

Abstract
A long-standing question in cancer biology has been why oxygenated tumors ferment the majority of glucose they consume to lactate rather than oxidizing it in their mitochondria, a phenomenon known as the ‘Warburg effect’. An abundance of evidence shows not only that most cancer cells have fully functional mitochondria but also that mitochondrial activity is important to proliferation. It is therefore difficult to rationalize the metabolic benefit of cancer cells switching from respiration to fermentation. An emerging perspective is that rather than mitochondrial metabolism being suppressed in tumors, as is often suggested, mitochondrial activity increases to the level of saturation. As such, the Warburg effect becomes a signature of excess glucose being released as lactate due to mitochondrial overload.
Keywords: the Warburg effect, aerobic glycolysis, aerobic fermentation, cancer metabolism, mitochondrial metabolism
Warburg’s paradox
Glucose is primarily metabolized via glycolysis in the cytosol of mammalian cells, which produces two moles of pyruvate and two moles of NADH for every mole of glucose. The pyruvate and NADH derived from glucose have two major fates. Either they can be transported into mitochondria to support respiration, or pyruvate can be reduced by NADH to produce lactate and NAD+. The former is generally referred to as ‘glucose oxidation’ and yields on the order of 30–38 ATP per molecule of glucose [1]. The latter can occur in the absence of oxygen and is called ‘fermentation’. The transformation of glucose into lactate produces only a net gain of 2 ATP and, because lactate is generally assumed to be excreted from the cell, prevents the use of glucose carbon for biomass.
Proliferation is a defining characteristic of cancer development and progression [2]. For a cell to proliferate, it must replicate its contents. Amino acid synthesis is required for proteins, lipid synthesis for membranes, and nucleotide synthesis for genetic material. This anabolic burden and its associated energetic demands would appear to be best supported by glucose oxidation. A century ago, however, Otto Warburg made the seminal discovery that oxygenated slices of tumor tissue ferment most of the glucose they consume rather than oxidizing it [3]. Identifying biochemical mechanisms to explain Warburg’s seemingly counterintuitive observations has been the focus of much research and discussion. One argument put forward is that, although the yield is lower, fermentation of glucose produces ATP faster than oxidation of glucose in mitochondria [4,5]. Alternatively, the limited physical volume of a cell might make fermentation of glucose a more spatially efficient way to produce ATP than oxidation of glucose [6]. These possibilities and many others have been reviewed in detail elsewhere [7,8]. The goal of the current article is to highlight findings from recent years that support the idea of a ‘respiratory bottleneck’ [9], where glucose fermentation occurs in oxygenated cancer cells as a result of mitochondrial pathways being saturated.
The Warburg anachronism
Warburg initially used the term ‘aerobic glycolysis’ to describe the tendency of tumors to transform glucose into lactate in the presence of oxygen [3], a phenomenon that was later called the ‘Warburg effect’ [10]. Although the expression aerobic glycolysis continues to be widely used today, it should be recognized as an anachronism. At the time of Warburg’s early studies using Flexner-Jobling rat carcinoma in the 1920s [3], the molecular details of glycolysis and respiration had not yet been elucidated [11]. The word “glycolysis” was used to indicate production of lactate from glucose, a process that would more specifically be called fermentation in most modern biochemistry textbooks. Accordingly, we refer to the Warburg effect as ‘aerobic fermentation’ here rather than ‘aerobic glycolysis’. Indeed, the reduction of glucose-derived pyruvate in the presence of oxygen is commonly called ‘aerobic fermentation’ in other fields [12] and, by 1956, Warburg himself was using the term [13].
