

Structured Abstract
INTRODUCTION:
Cerebrovascular pathology is an early and causal hallmark of Alzheimer’s disease (AD), in need of effective therapies.
METHODS:
Based on the success of our previous in vitro studies, we tested for the first time in a model of AD and cerebral amyloid angiopathy (CAA), the carbonic anhydrase inhibitors (CAIs) methazolamide and acetazolamide, FDA-approved against glaucoma and high-altitude sickness.
RESULTS:
Both CAIs reduced cerebral, vascular and glial Amyloid-β (Aβ) accumulation and caspase activation, diminished gliosis and ameliorated cognition in TgSwDI mice. The CAIs also improved microvascular fitness and induced protective glial pro-clearance pathways, resulting in the reduction of Aβ deposition. Notably, we unveiled that the mitochondrial carbonic anhydrase-VB (CA-VB) is upregulated in TgSwDI brains, CAA and AD+CAA human subjects, and in endothelial cells upon Aβ treatment. Strikingly, CA-VB silencing specifically reduces Aβ-mediated endothelial apoptosis.
DISCUSSION:
This work substantiates the potential application of CAIs in clinical trials for AD and CAA.
Keywords: Amyloid β, cerebral amyloid angiopathy, cerebrovascular dysfunction, neuroinflammation, clearance, endothelial cells, astrocytes, microglia, carbonic anhydrase inhibitors, Alzheimer’s disease
Background
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disorder, where morpho-functional alterations of the vasculature are critical and early pathological hallmarks1–7. It is now accepted that cerebrovascular dysfunction (CVD) may contribute to the initiation and progression of the neuronal demise, with a tight association between CVD, neuroinflammation, neurodegeneration and cognitive impairment1,8–13. In addition to parenchymal amyloid beta (Aβ) plaques and tau neurofibrillary tangles (NFT), about 90% of AD cases exhibit cerebral amyloid angiopathy (CAA), the deposition of Aβ aggregates at the vessel walls of the brain vasculature4,14–21. CAA is also found in many non-AD older adults, in a proportion correlating with age. Current immunotherapeutic approaches may also increase CAA, inducing Amyloid Related Imaging Abnormalities (ARIA)22,23. Endothelial and glial cells actively co-operate in mediating vascular fitness and brain clearance, promoting the removal of toxic material from the brain, including Aβ. Mechanisms implicated in Aβ perivascular clearance have recently aroused high interest in regard to their therapeutic impact4,19,24–26. Indeed, if on one hand vascular Aβ deposition triggers CVD and affects brain clearance, on the other hand, CVD and dysregulated clearance exacerbate vascular Aβ deposition, leading to a vicious cycle which boosts cellular stress, blood-brain barrier (BBB) permeability, metabolic waste accumulation, neuroinflammation27–29, and ultimately leads to neurodegeneration and cognitive dysfunction1,30,31. Yet, the molecular players mediating these harmful effects are not fully elucidated, and the scientific community urges to find novel therapeutic strategies to prevent neurovascular impairment and improve brain clearance in AD and CAA. Our lab and others have shown that Aβ induces mitochondrial dysfunction and caspase-mediated apoptosis in endothelial, glial and neuronal cells15,16,32–35. Carbonic anhydrases (CAs) catalyze the reversible hydration of CO2 to bicarbonate and a proton and are essential for pH regulation and other physiological functions. Humans express 15 CA isoforms with different cellular/tissue distribution patterns, including CA-VA and VB in the mitochondria. Acetazolamide (ATZ) and methazolamide (MTZ) are FDA-approved CAIs used for glaucoma, high-altitude sickness, cerebral edema and seizures. Their pharmacokinetics and side effects are reported, they are safe for long-term administration and can cross the BBB36–40. Acute inhibition of CAs promotes cerebral blood flow (CBF), vasoreactivity and neuronal excitability in humans41,42. Our recent studies demonstrated that ATZ and MTZ suppress mitochondrial dysfunction and apoptosis induced by Aβ in vascular and neural cells by reducing mitochondrial ROS production and loss of mitochondrial membrane potential43,44. However, the effects of CAIs have never been dissected in vivo in a transgenic animal model of amyloidosis and cerebrovascular dysfunction. In this study we tested for the first time in the AD/CAA field, a chronic therapeutic regimen employing the CAIs MTZ and ATZ in TgSwDI mice, which express human Amyloid-β Precursor Protein (hAPP), carrying the Swedish, Dutch and Iowa mutations. This model is characterized by extensive vascular Aβ deposition, starting at about 6 months of age, diffused parenchymal Aβ deposits and neuroinflammation. We assessed cognitive function and Aβ pathology following ATZ or MTZ treatment. Additionally, we tested the hypothesis that CAIs can halt or reduce Aβ cerebrovascular/neurovascular toxicity and promote glial cell fitness. Importantly, we determined that a specific mitochondrial CA isoform mediates these effects.
Overall, this work demonstrates for the first time that CAIs can foster cerebrovascular fitness, revert the neuroinflammatory state, promote Aβ clearance and prevent cognitive impairment in a model of cerebral amyloidosis. Additionally, we provide pioneer evidence pointing to CA-VB as a key player in CAA and AD.
Methods
Animals and treatment
TgSwDI (APP-Swedish, Dutch, Iowa) mice (C57BL6/6 background) were obtained from Dr. Thomas Wisniewski (New York University, NYU), and bred internally. These animals carry the human APP gene (isoform 770) with the Swedish (K670N/M671L), Dutch (E693Q) and Iowa (D694N) mutations, under the control of the mouse neuronal Thy1 promoter, triggering an enhanced abnormal cerebral Aβ production and deposition. In particular, mutations within Aβ peptide sequence, such as the Dutch and the Iowa (at position 22 and 23, respectively), are mainly associated with fibrillar Aβ burden in the brain microvasculature and diffused parenchymal deposits. The animals were housed in accordance with Institutional and National Institutes of Health (NIH) guidelines, and the animal protocol was approved by the Animal Studies Committee. Mice were maintained under controlled conditions (~22°C, and in an inverted 12hr light/dark cycle, lights OFF 10am-ON 10pm) with unrestricted access to food and water. To determine the impact of CAIs on Aβ-mediated pathology in TgSwDI mice, we fed animals for 8 months (8–16 months) or for 4 months (12–16 months) with a CAI-supplemented diet. MTZ or ATZ (20mg/kg/day, corresponding to 100 ppm), were incorporated in a control rodent diet (5053 by TestDiets, Quakertown, PA). To monitor for potential toxicity, the weight of each animal was measured every 2 weeks for the first 2 months, and before behavioral analysis, and skin/fur appearance was observed weekly.
Behavioral analysis
Mice were tested in a behavioral test battery performed during the dark phase of the light cycle. The animals were transported and acclimated to the testing room, at least 30’ prior to each testing session. All behavioral testing was conducted in accordance with the NYU School of Medicine’s Institutional Animal Care and Use Committee, NYU School of Medicine.
Rotarod
Locomotor function, coordination, balance and motor learning were measured using an accelerating rotarod procedure across 3 trials spaced by a 40’ ITI. 5 mice were run simultaneously on 9.5cm diameter rods of a 5-lane rotarod apparatus (IITC Life Science Inc.). All mice were first given a habituation trial in which they were placed on static horizontal rods, and required to stay there, without falling, for 1’. Animals were then tested in 3 experimental trials, during which the rod was rotating and accelerating steadily from 4 to 40rpm over the course a 5’ period, and the latency to fall (sec) from the rods was recorded, and plotted as shown.
Grip strength
Paw/limb grip strength was assessed as the maximal horizontal force (gr) generated by the subject while grasping a specialized 6×10cm stainless steel grid platform connected to a sensitive force sensor (Bioseb). Two different grip strength indices were collected: forelimbs and all limbs (combined fore- and hind-limbs), and both were adjusted to the body weight. For both indices, each mouse was subjected to 6 testing trials with an inter-trial interval (ITI) of 10–20”, and a 40’ interval between forelimb and all-limb measurements. In each trial, the mouse was put onto the grid platform, allowing only its forepaws (forelimbs) or all four paws (all limbs) to clasp onto the central top-half portion of the grid. Once the paws were grasping the grid and the mouse’s torso was in horizontal position, the animal was moved to the cage by the operator. The truncated mean (highest and lowest scores removed) of 6 consecutive trials was taken as the index of grip strength. The body weight was measured after grip strength testing.
Barnes Maze
The Barnes maze procedure provides an index of visuospatial learning and memory in mice to navigate and escape an aversive, open area. The maze apparatus consists of a beige, textured plastic platform surface, 36” in diameter, elevated 91.5cm from the floor (San Diego Instruments, San Diego, CA, USA). The platform has 20 holes (5cm dimeter), equally spaced around the periphery of the platform (~2.5cm from the edge). A gray plastic escape box (~10×8.5×4cm) was located under one of the holes (target/escape hole), and kept consistent across trials. The spatial location of the target hole was counter-balanced across subjects/groups. Visible distal cues were placed around the room and remained constant throughout the testing period. The maze was illuminated by an overhead lamp (~600 lux) above the center of the platform. Mice were tested for 12 trials across 6 consecutive days: one habituation trial (day 1), 10 training trials (days 1–5, 2 trials/day), and one probe trial (day 6). For the habituation trial, each mouse was placed into the center of the maze under an inverted, clear 500ml beaker for 1’. A white noise generator (~80–90dB at platform level) was turned ON and, after 1’ period, the beaker was slowly moved to the target hole with the animal still inside. The mouse was allowed to enter the escape box and explore it for 2’. After the habituation (2hr ITI), each mouse was given 2 standard training trials each day, during which the animal was placed into the center of the maze in a plastic 15×15×20cm start box. The white noise was turned ON and, after 10/15”, the start box was lifted. The mouse was given a maximum of 3’ to find the designated hole and escape box. Once the animal entered the escape box, the white noise was turned OFF, and the animal left inside the box for 30’, and then moved to the home cage. In the case of failure of finding the escape hole within the 3’ limit, the mouse was slowly guided to the target hole under the inverted beaker, as described above. Each day, trials were spaced by 1.5/2hr ITI and, for each trial, the maze platform was rotated. On day 6, a 2’ probe trial was conducted, similarly to the training trials, except that the escape box was removed. Behavior was recorded using an overhead camera for later tracking, and the analysis performed by Noldus Ethovision software (v11.5). Distance travelled, number of mistakes (non-target holes visited) and latency to find the target hole were measured as indices of spatial memory, and then plotted as shown.
