

Abstract

Receptor-selective peptides are widely used as smart carriers for specific tumor-targeted delivery. A remarkable example is the cyclic nonapeptide iRGD (CRGDKPGDC, 1) that couples intrinsic cytotoxic effects with striking tumor-homing properties. These peculiar features are based on a rather complex multistep mechanism of action, where the primary event is the recognition of RGD integrins. Despite the high number of preclinical studies and the recent success of a phase I trial for the treatment of pancreatic ductal adenocarcinoma (PDAC), there is little information available about the iRGD three-dimensional (3D) structure and integrin binding properties. Here, we re-evaluate the peptide’s affinity for cancer-related integrins including not only the previously known targets αvβ3 and αvβ5 but also the αvβ6 isoform, which is known to drive cell growth, migration, and invasion in many malignancies including PDAC. Furthermore, we use parallel tempering in the well-tempered ensemble (PT-WTE) metadynamics simulations to characterize the in-solution conformation of iRGD and extensive molecular dynamics calculations to fully investigate its binding mechanism to integrin partners. Finally, we provide clues for fine-tuning the peptide’s potency and selectivity profile, which, in turn, may further improve its tumor-homing properties.
1. Introduction
Nowadays, the clinical efficacy of chemotherapeutic drugs is frequently hampered either by a lack of selectivity over healthy cells or by poor pharmacokinetic properties, including cancer homing and penetration. This is especially true for solid tumors, which are frequently characterized by the upregulation of junction proteins such as desmoglein 2 (DSG2) and E-cadherin, and extracellular matrix (ECM) components (i.e., fibrinogen and collagen), which form a physical barrier against the intracellular transport of exogenous molecules.1 For these reasons, anticancer compounds often need to be administered at high doses to exert relevant pharmacological effects, with the rise of serious adverse reactions.2−4 A feasible solution to the tissue penetration problem is represented by smart carriers that can vehicle the desired drug to extravascular cancer tissue. Carriers of different natures have been developed such as gold nanoparticles,5−7 liposomes,8,9 polymer micelles,10 or receptor-selective peptides.11−15 In this context, Ruoslahti and co-workers identified an RGD integrins-targeting cyclic nonapeptide, namely, iRGD (internalizing RGD, CRGDKGPDC, 1—Chart 1), endowed with remarkable tumor-homing properties.16,17 Notably, this peptide can improve the tumor penetration and efficacy of chemotherapeutics through two alternative mechanisms.1,18−32 In fact, iRGD can be either covalently bioconjugated—usually functionalizing the C-terminal Cys9—to organic and peptidic drugs or attached to the surface of other delivering systems like nanoparticles, liposomes, or oncolytic viruses.18−32 On the other hand, the tumor endocytosis of cytotoxic agents such as cisplatin, gemcitabine, doxorubicin, nab-paclitaxel, and trastuzumab is enhanced by the simple coadministration of 1.17,33,34 As a result, hundreds of distinct applications involving iRGD have been published during the past decade,18−35 claiming the potential of this peptide as a game changer in the anticancer field.36 Worth of special note are the clinical results obtained by the combination of 1 with nab-paclitaxel and gemcitabine: a phase I trial reported a safe tolerability profile and a longer progression-free survival in the treatment of metastatic pancreatic ductal adenocarcinoma (PDAC), a neoplasia that is usually poorly susceptible to both traditional chemotherapy and immunotherapy.34,37
Chart 1. 2D Chemical Structure of iRGD (1).

The striking iRGD’s tumor-homing activity is linked to the marked overexpression of RGD integrins on the cancer cell membrane.38−40 Notably, many peptides, peptidomimetics, and small molecules targeting these receptors have been used over the years, but none of them have shown tumor-penetrating properties comparable to 1. This can be correlated to the peculiar multistage internalization process of iRGD, where the binding to integrins represents only the first step.16 In fact, once the peptide binds to the integrin receptor, it experiences a proteolytical cleavage at the Lys5–Gly6 bond that results in exposure and the following release of the cryptic C-terminal CRGDK sequence (CendR motif). The latter is a common recognition pattern of neuropilin-1 (NRP-1), a tyrosine kinases’ coreceptor playing multiple roles in angiogenesis, cell migration, and invasion.41 The binding of the CendR motif to NRP-1 triggers the internalization of the peptide–receptor complex and is responsible for the iRGD’s intrinsic cytotoxic effects.42 In this regard, in vitro experiments proved that iRGD can inhibit tumor migration and induce chemorepulsion based on a CendR- and NRP-1-dependent mechanism of action.43 The tropism and selectivity of 1 for cancer tissues are, however, ruled by its affinity for RGD integrins.16 So far, only the binding to the αvβ3 and αvβ5 isoforms has been demonstrated, whereas to our knowledge, no direct interaction data are currently available for other clinically relevant subtypes such as αvβ6. The latter recently came to the limelight for its involvement in the development of idiopathic pulmonary fibrosis (IPF) as well as several malignancies such as colorectal cancers and PDAC.44−47 Indeed, αvβ6 promotes in vitro PDAC cell growth, survival, migration, and invasion. Accordingly, the treatment of either αvβ6-positive human PDAC xenografts or transgenic mice with an αvβ6 blocking antibody combined with gemcitabine was shown to significantly reduce tumor growth while increasing the survival rate.48 Thus, we wondered if at least part of the peptide’s efficacy may be due to a still undetected affinity to αvβ6. To answer this question, we here extended the in vitro characterization of the iRGD’s integrin selectivity profile, repeating the IC50s measurements for the known cognate receptors αvβ3 and αvβ5 and demonstrating for the first time a mid-low nanomolar potency toward the αvβ6 isoform. Furthermore, the present article fills the knowledge gap on the structural basis of the iRGD–integrin interaction. Indeed, despite the potential clinical relevance of 1,34,37 its solution conformation, interaction mode with integrin receptors, and structure–activity relationships are still unknown. In this perspective, we used a combination of bioinformatics and advanced biosimulations to provide a high-resolution description of the iRGD’s folding and mode of binding to the αvβ3, αvβ5, and αvβ6 integrins. The peptide’s affinity and selectivity profile were thus rationalized, also explaining how its proteolytic cleavage and bioconjugation (with cargos of different natures) can take place without altering the affinity for the target receptors. Our results help to understand in more detail the integrin binding properties of iRGD and provide valuable clues for fine-tuning its selectivity profile and, in turn, its tumor-homing properties.
2. Materials and Methods
2.1. Solid-Phase Integrin Binding Assay
iRGD and cilengitide49,50 were purchased as pure substances from MedChemExpress, while [RGD-Chg-E]-CONH2 (2) was synthesized according to a previously reported protocol.51 Purified αvβ3 (Sino Biological Europe GmbH) and αvβ5 (R&D Systems, Inc.) were diluted to 0.5 μg/mL, while αvβ6 (Sino Biological Europe GmbH) was diluted to 1 μg/mL in coating buffer containing Tris–HCl (20 mmol/L; pH 7.4), NaCl (150 mmol/L), MnCl2 (1 mmol/L), CaCl2 (2 mmol/L), and MgCl2 (1 mmol/L). An aliquot of diluted receptors (100 μL per well) was added to 96-well microtiter plates (Nunc MW96F Maxisorp Straight) and incubated overnight at 4 °C. The plates were then incubated with blocking solution (coating buffer plus 1% bovine serum albumin) for an additional 2 h at room temperature to block nonspecific binding, followed by 3 h incubation at room temperature with various concentrations of test compounds in the presence of, respectively, vitronectin (1 μg/mL, Sigma-Aldrich) for αvβ3 and αvβ5 and fibronectin for αvβ6 (1 μg/mL, Sigma-Aldrich) biotinylated using the EZ-Link Sulfo-NHS-Biotinylation kit (Thermo Fisher). After washing, the plates were incubated for 1 h at room temperature with the streptavidin–biotinylated peroxidase complex (GE Health), followed by 15 min incubation with substrate reagent solution (100 μL; R&D Systems) before stopping the reaction by addition of H2SO4 (0.25 M, 50 μL). Absorbance at 415 nm was read with a BMG Labtech Fluostar Optima microplate reader. All of the experiments were performed in triplicates, and the data collected were analyzed using the GraphPad 5.0 Software Package (GraphPad Prism, San Diego, CA).
2.2. PT-WTE Calculations
Parallel tempering in the well-tempered ensemble (WTE)52 is a powerful enhanced sampling method based on the combination of parallel tempering (PT) and well-tempered metadynamics (WT-MetaD).53,54 In PT, n copies of the system are simulated at different temperatures, with periodic coordinate exchanges attempted between adjacent replicas and ruled by the Metropolis–Hastings criterion. The general idea is that medium–high free energy barriers that trap low-temperature replicas in local energy minima can be crossed at higher temperatures. On the other hand, in MetaD, the sampling of the simulation is boosted by a history-dependent bias potential (VG), made of Gaussians deposited on a selected number of reaction coordinates (i.e., slow degrees of freedom) referred to as collective variables (CVs)
| 1 |
where Si is the value of the ith CV, σi is the width of the Gaussian function, and ω is the rate at which the bias is deposited. In WT-MetaD, the bias deposition rate ω is exponentially rescaled over time depending on how much potential has already been added in the same region of the CV phase space, according to the following formula
| 2 |
where W is the Gaussian height, kB is Boltzmann’s constant, ω0 is the initial energy rate, τG is the Gaussian deposition stride, and VG (S, t) is the bias potential accumulated in S over time t. ΔT is an input parameter with the dimension of a temperature, which controls how quickly the Gaussian height is decreased and is often written in terms of a so-called bias factor γ = (T + ΔT)/T. At the end of a WT-MetaD simulation, the deposited bias potential VG asymptotically converges to the inverse value of a fraction of free energy F
| 3 |
When the potential energy is used as CV in WT-MetaD simulations, a well-defined distribution known as the well-tempered ensemble (WTE) is sampled. In WTE, the system experiences enhanced energy fluctuations that can be used to facilitate the exchange processes and reduce the number of replicas required for PT. In our case, the metadynamics Gaussians were deposited every 0.5 ps with a width of 145 kJ/mol and an initial height of 2.5 kJ/mol, which was gradually decreased based on a bias factor γ = 24. Then, 6 replicas were distributed according to the formula proposed by Prakash et al.55 to span the temperature interval 300–450 K. Each replica was simulated for 140 ns in the NVT ensemble using the stochastic rescaling thermostat.56 The coordinate exchanges were attempted every 0.5 ps, obtaining an average acceptance probability of 25% between all of the neighbor replicas. A further advantage of the WTE ensemble is that the canonical energy average is conserved, and all of the other canonical observables can be estimated a posteriori. Thus, the Tiwary–Parrinello reweighting scheme57 was employed to compute the free energy surface (FES) associated with the folding of 1 as a function of two selected CVs. The first one is the dihedral correlation (Dihcor) between all of the torsion angles of the peptide backbone, also including the peptide disulfide bridge
| 4 |
where the ϕi and ψi values are the instantaneous values for the torsion angles of interest. This function measures the degree of similarity between adjacent dihedral angles and, if extended to the entire backbone, can describe global conformational changes. The second CV, Hbonds, estimates the number of intramolecular backbone–backbone H-bonds
 |
5 |
where i and j are defined as all of the possible combinations between the amide hydrogen and oxygen atoms (except the C-terminal carboxyl oxygens) of the peptide backbone; d0 and r0 distances were set to 0 and 2.5 Å, while the n and m exponentials were modulated to 10 and 26, respectively. For the designed virtual compounds 3–11 (see Supporting Table S1 and Section 3 for further details), the WTE bias was reweighted alternatively combining Dihcor and Hbonds with an additional CV: the root-mean-square distances (RMSD) from the coordinates of the backbone heavy atoms of the iRGD lowest-energy conformation predicted by PT-WTE.
