 Overexpression of PLCB4 suggests a potential contribution to the activation of Gαq pathway in the absence of PLCB4 activating mutations. RASGRP3 has been shown to mediate MAPK pathway activation in UM.8,9 We identified an additional GPCR downstream pathway gene, RAPGEF4, which was significantly upregulated in both models.
Ranking the consistent DEGs by fold change indicated that the top 50 most upregulated genes in MP41 and MP46 were quite similar (Table 1). Several ncRNAs, including HAGLR and TRPM2-AS, which have been previously reported to participate in oncogenesis,26–30 were also found to be overexpressed. This suggests that other consistently overexpressed genes may also play functional roles in UM. The overlap between differentially downregulated genes in MP41 and MP46 was less pronounced (Table 1). Only eight of the top 50 most downregulated genes were shared between models. Consistent with the IHC result, BAP1 was the most downregulated gene in MP46.
Table 1.
Genes consistently in top 50 up- or downregulated in UM models vs. NM
| Gene name | Consistently up- or downregulated | Ranked Log2FC in MP46 | MP41 vs. NM: Log2FC | MP41 vs. NM: padj | Ranked Log2FC in MP46 | MP46 vs. NM: Log2FC | MP46 vs. NM: padj |
|---|---|---|---|---|---|---|---|
| HAGLR | up | 1 | 12.16 | 2.44E-63 | 1 | 12.48 | 3.56E-62 |
| TRPM2-AS | up | 2 | 12.12 | 9.08E-29 | 14 | 9.33 | 3.29E-17 |
| PRAME* | up | 3 | 12.11 | 3.78E-138 | 2 | 11.92 | 1.46E-113 |
| MT1M | up | 4 | 11.7 | 1.77E-25 | 16 | 8.93 | 6.35E-15 |
| ASB11 | up | 5 | 11.24 | 5.77E-55 | 6 | 10.59 | 3.51E-47 |
| KLHL30 | up | 6 | 10.73 | 8.10E-118 | 8 | 10.16 | 9.85E-100 |
| NRN1 | up | 7 | 10.56 | 2.21E-19 | 23 | 7.68 | 3.35E-09 |
| TRPM2 | up | 8 | 10.44 | 5.56E-16 | 22 | 7.73 | 5.83E-08 |
| CCSER1 | up | 9 | 9.98 | 9.12E-53 | 10 | 9.74 | 1.10E-48 |
| RASGRP3* | up | 10 | 9.96 | 1.95E-77 | 4 | 10.84 | 5.23E-77 |
| HIF3A | up | 11 | 9.46 | 1.41E-57 | 25 | 7.48 | 1.31E-34 |
| HOXA9//HOXA10 | up | 12 | 9.45 | 2.59E-87 | 3 | 10.85 | 9.40E-108 |
| PAQR5 | up | 13 | 9.38 | 7.74E-33 | 29 | 7.14 | 7.21E-19 |
| LMX1B | up | 14 | 9.31 | 3.28E-30 | 19 | 8.09 | 2.54E-22 |
| HOXA11 | up | 15 | 9.29 | 8.02E-38 | 9 | 10.1 | 5.99E-42 |
| EPHA4 | up | 16 | 9.04 | 7.55E-124 | 18 | 8.1 | 5.07E-90 |
| HOXC4//HOXC5//HOXC6 | up | 17 | 8.91 | 1.65E-09 | 21 | 7.75 | 1.82E-06 |
| IGLL3P | up | 18 | 8.79 | 4.08E-24 | 12 | 9.59 | 7.85E-26 |
| LINC00482 | up | 19 | 8.6 | 2.51E-103 | 36 | 6.4 | 1.90E-49 |
| PLCB4* | up | 21 | 8.59 | 1.43E-13 | 7 | 10.28 | 3.01E-16 |
| PNLDC1 | up | 22 | 8.56 | 1.19E-99 | 42 | 6.2 | 2.55E-46 |
| MAL2 | up | 23 | 8.43 | 3.04E-24 | 5 | 10.6 | 2.43E-32 |
| TTC39A | up | 24 | 8.17 | 1.72E-164 | 26 | 7.42 | 2.68E-120 |
| SERPINB9P1 | up | 25 | 8.01 | 1.98E-65 | 32 | 6.69 | 2.66E-41 |
| LINC01531 | up | 27 | 7.77 | 5.71E-106 | 11 | 9.65 | 9.41E-141 |
| RAPGEF4* | up | 28 | 7.71 | 3.11E-39 | 15 | 9.09 | 5.34E-46 |
| CACNA2D2 | up | 32 | 7.58 | 6.51E-70 | 43 | 6.09 | 1.45E-38 |
| EPHA8 | up | 33 | 7.51 | 6.15E-53 | 24 | 7.52 | 1.83E-48 |
| TWIST1 | up | 34 | 7.46 | 2.18E-48 | 20 | 8.01 | 1.59E-52 |
| LRRN2 | up | 35 | 7.12 | 1.89E-38 | 46 | 5.93 | 1.01E-22 |
| PITX2 | up | 37 | 6.82 | 5.05E-19 | 30 | 6.89 | 1.88E-16 |
| CYP26B1 | up | 38 | 6.79 | 1.94E-35 | 17 | 8.34 | 3.88E-49 |
| ITGB2 | up | 39 | 6.67 | 7.50E-31 | 27 | 7.37 | 5.36E-32 |
| LTK | up | 43 | 6.57 | 1.09E-19 | 33 | 6.55 | 1.25E-16 |
| GABRA3 | up | 44 | 6.56 | 2.94E-05 | 41 | 6.21 | 2.80E-04 |
| MGAT4A | up | 47 | 6.38 | 3.23E-44 | 50 | 5.71 | 7.25E-30 |
| HAS2 | up | 50 | 6.17 | 5.40E-44 | 38 | 6.32 | 8.15E-39 |
| ZNF667 | down | 10 | −9.6 | 2.32E-44 | 19 | −8.96 | 1.89E-50 |
| GFRA3 | down | 23 | −8.52 | 1.13E-13 | 24 | −8.56 | 1.58E-13 |
| ABCA8 | down | 24 | −8.4 | 2.02E-47 | 10 | −9.91 | 3.14E-43 |
| SCN1B | down | 25 | −8.31 | 5.38E-97 | 26 | −8.45 | 2.62E-86 |
| PMEPA1 | down | 28 | −8.13 | 2.64E-72 | 28 | −8.43 | 1.34E-64 |
| FAM19A5 | down | 29 | −8.1 | 1.39E-65 | 3 | −12.13 | 1.92E-49 |
| HS3ST1 | down | 36 | −7.56 | 3.29E-21 | 8 | −10.2 | 2.13E-15 |
| MGMT | down | 49 | −7.11 | 1.78E-28 | 20 | −8.86 | 7.47E-35 |
Genes found in top 50 overexpressed or underexpressed in both MP41 and MP46 compared to normal uveal melanocytes. Genes are ranked by log2 fold changes for each comparison.
Genes previously identified in UM studies are indicated with.
We identified 101 cytobands that contained shared DEGs (Figure S1D) most frequently located on 8q (18%). Upregulated genes were significantly associated with copy number increases on chromosomes 8q, 1q, and 21q in MP41 and MP46. We also found upregulated genes on cytobands from 5q and 4q, which had normal copy numbers in MP41 and MP46, and on cytobands from 2p/2q and 7q, which were gained only in MP46. Downregulated genes were associated with loss of 1p, 3p, 8p, and 16q in MP41 and MP46. Yet, we also found significantly downregulated genes in 9q, 10q, and 19p/q, which are lost only in MP41, and on 12q and 17q that were lost only in MP41 and MP46, respectively.
We next used Reactome31 analysis to identify enriched pathways in the DEG. The top 50 dysregulated pathways are shown in Figure 2G. These pathways include proliferation-related pathways (cell cycle, mitosis, checkpoints) as well as chromatin maintenance and DNA repair pathways (DNA double-strand break repair, Fanconi anemia [FA]) (Figure S2). DNA damage and repair (DDR) pathways were found to be enriched, and within DDR pathways, the homologous recombination (Figure S2B) pathway was the most significantly enriched (p: 5.99E-0) based on the enrichment of DNA damage sensors and repair enzymes genes. Additionally, 13 genes from within the FA (Figure S2C) were consistently enriched after comparing UM to NM (min p value: 2.94E-4).
In summary, these analyses revealed a set of DEGs from two UM models compared to NMs with notably the overexpression of two GNA11/GNAQ pathway downstream genes, RAPGEF4 and PLCB4. Interestingly, DNA damage repair pathways were significantly enriched, and PRAME, a marker of aggressiveness in UM, was among the most upregulated genes in both MP41 and MP46.
Optical mapping and FISH analyses reveal major chromosomal aberrations
To further investigate genomic aberrations related to UM, we performed optical mapping with the Bionano platform32,33 as well as telomere and centromere staining followed by M-FISH (TC + M-FISH34) on MP41 and MP46. The optical mapping achieved 500-bp resolution and a minimum coverage of 97x per sample, which revealed long-range DNA alterations including translocations, insertions, duplications, and small deletions in both models (Figures 3A–3C). In MP41, optical mapping revealed both intra- and inter-chromosomal translocations (t(19; 19) t(1; 12) and t(6; 8)). In MP46, intra-chromosomal translocations for chr19 and the inter-chromosomal translocation t(1; 22) were identified. Structural variants (SVs), including deletions, insertions, and duplications, were also identified (Figure 3C).
Figure 3. DNA optical mapping and FISH analysis of MP41 and MP46.
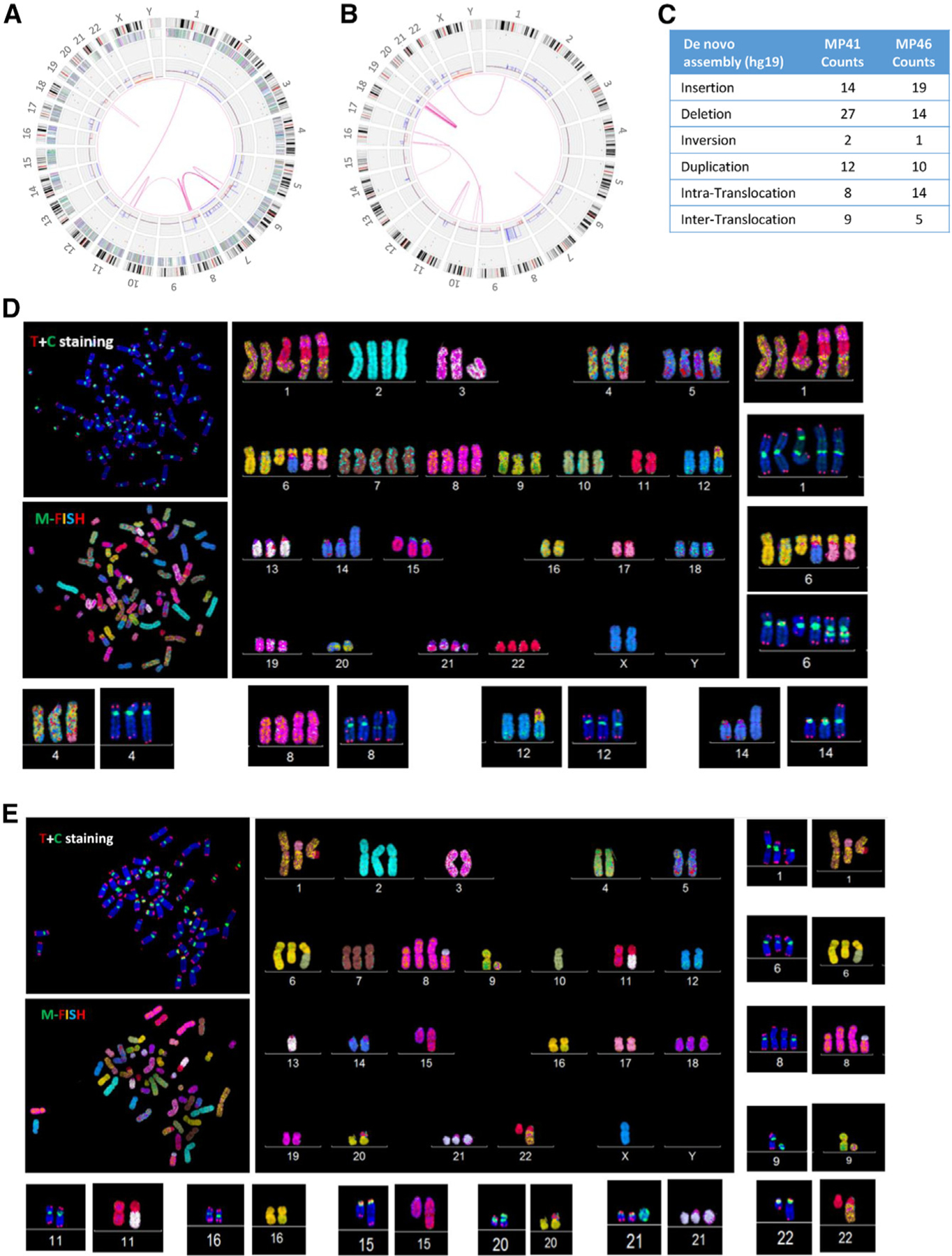
(A and B) Circos plot of aberrations in (A) MP41 and (B) MP46. From the central to the periphery of the circos plot: whole-genome view summarizes intra- and inter-chromosomal translocations (pink lines), copy number gains and losses are listed on the first internal layer of the circos, and SVs (insertion, deletion inversion, and duplication) are labeled as colored dots in the intermediate layer of the circos. Gene density, cytobands, and chromosomes comprise the outer layers of the circos.