Mouse brain processing
After behavioral analysis, brains were harvested for biochemical and immunohistochemical (IHC) assessments. Briefly, animals were anesthetized with pentobarbital, transcardially perfused with either solely ice-cold saline solution (0.9% NaCl) to wash out blood from vasculature, or with saline solution first, and then with 4% paraformaldehyde (PFA) to fix tissues, based on the experimental purpose44. Unfixed brains were removed, flash frozen in liquid N2, and stored at −80°C until their processing for the biochemical assessment. Fixed brains were left O/N at 4°C in 4% PFA, and incubated at 4°C one day in 15% sucrose solution, followed by additional 2 days at 4°C in 30% sucrose, as cryoprotectant. They were then washed in PBS, assembled in a plastic mold with Tissue-Tek O.C.T. compound (Fisher Scientific), and frozen with a mixture of liquid N2 and isopentanol. Brains were stored at −80°C until sectioning. Serial cryostat sections of 8μm thickness were collected on positively charged microscope slides (Fisher Scientific), and stored at −80°C until further immunohistochemical analysis.
Thioflavin S staining
Brain slices were washed in dH2O and incubated in 0.015μM Thioflavin S for 30’, following which, sections were washed in 80% Et-OH. After washing in 1xPBS, slices were dried, and dipped into water-based medium before mounting45. All chemicals were from Sigma (St. Louis, MO). Thioflavin S staining was quantified in at least 4 evenly spaced coronal sections/animal and 5 animals/group using the ImageJ fluorescence analysis tool. Cortical (RSC and gRSC), hippocampal (dentate gyrus, CA1 and CA3) and hypothalamic Aβ fibrillar burden (defined as the number of Thioflavin+ deposits) was quantified separately.
Extraction and quantification of soluble and insoluble Aβ
Mouse brains were homogenized in ice-cold homogenization buffer containing 20mM Tris pH 7.4, 1mM EDTA, 1mM EGTA, and 250mM sucrose. After homogenization, equal amount of protein was used to obtain the Aβ soluble and insoluble fraction. To obtain the soluble fraction, which contains Aβ monomers, oligomers, and protofibrils, a diethylamine (DEA) extraction was performed46,47. Briefly, 0.4 % DEA buffer (v/v) was added to each sample and centrifuged at 50,000xg for 1hr at 4°C. The supernatant was transferred to a new tube, and 0.5M Tris-HCl was added at a 1:10 dilution. The resulting soluble fraction was then stored at −80°C for further analyses. To obtain the insoluble fraction, which represents the fibrillar Aβ associated to plaques/CAA, a formic acid (FA) extraction was performed. The resulting pellet was homogenized in 99% FA buffer. The sample was then centrifuged at 50,000xg for 1hr at 4°C, followed by the addition of the neutralization buffer containing 1M Tris base, 0.5M Na2HPO4, and 0.05% NaN3, and then stored at −80°C for further analyses. The soluble and insoluble fractions were used to measure the levels of human Aβ40 (Invitrogen) and Aβ42 (Invitrogen) by solid-phase sandwich ELISA according to manufacturer’s instructions. Aggregated Aβ within the soluble fraction was measured using a conformation-specific ELISA assay (Invitrogen).
Caspase-3 activity measurement
Brains were homogenized in 25mM HEPES, 5mM MgCl2, 1mM EGTA buffer, containing 1X Halt protease inhibitors (Thermo Fisher), following the 100mg tissue/1ml homogenization buffer proportion. Cerebral homogenates were centrifuged at 13.000rpm for 15’ at 4°C, and supernatants were collected and stored at −80°C until usage. Following protein content quantification via BCA method, 10μg protein/sample were added to each well of a white-walled 96-well luminometer plate, in combination with Caspase-Glo® 3/7 Reagent (Promega)33. After incubating for 1hr at RT, the caspase-3 activation-dependent luminescence of each sample was read in a plate-reading luminometer, and plotted as percentage of caspase-3 activity of WT animals.
Immunohistochemical assessment
For immunostaining evaluation, brain sections were blocked with 10% NGS, 1% BSA solution for 2hrs at RT, and then incubated O/N at 4°C with primary antibodies diluted in 0.1% Triton X-100 (Sigma) blocking solution. Slices were stained with the following primary antibodies: Rt anti-CD31 (BD Pharmigen, 1:200), Ms anti-Aβ (BioLegend, clone 6E10, 1:500), Chk anti-GFAP (Aves, 1:2000), Rb anti-cleaved (active) caspase-3 (Cell Signaling, 1:500), Rb anti-CD68 (Abcam, 1:500), Gt anti-IBA1 (Abcam, 1:500), Rt anti-TREM2 (Abcam, 1:200). The following day, the species-appropriate secondary antibodies Alexa Fluor-conjugated (Thermo Fisher, 1:1000) were employed (2hrs at RT), following which 1.5μg/ml DAPI (Invitrogen, D21490) was used as nuclear staining (10’ incubation at RT). Stained sections were imaged with a Nikon Ti2-E fluorescence deconvolution microscope equipped with 340/380, 465/495, 540/580 and 590/650 nm excitation filters, keeping identical settings within each session, and using either 10x or 60x zoom objectives. 60x images were acquired with a 0.5μm Z-stack. In order to have a consistent examination, for each animal, 2 or 3 different images were acquired in the same brain area of interest. To eliminate out of focus signals, all images were deconvolved using the same deconvolution parameters. The analysis was performed in equally thresholded ten–slice maximal intensity projection images, using Fiji, an open source image processing software. Aβ, active caspase-3, GFAP, IBA1, CD68 and TREM2 were measured as positive staining area (number of positive pixels) per acquisition field. Colocalization was analyzed with JaCoP (Just another Colocalization) ImageJ plug-in, calculating Manders’ coefficients (M1 and M2) which imply the actual overlap of the signals (A over B, and B over A, respectively), and represent the true degree of colocalization48. M1 and M2 coefficients were scored from 0 to 1 [e.g., M1=1.0 and M2=0.7, in red (signal A)- green (signal B) pair, indicate that 100% of red pixels colocalize with green, and 70% of green pixels colocalize with red]. The M1 and M2 coefficient values were then multiplied by the percentage area of Aβ, active caspase-3, or CD68, accordingly, and plotted.
Vessel width measurement
Vessels were stained using antibodies against CD31, as described above, in WT and CAI-treated or untreated TgSwDI mice. The average number of visible vessels for quantification in each image of DG and cortex per mouse was 16. To measure the width of the BVs, a line perpendicular to the long axis of the vessel was delineated at its widest point (corresponding to the widest diameter of every visible vessel), and analyzed using NIS Elements Analysis software from Nikon. Histograms show the distribution and frequency of the vessel width (μm) in each group.
Microhemorrhage staining and quantification
Cerebral slices were blocked for 2hrs at RT in blocking solution (10% NGS-Normal Goat Serum, 1% BSA in PBS), following incubation O/N at 4°C with CD31 primary antibody diluted in blocking solution plus 0.1% Triton-X 1:200. The day after, sections were washed in ice-cold PBS and incubated in anti-Rat 647 secondary antibody (1:1000 in blocking buffer), for 2hrs at RT, to identify vessels. Perls Prussian Blue Staining was then performed on the same slices to detect microhemorrhages, as previously published49–51. Briefly, brains were incubated in a solution containing 5% C₆FeK₄N₆ in dH2O (Macron) and 10% HCl in dH2O, mixed in a ratio 1:1, for 45’ at RT. Following, slices were incubated for 15’ at RT in 0.1% DAB (3,3′ Diaminobenzidine) in 1xPBS (Acros) solution, and then in 0.033% H2O2 in 0.1% DAB, for another 15’ at RT. DAB enhancement of Perls Prussian Blue staining, as described above, highlights iron accumulation in the cerebral tissue. Samples were mounted and processed in light and fluorescence microscopy. Images were assessed for number of microhemorrhages (DAB+ Prussian blue staining adjacent to a CD31+ blood vessel), in the cortex and DG areas, as well as in meningeal arteries. For each animal, microbleeds were counted in the 10x images in two different sequential slices (in both hemispheres), in 5animals/group, and the numbers were plotted as shown.
Cerebral blood volume and cerebral blood flow measurement
Animals and treatment
CBF and CBV measurements were performed in parallel animal groups at the University of Aarus, Denmark. The experimental procedures were performed according to the regulations of the Danish Ministry of Justice and Animal Protection Committees, with the permit 2017–15-0201–01241. Mice were fed a standard diet, or an ATZ- or MTZ-diet, as described above, following which randomize group scans were performed by researchers blinded to the treatment. As vascular impairment52 starts early in TgSwDI mice, vascular functionality was assessed in 10/11-month-old mice after a 6-month treatment with CAIs.
Surgical preparation and training for awake imaging
For cerebral blood volume (CBV) and cerebral blood flow (CBF) assessment, a chronic cranial window was implanted onto the somatosensory area of the barrel cortex (S1BC), as previously described53. Briefly, mice were handled 5 days before surgical preparation to reduce stress during the scanning sessions. All surgical procedures were performed under anesthesia with 1.75/2% isoflurane (induction with 3%) with 100% O2 flow. To avoid brain edema, 4.8mg/kg dexamethasone was injected subcutaneously before the surgical procedure. The cranial window of ~3mm was placed on the cortical area +1.5/2mm antero-posterior and +3mm medio-lateral from bregma. The window was closed with a glass plug, and the edges fixed with cyanoacrylate glue (Loctite Super Glue gel, Loctite®). For restraining the mouse during the imaging session, an in-house metal bar was fixed on the frontal bone using dental acrylic (Meliodent, Germany). Ampicillin (200mg/kg) and carprofen (10mg/kg) were provided for 5 days post-surgery. To avoid stress and improve training sessions, handling continued after surgical preparations. For awake imaging, mice underwent daily training sessions until they reached a total training time of ~2.5hrs (15’/training session, and for each next session, 15’ are added, until ~2.5hrs were reached). The training sessions were performed using a replica of the custom-built frame used for optical imaging. The movement was tracked in x-y-z directions with an accelerometer to discard any data with excessive movement. Excessive movement was defined as a signal above or below the mean ± 3 SD in the 3 recorded directions. The accelerometer signal detection was performed using a Powerlab acquisition unit and LabChart 8 software (ADInstruments). The scans for CBF and CBV analysis were performed on the same day and optical table. Before each recording trial, we performed an acclimation test for each mouse to get it used to the stimulation paradigm.
Intrinsic optical imaging
Relative change in cortical CBV (rCBV) during functional activation was estimated using intrinsic optical signal imaging (IOSI). This scanning methodology relies on hemoglobin (Hb) absorption54. An isosbestic point (Greenlight, 570nm LED) for both oxy- and deoxy-Hb was selected to yield total Hb, considered equivalent to CBV. The window was illuminated with a cold LED light source equipped with a 570 ± 2nm bandpass filter (FB570–10, Thorlabs). Images were collected with a CMOS camera (UI-3280CP Rev. 2, IDS Imaging Development Systems GmbH) at 5 frames per second (fps), with a resolution of 2054 × 2054 pixels (3.45μm/pixel). Subsampling was performed by a factor 2 in both horizontal and vertical dimensions to reduce the file size. Two 20 epochs were performed in each mouse, after the acclimation trial (20 epochs). The methodology for processing and generating the intrinsic signal has been previously reported55. Briefly, using a custom-written script in MATLAB (MathWorks), the acquired videos were downsampled to 512 × 512 pixels. First, motion correction of the videos was performed using the SPM12 toolbox, re-aligning all frames to the first frame acquired. Next, the intensity of each pixel was estimated over the entire time course. Finally, the baseline for each pixel was calculated as the mean intensity during the first 5” of each epoch, and whisker stimulation evoked response was calculated as relative intensity change to the baseline.
