

Abstract
Cis-regulatory elements (CREs) are important sequences for gene expression and for plant biological processes such as development, evolution, domestication, and stress response. However, studying CREs in plant genomes has been challenging. The totipotent nature of plant cells, coupled with inability to maintain plant cell types in culture and the inherent technical challenges posed by the cell wall have limited our understanding of how plant cell types acquire and maintain their identities and respond to the environment via CRE usage. Advances in single-cell epigenomics have revolutionized the field identifying cell-type-specific CREs. These new technologies have the potential to significantly advance our understanding of plant CRE biology, and shed light on how the regulatory genome gives rise to diverse plant phenomena. However, there are significant biological and computational challenges associated with analyzing single-cell epigenomic datasets. In this review, we discuss the historical and foundational underpinnings of plant single-cell research, challenges and common pitfalls in analysis of plant single-cell epigenomic data, and highlight biological challenges unique to plants. Additionally, we discuss how the application of single-cell epigenomic data in various contexts stands to transform our understanding of the importance of CREs in plant genomes.
Introduction:
Multicellular eukaryotes arose due to evolutionary pressures driving the sub-functionalization of cells into dedicated roles, allowing organisms to have functions that are more advanced than its cellular components. Cellular specialization results from differential use of the genome between cell types, which is partly driven by variable use of cis-regulatory elements (CREs) that are important for gene transcription and silencing. In plants, cell types have evolved specialized metabolisms and unique cell wall morphologies that link form to function enabling cells to fill their structural and physiological roles in planta (Alberts et al., 2002). Plant cell structures fascinated early plant scientists; the observation of microscopic cell wall ‘cages’ within onion leaves led to Robert Hooke to develop the term ‘cell’ and variable cell wall morphologies were first used to classify plant cell types (Hooke, 1665). However, plant cell-type definitions have been continually refined by sequential scientific breakthroughs such as the increased resolution of microscopy and advances in molecular genetic techniques. Advances in single-cell genomics allow measurement of cell-type-specific transcripts and CRE chromatin accessibility, which is bettering understanding of the gene regulatory networks present in cells, and how they impact all manner of phenomena in planta. However, there are numerous technical and biological challenges associated with single-cell genomics data that must be overcome before these questions can be addressed.
In this perspective, we discuss the historical ways plant cell types have been described and how cell-type definitions have evolved. We examine how cell types have been defined genetically, and how this identified marker genes critical for cell-type function. We discuss current biological and technical challenges associated with the single-cell genomics identification of cell-type-specific regulatory sequences. Lastly, we highlight how emerging technologies will overcome some of these challenges, improving the ability to study the cellular context in which molecular processes affect plant phenotypes.
Plant Cell Types - A Historical Perspective:
Scientists have been describing the cellular make-up of plants for centuries. Plant cell biology began with the advent of microscopy and histology, with early descriptions of stomata and guard cells dating back to 1671 (“Anatomie des plantes,” n.d.). This research laid the groundwork of plant anatomy and established early models of plant cell types. Cells were classified based on the structure of their cell walls, with parenchyma having thin non-lignified cell walls, collenchyma having thick non-lignified cell walls, and sclerenchyma having lignified cell walls (Imperatorskai︠a︡ akademīi︠a︡ nauk (Russia) et al., 1868). Although critical, these early descriptions of plant “cell types” had limited resolution and overlooked cells with unique structure and function. Advances in microscopy in the following centuries facilitated more accurate descriptions of plant cell types. Increasing microscopy resolution produced descriptions of cell-type subclasses within the classical definitions of parenchyma, collenchyma and sclerenchyma (Leroux, 2012). This led to the first described companion cells, sieve tube elements, and bundle sheath cells (Strasburger, 1888; Wilhelm, 1880). These newly described cell types were not just categorized but were described in their developmental and gross anatomical contexts within the plant. Foundational work by Esau and Sharman combined microscopy with serial sectioning experiments, describing vascular development in multiple plant species (Esau, 1954, 1939; Sharman, 1942). This combination of techniques revealed the cellular patterns in mature tissue, and how these arrangements emerge from their cellular precursors (Esau, 1943; Sharman, 1942). Further work focused on the meristem, a collection of plant stem cells that divide to produce new growth. Tracking cellular division and maturation from meristems provided an early understanding of plant cell-type differentiation, revealing how anatomical patterns are established by development (Evert et al., 2006).
Early on it was understood that DNA encoded the genetic instructions which give rise to plant form, but our understanding the genetic processes that controlled cell-fate decisions were limited. Initial genetic analysis exploited the clonal development of mutant sectors with visible phenotypes. In brief, these studies used mutagens, like X-rays, to induce somatic mutations in progenitor cells to determine the cells contributions to organismal phenotype. In plants, mutant based studies demonstrated that manipulation of DNA sequence could radically change plant phenotypes and cell fates(Hake and Freeling, 1986; Sinha and Hake, 1990). For instance, stable mutagenesis gave rise to liguleless-1 mutants that have radically different leaf morphology with a misplaced ligule on the margin of the leaf blade (Becraft and Freeling, 1991). However, these studies were limited in their capacity to identify the sequence causing these morphological alterations. However, this changed with advances in molecular genetic techniques that allowed for pinpointed manipulation of plant DNA.
In the 1990’s molecular genetic techniques allowed fine scale alteration of DNA and inquiry into the genetic processes driving the emergence of specific cell types. Early genetic screens found that cell identity could be ablated by single-gene knockouts. One excellent example is shortroot (shr) in Arabidopsis thaliana (Benfey et al., 1993). In shr mutant plants, root endodermis cells fail to form, resulting in significantly stunted root growth and illustrating that SHR is indispensable for endodermis cell-type identity. Further analysis of SHR revealed it is a mobile transcription factor critical for cell fate differentiation (Helariutta et al., 2000). The identification of SHR, and and other transcription factors that defined cell identity generated questions aimed at how cell fates were encoded within the genome. These questions remain the subject of active investigation, with ongoing experiments continually offering deeper insights into the molecular events that drive plant cell-fate decisions.
The Genetic Underpinnings of Plant Cell Identity:
Plant cell development and function result from a complex interplay of genes responsible for determining cell fate and maintaining cellular identity. Identification of key developmental regulators, like SHR, demonstrated that the development of entire cell lineages depended on the expression of a few genes. Determining how and where these essential “marker” genes of cell identity were expressed became a central question in plant genetics. Subsequently, molecular genetic approaches such as mutagenesis screens, and reporter gene assays, were developed to assess the cellular context in which these marker genes were expressed. These advancements resulted in the identification of many other genes critical in cell-type identity. For example, GLABRA1 (GL1) in A. thaliana controls trichome fate, as gl1 null plants generated by T-DNA insertion had no trichomes on the leaf and stem (Figure 1A) (Herman and Marks, 1989; Oppenheimer et al., 1991). Despite establishing the necessity of GL1 for trichome formation, this finding did not elucidate its expression pattern or how GL1 facilitated trichome development. This knowledge gap led to the creation of promoter reporter lines, in which a gene’s transcriptional regulatory sequences (promoters and CREs) are fused to a reporter (e.g., GUS, GFP) to illuminate where and when the gene is expressed (Birnbaum et al., 2003; Brady et al., 2003; Helariutta et al., 2000; Stadler et al., 2005). In GL1 reporters, expression was found to change throughout development; in early development, GL1 is expressed throughout the early leaf primordia, but, as the epidermis matures, only trichomes precursors maintain high GL1 expression (Kirik et al., 2001; Larkin et al., 1994; Oppenheimer et al., 1991). Research into genes crucial for cell development expanded reporter methods by combining cell-type reporters with protoplast isolation to isolate cell populations and conduct genome-wide identification of transcription factors associated with specific cell types (Birnbaum et al., 2005; Toufighi et al., 2005). Application of these genome-wide assays identified genes crucial for the development of particular cell types. Presently, cell-type-specific genetic inquiry in plants has the potential to be significantly enhanced through single-cell methodologies, allowing for refined discrete measurement from individual cells empowering our understanding of plant cell fate decisions.
Figure 1: Plant cell-type markers define either unique developmental, metabolic or physiological states.