(C) Number of insertions, deletions, inversions, and duplications and intra- and inter-translocations are detailed for MP41 and MP46 defined by Bionano optical mapping.
(D and E) Telomere and M-FISH from analysis of (D) MP41 and (E) MP46 are derived from two different FISH analyses. Upper left panels show telomere (red signal) and centromere (green signal) staining and are counter labeled with DAPI (blue). Lower left panels show M-FISH analysis. Main panels correspond to the karyotype view.
TC + M-FISH revealed a hyper-triploid genome for MP41 (Figure 3D) with dicentric chromosomes (dic(14;16), i(8q), dic(1;11;8)). We also identified four telomeric-related losses dic(1;11;6;8), dic(1;11), dic(6;8;14), and dic(6;8;17), and one interstitial telomeric sequence, dic(6;8;17) (Figure S3). Chromosomal end-to-end fusion is often associated with dicentric chromosomes and aberrant chromosomal structures. MP46 also displays a complex karyotype (Figure 3E): a hyper-diploid genome with multiple dicentric chromosomes: dic(1;17), dic(6;10), dic(8;21), dic(13;22), dic(16;20), and dic(20;22). Two translocations were identified in MP46: t(1;22) and der(15)t(11;15). In addition to chromosome structural alterations, we repeatedly found ring chromosomes derived from chromosomes 1, 8, and 11 in MP41 and from chromosomes 9 and 21 in MP46 (Figure S3E).).
We next analyzed the RNA-seq data for potential fusions using multiple algorithms and confirmed them using RT-PCR and Sanger sequencing. In most cases, SVs were directly associated with the fusion RNAs. For example, an insertion-duplication on 12q24 in both MP41 and in MP46 led to the fusion of MAPKAPK5-ACAD10 in MP41 and KDM2B-RHOF in MP46. Both events occurred within the replication fragile site FRA12E,35 which has also been associated with germline structural polymorphisms.36 In MP46, a t(1; 22) translocation between CABIN1 and MPRS21 led to the fusion RNA CABIN1-MPRS21.
Interestingly, complex translocation t(6; 8) in MP41 resulted in two fusion RNAs: GPAT4-NCOA7 and POMK-RSPO3. From FISH, we determined that the fusion was located on derivative chromosomes 6: der(6)t(6;8); dic(der(6)t(6;8);14); dic(der(6) t(6;8);16); dic(der(6)t(6;8);17), and derivative chromosomes 8: der(8)t(6;8), ider(8)(q10)t(6;8)x2, dic(1;11;8;6)x2, and on a ring chromosome r(dic1;11;8;6). In MP41, we specifically probed NCOA7 and RSPO3 (both on chromosome 6), as well as GPAT4 and POMK (both on chromosome 8) (Figure S3G). Contrary to dicentric chromosome 6, which was labeled with all four probes, normal chromosomes 6 only stained with NCOA7 and RSPO3 probes (Figure S3H). On chromosome 8, we could stain the native genes GPAT4 and POMK as well as chromosome 6-derived NCOA7 (but not RSPO3) (Figure S3H). Isochromosome 8 was labeled only with GPAT4 and NCOA7, as dic(1;11;8;6) (Figure S3H). A potential model for the generation of these complex patterns is in Figure S3I.
To verify if genomic instability is recurrent in UM, we investigated structural genomic aberrations in additional models of aggressive UM (Mel202, MM66, OMM1, and OMM2.3) through optical mapping and FISH analyses (Figure S4). Major SVs were detected in all tested UM models (Figure S4B). Telomere aberrations were present in all tested UM cell lines (MP41, MP46, Mel202, MM66, OMM1, and OMM2.337,38) and absent in normal controls (Figure S4C). Translocations and dicentric chromosomes were detected in the cellular models (Mel202, MM66, OMM1, OMM2.3, Figures S4D–S4G). Derivative chromosomes were also associated with complex SVs identified with optical mapping.
In summary, high-resolution DNA optical mapping combined with TC + M-FISH shows that high levels of genomic instability are a recurrent pattern in diverse aggressive UM models.
DNA methylation analysis reveals differences in CpG island (CGI) patterns and identifies BAP1 promoter deletion
To characterize DNA methylation patterns, OxBS sequencing (Cambridge Epigenetix) was performed on both UM and NM. Oxidative bisulfite sequencing39 was more robust in our samples, particularly for the NMs, most probably due to the abundance of melanin. First, a random forest analysis placed MP41 and MP46 in the TCGA methylation groups 2 (corresponding to BAP1 proficient) and 4 (BAP1 deficient) respectively. We next categorized methylation levels with respect to the following genomic localizations: CGI promoters, non-CGI promoters, exons, introns, intergenic regions, and repeat elements (identified using RepeatMasker annotations40) (Figure 4A). While CGI promoters are generally equally demethylated in UM samples and NMs, tumor samples are globally less methylated in non-CGI promoters compared to normal samples across all genomic localization categories.41,42
Figure 4. Whole-genome DNA methylation analysis of UM models and normal melanocytes.
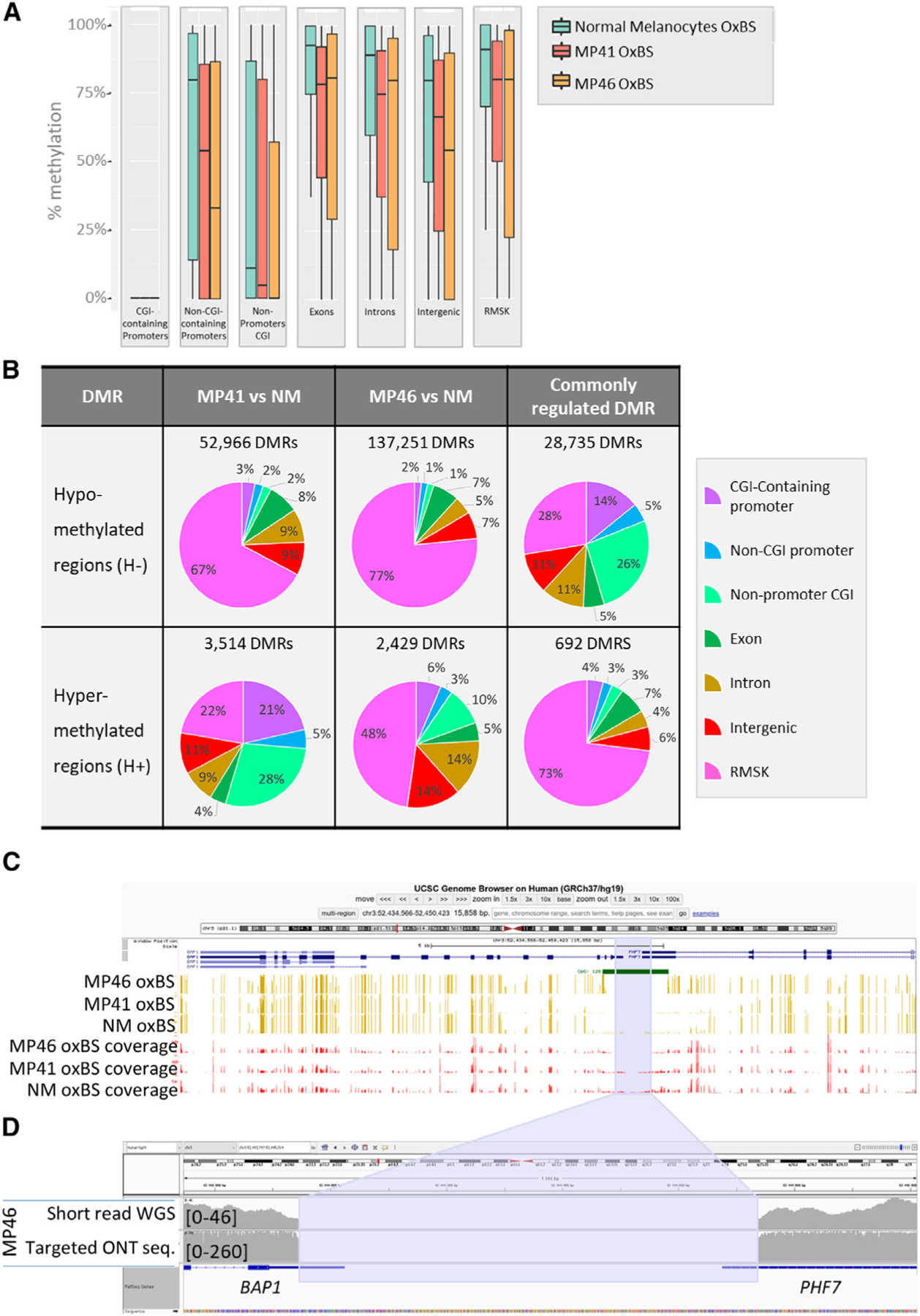
(A) DNA methylation levels based on oxidative bisulfite DNA treatment followed by whole-genome sequencing are shown at CGI promoters, non-CGI promoters, non-promoters CGI, exons, introns, intergenic regions, and on repeats, in normal melanocytes, MP41, and MP46.
(B) Differentially methylated regions (DMRs) as hypo- and hyper-DMRs (H− and H+) in 300-kb window in MP41 vs. NM and MP46 vs. NM. Commonly regulated DMRs correspond to DMRs identified in MP41 vs. NM and MP46 vs. NM comparisons. DMRs were considered as commonly regulated where sharing the same variation (H− or H+) and when their coordinates were identical or overlapping.
(C) Percentage of CpG methylation in MP41, MP46, and NM in BAP1 locus through UCSC Genome Browser are represented in yellow, and sequencing coverages are represented in red bars. CpG island 129 overlaps the BAP1/PHF7 promoter. The blue area highlights the deletion detected in MP46.
(D) IGV view of MP46 short-read sequencing (first line) and targeted ONT sequencing (second line) illustrate the boundaries of promoter/5′ UTR deletion in BAP1 and PHF7 genes.
