

Summary:
The development of CRISPR-based barcoding methods creates an exciting opportunity to understand cellular phylogenies. We present a compact, tunable, high-capacity Cas12a barcoding system, Dual Acting Inverted Site arraY (DAISY). We combined high-throughput screening and machine-learning to predict and optimize the 60-bp DAISY barcode sequences. After optimization, top-performing barcodes had ~10-fold increased capacity relative to the best random-screened designs, and performed reliably across diverse cell types. DAISY barcode arrays generated ~12-bits of entropy and ~66,000 unique barcodes. Thus, DAISY barcodes – at a fraction of the size of Cas9 barcodes – achieved high-capacity barcoding. We coupled DAISY barcoding with single-cell RNA-seq to recover lineages and gene expression profiles from ~47,000 human melanoma cells. A single DAISY barcode recovered up to ~700 lineages from one parental cell. This analysis revealed heritable single-cell gene expression and potential epigenetic modulation of memory gene transcription. Overall, Cas12a DAISY-barcoding is an efficient tool for investigating cell state dynamics.
INTRODUCTION
Uncovering the relationship between single-cell lineage and gene expression state has contributed to our understanding of the dynamics of multicellular systems (Cao et al., 2020; Neftel et al., 2019; Regev et al., 2017; Tabula Muris Consortium et al., 2018; Tirosh et al., 2016; Travaglini et al., 2020). Mathematical inference has been widely used to estimate cellular trajectories from measurements of the gene expression profiles of cells (Wolf et al., 2019; Setty et al., 2019; Manno et al., 2018; Saelens et al., 2019; Kester and van Oudenaarden, 2018). These approaches are limited due to the requirement for mathematical assumptions, like irreversibility of trajectories, that may not match the biological ground truth. An alternative approach is to integrate single-cell transcriptomic profiling with methods that allow direct analysis of cell lineages. Lineage history can be determined by cellular barcoding, where each cell is given diverse DNA sequences as molecular barcodes (Weinreb et al., 2020; Biddy et al., 2018; Rogers et al., 2017; Kebschull and Zador, 2018; Kalhor et al., 2017; Perli et al., 2016). Recently, CRISPR-Cas9 has been utilized to develop evolvable barcoding systems where editing of a barcode can generate diverse insertions-deletions (indels) that accumulate over time (Alemany et al., 2018; Chan et al., 2019; Raj et al., 2018). In this way, a barcode sequence can evolve over time into many distinct outcomes (edited sequences, or states), allowing computational reconstruction of subclonal lineages (Jones et al., 2020). The lineage information from CRISPR barcoding coupled with single-cell gene expression profiles from RNA-seq has allowed interrogation of important biological questions in mammalian (Chan et al., 2019; Bowling et al., 2020) and zebrafish development (Raj et al., 2018), as well as cancer metastasis (Quinn et al., 2021; Simeonov et al., 2021). Although they are sufficient to yield biological insights, existing CRISPR barcodes have not been thoroughly optimized to enable high capacity and tunable lineage tracking within complex biological systems. Importantly, unlike endogenous genome editing, editing of CRISPR barcodes could theoretically use any synthetic sequence, a vast space for this design optimization problem.
Here, we harnessed the compactness and multi-target gene-editing ability of CRISPR-Cas12a to enable high capacity and tunable molecular barcoding (Figure 1A) (Zetsche et al., 2015; Liu et al., 2019). Barcode capacity is the entropy of editing outcomes generated within the barcode. Entropy, measured as the complexity of the evolvable barcode’s lineage tree, is a proxy for the number of uniquely trackable lineages (Shannon, 1948). CRISPR barcode entropy directly correlates with its lineage tracking capacity (Kalhor et al., 2017) as well as the accuracy of recovered lineages (Jones et al., 2020). Barcode tunability is a feature that allows programmable editing kinetics of a barcode sequence to match the underlying biological process that is being recorded. CRISPR barcodes evolve continuously, and the speed of barcoding determines the time span for recording a biological process, like lineage commitment. This could be measured via the change rate of the entropy as the barcode evolves, which is analogous to the sampling rate in information theory (Shannon, 1948).
Figure 1. Overview of Cas12a-based DAISY barcodes and pipeline to couple lineage information with single-cell transcriptomic profiling.
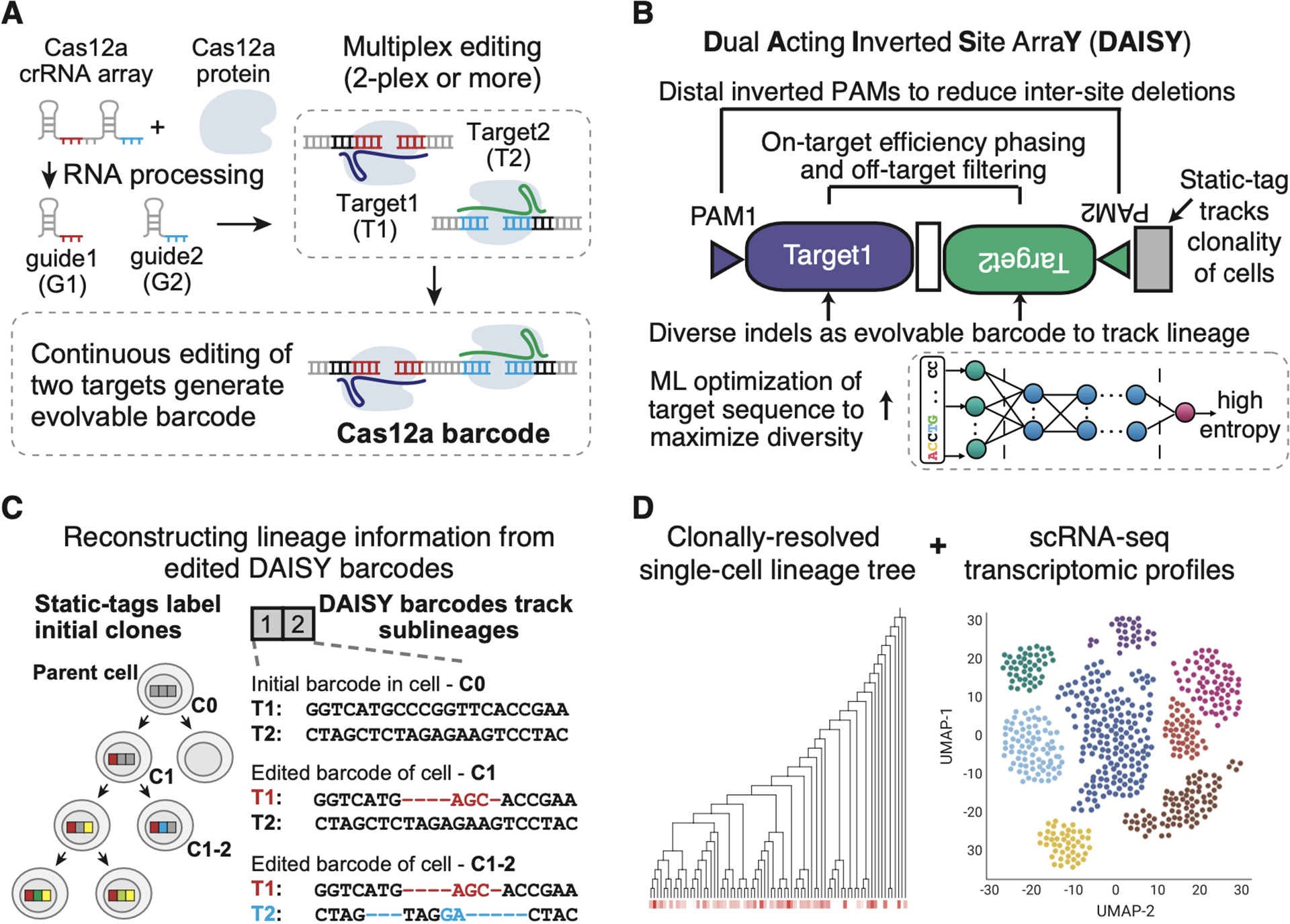
(A) Design of Cas12a-based barcode system, in which a single crRNA array with two guides (G1/G2) could be processed to edit two target sites within a barcode. (B) Dual acting inverted site array (DAISY) barcode design with two crRNA-target pairs. The guide sequences were selected to have phased editing efficiency (Seq-deepCpf1) and low off-target scores (FlashFry), see Methods for details. (C) Editing outcomes at the target sites (T1/T2) within a barcode are used to place cells within a lineage tree. Here, an initial edit in T1 allows for grouping of descendent daughter cells that contain differentiating edits in T2. (D) Simultaneous recovery of the transcriptome of a cell and an expressed DAISY barcode enables lineage tracking and cell state classification.
Design
Existing Cas9-based evolvable barcode systems present several challenges. First, there has been no systematic effort to optimize the diversity of editing outcomes generated by CRISPR barcodes to improve its lineage tracking capability. Moreover, Cas9-based systems present challenges in scalability due to the difficulty of delivering multiple guideRNAs (gRNAs) with portable delivery systems like lentivirus. Compared to Cas9 barcodes, Cas12a allows a substantially more compact barcoding system thanks to the much shorter CRISPR RNA (crRNA), and the ability to edit multiple target sites via a single crRNA array. Moreover, the Cas12a system has higher targeting specificity than Cas9, which could reduce toxicity from off-target cleavage in barcoding experiments (Kim et al., 2016).
Current evolvable barcode systems often contain more than one target site to increase barcode capacity. However, inter-site deletions that span two or more target sites within a barcode are frequent, destroy at least one PAM sequence, and remove a large region of the barcode (Bowling et al., 2020; McKenna et al., 2016). These deletions also prevent further editing, so that many descendant cells end up with undistinguishable barcode sequences, thus reducing the barcode capacity (Bowling et al., 2020; McKenna et al., 2016). To overcome this problem, we designed Cas12a barcodes to have two target sites with phased editing efficiencies to reduce the chance of barcode collapse, which we refer to as Dual Acting Inverted Site ArraY (DAISY) barcodes. DAISY features an inverted-two-target-sites design (Figure 1B), where the Cas12a PAM sequences are at the ends of the barcode region. Thus, the Cas12a enzyme cleavage sites are centered to minimize PAM removal due to inter-site deletion.