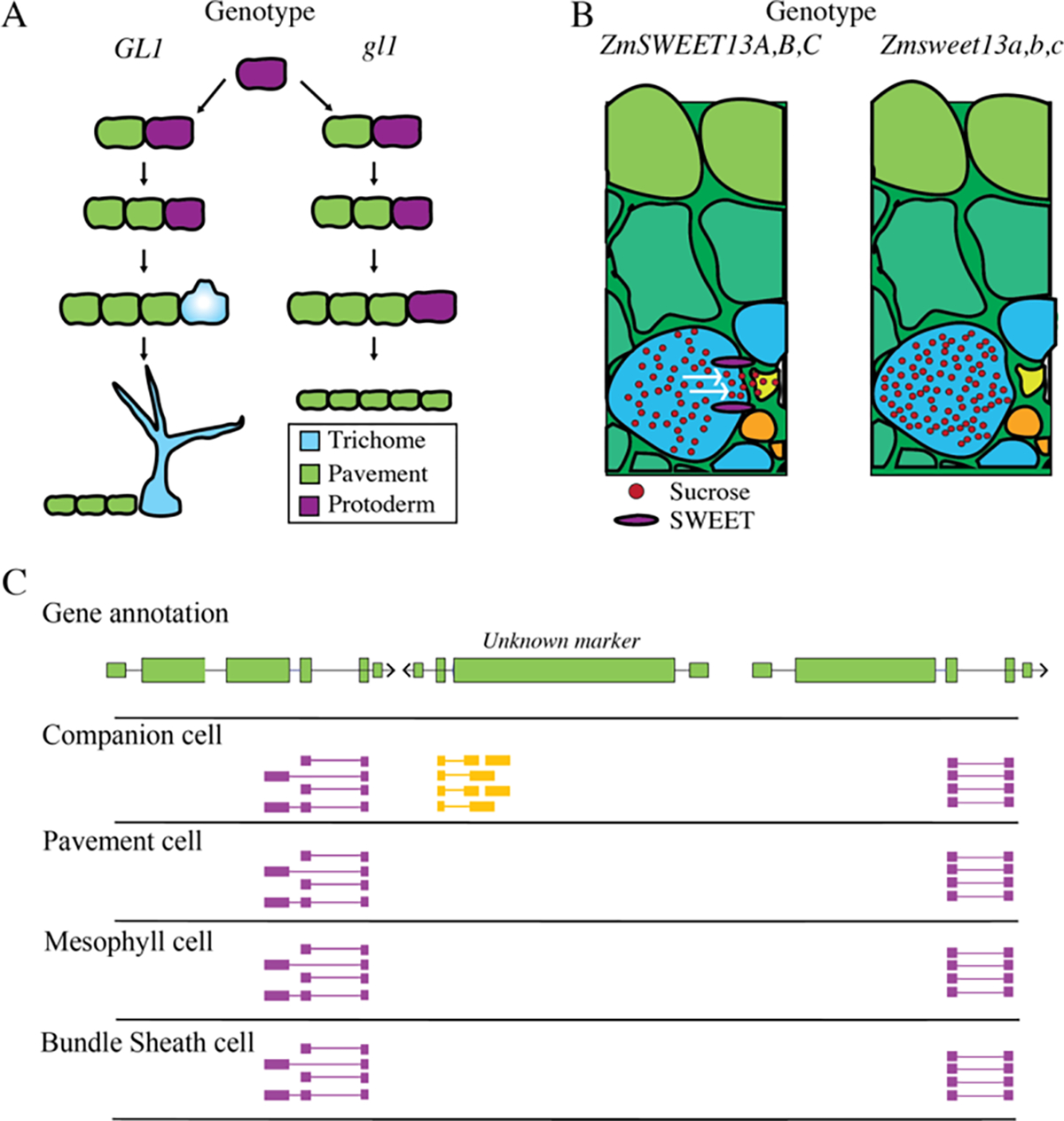
A) Model for proper function of GLABRA1 in A. thaliana (GL1), which promotes trichome development (left). Knockouts of gl1 removes the capacity for protoderm cells to differentiate into trichomes, generating additional pavement cells (right). B) ZmSWEET13s (purple) are required for transport of sucrose from bundle sheath cells into the vasculature in Zea mays. zmsweet13 knockouts raise sucrose concentrations in bundle sheath cells. C) Hypothetical example of a de novo discovered marker gene identified by single-cell RNA-seq. Expression of the de novo discovered marker, Unknown, is limited to companion cells, as opposed to pavement cells, mesophyll cells, and bundle sheath cells. Single cell RNA-seq reads are colored by their strand, with purple reads representing the positive strand and yellow reads representing the negative strands.
While genes important in the development of cell types are critical to our understanding of how cell types differentiate, they do not reveal much about plant cell-type function. This has led to researchers looking for genes which are important to the function of mature cell-types. For instance, genes such as SUGARS WILL EVENTUALLY BE EXPORTED TRANSPORTER 13 (ZmSWEET13), a sucrose transporter, is expressed specifically bundle sheath cells and phloem parenchyma (Bezrutczyk et al., 2021, 2018). Knockouts of ZmSWEET13 impair phloem loading increasing sucrose concentrations in leaves (Figure 1B). Although not required for abaxial bundle sheath cell development, ZmSWEET13 represents a key gene required for cell-type-specific function. Similarly, Arabidopsis SUCROSE-PROTON SYMPORTER 2 (SUC2) drives sieve element sucrose loading through companion cell-specific expression (Stadler and Sauer, 1996). suc2 knockout plants are stunted due to impaired sucrose transport, but companion cell identity is unaffected (Gottwald et al., 2000). Genes such as SUC2 and ZmSWEET further our understanding of the genetic partitioning of functions between plant cell types. This genetic division enhances our perspective of what constitutes a plant cell type, transitioning from definitions based on histology, to those based on gene expression and function of discrete genetic loci. Although finding cell-type-specific functional genes is valuable, their identification is generally done by investigating a single gene at a time, requiring significant investment of time and resources. Single-cell genomics provides an opportunity to discover additional genes important to function of specific cell types on a genome-wide scale, across all cell types sampled at a single time. This influx of information will quickly evolve our understanding of cell types from a few key loci, to combinations of genes critical for both development and function.
Single-cell genomics allows measurement of chromatin states and mRNAs in thousands of individual cells (Buenrostro et al., 2015b; Cusanovich et al., 2015; Jaitin et al., 2014). Plant single-cell genomics is especially exciting given the lack of cell-type-specific genomic measurements outside of model plant systems. The information-richness and high-complexity of single-cell datasets are useful because it allows for a detailed understanding of how different cell types utilize the genome. However, single-cell technologies remove cells from the sampled tissue, erasing any knowledge about position or identity, and complicating the identification of each cell’s cell type. Therefore, annotation relies on molecular marker genes to reveal cell identity post hoc. This annotation is confounded by the gradient of transitional cell identities that underpin differentiation. For instance, in A. thaliana guard cell differentiation from protoderm involves five state transitions, necessitating additional markers to accurately delineate cellular states (Chen et al., 2020). These transitory states make having well-established developmental marker genes critical to accurate annotation of single-cell datasets. For this reason, the first plant single-cell RNA-seq (scRNA-seq) analysis was conducted on A. thaliana roots because root cell types have well described genes associated with specific cell types and developmental stages (Ryu et al., 2019; Shulse et al., 2019). Once single-cell datasets are accurately annotated, they can be leveraged in powerful ways. Testing for differentially expressed genes in annotated cell types allows for the identification of novel genes potentially critical in proper cell-type function. One scRNA-seq A. thaliana study used annotated root cell types to discover 50 genes with cell-type-specific expression patterns (Figure 1C) (Zhang et al., 2019). This “de novo “ discovery of cell-type-specific genes provides a wealth of candidate genes to target and study, which will further reveal their importance in specific cell types of interest. Single-cell genomics will increase the speed of cell-type-specific gene identification, improving our understanding of which loci are critical for proper cell-type function and development in plants.
The Regulatory Genome Specifies How Cell types are Established
Although scRNA-seq will improve our knowledge of cell-type-specific gene expression, our understanding of the processes driving these expression patterns remains poor. Pairing of single-cell technologies with assays identifying the regulatory genome stand to greatly enhance our understanding of how the genome can regulate the expression of both developmental and functionally important genes. Cell-type-specific expression is the result of different cells using the same genetic blueprint encoded in the genome in different ways. Cell fates are determined by the interpretation, enhancement, or silencing of instructions encoded in DNA which are driven by CREs (Andersson et al., 2015). CREs are non-coding sequences of DNA composed of transcription factor (TF) binding sites. TFs bind CREs within nucleosome depleted sequences, to recruit co-factors, remodel chromatin, and regulate gene transcription (Lai et al., 2019). This cis-regulation has implications on plant development, environmental response, and evolution (Cramer, 2019).