Next, we identified differentially methylated regions (DMRs) in each PDX compared to NMs (Figure 4B). Most DMRs are hypomethylated (H−) in UM, in agreement with global methylation patterns (Figure 4A). We focused our analysis on the DMRs, which are consistent in MP41 and MP46. As expected, most shared DMRs are H− (28,735/29,427, 98%). Notably, half of MP41 DMRs were shared with MP46, but MP46 shared less than a third of its DMR with MP41 (H−: 21%, H+: 28%). Interestingly, while H− genomic localizations were strongly enriched at repeat elements in each model independently, the shared H− ones were strongly depleted at repeat elements (Figure 4B). Genomic localizations of H+ DMRs are differentially distributed in MP41 and MP46 with the largest differences occurring in repetitive elements and CpG islands. The localization of the 692-shared H+ ones were primarily in repeat elements, and a subset was located in CGI and non-CGI promoters (Figure 4B).
As mentioned before, MP46 clusters with the DNA methylation group 4 of TCGA,18 which includes BAP1-deficient and monosomy 3 UM tumors. We analyzed the status of BAP1 promoter methylation because we did not find BAP1 coding mutations in MP4621 despite the absence of BAP1 protein. Although MP41 displayed a hypomethylated promoter similar to NMs, consistent with expression levels (Figure S5A), MP46 shows a specific hyper-methylation pattern in the promoter (CpG129, UCSC Genome Browser [hg19; chr3:52,443,678-52,445,104]) that co-localized with the boundary of an 809-bp deletion identified in the whole-genome OxBS data (Figure 4C). The BAP1 promoter is a bidirectional promoter shared between BAP1 and PHF7. In MP46, the BAP1 promoter was deleted, and deletion boundaries were hyper-methylated. Furthermore, both BAP1 and PHF7 were not expressed in MP46 (Figures S5A and S5B). This large deletion was confirmed in DNA labeling, optical mapping, and WGS data.
BAP1 promoter deletion has not been described in the 1346 ClinVar records or in the Cosmic database. To investigate if promoter deletion explains other cases of BAP1 deficiency in UM, a targeted next-generation sequencing (NGS) approach based on tiling amplicon sequencing covering BAP1 was performed on 53 tumor samples (Figure S5C). We identified two additional cases with similar deletions in the BAP1 promoter (Figure S5D) for which immunohistochemistry confirmed the absence of BAP1 expression (Figure S5E). A recent UM case has also been identified internally harboring a 2.2-kb deletion in BAP1/PHF7 5′ UTR/promoter. In the tumor, BAP1 could not be detected by IHC, and the promoter deletion was confirmed by long-range PCR (Figures S5F–S5H). This recent UM case was analyzed as part of the French initiative “France Medecine Genomique 2025” and made possible by the SeqOIA platform (https://pfmg2025.aviesan.fr/en/).
Next, we analyzed the methylation status of the consistent DEGs in MP41 and MP46. Most of the DEGs did not have a significant methylation switch, since only 5% of the DEGs displayed hypo- or hyper-methylation of their promoters. However, several of the greatest DEGs, such as RASGRP3 and PRAME discussed above, were part of this minority.
DNA topology analysis reveals stable compartments and topologically associated domains containing most differentially expressed genes
The spatial organization of melanocyte genomes, particularly their physical interactions, may contribute to the regulation of gene expression during transformation. We performed chromosome conformation capture (Hi-C) to elucidate whether gene expression changes are associated with chromatin organization and DNA folding in MP41 and MP46 (Figure 5A). To account for background from copy number alterations, we first compared multiple computational approaches for normalization (Figure S6). We found that CAIC was less sensitive to copy number variations than ICE and LOIC (Figure S6C), similar to previously analyzed breast cancer cell lines.43 Genome folding presents itself at multiple length scales, and chromosome territories contain physically separated euchromatic and heterochromatic regions known as A and B compartments44 and topologically associated domains (TADs) that result from loop extrusion.45–48
Figure 5. Compartments and TADs in NMs and Ums.
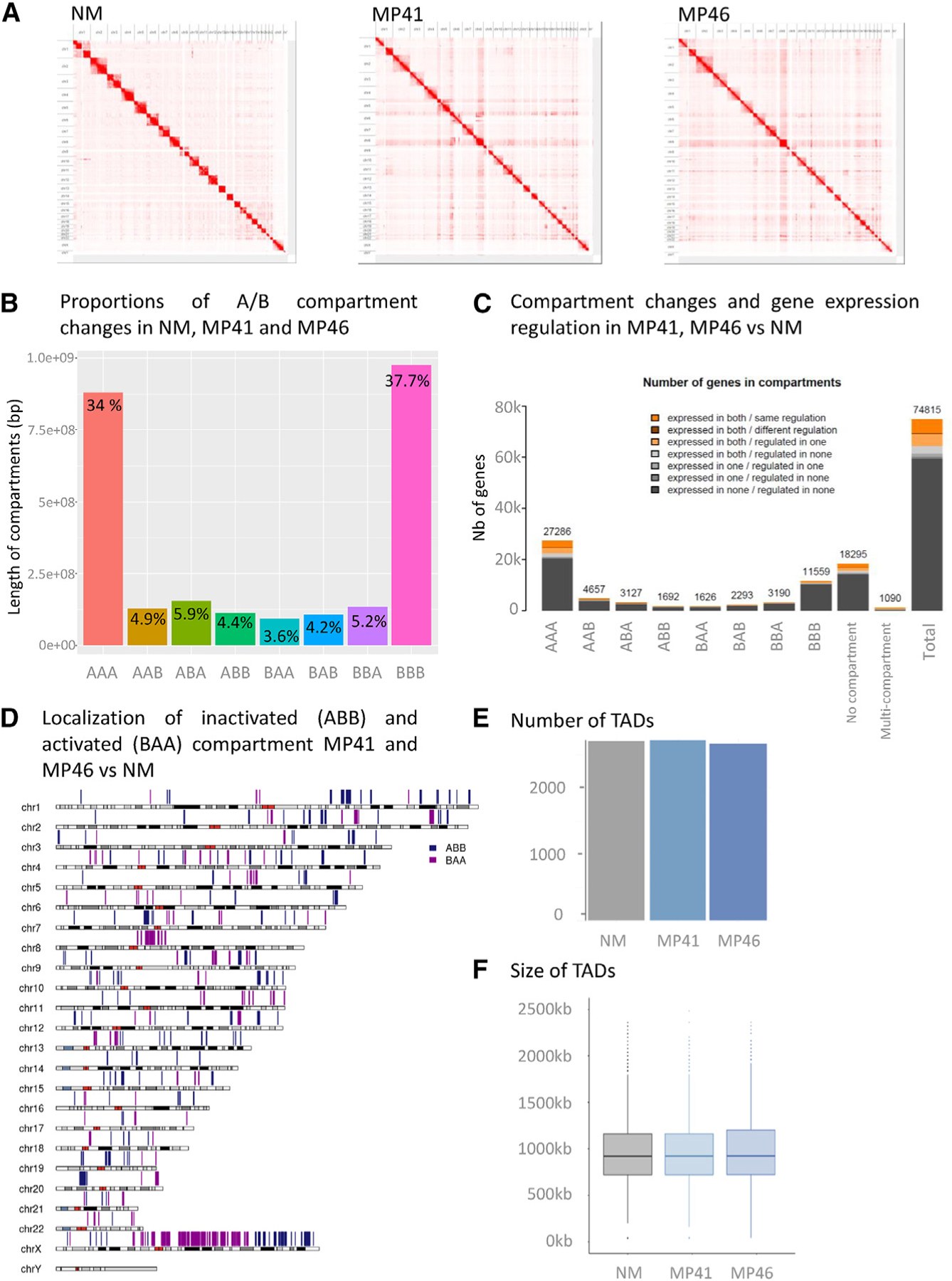
(A) Contacts maps derived from in situ Hi-C at the whole genome level for NM, MP41, and MP46.
(B) Histogram of compartment changes in NM, MP41, and MP46. A and B compartments identified at 250-kb resolution.
(C) Localization of inactivated (ABB) and activated (BAA) compartment in MP41 and MP46 vs. NM on a whole-genome view.
(D) Integration of compartment changes and gene expression among NM, MP41, and MP46.
(E and F) Number (E) and size (F) of TADs in NM, MP41, and MP46.
First, we performed a compartment analysis at 250-kb resolution,44 in NM, MP41, and MP46 (and refer to each window with a three-letter code for its compartment status in NM, MP41, and MP46 respectively). Most compartments (~72%) shared the same status between our three models: A compartments corresponding to euchromatic regions and active regions (labeled as “AAA,” 34.04%) and B compartments corresponding to heterochromatic regions and inactive regions (BBB: 37.72%) (Figure 5B). Notably, CAIC normalization allowed equal detection of A/B compartments irrespective of copy number status (Figure S6C). Other compartment patterns were roughly frequent, accounting for 3.5%–6% of windows (Figure 5B). A karyotype view was used to illustrate the position of compartment assignment changes in NM and UM models (Figure 5C).
We further analyzed whether changes in compartment status overlapped with gene content and expression (Figure 5D). Most of the DEGs were located in the constitutively active A compartments (AAA), this enrichment was not significant when corrected for the total number of genes and the number of genes per compartment. The ABB and BAA compartments, corresponding respectively to specific inactive and active compartments in UM models, contained 159 DEGs (same regulation, 96 in ABB and 63 in BAA); however, this was not statistically significant (Table S6). Second, TADs were analyzed in both UM models using insulation score analysis in 40-kb bins.49 No significant differences in the number or size of TADs were observed between UM and NM (Figures 5E and 5F).
In summary, no differences in TAD structures were found, and most DEGs (83%) were found in compartments that did not change status from NM to UM (Figure 4C). However, 159 DEGs (~3%) were associated with changes in compartment status: 63 DEGs belong to active compartments in MP41 and MP46, and 96 DEGs belong to inactive compartments, indicating that differences in topology could underly differences in gene expression.
Chromatin topology and histone mark changes are associated with upregulation of PRAME
To further enrich our understanding of chromatin organization and gene expression regulation in NM, MP41, and MP46, ChIP and sequencing analysis were carried against the active epigenetic mark H3K4me3 and repressive marks H2AUb and H3K27me3. As above with RNA-seq, the profiles of these three histone marks in cell lines of MP41 and MP46 consistently resembled the matched PDX (Table S7). Additionally, in MP41 and MP46, we used H3K27Ac to find active enhancers and CTCF to study cohesin-mediated loop extrusion.50 Due to limitation amounts of DNA available for NM, H3K27Ac and CTCF were not profiled.
As depicted in Figure 5D, 371 active compartments were identified by Hi-C specifically in PDX samples (BAA) containing 63 consistently DEGs (37 higher and 26 lower) in MP41 and MP46 versus NM. Among the 37 upregulated genes in activated compartments, PRAME and ZNF280A were enriched in H3K4me3. Also, 33 genes (including PRAME, ZNF280 A/B, EZH2) display H3K27Ac peaks, two genes lost H3K27me3 marks (PITX2 and COL4A5), and four genes were demethylated in their promoters. Among the 26 downregulated genes, none were enriched in H3K27me3 marks in both UM models, only one gene (ZC4H2) lost H3K4me3, 20 genes contain H3K27Ac marks, and no gene displays a DNA hyper-methylated promoter.