While there have been efforts to predict CRISPR editing outcomes for other purposes such as precise therapeutic gene editing (Chen et al., 2019; Allen et al., 2018; Shen et al., 2018), optimizing an evolvable CRISPR barcode is more difficult due to the vast potential sequence choices (a 20-base-pair CRISPR target sequence has 420 or ~1 trillion possible sequences) (Barrangou and Doudna, 2016). To address this challenge, we coupled high-throughput DAISY barcode screening with machine learning (ML)-guided search processes to design an iterative experiment-computation workflow (Figure 1B), which we termed CRISPR Learning and Optimization via Variants Exploration with Regression (CLOVER). CLOVER was able to generate a collection of high-capacity DAISY barcodes that demonstrated robust performances across multiple cell lines. Next, we concatenated multiple DAISY barcodes into a DAISY chain barcode that contained minimal inter-site deletions to enable exponentially higher lineage-tracking capacity. Finally, as an initial biological showcase, we integrated DAISY barcoding with single-cell RNA-seq in two experiments that contained ~47,000 human melanoma cells. We harnessed the joint lineage and gene expression profile data from these melanoma cells to investigate transcriptional memory and nominate EZH2 as a putative epigenetic regulator (Shaffer et al., 2020).
RESULTS
Developing a Cas12a-based barcoding tool and benchmarking with Cas9 barcodes
Cas12a, a class II type V CRISPR-Cas enzyme, has dual RNase and DNase activities (Zetsche et al., 2015). Cas12a binds to the ~20 palindromic nucleotide scaffold sequences (direct repeat, DR) and processes a crRNA array to generate multiple crRNAs (Zetsche et al., 2017). The processed crRNAs can have distinct guide sequences that allow Cas12a to edit multiple target sites (Figure 1A). Cas12a also has significantly improved specificity versus Cas9, leading to lower cellular toxicity from off-target effects (Kim et al., 2016) (Figure S1). As Cas12a editing occurs, the initial barcode sequence evolves and branches into multiple lineages (Figure 1C). When coupled with single-cell RNA-Seq (scRNA-Seq), a Cas12a barcode enables reconstruction of cellular phylogeny and states from readouts of edited barcodes and transcriptomes, respectively (Figure 1D) (McKenna et al., 2016).
To initially compare the entropy of the repair outcomes generated by Cas9 versus those generated by Cas12a, we evaluated the editing at 7 endogenous genomic loci (Figure 2A). We tested Cas9 and Cas12a target sequences that are proximal to one another in order to control for sequence-based editing outcome biases (Figure 2A and 2C). After transient expression of Cas12a or Cas9, we quantified the editing entropies at each genomic target (Methods). Across 6 of the 7 loci, Cas12a led to significantly higher entropy and more diverse editing outcomes, while Cas9 often led to outcomes that were dominated by the most frequent indels (Figure 2D, 2E and S2D). Importantly, the higher entropy was not due to differential editing efficiency between Cas12a and Cas9 (Figure 2B).
Figure 2. Comparison of Cas9 and Cas12a for gene editing-based cell barcoding.
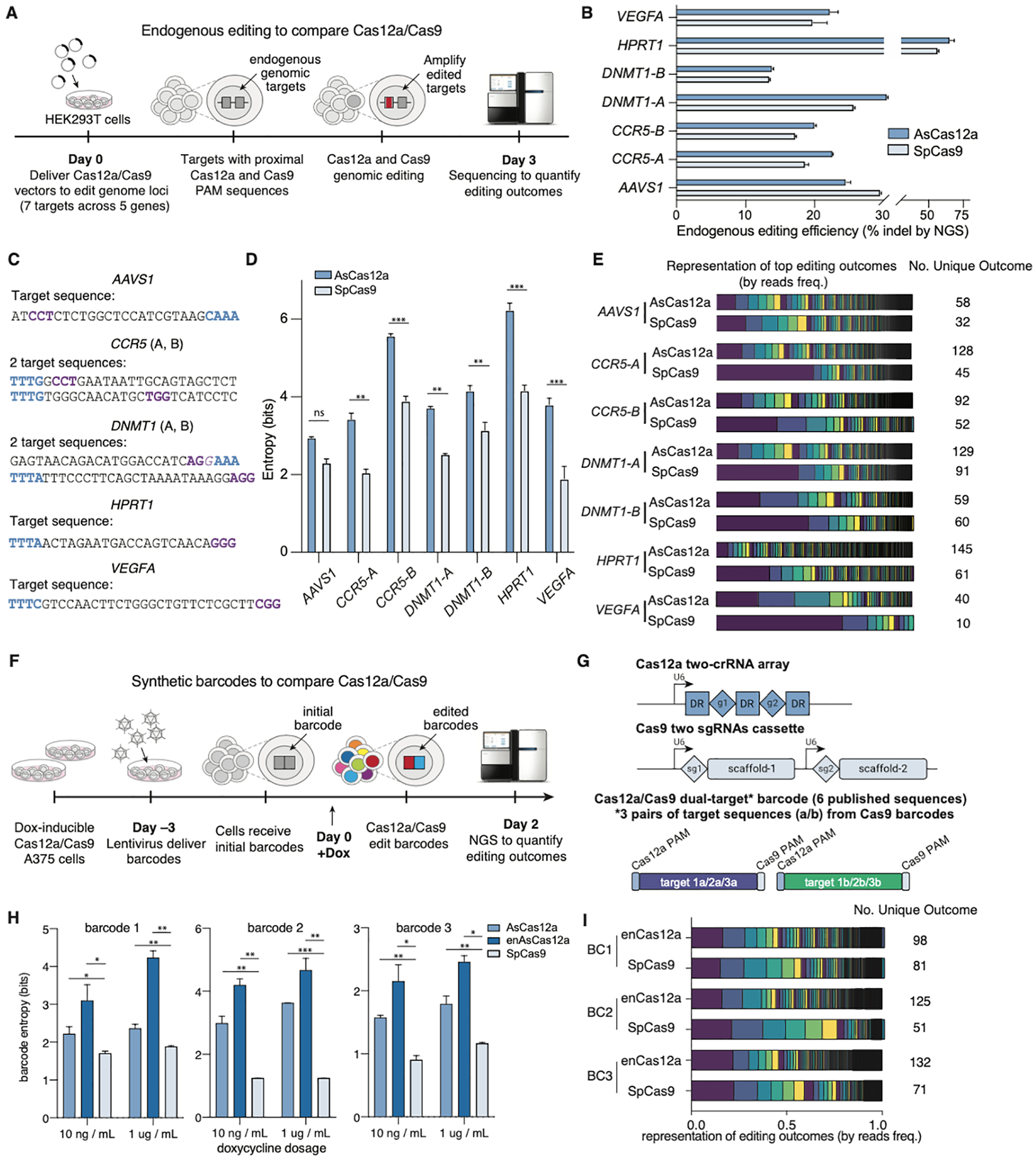
(A) Design of the endogenous editing experiment to compare Cas12a/Cas9 editing outcomes using transient transfection. (B) Gene-editing efficiencies across endogenous targets showing comparable levels of indel formation between Cas12a/Cas9. (C) Endogenous target sequences indicating the proximal PAM sequences (Cas12a in blue, Cas9 in purple). (D) Entropy of Cas12a and Cas9-based editing outcomes at endogenous targets. (E) Stacked bar chart comparing the editing outcome distribution of Cas12a- vs. Cas9-based editing outcomes. Bar areas correspond to the sequencing reads frequency of each unique indel outcome. (F) Design of synthetic barcode experiments to compare Cas12a/Cas9 using lentiviral vectors and doxycycline-inducible cell lines. (G) Vector designs for Cas12a editing (top) and Cas9 editing (middle) of a common two-target barcode (bottom). We picked 3 published barcodes from a published Cas9 study (Bowling et al., 2020). (H) Entropy of editing outcomes within each barcode after doxycycline-induced Cas12a/Cas9 expression. (I) Stacked bar chart comparing editing outcome distribution as in panel e. Unless otherwise noted, all statistical comparison in this and following figures were performed via a t-test with 1% false-discovery rate (FDR) using a two-stage step-up method of Benjamini, Krieger and Yekutieli, * (p < 0.05); ** (p < 0.01); *** (p < 0.001).
To compare the entropy of Cas9 and Cas12a editing within exogenous barcodes, we generated cell lines with doxycycline-inducible Cas12a or Cas9 (Figure S2A–C) and compared the resulting entropies across three barcode sequences extracted from a published study (Bowling et al., 2020) (Figure 2F and 2G). We evaluated the entropy of editing outcomes in each lentivirally delivered barcode after induced expression of Cas12a (AsCas12a), enhancedCas12a (enAsCas12a-HF) (Kleinstiver et al., 2019) or Cas9 (SpCas9) (Figure 2F and 2G). Cas12a-based editing consistently led to higher barcode entropy than that of Cas9 (Figure 2H). Cas12a yields a higher number of outcomes, and their distribution is closer to uniform compared with Cas9 (Figure 2I). Collectively, our data indicate that Cas12a generates a wider range of indels and thus higher entropy evolving barcodes. These results motivated us to further develop a Cas12a-based evolvable barcode system.
Two-site inverted Cas12a barcode system reduces inter-site deletions, improves barcoding capacity, and allows high-throughput screening
Cas9-based barcodes often contain more than one target site to increase barcode capacity. However, inter-site deletions that span two or more target sites within a barcode are frequent, destroy at least one PAM sequence, and remove a large region of the barcode (Bowling et al., 2020; McKenna et al., 2016). These deletions also prevent further editing, so that many descendant cells have undistinguishable barcode sequences, thus reducing the lineage-tracking capacity (Bowling et al., 2020; McKenna et al., 2016). Analysis from our initial test (Figure 2) also confirmed that levels of inter-site deletion correlate with lower barcoding efficiencies (up to 3-fold reduction; Figure S2E). To overcome this deletion issue, we redesigned Cas12a barcodes to have two sites with different editing efficiencies, or phased efficiency, to reduce barcode collapse (Jones et al., 2020), as Dual Acting Inverted Site ArraY (DAISY) barcodes (Figure S3A–C). Phased efficiency theoretically reduces the probability of barcode collapse by minimizing the chances of two simultaneous double-stranded breaks (DSBs) forming within the barcode that would result in a large deletion during non-homologous end joining (NHEJ) between the flanking DNA (McKenna et al., 2016). Moreover, a key attribute of DAISY barcodes is the inverted design in which the Cas12a PAM sequences are at the distal ends of the barcode, thus helping to minimize the chance of PAM removal due to barcode deletion.