Laser Doppler flowmetry
The whisker stimulation evoked changes in cortical CBF were determined using a Laser Doppler Monitor MOORVMS-LDF (Moor Instruments) for laser-Doppler flowmetry (LDF) through the cranial window. The tip of the LDF probe was positioned ~0.5mm above the cranial window. 2recordings of 20 epochs were recorded at 10Hz. The signal was processed with a Chebyshev filter using a MATLAB custom-written script. The relative change to the baseline (5”) was calculated for each time point. An average was computed for each trial and considered for statistical analysis.
Stimulation paradigm
Functional activation consisted of 10” series of gentle air-puffs (~1bar) delivered by a custom-built air-puff system. Each puff lasted ~155ms and was delivered to the contralateral whisker pad at 3Hz.
Astrocyte area, microglia shape quantification and count
Astrocyte cell area measurement and morphological classification of microglia were performed using Nikon NIS Elements Analysis software. Planes containing GFAP+ and IBA1+ cells were identified by manually scrolling through the Z-stack. Only slices containing identified glial cells with nucleus were processed as a maximum intensity projection to avoid overlapping with cells located in more superficial or deeper layers. Maximum intensity projections of both astrocytes and microglia were thresholded to create a binary mask, and the perimeter was delineated to measure the cell surface area. IBA1+ microglia were counted, and morphologically classified as resting, bushy or amoeboid, based on previously published descriptions56, using NIS Elements Analysis software from Nikon. Briefly, resting microglia are identified as IBA1+ cells with thin processes and small cell bodies. Amoeboid microglia are characterized by large cell bodies, rounded macrophage-like morphology with no or few processes and are associated with maximal proinflammatory activation, oxidative-free radicals, and microglial apoptosis57,58. They can also actively remove endangered but potentially viable neurons, contributing to brain pathology and neurodegeneration59,60. Bushy microglia present an intermediate activation state and a shape between resting and amoeboid, with intermediate/large cell bodies and thick projections, and is typically correlated with a pro-healing phenotype61,62.
Immunoblotting assessments
For WB assay, cerebral tissue was homogenized in RIPA buffer containing 1X Halt protease inhibitors (Thermo Fisher) (100mg brain/1ml homogenization buffer, 20mM Tris Base, 0.25M sucrose, 5mM EDTA, 1mM EGTA). Homogenates were kept in ice for 30’, and spun at 21000xg for 30’ at 4°C. Supernatants were collected and quantified with BCA method. 20, 50 and 35μg of proteins (for cell lysates, mouse brains and human cortices, respectively) were added with 1x Sample buffer (Thermo Fisher) and 1x Sample Reducing Agent (Thermo Fisher), and boiled (100°C) for 5’. Proteins were fractionated by SDS-PAGE in reducing condition, transferred to 0.45μm nitrocellulose membrane (Bio-Rad), and probed with the following primary antibodies: Rb anti-Carbonic Anhydrase-VB (NovusBio, NBP1–86090, 0.4μg/ml), Rb anti-Carbonic Anhydrase-VA (Invitrogen, PA5–36931, 1:500), Rb anti-Carbonic Anhydrase-II (Invitrogen, PA5–51598, 0,4μg/ml), Ms anti-APP (Millipore, clone 22C11, MAB348, 1:500), Rb anti-APH-1 (Sigma, PRS4003, 1:250), Rb anti-Nicastrin (Cell Signaling, 3632s, 1:200), Rb anti-ADAM10 (Millipore, AB19026, 1:500), and Rb anti-CD68 (Cell Signaling, 97778, 1:500). As internal loading controls, Rb anti-ATP5a (NovusBio, NBP2–15512, 1:300) was used for mitochondrial proteins, and Ms anti-GAPDH (St Cruz, sc32233, 1:500) was used for non-mitochondrial/total proteins. Membranes were then incubated with the appropriate IRDye secondary antibody (LI-COR, 1:10000) for 1hr at RT, and bands acquired with the Odyssey CLX Infrared Imager (LI-COR). The ratios (protein/GAPDH or mitochondrial protein/ATP5a) were plotted as % of WT (or % of Tg, for hAPP), following quantification (Image Studio Lite Vers 5.2).
Human subjects
For this study, post mortem human brains were provided by the Newcastle Brain Tissue Resource, which is funded- in part- by a grant from the UK Medical Research Council (G0400074), by NIHR Newcastle Biomedical Research Centre awarded to the Newcastle upon Tyne NHS Foundation Trust and Newcastle University, and as part of the Brains for Dementia Research Program jointly funded by Alzheimer’s Research UK and Alzheimer’s Society. Based on pathological post mortem examination, the brains were evaluated by the brain bank neuropathologist as CAA, AD with CAA, or healthy (if pathology was absent). We analyzed occipital cortices, the most frequently and severely affected brain areas in CAA, which were flash frozen and stored at −80°C until the biochemical analysis described here.
hCMEC/D3 cells
Immortalized human cerebral microvascular ECs (hCMEC/D3) were obtained from Babette Weksler (Cornell University)63. Cells were grown in endothelial basal medium (EBM-2, Lonza), supplemented with growth factors (Hydrocortisone, hFGF-B, VEGF, R3-IGF-1, ascorbic acid, hEGF, and GA-1000) and 5%FBS, and maintained in a humidified cell culture incubator at 37°C and 5%CO2.
Aβ peptides and treatment
Aβ40, Aβ40-Q22 and Aβ42 were synthesized by Peptide 2.0 (Chantilly, VA), as previously described64. Peptides were dissolved in hexafluoroisopropanol (HFIP) at a 1mM concentration, incubated O/N to allow the breakdown of secondary structures and obtain monodisperse preparations,16 and then lyophilized using a Benchtop Freeze Dryer (LABCONCO, Kansas City, MO, USA). Lyophilized peptides were resuspended to a 10mM concentration in DMSO, and dH2O was added to achieve a final concentration of 1mM. Prior to the cell culture experimental procedures, peptides were further diluted to the final concentration in 1% FBS EBM-2 for cell treatment.
Carbonic anhydrase silencing in endothelial cells
Downregulation of CA-VB, -VA or -II was obtained using Ambion® Silencer® siRNA, according to manufacturer’s recommendation. Briefly, hCMEC/D3 were seeded to reach a confluency of 60–70% by 24hrs. On the day of transfection, cells were treated with Lipofectamine RNAiMAX (Invitrogen) and Ambion® Silencer® siRNA (Life Technologies) at a final concentration of 10μM in Opti-MEM™ Reduced Serum Medium (Gibco, Life Technologies). After 4hrs of transfection, cells were supplemented with complete media for 24hrs. After 24hrs, the transfection media was removed, and the cells were grown in complete medium until the experimental endpoint. The efficiency and specificity of CA-VB, -VA or -II downregulation were tested by quantitative RT-PCR and normalized to Cyclophilin-B expression (Cyp-B). Briefly, 48hrs post-transfection, RNA was extracted using the miRNeasy (Qiagen), and cDNA was obtained using the QuantiTect® Reverse Transcription Kit (Qiagen) according to manufacturer’s instructions.
qRT-PCR
Quantitative RT-PCR was performed with SYBR™ Green (applied biosystems, Thermo Fisher Scientific) and custom synthesized oligonucleotide primers from Gene Link, using the QuantiStudio 3 system (applied biosystems, Thermo Fisher Scientific). CA-VB: 5’TTCGTTCATCCTTCCGGCAT3’ (F) and 5’TTTTAGGGGGTTGCTTGGCT3’ (R). CA-VA: 5’ACTATCGCCCACTTCAACCC3’ (F) and 5’TCTCTAGGACCTTGTGCCCT3’ (R). CA-II: 5’GAGGGTGAACCCGAAGAACT3’ (F) and 5’GGAAGCTTTGATTTGCCTGT3’ (R). Cyp-B: 5’GATGGCACAGGAGGAAAGAG3’ (F) and 5’AGCCAGGCTGTCTTGACTGT3’ (R). Relative gene expression was calculated utilizing the ΔΔCt method vs. scrambled siRNA (siScr).
Cell Death ELISA
To assess the extent of apoptosis induced by Aβ in the presence or downregulation of CA-VB, CA-VA and CA-II, fragmented nucleosomes formation was quantified using the Cell Death ELISAPLUS assay (Roche Applied Science), as previously published43. Briefly, hCMEC/D3 were transfected with siRNA for 48hrs, at which point the cells were challenged with Aβ42 (10μM), Aβ40 (25μM) or Aβ40-Q22 (25μM), for 24hrs. After the 24hr-treatment with Aβ, the plates were centrifuged for 10’ at 200xg, the cells lysed, and fragmented DNA-histone complexes (nucleosomes, indicating apoptosis) quantified by Cell Death ELISA, according to manufacturer’s instructions.
Statistical analysis
Prior to statistical analysis, outliers were identified and removed from the original dataset using ROUT method with GraphPad Prism 9.1.0 software. Data were analyzed using ordinary unpaired two-tailed t-test or one-way analysis of variance (ANOVA) test, followed by Tukey’s post hoc test (GraphPad Prism 9.1.0). Differences between groups were considered statistically significant when p≤0.05. In all figures and legends, asterisk (*) and plus (+) symbols denote statistically significant differences. If not differently specified, significant differences versus WT animals are indicated with + symbols, graphs are representative of at least 3 independent experiments and data are represented as means ±SEM. In the legends, the number of experiments in vitro or the number of mice per condition (or group) are indicated with “N”, while “n” indicates, for in vitro treatments, the number of replicates, and in vivo, i) the number of technical replicates (e.g. in WB), ii) the counts (e.g. in MH analysis), or iii) the number of measurements acquired (e.g. in IHC).
Behavioral data were analyzed using two-way ANOVA with group (WT, TgSwDI, Tg+ATZ, Tg+MTZ) and sex (females, males), as between-subject factors. For the analysis of rotarod, body weight was used as covariate. Whenever data were judged to be non-normal by inspection of the quantile-quantile (Q-Q) plot and the Kolmogorov-Smirnov (K-S) test, data were transformed using a transform appropriate to the metric (e.g. log of distance, square root of errors). If transformation did not yield normality, differences between groups were analyzed using the nonparametric Kruskal-Wallis test. Significant main effects or interactions were decomposed using simple main effects and Tukey’s post-hoc comparisons for ANOVA or Dunn’s multiple comparisons for nonparametric tests. All statistical analyses were performed using IBM SPSS Statistics v25. Power analysis for the behavioral tests (www.stat.ubc.ca) was performed assuming 2-sided students t tests, α=0.05, power=0.8 and a large effect size (Cohen’s d >1.15), based on our preliminary studies. This yielded a reasonable sample sizes of 13 mice/ Tg group (and 10 mice/WT group) for behavioral analysis, consistent with our compelling data; it was also consistent with the goal of using the smallest number of animals required to carry out the research.