CREs often work in concert and are then referred to as cis-regulatory modules (CRMs) (Figure 2) (Schmitz et al., 2022; Shlyueva et al., 2014). CRMs are further subdivided as “enhancers,” or “silencers,” based on the ability to recruit co-activators or co-repressors to genes (Gisselbrecht et al., 2020; Pang and Snyder, 2020; Shlyueva et al., 2014). Identification of CRMs genome wide is routinely performed with assays that measure accessible chromatin environments, as these are the regions that are open to TF binding. Methods such as DNase-seq, MNase-seq, as well as FAIRE-seq have been used to study CRMs genome wide (Boyle et al., 2008; Giresi et al., 2007; Johnson et al., 2006). Currently, the most widely adopted method to investigate accessible chromatin is Assay for Transposase-Accessible Chromatin followed by sequencing (ATAC-seq) (Buenrostro et al., 2015a). In brief, ATAC-seq works by utilizing a hyperactive Tn5 transposase to directly insert sequencing adapters into accessible chromatin regions of DNAs (Figure 2). The fragments generated are then amplified, sequenced, aligned to the genome, and areas more accessible than genomic background are computationally identified (Figure 2) (Yan et al., 2020). These peaks, named accessible chromatin regions (ACRs), are well accepted proxies for CRMs, and thus collections of CREs (Bajic et al., 2018; Lu et al., 2017).
Figure 2: Deciphering the regulatory genome with ATAC-seq.

Top: Gene annotation TF Motifs: Transcription factor binding motifs. Colors represent different motifs. Motifs occur in clusters referred to as cis-regulatory modules (CRMs). Gray arrow highlights a specific TF motif acting as a CRE. ATAC-seq: Example of aligned reads from an ATAC-seq experiment. Note that reads align heavily to CRM regions. Depth: Histograms of the above read depth. ACRs: Identified accessible chromatin regions from the read depth above. Chromatin looping: An example of how ACRs do not always operate on the closest gene. The line represents chromatin interaction between two ACRs.
With the widespread adoption of ACR identification, numerous discoveries have been made about the regulatory nature of DNA in plant genomes. For instance, it has recently been revealed that ACRs frequently operate on genes >50 kilobases away in plants with large genomes (Ricci et al., 2019). Additionally, variable ACR usage has been implicated in biotic and abiotic stress responses, providing more insights into how the genome tunes expression to the environment. (Han et al., 2020; Raju, 2020; Zeng et al., 2019; Zhou et al., 2022). However, ACRs provide no information about whether these regions are enhancing or repressing transcription. This can be predicted by overlaying ACRs with ChIP-seq data which measures the histone modifications nearby (Lu et al., 2019; Oka et al., 2017; Ricci et al., 2019). ACRs that are active are flanked by histone acetylation, whereas those that are actively repressing a target gene are flanked by histone methylation and Polycomb silencing (Lu et al., 2019; Ricci et al., 2019). Recently, the ability to apply ATAC-seq to single cells (scATAC-seq) was developed, allowing for cell-type-specific ACR identification and measurement (Buenrostro et al., 2015b; Cusanovich et al., 2015).
Single Cell ATAC-seq, Emerging Paradigms and Tangible Value
ScATAC-seq has revealed differential usage of ACRs in cellular identity and development in plant models (Dorrity et al., 2020; Farmer et al., 2021; Marand et al., 2021). Although the application of scATAC-seq techniques to plants stands to teach us much about cis-regulatory biology, implementing these techniques are non-trivial and come with a series of caveats and challenges. The ability to deconvolute cellular heterogeneity in plant tissue allows for the identification of cell-type-specific ACRs, which can be used to identify TFs and CREs important to cell function.(Marand et al., 2021). Intriguingly, scATAC-seq also offers a method to study developmental trajectories within cell types. Key genes or CREs that operate differently through development can be identified by ordering cell lineages from progenitor to mature cell type (Nelms and Walbot, 2019; Trapnell et al., 2014). This “pseudo time” method was applied to root hair development in Oryza sativa, as well as phloem companion cell development in Z. mays root. In O. sativa, pseudotime analysis found 13,000 ACRs and 3,000 genes important in the transition into root hair cell-type identity (Zhang et al., 2021). In Z. mays, it was found that as cells differentiate from quiescent center cells to phloem companion cells the fractions of ACRs which were accessible decreased significantly (Marand et al., 2021). Pseudotime analyses are just the beginning as application of scATAC-seq to plants will reveal roles of cis-regulation in evolution, stress responses, and adaptation. While exciting, the analysis and annotation of these datasets is computationally challenging and requires awareness of current limitations.
The computational challenges associated with scATAC-seq data analysis are primarily due to the low number of Tn5 integration events per cell. For example, upwards of 99% of the chromatin accessibility measurements genome wide are often missing from any particular cell (Buenrostro et al., 2015b). This data scarcity has significant ramifications in scATAC-seq analysis. The first step of scATAC-seq analysis is isolating high quality cells. One way of doing this is by “pseudo-bulking,” which mimics bulk ATAC-seq by aggregating the reads from all nuclei, to identify peaks (ACRs) (Chen et al., 2019). Then broken nuclei are removed by estimating the Fraction of Reads in Peaks (FRiP) per nucleus, and removing nuclei with Tn5 integration events below a FRiP threshold (generally >.25). Next, doublets, which are instances where two cells are mistakenly sequenced as one are removed by comparing them against an in-silico generated doublet set of cells (Wolock et al., 2019). Based on the single cell technologies used, the top 5–10% of cells with the highest “doublet score” are removed. The next steps annotate similar cells into cell types. Annotation starts by generating a binary matrix of Tn5 insertions in ACRs by cells, which is fed into dimensionality reduction algorithms. These algorithms, such as singular value decomposition (SVD) or principal component analysis (PCA), cluster cells into similar groups by identifying correlated features (ACR presence/absence), which reveal underlying patterns and relationships among the cells (Figure 3A–B). The resulting principal components, or meta-features, represent high-dimensional data (ACR accessibility) in low-dimensional space.. Cell proximity in this low dimensional is a proxy for cell relatedness, either biological or technical (Figure 3B). Presently, either Uniform Manifold Approximation and Projection (UMAP) or t-Distributed Stochastic Neighbor Embedding (t-SNE) are used to visualize scATAC-seq data. These techniques plot cells in 2D, while trying to preserve the high dimensional space computed above (Figure 3B) (Maaten and Hinton, 2008; McInnes et al., 2020). One should not make biological conclusions about relative distance and space between cells, as recent evidence points to the inherent flaws in this approach (Chari et al., 2021). For instance when using three dimensional datasets with known spatial relationships between points, t-SNE or UMAP processing scrambles the relative distance between points, indicating that the 2D distances generated are artifactual (Chari et al., 2021). From this embedding, discrete cell clusters are assigned using community detection methods such as Louvain or Leiden algorithms (Blondel et al., 2008; Waltman and van Eck, 2013). In brief, these methods work by trying to identity clusters of cells in high-dimensional space, which maximizes the differences between groups and minimizes the differences within groups based off a given parameter set. Clusters are then analyzed with the assumption that they are representative of roughly homogenous cell types. Annotating clusters to a cell type involves approximating gene expression of cell-type markers by summing gene body and promoter chromatin accessibility (Cusanovich et al., 2018a). This approximation, while valuable, is imperfect, as chromatin accessibility does not always correlate with gene expression (Figure 3D). Based on the specific chromatin accessibility patterns of known marker genes, clusters are assigned cell types (Figure 3C). The clustering and annotation of cell types remains one of the most time consuming and difficult steps in scATAC-seq analysis. Current heuristic methods rely on user based decisions that are often difficult to replicate (Gibson, 2022). As the field matures, more consistent annotation metrics are needed to ensure proper and timely cell type assignment.
Figure 3: Schematic of analysis paradigms and challenges of single-cell ATAC-seq data.

A) An example binary matrix being transformed into a UMAP by means of SVD where rows are ACRs identified from a bulked dataset and columns are cells. The input matrix is binary if a cell either has a Tn5 integration event in that ACR or not B) Left) An example of a good UMAP embedding where Tn5 insertion density is not driving the clustering of cells. Right) An example of a poor UMAP embedding where technical artifacts due to Tn5 insertion density are driving the clustering C) An instance where chromatin accessibility of a gene (Gene Acc.) does not align with expectations based on gene expression (Gene Expression). Here, the upstream region of the gene depicted could harbor a silencer that recruits TFs to repress its target gene. D) Top) An example of where the UMAP embedding correlates well with the chromatin accessibility of multiple marker genes for a specific cell type. Bottom) Example of a poor UMAP embedding where markers do not agree with each other. Whether this is due to poor markers or a poor selection of features to generate the embedding requires additional inquiry. E) Challenges associated with the assignment of single-cell ACRs. Whether the middle ACR is cell-type specific or restricted to a few key cell types is challenging to determine. This potentially reflects issues in annotation, or that related cell-types share similar chromatin environments. The assignment of cell-type specific ACRs is non-trivial and requires careful considerations by the researcher of both biological and technical challenges.
ScATAC-seq analysis is a deeply iterative process. For instance, selecting different ACRs to include in dimensionality reduction can drastically alter cluster membership and generate different results. This requires researchers to try different selections of ACRs to find a set that reduces technical artifacts but maximizes biological interpretation. Additionally, technical artifacts can have significant effects on annotations and interpretation of results. For example, cells with a high density of Tn5 insertion events per cell can cluster together, thus the underlying embedding doesn’t represent one of biological variation, but technical (Figure 3B). Technical artifacts can be even more misleading, with cells being assigned specific clusters due to the lack of data, rather than the presence of genuine differences.