The compactness of DAISY barcodes enables high-throughput barcode design screening. While prior work has leveraged pooled screening to measure CRISPR editing (Leenay et al., 2019), there have not been large-scale efforts to uncover optimal CRISPR barcode sequences. Leveraging the Cas12a editing system, we evaluated the capacity of more than 14,000 DAISY barcodes (Figure 3A, S3A–C) We generated a pool of oligos that contained Cas12a DAISY barcodes, next to a crRNA array to edit the target sites, and a static tag to uniquely identify each initial DAISY barcode sequence (Figure 3A). To measure the entropy of diverse barcode designs, we generated 5,000 random CRISPR target sites, pair-wisely assembled all 25 million combinations, and filtered the resulting pairs to prioritize those with phased efficiency (Figure S3A–C, Methods). We constructed a lentiviral library that contained 14,358 unique DAISY barcodes and delivered them into our inducible Cas12a cell lines (Figure 3A). After initiation of Cas12a editing, we sequenced the DAISY barcodes across multiple time points, aligned them to the original barcode reference, and quantified the entropy of the editing outcomes (Figure 3A, Methods).
Figure 3. High-throughput screening with machine learning optimization to generate high-capacity DAISY barcodes.
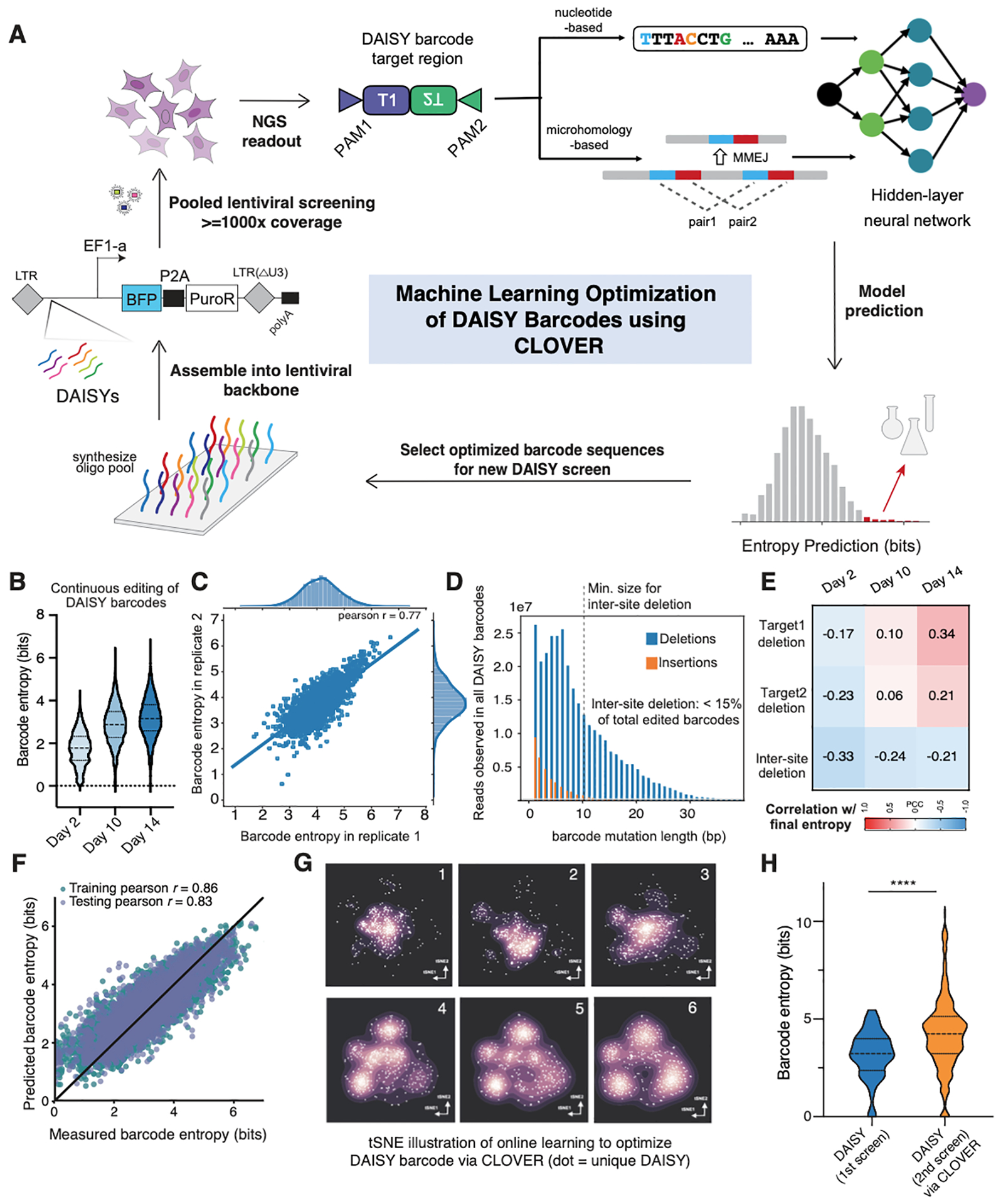
(A) Overall design of CLOVER pipeline to optimize DAISY barcode sequences via iterative pooled screening and machine learning modeling. (B) Distribution of barcode entropies across all DAISY barcodes at each timepoint. (C) Barcode entropy measured at Day-14 from two biological replicates, showing consistent results from separate lentiviral transductions. (D) Indel length distribution across all barcodes where the minimum inter-site deletion length is indicated. (E) Pearson Correlation Coefficients (PCC) between indel outcome types at each timepoint and the final barcode entropy across all DAISY barcodes. (F) Neural network model accurately predicts entropy of DAISY barcodes. (G) 6 rounds of path-regularized online learning were performed (round indicated at top right of each panel). 96 designs are chosen through path regularization (see Methods) in each round (5 simulations total). Therefore, each plot contains 96×5 designs, where the Kernel Density Estimation (KDE) is based on the first two tSNE coordinates. The exploration converges on 4 local maxima as indicated by increased point density after 6 rounds. (H) Distributions of barcode entropy from DAISY barcodes in 1st screen (initial pool) and from 2nd screen (CLOVER-optimized) in A375 cells. * (p < 0.05); ** (p < 0.01); *** (p < 0.001), **** (p < 0.0001).
DAISY barcodes have consistent, evolvable barcoding activities, with low inter-site deletions
Accumulated editing within the DAISY barcodes led to barcode entropy increase over time, with the median rising from ~2 bits at Day-2 to ~3.5 bits at Day-14 (Figure 3B). We confirmed reproducibility of barcode entropy across two biological replicates (Figure 3C). Also, we observed high variability in the barcode entropies (Figure 3B and 3C), consistent with initial barcode sequences influencing the barcode capacity. Notably, we found that the barcode entropy of DAISY barcodes did not correlate with the Seq-deepCpf1 prediction of editing efficiency (Figure S3D). This indicates that the barcode editing process over two adjacent target sites (a unique feature of our DAISY library) cannot be predicted using existing models based on single-site Cas12a editing data. Furthermore, ~85% of observed indels were fewer than 10 nucleotides in length, providing evidence that DAISY reduces inter-site deletions (given the 10bp linker between target sites, any inter-site deletion would be expected to be at least 10 nucleotides in length) (Figure 3D).
Temporal dynamics of barcode editing and their effects on barcoding capacity
Next, we analyzed how the temporal dynamics of barcode editing influenced the final barcode capacity. We calculated the pairwise correlation between the three major types of deletions and the measured barcode entropy, across all barcode sequences over time (Figure 3E). Several trends were immediately apparent. First, early deletion events of all types (on Day-2) reduced the chance of further editing and correlated negatively with the final barcode entropy (Figure 3E, Day2 column). Second, there was a strong negative correlation across all time points between inter-site deletion and barcode entropy (Figure 3E, bottom row). In particular, early inter-site deletions at Day-2 had the strongest negative correlation. Third, single-site editing at later time points (Day-10,14) was positively correlated with barcode entropy, which was notable on Day-10 and significantly stronger on Day-14 (Figure 3E, top two rows). Moreover, we assessed whether Cas12a could retarget a previously edited site, allowing for a continuous increase in barcode entropy. As opposed to Cas9, Cas12a makes PAM-distal cuts that may leave the seed sequence intact, which is required for cleavage (Swarts et al., 2017). We found evidence of disjointed indels that occur within a single target site, which may be a hallmark of retargeting (Figure S3E). The frequency of these relatively rare events increased from ~0.2% to ~3% over the course of the experiment to contribute to the continuous increase in barcode entropy (Figure S3F). Together, these observations suggest that preventing inter-site deletion and promoting a continuous increase in barcode entropy are critical to high-capacity Cas12a barcoding.
Machine learning modeling predicts Cas12a barcode entropy and allows optimization over vast sequence space to generate high-entropy DAISY barcodes
Our initial DAISY library screen suggested that the choice of the initial barcode sequence significantly influences the barcode entropy. Thus, optimal choices of the barcode sequence should maximize the capacity for lineage tracking. Exhaustively testing all possible barcode sequences would require the analysis of trillions of possibilities. To address this challenge, we harnessed the predictive power of machine learning (ML)-guided search processes, to design an iterative experiment-computation workflow, which we termed CRISPR Learning and Optimization via Variants Exploration with Regression (CLOVER) (Figure 3A). The aim of CLOVER was to use the data from DAISY barcode screening to build a ML model that could predict the entropy of untested DAISY barcodes followed by focused experimental testing to identify top barcode sequences. The results of the tests can then be added to the data pool to improve the prediction model, thus enabling an iterative pipeline of barcode optimization (Figure 3A, Methods).
Feature selection and model building of CLOVER for optimizing barcode sequence
The CLOVER pipeline consists of three modules: feature engineering, entropy prediction, and path-regularized online learning (Figure 3A, Methods). The first module is a library of features for predictive ML. Inspired by existing machine learning models for single-target CRISPR editing (Allen et al., 2018; Chen et al., 2019; Kim et al., 2018; Shen et al., 2018), we constructed a collection of features for the DAISY barcodes, which are based on one-hot-encoding of nucleotides, GC content, and a Jaro-Winkler-based distance feature that encoded the process of microhomology-mediated end joining (MMEJ). The Jaro-Winkler gives more weight to the common prefix of two sequences that flank the predicted cut sites. Therefore, it appropriately weighs the increased prevalence of MMEJ-driven editing outcome events as a function of the distance of microhomology tracts from the predicted cut site (Figure 3A).