For CBV and CBF, the following parameters were estimated from the time-series: i) maximum response during stimulation (peak), ii) time-to-peak response, iii) area under the curve during the 10” stimulation (A.U.C. Stimulation). We used R version 4.03 to compile the database, to perform statistical analysis and plots. All the statistical analysis of time-series parameters was performed constructing linear mixed models with the package ‘lme4’. To analyze the differences in the estimated parameters, we applied a linear mixed model including group (categorical) as a fixed effect. To account for the correlation in the repeated measurements (2 trials) as well as possible drops out of the data for some mice due to corrupted data, we used an indirect specification of covariance using intercepts for the subject (1|mouse). Gender variability was examined by adjusting each model for gender as a fixed effect. The model to use was chosen using the Maximum likelihood ratio test between the model with the fixed effect gender and the one without the effect in question. Finally, a comparison between groups was performed, changing the reference level of the fixed effect group. ** p<0.01. For rCBV, 11 videos were discarded from analysis due to file corruption (WT = 2, Tg-Ctrl = 2, Tg-ATZ = 2, Tg-MTZ =2). A total of 7 ± 3 epochs of CBV and 7 ± 2 epochs of CBF time series were excluded from analysis due to excessive movement during the data acquisition.
Results
Chronic CAI treatment ameliorates spatial memory, reduces brain Aβ deposits and decreases caspase-3 activation
We have recently demonstrated, for the first time in the AD field, that CAIs reduce mitochondrial dysfunction and cell death pathways induced by Aβ in vascular and neural cells in culture, as well as in brain cells in vivo after intra-hippocampal Aβ injection39,43,44. Considering that mitochondrial health is essential for proper brain cell function, and that both mitochondrial impairment and CVD are very early events in AD pathology, we hypothesized that a treatment with CAIs may improve cognitive and pathological outcomes in a transgenic mouse model of cerebral amyloidosis with CAA, the TgSwDI mice. The FDA-approved CAIs ATZ or MTZ (20mg/kg/day) were chronically administered in the chow, which was otherwise identical to the control mouse diet (TestDiets). The treatment with ATZ or MTZ was started when Aβ deposition is mild in this model (7/8 months of age), to replicate a clinical treatment in MCI (Mild Cognitive Impairment) patients, or at a later age (12 months), to replicate clinical treatment in mild/moderate AD patients.
Behavioral studies
Once the mice reached a stage of advanced pathology (15/16 months), they all underwent a battery of behavioral tests. Before the cognitive assessment, weight was measured, and all study groups were subjected to motor activity tests, to verify that the CAI treatment was not toxic and did not affect motor skills. Rotarod performance (Fig. 1A) was impaired in all Tg groups compared to WT animals (TgSwDI p=0.016, Tg+ATZ p=0.012, Tg+MTZ p=0.006). Untreated TgSwDI mice had lower all-limb grip strength than WT control mice (p=0.003) (Fig.1A). However, grip strength in CAIs-treated Tg animals was not statistically different from WT animals. Forelimb (only) grip strength showed a similar pattern to the all-limb results (Supplementary Fig.1A). No significant differences in body weight due to the genotype or CAI treatment were observed between groups (Fig. 1A) or between same-gender groups (Supplementary Fig.1A), although as expected, males were heavier than females. Death rates also did not change between untreated Tg and CAI-treated Tg animals (Supplementary Fig. 1A), confirming that chronic CAI treatment was not toxic. Spatial memory was assessed using the Barnes maze test. First, all treated animals were compared to untreated Tg group and WT animals. The Barnes maze probe test data are presented in Fig 1B. Post-hoc comparisons between groups on each measure revealed that untreated TgSwDI mice ran a longer distance (p=0.0104) and made more mistakes (p=0.0117) than the WT control mice, indicating cognitive impairment. Tg mice treated with CAIs were not statistically different from WT animals, indicating that CAI treatment preserves spatial memory in TgSwDI mice (Fig. 1B). The same pattern of results was observed with ANOVA applied to the untransformed data or when using nonparametric Kruskal Wallis analysis. The results were similar after splitting the groups treated for 8 and 4 months (Supplementary Fig. 1B). To help rule out the potential contribution of confounding factors on Barnes maze performance measures, we also performed ANOVAs including body weight, distance travelled in the open field arena and percentage time spent in the center of the open field (not shown) as covariates. These covariates were not significant predictors of performance in any of the analyses (p > 0.05).
Figure 1: CAI treatment attenuates cognitive impairment, reduces brain Aβ pathology and decreases caspase-3 activation in TgSwDI mice.
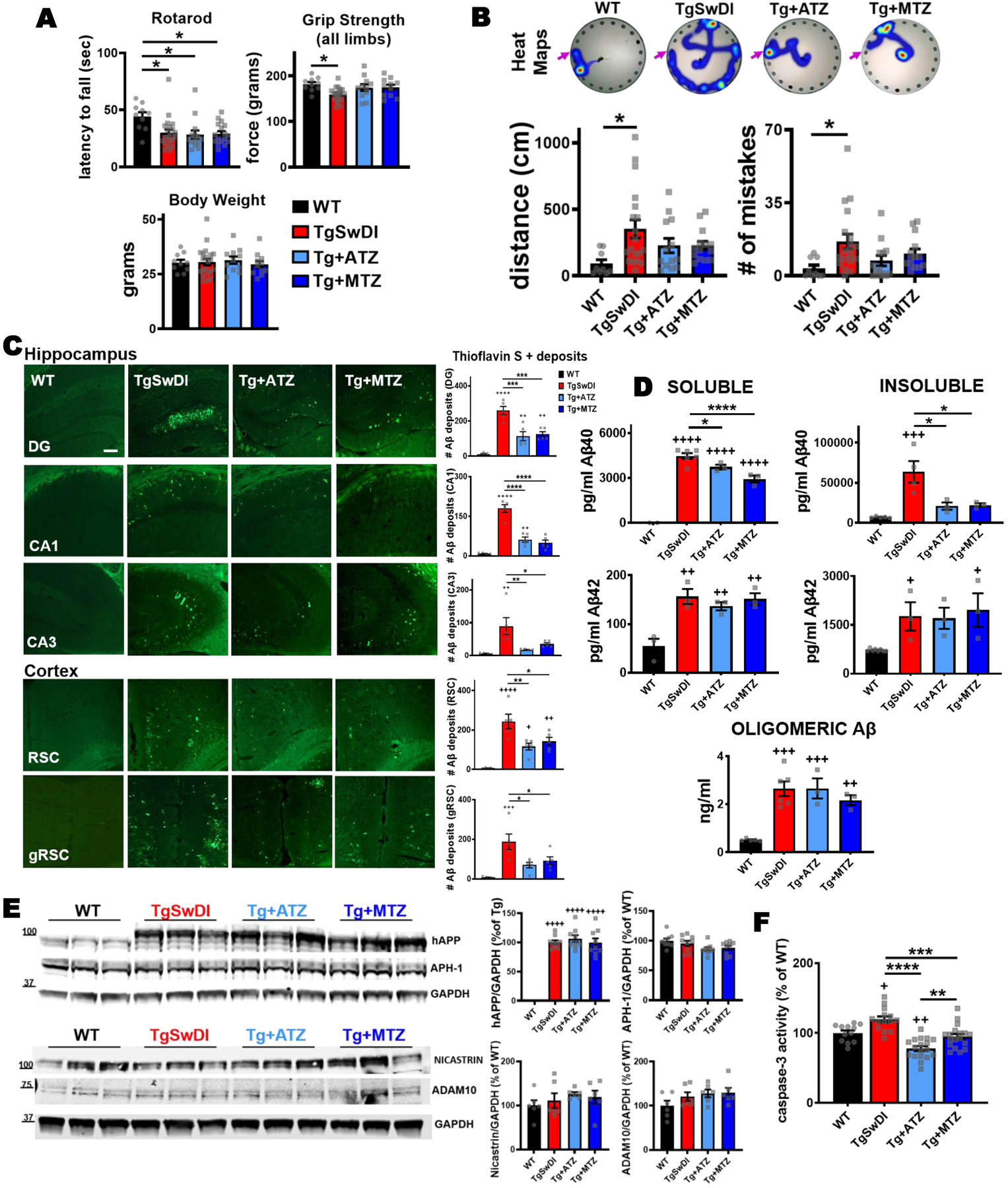
A) CAIs did not affect motor coordination tested with rotarod. In the rotarod performance, there was a significant main effect of group (F(1,57)=4.863, p=0.004) with body weight significant as covariate (F(1,57)=6.059, p=0.017). Post-hoc tests showed that all Tg groups were significantly impaired relative to WT control mice (TgSwDI p=0.016, Tg+ATZ p=0.012, Tg+MTZ p=0.006). The analysis of motor function tested with grip strength (all limbs) showed that there was a significant main effect of group (F(3,57)=5.449, p=0.002) on grip strength with body weight significant as covariate (F(1,57)=29.302, p=0.000). Post-hoc comparisons revealed that untreated TgSwDI mice had lower all-limb grip strength than WT mice (p=0.003). Body weight did not change between groups. B) Spatial memory tested via Barnes maze task in 15/16-month-old WT and TgSwDI mice, in the presence or absence of MTZ- or ATZ-treatment. The heat maps show the path (in blue) covered by the animals to reach the escape hole (indicated with pink arrows) in the probe test. The plots represent the distance covered (cm) and the number of mistakes made before finding the escape hole, during the probe test. There was a significant main effect of group on appropriately transformed indices of distance (F(3, 48)=4.456, p=0.008) and mistakes (F(3, 48)=5.059, p=0.004). Post-hoc comparisons between groups on each measure revealed that only untreated Tg mice were impaired in distance (p=0.0104) and mistakes (p=0.0117), compared to WT animals. Same pattern of results was observed with ANOVA applied to the untransformed data or when using nonparametric Kruskal Wallis analysis. WT: N=10, TgSwDI: N=19, ATZ: N=13 and MTZ: N=14. Two-way ANOVA and Dunn’s post-hoc test. Data are expressed as mean ± SEM. C) Representative images of cerebral Aβ deposits stained with Thioflavin S. 16-month-old untreated TgSwDI mice exhibit a greater amount of Aβ, in both hippocampus (DG, CA1 and CA3 areas) and cortex (retrosplenial and granular retrosplenial cortex, RSC and gRSC), compared to WT animals. CAI treatment significantly decreases Aβ deposits in all areas. Original magnification, 20x. Scale bar 150μm. On the right, relative quantification. WT, TgSwDI, ATZ and MTZ: N=5, n≥10 measurements acquired/group. D) Brain Aβ content measured by ELISA in soluble and insoluble fractions. Compared to age-matched WT, 16-month-old TgSwDI animals show significantly higher concentration (pg/ml) of both soluble and insoluble Aβ40 and Aβ42. CAI treatment diminishes soluble Aβ40, and even more considerably insoluble Aβ40, compared to Tg mice. Oligomeric Aβ content (ng/ml) within the soluble fraction does not significantly change among Tg groups. Graphs represent N=3–8 animals/group, n=2 measurement/animal. E) CAI treatment does not change the expression of hAPP and APP metabolism enzymes APH-1 and nicastrin (γ-secretase subunits), and ADAM10 (α-secretase). Quantification (right side), normalized vs GAPDH, and expressed in percentage vs WT mice (or % vs Tg in hAPP graph). N=3 mice/group, n=9 technical replicates/group. F) Caspase-3 activity measured in brain homogenates. ATZ and MTZ treatments significantly reduce total cerebral caspase-3 activation, compared to untreated TgSwDI mice. N=3–5 mice/group, n ≥12 measurements/group. In (C-F), one-way Anova and Tukey’s post-hoc test: * (vs Tg) and + (vs WT) p<0.05, ** and ++ p<0.01, *** and +++ p<0.001, **** and ++++ p<0.0001. Data are expressed as mean ± SEM.