Once annotations are finalized, cell-type-specific ACRs are identified. Combining cells of the same cell type via “pseudo-bulking” allows for the robust identification of ACRs for individual cell types (Cusanovich et al., 2018b; Domcke et al., 2020). This deconvolution of tissue-level chromatin accessibility to cell-type resolved accessibility is where the power of single cell lies. While identifying ACRs from cell-type-level data is straightforward, classifying these ACRs as cell-type-specific or broadly accessible is challenging and is heavily impacted by the statistical approach chosen (Figure 3E). Making this categorization more opaque is cell types which share developmental origins often have similar chromatin accessibility patterns (Cusanovich et al., 2018a; Domcke et al., 2020). This leads to an additional class of ACRs that are cell-type-restricted or limited in their chromatin accessibility to a few cell types. Recent plant scATAC-seq studies have found between 23–27% cell-type-restricted ACRs in given species (Marand et al., 2021; Zhang et al., 2021). However, the number and proportion of ACRs that are cell-type specific is unknown, and being established in model systems with more exhaustive sampling (Chen et al., 2018; Domcke et al., 2020). Whether these cell-type-specific ACRs are critical to cell-type function is uncertain and requires follow-up molecular genetics studies. Finally, scATAC-seq provides exciting opportunities to begin deciphering both cis and trans regulators of the genome. Recent studies have shown the ability to link TFs with their likely binding sites in a cell-type-specific manner, by correlating the chromatin accessibility of transcription factor gene bodies with the accessibility of their corresponding binding sites (Marand et al., 2021). This allows for the identification of cell-type-specific gene regulatory networks which have been long elusive. While the computational workflow and challenges labeled here may seem daunting, rigorous data analysis avoids many of these pitfalls. However, it should be noted that these specific computational challenges aren’t the only issue. Quirks associated with evolution, genome structure, and the unique ways plant cell-type identity can be modified also need to be considered.
Biological Challenges Associated with the Analysis of scATAC-seq in Plants
While the computational challenges associated with scATAC-seq are laid out and addressable, certain unique features of plant biology complicate analysis. Variable genome sizes, rapid changes in gene function caused by molecular evolution, and the totipotent nature of plant cells all alter the interpretation of plant scATAC-seq data. However, while analytically challenging, these features offer unique opportunities to study CREs and their relationship to plant biology generally.
Significant variations in genome size can add additional hurdles to analyzing scATAC-seq data. For instance, genome size affects the use of gene proximal chromatin accessibility as a proxy for gene expression. In compact genomes, the reduced proximity between transcriptional start sites (TSSs) means ACRs often encompass two promoters, which convolutes correlating chromatin accessibility with gene expression (Figure 4A). This is in stark contrast to larger genomes which result from the expansion of intergenic and intronic gene space often as a result of increased transposon load (Lee and Kim, 2014; Lu et al., 2019). This size expansion moves CRMs important for gene expression further upstream of the TSS, increasing the prevalence of gene-distal ACRs (Lu et al., 2019; Oka et al., 2017; Ricci et al., 2019; Zhao et al., 2018). Linking these distal ACRs to their target genes is challenging and requires additional data from proximity-ligation based methods. Hi-C is the most commonly used, as it captures chromatin interactions ranging from 1 kb to >100kb depending on the experimental setup and sequencing depth. (Figure 2) (Eagen et al., 2015; Lieberman-Aiden et al., 2009; Mifsud et al., 2015). However, Hi-C remains restricted to bulk tissues, limiting the detection or confirmation of gene-ACR interactions in rare cell types. Predictions about chromatin contacts can be made from scATAC-seq itself, but requires further experimentation for validation (Marand et al., 2021; Pliner et al., 2018). Genome size variation necessitates adapting analysis strategies on a per-genome basis, impeding the standardization of scATAC-seq analysis between species.
Figure 4: Biological challenges in single-cell ATAC-seq data take many forms and unique situations.

A) The gene SHORT SUBUNIT (SSU) is known to have specific function in bundle sheath cells in Z. mays. However, SSU may not behave as a useful marker for annotation via scATAC-seq due to the proximity of its promoter to the start of an uncharacterized gene. B) Leaf cross sections of two species, Z. mays, and O. sativa. Cell types are color coded, and important marker genes are labeled in gray circles. Red lines point to the cell types these genes are active in. Although SHR1 is expressed in bundle sheath and vascular cells in Z. mays, it is not found in O. sativa bundle sheath cells. C) Leaf laid out from proximal to distal ends with developmental gradient overlaid on top, with the oldest cells being at the tip and newest cells at the base. It is currently unknown whether different regions of the leaf might have different regulatory chromatin environments and subfunctionalization (bottom). D) Leaf cross sections where colors indicate cells originating from either parenchyma (ground tissue) or procambium. Left) Normal development Right) Abnormal development with position dependent effects altering the origin of a mesophyll cell derived from the procambium.
Although variation in plant genome size complicates scATAC-seq, the lack of high-quality markers for plant species remains the biggest challenge in the annotation and analysis of single-cell ATAC-seq datasets. In non-model plants, the specificity of most markers is inferred from the cell-type specificity of orthologs in model plants. However, it is known that gene expression changes rapidly due to molecular evolution (Hill et al., 2020). Gene duplication followed by neo-functionalization, whole genome duplications rewiring large scale expression patterns, and rapid gene family expansion are a few of the many ways molecular evolution can reshape gene function, and expression (Birchler and Yang, 2022; Hughes et al., 2014; Panchy et al., 2016). Even in a relatively short evolutionary time span of 65–70 million years key developmental genes can shift their cell-type expression context, complicating their use in a cross species context (Hughes and Langdale, 2022). For example, SHR has different cell-type specificity in Z. mays and O. sativa leaves; in Z. mays, expression of ZmSHR1 is limited to vasculature and bundle sheath cells, whereas the O. sativa ortholog, OsSHR2, has limited vasculature expression, and is absent from bundle sheath cells (Figure 4B) (Schuler et al., 2018). Due to the unreliability of individual markers, non-model systems need to use sets of markers to annotate cell types in order to Minimize incorrect cell annotation. However, to date the number of markers per cell type is limited, restricting the ability to apply single cell techniques to non-model plants.
Plants cells have a unique relationship between cell identity by descent, and cell position in the plant. Plant cell fates are not genetically hardwired based off precursors. For instance, although plant cell types generally develop in predictable lineages, such as the procambium giving rise to the vasculature, exceptions can occur. Instead, the location of a plant cell during development can have greater impacts on cell fate than their stem cell niche of origin (Reinhardt et al., 2003). This has been shown in Z. mays, where the mesophyll cells neighboring the bundle sheath lineage may be derived from procambial cells, or ground meristem cells (Figure 4D) (Esau, 1943; Langdale et al., 1989; Sharman, 1942). This position-dependent effect is well documented for vasculature and epidermal cell types in species including cotton, tobacco, and sunflower (Dolan and Poethig, 1998; Esau, 1954; Hung et al., 1998; Jegla and Sussex, 1989; R. S. Poethig and Sussex, 1985; R.S. Poethig and Sussex, 1985). This poses a challenge, as these cells which have undergone position dependent effects are likely to cluster with cells that share the same precursor, and not with cells with the same terminal identity. This makes annotation more difficult and increases the heterogeneity in identified cell types. Finally, since these events are rare, isolating and studying these populations is challenging, but could pose a valuable study system to understand the role of how variable precursors alter the chromatin environment of differentiated cells.
Finally, the gradient nature of plant growth provides additional challenges. Plant organs grow in a gradient of development, with younger cells found closer to the dividing meristem, and older cells further away. Continual organogenesis and development results in gene expression profiles that are dependent on the section sampled within an organ (Figure 4C). In Z. mays this developmental progression yields differences in expression of key carbon metabolism enzymes at different sections of the leaf (Li et al., 2010; Pick et al., 2011; Wang et al., 2013). Whether these different sections of the leaf, and the cell types found within, constitute different cell types or specific sub-functionalization is up for debate, and further complicates placing cells into discrete categories (Zeng, 2022). This heterogeneity has already been hinted at in some studies. A combinatorial scATAC-seq and scRNA study of A. thaliana roots, found unique genetic and epigenomic markers in three different clusters of endodermal cells, illustrating that discrete sub-functionalization of may happen within previously described cell types (Dorrity et al., 2020). The extent to which these clusters represent unique sub-functional cell types remains open and requires further exploration.