The second module is a ML-based model to predict a sequence’s entropy. We trained a ridge regression model to test the predictive power of our feature space and found that the model was highly predictive of barcode entropy, with a testing Pearson r of 0.80. To further obtain entropy-guided representations, we trained a neural network using deep learning to arrive at representative features with testing Pearson r of 0.83, which was used for subsequent modeling and design exploration tasks (Figure 3F and S3G, Methods).
The third module enables adaptive search and dynamic exploration of the design space via in silico mutagenesis (Figure 3G, Methods). We developed a path-regularized online learning method using a bandit optimization formulation: at each round of optimization, a learning agent chooses an arm – a combination of designs to experiment on – and receives a stochastic reward. The difference between this reward and the maximal reward at this round, assuming it exists, is defined as instantaneous regret. In the context of our DAISY barcode optimization, the rewards were defined as the average barcode entropy of the identified barcodes and the instantaneous regret is the difference between this value and its maximal-possible value in the pool of sequences. Minimizing the instantaneous regret is equivalent to maximizing the barcode entropy of sequences that we chose for new experiments. We chose an upper-confidence bandit learning approach for recommending new designs utilizing a probabilistic surrogate model (Abbasi-yadkori et al., 2011). The approach would recommend random new design sequences with probability proportional to a “potential” score, where the score is a combination of the design’s predicted entropy and the prediction’s level of uncertainty. This would encourage exploring new designs that are highly dissimilar to tested barcode sequences, which enables fast exploration of large sequence space and fast convergence to the optimal solution (Abbasi-yadkori et al., 2011; Auer, 2002; Rusmevichientong and Tsitsiklis, 2010).
High lineage tracking capacity and tunability of optimized DAISY barcodes
Based on the first DAISY barcode screen, we employed the CLOVER pipeline to generate a new set of 2000 optimized barcode sequences to test in a second pooled screen (Methods). We generated a lentiviral library with new optimized barcodes (DAISY 2nd screen) as well as a set of controls from the original screen (DAISY 1st screen) to serve as internal benchmarks (Figure 3H). The ML-optimized DAISY barcodes significantly increased the average barcode entropy compared with the 1st screen, with top barcodes achieving an entropy increase of over 3 bits, suggesting an ~10-fold increase of the number of lineages that the barcode is capable of tracking (Figure 3H, Table S7) We tested the 2nd screen DAISY barcodes in both human melanoma and lung adenocarcinoma cell lines and found that barcode entropies were comparable between the two cell types. Thus, our pooled second screen validated the exploratory and predictive power of CLOVER to identify optimal barcode sequences within the vast target sequence space (Figure 4A).
Figure 4. ML-optimized DAISY barcodes have robust performance across cell lines with doxycycline-controllable tunability.
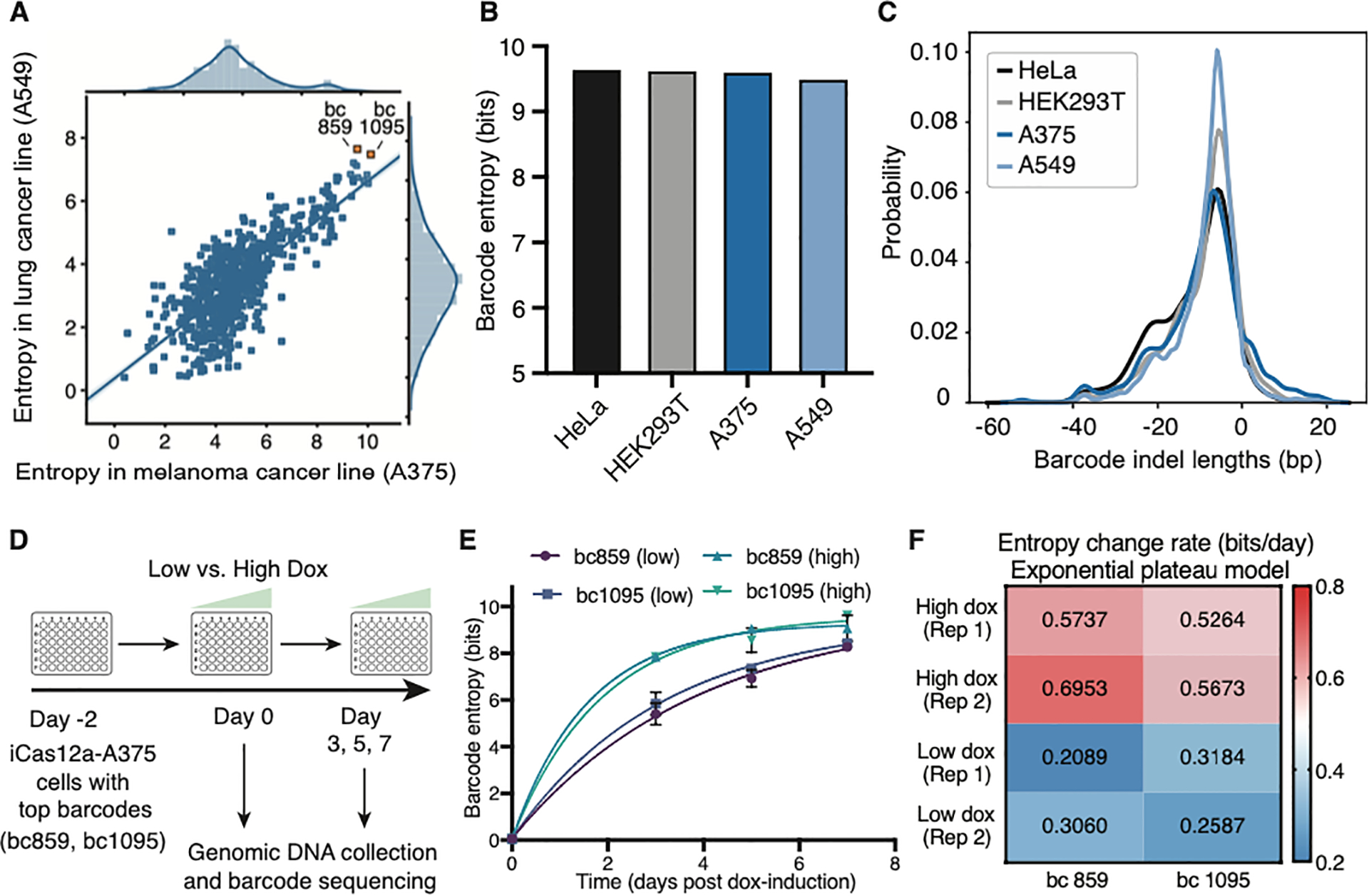
(A) Comparison of barcode entropy demonstrating consistent performance of CLOVER-optimized DAISY barcodes in A375 melanoma and A549 lung adenocarcinoma cell lines. Top barcodes used in later experiments are highlighted. (B) Comparison of total barcode entropy across all clones within each indicated cell type. (C) Consistent indel mutation length distributions of editing outcomes within the DAISY barcode (bc859) across cell lines (D) Experiment design to measure doxycycline-dependent tunability of top DAISY barcodes in A375 cells. Low and High-dox were 40 and 1000 ng/mL. (E) Change in the barcode entropy over time using low and high-dox. (F) Rate kinetics of barcode entropy (based on the Exponential plateau model) across doxycycline dosages and biological replicates.
To further confirm the portability of DAISY barcodes, we tested a top barcode after CLOVER optimization (bc859) in additional cell types, in which we included a static tag that marks clonal cell populations. We delivered this barcode using a lentiviral vector to four different cell lines (A375, A549, HeLa, and HEK293T) derived from lung epithelial, cutaneous skin, cervical, and kidney tissues, respectively that expressed Cas12a. The total barcode entropy across each cell type was consistently ~9.5 bits after 10 days of editing (Figure 4B and S4A). Furthermore, the indel length distributions showed an enrichment of small deletions (-6 bp) across cell lines, suggesting that DNA repair activity did not skew the evolution of evolved bc859 sequences in a cell-type-specific fashion (Figure 4C). DAISY barcodes performed robustly across cell types, supporting that it is a portable tool for lineage tracking.
Finally, we tested the tunability of two top-performing DAISY barcodes, bc859 and bc1095, using a doxycycline-inducible Cas12a cell line (Figure 4D). Similar to a recently described one-phase exponential decay model (Park et al., 2021), we derived the entropy change rate of these top DAISY barcodes based on our longitudinal measurements (Figure 4E and 4F). We found that by varying the doxycycline concentration, the entropy change rate of the barcodes could be tuned to range from ~0.25 bits/day (low dosage, slower barcode evolution) to ~0.5 bits/day (high dosage, faster barcode evolution). We further demonstrated doxycycline-dependent tunability of DAISY barcoding across additional cellular contexts (Figure S4C–F). The tunable feature of DAISY barcodes could facilitate applications in which the rate of barcode evolution needs to match the biological processes under investigation (Wagner and Klein, 2020). Taken together, these results demonstrated that the optimized Cas12a DAISY barcoding system is compact, high-capacity, and tunable.
Combining multiple DAISY barcodes into a high-capacity barcode array (DAISY chain)
While the basic DAISY system uses a two-target design, many published CRISPR barcode systems use 8–10 Cas9 target sites, or deliver more than 20 copies of the same Cas9 barcode to increase tracking capacity (Bowling et al., 2020; Quinn et al., 2021; Simeonov et al., 2021). Such designs are expected to have higher capacity, as multiple target sites could evolve independently and generate more unique outcomes for lineage tracking. To this end, we concatenated top DAISY barcode sequences (bc859 and bc1095) into a DAISY chain barcode (Figure 5A). We measured the capacity of the DAISY chain as in previous experiments (Supplementary Protocol Figure 2). Over the course of 9 days, indels accumulated at the expected cut sites (Figure 5B). Overall, this 120-bp DAISY chain generated ~66,000 unique edits, reaching over ~12 bits of entropy (Figure 5C). Strikingly, the indel profiles and length distribution demonstrated the rarity of inter-site deletions (Figure 5D). This contrasts with Cas9 barcode arrays that usually generated large deletions (Bowling et al., 2020; McKenna et al., 2016), and helps to explain the high capacity of the DAISY chain barcode. Further, we profiled the sequence evolution of the DAISY chain by assigning alleles to clonal populations using the associated static tag. Diverse barcode alleles accumulated over time, with most alleles assigned to a single clone, demonstrating the capacity of the DAISY barcode to uniquely label subclonal lineages (Figure 5E). Additionally, we designed a competition assay, between cells with and without barcode editing, to measure genotoxicity from multiplexed Cas12a cleavage of the DAISY barcodes. We did not observe competitive advantage of unedited cells, supporting that DAISY barcoding is not genotoxic (Figure S4B). Taken together, these results support the scalability and low toxicity of the DAISY barcoding method (Supplementary Protocol Figure 1).