Mitochondria support proliferation
Although Warburg originally postulated that tumors have impaired respiration due to dysfunctional mitochondria, we now know that not to be the case for the majority of cancers [14]. In fact, the body of evidence supporting functional mitochondria in cancer cells is so extensive that we are unable to provide a complete review of all relevant studies here and point those who wish to obtain additional information to other resources [14–18]. Most directly, respirometry experiments dating back to Warburg show that cancer tissues do consume oxygen from their environment [19]. Similarly, by administering isotopically labeled tracers to cancer cells in culture, carbon from glucose has been tracked into mitochondrial pathways such as the tricarboxylic acid (TCA) cycle [20,21]. In recent years, isotopically labeled tracers have been given to animal models and human patients with cancer, with results similar to in vitro experiments being obtained from mass spectrometry and NMR analysis of resected tumors [22,23]. These studies and many others have demonstrated that cancer cell mitochondria oxidize not only glucose but also other nutrients, such as glutamine [24–27], fatty acids [28,29], acetate [30,31], and lactate [32–34]. The data are consistent with a multitude of other reports that indirectly support mitochondrial activity in cancer cells on the basis of changes in transcriptional networks and mitochondrial dynamics [35,36].
Interestingly, for some proliferating cells, recent data indicate that mitochondrial activity might be more important for the production of macromolecular precursors than for the generation of ATP [37]. Pathways for the biosynthesis of fatty acids, cholesterol, amino acids, and nucleotides all have one or more nodes in mitochondria [38]. Aspartate, in particular, is made through transamination of the TCA cycle intermediate oxaloacetate and is required for the synthesis of proteins, purines, and pyrimidines. Aspartate could theoretically be consumed from the tumor microenvironment, but levels of free aspartate in the circulation are low, and aspartate transport is inefficient in most mammalian cells [39–41]. As such, an essential function of mitochondrial respiration in proliferating cancer cells is to support aspartate synthesis [37,39]. The sensitivity of cancer cells to respiratory inhibitors is inversely correlated with intracellular aspartate levels, and supplying supraphysiological concentrations of exogenous aspartate to cancer cells in which respiration has been impaired is sufficient to restore normal proliferation rates [37,42]. Moreover, in vivo studies suggest that reduced aspartate availability might limit the growth of some tumors during hypoxia [42].
Although there is increasing recognition that oxidative metabolism persists in most cells exhibiting the Warburg effect, the amount of mitochondrial activity that is present has remained less clear. Conventionally, based on the pioneering work of Louis Pasteur in the 19th century, it is assumed that introducing oxygen to cells in a previously anaerobic environment will inhibit fermentation so that the majority of glucose is oxidized [15]. This raises the question of why most of the glucose consumed by proliferating cells undergoes fermentation in oxygenated conditions. One argument often made is that tumors reprogram their metabolism such that mitochondrial activity is suppressed, thereby reversing the regulatory mechanisms underlying Pasteur’s observations. Accordingly, even though cancer cells theoretically have the capacity to oxidize all of the glucose consumed, they do not. The rate of respiration that is needed to produce sufficient levels of ATP and/or macromolecular precursors during proliferation is not well defined. High levels of respiration may not be required to fulfill the energetic and anabolic demands of a cancer cell, thus enabling the majority of glucose consumed to be diverted away from mitochondrial oxidation without decreasing tumor growth. A potential outcome of reduced respiration might be that cancer cells are less reliant on oxygen and therefore can better endure intermittent hypoxia, which is a common feature of most tumors [43,44]. This model of functional but deactivated mitochondria is consistent with experimental evidence of respiration, and it also aligns with elements of Warburg’s original thinking by invoking a “switch” to a glycolytic phenotype where proliferating cells ‘prefer’ to ferment glucose rather than oxidize it.
Saturated mitochondrial activity
Starting with Warburg a century ago, researchers have sought to explain why tumors employ distinct mechanisms of metabolic regulation compared with most other cells and tissues. Specifically, investigators have aimed to understand why the effect first observed by Pasteur, namely inhibition of fermentation by oxygen, appears to be reversed in proliferating cells [15]. A number of intriguing theories have been proposed to rationalize why such an adaptation, though wasteful, might benefit cancer cells. Here, we offer a different opinion. We maintain that, rather than actively suppressing glucose oxidation as a means to support proliferation, the rate of glycolysis in cancer cells exceeds the maximum rate at which glucose can be oxidized. Hence, pyruvate and NADH are generated in the cytosol faster than they can be transported into and used by mitochondria. The result is fermentation, which is not a preferred metabolic phenotype of cancer cells but instead a secondary consequence of mitochondrial saturation. Lactate production is used as an overflow channel to allow NAD+ levels to be replenished (Figure 1).