The GROMACS 2018.858 code patched with the PLUMED 2.5.6 plugin59,60 was used to run the PT-WTE simulations. The peptide was parametrized using the ff14SB Amber force field61 and then solvated in a 12.0 Å layer rhombic dodecahedron box using the TIP3P water model parameters.62 Prior to metadynamics, each replica of each system was equilibrated through 5 ns of MD under NPT conditions at 1 atm and 300 K. A time step of 2 fs in a leapfrog integrator was used. All covalent bonds were constrained to their equilibrium value using the LINCS algorithm.63 The Lennard–Jones potential was used to compute atom–pair interactions, with a cutoff of 10.0 Å. The simulations were carried out in periodic boundary conditions using the particle mesh Ewald (PME) to treat long-range electrostatic (grid spacing = 1.0 Å) interactions. The clustering of the peptides’ conformations corresponding to the various free energy minima was performed with the gmx cluster tool. Specifically, the clustering was performed with the GROMOS algorithm by considering the RMSD of the peptide cyclic backbone atoms (including the disulfide bridge) using a cutoff of 1.5 Å.
2.3. Convergence and Error Analysis
The convergence of the PT-WTE calculations was carefully assessed in both quantitative and qualitative ways. As for compound 1, the computation of the FES at regular time intervals (Figure S1A) highlighted that after the first 80 ns (per replica) of simulations, the overall shape of the free energy landscape is conserved. Then, a block averaging analysis estimated the error associated with the ΔG computation (Figure S1B) at the acceptable value of ≈1 kJ/mol. In parallel, the convergence of the parallel tempering scheme was also evaluated. To further prove the reliability of our results, we monitored the values of the CVs (Figure S1C) employed for reweighting in the continuous (demuxed) trajectories of each replica. The plots in Figure S1C show a diffusive behavior of both CVs in all of the replicas of compound 1, suggesting that no simulation was stuck in a particular region of the phase space (that is, no observed hysteresis). The average exchange acceptance ratio was ≈25%, which testifies to a good diffusion of all of the replicas over the entire temperature range (Figure S2).
2.4. Homology Modeling
Due to the lack of any experimental three-dimensional (3D) structure in the Protein Data Bank (PDB), a homology model of the αvβ5 head was built. Since the αv subunit had already been solved in the X-ray structures of the αvβ3, αvβ6, and αvβ8 isoforms,64−68 only the β5 subunit was actually modeled. First, a multiple sequence alignment between the heads (region corresponding to β3 residues 109–353) of all of the human RGD β subunits (β1, β3, β5, β6, β8) was performed with ClustalOmega69,70 (Figure S3). This analysis showed that two isoforms, namely, β3 and β6, possess the highest identity rate with β5 (65 and 58%, respectively). Unsurprisingly, most of the mutations and all of the amino acidic gaps occur at the specificity determining loop (SDL) region, which is at least two residues longer in β5 than in any other RGD integrin. Given the importance of the SDL for the ligand binding to the integrins orthosteric site, particular attention was paid to the modeling of this portion. Therefore, a further sequence alignment restricted to the SDL region was performed (Figure S4). In this comparison, β6 showed a higher (44%) local (SDL region) identity value than β3 (41%); thus, the crystal structure of αvβ6 in complex with the LAP peptide of TGF-β (PDB code: 4UM9(66)) was chosen as a template.66 Then, the knowledge-based method implemented in Prime71 was used to build the 3D receptor model. Furthermore, a refinement was carried out for loops bearing amino acids with missing coordinates (i.e., not coming from the template) by means of the Maestro “Refine Loops” panel.72 Specifically, short loops were refined using default sampling rates, whereas the folding of the SDL residues comprised between the conserved disulfide bridge C176–C185 was refined using the Extended protocol implemented in Prime.71 Finally, the coordinates of all of the nonconserved side chains were optimized using an energy cutoff of 10 kcal/mol. For the numbering of the β5 and β6 residues over the text, we followed the common practice to align both the receptors to a reference, represented by the first-published αvβ3 crystal structure (PDB code: 1L5G(64)). The resulting model was validated by computing the protein Ramachandran plot (Figure S5A) with the aid of the MolProbity Web server (http://molprobity.biochem.duke.edu) and evaluating the stability of the secondary structure elements in a 2 μs long MD simulation (Figure S5B).
2.5. Molecular Docking
Docking of the PT-WTE-predicted conformation of 1 was performed in the integrin head of αvβ5 (homology model) as well as of αvβ3 and αvβ6 (crystal structures: 4MMX(65) and 4UM9,66 respectively). Both the ligand and the receptors were prepared using the Protein Preparation Wizard tool, implemented in the Maestro Suite 2019.73 The cocrystallized Mg2+ and Ca2+ divalent cations at the protein “metal ion-dependent adhesion site” (MIDAS), “adjacent to MIDAS” (ADMIDAS), and “ligand-associated metal ion-binding site” (LIMBS) were retained and treated using the default parameters. Correct bond orders were assigned, missing hydrogen atoms were added, and all of the water molecules were deleted from the receptor structure. Then, protonation and tautomeric states at pH 7.4 were assigned to the side chains using Epik.74,75 Finally, the positions of all of the hydrogens were minimized. A virtual box of 30 Å × 30 Å × 30 Å, surrounding the typical RGD binding site, was selected as the search area by the means of the Receptor Grid Generator tool of Glide 8.5.76,77 Docking calculations were performed employing the Glide SP-peptide protocol and the OPLS3A force field.78 The peptide backbone was kept fixed in order to preserve the conformation obtained from the PT-WTE simulation, while all of the other parameters were kept to default values. Thus, the obtained solutions were clustered based on the ligand RMSD (default parameters) and ranked according to the Glide SP scoring function.76,77 The designed virtual compounds 4, 6, 8, and 11 (see Supporting Table S1 and Section 3) were docked in the energy-minimized averaged MD structure of αvβ6 using the same protocol. In this case, however, positional restraints were used to discard all of the solutions, not respecting the typical RGD binding pattern.
2.6. Molecular Dynamics
All of the proteins and the peptide were parametrized using the ff14SB Amber force field.61 The divalent cations present in the integrins structures were treated with the parameters developed by Panteva et al.79 The PMEMD engine (GPU version) of AMBER 1880 was used to perform the simulations. The short-range interactions were defined as all possible contacts within a cutoff of 10 Å from every simulated atom. The long-range electrostatic interactions were computed through the particle mesh Ewald method81 using a 1.0 Å grid spacing in periodic boundary conditions. The iterative SHAKE algorithm82 was applied to constraint all bonds containing hydrogens, allowing for a 2 fs integration time step. In order to solve all of the steric clashes, each system underwent 30,000 steps of mixed steepest descent/conjugated gradient energy minimization. Then, each complex was equilibrated and heated up to 300 K, alternating NPT and NVT cycles (125,000 steps each) with the Langevin coupling bath83 and the Berendsen barostat,84 while applying gradually decreasing harmonic constraints on the heavy atoms of the protein and ligand. Finally, a production run of 2 μs was performed for each ligand–protein complex in the NPT ensemble with a target pressure and temperature of 1 atm and 300 K, respectively.
3. Results and Discussion
3.1. In Vitro Binding Assay
To provide a more exhaustive picture of the integrin affinity of iRGD, direct solid-phase binding assays were performed between 1 and its known target receptors αvβ3 and αvβ5 as well as the clinically important αvβ6 isoform (Table 1 and see Section 2 for details).
Table 1. Binding Affinity of 1 toward αvβ3, αvβ5, and αvβ6.
In line with the previously reported data,16iRGD showed a mid-low nanomolar potency toward both αvβ3 and αvβ5. More interestingly, we here detected for the first time the peptide’s affinity for the αvβ6 isoform, albeit with an IC50 higher than those measured for the αvβ3 and αvβ5 subtypes. The renewed integrin binding profile of 1 introduces intriguing questions and new perspectives. In particular, what is the molecular basis for this specificity? Can iRGD be modified to selectively shift its affinity toward each of the three isoforms? In an attempt to answer these queries, we carried out a full characterization of the iRGD binding mechanism. In the first stage, we investigated the intrinsic folding properties of the peptide, leading to the identification of its in-solution conformation.