The upregulated genes associated with activated compartments include EZH2, EPHA4, and PRAME. Among the major regulated genes, PRAME is associated with a particularly high fold change from undetectable in NM to highly expressed in both PDXs (log2 FC ~ 12.1 in MP41 and 11.9 in MP46 vs. NM) (Figure 6A). The PRAME gene is located on 22q11.22 (hg19 chr22:22,890,123-22,900,022) between the tandem Zinc finger proteins ZNF280A and ZNF280B downstream of PRAME and a gene encoding a putative membrane glycoprotein (POM121L1P) upstream of PRAME, as illustrated on Figure 6D. In this locus, only ZNF280A, ZNF280B, and PRAME are upregulated in a compartment being activated in our UM models compared with NM (Figure 6A). We also observed hyper-methylation of the PRAME promoter in NMs and hypo-methylation in MP41 and MP46 tumor models (Figure 6B), which correlated with the activation of PRAME expression in UM (Figures 6A and 6D).51 The PRAME promoter methylation status has previously been described in UM as a marker of aggressiveness.52
Figure 6. Multi-omics analysis of PRAME locus.
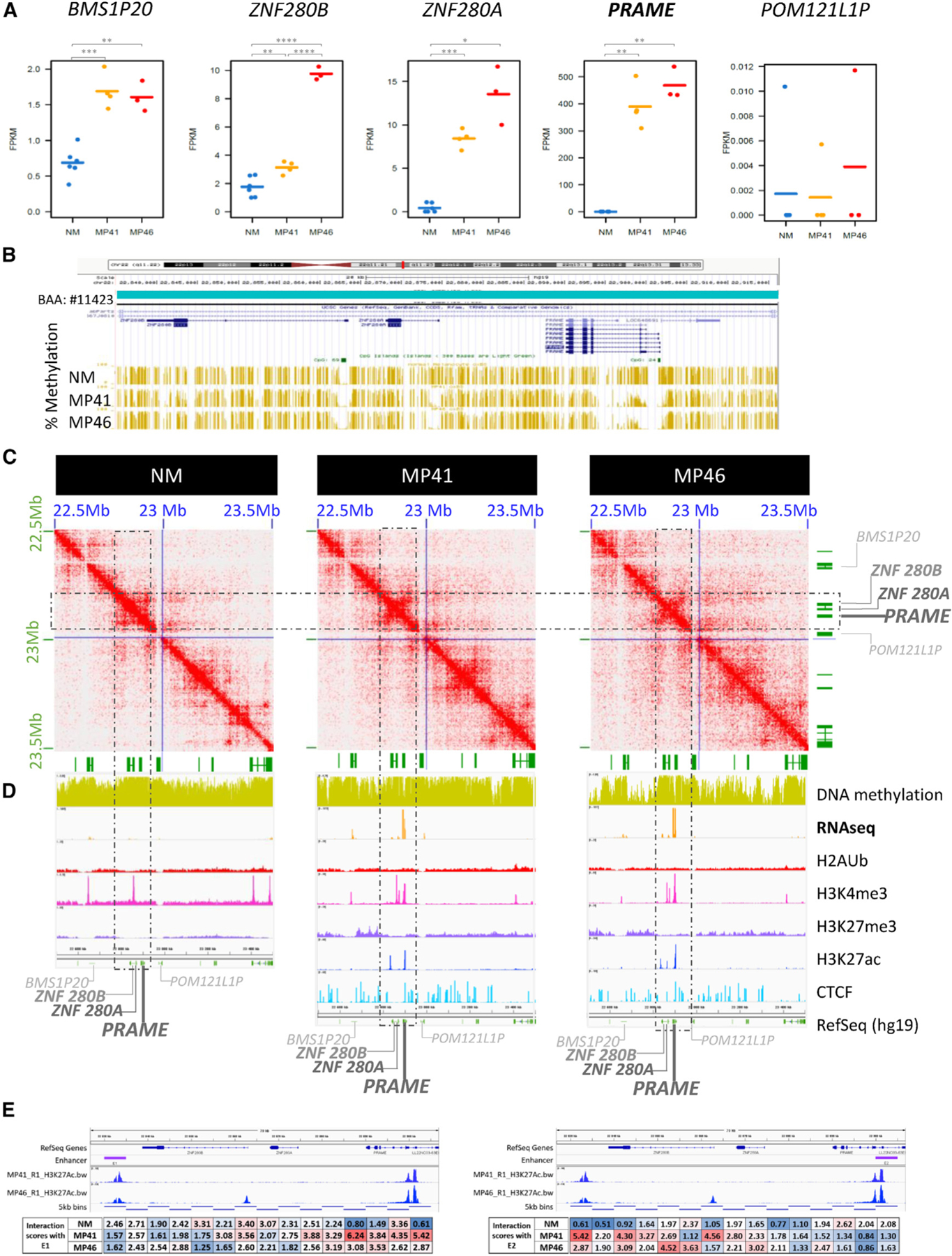
(A) Gene expression of PRAME and its neighbors as BMS1P20, ZNF280B, and ZNF280A upstream genes and POM121LP downstream. RNA-seq data of NM, MP41, and MP46 replicates (FPKM).
(B) UCSC Genome Browser view (hg19) of percentage of DNA methylation (golden bars).
(C) DNA contacts maps of NM, MP41, and MP46 at 5-kb resolution in PRAME TAD (blue square).
(D) UCSC Genome Browser view of the PRAME locus showing DNA methylation, RNA-seq (log2), H2AUb, H3K4me3, H3K27me3, H3K27Ac, CTCF, and RefSeq genes.
(E) H3K27Ac marks and HiC interaction of PRAME promoter with potential distal enhancer E1. HiC interactions anchored in E1 (left) and E2 (right) quantified in boxes under genomic locus.
Chromatin conformation analysis at the PRAME locus revealed a different largescale pattern in NM compared to UM MP41 and MP46 models. In NMs, the contacts are densely connected throughout the whole locus (Figure 6C, dashed box), while in both tumor models, overall contact density is reduced and an “anti-diagonal” pattern is apparent, consistent with opening of the chromatin and anchored by a restricted set of interactions (Figure 6C).
Our analysis of histone marks at the PRAME locus in MP41 and MP46 revealed the presence of active histone marks (H3K4me3 and H3K27Ac) and the absence of repressed histone marks such as H3K27me3. In NMs, H3K4me3 and H3K27me3 peaks were not observed on the PRAME promoter (Figure 6D). Importantly, the H3K27Ac profiles revealed consistent peaks in both MP41 and MP46 proximal to the PRAME promoter as well as distally, just downstream of ZNF280B. To investigate whether these H3K27Ac regions physically interact, we analyzed the Hi-C interactions at high resolution. Anchoring the analysis either at the distal peak “Enhancer 1” or the PRAME promoter proximal peak “Enhancer 2” revealed a clear enrichment for interactions in the two tumor models and not in NMs (Figure 6E), confirming a three-dimensional looping interaction with this distal regulatory site.
Analyzing the ENCODE resource of transcription factor ChIP-seq experiments,53,54 128 transcription factor recognition sites were identified between ZNF280A/B and PRAME, corresponding to 85 different DNA binding factors involved in chromatin and transcription regulation. Among these, 20 are also significantly upregulated in our gene expression analysis (Figure S7). Expressed and upregulated PRAME locus DNA binding factors include the cohesin components RAD21 and SMC3, which may contribute to the changes in chromatin conformation. Chromatin organization modifiers such as DNA binding helicases CHD1 and CHD2 were also found upregulated in our analysis. All of these proteins are associated with chromatin remodeling and may contribute to PRAME overexpression in our UM models. Overall, we identified changes in chromatin conformation and long-range looping with a putative distal enhancer associated with striking overexpression of PRAME in UM.
DISCUSSION
Here, we report an extensive multi-omics approach comparing two aggressive UM models with short-term cultures of NMs. The multi-omics analysis includes whole-genome somatic mutations, transcriptome, copy number, methylome, DNA optical mapping, FISH, histone modifications, and DNA topology analysis by Hi-C. The MP41 tumor did not display any identifiable BSE event, expresses BAP1, and is classified in the TCGA copy number group enriched in the SF3B1-mut UM. The second tumor sample, MP46, belongs to the TCGA high-risk group and did not express BAP1 despite the absence of mutations in the BAP1 coding sequence.
Although UM patients with disomy 3 display a favorable outcome overall, a proportion do develop metastases. This was the case of the patient from whom MP41 was derived. Interestingly, in the TCGA cohort, one-third of disomy 3 UM samples (13 out of 38) do not display a BSE event, whereas the absence of BSE event is rare in the monosomy samples (3 out of 42). Given the absence of a BSE mutational event in MP41, we carefully analyzed WGS data for mutations potentially explaining tumor progression in MP41. In addition to GNA11 mutation, a nonsense truncating mutation in KMT2C was predicted as an oncogenic driver. This gene is altered in about 5% of cancers, but KMT2C mutations have not been described in UM.55 The RNA level of KMT2C in MP41 is not significantly different of that in NM, and we observed a very high expression of this gene in MP46. Further functional studies restoring the wild-type allele of KMT2C in cell lines derived from MP41 need to be performed to explore the potential contribution of the nonsense KMT2C mutation to UM. MP41 may thus serve as an informative model of aggressive UM outside of the classical BSE context.
Our gene expression profiling revealed several interesting changes compared with those in normal uveal melanocytes. Notably, PLCB4 and RASGRP3 were among the top 50 upregulated genes in both tumor models. Whereas activating mutations of PLCB4 have been described in UM, our data suggest that high expression can also contribute to over-activation of the PKC pathway. The increase in expression of RASGRP3 in UM samples is also of particular interest given that this protein has been shown to mediate MAPK pathway activation in UM.8,9 In the current study, we have identified an additional GPCR downstream pathway gene, the Rap guanine nucleotide exchange factor RAPGEF4, which was significantly upregulated in both UM PDXs compared to NMs. Further functional studies must be conducted to evaluate the potential role of this protein in the UMs.
UM is classically considered to display a relatively simple pattern of karyotypic alterations in comparison with other solid tumors. Interestingly DNA repair is among the most deregulated pathways in the expression dataset. The TCGA consortium described an activation of DNA damage repair in transcription-based cluster 4, which is enriched in patients with the poorest prognosis. Consistent with activation of these pathways at the gene expression level, our structural DNA analysis combining optical mapping and FISH revealed multiple chromosomal aberrations including intra and inter-chromosomal translocations, insertion-duplications, telomere shortening, and telomere aberrations. Importantly, we observed chromosomal aberrations using optical mapping and FISH approaches in six other UM cell lines strongly suggesting that genomic/chromosomal instability is a hallmark of aggressive UM.
Biallelic inactivation of BAP1 is associated with an increased risk of metastasis in patients with UM. In this work, we describe a recurrent genetic mechanism of BAP1 deficiency consisting of a deletion on the BAP1 promoter and boundary hyper-methylation. A targeted NGS approach identified two other cases of UM displaying deletion in the same genomic area resulting in a lack of BAP1 expression indicating recurrence of this mechanism. Given the correlation of BAP1 deficiency with the risk of developing metastases, our results indicate that in UM patients, it will be informative to look at the BAP1 promoter when mutations or indels in the coding sequence are not detected. This has been recently implemented in Institut Curie where NGS is performed on UMs as part of the national initiative “France Medecine Genomique 2025.” Within this initiative, we have recently detected an additional UM case that displayed the deletion of the BAP1 promoter.
DNA topology was investigated by using an in situ Hi-C approach. Most changes in gene expression between NM and our two UM models were not accompanied with changes in compartment status. An important exception to this general pattern could be observed at the PRAME locus, where striking overexpression was accompanied by compartment switching and a qualitative change in the intra-TAD contact pattern with an overall opening of the interactions but dedicated interactions between the PRAME promoter and a distal peak of H3K27Ac. This finding highlights the relevance of using a multi-omics approach that combines chromatin confirmation alterations and RNA expression. Further investigation of repositioned compartments identified in our comparison could help address the questions of compartment specificity recently identified in other tumor types.56
In summary, our study illustrates how multi-omics integrative approaches conducted in a limited number of samples can improve our understanding of tumorigenesis and reveals two distinct mechanisms of gene expression dysregulation in UM with potential clinical implications.