The Age of Single Cell Regulatory Genomics:
scATAC-seq enables the genome-wide investigation into the function and importance of plant cell-type-specific CREs. Although we can now identify cell-type-specific CREs in plant genomes, our understanding of how these regions interact with the coding genome is still quite limited. Leveraging intra- and inter-genetic diversity, along with treatment conditions, stands to greatly improve our understanding of CREs in plant biology and their role in responding to environmental stimuli, population adaptation and diversity, as well as reveal their importance over evolutionary time.
Performing scATAC-seq on a phenotypically diverse intra-specific population will clarify the influence of genetic CRE variation on phenotypes with cell-type resolution. Genetic variation in regulatory sequences can result in species adaptation to novel environments in both plant and animal systems (Cleves et al., 2014; Studer et al., 2011; van der Burg et al., 2020; Wucherpfennig et al., 2022). In plants, CRE variation in the flowering time gene CONSTANS underlies flowering time diversity in natural accessions of A. thaliana (Rosas et al., 2014). However, most studies addressing CRE genetic variation lack cell-type resolved data and therefore may overlook genetic variance in rare cell-type CREs that underpins local adaptation. Combining quantitative genetic approaches, like genome-wide association (GWA), with scATAC-seq, phenotypic associations and chromatin accessibility variation can be correlated, potentially identifying the CREs, and cell types, underpinning trait variation within distinct populations (Das et al., 2022). Although a nascent area of study, the combination of scATAC-seq and population diversity may reveal how CRE genetic diversity alters the regulatory epigenome to shape species adaptation.
Beyond applying scATAC-seq to single species populations, comparative genomics focused on diverse species offers the opportunity to examine plant CRE evolution at a deeper timescale. Plant genomes exhibit a high rate of structural variation and sequence turnover as compared to animal genomes, causing rapid CRE turnover (Paterson et al., 2010). Highlighting the high rate of CRE change between even closely related species, a study comparing distal CREs between sister species Z. mays and S. bicolor, found approximately one-third of CREs were shared and accessible in the same tissue, one-third were novel to each lineage, and one-third shared sequence similarity but were not within accessible chromatin in the tissue examined (Lu et al., 2019). While CREs sequences change quickly, the gene regulatory networks they influence may be more stable. Investigating root hair cell type development in four eudicots found that although few orthologous CREs were conserved across all species, TF binding at key genes was preserved (Maher et al., 2018). Pairing comparative genomics analyses with scATAC-seq will allow investigation into the pace of CRE sequence changes in specific cell types within individual plant lineages. This approach will enhance both our understanding of plant CRE evolution and uncover conserved mechanisms underpinning plant adaptation and resilience to environmental changes.
Finally, CREs drive responses to environmental stimuli. Differential CRE usage is vital in response to disease, cold, drought, and hormonal signals (Azodi et al., 2020; Moore et al., 2022; Reynoso et al., 2019; Zou et al., 2011). One comparative genomics study examined CRE usage with a flooding treatment and identified root-specific CREs associated with flooding, which revealed shared motifs within flood-responsive CREs across four species studied, representing 123 million years of evolutionary divergence (Reynoso et al., 2019). This flooding research suggests that regulatory networks behind abiotic stress responses may be conserved for millions of years. Integrating scATAC-seq with environmental treatments will identify the CREs crucial for cell-type-specific environmental responses. Beyond discovering environmentally dynamic CREs, this approach will find the cell types with the most responsive CRE usage in different conditions, revealing which cell types drive stress adaptation. This focus on cell-type responses could have far-reaching implications for our understanding of environmental response in plants, as previous study has traditionally been restricted to organismal response.
Plant Cell Types – Definitions in Flux:
While the age of single-cell genomics stands to alter our understanding of plant cell biology, it is important to acknowledge that the definition of a cell type is in flux. In this perspective, we define a “cell-type” as a cell with unique molecular signatures, and that alteration of this signature modifies the form or function of a given cell type. However, although valuable, this definition has limitations. For instance, what is the threshold of molecular changes needed to separate related cells into distinct cell types? How many differentially expressed genes or differentially accessible CREs are needed to constitute a novel cell type? This problem becomes especially acute when trying to delineate plant cells in transitional identities, as developing plant cells exist along a continuum of maturity with few discrete stages. While the discussions surrounding plant cell-type classifications may appear semantical, it underpins real biological questions. How we define “cell types” will have real implications for biologist moving forward (Efroni, 2018; Zeng, 2022).
Despite their immense development, maturity, and anatomical differences, inevitably, knowledge of plant cell types will be compared to what we know about animal cell types. IN plants there is wide variation in the number of identified cell types, with 55 being identified in Z. mays and 180 in A. thaliana (Lee et al., 2023; Marand et al., 2021). This contrasts significantly with animals, as in mouse brains alone there exists 45 types of inhibitory neurons (Hodge et al., 2019). The definition of fewer plant cell types could be explained by more technological limitations and less intensive study than that found in animal models. Alternatively, the paucity of plant cell types may reflect real biological differences between plants and animals. Unlike mammals, plant cell divisions result in the incomplete separation of nuclei; cytokinesis ends with the deposition of a new cell wall (cell plate) that contains plasmodesmata pores that retain cytoplasmic, and endoplasmic reticulum, connections between the daughter cells (Burch-Smith and Zambryski, 2012). The interconnectedness of plant cells through plasmodesmata has large implications in plant biology and may fuel the differences between plant and animal cell types, as plant cells exist as a connected community not individuals. This interconnectedness has led some to propose a more holistic ‘organism-level’ view. Instead of focusing on cells or cell types as the biologically meaningful units of study, the organismal theory proposes to focus on the entire organism, as plant cells rarely work in isolation (Kaplan and Hagemann, 1991). However, this organism-level perspective conflicts with the severe phenotypic alterations caused by mutants that eliminate specific cell types as detailed above. In either case, single-cell (epi)genomics will reveal more about why plant cell types are less numerous than their mammalian counterparts. These techniques provide unprecedented cellular resolution, and if the lack of plant cell types is driven by past technical limitations, single-cell genomics will usher in an era of discovery wherein many new discrete plant cell types will be unveiled. Alternatively, if these new techniques confirm a relatively small number of more homogenous plant cell types, it may provide credence to the notion that plant cells should be studied as a physiological unit, highlighting the importance of intercellular cooperation in plant biology. Single-cell regulatory genomics stands to enliven plant research and provides the toolset to address these basic questions about the cell-type composition of plants.
Acknowledgments
We would like to thank KD for feedback on this perspective. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institute of Health under award number T32GM007103. Additionally this work was supported by the National Science Foundation (IOS-1856627, IOS-2026554 and MCB-2120132) to RJS.
References
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, 2002. The Plant Cell Wall. Mol. Biol. Cell 4th Ed. [Google Scholar]
- Anatomie des plantes [WWW Document], n.d. URL https://bibdigital.rjb.csic.es/records/item/13576-redirection (accessed 12.19.22).