3.2. Folding of iRGD
Peptide and protein folding are events that naturally occur in time scales (tens of μs to ms) not accessible to standard molecular simulations. For this reason, we adopted an enhanced sampling approach, namely, PT-WTE metadynamics, to study the conformational behavior of iRGD in water. PT-WTE is a theoretical method widely employed for predictions of small proteins’ and peptide’s folding,85−93 which requires no prior knowledge of the system under study: the sampling is boosted by the combination of a typical parallel tempering scheme with a metadynamics (MetaD) bias potential deposited on the potential energy of the system (WTE ensemble). Once the simulation is converged, the user can define some collective variables (CVs) to compute the free energy surface (FES) through the desired reweighting scheme. In our case, 6 parallel replicas were employed to span the temperature range going from 300 to 450 K in the WTE ensemble. Each replica was simulated for 140 ns for a total simulation time of 840 ns, in which the conformational space of the peptide was widely explored (Figure S1C). At the end of the calculation, the MetaD bias of the 300 K replica was reweighted according to the Tiwary–Parrinello57 algorithm. Indeed, the FES was computed as a function of two CVs specifically selected for describing the folding (see Section 2 for details): (i) the degree of similarity between contiguous dihedral angles of the backbone (dihedral correlation, Dihcor), which can help to describe backbone conformational changes94 and (ii) the number of intramolecular backbone–backbone hydrogen bonds (Hbonds) that can be indicative of the presence of specific secondary structure elements. Looking at the resulting FES (Figure 1), a single energy minimum can be identified. The structures contained in this energy basin were clustered based on the RMSD of all of the backbone heavy atoms, highlighting the presence of a unique predominant conformation (>90% of occurrence). In the latter, 1 is folded in a peculiar horseshoe-like shape, characterized by two hydrogen bonds formed by (i) the carbonyl oxygen of Arg2 with the amide nitrogen of Gly6 and (ii) the carbonyl group of Pro7 with the amide nitrogen of Arg2. The reliability of this result is strongly supported by the high convergence reached by the calculation and the low computed error (≈1 kJ/mol) associated with the ΔG prediction (Figure S1 and Section 2 for details). The identification of the low-energy horseshoe-like conformation of iRGD represents an important achievement, as it is known that conformations of peptides in aqueous environments often overlap with the receptor-bound one.51,95,96
Figure 1.

Free energy surface (FES) of the folding process of 1 as a function of Dihcor and Hbond CVs with isosurfaces displayed every 1.5 kJ/mol. The conformation representing the main free energy minimum is shown as the inset.
3.3. Binding Mode Studies
Starting from the PT-WTE results, extensive computational studies were carried out to unravel the binding modalities of iRGD toward αvβ3 and αvβ5 as well as the newly discovered biological partner αvβ6. An initial guess of the ligand binding poses was obtained by performing molecular docking in the X-ray structures of αvβ3 and αvβ6, as well as in the newly built αvβ5 homology model (see Section 2 for details). The PT-WTE-predicted conformation of iRGD was used as the starting point for the docking calculations. According to the employed protocol, the macrocyclic backbone of 1 was kept rigid, while sampling of the side chains’ orientation was allowed. Docking in both αvβ3 and αvβ5 converged toward a well-defined binding pose (Figure S6A,B) in which 1 adopts the typical RGD binding pattern. In particular, the peptide’s Arg2 forms a salt bridge with the conserved (αv)-D218 and a cation–π interaction with (αv)-Y178, whereas the carboxylate group of Asp4 chelates the Mg2+ cation at the protein MIDAS (‘metal ion-dependent adhesion site’) in the β subunit. Notably, the most relevant difference between the two docking poses consists in the position of the peptide’s residues flanking the RGD motif (a.a. 5–9) with respect to the region defined by the specificity determining loop (SDL) that is distinctive of the various integrin subtypes.51,96−99 Indeed, in αvβ3, the flanking amino acids of 1 approximate this region, while in αvβ5, they are predicted to point in the opposite direction, establishing two H-bonds with the side chains of (β5)-T315 and (β5)-N317.
The meaning of such small differences together with the energetics and the overall stability of the docking complexes was then evaluated in 2 μs long MD simulations. A refinement of the binding poses was indeed desirable to optimize potential clashes or small artifacts due to the use of both a rigid receptor and restraints on the ligand’s peptide backbone upon docking. In this perspective, long MD trajectories can allow the system to escape from relative energy minima in which it might be trapped, fully considering the receptor flexibility, the solvent effects, and the entropic contributions, which are neglected during docking calculations. The MD results showed that, in both αvβ3 and αvβ5, iRGD slightly rearranges during the first 20–100 ns of the simulation to assume a binding mode that is mostly conserved for the rest of the trajectory (Figure 2). This trend is confirmed by the ligand’s RMSD with respect to either the first frame (Figure 3A,B) or the peptide’s average position in each MD trajectory (Figure 3D,E). Notably, the ligand’s rearrangement is more evident in the iRGD/αvβ5 complex, where 1 loses the interactions with (β5)-T315 and (β5)-N317 predicted by docking (Figure S7) to get closer to the SDL pocket (Figure S8) as in αvβ3. The overall stability of the final peptide binding conformation was further assessed by the analysis of the ligand’s root-mean-square fluctuation (RMSF), showing very low per-residue fluctuations over the 2 μs time scale (Figure 4). A detailed analysis of the ligand–receptor interactions throughout the MD trajectories was thus performed. First, it was shown that the typical interaction scheme involving the RGD motif is highly conserved over time in both αvβ3 and αvβ5 (Figures S9 and S10). Then, we observed that during the MD runs, new contacts are established by 1 in each of the investigated complexes. In detail, the Asp4 side chain and the amide backbone form hydrogen bonds with (β3)-S121 and (β3)-R216 in αvβ3 and with (β5)-S126 and (β5)-D221 in αvβ5. Additionally, van der Waals interactions are formed by the peptide Pro7 and the aromatic ring of (αv)-Y178. Finally, in the iRGD/αvβ3 complex, a transient H-bond is detected between the backbone of the ligand’s Lys6 with the side chains of (β3)-R214 (Figure S9). It is also interesting to note that the highly compact folding assumed by the peptide is conserved throughout the simulations. In this regard, we report that very low ligand’s backbone RMSD values were computed with respect to the horseshoe-like conformation predicted by PT-WTE, especially in the most affine receptor αvβ3, and that the two stabilizing intramolecular H-bonds are retained in more than 90% of both the entire trajectories (Figures S11 and S12). These outcomes are further indicative of the similarity between the in-solution and receptor-bound conformations of iRGD.
Figure 2.
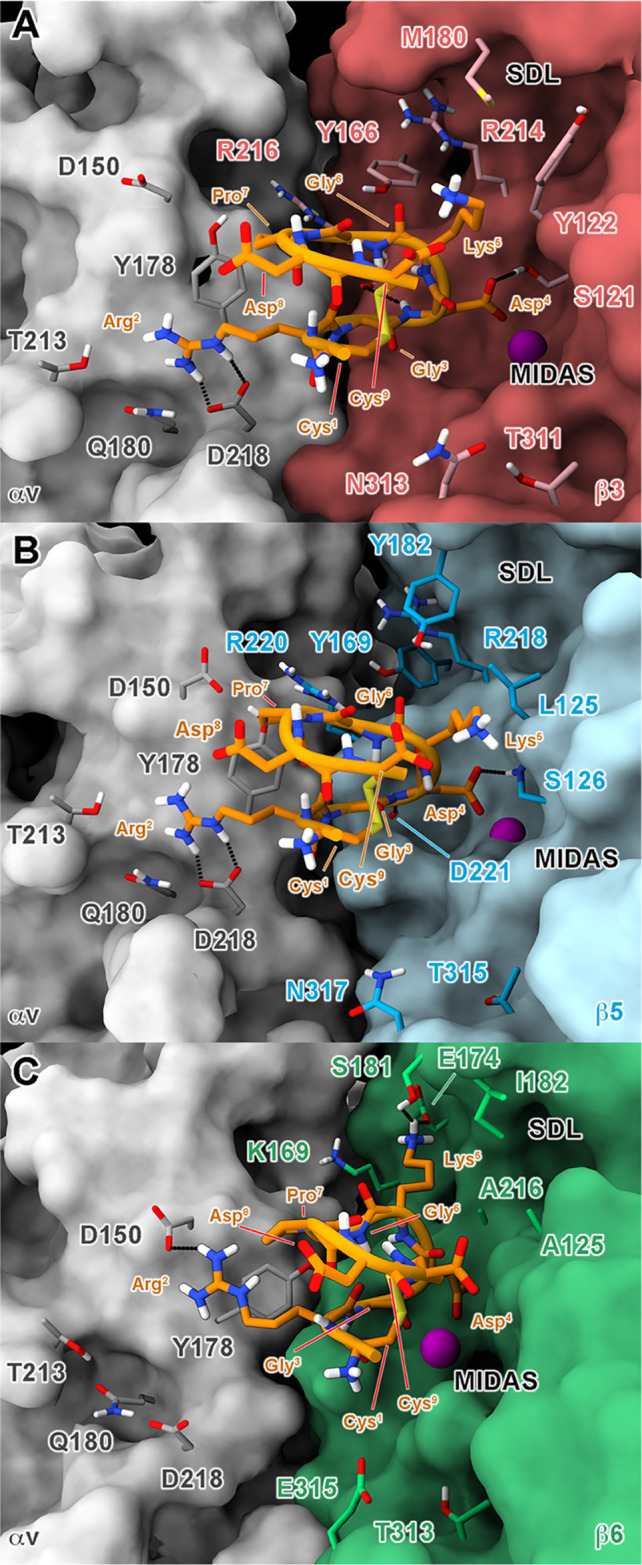
MD-predicted binding mode of iRGD at the orthosteric binding site of (A) αvβ3, (B) αvβ5, and (C) αvβ6 integrins. The different receptor subunits are depicted as colored surfaces (αv = gray, β3 = red, β5 = cyan, and β6 = green). Amino acids important for peptide binding are highlighted as sticks, while the Mg2+ ion in MIDAS is shown as a purple sphere. The ligand is represented as orange ribbons and sticks; nonpolar hydrogens are omitted for the sake of clarity; and H-bonds are shown as black dashed lines.
Figure 3.

Ligand’s RMSD plots for the three simulated binding complexes. Prior to the RMSD computation, the trajectories were aligned on the Cα-atoms of the most stable secondary structure elements. Two different ligand’s reference conformations were used: (i) the first frame (equilibrated docking pose) of each MD complex (first row: A–C) and (ii) the average binding pose observed during each MD run (second row: D–F). The RMSD values (bolded lines) are smoothed with a rolling window of 5 ns, while the actual fluctuations are shown with a slight transparency.
Figure 4.
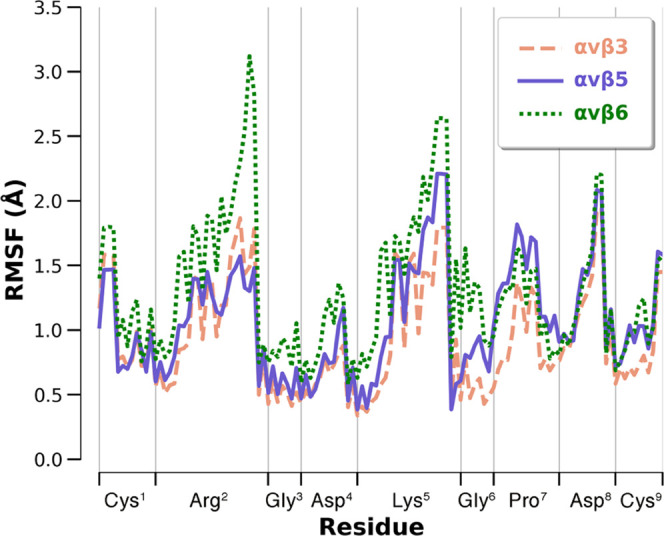
iRGD (1) residues RMSF in three binding complexes. The computation was performed on all of the ligand’s heavy atoms.