Limitations
Our study has several limitations. While we integrated a large number of diverse genome-scale assays, we focused primarily on two tumor models. Particular results were confirmed in additional samples, but the full collection of assays would need to be performed in larger cohorts to definitely determine recurrence frequencies. Although we were able to generate important datasets in a rare resource of primary uveal melanocytes as a proxy for the cell of origin, it was essential to grow them as short-term cultures to generate pure populations of sufficient cell numbers for our assays. Since growth conditions are known to have effects on chromatin regulation and gene expression, we also profiled PDX-derived cell lines to compare with the PDXs and NMs. Determining if in vivo uveal melanocyte profiles match these short-term cultures will be important to verify in further studies. Finally, the distal enhancer at the PRAME locus remains to be studied in detail to identify the trans factors binding and driving looping with the PRAME promoter to fully dissect the mechanism driving such elevated expression.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Joshua J Waterfall (joshua.waterfall@curie.fr)
Materials availability
This study did not generate new unique reagents.
Data and code availability
Sequencing data generated in this work have been deposited in the GEO repository, under the accession GSE199679 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE199679). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
PDX models
UM models MP41 and MP46 were obtained by engrafting biopsies from primary UM of patients. The protocol was approved by the Institut Curie Hospital committee (CRI: Comité de Revue Institutionnel). Primary tumor biopsies were engrafted with informed consent from the patient into the interscapular fat pad of female SCID mice (Charles River Laboratories), 5 to 7 weeks old, without any extracellular matrix preparation under total xylazine/ketamine anesthesia. Mice were maintained under specific pathogen-free conditions. Their care and housing were in accordance with institutional guidelines and the rules of the French Ethics Committee: CEEA-IC (Comité d’Ethique en matiè re d’expérimentation animale de l’Institut Curie, National registration number: #118). The project authorisation no. is APAFIS#22337-2019100820381537. The housing facility was kept at 22°C (±2°C) with a relative humidity of 30–70%. The light/dark cycle was 12 h light/12 h dark. MP41 and MP46 models were obtained from engrafting of biopsies from a primary tumor respectively of a 50-year-old female patient and a 69-year-old male patient. Patients at Institut Curie were informed that their tumor samples might be used for scientific purposes and had the opportunity to decline. This study protocol was approved by the local ethical committee: Uveal Melanoma Group of Institut Curie Hospital. The patient follow-up was carried out at Institut Curie hospital.
Cell lines
UM cell lines in our study were derived from 3 female patients (MP41, MM66 and Mel202) and 3 male patients (MP46, OMM1, and OMM2.3). Mel202 (Ksander et al., 1991), were purchased from The European Searchable Tumor Line Database (Tubingen University, Germany). OMM1 and OMM2.3 (Luyten et al., 1996; Chen et al., 1997) were kindly provided by P.A. Van Der Velden (Leiden University, The Netherlands). Cell lines were cultured in RPMI-1640 supplemented with 20% (MP41, MP46, and MM66) or 10% (Mel202, OMM1, and OMM2.3) FBS (Life Technologies), Penicillin 100 U/ml and Streptomycin 100 mg/mL (Life Technologies). All cell lines were tested for Mycoplasma and proved Mycoplasma free. Cell lines were maintained in a humidified atmosphere with 5% CO2 at 37°C. Cell authentication was verified before whole genome analysis based on STR profiling (Powerplex 16HS kit, Promega). The STR profiles and sex determination markers were analyzed with GeneMapper 5.0 (Life technologies) and results were controlled with the help of Cellosaurus (https://www.cellosaurus.org) and ATCC databases (https://www.atcc.org/search-str-databasedatabases).
Normal uveal melanocytes
Normal uveal melanocytes derive from one female healthy donor and the distal enucleation material of three UM patients (two female and one male). Cells were dissociated and maintained in culture from choroidal membranes obtained from Pr Simon Saule and Dr Geraldine Liot (Institut Curie). After enzymatic (with collagenase) and mechanical dissociation, primary cells were cultured in HAM/F12 supplemented with 10% FBS, Penicillin 100 U/mL, Streptomycin 100 U/mL, 2mM L-glutamine, 2.5μg/mL Amphotericin B. Extemporaneously completed media was supplemented with 0.1mM IBMX, 10 ng/μL of Cholera toxin (10 ng/mL final), and 10 mg/mL βFGF and filter with a 0.22μm filter. Cells were maintained in a humidified atmosphere with 5% CO2 at 37°C, and culture medium was exchanged twice per week.
METHOD DETAILS
Isolation of UM cells from PDX
As described before,19,20,22 MP41 and MP46 xenograft tumors were harvested before they reached a volume of 1 cm3, following ethical rules, and processed immediately for dissociation, immunolabelling and sorting based on Petit et al. protocol.57 To avoid isolation of heterogeneous UM cell populations in batches of experiments, immunostaining was conducted with anti Muc18 containing a rabbit Fc: clone 8H2rFc, also with anti CEACAM1 (8G5hFc) and anti NG2 (14A7hFc) as characterized previously.22 Secondary antibodies used were anti rabbit FC-AF647nm to reveal Muc18 labeling, and anti-human Fc to reveal at the same time CEACAM1 and NG2 labeling. Cell sorting was conducted on single live cells with the help of cytometry platforms of Institut Curie. In total, 15–20 mice were grafted per model, generating 225*106 cells for MP41 and 133*106 cells for MP46. Twelve batches of dissociation and cell sorting were carried out for MP41 and 10 batches for MP46 generating in total 225×106 and 133×106 cells respectively for MP41 and MP46. After cell sorting, a fraction of cells was kept for DNA and RNA extraction, and most cells were fixed according to Rao et al. protocol.
Quality control of isolated cells
To monitor each fraction of MP41/MP46 isolated cells, DNA and RNA were extracted as described before, and tested for chromosomal copy number alterations and gene expression using Affymetrix microarrays respect to previous analysis done on primary tumors or PDX samples.22
Based on this analysis, pools of MP41 and MP46 cells were used for whole genome sequencing, RNAseq, DNA methylation, in situ Hi-C, ChIPSeq experiments, and to allow replicates of in situ Hi-C and ChIPSeq analyses.
Whole genome analysis
Genomic DNA was extracted using QIAamp DNA Mini kit, and quality control was achieved using a Nanodrop ND1000 to evaluate DNA purity and Qubit dsDNA BR/HS Assays to evaluate the double strand DNA concentration. 250ng of gDNA from MP41 and MP46 sorted cells were characterized using Affymetrix/Thermo Cytoscan HD microarrays to monitor copy number and LOH. Next, two micrograms of MP41 and MP46 DNAs were used to prepare paired-end 100bp Illumina libraries for whole genome sequencing. Genomic DNA from healthy surrounding tissues was sequenced according to approval by ethic committee of Institut Curie to filtered germline mutations.
Whole genome sequencing was conducted on the genomic DNA extracted from PDX derived UM cells and from normal tissue preserved from enucleation. Illumina short read sequencing was achieved in two-separated runs due to the availability of DNAs. Alignment of the sequenced reads to hg19 was performed with Burrows-Wheeler Aligner (BWA-MEM v0.7.10). MuTect2 from the Genome Analysis Toolkit (GATK v3.5) was used in “Tumor with matched normal” mode to call somatic variants. Somatic point mutation analysis of whole genome sequencing revealed 1348 and 1186 single nucleotide variants in MP41 and MP46 respectively representing less than one somatic mutation per Mb (0.42 and 0.37 SNV/Mb) as observed in UM.18 Detected variants with a frequency in the normal sample greater than 20% were filtered out. Variants in UM models were selected according coverage (>10), read counts (>20) and allele frequency (>20%) in UM models. Cancer Genome Interpreter (CGI,23) and VarSome tools (containing 10 pathogenic predictions,24) were combined to characterize variants.
Targeted long read sequencing
To exclude false positive results from WGS on known UM genes as CYSLTR2, PLCB4, SF3B1, SRSF2, EIF1AX, SUGP1, and MBD4 we apply a complementary sequencing analysis with Oxford Nanopore Technologies tools, and more precisely a targeted CRISPR/Cas9 targeted Nanopore sequencing using an excision approach. First, guide RNAs were designed via IDT tools for the analysis of known hotspots described in COSMIC and TCGA databases occurring in CYSLTR2, PLCB4, SF3B1, SRSF2, EIF1AX, SUGP1, and MBD4 (https://eu.idtdna.com/site/order/designtool/index/CRISPR_CUSTOM). Guide RNA sequences are listed on Table S3.
To achieve the excision approach, two guide crRNA were designed per region of interest of 5kb window. A 1kb area upstream and downstream of the region of interest was taken into account for the design of gRNA and gDNA hybridization. An equimolar pool of crRNA was realized (100 μM). Five microgramms of MP41 and MP46 DNAs were dephosphorylated with Phosphatase (ONT, Oxford, UK), and next mixed with the RNP complex pool including the crNRA pool for a 30-min incubation at 37°C. A polyA tail was then ligated for 5 min at 72°C with a Taq DNA polymerase (ONT, Oxford, UK). Sequencing adaptors were ligated in ligation buffer from the SQK-CS9109 cas9 sequencing kit (ONT, Oxfrod, UK) and T4 DNA Ligase (ONT, Oxford, UK). DNAs were purified with 0.3X Ampure beads and washed with Long Fragment Buffer. Libraries were sequenced with R9.4.1 flowcells on a MinION, up to 48 h to generate a maximum reads respect to active pores. Raw data were processed with the following parameters: the base caller used was Guppy (5.0.17) using a super high accuracy model. Variant calling was performed with Sniffes2.0.5, cuteSV 1.08, SVIM1.4.2, and SURVIVOR 1.0.7 was used to synthetize variant calling. SNP caling was achieved with Pepper-margin-Deepvariant 0.8. Reads with a PHRED score higher than 7 were selected for further analysis in FATSQ files under hg38 human genome.
Whole genome methylation analysis
Whole genome DNA methylation analysis was performed with a Cambridge Epigenetics kit (TrueMethyl kit) that corresponds to an oxidative bisulfite reaction, to identify and analyze only 5-methylcytosine (5-mC). Briefly 400ng of genomic DNA were used to first perform an oxidation reaction, that converts 5-hydroxymethylcytosine (5-hmC) to 5- formylcytosine(5fC). After bisulfite conversion, unmodified C and 5fC will be converted into uracils and sequenced as thymines contrary to 5mC that remains a cytosine sequence. Libraries were sequenced on an Illumina Hiseq as paired end 100bp. Paired-end reads were trimmed with fastx toolkit v0.0.13 with these parameters: -f 8 -Q 33. Adapters were removed using Cutadapt v1.8-2. Cleaned reads were aligned with bismark v0.12.5, with default parameters on the Human reference Genome Hg19. Only reads mapping uniquely on the genome were conserved. Methylation calls were extracted after duplicate removal. Only CG dinucleotides covered by a minimum of 5 reads were conserved for the rest of the analysis. Differentially methylated regions (DMR) were assigned when a methylation difference of 30% occurs at least on 10 CpGs in a minimum of 500bp windows; windows are merged if distant between two DMRs is less than 500bp. DMR calling was performed using the Bioconductor package DSS.58
Chromatin structure analysis
We performed in situ Hi-C in duplicate for 3 normal melanocytes (NM) and 2 UM models (MP41 and MP46) as described by Rao et al.59 Briefly, after cell sorting of MP41 and MP46 cells, 3*106 NM and UM cells were fixed in 1% formaldehyde and stored at −80°C until further processing. Fixed cells were permeabilized, and an overnight digestion with MboI was conducted. DNA over-hangs were filled in the presence of biotin-14-dATP before proximal ends were ligated with T4 DNA ligase for 4 h. After crosslink reversal with Proteinase K, the DNA was purified using phenol/chloroform, quantified and sheared to a size of 400-500bp. Next, biotinylated DNA was pulled down with Dynabeads MyOne streptavidin T1 beads (Thermo Fisher) and DNA was repaired for 30 min with a mixture of T4 DNA ligase (NEB), T4 DNA polymerase I (NEB), large fragment of DNA polymerase I (NEB), and T4 Polynucleotide Kinase (NEB). The beads were washed and separated on a magnet before dA-tailing with Klenow exo minus (NEB). A final wash was performed before Illumina adaptor ligation. PCR amplification with Illumina primers was performed for 12 cycles and product was collected with AMpure. An equimolar pool of libraries was sequenced on the Illumina HiSeq - rapid Run, generating 250–436*106 read pairs. Hi-C data were processed using HiC-Pro60 before comparing the three different normalization algorithms (CAIC, LOIC and ICE methods,43).