Figure 1:
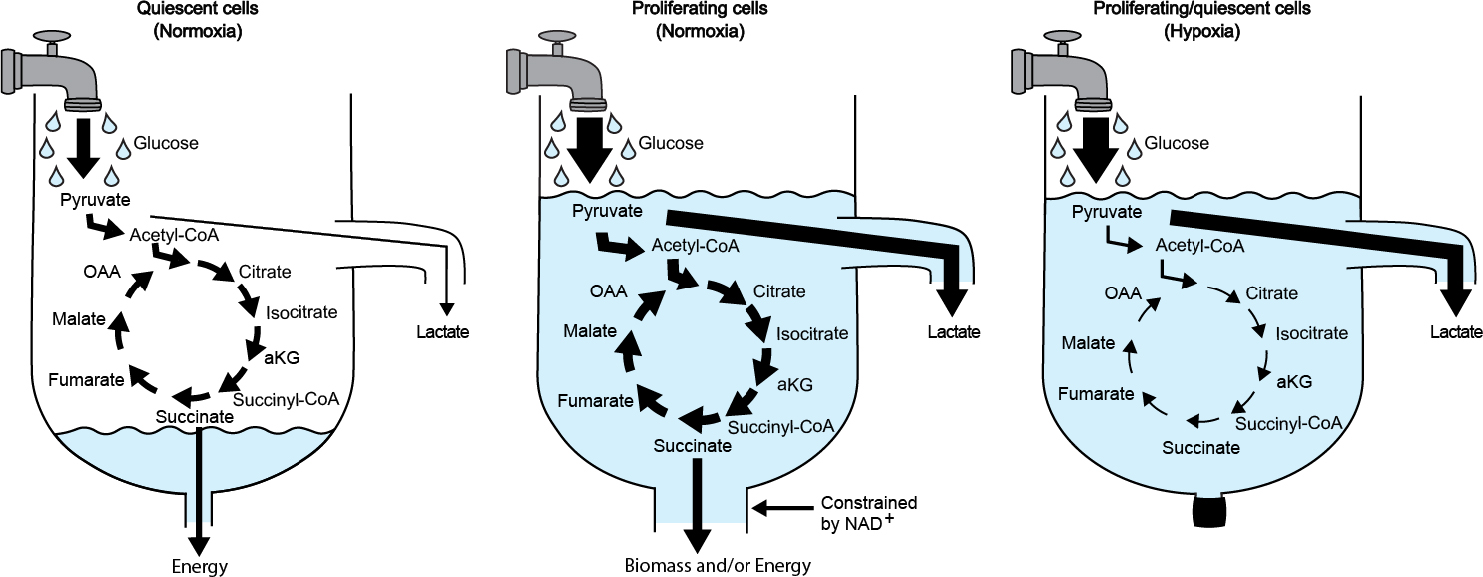
Sink overflow drain analogy. Water dripping out of the faucet represents glucose consumption and glycolytic activity. When the rate of glycolysis is slow in quiescent cells, pyruvate is oxidized by the tricarboxylic acid cycle to produce energy. This is equivalent to a slow drip, where the water is drained through the bottom of the sink (left). When the rate of glycolysis produces NADH faster than mitochondrial pathways can regenerate NAD+ in proliferating cells, then lactate is produced (middle). This is equivalent to a fast drip, where the water accumulates in the sink faster than the bottom drain can remove it, and the overflow drain goes into effect. In this analogy, the diameter of the drain at the bottom of the sink is determined by the rate NAD+ can be turned over by mitochondrial pathways (i.e., the higher the turnover, the wider the drain). During hypoxia, by extension, the drain is closed (right). Abbreviations: αKG, alpha-ketoglutarate; CoA, coenzymeA; OAA, oxaloacetate.