As concerns the αvβ6 receptor, the initial docking mode was generally similar to those observed in αvβ3 and αvβ5. (Figure S6C). However, during the following MD simulation, the peptide’s pose as well as its overall folding was less conserved than those in the other two investigated subtypes, as testified by the ligand’s RMSD (Figure 3) and RMSF (Figure 4). The latter shows that especially the RGD motif has higher fluctuations than observed in αvβ3 and αvβ5, suggesting a lower stability of the typical RGD interactions.
In detail, a certain tendency of Arg2 to break its ionic contact with (αv)-D218 and to bind, in turn, the side chain of (αv)-D150 (Figure S13) is observed; furthermore, the Mg2+ chelation is not fully conserved over the simulation (Figure S14), as also testified by the higher changes in the Gly2-ψ and Asp3-φ dihedral angles with respect to the MD trajectories on αvβ3 and αvβ5 (Figure S15).
Likewise, even the backbone of 1 is more fluctuating than in the other two systems, as shown by the RMSD plots computed against the predicted PT-WTE conformation (Figure S16). These observations, along with the general trend observed in the three simulations, agree with the selectivity profile of the in vitro assays and with the lower binding affinity of 1 for αvβ6 with respect to αvβ3 and αvβ5.
Notably, our in silico predictions are coherent with both the bioactivation mechanism and the chemical functionalization of iRGD. Indeed, in the MD-refined complexes, both the ligand’s Lys5 and Gly6 residues and the terminal Cys9 carboxylate group are solvent-exposed; moreover, the latter is not involved in any specific interaction with the receptor. Thus, the Lys5–Gly6 bond is easily accessible for proteolytic cleavage, which releases the NRP-1-recognizing CendR/K sequence (CRGDK). On the other hand, the solvent exposure of the Cys9 carboxylate explains why the bioconjugation of this group with bulky molecules, for either therapeutic or diagnostic purposes, is safely permitted.18−19,20,25,26,35 Both of these findings strengthen the reliability and increase the scientific impact of the obtained atomistic iRGD–integrin binding complexes.
3.4. Structural Basis of Integrin Selectivity and Hints for Drug Design
The range of theranostic applications of RGD integrin ligands highly depends on their activity and selectivity profiles. For this reason, the development of potent and subtype-specific compounds is a desirable, albeit challenging, task.100,101 In this perspective, the presented interaction models can help to rationalize the molecular basis of the selectivity of 1 for αvβ3, αvβ5, and αvβ6 and, in turn, provide hints to modulate the affinity toward each single isoform. In fact, the MD-predicted complexes suggest that the trend observed in the experimental binding assays can be ascribed to the punctiform mutations occurring in the SDL cavities (Figure S17) of the three receptors. These, in turn, cause changes in the steric and electrostatic requirements for the binding of RGD-featured ligands. As previously reported,51,96−99 the core of the (β3)/(β5)-SDL regions is occupied by the bulky side chains of (β3)-Y166/(β5)-Y169 (β2-β3 loop), (β3)-R214/(β5)-R218, and (β3)-R216/(β5)-R220 (α2-α3 loop), which are kept in place by a π–cation network where the tyrosine ring is interposed between the guanidinium groups of the two arginines (Figure S18 A,B). For this reason, high potency toward αvβ3 and αvβ5 is obtained by ligands able to orient the residues flanking their RGD motif in opposite direction to the bulky SDL pockets of these integrins.64,102 A prominent example is the most famous αvβ3/αvβ5 dual modulator cilengitide,49,50 in which the residues next to the RGD motif (d-Phe4 and NMe-Val5) face away from the α/β protein interface (Figure S19). Similarly, 1 avoids steric clashes with the hindered SDL cavities of αvβ3 and αvβ5, thus retaining the affinity for both receptors. This is allowed by the compact bound conformation assumed by iRGD in the two isoforms, as predicted by our MD simulations. Indeed, the peptide exposes toward the SDL region the small Gly6 and the flexible Lys5, whose side chain assumes an orientation closely resembling that of the cilengitide’s phenyl ring (Figure S19). Nonetheless, the potency of iRGD toward both these integrins could be further improved by increasing the number of interactions played with the αv subunit. For instance, our binding models suggest that the substitution of standard Pro5 with hydroxy-functionalized forms of the same amino acid, such as 4- or 5-hydroxyproline, could allow the compound to form an additional H-bond with (αv)-Y178.
On the other hand, achieving αvβ3/αvβ5 selectivity is far from being trivial due to the very similar steric and electrostatic features of the SDL region in the two receptors. Indeed, to our knowledge, most of the known medium-sized peptides targeting αvβ5 also retain activity toward the αvβ3 isoform.103 However, in the case of iRGD, our simulations propose that a receptor area different from the SDL could be targeted to discriminate between the αvβ3 and αvβ5 subtypes.
Particularly, the N-terminal amine group of 1 is directed toward the bottom part of the RGD binding site, where the integrins’ molecular surfaces delineate a small cleft (Figure 5). At this level, β3 and β5 are distinguished by three mutations, namely, (β3)-A252/(β5)-D256, (β3)-K253/(β5)-V257, and (β3)-V314/(β5)-H318, which could be taken into account to apply isoform-specific chemical modifications at the peptide’s N-terminus.
Figure 5.
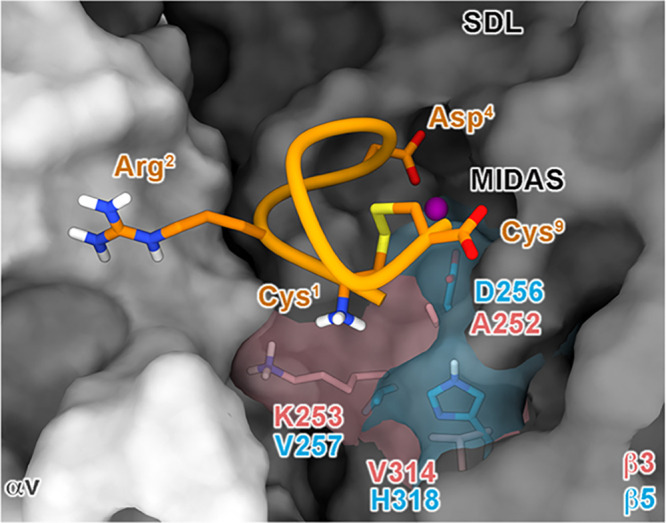
Possible hints for achieving αvβ3/αvβ5 selectivity. The N-terminus of 1 in its predicted binding modes at αvβ3/αvβ5 is close to a subpocket where three key mutations occur. The αv and β3/β5 subunits are depicted as light and dark gray surfaces, respectively. The key mutations between the two receptors are highlighted as red (β3) and cyan (β5) sticks. The ligand is represented as orange ribbons and sticks; nonpolar hydrogens are omitted for the sake of clarity.
In this regard, recent studies have shown that the functionalization of this moiety can be safely performed without affecting the iRGD biological properties. Indeed, a variant of the peptide in which the Cys1 N-terminus and the Cys9 C-terminus are capped with acetyl and N-methyl groups, respectively, has been recently developed, showing striking tumor-homing and -penetrating features.104
At variance with the β3 and β5 subunits, β6 displays a wider and more lipophilic SDL cavity. Here, (β3)-R214/(β5)-R218 and (β3)-R216/(β5)-R220 (α2-α3 loop) are replaced, respectively, by the smaller (β6)-A216 and by the hydrophobic (β6)-I218, while (β3)-Y66/(β5)-Y169 is mutated in (β6)-K169 (Figures 6 and S18C). Notably, in our simulation on the 1–αvβ6 complex, this residue is engaged in stable salt bridges with the carboxylic groups of (αv)-E121 and (αv)-D123; these interactions attract its side chain toward the αv subunit, further increasing the ligand-accessible volume within the SDL cavity (Figure S18C). In this scenario, iRGD is prompted to rearrange, as testified by the ligand’s RMSD evolution (Figure 3C,F), and occupy the cleft forming H-bonds with (β6)-E174 and (β6)-S181 through its Lys5 side chain (Figures 2 and S13). These movements can partially impair both the peptide’s folding (Figure S16) and the interaction scheme of the RGD motif (Figures S13–S15), contributing to explaining the lower affinity of 1 for αvβ6. In fact, higher potency toward this integrin is displayed by peptides that occupy the SDL through bulky lipophilic moieties while still preserving the correct RGD binding pattern. Representative examples are compounds showing a helical DLXXL/I motif like the endogenous αvβ6 ligand latency-associated peptide (LAP) of the transforming growth factor β (TGF-β)66 or the small selective cyclic pentapeptide developed by us, namely, [RGD-Chg-E]-CONH2 (2).51,105 The different αvβ6 interactions between these peptides and iRGD are highlighted by the superposition of the binding pose of 1 and 2 in this integrin receptor (Figure 6), which provides potential clues for the rational modification of the peptide. A possible strategy to selectively increase the iRGD’s affinity for αvβ6 might thus consist of the insertion of properly oriented bulky moieties able to target the wide and lipophilic SDL region of this integrin while being not tolerated by both the αvβ3 and αvβ5 isoforms. In this regard, the comparison of our interaction model to the 3D complex of αvβ6 and our selective pentapeptide 2 (Figure 6) shows that the Lys5 and Gly6 of iRGD partly overlap with the cyclohexylglycine (Chg4) of the reference compound, being prone to interact with the SDL groove.
Figure 6.

Superposition of the MD-predicted binding pose of iRGD at αvβ3 (A) and αvβ5 (B) with that of [RGD-Chg-E]-CONH2 at αvβ6. The αv and β subunits are depicted as light and dark gray surfaces, respectively. The key mutations between the three receptors are highlighted as red (β3), cyan (β5), and green (β6) sticks, contoured by transparent surfaces. Peptides 1 and 2 are depicted as orange and magenta sticks, respectively; nonpolar hydrogens are omitted for the sake of clarity.