Interaction analysis at the PRAME locus
We compared the interactions between enhancer and other regions at a resolution of 5kb in the PRAME locus (coordinates: hg19 chr22: 22,830,000-22,905,000). Specifically, we focused on two enhancer regions, namely E1 and E2, which were identified by the presence of H3K27ac peaks in the melanoma dataset (Enhancer 1: hg19 chr22: 22,830,000-22,835,000; Enhancer 2 hg19 chr22: 22,900,000-22,905,000). To quantify the strength of interactions, we employed distance-normalized contact values, also known as observed/expected ratios. A value above one indicated that an interaction was stronger than the expected average across the entire chromosome. These interaction strengths were then compared between normal samples and samples from individuals with melanoma.
DNA optical mapping and cytogenetics analysis
SVs analysis was conducted with Bionano DNA optical mapping from 1.5×106 MP41 and MP46 cell pellets. A direct labeling on CTTAAG motif (DLE1) was conducted according to Bionano recommendations. Labeled DNA were analyzed on the Saphyr system. De novo assembly was performed using the Bionano serve (version 1.6). Molecule N50 was 407.8kbp for MP41, label density was 16.3 per 100kbp and effective coverage of the assembly was 71.9X. For MP46, molecule N50 was 325.6kbp, label density was 16.9/100kbp and effective coverage of assembly was 84.5X.
Telomere and centromere staining followed by M-FISH technique were applied on cytogenetic slides after colcemid (0.1μg/mL) treatment of MP41 and MP46 cells as described previously34,61 to identify numerical and structural chromosomal alterations as well as telomere instability. Briefly, UM cells were cultured in T75 in DMEM with 10–20% SVF depending on models (10% SVF: MP41, Mel202, OMM1, OMM2.3; 20%: MP46). Forty-eight hours after passage, medium was supplemented with colcemid (0.1 μg/mL) for a 3h incubation to arrest cell division during metaphase. The cells were harvested, washed, suspended in 10mL DMEM with 0.075 M potassium chloride (Merck, Kenilworth, NJ, USA) and incubated for 20 min in a 37°C water bath (hypotonic shock) and fixed as previously described.34 Next a telomeres and centromeres staining followed by M-FISH technique (Metasystems Probes, Germany), were performed as previously described.62 The quantification of telomere FISH signal intensity in interphase cells was performed using Metacyte software (MetaSystems, version 3.9.1, Altlussheim, Germany) and TeloScore software (Cell Environment, Evry, France). The mean fluorescence intensity (FI) of telomeres was automatically quantified in 10,000 nuclei on each slide. The settings for exposure and gain remained constant between captures. The experiments were performed in triplicate.
Analysis of metaphase spreads allowed detection of telomere abnormalities and chromosomal aberrations using ChromoScore software (Cell Environment, Evry France) and Isis software (MetaSystems, Altlussheim, Germany). The images of metaphases were captured using automated acquisition module Autocapt software (MetaSystems, version 3.9.1) and a ZEISS Plan-Apochromat 63×/1.40 oil (Zeiss, Oberkochen, Germany), and CoolCube 1 digital high resolution CCD camera (MetaSystems, Altlussheim, Germany) with constant settings for exposure and gain.
For each UM model, telomere and chromosomal aberrations were analyzed automatically on 100 metaphases. The scored telomere abnormalities were: (i) sister telomere loss, likely occurring in G2, and defined as a telomere signal-free end in a single chromatid, (ii) telomere deletion defined as the loss of two telomere signals on the same chromosome arm (likely resulting from the loss of one telomere in G1/S), an aberration considered to represent double-strand breaks, leading to activation of DNA damage response. The scored chromosomal aberrations were dicentric chromosomes, centric rings, translocations, insertions and deletions.
Whole transcriptome analysis
Total RNA was extracted using miRNeasy kits following the manufacturer’s recommendations, including a DNAse step. Quality controls were achieved using Nanodrop ND1000 to evaluate RNA purity and concentration, and on automated electrophoresis to monitor RNA integrity (Bioanalyzer RNA 6000 Nano/Pico kits). PolyA RNASeq was conducted on total RNA (RIN>7), treated with DNase. An absolute fold change higher than 1.5 and a p value below 0.05 were selected as parameters for detecting differentially expressed genes. Splicing analysis was conducted using five different pipelines: deFuse, SOAPfuse, JAFFA, FusionCatcher, TopHat-Fusion. Fusion RNAs were identified present in at least 2 algorithms, and found in at least 2 replicates per model.
Histone modifications
ChIPSeq against H2AUb, H3K4me3 and H3K27me3 were conducted in simplicate in NM, MP41 and MP46 as described [52]. ChIPSeq against H3K27Ac and CTCF was conducted in duplicated in MP41 and MP46 to implement multi-omics analysis. 5 million cells were fixed according to the protocol used for the in situ Hi-C experiments for H2AUb, H3K4me3, and H3K27me3, and for H3K27ac and CTCF. The chromatin was prepared using the iDeal ChIP seq kit from Diagenode, according to the transcription factor protocol. The shearing conditions were set as 10 min using the following settings: 10 cycles of 30” [ON] 30” [OFF]. The shearing efficiency was monitored after reversion of the crosslinking and DNA purification. To increase sensitivity, an automated capillary electrophoresis system Fragment Analyser was used for chromatin shearing assessment (High sensitivity NGS fragment kit). ChIP assays were performed as defined in the optimizations using 10 μg or 1μg of chromatin per IP with the optimal antibody quantity resulting in the higher enrichment and lower background (CTCF 1μg, H3K27ac 1μg) IPs with a negative control isotype (IgG) were performed in parallel. For each sample, a library preparation was performed on ChIP and input DNA using the MicroPLEX v3 protocol. A control library was processed in parallel with the samples using a control Diagenode ChIP DNA. Five cycles of pre-amplification were performed and 1 μL of each library was analyzed by qPCR to determine the optimal number of amplification cycles required to obtain enough DNA for sequencing. Libraries were then loaded on Fragment Analyzer to determine whever enough material was generated. After the amplification, the libraries have been purified using AMPure beads and eluted in Tris. Then, the purified libraries were quantified using the Qubit ds DNA HS kit and analyzed on the Fragment Analyzer to assess their size. The molar concentration of each library was calculated using the quantification values from the Qubit and the size measurement generated by the Fragment Analyzer.
Quality control of FASTQ files was performed using the FastQC software. The sequences were aligned on hg19 assembly using bowtie2. The duplicates were marked and filtered out using Picard tools MarkDuplicates and samtools. The peak calling was performed using macs2 callpeak function. The parameter –broad was used for the Histone samples, not for the transcription factor samples. The affinity binding scores were obtained using DiffBind package in R, TMM normalization was applied. Peaks found in at least 50% of the samples from the same condition were kept. The peaks were annotated using FAST DB.
CUT&RUN-seq
CUT&RUN-seq was performed according to Skene and Henikoff, 2017 with minor adaptations. For each sample, 600,000 human cells (uveal melanoma cells MP41 or MP46) and 300,000 Drosophila melanogaster Schneider 2 cells were pelleted at 600 g for 5 min, washed twice with 1 mL of wash buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 500 μM spermidine and protease inhibitors) and resuspended in 1 mL of wash buffer. 10 μL of BioMagPlus Concanavalin A beads (Polysciences, Inc.) was washed twice with 1 mL of binding buffer (20 mM HEPES-KOH pH 7.9, 10 mM KCl, 1 mM CaCl2, 1 mM MnCl2), each time placed on the magnet stand to remove the buffer. In the end, 10 μL of binding buffer was used to resuspend the beads, the final slurry was then added to the cells and the mixture was placed on a rotator for a 10 min incubation. After a quick spin-down, the tubes were placed on the magnet stand and the liquid was removed. The antibody mix was prepared in a 1:100 dilution of the antibody in the antibody buffer (wash buffer with the addition of 0.1% of digitonin and 2 mM EDTA). The antibodies used were: H3K27me3 (CST C36B11), H3K4me3 (CST C42D8) and H2AK119ub (CST D27C4). The cells were incubated in the primary antibody for an hour at room temperature with mild agitation. Cells were then washed twice with 1 mL of dig-wash buffer (wash buffer completed with 0.1% of digitonin). 50 μL of 0.7 ng/μL pA-MNase in dig-wash buffer was added to the washed cells and placed on the rotator on mild agitation for 10 min at RT. Cells were washed twice with dig-wash buffer, resuspended in 100 µL of dig-wash buffer and placed in a heat block immersed in wet ice to cool down to 0°C. 2 µL of 100 mM CaCl2 was then added and the tubes were incubated on the heat block at 0°C for 30 min 100 μL of 2X STOP buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 0.02% digitonin, 0.5 μg/µL RNase A, 10 μg/mL glycogen) was added, and fragments were released by a 10 min incubation at 37°C. After centrifugation at 4 °C at 16,000 g for 5 min, DNA fragments were recovered by using a NucleoSpin kit (Macherey Nagel). The CUT&RUN was validated by quantitative PCR (qPCR). Primers sequences are as follow: MYT-1 (forward: AGGCACCTTCTGTTGGCCGA, reverse: AGGCAGCTGCCTCCCGTACA) and hGAPDH (forward: ATTTCTCCTCCGGGT GATGCTTTTC, reverse: GACCTCCATAAACCCACTTCTTTGA). Libraries were prepared and sequenced on an Illumina NovaSeq S1 (PE100) at the Institut Curie Next Generation Sequencing Platform. Reads were mapped to the Homo sapiens hg19 and to the Drosophila melanogaster dmelr6.28 genomes.