Overall, these results indicate that spatial memory and grip strength are improved in CAI-treated Tg mice, without toxicity of the chronic treatment or effects on motor coordination.
Quantification of Aβ burden and APP processing
We next asked whether CAI-driven cognitive amelioration was associated with a reduction in cerebral Aβ burden. The mice were sacrificed after behavioral testing at 16 months, and brain sections were stained with Thioflavin S, which binds to fibrillar Aβ deposits. Fibrillar Aβ burden in hippocampal, cortical and hypothalamic regions was quantified using an unbiased sampling scheme and a semiautomated image analysis system. As expected, we found that 16-month-old TgSwDI animals presented extensive Thioflavin S+ Aβ deposits in the hippocampus (dentate gyrus [DG], CA1 and CA3 areas), and cortex (retrosplenial [RSC] and granular retrosplenial cortex [gRSC]) (Fig. 1C), as well as in the hypothalamus (Supplementary Fig. 1C). Aβ deposits were drastically reduced in all areas when Tg animals were treated with ATZ and MTZ from 8 to 16 months (in DG, ATZ −56% and MTZ −52%; in CA1, ATZ −65.6 % and MTZ −72.6%; in CA3, ATZ −80.7% and MTZ −61%; in RSC, ATZ −52.4% and MTZ −41.6%; in gRSC, ATZ −62.5% and MTZ −51.4%; in hypothalamus, ATZ −44% and MTZ −32%, vs Tg). Differences in Aβ burden were also present in Tg mice treated for shorter time (from 12 to 16 months) with CAIs (Supplementary Fig.1D). However, longer treatment with ATZ and MTZ induced a much more evident reduction of cerebral Aβ pathology. Therefore, we focused our next analyses on the long-treatment group. We separated soluble and insoluble cerebral Aβ and measured the respective levels by ELISA (Fig. 1D). TgSwDI mice showed significant increase in all Aβ40 and Aβ42 species, compared to WT animals. In contrast to untreated Tg animals, CAI treatment significantly decreased Aβ40 content (the main peptide deposited in the vasculature, both in human AD and in this model), and particularly reduced insoluble Aβ40 (soluble Aβ40 reduction: ATZ −16% and MTZ −54.7%; insoluble Aβ40 reduction: ATZ −67.3% and MTZ −65%, vs Tg animals), suggesting that CAIs may reduce aggregated Aβ40 deposits around the cerebral vasculature (CAA). Aβ oligomers within the soluble fraction were also measured using a specific Aβ ELISA assay, and did not show differences (Fig 1D bottom panel). To rule out the possibility that this reduction was due to effects of CAIs on brain Aβ production through modulation of the expression of APP and/or its processing enzymes, we performed immunoblot analysis of these proteins. We found that CAIs did not alter the expression of human APP (hAPP) (Fig. 1E), or its amyloidogenic and non-amyloidogenic pathway enzymes, such as APH-1 and nicastrin (γ-secretase complex subunits) and ADAM10 (α-secretase), respectively (Fig. 1E), indicating that CAI-mediated reduction of cerebral Aβ deposition was not dependent on APP metabolism and Aβ production. This evidence, along with the knowledge that brain Aβ exists in a finely tuned balance between production and clearance, suggests that CAIs may promote Aβ clearance.
Total caspase-3 activation
To test whether the drastic cerebral Aβ reduction observed in CAI-treated mice was associated with a reduction in apoptotic cells, we measured the activity of caspase-3 in total brain lysates using a luminometric Caspase-Glo assay (Fig. 1F). We observed an increase in active caspase-3 in TgSwDI mice compared to WT, which was significantly decreased in ATZ- and MTZ-treated Tg animals, indicating that CAIs reduce the activation of apoptotic pathways in brain cells.
ATZ and MTZ mitigate vascular Aβ burden, reduce endothelial active caspase-3, microhemorrhages, and revert vasoconstriction
To determine whether CAIs specifically reduce vascular Aβ deposition and Aβ-mediated endothelial apoptosis, we measured vascular Aβ and assessed caspase-3 activation in endothelial cells (ECs). First, immunofluorescence (IF) evaluation with an Aβ-specific antibody confirmed that ATZ- and MTZ-treated animals had massively decreased total cerebral Aβ burden, corroborating the Thioflavin staining. In addition, total caspase-3 activation, assessed by IF with an antibody that specifically binds the active form of caspase-3, was reduced in DG (Fig. 2A), cortex (Fig. 2B), and CA1 (Supplementary Fig. 2A), in CAI-treated animals (in DG, ATZ −72% and MTZ −59%; in cortex, ATZ −58.5% and MTZ −59.7%; in CA1s, ATZ −65.7% and MTZ −57%, vs Tg), confirming the results of the luminometric caspase-3 activation assay. We then observed that vascular Aβ deposits (Aβ signal colocalized with CD31) also colocalized with active caspase-3 signal in CD31+ ECs (Fig. 2A and 2B; arrows and magnified images), suggesting that vascular Aβ triggers apoptotic processes in ECs, and supporting our previous in vitro studies16,32–35,43. Remarkably, ATZ and MTZ attenuated both microvascular Aβ burden (measured as Aβ staining overlapping with CD31 signal), and endothelial caspase-3 activation (active caspase-3 signal overlapping with CD31), in DG (ATZ −74.2% and MTZ −89.4%, vs Tg; Fig. 2A) and to a lesser degree (ATZ −40.8%, p= 0.57, and MTZ −72.5%, p= 0.078, vs Tg) in the cortex (Fig. 2B), indicating that CAIs mitigate both vascular Aβ deposition and Aβ-driven endothelial cell apoptosis. The presence of Aβ around the brain vasculature, especially in familial disorders or animal models bearing the Dutch or analogue vasculotropic mutations, is typically associated with cerebrovascular lesions, including microhemorrhages (MH)65–68 . In TgSwDI mice, MH are present starting at 12 months of age69. Therefore, we tested whether the reduction of vascular Aβ and endothelial caspase-3 activation by CAIs could also result in a reduction of MH in the Tg mice. Compared to age-matched WT, 16-month-old Tg animals had a significantly higher number of MH in DG (Fig. 2C), cortex (Fig. 2D), and leptomeningeal arteries (Supplementary Fig. 2B). The number of MH was reduced in MTZ- and ATZ-treated groups (in DG, ATZ −37.4%, and MTZ −53.2%; in cortex, ATZ −41% and MTZ −59%; in leptomeningeal arteries, ATZ −90.9% and MTZ −93.7%, vs Tg), indicating that CA inhibition prevents Aβ-mediated vessel wall degeneration. It has also been shown that Aβ deposition reduces vessel size in AD patients70 and Tg mice66,71–73. Hence, we measured vessel width in WT, Tg and treated animals. As expected, 16-month-old TgSwDI mice presented constricted vessels in DG (Fig. 2E) and cortex (Fig. 2F), compared to WT animals. In contrast, vessel diameter in CAI-treated Tg mice was not different from WT mice, corroborating our hypothesis that CAIs regulate cerebrovascular tone74, besides promoting EC survival.
Figure 2: Vascular Aβ burden, endothelial caspase-3 activation, microhemorrhages and microvessel constriction are reduced by CAIs.