Since Lys5 is part of the CendR sequence (needed for the interaction with NRP-1), it could be replaced only by amino acids sharing analogous physicochemical properties (i.e., arginine). Therefore, a focused chemical optimization campaign could be performed by replacing Gly6 with natural (i.e., valine, leucine, isoleucine, phenylalanine, tryptophan) or non-natural (i.e., cyclohexylglycine, cyclohexylalanine, cyclopropylalanine, allylglycine) lipophilic residues with moderate bulkiness so as to fill the peculiar (more hydrophobic and wider with respect to αvβ3 and αvβ5) pocket mostly made up by the αvβ6 SDL. However, given the higher flexibility of glycine with respect to all of the other amino acids, the impact of such modifications on the peptide’s folding and ability to interact with the receptor should be carefully pre-evaluated. To this aim, we designed a small virtual library of nine designed iRGD derivatives, whose folding properties were predicted through additional PT-WTE calculations (compounds 3–11; see Table S1 and Figures 7A, S20, and S21). In particular, we investigated the capability of these compounds to adopt a conformation similar to that of 1 by computing their folding free energy surfaces as a function of two different sets of CVs.
Figure 7.
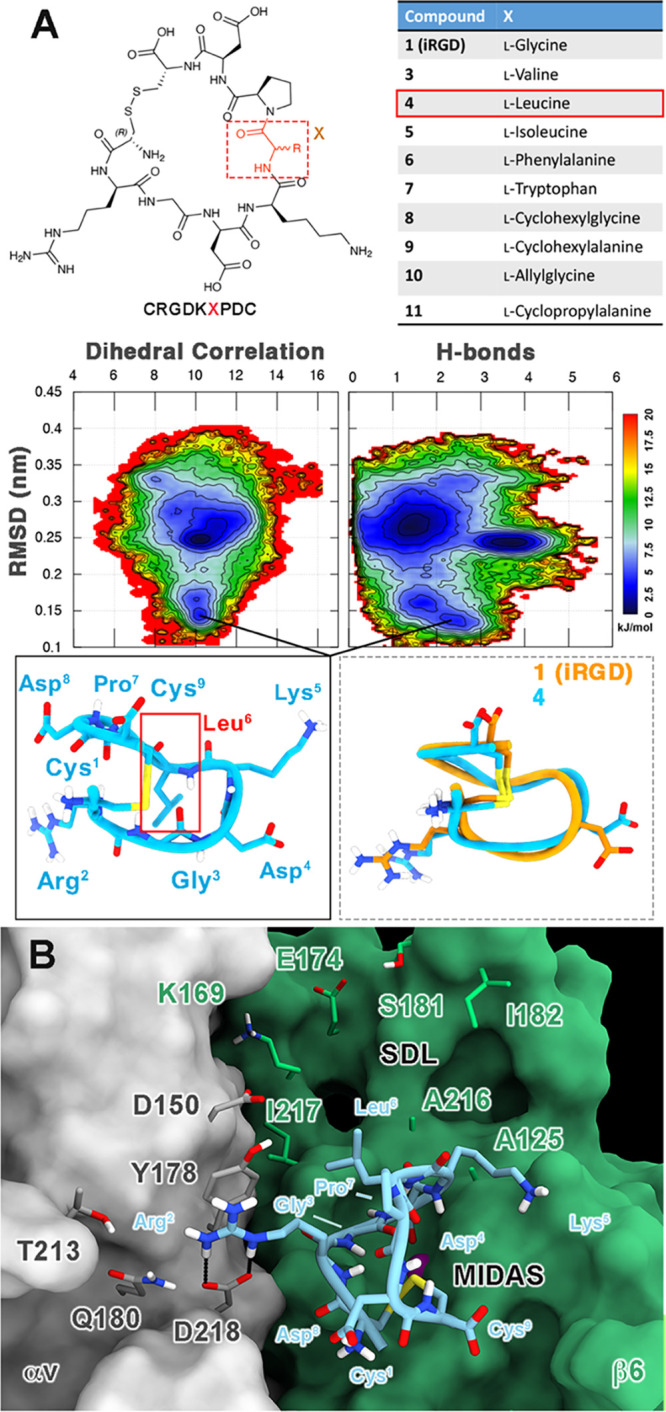
(A) Hints for increasing αvβ6 affinity and selectivity. A virtual library of nine compounds was designed by mutating Gly6 of 1 with natural and non-natural lipophilic amino acids (upper panel). Free energy surfaces (FESs) of the folding process of 4 with isosurfaces were displayed every 1.5 kJ/mol (lower panel). The horseshoe-like conformation of 4 and its superimposition with 1 are shown as insets. (B) Docking-predicted binding mode of compound 4 at the RGD binding site of the αvβ6 integrin. The different receptor subunits are depicted as colored surfaces (αv = gray and β6 = green). Amino acids important for peptide binding are highlighted as sticks, while the Mg2+ ion in the MIDAS is shown as a purple sphere. The ligand is represented as cyan ribbons and sticks; nonpolar hydrogens are omitted for the sake of clarity; and H-bonds are shown as black dashed lines.
Specifically, each of the CVs used to characterize the folding energy landscape of 1 (Dihcor and Hbond) was alternatively combined with the peptides’ RMSD values computed with respect to the horseshoe-like conformation of iRGD. The predicted FES showed that five out of the designed peptides (3, 5, 7, 9, and 10) have folding profiles quite diverging from the parent peptide (Figures S20 and S21), which may lead to a total loss or a reduction of integrin affinities. Nonetheless, the remaining four molecules (4, 6, 8, and 11) could be able to assume a conformation comparable to that of 1. In fact, although these peptides seem to have a more complex conformational ensemble than iRGD, their folding FESs present a clear energy basin at low RMSD values (1.2–1.7 Å) with respect to the horseshoe-like shape of 1. This is particularly evident for 4, where Gly6 is replaced by a leucine, showing a low-energy conformation that almost perfectly overlaps with iRGD (Figure 7A). To verify whether these peptides could fit into the wide and lipophilic SDL pocket of αvβ6, we performed additional semiflexible docking calculations on this receptor. Particularly, the docking pose of 4 shown in Figure 7B is representative of how the new lipophilic residue at the 6 position—here Leu6—could be oriented toward the hydrophobic SDL cavity and may thus contribute to increasing the αvβ6 affinity and specificity.
4. Conclusions
Over the past decade, the tumor-homing iRGD (1) peptide has emerged as a powerful tool for anticancer therapy,1,17,21,25−27 due to its high tropism for cancer tissues and the possible coadministration with a plethora of chemotherapeutic agents. In this article, we provide unprecedented data about the biological and structural aspects of iRGD’s mechanism of action. First, we discovered that 1 has affinity not only for the known targets αvβ3/αvβ5 integrins but also for the αvβ6 subtype. These data have great clinical relevance considering that the latter receptor is overexpressed in many malignancies, including pancreatic ductal adenocarcinoma, against which iRGD has shown to be a promising therapeutic weapon in combination with paclitaxel and gemcitabine.34,37 Second, by means of advanced metadynamics simulations, we discovered that in an aqueous environment, 1 mainly adopted a peculiar horseshoe-like conformation, which was then employed as a starting point for accurate interaction studies with the three integrin receptors through molecular dynamics simulations. Notably, our in silico binding predictions are in agreement with the peptide’s activation mechanism, relying on the proteolytical cleavage of its Lys5–Gly6 bond when bound to the integrin surface and the following release of the internalizing CendR motif. Moreover, they allowed us to rationalize at the atomic level the experimentally measured potency and selectivity trend of 1 (αvβ3 ≥ αvβ5 > αvβ6). In particular, the compact horseshoe-like shape allows the peptide to fit the peculiar features of the integrins’ SDL cleft and seems thus fundamental to achieving nanomolar binding affinity. This requirement is better satisfied in the sterically hindered αvβ3/αvβ5 integrins than in αvβ6, where this cleft is wider and lipophilic due to the presence of specific mutations that were clearly described over text. On this basis, we set up a few possible strategies to fine-tune the affinity/selectivity of iRGD toward each of the investigated integrins, especially designing new peptides potentially able to selectively recognize the newly emerged anticancer target αvβ6. We are confident that these clues can pave the way for a new successful drug design campaign aimed at producing iRGD derivatives with optimized and fine-tuned pharmacodynamic properties.
Acknowledgments
This research was funded by Regione Campania–POR Campania FESR 2014/2020 (B61G18000470007).
Glossary
Abbreviations
- DSG2
desmoglein 2
- ECM
extracellular matrix components
- IPF
idiopathic pulmonary fibrosis
- iRGD
internalizing RGD
- PDAC
pancreatic ductal adenocarcinoma
- NRP-1
neuropilin-1
- MetaD
metadynamics
- PT
parallel tempering
- PT-WTE
parallel tempering in the well-tempered ensemble
- WTE
well-tempered ensemble
Data Availability Statement
All of the input files and trajectory data sets are published on a public Zenodo folder and freely available at the following link: https://zenodo.org/record/8089457.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.3c01071.