- Andersson R, Sandelin A, Danko CG, 2015. A unified architecture of transcriptional regulatory elements. Trends Genet. 31, 426–433. 10.1016/j.tig.2015.05.007 [DOI] [PubMed] [Google Scholar]
- Azodi CB, Lloyd JP, Shiu S-H, 2020. The cis-regulatory codes of response to combined heat and drought stress in Arabidopsis thaliana. NAR Genomics Bioinforma. 2, lqaa049. 10.1093/nargab/lqaa049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajic M, Maher KA, Deal RB, 2018. Identification of Open Chromatin Regions in Plant Genomes Using ATAC-Seq, in: Bemer M., Baroux C. (Eds.), Plant Chromatin Dynamics. Springer New York, New York, NY, pp. 183–201. 10.1007/978-1-4939-7318-7_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becraft PW, Freeling M, 1991. Sectors of liguleless-1 tissue interrupt an inductive signal during maize leaf development. Plant Cell 3, 801–807. 10.1105/tpc.3.8.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfey PN, Linstead PJ, Roberts K, Schiefelbein JW, Hauser M-T, Aeschbacher RA, 1993. Root development in Arabidopsis : four mutants with dramatically altered root morphogenesis. Development 119, 57–70. 10.1242/dev.119.1.57 [DOI] [PubMed] [Google Scholar]
- Bezrutczyk M, Hartwig T, Horschman M, Char SN, Yang J, Yang B, Frommer WB, Sosso D, 2018. Impaired phloem loading in zmsweet13a,b,c sucrose transporter triple knock-out mutants in Zea mays. New Phytol. 218, 594–603. 10.1111/nph.15021 [DOI] [PubMed] [Google Scholar]
- Bezrutczyk M, Zöllner NR, Kruse CPS, Hartwig T, Lautwein T, Köhrer K, Frommer WB, Kim J-Y, 2021. Evidence for phloem loading via the abaxial bundle sheath cells in maize leaves. Plant Cell 33, 531–547. 10.1093/plcell/koaa055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Yang H, 2022. The multiple fates of gene duplications: Deletion, hypofunctionalization, subfunctionalization, neofunctionalization, dosage balance constraints, and neutral variation. Plant Cell 34, 2466–2474. 10.1093/plcell/koac076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum K, Jung JW, Wang JY, Lambert GM, Hirst JA, Galbraith DW, Benfey PN, 2005. Cell type–specific expression profiling in plants via cell sorting of protoplasts from fluorescent reporter lines. Nat. Methods 2, 615–619. 10.1038/nmeth0805-615 [DOI] [PubMed] [Google Scholar]
- Birnbaum K, Shasha DE, Wang JY, Jung JW, Lambert GM, Galbraith DW, Benfey PN, 2003. A Gene Expression Map of the Arabidopsis Root. Science 302, 1956–1960. 10.1126/science.1090022 [DOI] [PubMed] [Google Scholar]
- Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E, 2008. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, P10008. 10.1088/1742-5468/2008/10/P10008 [DOI] [Google Scholar]
- Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE, 2008. High-Resolution Mapping and Characterization of Open Chromatin across the Genome. Cell 132, 311–322. 10.1016/j.cell.2007.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady SM, Sarkar SF, Bonetta D, McCourt P, 2003. The ABSCISIC ACID INSENSITIVE 3 (ABI3) gene is modulated by farnesylation and is involved in auxin signaling and lateral root development in Arabidopsis. Plant J. 34, 67–75. 10.1046/j.1365-313X.2003.01707.x [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, Greenleaf WJ, 2015a. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide: ATAC-seq for Assaying Chromatin Accessibility, in: Ausubel FM., Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. (Eds.), Current Protocols in Molecular Biology. John Wiley & Sons, Inc., Hoboken, NJ, USA, p. 21.29.1–21.29.9. 10.1002/0471142727.mb2129s109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ, 2015b. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490. 10.1038/nature14590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch-Smith TM, Zambryski PC, 2012. Plasmodesmata Paradigm Shift: Regulation from Without Versus Within. Annu. Rev. Plant Biol. 63, 239–260. 10.1146/annurev-arplant-042811-105453 [DOI] [PubMed] [Google Scholar]
- Chari T, Banerjee J, Pachter L, 2021. The Specious Art of Single-Cell Genomics (preprint). Genomics. 10.1101/2021.08.25.457696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lareau C, Andreani T, Vinyard ME, Garcia SP, Clement K, Andrade-Navarro MA, Buenrostro JD, Pinello L, 2019. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 20, 241. 10.1186/s13059-019-1854-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Wu Z, Hou S, 2020. SPEECHLESS Speaks Loudly in Stomatal Development. Front. Plant Sci. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Miragaia RJ, Natarajan KN, Teichmann SA, 2018. A rapid and robust method for single cell chromatin accessibility profiling. Nat. Commun. 9, 5345. 10.1038/s41467-018-07771-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleves PA, Ellis NA, Jimenez MT, Nunez SM, Schluter D, Kingsley DM, Miller CT, 2014. Evolved tooth gain in sticklebacks is associated with a cis -regulatory allele of Bmp6. Proc. Natl. Acad. Sci. 111, 13912–13917. 10.1073/pnas.1407567111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P, 2019. Organization and regulation of gene transcription. Nature 573, 45–54. 10.1038/s41586-019-1517-4 [DOI] [PubMed] [Google Scholar]
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, Shendure J, 2015. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914. 10.1126/science.aab1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, Filippova GN, Huang X, Christiansen L, DeWitt WS, Lee C, Regalado SG, Read DF, Steemers FJ, Disteche CM, Trapnell C, Shendure J, 2018a. A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174, 1309–1324.e18. 10.1016/j.cell.2018.06.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Reddington JP, Garfield DA, Daza R, Aghamirzaie D, Marco-Ferreres R, Pliner H, Christiansen L, Qiu X, Steemers FJ, Trapnell C, Shendure J, Furlong EEM, 2018b. The cis-regulatory dynamics of embryonic development at single cell resolution. Nature 555, 538–542. 10.1038/nature25981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AC, Foroutan A, Qian B, Hosseini Naghavi N, Shabani K, Shooshtari P, 2022. Single-Cell Chromatin Accessibility Data Combined with GWAS Improves Detection of Relevant Cell Types in 59 Complex Phenotypes. Int. J. Mol. Sci. 23, 11456. 10.3390/ijms231911456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan L, Poethig RS, 1998. Clonal Analysis of Leaf Development in Cotton. Am. J. Bot. 85, 315–321. 10.2307/2446322 [DOI] [PubMed] [Google Scholar]
- Domcke S, Hill AJ, Daza RM, Cao J, O’Day DR, Pliner HA, Aldinger KA, Pokholok D, Zhang F, Milbank JH, Zager MA, Glass IA, Steemers FJ, Doherty D, Trapnell C, Cusanovich DA, Shendure J, 2020. A human cell atlas of fetal chromatin accessibility. Science 370. 10.1126/science.aba7612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrity MW, Alexandre C, Hamm M, Vigil A-L, Fields S, Queitsch C, Cuperus J, 2020. The regulatory landscape of Arabidopsis thaliana roots at single-cell resolution (preprint). Genomics. 10.1101/2020.07.17.204792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagen KP, Hartl TA, Kornberg RD, 2015. Stable Chromosome Condensation Revealed by Chromosome Conformation Capture. Cell 163, 934–946. 10.1016/j.cell.2015.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni I, 2018. A Conceptual Framework for Cell Identity Transitions in Plants. Plant Cell Physiol. 59, 696–706. 10.1093/pcp/pcx172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esau K, 1954. Primary Vascular Differentiation in Plants. Biol. Rev. 29, 46–86. 10.1111/j.1469-185X.1954.tb01397.x [DOI] [Google Scholar]
- Esau K, 1943. Ontogeny of the vascular bundle in Zea Mays. Hilgardia 15, 325–368. [Google Scholar]
- Esau K, 1939. Development and Structure of the Phloem Tissue. Bot. Rev. 5, 373–432. [Google Scholar]
- Evert RF, Esau K, Esau K, 2006. Esau’s Plant anatomy: meristems, cells, and tissues of the plant body: their structure, function, and development, 3rd ed. ed. Wiley-Interscience, Hoboken, N.J. [Google Scholar]
- Farmer A, Thibivilliers S, Ryu KH, Schiefelbein J, Libault M, 2021. Single-nucleus RNA and ATAC sequencing reveals the impact of chromatin accessibility on gene expression in Arabidopsis roots at the single-cell level. Mol. Plant 14, 372–383. 10.1016/j.molp.2021.01.001 [DOI] [PubMed] [Google Scholar]
- Gibson G, 2022. Perspectives on rigor and reproducibility in single cell genomics. PLoS Genet. 18, e1010210. 10.1371/journal.pgen.1010210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD, 2007. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17, 877–885. 10.1101/gr.5533506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisselbrecht SS, Palagi A, Kurland JV, Rogers JM, Ozadam H, Zhan Y, Dekker J, Bulyk ML, 2020. Transcriptional Silencers in Drosophila Serve a Dual Role as Transcriptional Enhancers in Alternate Cellular Contexts. Mol. Cell 77, 324–337.e8. 10.1016/j.molcel.2019.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwald JR, Krysan PJ, Young JC, Evert RF, Sussman MR, 2000. Genetic evidence for the in planta role of phloem-specific plasma membrane sucrose transporters. Proc. Natl. Acad. Sci. 97, 13979–13984. 10.1073/pnas.250473797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hake S, Freeling M, 1986. Analysis of genetic mosaics shows that the extra epidermal cell divisions in Knotted mutant maize plants are induced by adjacent mesophyll cells. Nature 320, 621–623. 10.1038/320621a0 [DOI] [Google Scholar]