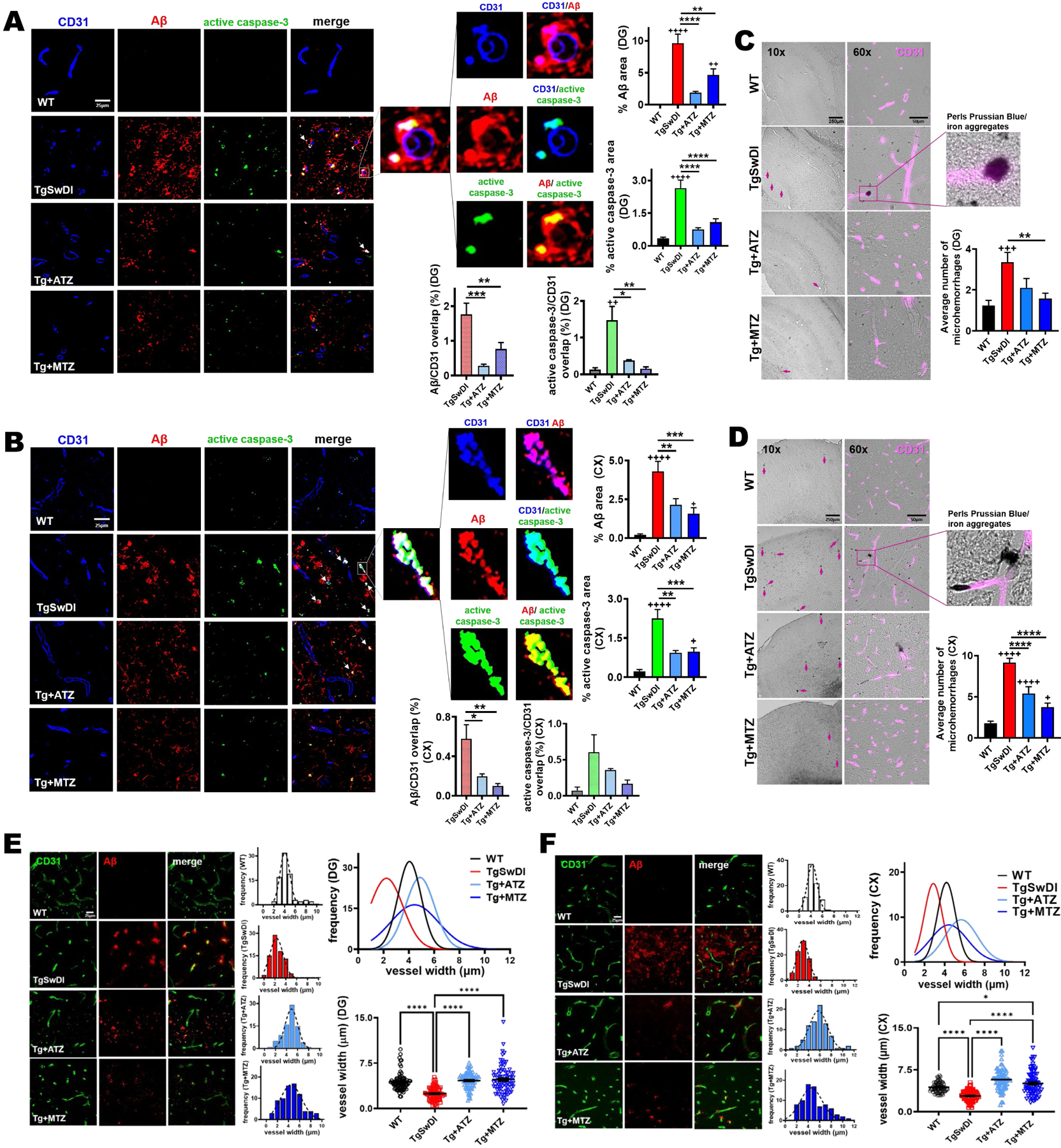
A, B) Representative immunofluorescence images of DG (A) and cortex (B) of 16-month-old mice. TgSwDI mice present highly elevated cerebral Aβ (red) and active caspase-3 (green) staining, which are significantly decreased by CAI treatment. Original magnification, 60x. Scale bar, 25μm. On the right, relative quantification plotted as the percentage of area occupied by Aβ or active caspase-3 per acquisition field. For %Aβ graph, WT, TgSwDI and MTZ: N=5, ATZ: N=4, n≥12 measurements acquired /group. For % active caspase-3 graph, WT, TgSwDI and MTZ: N=5, ATZ: N=3, n≥9 measurements acquired /group. In the merged images, arrows point to Aβ deposits surrounding ECs (CD31+ ECs in blue). The magnified images illustrate overlap between the signals: ECs and Aβ (signal overlap in magenta), ECs and active caspase-3 (signal overlap in cyan), and Aβ overlapping active caspase-3 (yellow). Below, the graphs depict the percentage of Aβ and active caspase-3 signals, respectively, overlapping with CD31. For Aβ/CD31 overlap, N=5 for TgSwDI and MTZ, N=4 for ATZ, n≥12 measurements/group. For active caspase-3/CD31 overlap, N=3–5 mice/group, n ≥5 measurements/group. C) Representative 10x and 60x images of DAB-enhanced Perls Prussian Blue staining in DG of 16-month-old mice, showing higher number of microhemorrhages (MH) (indicated with arrows) in TgSwDI mice, vs WT animals, which are reduced in CAI-treated groups. Colocalization of iron aggregates with CD31+ BVs (magenta) is shown in the 60x and magnified images. Scale bars are 250μm and 50μm, respectively for 10x and 60x magnification. The plot on the right represents the average number of MH counted. WT, TgSwDI and MTZ: N=4, ATZ: N=3, n≥10 counts/group. D) Representative 10x and 60x images of Perls Prussian Blue staining, in cortex. MH presence (indicated with arrows) in TgSwDI mice, significantly decreased by CAI-diet. DAB-enhanced iron aggregates colocalizing with CD31+ BVs (magenta) shown in the magnified image. For magnification 10x and 60x, scale bar 250μm and 50μm, respectively. Relative quantification plotted on the right as the average number of MH. WT, TgSwDI and MTZ: N=4, ATZ: N=3, n≥10 counts/group. E) CAI treatment diminishes Aβ deposition (red) in DG, in TgSwDI mice, affecting vessel diameter (CD31, vascular marker, green). Original magnification, 60x. Scale bar, 25μm. On the right, vessel width frequency quantification in each group. On the top right, the frequency of all groups is plotted. On the bottom right, vessel diameter quantification, showing that CAI-fed mice, similarly to WT animals, displayed BVs with wider average diameter compared to untreated TgSwDI animals. WT, TgSwDI and MTZ: N=5, ATZ: N=4, n=80 vessels/group. F) CAI-diet increased blood vessel width in cortex, in 16-month-old TgSwDI mice. Original magnification 60x, scale bar 25μm. WT, TgSwDI and MTZ N=5, ATZ: N=4, n=80 vessels/group. In (A-F), one-way ANOVA and Tukey’s post-hoc test: * and + p<0.05, ** and ++ p<0.01, *** and +++ p<0.001, **** and ++++ p<0.0001. Data are expressed as mean ± SEM.
Analysis of cerebral blood volume and flow in living animals
Previous studies have shown an age-dependent impairment in neurovascular coupling (NVC) in the TgSwDI model52. Therefore, we tested whether CAIs affected NVC impairment by measuring the relative changes in cerebral blood volume (rCBV) and cerebral blood flow (rCBF) in 10/11-month-old awake-restrained mice following whisker stimulation (Fig. 3 and Table 1). Based on a time-series recording, we estimated the maximum evoked-response in CBV (rCBV Peak) and CBF (rCBF Peak), and extracted the area under the curve (rCBV A.U.C. and rCBF A.U.C.), which is considered as the total CBV and CBF change during functional activation/stimulation. Upon activation, we observed the expected increase in rCBV and rCBF in all groups (Fig. 3B and 3E), but no significant difference in rCBV Peak (Fig. 3C), rCBV A.U.C. (Fig. 3D) and rCBF Peak (Fig. 3F), between Tg mice and WT animals, was evident at this age. Interestingly, we observed a significant increase in rCBF A.U.C. in the MTZ-treated animals compared to the WT mice (Slope ≈ 0.42 ± 0.13, t(66)=3.16, p=0.002) (Fig. 3G).
Figure 3: Analysis of cerebral blood flow and volume during functional activation.
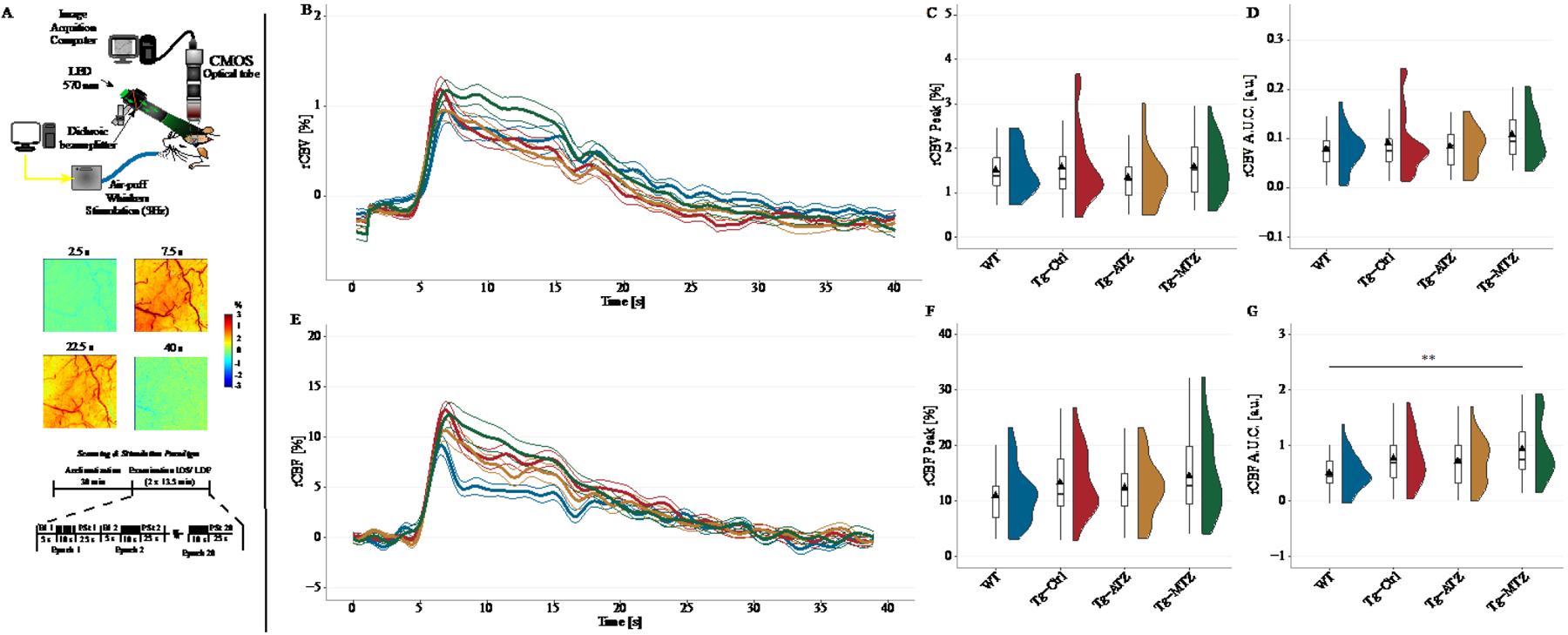
A) Experimental setup for IOS to measure total Hemoglobin (Hb), a proxy for cerebral blood volume (CBV). A LED constantly illuminated the cranial window with a spectrum of 570 ± 2nm (top), while AVI videos were recorded using a CMOS camera at 5-fps. IOS relies on the reflectance of light from the illuminated cortex. At 570nm, both oxy- and deoxy-Hb are absorbed at the same rate, equivalent to total Hb (rCBV). Whisker stimulation increases CBV, reducing the reflectance, hence the signal. In the postprocessing, the signal was inverted for better representation (middle). For IOS and LDF, whisker stimulation occurred from the second 5 to the second 15 in each epoch (bottom). B) Relative changes in CBV to the baseline (rCBV [%]) were estimated and plotted as time-series. C) No difference between the groups in the maximum CBV response (rCBV Peak [%]) to stimulation, and area under the curve (rCBV A.U.C. [a.u.]) (D), during the stimulation period (10’). E) Estimated changes in regional cerebral blood flow (CBF) during functional activation, using a commercial LDF. F) No difference between groups in the maximum response to functional activation (rCBF Peak [%]). G) Analysis of the A.U.C. for regional CBF (rCBF A.U.C. [a.u.]). Tg+MTZ mice showed an overall increase in CBF response during the stimulation period, compared to the WT mice. N values: WT = 16 mice (8 males and 8 females); TgSwDI, N=17 (6 males and 11 females); Tg+ATZ, N=17 (11 males and 6 females); Tg+MTZ, N=16 (8 males and 8 females). ** p<0.01. Abbreviations: Bl: Baseline, PSt: poststimulation. Statistics: Cluster analysis using linear mixed-models predicting the changes in peak and A.U.C. by stimulation and adjusted for gender.
Table 1.