Figure 5. Concatenation of DAISY barcode into a high-capacity DAISY chain barcode array.
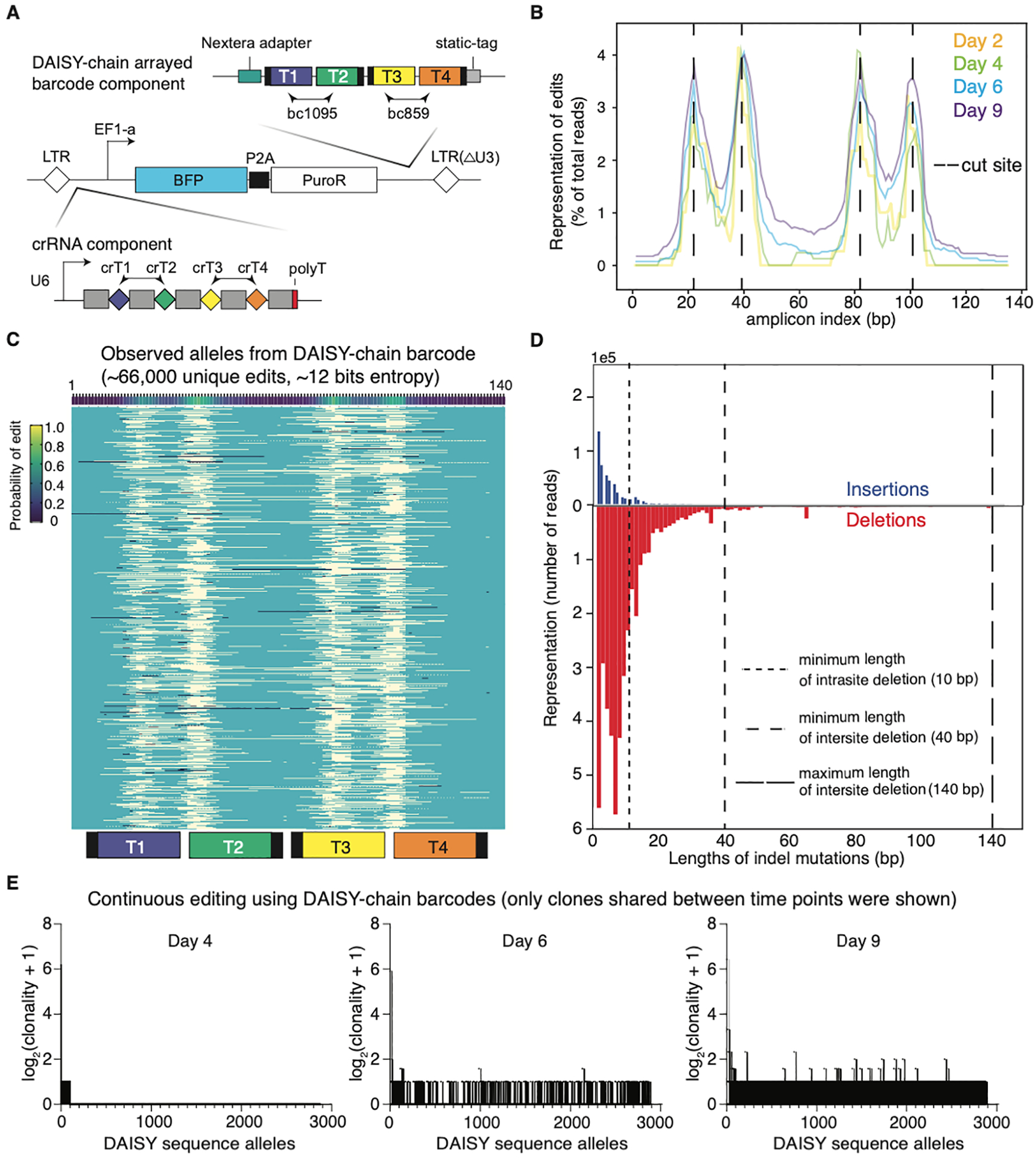
(A) Design of a two-DAISY barcode array using top optimized DAISY designs (bc859 and bc1095), encoded in a lentiviral vector. (B) Editing events distribution within the DAISY barcode array over the 9-day experiment. (C) Observed barcode alleles generated by the 120-bp DAISY barcode array, with light yellow showing deletions, and dark blue showing insertions. The probability of editing derived from all alleles are shown on top, and the position of four target sites are shown at bottom. (D) Lengths of indel mutations from all alleles using DAISY chain barcode array. Dash lines marked inter-site deletion limits. (E) The number of clones associated with each DAISY sequence allele is plotted on the y-axis for three different timepoints (Day-4, Day-6, and Day-9). Each allele is given an index on the x-axis.
Cas12a-based single-cell barcoding-profiling recovers lineage history and gene expression profiles at scale, revealing high-memory genes in human cancer cells
To use DAISY barcoding to uncover lineage information and single cell gene expression, we cloned a top DAISY barcode into a lentiviral vector in which edited barcodes would be transcribed and captured by single-cell RNA-seq (Figure 6A). Our single-cell DAISY barcode (scDAISY-seq-v1) vector contained a single cassette in which the crRNAs targeting the DAISY barcode were followed by the target sequences, with a static tag to label the founding clonality of cells (Supplementary Protocol Figure 3A). We transduced melanoma cells with doxycycline-inducible Cas12a and the scDAISY-seq-v1 vector. We bottlenecked the cells to have approximately 5 parental cells, induced Cas12a to initiate editing of the DAISY barcodes, and harvested cells for single-cell RNA-seq (Figure 6A).
Figure 6. Single-cell demonstration with optimized DAISY barcodes recovers lineage history and transcriptomic information.
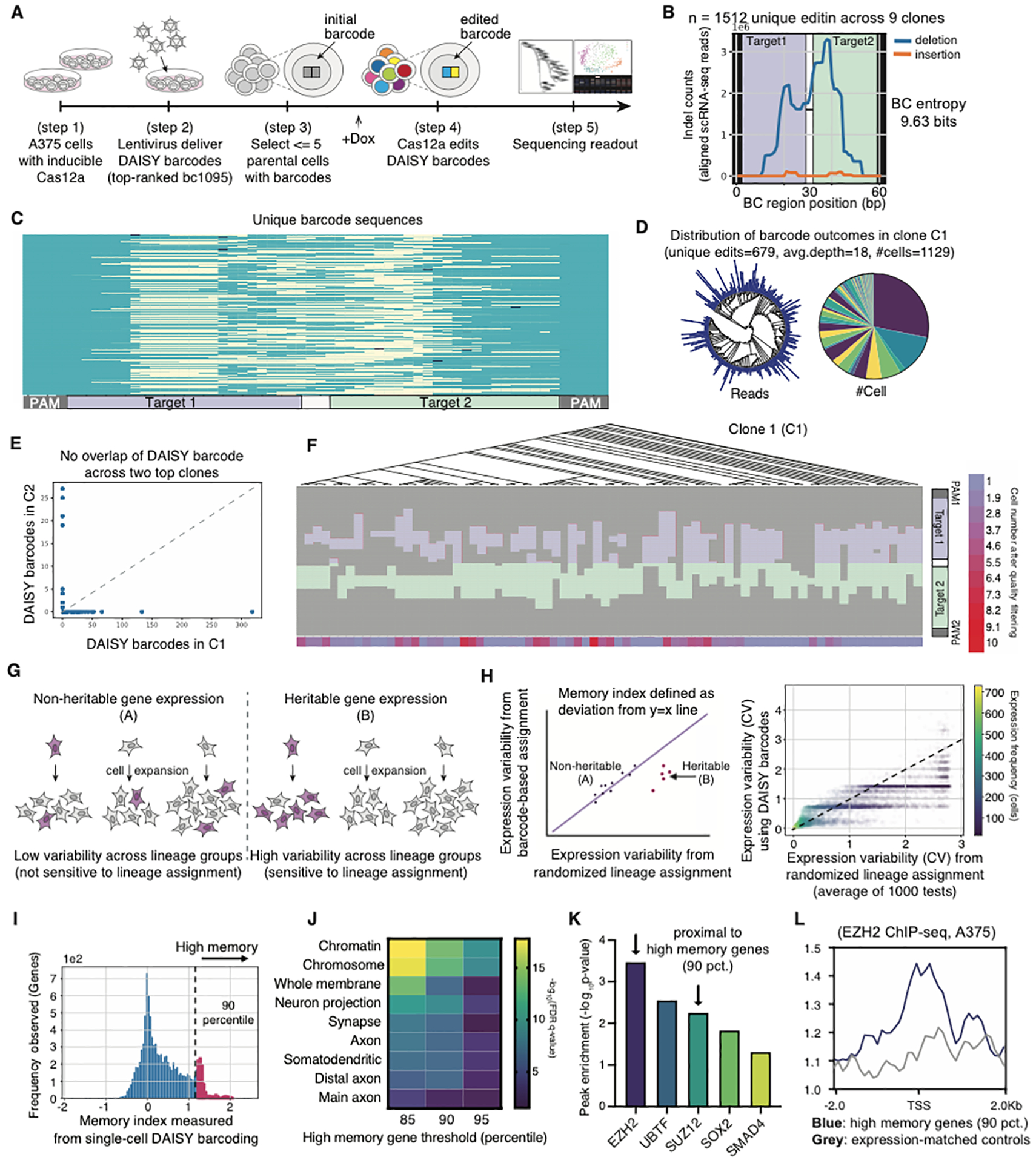
(A) Design of single-cell experiment using lentiviral delivery of an optimized DAISY barcode (scDAISY-seq). (B) Distribution of editing outcomes within the DAISY barcode (BC) region. Barcode entropy from single-cell data shown on right. (C) Unique barcode sequences recovered from scRNA-seq with yellow marks deletions and dark blue marks insertions. (D) Lineage tree reconstructed from single-cell barcode sequences of largest Clone 1 (C1), read counts shown in log scale. Pie charts on the right showing the cell distribution of identified unique lineages. (E) Homoplasy check showing no overlap between DAISY barcode sequences recovered from the largest two clones C1 and C2. (F) Reconstructed lineage tree from C1 using DAISY barcodes. Observed edits are illustrated below leaves of the tree. Purple and green bars indicate edits within two target sites. Heatmaps indicate cell numbers after quality filtering. (G) Illustration of transcriptional memory showing that an expressed gene (amber) can exhibit non-heritable/heritable expression patterns depending on if its expression level persists within certain lineages. (H) (Left) Quantitative definition of a memory index using single-cell transcriptomic data with randomized (x-axis) vs. barcode-defined (y-axis) lineage assignments. (Right) Data from scDAISY-seq were analyzed to calculate memory index for each gene. CV is the coefficient of variation of gene expression (see Methods). (I) The distribution of memory index values across all genes. (J) Top significantly enriched gene sets from found high memory genes. (K) Top 5 proteins enriched proximally to the high memory genes (90 percentile) based on ENCODE data. (L) ChIP-Seq peak profiles of high memory genes (90 percentile) in blue versus control genes (expression-matched, see Methods) in grey.