The idea that mitochondrial activity is saturated during proliferation is conceptually straightforward but challenging to prove experimentally. A complication is that tumors are composed of a mixed population of cells, some of which are not proliferating. Additionally, tumors often contain regions of necrosis and hypoxia. It is therefore difficult to discern the absolute level of respiration originating from oxygenated cancer cells in the proliferative state by metabolic analysis of bulk tumor tissue, and experimental purification of specific cell types from the tumor is not feasible, because processes such as cell sorting cause major perturbations in metabolism [45]. A simpler strategy is to apply in vitro models where proliferation can be controlled as an independent variable. Using fibroblasts that undergo reversible quiescence due to contact inhibition, respiration was found to be almost twofold higher during proliferation than in the quiescent state [35]. Transformation of the cells with H-Ras caused respiration to increase even further during proliferation [35]. Analysis of cultured T cells has led to similar conclusions [46]. In one notable study, when resting T cells were stimulated to induce proliferation, respiration increased by ~70% [47].
It is important to point out that glucose is not the only substrate that fuels respiration in proliferating cells. Thus, changes in respiration alone cannot be used to assess changes in glucose oxidation. It is possible, for example, to have increased oxidation of glucose without an accompanying increase in respiration when oxidation of a nonglucose substrate simultaneously decreases. An approach better suited to quantifying the flux of glucose oxidation is to trace labels from 13C-glucose into the TCA cycle. Using this technique, cell-culture studies have shown increased oxidation of glucose during proliferation relative to quiescence [48]. More impressively, multiple investigations of human lung tumors from patients administered 13C-glucose have yielded consistent results [49,50]. Despite the potential limitation of labeling in the TCA cycle being diluted by nonproliferating, necrotic, and hypoxic cells from bulk tumors, these cancerous tissues still show enhanced glucose oxidation compared with adjacent benign tissue.
The above reports demonstrate that cell proliferation promotes glucose oxidation in various settings. Nevertheless, most of the glucose consumed by proliferating cells is still fermented in the presence of oxygen. Assuming that lactate production is not a metabolic driver of cell proliferation and that oxidation is the preferred fate of glucose, what, then, constrains the amount of glucose that is used to fuel respiration? One possibility that has been suggested is that glycolysis produces pyruvate faster than the pyruvate dehydrogenase complex (PDHc) can oxidize it [51]. PDHc catalyzes the conversion of pyruvate to acetyl-coenzyme A, which is used as an intermediate to transfer glucose carbon into the TCA cycle. In some cells, the maximum rate of glycolysis may exceed the Vmax of PDHc by as much as 20-fold. Intriguingly, activation of PDHc reduces cancer cell proliferation and tumor growth [52–54]. Until recently, these findings seemed to reinforce the notion that suppressing oxidative metabolism benefits cellular proliferation. A key discovery was made by Luengo and colleagues in 2021, however, that helped shed new light on the interpretation of these results [55]. The new work shifted the focus from glucose carbon to redox cofactors. In addition to decarboxylating pyruvate, PDHc reduces NAD+ to NADH. The electron transport chain (ETC) recycles NADH back to NAD+ through a series of redox reactions that are coupled to ATP synthesis. Both NAD+ and ATP are important to support proliferation [56]. The issue is that NADH production may outpace ATP synthesis in proliferating cells, especially upon PDHc activation. Luengo and colleagues argue that glucose oxidation in proliferating cells is limited by the activity of ATP synthase, and they suggest that lactate is produced as a compensatory mechanism to recycle NAD+ faster than ATP synthase allows.
One factor that constrains ATP synthase activity is slow turnover of ATP to ADP. In other words, when the rate of ATP synthesis is faster than the rate of ATP hydrolysis, eventually ADP levels become depleted and ATP synthesis can no longer occur. Relative to their corresponding healthy tissues, some tumors do have slower rates of ATP synthesis [57]. Decreased ATP production occurs because tumors downregulate ATP-demanding processes that are characteristic of the tissue of origin (e.g., pancreatic tumors decrease synthesis of digestive enzymes). Although shedding ATP demands associated with healthy tissue function could enable cancer cells to continue proliferating at times when nutrients are scarce, the trade-off might be that it leads to constrained ATP synthase activity and supercharged mitochondria at times when nutrients are abundant.