Time series analysis of OISI (rCBV) and LDF (rCBF)
| Variable | WT | Tg-Ctrl | Tg-ATZ | Tg-MTZ |
|---|---|---|---|---|
| Mice included | 16 mice (8 M and 8 F) |
17 mice (6 M and 11 F) |
17 mice; (11 M and 6 F) |
16 mice (8 M and 8 F) |
| OISI (N value) | N = 30 | N = 32 | N = 27 | N = 30 |
| rCBV Peak [%] | 1.49 ± 0.49 | 1.56 ± 0.81 | 1.33 ± 0.62 | 1.57 ± 0.60 |
| AUC [a.u.] | 0.07 ± 0.04 | 0.09 ± 0.06 | 0.08 ± 0.04 | 0.1 ± 0.04 |
| LDF (N value) | N = 32 | N = 34 | N = 32 | N = 34 |
| rCBF Peak [%] | 10.87 ± 4.10 | 13.19 ± 5.44 | 12.27 ± 4.90 | 14.37 ± 6.27 |
| AUC [a.u.] | 0.50 ± 0.26 | 0.75 ± 0.42 | 0.70 ± 0.36 | 0.92 ± 0.45** |
M: Males, F: Females.
p < 0.01
CAI treatment reduces astrocytic Aβ accumulation and caspase-3 activation, rescuing Aβ-dependent astrogliosis
Astrocytes exert multiple functions in the brain, including mediating vascular fitness and permeability. They are physically in close association with both blood vessels (BVs) and neuronal cells75, they assure the modulation of vascular response based on neuronal activity (NVC), and mediate perivascular clearance pathways4,26,76,77. Moreover, both astrocytes and microglia act as scavenger cells, capable of internalizing and degrading brain waste molecules, including Aβ. Dysfunctional glial cells result in impaired Aβ clearance, and exacerbate cerebral Aβ overload and inflammatory state, also contributing to vascular Aβ deposition and loss of cerebrovascular functionality. In turn, vascular alterations, such as the observed Aβ-induced endothelial cell stress, can mediate glial activation and inflammatory state, and interfere with glial and perivascular clearance. Accordingly, we observed that severe Aβ accumulation in untreated Tg mice was associated with substantial astrogliosis in the hippocampus. Notably, both Aβ and Glial Fibrillary Acidic Protein (GFAP) stainings were reduced by ATZ and MTZ treatment (Fig. 4A). Colocalization of Aβ and GFAP signals showed that in Tg mice Aβ abundantly accumulated inside the astrocytes, while CAI treatment prevented the astrocytic Aβ overload (Fig. 4B). As expected, TgSwDI animals had an increased percentage of GFAP+ fluorescent area in the DG (Fig. 4C), indicating astrogliosis and hypertrophy of astrocytic processes. This pro-inflammatory phenotype was prevented by CAI treatment, in association with a drastic reduction of Aβ accumulation within astrocytes (quantified as colocalization of Aβ signal overlapping with GFAP signal). Concurrently, we observed a significant mitigation of active caspase-3 signal within GFAP+ astrocytes (Fig. 4C), particularly with ATZ treatment. ATZ and MTZ treatment resulted in a similar reduction of GFAP+ area, astrocytic Aβ accumulation and astrocytic caspase-3 activation in the cortex (Fig. 4D) and in the CA1 hippocampal area (Supplementary Fig. 3A). These results suggest that CAIs limit the activation of apoptotic pathways in astrocytes, facilitate Aβ removal and reduce astrogliosis. To corroborate these findings, we measured astrocytic hypertrophy, as the average area (μm2) of single GFAP+ cells in the hippocampus, cortex (Fig. 4E), and hypothalamus (Supplementary Fig. 3B). TgSwDI mice displayed a dramatic increment in the average single astrocyte area (in DG, +260%; in CA1, +310%; in CA3, +322%; in RSC, +287%; in gRSC, +290%; in hypothalamus, +303%, vs WT), denoting a typical astrocytic reactive phenotype, which was significantly reverted by CAI treatment (in DG, ATZ −34% and MTZ −46.6%; in CA1, ATZ −57% and MTZ −67%; in CA3, ATZ −39% and MTZ −37%; in RSC, ATZ −50% and MTZ −36.8%; in gRSC, ATZ −57% and MTZ −35%; in hypothalamus, ATZ −59.5% and MTZ −37%, vs Tg mice). Overall, this evidence highlights the ability of CAIs to mitigate Aβ-initiated cerebral astrocytosis, and Aβ-driven apoptotic pathways in astrocytes, indicating that CAIs may improve glial health, favoring Aβ clearance.
Figure 4: Astrocytosis, astrocytic Aβ content and caspase-3 activation are reduced in CAI-treated TgSwDI mice.
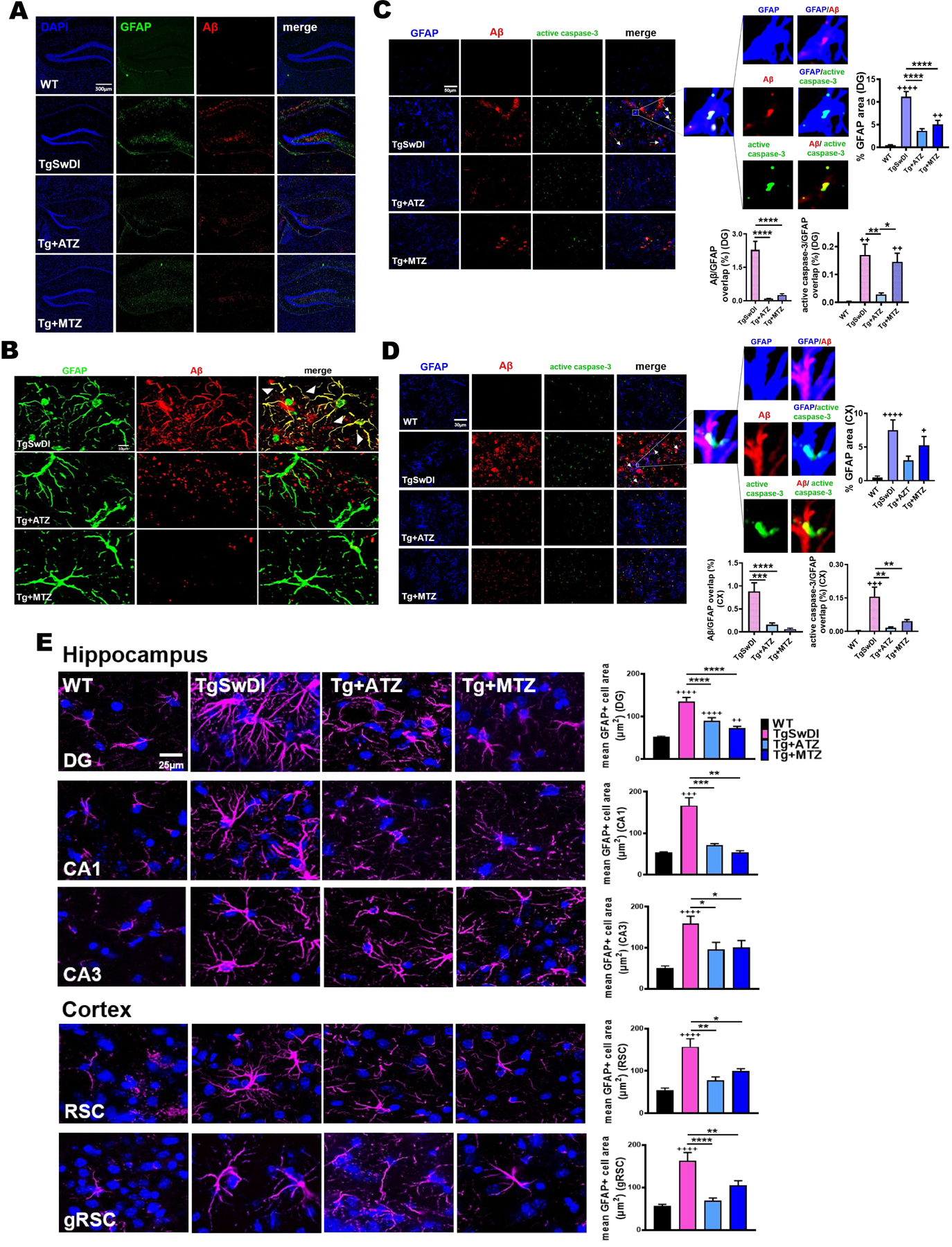
A) 10x images of DG of 16-month-old TgSwDI mice show augmented GFAP expression (green) and Aβ overload (red). Nuclei stained with DAPI (blue). CAI treatment reduces both astrogliosis and Aβ deposits. Original magnification, 10x. Scale bar, 300μm. B) In TgSwDI mice, Aβ (red) is trapped within astrocytes (green), as shown by the colocalization (yellow) in the merged image. ATZ and MTZ treatment reduces astrocytic Aβ content. C, D) Representative immunofluorescence images of DG (C) and cortex (D). Compared to WT, untreated Tg animals exhibit greater expression of the astrocytic marker GFAP (blue), as well as Aβ (red) and active caspase-3 (green), all decreased by treatment with CAIs. Original magnification, 60x. Scale bar, 30μm. On the right, astrogliosis is plotted as %GFAP area per acquisition field. WT, TgSwDI and MTZ: N=5, ATZ: N=3, n≥9 measurements acquired/group. Arrows indicate colocalization of Aβ and active caspase-3 in astrocytes. The magnified images display Aβ within GFAP+ cells (signals overlap in magenta), astrocytic caspase-3 activation (signals overlap in cyan), and Aβ colocalizing with caspase-3 (yellow). Below, graphs represent the percentage of Aβ and active caspase-3 signals overlapping with GFAP+ cells, indicating that CAIs significantly reduced astrocytic Aβ accumulation and caspase-3 activation (significant for ATZ in DG, and for both CAIs in cortex) in astrocytes. WT, TgSwDI and MTZ: N=5, ATZ: N=3, n≥9 measurements/group. E) 60x magnified images representing astrogliosis in 16-month-old mice. TgSwDI brains are characterized by significantly increased GFAP+ average cell area (μm2) (GFAP in magenta), in both the hippocampus (DG, CA1 and CA3 areas) and cortex (RSC and gRSC). Nuclei are stained with DAPI (blue). 8-month-CAI-diet attenuates astrogliosis. Original magnification, 60x. Scale bar, 25μm. On the right, relative quantification of the mean area of one GFAP+ cell, for each brain area analyzed. For both hippocampus and cortex, WT, TgSwDI and MTZ: N=5, ATZ: N=4 animals/group. In (A-E), one-way ANOVA and Tukey’s post-hoc test: * and + p< 0.05, ** and ++ p<0.01, *** and +++ p<0.001, **** and ++++ p<0.0001. Data are expressed as mean ± SEM.