Recovery of lineage trees from barcoded single-cell profiles
From the single-cell RNA-seq data, we recovered sequencing reads corresponding to the DAISY barcodes in ~2000 cells, or ~70% of all cells that passed initial filtering to remove those with poor sequencing quality (Figure 6A, S5A–C, Methods). These single-cell DAISY barcode reads harbored a total of 1512 unique editing outcomes (Figure 6B). The bimodal distribution of the editing events within the barcode region (Figure 6B) demonstrated that most indels were within the target sites (T1, T2). Consistent with our bulk measurements, the DAISY barcode reached an entropy of more than 9-bits across the two dominant clonal populations. We examined the largest and dominant clonal population (Clone 1, or C1) defined by the static tag, which contained 1129 cells, with 679 unique edited barcodes (Figure 6C and 6D). Further, 60% of the edited barcode sequences uniquely labeled one descendant cell per sequence (Figure 6D). These unique editing outcomes had no observed overlap with barcode sequences from the second largest clone (Figure 6E). This means that, despite its small size, the DAISY barcode tracked a significant portion of cell lineages at single-cell resolution. This optimized DAISY barcode has comparable tracking capacity as several Cas9 barcodes, which are often longer and need multiple copies of the same barcode via transposition or repeated lentiviral transduction to boost their capacity (Supplementary Protocol Figure 1).
Identification of high-memory genes with heritable expression and potential biological significance
We integrated the lineage history recovered from the DAISY barcode, together with single-cell transcriptional profiles, to investigate the inheritance of gene expression (Shaffer et al., 2020) (Figure 6F and 6G). Using the scDAISY-seq data, we calculated the variability of gene expression within DAISY-barcode-defined lineage groups and compared them with a baseline averaged from randomized groups (Figure 6H, Supplementary Protocol Figure 4). We measured the strength of heritable gene expression, or transcriptional memory, by computing a memory index for each gene (Shaffer et al., 2020) (Figure 6H and 6I, Methods). Then, by ranking genes according to their memory indices, we identified a subset of high-memory genes exhibiting heritable expression patterns across cells (Figure 6I and Table S6).
We examined gene sets enriched within high-memory genes and identified an enrichment for neuronal and chromatin-related pathways (Ashburner et al., 2000; Subramanian et al., 2005) (Figure 6J). The association with neuronal genes in melanoma is intriguing as they arise from melanocytes that originate from neural crest cells (Zabierowski et al., 2011). While further investigation will be needed, a rare neural crest stem cell state has been associated with therapeutic resistance in melanoma (Rambow et al., 2018). In addition, the chromatin gene enrichment suggested that a feedback mechanism may be involved in maintaining heritable gene expression through epigenetic regulation (Shaffer et al., 2020; Takei et al., 2021). To assess this possibility, we conducted meta-analysis using ENCODE data (ENCODE Project Consortium, 2012) and identified enriched proteins that bound proximally to these high memory genes (Figure 6K). Intriguingly, two top proteins, EZH2 and SUZ12, are members of the polycomb repressive complex 2 (PRC2), which plays key roles in epigenetic regulation (Kim and Roberts, 2016; Holoch and Wassef et al., 2021) (Figure 6K). In support, we observed strong enrichment of EZH2 peaks at the transcriptional start sites (TSSs) of identified high-memory genes, in contrast to control genes with similar expression levels, using ChIP-seq data from melanoma cells (Su et al., 2019) (Figure 6L).
Investigating clonal dynamics through a time-course scDAISY-seq experiment
We next performed a time-course scDAISY-seq experiment to characterize clonal dynamics with Cas12a barcoding. In this experiment, we transduced A375 melanoma cells expressing doxycycline-inducible Cas12a with lentivirus containing the DAISY chain barcode placed within the UTR of eGFP for transcript recovery from single cells (scDAISY-seq-v2, Figure 7A, Supplementary Protocol Figure 3B). We bottlenecked the population such that subclones would be composed of ~10-to-50 cells by the first analysis at 7 days after Cas12a-induction. Furthermore, half of the population would then be used to re-seed wells for a final analysis at 14 days after Cas12a-induction. In total, we recovered 45914 cells in this experiment and ~700 clonal populations across all samples (Table S8). After filtering for high-quality cells (see Methods and Supplementary Protocol Figure 4) we recovered 9 clones well-represented across Day-7 and Day-14 timepoints. These clones showed stable barcode expression and variable population growth over time (Figure 7B–D, Table S9). Cells broadly clustered within transcriptional space consistent with the expectation that cell cycle-specific gene expression largely drives cell state heterogeneity across these melanoma cells (Figure S5A–F).
Figure 7. Clonal resampling over time using scDAISY-seq reveals features of transcriptional memory dynamics.
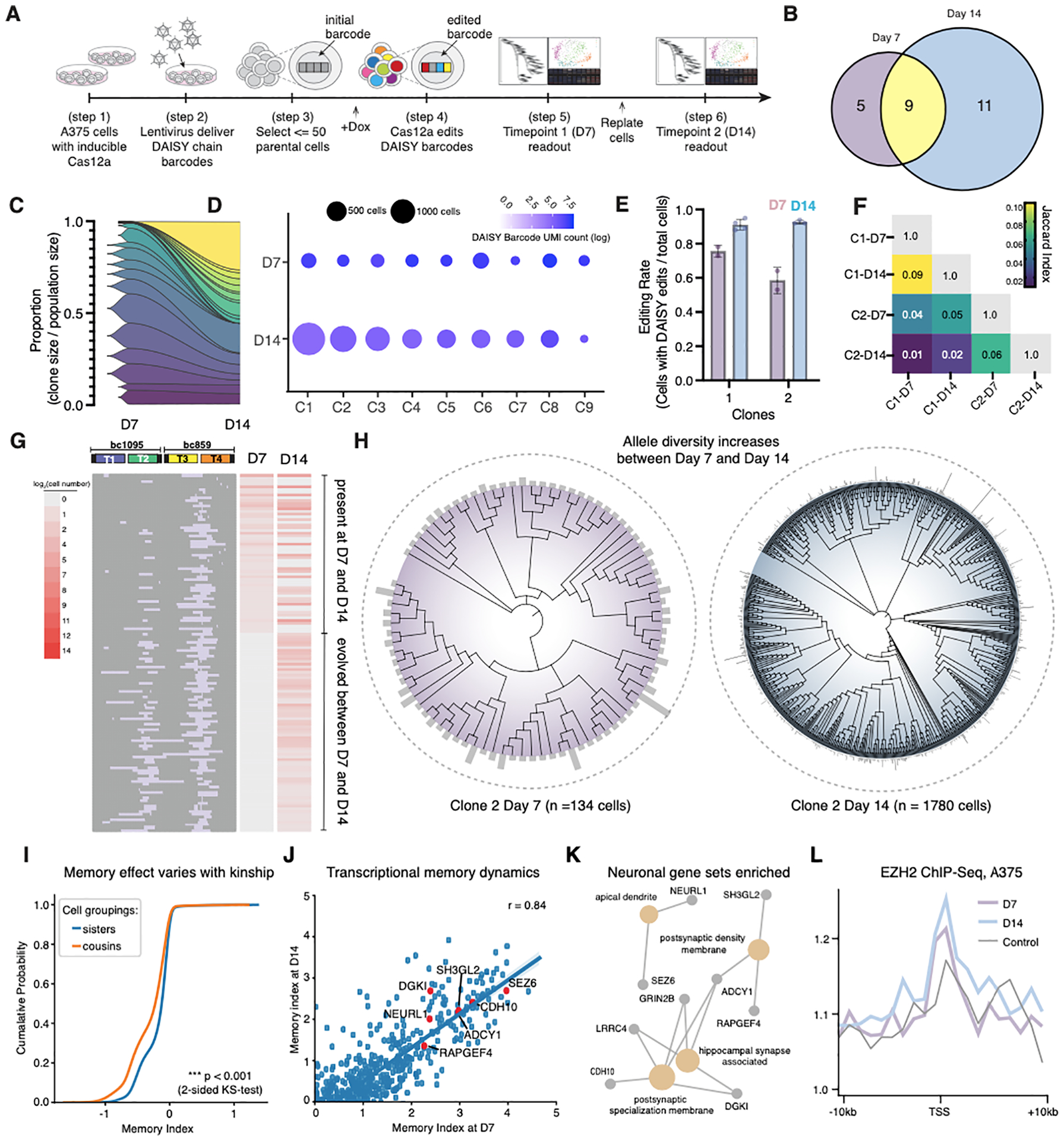
(A) Design of the time course scDAISY-seq experiment with clonal resampling. A375 cells expressing inducible AsCas12a were transduced with lentivirus containing DAISY barcodes. Cells were bottlenecked and allowed to proliferate for collections 7 and 14-days post doxycycline induction. (B) Venn diagram of the resampling of top-ranked clones by population size. (C) Fish plot of the change in proportions of the top-ranked clone sizes between Day-7 and Day-14. (D) Dot plot of the size and expression level across the top-ranked clones (E) Measurement of editing rate within two top represented clones over time. (F) Sets of alleles within two top represented clones were compared to each other using the Jaccard Index of similarity, where complete intersection of sets is 1.0 and complete independence of sets is 0.0. (G) Representative profile of indel formation within DAISY chain barcode from one biological replicate. Indels marked with purple, and cell numbers marked with a heatmap. (H) Phylogenetic reconstructions of a dominant clonal population at Day-7 and Day-14. Subclonal lineages defined by the DAISY barcode state are at the leaves of the tree and their population sizes are indicated by the adjacent bar heights with the maximum height of 10 cells (left) and 50 cells (right). The height of the bar scales linearly with population size. (I) Change in the distribution of the memory index within a clone (C2) when grouping cousins together versus sister cell groupings. (J) Memory index of genes with positive indices (averaged across all top represented clones) at Day-7 versus Day-14 (Pearson Correlation Coefficient is shown at the top right). A representative group of high memory genes is highlighted in red. (K) Gene set enrichment analysis of high memory genes reveals neuronal gene sets that include dendritic and synaptic biological components. (L) EZH2 ChIP-Seq of high memory genes across time using genes within the top 85th percentile of the memory index distribution.