PT-WTE convergence assessment and additional analysis on all of the simulated systems (PDF)
Author Contributions
# V.M.D. and G.D. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Kang S.; Lee S.; Park S. iRGD Peptide as a Tumor-Penetrating Enhancer for Tumor-Targeted Drug Delivery. Polymers 2020, 12, 1906 10.3390/polym12091906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould D. R.; Hutson P. R. Critical Considerations in Anticancer Drug Development and Dosing Strategies: The Past, Present, and Future. J. Clin. Pharmacol. 2017, 57, S116–S128. 10.1002/jcph.983. [DOI] [PubMed] [Google Scholar]
- Hambley T. W. Is Anticancer Drug Development Heading in the Right Direction?. Cancer Res. 2009, 69, 1259–1261. 10.1158/0008-5472.CAN-08-3786. [DOI] [PubMed] [Google Scholar]
- Bartelink I. H.; Jones E. F.; Shahidi-Latham S. K.; Lee P. R. E.; Zheng Y.; Vicini P.; van ‘t Veer L.; Wolf D.; Iagaru A.; Kroetz D. L.; Prideaux B.; Cilliers C.; Thurber G. M.; Wimana Z.; Gebhart G. Tumor Drug Penetration Measurements Could Be the Neglected Piece of the Personalized Cancer Treatment Puzzle. Clin. Pharmacol. Ther. 2019, 106, 148–163. 10.1002/cpt.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit S.; Novak T.; Miller K.; Zhu Y.; Kenney M. E.; Broome A. M. Transferrin Receptor-Targeted Theranostic Gold Nanoparticles for Photosensitizer Delivery in Brain Tumors. Nanoscale 2015, 7, 1782–1790. 10.1039/C4NR04853A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.; Hwang H. S.; Lee S.; Kim B.; Kim J. O.; Oh K. T.; Lee E. S.; Choi H. G.; Youn Y. S. Rabies Virus-Inspired Silica-Coated Gold Nanorods as a Photothermal Therapeutic Platform for Treating Brain Tumors. Adv. Mater. 2017, 29, 1605563 10.1002/adma.201605563. [DOI] [PubMed] [Google Scholar]
- Ruan S.; Hu C.; Tang X.; Cun X.; Xiao W.; Shi K.; He Q.; Gao H. Increased Gold Nanoparticle Retention in Brain Tumors by in Situ Enzyme-Induced Aggregation. ACS Nano 2016, 10, 10086–10098. 10.1021/acsnano.6b05070. [DOI] [PubMed] [Google Scholar]
- Ying M.; Zhan C.; Wang S.; Yao B.; Hu X.; Song X.; Zhang M.; Wei X.; Xiong Y.; Lu W. Liposome-Based Systemic Glioma-Targeted Drug Delivery Enabled by All-D Peptides. ACS Appl. Mater. Interfaces 2016, 8, 29977–29985. 10.1021/acsami.6b10146. [DOI] [PubMed] [Google Scholar]
- Gu Z.; Da Silva C. G.; van der Maaden K.; Ossendorp F.; Cruz L. J. Liposome-Based Drug Delivery Systems in Cancer Immunotherapy. Pharmaceutics 2020, 12, 1054 10.3390/pharmaceutics12111054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran D.; Mao J.; Shen Q.; Xie C.; Zhan C.; Wang R.; Lu W. GRP78 Enabled Micelle-Based Glioma Targeted Drug Delivery. J. Controlled Release 2017, 255, 120–131. 10.1016/j.jconrel.2017.03.037. [DOI] [PubMed] [Google Scholar]
- Hoppenz P.; Els-Heindl S.; Beck-Sickinger A. G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571 10.3389/fchem.2020.00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig B. S.; Tomassi S.; Di Maro S.; Di Leva F. S.; Benge A.; Reichart F.; Nieberler M.; Kühn F. E.; Kessler H.; Marinelli L.; Reuning U.; Kossatz S. The Organometallic Ferrocene Exhibits Amplified Anti-Tumor Activity by Targeted Delivery via Highly Selective Ligands to αvβ3, αvβ6, or α5β1 Integrins. Biomaterials 2021, 271, 120754 10.1016/j.biomaterials.2021.120754. [DOI] [PubMed] [Google Scholar]
- Han J.; Räder A. F. B.; Reichart F.; Aikman B.; Wenzel M. N.; Woods B.; Weinmüller M.; Ludwig B. S.; Stürup S.; Groothuis G. M. M.; Permentier H. P.; Bischoff R.; Kessler H.; Horvatovich P.; Casini A. Bioconjugation of Supramolecular Metallacages to Integrin Ligands for Targeted Delivery of Cisplatin. Bioconjugate Chem. 2018, 29, 3856–3865. 10.1021/acs.bioconjchem.8b00682. [DOI] [PubMed] [Google Scholar]
- Hovlid M. L.; Steinmetz N. F.; Laufer B.; Lau J. L.; Kuzelka J.; Wang Q.; Hyypiä T.; Nemerow G. R.; Kessler H.; Manchester M.; Finn M. G. Guiding Plant Virus Particles to Integrin-Displaying Cells. Nanoscale 2012, 4, 3698–3705. 10.1039/c2nr30571b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi-Artzner V.; Lorz B.; Hellerer U.; Kantlehner M.; Kessler H.; Sackmann E. Selective Adhesion of Endothelial Cells to Artificial Membranes with a Synthetic RGD-Lipopeptide. Chem. - Eur. J. 2001, 7, 1095–1101. . [DOI] [PubMed] [Google Scholar]
- Sugahara K. N.; Teesalu T.; Karmali P. P.; Kotamraju V. R.; Agemy L.; Girard O. M.; Hanahan D.; Mattrey R. F.; Ruoslahti E. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara K. N.; Teesalu T.; Karmali P. P.; Kotamraju V. R.; Agemy L.; Greenwald D. R.; Ruoslahti E. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science 2010, 328, 1031–1035. 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Zhang Q.; Yang J.; Wang H.; Xu J.; Zheng J. iRGD as a Tumor-Penetrating Peptide for Cancer Therapy (Review). Mol. Med. Rep. 2017, 15, 2925–2930. 10.3892/mmr.2017.6419. [DOI] [PubMed] [Google Scholar]
- Liu C.; Yao S.; Li X.; Wang F.; Jiang Y. iRGD-Mediated Core-Shell Nanoparticles Loading Carmustine and O6-Benzylguanine for Glioma Therapy. J. Drug Targeting 2017, 25, 235–246. 10.1080/1061186X.2016.1238091. [DOI] [PubMed] [Google Scholar]
- Lu L.; Zhao X.; Fu T.; Li K.; He Y.; Luo Z.; Dai L.; Zeng R.; Cai K. An iRGD-Conjugated Prodrug Micelle with Blood-Brain-Barrier Penetrability for Anti-Glioma Therapy. Biomaterials 2020, 230, 119666 10.1016/j.biomaterials.2019.119666. [DOI] [PubMed] [Google Scholar]
- Shi X.; Ma R.; Lu Y.; Cheng Y.; Fan X.; Zou J.; Zheng H.; Li F.; Piao J. G. iRGD and TGN Co-Modified PAMAM for Multi-Targeted Delivery of ATO to Gliomas. Biochem. Biophys. Res. Commun. 2020, 527, 117–123. 10.1016/j.bbrc.2020.04.064. [DOI] [PubMed] [Google Scholar]
- Hou G.; Li Y.; Wang Q.; Zhang H.; Liang S.; Liu B.; Shi W. iRGD-Grafted N-Trimethyl Chitosan-Coated Protein Nanotubes Enhanced the Anticancer Efficacy of Curcumin and Melittin. Int. J. Biol. Macromol. 2022, 222, 348–359. 10.1016/j.ijbiomac.2022.09.171. [DOI] [PubMed] [Google Scholar]
- Paul B.; Gaonkar R. H.; Dutta D.; Dasi R.; Mukherjee B.; Ganguly S.; Das S. K. Inhibitory Potential of iRGD Peptide-Conjugated Garcinol-Loaded Biodegradable Nanoparticles in Rat Colorectal Carcinoma. Biomater. Adv. 2022, 134, 112714 10.1016/j.msec.2022.112714. [DOI] [PubMed] [Google Scholar]
- Alghamri M. S.; Banerjee K.; Mujeeb A. A.; Mauser A.; Taher A.; Thalla R.; McClellan B. L.; Varela M. L.; Stamatovic S. M.; Martinez-Revollar G.; Andjelkovic A. V.; Gregory J. V.; Kadiyala P.; Calinescu A.; Jiménez J. A.; Apfelbaum A. A.; Lawlor E. R.; Carney S.; Comba A.; Faisal S. M.; Barissi M.; Edwards M. B.; Appelman H.; Sun Y.; Gan J.; Ackermann R.; Schwendeman A.; Candolfi M.; Olin M. R.; Lahann J.; Lowenstein P. R.; Castro M. G. Systemic Delivery of an Adjuvant CXCR4-CXCL12 Signaling Inhibitor Encapsulated in Synthetic Protein Nanoparticles for Glioma Immunotherapy. ACS Nano 2022, 16, 8729–8750. 10.1021/acsnano.1c07492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K.; Lin C.; Li M.; Xu K.; He Y.; Mao Y.; Lu L.; Geng W.; Li X.; Luo Z.; Cai K. Multienzyme-like Reactivity Cooperatively Impairs Glutathione Peroxidase 4 and Ferroptosis Suppressor Protein 1 Pathways in Triple-Negative Breast Cancer for Sensitized Ferroptosis Therapy. ACS Nano 2022, 16, 2381–2398. 10.1021/acsnano.1c08664. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Wang Z.; Wang T.; Wang J.; Zhang H.; Li Z.; Wang S.; Sheng F.; Yu J.; Hou Y. Biodegradable MnO-Based Nanoparticles with Engineering Surface for Tumor Therapy: Simultaneous Fenton-Like Ion Delivery and Immune Activation. ACS Nano 2022, 16, 11862–11875. 10.1021/acsnano.2c00969. [DOI] [PubMed] [Google Scholar]
- Pei P.; Chen L.; Fan R.; Zhou X.-R.; Feng S.; Liu H.; Guo Q.; Yin H.; Zhang Q.; Sun F.; Peng L.; Wei P.; He C.; Qiao R.; Wang Z.; Luo S.-Z. Computer-Aided Design of Lasso-like Self-Assembling Anticancer Peptides with Multiple Functions for Targeted Self-Delivery and Cancer Treatments. ACS Nano 2022, 16, 13783–13799. 10.1021/acsnano.2c01014. [DOI] [PubMed] [Google Scholar]
- Liang H.; Wu X.; Zhao G.; Feng K.; Ni K.; Sun X. Renal Clearable Ultrasmall Single-Crystal Fe Nanoparticles for Highly Selective and Effective Ferroptosis Therapy and Immunotherapy. J. Am. Chem. Soc. 2021, 143, 15812–15823. 10.1021/jacs.1c07471. [DOI] [PubMed] [Google Scholar]
- de Mendoza T. H.; Mose E. S.; Botta G. P.; Braun G. B.; Kotamraju V. R.; French R. P.; Suzuki K.; Miyamura N.; Teesalu T.; Ruoslahti E.; Lowy A. M.; Sugahara K. N. Tumor-Penetrating Therapy for β5 Integrin-Rich Pancreas Cancer. Nat. Commun. 2021, 12, 1541 10.1038/s41467-021-21858-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Dai Q.; Yang Q. Y.; Bao X.; Zhou Y.; Zhong H.; Wu L.; Wang T.; Zhang Z.; Lu Y.; Zhang Z.; Lin M.; Han M.; Wei Q. Therapeutic Nucleus-Access BNCT Drug Combined CD47-Targeting Gene Editing in Glioblastoma. J. Nanobiotechnol. 2022, 20, 102 10.1186/s12951-022-01304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoodi Z.; Shafiee F. Internalizing RGD, a Great Motif for Targeted Peptide and Protein Delivery: A Review Article. Drug Delivery Transl. Res. 2022, 12, 2261–2274. 10.1007/s13346-022-01116-7. [DOI] [PubMed] [Google Scholar]
- Gregory J. V.; Kadiyala P.; Doherty R.; Cadena M.; Habeel S.; Ruoslahti E.; Lowenstein P. R.; Castro M. G.; Lahann J. Systemic Brain Tumor Delivery of Synthetic Protein Nanoparticles for Glioblastoma Therapy. Nat. Commun. 2020, 11, 5687 10.1038/s41467-020-19225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Li M.; Tang Z.; Li Q.; Yang Y.; Liu H.; Duan T.; Hong H.; Chen X. Methoxypoly(Ethylene Glycol)-Block-Poly(L-Glutamic Acid)-Loaded Cisplatin and a Combination With iRGD for the Treatment of Non-Small-Cell Lung Cancers. Macromol. Biosci. 2012, 12, 1514–1523. 10.1002/mabi.201200145. [DOI] [PubMed] [Google Scholar]