To quantitatively compare the cell line peaks with the PDX and NM peaks, the original peak calls were merged and corresponding coverage from NM and PDX datasets were calculated with deeptools multiBamSummary. A random forest classifier was then trained on these datasets for each mark and each PDX to classify PDX vs. NM using RandomForestClassifier from scikit learn, with n_estimators = 1000. This classifier was then applied to the cell line profiles with predict_proba function.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was calculated using R software. Statistical significance of differentially methylated regions (DMRs) is based on a Wald test applied to a beta-binomial regression as applied in the Bioconductor package DSS.58 Statistical significance of differential gene expression was calculated by two-sided t test. Significant pathway enrichment was calculated by over-representation analysis based on a hypergeometric distribution.31
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| anti Muc18, clone 8H2rFc (for the isolation of UM cells from PDX) | Crépin, Gentien et al.22 | N/A |
| anti CEACAM1, clone 8G5hFc (for the isolation of UM cells from PDX) | Crépin, Gentien et al.22 | N/A |
| anti NG2, clone 14A7hFc (for the isolation of UM cells from PDX) | Crépin, Gentien et al.22 | N/A |
| anti H3K27me3, clone C36B11 | Cell Signaling Technology | Cat# 9733; RRID: AB_2616029 |
| anti H3K4me3, clonce C42D8 | Cell Signaling Technology | Cat# 9751; RRID: AB_2616028 |
| anti H2AK119ub, clone D27C4 | Cell Signaling Technology | Cat# 8240; RRID: AB_10891618 |
| anti H3K27Ac | Diagenode | Cat# C15410196; RRID: AB_2637079 |
| anti CTCF | Diagenode | Cat# C15410210; RRID: AB_2753160 |
|
| ||
| Biological samples | ||
|
| ||
| MP41 PDX | Institut Curie | N/A |
| MP46 PDX | Institut Curie | N/A |
| Normal melanocytes | This paper | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Affymetrix/Thermo Cytoscan HD assay | Thermo | #901835 |
| TrueMethyl kit | Cambridge Epigeneics | #CEGXTMS |
| Bionano Prep SP-G2 Blood & Cell kit | Bionano | #90154 |
| M-FISH | Cell Environement | #A000867-000478 |
| iDeal ChIP-seq kit | Diagenode | #C01010170 |
| High sensitivity NGS fragment kit | Agilent Technologies | #DNF-474-0500 |
| Qubit ds DNA HS kit | Thermo Fisher Scientific | #Q33230 |
| NucleoSpin kit | Macherey Nagel | #740952.5 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed data | This paper | GEO: GSE199679 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MP41 cell line | Institut Curie, and ATCC | CRL-3297 |
| MP46 cell line | Institut Curie | CRL-3298 |
| Mel202 cell line | European Collection of Authenticated Cell Cultures (ECACC) | 13012457 |
| MM66 cell line | Institut Curie | N/A |
| OMM1 cell line | kindly provided by P.A. Van Der Velden (Leiden University, The Netherlands) | N/A |
| OMM2.3 cell line | kindly provided by P.A. Van Der Velden (Leiden University, The Netherlands) | N/A |
| Drosophila melanogaster Schneider 2 (see Cut and RUN method) | ATCC | CRL-1963 |
|
| ||
| Oligonucleotides | ||
|
| ||
| guide RNAs (sequence listed on Table S5) | IDT design and synthesis | N/A |
| SQK-CS9109 cas9 sequencing kit | Oxford Nanopore Technologies | SQK-CS9109 |
|
| ||
| Software and algorithms | ||
|
| ||
| Cancer Genome Interpreter | https://www.cancergenomeinterpreter.org/analysis | N/A |
| VarSome | https://varsome.com/ | N/A |
| Guppy (5.0.17) | Oxford Nanopore Technologies | N/A |
| Sniffles2.0.5 | https://github.com/fritzsedlazeck/Sniffles | N/A |
| cuteSV 1.08 | https://github.com/tjiangHIT/cuteSV | N/A |
| SVIM1.4.2 | https://github.com/eldariont/svim | N/A |
| SURVIVOR 1.0.7 | https://github.com/fritzsedlazeck/SURVIVOR | N/A |
| Pepper-margin-Deepvariant 0.8 | https://github.com/kishwarshafin/pepper | N/A |
| Cutadapt v1.8-2 | https://pypi.org/project/cutadapt/ | N/A |
| bismark v0.12.5 | https://github.com/FelixKrueger/Bismark | N/A |
| HiC-Pro | Servant et al.60 | N/A |
| CAIC method | Servant et al.43 | N/A |
| LOIC method | Servant et al.43 | N/A |
| ICE method | Servant et al.43 | N/A |
| Bionano ser, version 1.6 | Bionano | N/A |
| Metacyte, version 3.9.1 | MetaSystems | N/A |
| TeloScore | Cell Environment | N/A |
| ChromoScore | Cell Environment | N/A |
| Isis | MetaSystems | N/A |
| Autocapt | MetaSystems | N/A |
| FastQC | https://github.com/s-andrews/FastQC | N/A |
| Picard tools MarkDuplicates | https://broadinstitute.github.io/picard/ | N/A |
| samtools | http://www.htslib.org/ | N/A |
| DiffBind package | https://bioconductor.org/packages/release/bioc/html/DiffBind.html | N/A |
| Juicebox | https://github.com/aidenlab/Juicebox/wiki/Download | N/A |
| FAST DB | Genosplice | N/A |
Highlights.
Aggressive uveal melanomas display genomic instability
Promoter deletions are a recurrent mechanism of BAP1 deficiency
Looping and chromatin marks identify distal regulatory elements for PRAME expression
ACKNOWLEDGMENTS
The authors thank the members of the Institut Curie animal facility involved in UM projects, the members of the cytometry platform of Institut Curie and Hospital Saint Louis in Paris for their support with UM cell sorting, the members of the recombinant protein and nanobodies platforms of Institut Curie, the Pathex platform of Institut Curie for BAP1 immunochemistry, and the biological resources center for providing UM samples for the BAP1 analysis. The authors would like to thank the Unit of Somatic Genetic at the hospital of Institut Curie for the analysis done on NM. The authors thank the genomics platform of Institut Curie for assistance with STR profiling, copy number analysis, and DNA optical mapping of UM samples (Audrey Rapinat, Matéo Bazire), with the support of the Region Ile France (SESAME 2019 grant). High-throughput sequencing was performed by the ICGex NGS platform of the Institut Curie supported by the grants ANR-10-EQPX-03 (Equipex) and ANR-10-INBS-09-08 (France Génomique Consortium) from the Agence Nationale de la Recherche (“Investissements d’Avenir” program), by the ITMO-Cancer Aviesan (Plan Cancer III), and by the SiRIC-Curie program (SiRIC grant INCa-DGOS-4654). This work was supported too with a grant from the National Human Genome Research Institute to J.D. (HG003431). J.D. is an investigator of the Howard Hughes Medical Institute. The authors also thank the Bionano Genomics Company and the GENTYANE platform in Clermont Ferrand for the first analysis of MP41 and MP46 models using optical mapping.
Footnotes
DECLARATION OF INTERESTS
P.d.l.G. is a co-founder of Genosplice technology. A.J. and R.G. are employees of Genosplice technology. R.M. is the founder of Cell Environment.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113132.
REFERENCES
- 1.Rodrigues M, Mobuchon L, Houy A, Alsafadi S, Baulande S, Mariani O, Marande B, Ait Rais K, Van der Kooij MK, Kapiteijn E, et al. (2019). Evolutionary Routes in Metastatic Uveal Melanomas Depend on MBD4 Alterations. Clin. Cancer Res 25, 5513–5524. 10.1158/1078-0432.CCR-19-1215. [DOI] [PubMed] [Google Scholar]
- 2.Krantz BA, Dave N, Komatsubara KM, Marr BP, and Carvajal RD (2017). Uveal Melanoma: Epidemiology, Etiology, and Treatment of Primary Disease. Clin. Ophthalmol 11, 279–289. 10.2147/OPTH.S89591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, and Bastian BC (2009). Frequent Somatic Mutations of GNAQ in Uveal Melanoma and Blue Naevi. Nature 457, 599–602. 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, et al. (2010). Mutations in GNA11 in Uveal Melanoma. N. Engl. J. Med 363, 2191–2199. 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore AR, Ceraudo E, Sher JJ, Guan Y, Shoushtari AN, Chang MT, Zhang JQ, Walczak EG, Kazmi MA, Taylor BS, et al. (2016). Recurrent Activating Mutations of G-Protein-Coupled Receptor CYSLTR2 in Uveal Melanoma. Nat. Genet 48, 675–680. 10.1038/ng.3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johansson P, Aoude LG, Wadt K, Glasson WJ, Warrier SK, Hewitt AW, Kiilgaard JF, Heegaard S, Isaacs T, Franchina M, et al. (2016). Deep Sequencing of Uveal Melanoma Identifies a Recurrent Mutation in PLCB4. Oncotarget 7, 4624–4631. 10.18632/onco-target.6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma J, Weng L, Bastian BC, and Chen X (2021). Functional Characterization of Uveal Melanoma Oncogenes. Oncogene 40, 806–820. 10.1038/s41388-020-01569-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen X, Wu Q, Depeille P, Chen P, Thornton S, Kalirai H, Coupland SE, Roose JP, and Bastian BC (2017). RasGRP3 Mediates MAPK Pathway Activation in GNAQ Mutant Uveal Melanoma. Cancer Cell 31, 685–696.e6. 10.1016/j.ccell.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore AR, Ran L, Guan Y, Sher JJ, Hitchman TD, Zhang JQ, Hwang C, Walzak EG, Shoushtari AN, Monette S, et al. (2018). GNA11 Q209L Mouse Model Reveals RasGRP3 as an Essential Signaling Node in Uveal Melanoma. Cell Rep 22, 2455–2468. 10.1016/j.celrep.2018.01.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harbour JW, Onken MD, Roberson EDO, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, and Bowcock AM (2010). Frequent Mutation of BAP1 in Metastasizing Uveal Melanomas. Science 330, 1410–1413. 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin M, Maßhöfer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, et al. (2013). Exome Sequencing Identifies Recurrent Somatic Mutations in EIF1AX and SF3B1 in Uveal Melanoma with Disomy 3. Nat. Genet 45, 933–936. 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harbour JW, Roberson EDO, Anbunathan H, Onken MD, Worley LA, and Bowcock AM (2013). Recurrent Mutations at Codon 625 of the Splicing Factor SF3B1 in Uveal Melanoma. Nat. Genet 45, 133–135. 10.1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. (2016). Cancer-Associated SF3B1 Mutations Affect Alternative Splicing by Promoting Alternative Branchpoint Usage. Nat. Commun 7, 10615. 10.1038/ncomms10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yavuzyigitoglu S, Koopmans AE, Verdijk RM, Vaarwater J, Eussen B, van Bodegom A, Paridaens D, Kiliç E, and de Klein A; Rotterdam Ocular Melanoma Study Group (2016). Uveal Melanomas with SF3B1 Mutations: A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology 123, 1118–1128. 10.1016/j.ophtha.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 15.van den Bosch T, van Beek JGM, Vaarwater J, Verdijk RM, Naus NC, Paridaens D, de Klein A, and Kiliç E (2012). Higher Percentage of FISH-Determined Monosomy 3 and 8q Amplification in Uveal Melanoma Cells Relate to Poor Patient Prognosis. Invest. Ophthalmol. Vis. Sci 53, 2668–2674. 10.1167/iovs.11-8697. [DOI] [PubMed] [Google Scholar]
- 16.Cassoux N, Rodrigues MJ, Plancher C, Asselain B, Levy-Gabriel C, Lumbroso-Le Rouic L, Piperno-Neumann S, Dendale R, Sastre X, Desjardins L, and Couturier J (2014). Genome-Wide Profiling Is a Clinically Relevant and Affordable Prognostic Test in Posterior Uveal Melanoma. Br. J. Ophthalmol 98, 769–774. 10.1136/bjophthal-mol-2013-303867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Royer-Bertrand B, Torsello M, Rimoldi D, El Zaoui I, Cisarova K, Pescini-Gobert R, Raynaud F, Zografos L, Schalenbourg A, Speiser D, et al. (2016). Comprehensive Genetic Landscape of Uveal Melanoma by Whole-Genome Sequencing. Am. J. Hum. Genet 99, 1190–1198. 10.1016/j.ajhg.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, Hess JM, Uzunangelov V, Walter V, Danilova L, et al. (2017). Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 32, 204–220.e15. 10.1016/j.ccell.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Némati F, Sastre-Garau X, Laurent C, Couturier J, Mariani P, Desjardins L, Piperno-Neumann S, Lantz O, Asselain B, Plancher C, et al. (2010). Establishment and Characterization of a Panel of Human Uveal Melanoma Xenografts Derived from Primary and/or Metastatic Tumors. Clin. Cancer Res 16, 2352–2362. 10.1158/1078-0432.CCR-09-3066. [DOI] [PubMed] [Google Scholar]