We next explored the gene editing efficiency and diversity within these clones. Editing efficiency increased from a range of 50–70% to >95% for top-represented clones (C1 and C2) (Figure 7E). To assess genotoxic effects of barcoding with single-cell resolution, we examined the gene expression of cells with varying levels of editing of the recovered DAISY barcodes (Figure S5G–I). We did not observe evidence of genotoxic effects using a panel of DNA damage response genes (Ihry et al., 2018). After confirming efficient and non-toxic editing within the DAISY chain barcode, we assessed the diversity of editing within the top C1 and C2 clones. We observed minimal overlap between the set of alleles that evolved across clones. Therefore, the barcode capacity of the DAISY chain was sufficient to prevent homoplasy or independent evolution of the same allele (Figure 7F). We then analyzed the editing outcomes within a top represented clone (C1) present at both timepoints. We observed accumulating indels within the DAISY chain barcode as the cells continued to proliferate within the clonal population (Figure 7G). These editing outcomes allowed us to perform phylogenetic reconstruction of top-represented clone at both timepoints (Figure 7H). The results showed that the number of subclones increased ~8-fold (from 93 at Day-7 to 746 at Day-14), while the population size increased by ~12-fold (Figure 7H). These data support that barcode editing of the DAISY chain captured lineage bifurcations between the two timepoints. Thus, DAISY chain barcodes enabled longitudinal single-cell lineage tracking.
Dynamics and reproducibility of transcriptional memory in A375 melanoma cells
We analyzed lineage information encoded within DAISY barcodes along with single-cell transcriptional profiles to further evaluate transcriptional memory. We calculated the memory index of each gene and visualized the distribution. The distribution was centered at 0 (no memory effect) and had a right skew populated by genes with a putative memory effect (Figure S6A and S6B). With the increased complexity of allelic information within the DAISY chain, we evaluated the relationship between lineage distance and the memory effect. First, we recalculated the memory index of each gene when grouping cousin cells together as opposed to restricting the cell groupings to sister cells that share a most recent common ancestor (MRCA) (see Methods). Interestingly, the memory effect was significantly weakened when grouping cousin cells together, as indicated by a shift in the memory index distribution relative to the distribution generated through sister cell groupings (Figure 7I). The weakened effect gives insight into the permanence of transcriptional memory. If we assume that the barcode was evolving at a rate of ~0.5 bits / day (Figure 4F) and that cousins are differentiable with 2-bits of information (encoding 4 states), then cousins are uniquely marked by a DAISY barcode edit within a subclone after ~4 days. Therefore, the memory effect may weaken within this timeframe, which is consistent with previous imaging studies that investigate the dynamics of transcriptional memory and H3K27me3 deposition (Shaffer et al., 2020; Takei et al., 2021).
Next, we analyzed the reproducibility of the memory effects across samples within our dataset. First, we observed a positive correlation in the memory effect when comparing across biological replicates (Figure S6E). Second, we assessed memory effect across different clones, and found strong evidence for inter-clonal correlation (Figure S6F), which reinforced results detected earlier from a single clone (Figure 6). Third, we assessed how stable the memory effects were over time. We observed a positive correlation in memory effects across Day-7, Day-14 for genes with a positive memory index (Figure 7J). Therefore, the consistency of the observed memory effect across biological replicates, clones, and timepoints supports that an underlying mechanism regulates transcriptional memory, as opposed to purely stochastic effects (Holoch et al., 2021). Consistent with this conclusion and our initial experiment, high-memory genes fall within dedifferentiated cellular states (Figure 7K, S7A and S7C). Notably, high memory genes were involved in neuronal functions like dendrite and synapse formation. Lastly, as before, we also observed evidence of EZH2 binding at the TSS of high memory genes at Day-7 and Day-14 (Figure 7L). Therefore, EZH2 may regulate the cell state transitions of melanoma cells into dedifferentiated cell states composed of high memory genes. Taken together, DAISY barcoding coupled with single-cell transcriptomic analysis demonstrated the ability to uncover gene expression dynamics that are otherwise not revealed by static gene expression measurements.
DISCUSSION
We present the Cas12a-based DAISY system as a compact, tunable, and high-capacity CRISPR barcoding method. Our data suggest that Cas12a editing results in intrinsically more diverse editing outcomes than Cas9. This phenomenon may be due to differences in the biochemical properties of Cas12a, which include its PAM-distal and staggered cutting sites, and its faster dissociation from a genomic locus than Cas9 (Strohkendl et al., 2018; Zetsche et al., 2015; Swarts et al., 2017; Hussmann et al., 2021). We initially profiled thousands of Cas12a DAISY barcode sequences. Using this unique dataset, we optimized the barcode sequence to maximize barcode entropy with a machine learning pipeline (CLOVER), whose predictions were experimentally validated and refined. We provide a detailed list of features used in our model, with feature gradients and potential biological interpretations to aid future work (Table S7). In addition, we expect that the iterative strategy utilized by our CLOVER model can be applied to other molecular biology optimization problems (Yang et al., 2019). Combining top machine-learning-optimized DAISY barcodes, we generated an arrayed DAISY chain to demonstrate the scalability of a Cas12a-based barcoding system (Supplementary Protocol Figure 1) (Bowling et al., 2020; Kalhor et al., 2017).
We demonstrated the utility of DAISY barcoding in single-cell experiments (scDAISY-seq). Our results suggest that a 60-bp DAISY barcode can label hundreds of descendants from a parent cell. Furthermore, we scaled up the scDAISY-seq approach using a DAISY chain barcode to perform a time-course experiment that enabled clonal resampling at subclonal resolution. These two independent experiments revealed heritable gene expression patterns and suggested EZH2 (PRC2 complex) as a potential epigenetic regulator of transcriptional memory. Our results may also have practical implications. Using The Cancer Genome Atlas (TCGA) datasets, we analyzed gene expression of skin cutaneous melanoma (SKCM) tumors from patients and observed that EZH2 expression levels were positively correlated with the tumor transcriptional heterogeneity, a property that relates to transcriptional memory, and negatively correlated with overall patient survival (Shaffer et al., 2020; Tiffen et al., 2016) (Figure S7B and S7D). Future studies using single-cell DAISY-seq could reveal adaptive mechanisms in cancer at the epigenetic level, extending our knowledge beyond the genetically encoded evolution of cancer (Bradner et al., 2017).
CRISPR-Cas9-based single-cell barcoding approaches have been utilized to study a wide breadth of biological processes (Alemany et al., 2018; Chan et al., 2019; Bowling et al., 2020; Spanjaard et al., 2018; Quinn et al., 2021). We expect that implementation of Cas12a-based barcoding methods will further contribute to the lineage tracking toolkit. For example, compact DAISY barcodes minimize the likelihood of genotoxic stress due to editing relative to larger Cas9-based arrayed barcodes (Meyers et al., 2017; Wang et al., 2017). Furthermore, given the simplicity of multi-target editing using the Cas12a system, we envision that combining genetic perturbations with scDAISY-seq will enhance our understanding of how genes control the development of cellular states (Adamson et al., 2016; Datlinger et al., 2017; Dixit et al., 2016). A combined measurement of cell state and ontogeny following a genetic perturbation will elucidate how genes control cell fate decisions during development and how these decisions go awry during disease.
Limitations of the technology
Our study introduces a portable, tunable, and scalable technology for lineage tracking using a machine learning optimized Cas12a barcode. The DAISY barcode system could still be improved in several ways. For example, additional rounds of CLOVER optimization to uncover better DAISY barcodes would, in theory, further increase the barcode entropy. This could help decrease the need for the delivery of multiple target sites that could result in genotoxic stress due to editing (Meyers et al., 2017; Wang et al., 2017). Second, DAISY barcodes could be coupled to functional inputs to record biological signals, like the activation of signal transduction pathways or cell cycles, in addition to the lineage information of a cell (Kempton et al., 2020; Tang and Liu, 2018). Finally, further advances in barcoding tunability would allow greater flexibility to record biological processes of different time scales, ranging from the rapid cell differentiation in embryonic development, to the gradual process of neurogenesis in the adult brain (Spalding et al., 2013; Ogawa, 1993).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to members of the Cong and Winslow laboratories for their support. We are grateful to Jess Hebert, Ravi Dinesh, Sarah Pierce, Feng Pan, Li Zhu, and Will Johnson for helpful discussions on the manuscript, and support on experiments. We thank the following scientists: pY108 (lenti-AsCpf1) was a gift from Dr. Feng Zhang (Addgene # 84739); pCAG-enAsCas12a-HF1 was a gift from Dr. Keith Joung (Addgene # 107942). This work was supported by the National Institutes of Health [R35-HG011316 to L.C.] and [R01-CA231253 to M.M.W.]; a Donald and Delia Baxter Foundation Faculty Scholar award [to L.C.]; National Science Foundation [NSF Awards 1953686 to M.W. and 1953415 to L.C.]. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. 2018261164 [to N.W.H.]. The computational analysis is supported by NIH grant 1S10OD023452 to Stanford Genomics Cluster (SCG).