A complementary line of reasoning is that glucose oxidation is limited in the presence of oxygen due to saturation of the malate-aspartate shuttle (MAS) and the glycerol 3-phosphate shuttle (G3PS) [58–62]. Glucose-derived NADH cannot be directly transported into mitochondria to fuel respiration. The MAS and G3PS provide an indirect mechanism to transfer its reducing equivalents to the mitochondrial ETC, while also replenishing cytosolic NAD+ so that glycolysis can continue. Prior work suggests that the rate of glycolysis in oxygenated tissues such as skeletal muscle [60], heart [59,61], and brain [62] can exceed the maximum speed at which the MAS and G3PS can operate. Computational and experimental analyses demonstrated that the same is true for oxygenated cancer cells [58,63]. Quantifying fluxes with glucose tracers, it was determined that glycolysis outpaces the activity of NADH shuttles by as much as tenfold in transformed and non-transformed proliferating cells. When the rate of glycolysis was reduced, the majority of the glucose consumed by cancer cells was oxidized. Only once the rate of glycolysis was increased to exceed the maximum activity of the MAS and GSPS did fermentation become highly activated to replenish cytosolic NAD+, indicating that lactate production is a secondary result of saturating the mitochondrial shuttle systems. Of note, the maximum activities of the MAS and G3PS varied between cell lines. Furthermore, the activity of the MAS directly correlated with the amount of glucose that was oxidized across more than 50 different human cancer cell lines, suggesting that MAS activity might limit glucose oxidation for a wide range of cell types [58].
Concluding remarks
Pasteur’s observations in the 19th century led to the idea that glucose fermentation is inhibited by the presence of oxygen. Warburg’s work on tumors was puzzling because it appeared contrary to Pasteur’s findings in that his cancer cells continued to ferment most of their glucose in the presence of oxygen. Warburg originally explained his results by arguing that mitochondria are damaged in cancer. Although it is now widely accepted that Warburg’s hypothesis on the origin of cancer does not hold in most cases, a legacy of his thinking seems to have been the notion that mitochondrial metabolism is suppressed or deactivated in proliferating cells. A number of impressive studies have highlighted that mitochondria serve key functions during proliferation, but the level of mitochondrial activity required to fulfill them has been largely unexplored, and statements that cancer cells prefer glucose fermentation over oxidation continue to appear in the literature. Here, we offer the opposite perspective, that glucose oxidation is preferred over glucose fermentation. Recent studies reveal that proliferating cells have increased glucose oxidation rates and overall mitochondrial activity compared with quiescent cells. Evidence from other reports demonstrates that glucose oxidation is constrained by the maximum flux at which mitochondrial pathways can recycle NAD+ from NADH. In proliferating cells, the rate at which glycolysis produces NADH is faster than the rate at which mitochondrial pathways can oxidize it. Lactate is produced during the Warburg effect, not because it is a requirement of proliferation, but because mitochondrial activity is saturated.
Some important questions still remain (see Outstanding questions). In particular, it is interesting to consider why proliferating cancer cells consume more glucose than their mitochondria can oxidize. One possibility is that glucose fermentation is beneficial to cancer cells because the accumulation of lactate modifies the tumor microenvironment [8,64]. Cancer cells with saturated mitochondria might have a fitness advantage because the lactate they excrete selectively disables the activation of cytotoxic cells, including natural killer cells. The challenge of exploring these types of mechanisms is that it requires assessing the metabolism of tumors within an organism. Historically, the most common approach to studying tumor metabolism in vivo has been to evaluate tissue in bulk. The results of such experiments, however, are difficult to interpret because nontransformed and hypoxic cells can produce lactate independent of mitochondrial saturation. As such, a critical step to advancing our knowledge of mitochondrial activity in cancer will be evaluating tumors with techniques that enable the metabolism of individual cells to be resolved. Already, we are making progress along that path. Using positron emission tomography tracers, for example, it was recently determined that myeloid cells within some tumors consume more glucose than cancer cells [65]. Moving forward, it will be insightful to superimpose metabolic phenotypes across a tumor with corresponding measurements of available and consumed nutrients at the same points. We expect that the amount of glucose oxidized by cancer cells is determined not just by the maximum capacity of mitochondria but also by how much of that capacity is filled by other respiratory substrates, such as glutamine.