- Dean A.; Gill S.; McGregor M.; Broadbridge V.; Jarvelainen H. A.; Price T. J. 1528P Phase I Trial of the First-in-Class Agent CEND-1 in Combination with Gemcitabine and Nab-Paclitaxel in Patients with Metastatic Pancreatic Cancer. Ann. Oncol. 2020, 31, S941 10.1016/j.annonc.2020.08.2011. [DOI] [Google Scholar]
- Qian J.; Zhou S.; Lin P.; Lei J.; Zheng S.; Xu W.; Wang Y.; Gao Z.; Yang J. Recent Advances in the Tumor-Penetrating Peptide Internalizing RGD for Cancer Treatment and Diagnosis. Drug Dev. Res. 2023, 84, 654–670. 10.1002/ddr.22056. [DOI] [PubMed] [Google Scholar]
- Springfeld C.; Neoptolemos J. P. CEND-1: A Game Changer for Pancreatic Cancer Chemotherapy?. Lancet Gastroenterol. Hepatol. 2022, 7, 900–902. 10.1016/S2468-1253(22)00197-2. [DOI] [PubMed] [Google Scholar]
- Dean A.; Gill S.; McGregor M.; Broadbridge V.; Järveläinen H. A.; Price T. Dual αv-Integrin and Neuropilin-1 Targeting Peptide CEND-1 plus Nab-Paclitaxel and Gemcitabine for the Treatment of Metastatic Pancreatic Ductal Adenocarcinoma: A First-in-Human, Open-Label, Multicentre, Phase 1 Study. Lancet Gastroenterol. Hepatol. 2022, 7, 943–951. 10.1016/S2468-1253(22)00167-4. [DOI] [PubMed] [Google Scholar]
- Teoh C.; Tan S.; Tran T. Integrins as Therapeutic Targets for Respiratory Diseases. Curr. Mol. Med. 2015, 15, 714–734. 10.2174/1566524015666150921105339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraamides C. J.; Garmy-Susini B.; Varner J. A. Integrins in Angiogenesis and Lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. 10.1038/nrc2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi H.; Ivaska J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. 10.1038/s41568-018-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuckran C. A.; Liu C.; Bruno T. C.; Workman C. J.; Vignali D. A. A. Neuropilin-1: A Checkpoint Target with Unique Implications for Cancer Immunology and Immunotherapy. J. Immunother. Cancer 2020, 8, e000967 10.1136/jitc-2020-000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teesalu T.; Sugahara K. N.; Kotamraju V. R.; Ruoslahti E. C-End Rule Peptides Mediate Neuropilin-1-Dependent Cell, Vascular, and Tissue Penetration. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16157–16162. 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara K. N.; Braun G. B.; De Mendoza T. H.; Kotamraju V. R.; French R. P.; Lowy A. M.; Teesalu T.; Ruoslahti E. Tumor-Penetrating iRGD Peptide Inhibits Metastasis. Mol. Cancer Ther. 2015, 14, 120–128. 10.1158/1535-7163.MCT-14-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay A.; Raghavan S. Defining the Role of Integrin αvβ6 in Cancer. Curr. Drug Targets 2009, 10, 645–652. 10.2174/138945009788680374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J.; Li Z. The Roles of Integrin αvβ6 in Cancer. Cancer Lett. 2017, 403, 128–137. 10.1016/j.canlet.2017.06.012. [DOI] [PubMed] [Google Scholar]
- Sheppard D. Roles of αv Integrins in Vascular Biology and Pulmonary Pathology. Curr. Opin. Cell Biol. 2004, 16, 552–557. 10.1016/j.ceb.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Henderson N. C.; Arnold T. D.; Katamura Y.; Giacomini M. M.; Rodriguez J. D.; McCarty J. H.; Pellicoro A.; Raschperger E.; Betsholtz C.; Ruminski P. G.; Griggs D. W.; Prinsen M. J.; Maher J. J.; Iredale J. P.; Lacy-Hulbert A.; Adams R. H.; Sheppard D. Targeting of αv Integrin Identifies a Core Molecular Pathway That Regulates Fibrosis in Several Organs. Nat. Med. 2013, 19, 1617–1624. 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reader C. S.; Vallath S.; Steele C. W.; Haider S.; Brentnall A.; Desai A.; Moore K. M.; Jamieson N. B.; Chang D.; Bailey P.; Scarpa A.; Lawlor R.; Chelala C.; Keyse S. M.; Biankin A.; Morton J. P.; Evans T. J.; Barry S. T.; Sansom O. J.; Kocher H. M.; Marshall J. F. The Integrin αvβ6 Drives Pancreatic Cancer through Diverse Mechanisms and Represents an Effective Target for Therapy. J. Pathol. 2019, 249, 332–342. 10.1002/path.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechantsreiter M. A.; Planker E.; Matha B.; Lohof E.; Holzemann G.; Jonczyk A.; Goodman S. L.; Kessler H. N-Methylated Cyclic RGD Peptides as Highly Active and Selective αvβ3 Integrin Antagonists. J. Med. Chem. 1999, 42, 3033–3040. 10.1021/jm970832g. [DOI] [PubMed] [Google Scholar]
- Mas-Moruno C.; Rechenmacher F.; Kessler H. Cilengitide: The First Anti-Angiogenic Small Molecule Drug Candidate. Design, Synthesis and Clinical Evaluation. Anti-Cancer Agents Med. Chem. 2010, 10, 753–768. 10.2174/187152010794728639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leva F. S.; Tomassi S.; Di Maro S.; Reichart F.; Notni J.; Dangi A.; Marelli U. K.; Brancaccio D.; Merlino F.; Wester H. J.; Novellino E.; Kessler H.; Marinelli L. From a Helix to a Small Cycle: Metadynamics-Inspired αvβ6 Integrin Selective Ligands. Angew. Chem., Int. Ed. 2018, 57, 14645–14649. 10.1002/anie.201803250. [DOI] [PubMed] [Google Scholar]
- Bonomi M.; Parrinello M. Enhanced Sampling in the Well-Tempered Ensemble. Phys. Rev. Lett. 2010, 104, 190601 10.1103/PhysRevLett.104.190601. [DOI] [PubMed] [Google Scholar]
- Laio A.; Parrinello M. Escaping Free-Energy Minima. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12562–12566. 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barducci A.; Bussi G.; Parrinello M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- Prakash M. K.; Barducci A.; Parrinello M. Replica Temperatures for Uniform Exchange and Efficient Roundtrip Times in Explicit Solvent Parallel Tempering Simulations. J. Chem. Theory Comput. 2011, 7, 2025–2027. 10.1021/ct200208h. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Tiwary P.; Parrinello M. A Time-Independent Free Energy Estimator for Metadynamics. J. Phys. Chem. B 2015, 119, 736–742. 10.1021/jp504920s. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; van der Spoel D.; van Drunen R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Tribello G. A.; Bonomi M.; Branduardi D.; Camilloni C.; Bussi G. PLUMED 2: New Feathers for an Old Bird. Comput. Phys. Commun. 2014, 185, 604–613. 10.1016/j.cpc.2013.09.018. [DOI] [Google Scholar]
- Bonomi M.; Bussi G.; Camilloni C.; Tribello G. A.; Banáš P.; Barducci A.; Bernetti M.; Bolhuis P. G.; Bottaro S.; Branduardi D.; Capelli R.; Carloni P.; Ceriotti M.; Cesari A.; Chen H.; Chen W.; Colizzi F.; De S.; De La Pierre M.; Donadio D.; Drobot V.; Ensing B.; Ferguson A. L.; Filizola M.; Fraser J. S.; Fu H.; Gasparotto P.; Gervasio F. L.; Giberti F.; Gil-Ley A.; Giorgino T.; Heller G. T.; Hocky G. M.; Iannuzzi M.; Invernizzi M.; Jelfs K. E.; Jussupow A.; Kirilin E.; Laio A.; Limongelli V.; Lindorff-Larsen K.; Löhr T.; Marinelli F.; Martin-Samos L.; Masetti M.; Meyer R.; Michaelides A.; Molteni C.; Morishita T.; Nava M.; Paissoni C.; Papaleo E.; Parrinello M.; Pfaendtner J.; Piaggi P.; Piccini G. M.; Pietropaolo A.; Pietrucci F.; Pipolo S.; Provasi D.; Quigley D.; Raiteri P.; Raniolo S.; Rydzewski J.; Salvalaglio M.; Sosso G. C.; Spiwok V.; Šponer J.; Swenson D. W. H.; Tiwary P.; Valsson O.; Vendruscolo M.; Voth G. A.; White A. Promoting Transparency and Reproducibility in Enhanced Molecular Simulations. Nat. Methods 2019, 16, 670–673. 10.1038/s41592-019-0506-8. [DOI] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. . [DOI] [Google Scholar]
- Xiong J. P.; Stehle T.; Zhang R.; Joachimiak A.; Frech M.; Goodman S. L.; Arnaout M. A. Crystal Structure of the Extracellular Segment of Integrin αVβ3 in Complex with an Arg-Gly-Asp Ligand. Science 2002, 296, 151–155. 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- Van Agthoven J. F.; Xiong J. P.; Alonso J. L.; Rui X.; Adair B. D.; Goodman S. L.; Arnaout M. A. Structural Basis for Pure Antagonism of Integrin αvβ3 by a High-Affinity Form of Fibronectin. Nat. Struct. Mol. Biol. 2014, 21, 383–388. 10.1038/nsmb.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X.; Hudson N. E.; Lu C.; Springer T. A. Structural Determinants of Integrin β-Subunit Specificity for Latent TGF-β. Nat. Struct. Mol. Biol. 2014, 21, 1091–1096. 10.1038/nsmb.2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X.; Zhao B.; Iacob R. E.; Zhu J.; Koksal A. C.; Lu C.; Engen J. R.; Springer T. A. Force Interacts with Macromolecular Structure in Activation of TGF-β. Nature 2017, 542, 55–59. 10.1038/nature21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Su Y.; Iacob R. E.; Engen J. R.; Springer T. A. General Structural Features That Regulate Integrin Affinity Revealed by Atypical αVβ8. Nat. Commun. 2019, 10, 5481 10.1038/s41467-019-13248-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W.; Lopez R.; McWilliam H.; Remmert M.; Söding J.; Thompson J. D.; Higgins D. G. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon M.; McWilliam H.; Li W.; Valentin F.; Squizzato S.; Paern J.; Lopez R. A New Bioinformatics Analysis Tools Framework at EMBL-EBI. Nucleic Acids Res. 2010, 38, 695–699. 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day T. J. F.; Honig B.; Shaw D. E. A Hierarchical Approach to All-Atom Protein Loop Prediction - Jacobson - 2004 - Proteins: Structure, Function, and Bioinformatics - Wiley Online Library. Proteins: Struct., Funct., Bioinform. 2004, 55, 351–367. 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Maestro, N. Schrödinger Release 2020–4; Schrödinger, LLC: New York, 2020.