Footnotes
REFERENCES
- Abbasi-yadkori Y, Pál D, and Szepesvári C (2011). Improved Algorithms for Linear Stochastic Bandits. Adv. Neural Inf. Process. Syst 24. [Google Scholar]
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alemany A, Florescu M, Baron CS, Peterson-Maduro J, and van Oudenaarden A (2018). Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112. [DOI] [PubMed] [Google Scholar]
- Allen F, Crepaldi L, Alsinet C, Strong AJ, Kleshchevnikov V, De Angeli P, Páleníková P, Khodak A, Kiselev V, Kosicki M, et al. (2018). Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer P (2002). Using Confidence Bounds for Exploitation-Exploration Trade-offs. J. Mach. Learn. Res 3, 397–422. [Google Scholar]
- Barrangou R, and Doudna JA (2016). Applications of CRISPR technologies in research and beyond. Nat. Biotechnol 34, 933–941. [DOI] [PubMed] [Google Scholar]
- Biddy BA, Kong W, Kamimoto K, Guo C, Waye SE, Sun T, and Morris SA (2018). Single-cell mapping of lineage and identity in direct reprogramming. Nature 564, 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling S, Sritharan D, Osorio FG, Nguyen M, Cheung P, Rodriguez-Fraticelli A, Patel S, Yuan W-C, Fujiwara Y, Li BE, et al. (2020). An Engineered CRISPR-Cas9 Mouse Line for Simultaneous Readout of Lineage Histories and Gene Expression Profiles in Single Cells. Cell 181, 1410–1422.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional Addiction in Cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, O’Day DR, Pliner HA, Kingsley PD, Deng M, Daza RM, Zager MA, Aldinger KA, Blecher-Gonen R, Zhang F, et al. (2020). A human cell atlas of fetal gene expression. Science 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan MM, Smith ZD, Grosswendt S, Kretzmer H, Norman TM, Adamson B, Jost M, Quinn JJ, Yang D, Jones MG, et al. (2019). Molecular recording of mammalian embryogenesis. Nature 570, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, McKenna A, Schreiber J, Haeussler M, Yin Y, Agarwal V, Noble WS, and Shendure J (2019). Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. Nucleic Acids Res. 47, 7989–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, Schuster LC, Kuchler A, Alpar D, and Bock C (2017). Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussmann JA, Ling J, Ravisankar P, Yan J, Cirincione A, Xu A, Simpson D, Yang D, Bothmer A, Cotta-Ramusino C, et al. (2021). Mapping the genetic landscape of DNA double-strand break repair. Cell 184, 5653–5669.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holoch D, Wassef M, Lövkvist C, Zielinski D, Aflaki S, Lombard B, Héry T, Loew D, Howard M, and Margueron R (2021). A cis-acting mechanism mediates transcriptional memory at Polycomb target genes in mammals. Nat. Genet 53, 1686–1697. [DOI] [PubMed] [Google Scholar]
- Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, Kommineni S, Chen J, Sondey M, Ye C, et al. (2018). p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med 24, 939–946. [DOI] [PubMed] [Google Scholar]
- Jones MG, Khodaverdian A, Quinn JJ, Chan MM, Hussmann JA, Wang R, Xu C, Weissman JS, and Yosef N (2020). Inference of single-cell phylogenies from lineage tracing data using Cassiopeia. Genome Biol. 21, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhor R, Mali P, and Church GM (2017). Rapidly evolving homing CRISPR barcodes. Nat. Methods 14, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull JM, and Zador AM (2018). Cellular barcoding: lineage tracing, screening and beyond. Nat. Methods 15, 871–879. [DOI] [PubMed] [Google Scholar]
- Kempton HR, Goudy LE, Love KS, and Qi LS (2020). Multiple Input Sensing and Signal Integration Using a Split Cas12a System. Mol. Cell 78, 184–191.e3. [DOI] [PubMed] [Google Scholar]
- Kester L, and van Oudenaarden A (2018). Single-Cell Transcriptomics Meets Lineage Tracing. Cell Stem Cell 23, 166–179. [DOI] [PubMed] [Google Scholar]
- Kim KH, and Roberts CWM (2016). Targeting EZH2 in cancer. Nat. Med 22, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Kim J, Hur JK, Been KW, Yoon S-H, and Kim J-S (2016). Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol 34, 863–868. [DOI] [PubMed] [Google Scholar]
- Kim HK, Min S, Song M, Jung S, Choi JW, Kim Y, Lee S, Yoon S, and Kim HH (2018). Deep learning improves prediction of CRISPR-Cpf1 guide RNA activity. Nat. Biotechnol 36, 239–241. [DOI] [PubMed] [Google Scholar]
- Kleinstiver BP, Sousa AA, Walton RT, Esther Tak Y, Hsu JY, Clement K, Welch MM, Horng JE, Malagon-Lopez J, Scarfò I, et al. (2019). Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nature Biotechnology 37, 276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT, Aghazadeh A, Hiatt J, Tse D, Roth TL, Apathy R, Shifrut E, Hultquist JF, Krogan N, Wu Z, et al. (2019). Large dataset enables prediction of repair after CRISPR–Cas9 editing in primary T cells. Nat. Biotechnol 37, 1034–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Srinivasan S, Li C-Y, Ho I-L, Rose J, Shaheen M, Wang G, Yao W, Deem A, Bristow C, et al. (2019). Pooled library screening with multiplexed Cpf1 library. Nat. Commun 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno GL, La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Findlay GM, Gagnon JA, Horwitz MS, Schier AF, and Shendure J (2016). Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nature Genetics 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, et al. (2019). An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M (1993). Differentiation and proliferation of hematopoietic stem cells. Blood 81, 2844–2853. [PubMed] [Google Scholar]
- Park J, Lim JM, Jung I, Heo SJ, Park J, Chang Y, Kim HK, Jung D, Yu JH, Min S, Yoon S, Cho SR, Park T, Kim HH. (2021). Recording of elapsed time and temporal information about biological events using Cas9. Cell 184, 1047–1063.e23. [DOI] [PubMed] [Google Scholar]
- Perli SD, Cui CH, and Lu TK (2016). Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science 353. [DOI] [PubMed] [Google Scholar]
- Quinn JJ, Jones MG, Okimoto RA, Nanjo S, Chan MM, Yosef N, Bivona TG, and Weissman JS (2021). Single-cell lineages reveal the rates, routes, and drivers of metastasis in cancer xenografts. Science 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, Wagner DE, McKenna A, Pandey S, Klein AM, Shendure J, Gagnon JA, and Schier AF (2018). Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol 36, 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, Karras P, Brown D, Chang YH, Debiec-Rychter M, et al. (2018). Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 174. [DOI] [PubMed] [Google Scholar]
- Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, et al. (2017). The Human Cell Atlas. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers ZN, McFarland CD, Winters IP, Naranjo S, Chuang C-H, Petrov D, and Winslow MM (2017). A quantitative and multiplexed approach to uncover the fitness landscape of tumor suppression in vivo. Nat. Methods 14, 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusmevichientong P, and Tsitsiklis JN. “Linearly Parameterized Bandits.” Mathematics of Operations Research 3, 5.2. [Google Scholar]
- Saelens W, Cannoodt R, Todorov H, and Saeys Y (2019). A comparison of single-cell trajectory inference methods. Nat. Biotechnol 37, 547–554. [DOI] [PubMed] [Google Scholar]
- Setty M, Kiseliovas V, Levine J, Gayoso A, Mazutis L, and Pe’er D (2019). Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol 37, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer SM, Emert BL, Reyes Hueros RA, Cote C, Harmange G, Schaff DL, Sizemore AE, Gupte R, Torre E, Singh A, et al. (2020). Memory Sequencing Reveals Heritable Single-Cell Gene Expression Programs Associated with Distinct Cellular Behaviors. Cell 182, 947–959.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE (1948). A mathematical theory of communication. The Bell System Technical Journal 27, 379–423. [Google Scholar]
- Shen MW, Arbab M, Hsu JY, Worstell D, Culbertson SJ, Krabbe O, Cassa CA, Liu DR, Gifford DK, and Sherwood RI (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonov KP, Byrns CN, Clark ML, Norgard RJ, Martin B, Stanger BZ, Shendure J, McKenna A, and Lengner CJ (2021). Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell 39, 1150–1162.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Boström E, Westerlund I, Vial C, Buchholz BA, et al. (2013). Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanjaard B, Hu B, Mitic N, Olivares-Chauvet P, Janjuha S, Ninov N, and Junker JP (2018). Simultaneous lineage tracing and cell-type identification using CRISPR-Cas9-induced genetic scars. Nat. Biotechnol 36, 469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohkendl I, Saifuddin FA, Rybarski JR, Finkelstein IJ, and Russell R (2018). Kinetic Basis for DNA Target Specificity of CRISPR-Cas12a. Mol. Cell 71, 816–824.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su D, Wang W, Hou Y, Wang L, Wang Y, Yang C, Liu B, Chen X, Wu X, Wu J, et al. (2019). Bimodal Regulation of the PRC2 Complex by USP7 Underlies Melanomagenesis. [DOI] [PMC free article] [PubMed]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabula Muris Consortium, Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis, Cell type annotation, Writing group, Supplemental text writing group, and Principal investigators (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Yun J, Zheng S, Ollikainen N, Pierson N, White J, Shah S, Thomassie J, Suo S, Eng C-HL, et al. (2021). Integrated spatial genomics reveals global architecture of single nuclei. Nature 590, 344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, and Liu DR (2018). Rewritable multi-event analog recording in bacterial and mammalian cells. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffen J, Wilson S, Gallagher SJ, Hersey P, and Filipp FV (2016). Somatic copy number amplification and hyperactivating somatic mutations of EZH2 correlate with DNA methylation and drive epigenetic silencing of genes involved in tumor suppression and immune responses in melanoma. Neoplasia 18, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, et al. (2020). A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DE, and Klein AM (2020). Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet 21, 410–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, and Sabatini DM (2017). Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 168, 890–903.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb C, Rodriguez-Fraticelli A, Camargo FD, and Klein AM (2020). Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, Rajewsky N, Simon L, and Theis FJ (2019). PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KK, Wu Z, and Arnold FH (2019). Machine-learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694. [DOI] [PubMed] [Google Scholar]
- Zabierowski SE, Baubet V, Himes B, Li L, Fukunaga-Kalabis M, Patel S, McDaid R, Guerra M, Gimotty P, Dahmane N, et al. (2011). Direct reprogramming of melanocytes to neural crest stem-like cells by one defined factor. Stem Cells 29, 1752–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, et al. (2017). Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol 35, 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.