Outstanding questions.
Why do cancer cells consume more glucose than they can oxidize in their mitochondria?
What intrinsic and extrinsic factors determine mitochondrial capacity in cancer cells? What is the physiological purpose of controlling mitochondrial capacity?
Does the mitochondrial capacity differ between various types of proliferating cells, such as T cells and stem cells?
Are there any other factors that limit mitochondrial oxidation in proliferating cells, aside from ATP synthesis and NADH shuttles?
What determines which substrates are used to fuel respiration in different tumors across different settings, and how does this affect mitochondrial capacity?
What additional insights might be obtained from technologies capable of measuring NAD+/NADH in specific cellular compartments from intact cells (e.g., the cytosol and mitochondria)?
Highlights.
Proliferating cells have increased mitochondrial activity relative to quiescent cells.
Metabolic pathways in mitochondria are saturated in proliferating cells exhibiting the Warburg effect.
Mitochondrial activity is constrained by the flux of NAD+ turnover in proliferating cells.
The Warburg effect occurs in proliferating cells because glycolysis outpaces the maximum rate of glucose oxidation.
Acknowledgments
Financial support was received for this work from a National Institutes of Health award R35ES028365 (to G.J.P.).)
Footnotes
Declaration of Interests
G.J.P. is the Chief Scientific Officer for Panome Bio and a scientific advisory board member for Cambridge Isotope Laboratories. The laboratory of G.J.P. has a collaborative agreement with Thermo Fisher Scientific, Agilent Technologies, and Bruker. Y.W. has no interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hinkle PC (2005) P/O ratios of mitochondrial oxidative phosphorylation. Biochim Biophys Acta 1706, 1–11. 10.1016/j.bbabio.2004.09.004 [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 3.WARBURG O (1925) THE METABOLISM OF CARCINOMA CELLS. The Journal of Cancer Research 9, 148–163 [Google Scholar]
- 4.Pfeiffer T et al. (2001) Cooperation and competition in the evolution of ATP-producing pathways. Science 292, 504–507. 10.1126/science.1058079 [DOI] [PubMed] [Google Scholar]
- 5.Kukurugya MA and Titov DV (2022) The Warburg Effect is the result of faster ATP production by glycolysis than respiration. bioRxiv, 2022.2012.2028.522160. 10.1101/2022.12.28.522160 [DOI] [Google Scholar]
- 6.Vazquez A et al. (2010) Catabolic efficiency of aerobic glycolysis: the Warburg effect revisited. BMC Syst. Biol. 4, 58. 10.1186/1752-0509-4-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shlomi T et al. (2011) Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Comput. Biol. 7, e1002018. 10.1371/journal.pcbi.1002018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liberti MV and Locasale JW (2016) The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41, 211–218. 10.1016/j.tibs.2015.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiechter A and Gmünder FK (1989) Metabolic control of glucose degradation in yeast and tumor cells. Adv. Biochem. Eng. Biotechnol. 39, 1–28. 10.1007/BFb0051950 [DOI] [PubMed] [Google Scholar]
- 10.Racker E (1972) Bioenergetics and the problem of tumor growth. Am. Sci. 60, 56–63 [PubMed] [Google Scholar]
- 11.Urbano AM (2021) Otto Warburg: The journey towards the seminal discovery of tumor cell bioenergetic reprogramming. Biochim Biophys Acta Mol Basis Dis 1867, 165965. 10.1016/j.bbadis.2020.165965 [DOI] [PubMed] [Google Scholar]
- 12.Grey EC et al. (1997) The enzymes of B. coli communis. Part V.—(a) anaerobic growth followed by anaerobic and aerobic fermentation. (b) the effects of aeration during the fermentation. Proceedings of the Royal Society of London. Series B, Containing Papers of a Biological Character; 92, 135–150. 10.1098/rspb.1921.0012 [Google Scholar]
- 13.Warburg O (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 14.DeBerardinis RJ and Chandel NS (2020) We need to talk about the Warburg effect. Nat Metab 2, 127–129. 10.1038/s42255-020-0172-2 [DOI] [PubMed] [Google Scholar]
- 15.Koppenol WH et al. (2011) Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337. 10.1038/nrc3038 [DOI] [PubMed] [Google Scholar]
- 16.Martins Pinto M et al. (2023) The Warburg effect and mitochondrial oxidative phosphorylation: Friends or foes? Biochim Biophys Acta Bioenerg 1864, 148931. 10.1016/j.bbabio.2022.148931 [DOI] [PubMed] [Google Scholar]