- Sastry G. M.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Shelley J. C.; Cholleti A.; Frye L. L.; Greenwood J. R.; Timlin M. R.; Uchimaya M. Epik: A Software Program for pKa Prediction and Protonation State Generation for Drug-like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- Greenwood J. R.; Calkins D.; Sullivan A. P.; Shelley J. C. Towards the Comprehensive, Rapid, and Accurate Prediction of the Favorable Tautomeric States of Drug-like Molecules in Aqueous Solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; Kaus J. W.; Cerutti D. S.; Krilov G.; Jorgensen W. L.; Abel R.; Friesner R. A. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Panteva M. T.; Giambaşu G. M.; York D. M. Comparison of Structural, Thermodynamic, Kinetic and Mass Transport Properties of Mg2+ Ion Models Commonly Used in Biomolecular Simulations. J. Comput. Chem. 2015, 36, 970–982. 10.1002/jcc.23881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Cheatham T. E.; Darden T.; Gohlke H.; Luo R.; Merz K. M.; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Kräutler V.; Van Gunsteren W. F.; Hünenberger P. H. A Fast SHAKE Algorithm to Solve Distance Constraint Equations for Small Molecules in Molecular Dynamics Simulations. J. Comput. Chem. 2001, 22, 501–508. . [DOI] [Google Scholar]
- Feller S. E.; Zhang Y.; Pastor R. W.; Brooks B. R. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys. 1995, 103, 4613–4621. 10.1063/1.470648. [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; Van Gunsteren W. F.; Dinola A.; Haak J. R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 1463–1472. 10.1063/1.448118. [DOI] [Google Scholar]
- Palazzesi F.; Barducci A.; Tollinger M.; Parrinello M. The Allosteric Communication Pathways in KIX Domain of CBP. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 14237–14242. 10.1073/pnas.1313548110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernetti M.; Masetti M.; Pietrucci F.; Blackledge M.; Jensen M. R.; Recanatini M.; Mollica L.; Cavalli A. Structural and Kinetic Characterization of the Intrinsically Disordered Protein SeV NTAIL through Enhanced Sampling Simulations. J. Phys. Chem. B 2017, 121, 9572–9582. 10.1021/acs.jpcb.7b08925. [DOI] [PubMed] [Google Scholar]
- D’Annessa I.; Di Leva F. S.; La Teana A.; Novellino E.; Limongelli V.; Di Marino D. Bioinformatics and Biosimulations as Toolbox for Peptides and Peptidomimetics Design: Where Are We?. Front. Mol. Biosci. 2020, 7, 66 10.3389/fmolb.2020.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.; McMullen P.; Yuan Z.; Pfaendtner J.; Jiang S. Elucidating Molecular Design Principles for Charge-Alternating Peptides. Biomacromolecules 2020, 21, 435–443. 10.1021/acs.biomac.9b01191. [DOI] [PubMed] [Google Scholar]
- Damas J. M.; Filipe L. C. S.; Campos S. R. R.; Lousa D.; Victor B. L.; Baptista A. M.; Soares C. M. Predicting the Thermodynamics and Kinetics of Helix Formation in a Cyclic Peptide Model. J. Chem. Theory Comput. 2013, 9, 5148–5157. 10.1021/ct400529k. [DOI] [PubMed] [Google Scholar]
- Burney P. R.; White N.; Pfaendtner J. Structural Effects of Methionine Oxidation on Isolated Subdomains of Human Fibrin D and AC Regions. PLoS One 2014, 9, e86981 10.1371/journal.pone.0086981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deighan M.; Bonomi M.; Pfaendtner J. Efficient Simulation of Explicitly Solvated Proteins in the Well- Tempered Ensemble. J. Chem. Theory Comput. 2012, 8, 2189–2192. 10.1021/ct300297t. [DOI] [PubMed] [Google Scholar]
- Levine Z. A.; Fischer S. A.; Shea J. E.; Pfaendtner J. Trp-Cage Folding on Organic Surfaces. J. Phys. Chem. B 2015, 119, 10417–10425. 10.1021/acs.jpcb.5b04213. [DOI] [PubMed] [Google Scholar]
- Barducci A.; Bonomi M.; Parrinello M. Linking Well-Tempered Metadynamics Simulations with Experiments. Biophys. J. 2010, 98, L44–L46. 10.1016/j.bpj.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piana S.; Laio A. A Bias-Exchange Approach to Protein Folding. J. Phys. Chem. B 2007, 111, 4553–4559. 10.1021/jp067873l. [DOI] [PubMed] [Google Scholar]
- Marelli U. K.; Frank A. O.; Wahl B.; Pietra V. La.; Novellino E.; Marinelli L.; Herdtweck E.; Groll M.; Kessler H. Receptor-Bound Conformation of Cilengitide Better Represented by Its Solution-State Structure than the Solid-State Structure. Chem. - Eur. J. 2014, 20, 14201–14206. 10.1002/chem.201403839. [DOI] [PubMed] [Google Scholar]
- Tomassi S.; D’Amore V. M.; Di Leva F. S.; Vannini A.; Quilici G.; Weinmüller M.; Reichart F.; Amato J.; Romano B.; Izzo A. A.; Di Maro S.; Novellino E.; Musco G.; Gianni T.; Kessler H.; Marinelli L. Halting the Spread of Herpes Simplex Virus-1: The Discovery of an Effective Dual αvβ6/αvβ8 Integrin Ligand. J. Med. Chem. 2021, 64, 6972–6984. 10.1021/acs.jmedchem.1c00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichart F.; Maltsev O. V.; Kapp T. G.; Räder A. F. B.; Weinmüller M.; Marelli U. K.; Notni J.; Wurzer A.; Beck R.; Wester H. J.; Steiger K.; Di Maro S.; Di Leva F. S.; Marinelli L.; Nieberler M.; Reuning U.; Schwaiger M.; Kessler H. Selective Targeting of Integrin αvβ8 by a Highly Active Cyclic Peptide. J. Med. Chem. 2019, 62, 2024–2037. 10.1021/acs.jmedchem.8b01588. [DOI] [PubMed] [Google Scholar]
- Maltsev O. V.; Marelli U. K.; Kapp T. G.; Di Leva F. S.; Di Maro S.; Nieberler M.; Reuning U.; Schwaiger M.; Novellino E.; Marinelli L.; Kessler H. Stable Peptides Instead of Stapled Peptides: Highly Potent αvβ6-Selective Integrin Ligands. Angew. Chem., Int. Ed. 2016, 55, 1535–1539. 10.1002/anie.201508709. [DOI] [PubMed] [Google Scholar]
- Kapp T. G.; Di Leva F. S.; Notni J.; Räder A. F. B.; Fottner M.; Reichart F.; Reich D.; Wurzer A.; Steiger K.; Novellino E.; Marelli U. K.; Wester H. J.; Marinelli L.; Kessler H. N -Methylation of isoDGR Peptides: Discovery of a Selective α5β1-Integrin Ligand as a Potent Tumor Imaging Agent. J. Med. Chem. 2018, 61, 2490–2499. 10.1021/acs.jmedchem.7b01752. [DOI] [PubMed] [Google Scholar]
- Nieberler M.; Reuning U.; Reichart F.; Notni J.; Wester H. J.; Schwaiger M.; Weinmüller M.; Räder A.; Steiger K.; Kessler H. Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers 2017, 9, 116 10.3390/cancers9090116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig B. S.; Kessler H.; Kossatz S.; Reuning U. RGD-Binding Integrins Revisited: How Recently Discovered Functions and Novel Synthetic Ligands (Re-)Shape an Ever-Evolving Field. Cancers 2021, 13, 1711 10.3390/cancers13071711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli L.; Gottschalk K. E.; Meyer A.; Novellino E.; Kessler H. Human Integrin αvβ5: Homology Modeling and Ligand Binding. J. Med. Chem. 2004, 47, 4166–4177. 10.1021/jm030635j. [DOI] [PubMed] [Google Scholar]
- Kapp T. G.; Rechenmacher F.; Neubauer S.; Maltsev O. V.; Cavalcanti-Adam E. A.; Zarka R.; Reuning U.; Notni J.; Wester H. J.; Mas-Moruno C.; Spatz J.; Geiger B.; Kessler H. A Comprehensive Evaluation of the Activity and Selectivity Profile of Ligands for RGD-Binding Integrins. Sci. Rep. 2017, 7, 39805 10.1038/srep39805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E.; Jarvelainen H.. Methods for Treating Pancreatic Cancer and Other Solid Tumors. WO Patent WO2021226148A1, 2021.
- Quigley N. G.; Tomassi S.; Di Leva F. S.; Di Maro S.; Richter F.; Steiger K.; Kossatz S.; Marinelli L.; Notni J. Click-Chemistry (CuAAC) Trimerization of an αvβ6 Integrin Targeting Ga-68-Peptide: Enhanced Contrast for in-Vivo PET Imaging of Human Lung Adenocarcinoma Xenografts. ChemBioChem 2020, 21, 2836–2843. 10.1002/cbic.202000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the input files and trajectory data sets are published on a public Zenodo folder and freely available at the following link: https://zenodo.org/record/8089457.