- Han J, Wang P, Wang Q, Lin Q, Chen Z, Yu G, Miao C, Dao Y, Wu R, Schnable J, Tang H, Wang K, 2020. Genome-wide Characterization of DNase I-hypersensitive Sites and Cold Response Regulatory Landscapes in Grasses. Plant Cell. 10.1105/tpc.19.00716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helariutta Y, Fukaki H, Wysocka-Diller J, Nakajima K, Jung J, Sena G, Hauser M-T, Benfey PN, 2000. The SHORT-ROOT Gene Controls Radial Patterning of the Arabidopsis Root through Radial Signaling. Cell 101, 555–567. 10.1016/S0092-8674(00)80865-X [DOI] [PubMed] [Google Scholar]
- Herman PL, Marks MD, 1989. Trichome Development in Arabidopsis thaliana. II. Isolation and Complementation of the GLABROUS1 Gene. Plant Cell 1, 1051–1055. 10.1105/tpc.1.11.1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MS, Vande Zande P, Wittkopp PJ, 2020. Molecular and evolutionary processes generating variation in gene expression. Nat. Rev. Genet. 1–13. 10.1038/s41576-020-00304-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, Yao Z, Eggermont J, Höllt T, Levi BP, Shehata SI, Aevermann B, Beller A, Bertagnolli D, Brouner K, Casper T, Cobbs C, Dalley R, Dee N, Ding S-L, Ellenbogen RG, Fong O, Garren E, Goldy J, Gwinn RP, Hirschstein D, Keene CD, Keshk M, Ko AL, Lathia K, Mahfouz A, Maltzer Z, McGraw M, Nguyen TN, Nyhus J, Ojemann JG, Oldre A, Parry S, Reynolds S, Rimorin C, Shapovalova NV, Somasundaram S, Szafer A, Thomsen ER, Tieu M, Quon G, Scheuermann RH, Yuste R, Sunkin SM, Lelieveldt B, Feng D, Ng L, Bernard A, Hawrylycz M, Phillips JW, Tasic B, Zeng H, Jones AR, Koch C, Lein ES, 2019. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. 10.1038/s41586-019-1506-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooke R, 1665. Micrographia : or, Some physiological descriptions of minute bodies made by magnifying glasses. With observations and inquiries thereupon [WWW Document]. Sci. Hist. Inst. Digit. Collect. URL https://digital.sciencehistory.org/works/9g54xj51s (accessed 4.20.23). [Google Scholar]
- Hughes TE, Langdale JA, 2022. SCARECROW is deployed in distinct contexts during rice and maize leaf development. Development 149, dev200410. 10.1242/dev.200410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TE, Langdale JA, Kelly S, 2014. The impact of widespread regulatory neofunctionalization on homeolog gene evolution following whole-genome duplication in maize. Genome Res. 24, 1348–1355. 10.1101/gr.172684.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung C-Y, Lin Y, Zhang M, Pollock S, David Marks M, Schiefelbein J, 1998. A Common Position-Dependent Mechanism Controls Cell-Type Patterning and GLABRA2 Regulation in the Root and Hypocotyl Epidermis of Arabidopsis. Plant Physiol. 117, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperatorskai︠a︡ akademīi︠a︡ nauk (Russia), nauk (Russia), akademīi︠ I., nauk (Russia), akademīi︠ I., 1868. Mémoires de l’Académie impériale des sciences de St.-Pétersbourg. L’Académie, St.-Petersburg. [Google Scholar]
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, Amit I, 2014. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science 343, 776–779. 10.1126/science.1247651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegla DE, Sussex IM, 1989. Cell lineage patterns in the shoot meristem of the sunflower embryo in the dry seed. Dev. Biol. 131, 215–225. 10.1016/S0012-1606(89)80053-3 [DOI] [PubMed] [Google Scholar]
- Johnson SM, Tan FJ, McCullough HL, Riordan DP, Fire AZ, 2006. Flexibility and constraint in the nucleosome core landscape of Caenorhabditis elegans chromatin. Genome Res. 16, 1505–1516. 10.1101/gr.5560806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Hagemann W, 1991. The Relationship of Cell and Organism in Vascular Plants. BioScience 41, 693–703. 10.2307/1311764 [DOI] [Google Scholar]
- Kirik V, Schnittger A, Radchuk V, Adler K, Hülskamp M, Bäumlein H, 2001. Ectopic Expression of the Arabidopsis AtMYB23 Gene Induces Differentiation of Trichome Cells. Dev. Biol. 235, 366–377. 10.1006/dbio.2001.0287 [DOI] [PubMed] [Google Scholar]
- Lai X, Stigliani A, Vachon G, Carles C, Smaczniak C, Zubieta C, Kaufmann K, Parcy F, 2019. Building Transcription Factor Binding Site Models to Understand Gene Regulation in Plants. Mol. Plant 12, 743–763. 10.1016/j.molp.2018.10.010 [DOI] [PubMed] [Google Scholar]
- Langdale JA, Lane B, Freeling M, Nelson T, 1989. Cell lineage analysis of maize bundle sheath and mesophyll cells. Dev. Biol. 133, 128–139. 10.1016/0012-1606(89)90304-7 [DOI] [PubMed] [Google Scholar]
- Larkin JC, Oppenheimer DG, Lloyd AM, Paparozzi ET, Marks MD, 1994. Roles of the GLABROUS1 and TRANSPARENT TESTA GLABRA Genes in Arabidopsis Trichome Development. Plant Cell 6, 1065–1076. 10.1105/tpc.6.8.1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-I, Kim N-S, 2014. Transposable Elements and Genome Size Variations in Plants. Genomics Inform. 12, 87–97. 10.5808/GI.2014.12.3.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TA, Nobori T, Illouz-Eliaz N, Xu J, Jow B, Nery JR, Ecker JR, 2023. A Single-Nucleus Atlas of Seed-to-Seed Development in Arabidopsis (preprint). Plant Biology. 10.1101/2023.03.23.533992 [DOI] [Google Scholar]
- Leroux O, 2012. Collenchyma: a versatile mechanical tissue with dynamic cell walls. Ann. Bot. 110, 1083–1098. 10.1093/aob/mcs186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR, Reidel EJ, Turgeon R, Liu P, Sun Q, Nelson T, Brutnell TP, 2010. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 42, 1060–1067. 10.1038/ng.703 [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J, 2009. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 326, 289–293. 10.1126/science.1181369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Hofmeister BT, Vollmers C, DuBois RM, Schmitz RJ, 2017. Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic Acids Res. 45, e41–e41. 10.1093/nar/gkw1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Marand AP, Ricci WA, Ethridge CL, Zhang X, Schmitz RJ, 2019. The prevalence, evolution and chromatin signatures of plant regulatory elements. Nat. Plants 5, 1250–1259. 10.1038/s41477-019-0548-z [DOI] [PubMed] [Google Scholar]
- Maaten L, van der, Hinton G., 2008. Visualizing Data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605. [Google Scholar]
- Maher KA, Bajic M, Kajala K, Reynoso M, Pauluzzi G, West DA, Zumstein K, Woodhouse M, Bubb K, Dorrity MW, Queitsch C, Bailey-Serres J, Sinha N, Brady SM, Deal RB, 2018. Profiling of Accessible Chromatin Regions across Multiple Plant Species and Cell Types Reveals Common Gene Regulatory Principles and New Control Modules. Plant Cell 30, 15–36. 10.1105/tpc.17.00581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marand AP, Chen Z, Gallavotti A, Schmitz RJ, 2021. A cis-regulatory atlas in maize at single-cell resolution. Cell 184, 3041–3055.e21. 10.1016/j.cell.2021.04.014 [DOI] [PubMed] [Google Scholar]
- McInnes L, Healy J, Melville J, 2020. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. [Google Scholar]
- Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, Herman B, Happe S, Higgs A, LeProust E, Follows GA, Fraser P, Luscombe NM, Osborne CS, 2015. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 47, 598–606. 10.1038/ng.3286 [DOI] [PubMed] [Google Scholar]
- Moore BM, Lee YS, Wang P, Azodi C, Grotewold E, Shiu S-H, 2022. Modeling temporal and hormonal regulation of plant transcriptional response to wounding. Plant Cell 34, 867–888. 10.1093/plcell/koab287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelms B, Walbot V, 2019. Defining the developmental program leading to meiosis in maize. Science 364, 52–56. 10.1126/science.aav6428 [DOI] [PubMed] [Google Scholar]
- Oka R, Zicola J, Weber B, Anderson SN, Hodgman C, Gent JI, Wesselink J-J, Springer NM, Hoefsloot HCJ, Turck F, Stam M, 2017. Genome-wide mapping of transcriptional enhancer candidates using DNA and chromatin features in maize. Genome Biol. 18, 137. 10.1186/s13059-017-1273-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer DG, Herman PL, Sivakumaran S, Esch J, Marks MD, 1991. A myb gene required for leaf trichome differentiation in Arabidopsis is expressed in stipules. Cell 67, 483–493. 10.1016/0092-8674(91)90523-2 [DOI] [PubMed] [Google Scholar]