- 17.Cassim S et al. (2020) Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers (Basel) 12. 10.3390/cancers12051119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zu XL and Guppy M (2004) Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun 313, 459–465. 10.1016/j.bbrc.2003.11.136 [DOI] [PubMed] [Google Scholar]
- 19.Warburg O et al. (1927) THE METABOLISM OF TUMORS IN THE BODY. J. Gen. Physiol. 8, 519–530. 10.1085/jgp.8.6.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.PORTAIS J-C et al. (1993) Metabolic flux determination in C6 glioma cells using carbon-13 distribution upon [1-13C]glucose incubation. European Journal of Biochemistry 217, 457–468. 10.1111/j.1432-1033.1993.tb18265.x [DOI] [PubMed] [Google Scholar]
- 21.Hiller K and Metallo CM (2013) Profiling metabolic networks to study cancer metabolism. Current Opinion in Biotechnology 24, 60–68. 10.1016/j.copbio.2012.11.001 [DOI] [PubMed] [Google Scholar]
- 22.Johnston K et al. (2021) Isotope tracing reveals glycolysis and oxidative metabolism in childhood tumors of multiple histologies. Med (N Y) 2, 395–410. 10.1016/j.medj.2021.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maher EA et al. (2012) Metabolism of [U-13C]glucose in human brain tumors in vivo. NMR in Biomedicine 25, 1234–1244. 10.1002/nbm.2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DeBerardinis RJ et al. (2007) Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proceedings of the National Academy of Sciences 104, 19345–19350. doi: 10.1073/pnas.0709747104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinberg F et al. (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107, 8788–8793. 10.1073/pnas.1003428107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altman BJ et al. (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nature Reviews Cancer 16, 619–634. 10.1038/nrc.2016.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen A et al. (2020) PET imaging of glutamine metabolism in a clinical trial of metastatic colorectal cancer. Journal of Nuclear Medicine 61, 630–63032238427 [Google Scholar]
- 28.Ma Y et al. (2018) Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett 435, 92–100. 10.1016/j.canlet.2018.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J-S et al. (2020) ATP Production Relies on Fatty Acid Oxidation Rather than Glycolysis in Pancreatic Ductal Adenocarcinoma. Cancers 12, 2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mashimo T et al. (2014) Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 159, 1603–1614. 10.1016/j.cell.2014.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Comerford SA et al. (2014) Acetate dependence of tumors. Cell 159, 1591–1602. 10.1016/j.cell.2014.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen YJ et al. (2016) Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 12, 937–943. 10.1038/nchembio.2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faubert B et al. (2017) Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e359. 10.1016/j.cell.2017.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hui S et al. (2017) Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. 10.1038/nature24057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao CH et al. (2019) Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife 8, pii e41351. 10.7554/eLife.41351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whitaker-Menezes D et al. (2011) Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 10, 4047–4064. 10.4161/cc.10.23.18151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sullivan LB et al. (2015) Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. 10.1016/j.cell.2015.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahn CS and Metallo CM (2015) Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab 3, 1. 10.1186/s40170-015-0128-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Birsoy K et al. (2015) An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551. 10.1016/j.cell.2015.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wuu JA et al. (1988) Amino acid concentrations in serum and aqueous humor from subjects with extreme myopia or senile cataract. Clin. Chem. 34, 1610–1613 [PubMed] [Google Scholar]