- 20.Laurent C, Gentien D, Piperno-Neumann S, Némati F, Nicolas A, Tesson B, Desjardins L, Mariani P, Rapinat A, Sastre-Garau X, et al. (2013). Patient-Derived Xenografts Recapitulate Molecular Features of Human Uveal Melanomas. Mol. Oncol 7, 625–636. 10.1016/j.molonc.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amirouchene-Angelozzi N, Némati F, Gentien D, Nicolas A, Dumont A, Carita G, Camonis J, Desjardins L, Cassoux N, Piperno-Neumann S, et al. (2014). Establishment of Novel Cell Lines Recapitulating the Genetic Landscape of Uveal Melanoma and Preclinical Validation of MTOR as a Therapeutic Target. Mol. Oncol 8, 1508–1520. 10.1016/j.molonc.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crépin R, Gentien D, Duché A, Rapinat A, Reyes C, Némati F, Massonnet G, Decaudin D, Djender S, Moutel S, et al. (2017). Nanobodies against Surface Biomarkers Enable the Analysis of Tumor Genetic Heterogeneity in Uveal Melanoma Patient-Derived Xenografts. Pigment Cell Melanoma Res 30, 317–327. 10.1111/pcmr.12577. [DOI] [PubMed] [Google Scholar]
- 23.Tamborero D, Rubio-Perez C, Deu-Pons J, Schroeder MP, Vivancos A, Rovira A, Tusquets I, Albanell J, Rodon J, Tabernero J, et al. (2018). Cancer Genome Interpreter Annotates the Biological and Clinical Relevance of Tumor Alterations. Genome Med 10, 25. 10.1186/s13073-018-0531-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, and Massouras A (2019). VarSome: The Human Genomic Variant Search Engine. Bioinformatics 35, 1978–1980. 10.1093/bioinformatics/bty897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Field MG, Decatur CL, Kurtenbach S, Gezgin G, van der Velden PA, Jager MJ, Kozak KN, and Harbour JW (2016). PRAME as an Independent Biomarker for Metastasis in Uveal Melanoma. Clin. Cancer Res 22, 1234–1242. 10.1158/1078-0432.CCR-15-2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang C, Shen S, Zheng X, Ye K, Sun Y, Lu Y, and Ge H (2019). Long Noncoding RNA HAGLR Acts as a MicroRNA-143-5p Sponge to Regulate Epithelial-Mesenchymal Transition and Metastatic Potential in Esophageal Cancer by Regulating LAMP3. FASEB J 33, 10490–10504. 10.1096/fj.201802543RR. [DOI] [PubMed] [Google Scholar]
- 27.Gillespie RF, and Gudas LJ (2007). Retinoid Regulated Association of Transcriptional Coregulators and the Polycomb Group Protein SUZ12 with the Retinoic Acid Response Elements of Hoxa1, RARb2, and Cyp26A1 in F9 Embryonal Carcinoma Cells. J. Mol. Biol 372, 298–316. 10.1016/j.jmb.2007.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E, Li E, Spence M, et al. (2013). Multiple Knockout Mouse Models Reveal LincRNAs Are Required for Life and Brain Development. Elife 2, e01749. 10.7554/eLife.01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orfanelli U, Wenke A-K, Doglioni C, Russo V, Bosserhoff AK, and Lavorgna G (2008). Identification of Novel Sense and Antisense Transcription at the TRPM2 Locus in Cancer. Cell Res 18, 1128–1140. 10.1038/cr.2008.296. [DOI] [PubMed] [Google Scholar]
- 30.Xiao J, Lin L, Luo D, Shi L, Chen W, Fan H, Li Z, Ma X, Ni P, Yang L, and Xu Z (2020). Long Noncoding RNA TRPM2-AS Acts as a MicroRNA Sponge of MiR-612 to Promote Gastric Cancer Progression and Radioresistance. Oncogenesis 9, 29. 10.1038/s41389-020-0215-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff-Ribeiro A, Griss J, Sevilla C, Matthews L, Gong C, et al. (2022). The Reactome Pathway Knowledgebase 2022. Nucleic Acids Res 50, D687–D692. 10.1093/nar/gkab1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, and Kwok P-Y (2012). Genome Mapping on Nanochannel Arrays for Structural Variation Analysis and Sequence Assembly. Nat. Biotechnol 30, 771–776. 10.1038/nbt.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan S, Lam E, Saghbini M, Bocklandt S, Hastie A, Cao H, Holmlin E, and Borodkin M (2018). Structural Variation Detection and Analysis Using Bionano Optical Mapping. Methods Mol. Biol 1833, 193–203. 10.1007/978-1-4939-8666-8_16. [DOI] [PubMed] [Google Scholar]
- 34.M’kacher R, Colicchio B, Borie C, Junker S, Marquet V, Heidingsfelder L, Soehnlen K, Najar W, Hempel WM, Oudrhiri N, et al. (2020). Telomere and Centromere Staining Followed by M-FISH Improves Diagnosis of Chromosomal Instability and Its Clinical Utility. Genes 11, 475. 10.3390/genes11050475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar R, Nagpal G, Kumar V, Usmani SS, Agrawal P, and Raghava GPS (2019). HumCFS: A Database of Fragile Sites in Human Chromosomes. BMC Genom 19, 985. 10.1186/s12864-018-5330-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holt R, Sykes NH, Conceição IC, Cazier J-B, Anney RJL, Oliveira G, Gallagher L, Vicente A, Monaco AP, and Pagnamenta AT (2012). CNVs Leading to Fusion Transcripts in Individuals with Autism Spectrum Disorder. Eur. J. Hum. Genet 20, 1141–1147. 10.1038/ejhg.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Griewank KG, Yu X, Khalili J, Sozen MM, Stempke-Hale K, Bernatchez C, Wardell S, Bastian BC, and Woodman SE (2012). Genetic and Molecular Characterization of Uveal Melanoma Cell Lines. Pigment Cell Melanoma Res 25, 182–187. 10.1111/j.1755-148X.2012.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jager MJ, Magner JAB, Ksander BR, and Dubovy SR (2016). Uveal Melanoma Cell Lines: Where Do They Come from? (An American Ophthalmological Society Thesis). Trans. Am. Ophthalmol. Soc 114, T5. [PMC free article] [PubMed] [Google Scholar]
- 39.Kernaleguen M, Daviaud C, Shen Y, Bonnet E, Renault V, Deleuze J-F, Mauger F, and Tost J (2018). Whole-Genome Bisulfite Sequencing for the Analysis of Genome-Wide DNA Methylation and Hydroxymethylation Patterns at Single-Nucleotide Resolution. Methods Mol. Biol 1767, 311–349. 10.1007/978-1-4939-7774-1_18. [DOI] [PubMed] [Google Scholar]
- 40.Tempel S (2012). Using and Understanding RepeatMasker. Methods Mol. Biol 859, 29–51. 10.1007/978-1-61779-603-6_2. [DOI] [PubMed] [Google Scholar]
- 41.Gama-Sosa MA, Slagel VA, Trewyn RW, Oxenhandler R, Kuo KC, Gehrke CW, and Ehrlich M (1983). The 5-Methylcytosine Content of DNA from Human Tumors. Nucleic Acids Res 11, 6883–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feinberg AP, and Vogelstein B (1983). Hypomethylation Distinguishes Genes of Some Human Cancers from Their Normal Counterparts. Nature 301, 89–92. 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 43.Servant N, Varoquaux N, Heard E, Barillot E, and Vert J-P (2018). Effective Normalization for Copy Number Variation in Hi-C Data. BMC Bioinf 19, 313. 10.1186/s12859-018-2256-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive Mapping of Long Range Interactions Reveals Folding Principles of the Human Genome. Science 326, 289–293. 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dixon JR, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardımcı GG, Chakraborty A, Bann DV, Wang Y, et al. (2018). Integrative Detection and Analysis of Structural Variation in Cancer Genomes. Nat. Genet 50, 1388–1398. 10.1038/s41588-018-0195-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. (2012). Spatial Partitioning of the Regulatory Landscape of the X-Inactivation Centre. Nature 485, 381–385. 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, and Mirny LA (2016). Formation of Chromosomal Domains by Loop Extrusion. Cell Rep 15, 2038–2049. 10.1016/j.celrep.2016.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao SSP, Huang S-C, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon K-R, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320.e24. 10.1016/j.cell.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, and Meyer BJ (2015). J. Condensin-Driven Remodelling of X Chromosome Topology during Dosage Compensation. Nature 523, 240–244. 10.1038/nature14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vos ESM, Valdes-Quezada C, Huang Y, Allahyar A, Verstegen MJAM, Felder A-K, van der Vegt F, Uijttewaal ECH, Krijger PHL, and de Laat W (2021). Interplay between CTCF Boundaries and a Super Enhancer Controls Cohesin Extrusion Trajectories and Gene Expression. Mol. Cell 81, 3082–3095.e6. 10.1016/j.mol-cel.2021.06.008. [DOI] [PubMed] [Google Scholar]
- 51.Schenk T, Stengel S, Goellner S, Steinbach D, and Saluz HP (2007). Hypomethylation of PRAME Is Responsible for Its Aberrant Overexpression in Human Malignancies. Genes Chromosomes Cancer 46, 796–804. 10.1002/gcc.20465. [DOI] [PubMed] [Google Scholar]
- 52.Field MG, Durante MA, Decatur CL, Tarlan B, Oelschlager KM, Stone JF, Kuznetsov J, Bowcock AM, Kurtenbach S, and Harbour JW (2016). Epigenetic Reprogramming and Aberrant Expression of PRAME Are Associated with Increased Metastatic Risk in Class 1 and Class 2 Uveal Melanomas. Oncotarget 7, 59209–59219. 10.18632/oncotarget.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. (2012). An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gab-dank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, et al. (2018). The Encyclopedia of DNA Elements (ENCODE): Data Portal Update. Nucleic Acids Res 46, D794–D801. 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang R, Wu H-X, Xu M, and Xie X (2020). KMT2A/C Mutations Function as a Potential Predictive Biomarker for Immunotherapy in Solid Tumors. Biomark. Res 8, 71. 10.1186/s40364-020-00241-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu Y, Nanni L, Sungalee S, Zufferey M, Tavernari D, Mina M, Ceri S, Oricchio E, and Ciriello G (2021). Systematic Inference and Comparison of Multi-Scale Chromatin Sub-Compartments Connects Spatial Organization to Cell Phenotypes. Nat. Commun 12, 2439. 10.1038/s41467-021-22666-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petit V, Massonnet G, Maciorowski Z, Touhami J, Thuleau A, Némati F, Laval J, Château-Joubert S, Servely J-L, Vallerand D, et al. (2013). Optimization of Tumor Xenograft Dissociation for the Profiling of Cell Surface Markers and Nutrient Transporters. Lab. Invest 93, 611–621. 10.1038/labinvest.2013.44. [DOI] [PubMed] [Google Scholar]
- 58.Feng H, and Wu H (2019). Differential Methylation Analysis for Bisulfite Sequencing Using DSS. Quant. Biol 7, 327–334. 10.1007/s40484-019-0183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P, Heard E, Dekker J, and Barillot E (2015). HiC-Pro: An Optimized and Flexible Pipeline for Hi-C Data Processing. Genome Biol 16, 259. 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.M’kacher R, Cuceu C, Al Jawhari M, Morat L, Frenzel M, Shim G, Lenain A, Hempel WM, Junker S, Girinsky T, et al. (2018). The Transition between Telomerase and ALT Mechanisms in Hodgkin Lymphoma and Its Predictive Value in Clinical Outcomes. Cancers 10, 169. 10.3390/cancers10060169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.M’kacher R, Maalouf EEL, Ricoul M, Heidingsfelder L, Laplagne E, Cuceu C, Hempel WM, Colicchio B, Dieterlen A, and Sabatier L (2014). New Tool for Biological Dosimetry: Reevaluation and Automation of the Gold Standard Method Following Telomere and Centromere Staining. Mutat. Res 770, 45–53. 10.1016/j.mrfmmm.2014.09.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data generated in this work have been deposited in the GEO repository, under the accession GSE199679 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE199679). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.