- Panchy N, Lehti-Shiu M, Shiu S-H, 2016. Evolution of Gene Duplication in Plants. Plant Physiol. 171, 2294–2316. 10.1104/pp.16.00523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang B, Snyder MP, 2020. Systematic identification of silencers in human cells. Nat. Genet. 52, 254–263. 10.1038/s41588-020-0578-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson AH, Freeling M, Tang H, Wang X, 2010. Insights from the Comparison of Plant Genome Sequences. Annu. Rev. Plant Biol. 61, 349–372. 10.1146/annurev-arplant-042809-112235 [DOI] [PubMed] [Google Scholar]
- Pick TR, Bräutigam A, Schlüter U, Denton AK, Colmsee C, Scholz U, Fahnenstich H, Pieruschka R, Rascher U, Sonnewald U, Weber APM, 2011. Systems Analysis of a Maize Leaf Developmental Gradient Redefines the Current C4 Model and Provides Candidates for Regulation. Plant Cell 23, 4208–4220. 10.1105/tpc.111.090324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A, Adey AC, Steemers FJ, Shendure J, Trapnell C, 2018. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 71, 858–871.e8. 10.1016/j.molcel.2018.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poethig RS, Sussex IM, 1985. The developmental morphology and growth dynamics of the tobacco leaf. Planta 165, 158–169. 10.1007/BF00395038 [DOI] [PubMed] [Google Scholar]
- Poethig RS, Sussex IM, 1985. The cellular parameters of leaf development in tobacco: a clonal analysis. Planta 165, 170–184. [DOI] [PubMed] [Google Scholar]
- Raju SKK, 2020. Comparative profiling examines roles of DNA regulatory sequences and accessible chromatin during cold stress response in grasses. Plant Cell. 10.1105/tpc.20.00471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt D, Frenz M, Mandel T, Kuhlemeier C, 2003. Microsurgical and laser ablation analysis of interactions between the zones and layers of the tomato shoot apical meristem. Development 130, 4073–4083. 10.1242/dev.00596 [DOI] [PubMed] [Google Scholar]
- Reynoso MA, Kajala K, Bajic M, West DA, Pauluzzi G, Yao AI, Hatch K, Zumstein K, Woodhouse M, Rodriguez-Medina J, Sinha N, Brady SM, Deal RB, Bailey-Serres J, 2019. Evolutionary flexibility in flooding response circuitry in angiosperms. Science 365, 1291–1295. 10.1126/science.aax8862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci WA, Lu Z, Ji L, Marand AP, Ethridge CL, Murphy NG, Noshay JM, Galli M, Mejía-Guerra MK, Colomé-Tatché M, Johannes F, Rowley MJ, Corces VG, Zhai J, Scanlon MJ, Buckler ES, Gallavotti A, Springer NM, Schmitz RJ, Zhang X, 2019. Widespread long-range cis -regulatory elements in the maize genome. Nat. Plants 5, 1237–1249. 10.1038/s41477-019-0547-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas U, Mei Y, Xie Q, Banta JA, Zhou RW, Seufferheld G, Gerard S, Chou L, Bhambhra N, Parks JD, Flowers JM, McClung CR, Hanzawa Y, Purugganan MD, 2014. Variation in Arabidopsis flowering time associated with cis-regulatory variation in CONSTANS. Nat. Commun. 5, 3651. 10.1038/ncomms4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KH, Huang L, Kang HM, Schiefelbein J, 2019. Single-Cell RNA Sequencing Resolves Molecular Relationships Among Individual Plant Cells. Plant Physiol. 179, 1444–1456. 10.1104/pp.18.01482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz RJ, Grotewold E, Stam M, 2022. Cis-regulatory sequences in plants: Their importance, discovery, and future challenges. Plant Cell 34, 718–741. 10.1093/plcell/koab281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler ML, Sedelnikova OV, Walker BJ, Westhoff P, Langdale JA, 2018. SHORTROOT-Mediated Increase in Stomatal Density Has No Impact on Photosynthetic Efficiency. Plant Physiol. 176, 757–772. 10.1104/pp.17.01005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharman BC, 1942. Developmental Anatomy of the Shoot of Zea mays L. Ann. Bot. 6, 245–282. 10.1093/oxfordjournals.aob.a088407 [DOI] [Google Scholar]
- Shlyueva D, Stampfel G, Stark A, 2014. Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet. 15, 272–286. 10.1038/nrg3682 [DOI] [PubMed] [Google Scholar]
- Shulse CN, Cole BJ, Ciobanu D, Lin J, Yoshinaga Y, Gouran M, Turco GM, Zhu Y, O’Malley RC, Brady SM, Dickel DE, 2019. High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types. Cell Rep. 27, 2241–2247.e4. 10.1016/j.celrep.2019.04.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha N, Hake S, 1990. Mutant characters of knotted maize leaves are determined in the innermost tissue layers. Dev. Biol. 141, 203–210. 10.1016/0012-1606(90)90115-y [DOI] [PubMed] [Google Scholar]
- Stadler R, Sauer N, 1996. The Arabidopsis thaliana AtSUC2 Gene is Specifically Expressed in Companion Cells. Bot. Acta 109, 299–306. 10.1111/j.1438-8677.1996.tb00577.x [DOI] [Google Scholar]
- Stadler R, Wright KM, Lauterbach C, Amon G, Gahrtz M, Feuerstein A, Oparka KJ, Sauer N, 2005. Expression of GFP-fusions in Arabidopsis companion cells reveals non-specific protein trafficking into sieve elements and identifies a novel post-phloem domain in roots. Plant J. 41, 319–331. 10.1111/j.1365-313X.2004.02298.x [DOI] [PubMed] [Google Scholar]
- Strasburger E, 1888. Histologische Beiträge. G. Fischer, Jena. 10.5962/bhl.title.24451 [DOI] [Google Scholar]
- Studer A, Zhao Q, Ross-Ibarra J, Doebley J, 2011. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 43, 1160–1163. 10.1038/ng.942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toufighi K, Brady SM, Austin R, Ly E, Provart NJ, 2005. The Botany Array Resource: e-Northerns, Expression Angling, and promoter analyses. Plant J. 43, 153–163. 10.1111/j.1365-313X.2005.02437.x [DOI] [PubMed] [Google Scholar]
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL, 2014. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386. 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg KRL, Lewis JJ, Brack BJ, Fandino RA, Mazo-Vargas A, Reed RD, 2020. Genomic architecture of a genetically assimilated seasonal color pattern. Science 370, 721–725. 10.1126/science.aaz3017 [DOI] [PubMed] [Google Scholar]
- Waltman L, van Eck NJ, 2013. A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 86, 471. 10.1140/epjb/e2013-40829-0 [DOI] [Google Scholar]
- Wang P, Kelly S, Fouracre JP, Langdale JA, 2013. Genome-wide transcript analysis of early maize leaf development reveals gene cohorts associated with the differentiation of C4 Kranz anatomy. Plant J. 75, 656–670. 10.1111/tpj.12229 [DOI] [PubMed] [Google Scholar]
- Wilhelm K, 1880. Beiträge zur Kenntniss des Siebröhrenapparates dicotyler Pflanzen. Verlag von Wilhelm Engelmann. [Google Scholar]
- Wolock SL, Lopez R, Klein AM, 2019. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 8, 281–291.e9. 10.1016/j.cels.2018.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig JI, Howes TR, Au JN, Au EH, Roberts Kingman GA, Brady SD, Herbert AL, Reimchen TE, Bell MA, Lowe CB, Dalziel AC, Kingsley DM, 2022. Evolution of stickleback spines through independent cis-regulatory changes at HOXDB. Nat. Ecol. Evol. 6, 1537–1552. 10.1038/s41559-022-01855-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Powell DR, Curtis DJ, Wong NC, 2020. From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 21, 22. 10.1186/s13059-020-1929-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, 2022. What is a cell type and how to define it? Cell 185, 2739–2755. 10.1016/j.cell.2022.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z, Zhang W, Marand AP, Zhu B, Buell CR, Jiang J, 2019. Cold stress induces enhanced chromatin accessibility and bivalent histone modifications H3K4me3 and H3K27me3 of active genes in potato. Genome Biol. 20, 123. 10.1186/s13059-019-1731-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T-Q, Chen Y, Liu Y, Lin W-H, Wang J-W, 2021. Single-cell transcriptome atlas and chromatin accessibility landscape reveal differentiation trajectories in the rice root. Nat. Commun. 12, 2053. 10.1038/s41467-021-22352-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T-Q, Xu Z-G, Shang G-D, Wang J-W, 2019. A Single-Cell RNA Sequencing Profiles the Developmental Landscape of Arabidopsis Root. Mol. Plant 12, 648–660. 10.1016/j.molp.2019.04.004 [DOI] [PubMed] [Google Scholar]
- Zhao H, Zhang W, Chen L, Wang L, Marand AP, Wu Y, Jiang J, 2018. Proliferation of Regulatory DNA Elements Derived from Transposable Elements in the Maize Genome. Plant Physiol. 176, 2789–2803. 10.1104/pp.17.01467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Enders TA, Myers ZA, Magnusson E, Crisp PA, Noshay JM, Gomez-Cano F, Liang Z, Grotewold E, Greenham K, Springer NM, 2022. Prediction of conserved and variable heat and cold stress response in maize using cis-regulatory information. Plant Cell 34, 514–534. 10.1093/plcell/koab267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C, Sun K, Mackaluso JD, Seddon AE, Jin R, Thomashow MF, Shiu S-H, 2011. Cis-regulatory code of stress-responsive transcription in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U. S. A. 108, 14992–14997. 10.1073/pnas.1103202108 [DOI] [PMC free article] [PubMed] [Google Scholar]