- 41.Sullivan LB et al. (2018) Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 20, 782–788. 10.1038/s41556-018-0125-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Bermudez J et al. (2018) Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol. 20, 775–781. 10.1038/s41556-018-0118-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muz B et al. (2015) The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 3, 83–92. 10.2147/hp.S93413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mendelsohn J et al. (2015) The Molecular Basis of Cancer Fourth Edition edn), [Google Scholar]
- 45.Llufrio EM et al. (2018) Sorting cells alters their redox state and cellular metabolome. Redox Biol 16, 381–387. 10.1016/j.redox.2018.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Almeida L et al. (2016) Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol. 28, 514–524. 10.1016/j.smim.2016.10.009 [DOI] [PubMed] [Google Scholar]
- 47.Sena LA et al. (2013) Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236. 10.1016/j.immuni.2012.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yao CH et al. (2016) Exogenous Fatty Acids Are the Preferred Source of Membrane Lipids in Proliferating Fibroblasts. Cell Chem Biol 23, 483–493. 10.1016/j.chembiol.2016.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fan TW et al. (2009) Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM). Mol Cancer 8, 41. 10.1186/1476-4598-8-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hensley CT et al. (2016) Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. 10.1016/j.cell.2015.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DeBerardinis RJ (2008) Is cancer a disease of abnormal cellular metabolism? New angles on an old idea. Genet. Med. 10, 767–777. 10.1097/GIM.0b013e31818b0d9b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hitosugi T et al. (2011) Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 44, 864–877. 10.1016/j.molcel.2011.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaplon J et al. (2013) A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112. 10.1038/nature12154 [DOI] [PubMed] [Google Scholar]
- 54.McFate T et al. (2008) Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem. 283, 22700–22708. 10.1074/jbc.M801765200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luengo A et al. (2021) Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol. Cell 81, 691–707.e696. 10.1016/j.molcel.2020.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Titov DV et al. (2016) Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352, 231–235. 10.1126/science.aad4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bartman CR et al. (2023) Slow TCA flux and ATP production in primary solid tumours but not metastases. Nature 614, 349–357. 10.1038/s41586-022-05661-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y et al. (2022) Saturation of the mitochondrial NADH shuttles drives aerobic glycolysis in proliferating cells. Mol Cell 82, 3270–3283.e3279. 10.1016/j.molcel.2022.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phypers B and Pierce JT (2006) Lactate physiology in health and disease. Continuing Education in Anaesthesia Critical Care & Pain 6, 128–132. 10.1093/bjaceaccp/mkl018 [DOI] [Google Scholar]
- 60.Spriet LL et al. (2000) An enzymatic approach to lactate production in human skeletal muscle during exercise. Med Sci Sports Exerc 32, 756–763. 10.1097/00005768-200004000-00007 [DOI] [PubMed] [Google Scholar]
- 61.O’Donnell JM et al. (2004) Limited transfer of cytosolic NADH into mitochondria at high cardiac workload. Am J Physiol Heart Circ Physiol 286, H2237–2242. 10.1152/ajpheart.01113.2003 [DOI] [PubMed] [Google Scholar]
- 62.Contreras L and Satrústegui J (2009) Calcium signaling in brain mitochondria: interplay of malate aspartate NADH shuttle and calcium uniporter/mitochondrial dehydrogenase pathways. J Biol Chem 284, 7091–7099. 10.1074/jbc.M808066200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dai Z et al. (2016) A Flux Balance of Glucose Metabolism Clarifies the Requirements of the Warburg Effect. Biophys J 111, 1088–1100. 10.1016/j.bpj.2016.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quinn WJ 3rd et al. (2020) Lactate Limits T Cell Proliferation via the NAD(H) Redox State. Cell Rep. 33, 108500. 10.1016/j.celrep.2020.108500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reinfeld BI et al. (2021) Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288. 10.1038/s41586-021-03442-1 [DOI] [PMC free article] [PubMed] [Google Scholar]