CA inhibition attenuates microglial Aβ accumulation and caspase-3 activation, fostering a microglial pro-healing phenotype
Microglia are the primary brain resident innate immune cells and the most active form of immune defense in the CNS78. As astrocytes, microglia have an important role in brain clearance. In a physiological state, microglia migrate towards the site of interest, remove dead cell debris, and promote both extracellular and intracellular degradation of unwanted toxic substances, including Aβ. A prolonged insult prompts the microglial pro-inflammatory phenotype, resulting in morphological and biochemical changes, such as augmented expression of ionized calcium binding adaptor molecule 1 (IBA1) protein. Indeed, we found that 16-month-old TgSwDI mice had an increased percentage of IBA1+ signal area (Fig. 5), indicative of microgliosis, compared to age-matched WT animals (in DG, +11fold change, F.C.; in cortex, +15 F.C.; in CA1, +8.3F.C., vs WT). Importantly, CAI treatment significantly reduced IBA1 staining in DG (Fig. 5A), cortex (Fig. 5B), and in the CA1 hippocampal area (Supplementary Fig. 4A) (in DG, ATZ −54% and MTZ −49%; in cortex, ATZ −64.7% and MTZ −56.7%; in CA1, ATZ −48% and MTZ −53%, vs Tg). In order to elucidate whether this change was associated with Aβ overload and active caspase-3 in microglia, we analyzed the amount of Aβ and active caspase-3 signals overlapping with IBA1 staining. We found that untreated Tg mice displayed a massive microglial Aβ accumulation in DG (Fig. 5A), cortex (Fig. 5B), and CA1 (Supplementary Fig. 4A), accompanied by a robust microglial-specific activation of caspase-3 (Fig. 5A, 5B and Supplementary Fig. 4A) (in DG, +63F.C.; in cortex, +49.5F.C.; in CA1, +100F.C., vs WT). Notably, CAIs attenuated both Aβ deposition and caspase 3 activation in microglia (Fig. 5A, 5B and Supplementary Fig. 4A) (for Aβ, in DG, ATZ −62% and MTZ −66%; in cortex, ATZ −71% and MTZ −48%; in CA1, ATZ −62.9% and MTZ −86%, vs Tg. For caspase-3 activation, in DG, ATZ −35% and MTZ −44.5%; in cortex, ATZ −65% and MTZ −67.6%; in CA1, ATZ −69% and MTZ −89%, vs Tg). We further investigated CAI impact on neuroinflammatory responses by evaluating microglial shape, which has been accepted as an indicator of their functional state79. Resting microglial cells present ramified morphology with some long and fine processes58. In the event of an insult, microglia become active and rapidly change their morphology from ramified to bushy cells with multiple short, thickened and sturdy processes57. At the site of injury, a pro-healing microglial phenotype contributes to neuroprotection80–82. Under excessive and chronic stress, microglia keep modifying their shape, retracting their processes, and adopting an amoeboid shape83, which is indicative of a severe pro-inflammatory state associated to the release of neurotoxic factors. In each treatment group, resting, bushy and amoeboid microglia subpopulations were present (Fig. 5C). As expected, resting microglia were the most represented subpopulation in the WT group (Fig. 5C). In contrast, in untreated TgSwDI brains, amoeboid microglia were the most abundant type. Notably, ATZ- and MTZ-treated mice were predominantly characterized by bushy microglia (Fig. 5C). These data suggest that CAIs reduce the toxic pro-inflammatory phenotype and promote the active but neuroprotective phenotype. When plotting resting, amoeboid and bushy microglia in the different treatment groups in DG (Fig. 5D), resting microglia were significant higher in WT compared to TgSwDI mice, and partially rescued by ATZ and MTZ. As expected, amoeboid microglia were higher in Tg mice, and partially reduced by MTZ and ATZ. Interestingly, the number of bushy microglia was significantly higher in CAI-treated animals, compared to both WT and untreated Tg mice, suggesting that CAIs may contribute to promote a microglial pro-clearance active phenotype. In the cortex, we found that TgSwDI animals had a significantly higher number of bushy microglia, compared to amoeboid, with similar levels in CAI-fed animals (Supplementary Fig. 4B), likely indicating a higher resilience of microglia in the cortical areas of these Tg mice. Both ATZ- and MTZ-treated groups presented a significantly higher number of resting microglia in the cortex compared to TgSwDI mice (Supplementary Fig. 4C), with similar counts as in WT animals, but the treatment did not affect the total number of amoeboid and bushy cells. To evaluate CAI effects on the total number of microglia, we counted IBA1+ cells. We found that TgSwDI mice had a trend to a reduced number of microglia in DG, compared to WT mice (although not significant), possibly due to microglial apoptosis, while CAIs rescued this loss, exhibiting microglial counts even higher than WT animals (Fig. 5E), suggesting a prevalence of healthy activated microglia in CAI-treated Tg mice. In the cortex, all Tg mice showed a higher number of microglial cells, compared to WT (Supplementary Fig. 4D), in line with the microglial phenotypes described above. These results indicate that CAIs reduce Aβ-mediated microglial toxicity and microglial caspase activation, promote microglial pro-healing phenotypes, and reduce the neurotoxic inflammatory phenotype, particularly in the hippocampus.
Figure 5: ATZ and MTZ reduce microglial Aβ overload and caspase-3 activity, and promote microglial pro-healing phenotype.
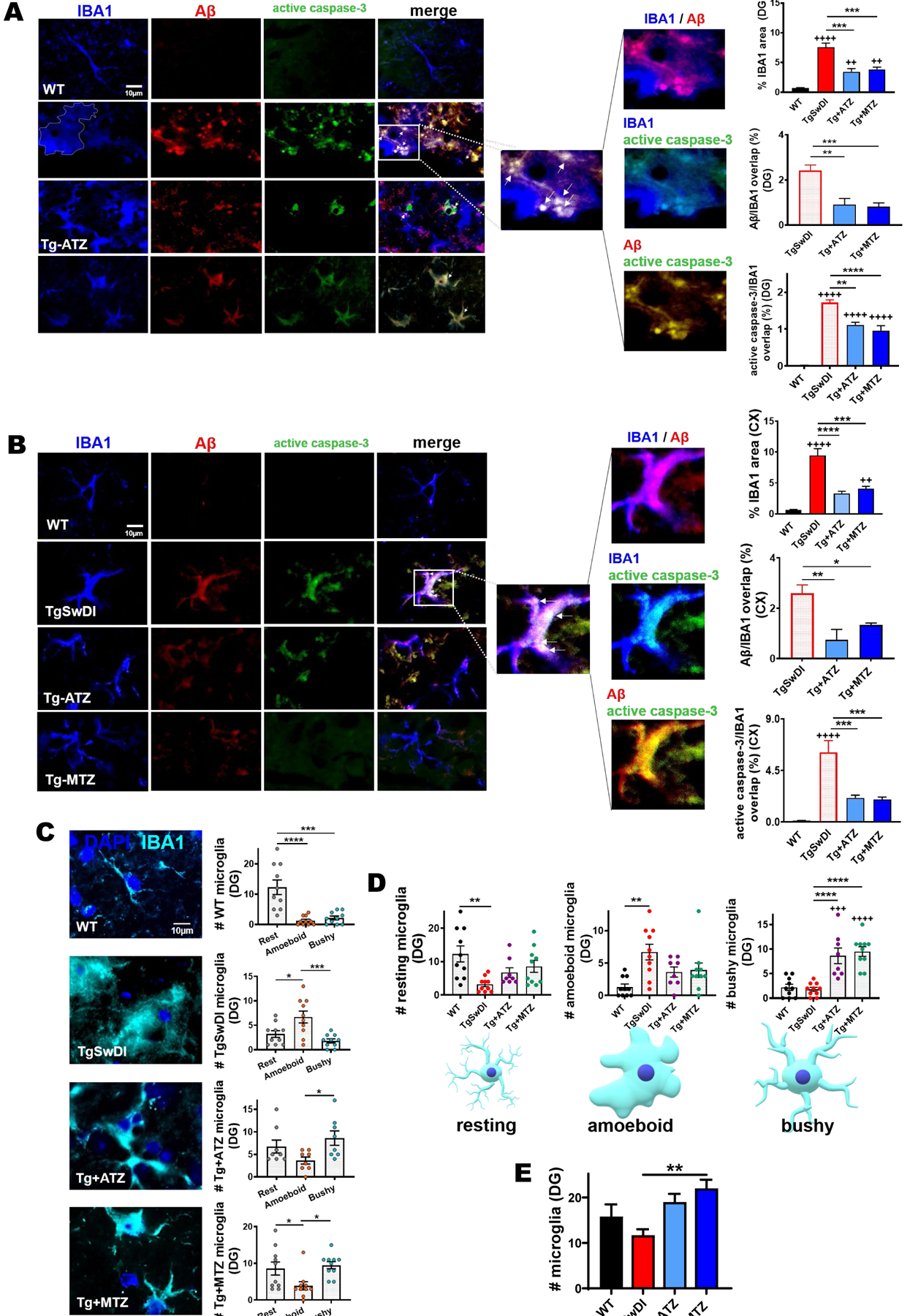
A, B) Representative immunofluorescence images of DG (A) and cortex (B), showing that Tg animals exhibit increased microgliosis (IBA1, microglia marker in blue), Aβ (red) accumulation and active caspase-3 (green) in microglia, all rescued by CAIs. Original magnification, 60x. Scale bar, 10μm. On the right, % of IBA1 area per acquisition field. WT, TgSwDI and MTZ: N=5, ATZ: N=4, n≥8 measurements acquired/group. Arrows indicate microglia presenting internalized Aβ and active caspase-3. The magnified images show Aβ within IBA1+ cells (signal overlap in magenta), active caspase-3 within IBA1+ cells (signal overlap in cyan), and Aβ colocalizing with caspase-3 (yellow). On the right, plots represent the percentage of Aβ and active caspase-3 signals overlapping with IBA1+ cells, indicating that CAIs significantly reduce microglial Aβ overload and caspase-3 activation. WT, TgSwDI and MTZ: N= 5, ATZ: N=4, n≥8 measurements/group). C) Representative immunofluorescence images of microglia (IBA1 marker, cyan) for analysis of resting, amoeboid and bushy morphology. On the right, plots represent the different microglial phenotypes in each treatment group, in DG. WT mice have resting microglia as the most numerous subpopulation. TgSwDI have amoeboid microglia as most represented microglial type, while ATZ- and MTZ-treated mice present more bushy and resting microglia than amoeboid. WT, TgSwDI and MTZ: N= 5, ATZ: N=4, n≥8 measurements/group. D) Comparison of DG resting, amoeboid or bushy microglia between the different groups. The number of resting microglia is significantly higher in WT mice compared to Tg mice, but is not significantly different from WT in MTZ- and ATZ-treated mice. The amount of amoeboid microglia is the highest in Tg mice, while bushy microglia is more abundant in CAI-treated mice compared to WT and Tg. WT, TgSwDI and MTZ: N=5, ATZ: N=4, n≥8 measurements/group. E) Microglial cell (IBA1+ cells) count in DG. TgSwDI mice have fewer microglia than WT animals, while CAIs increase microglial number in Tg mice. WT, TgSwDI and MTZ: N=5, ATZ: N=4, n≥8 measurements/group. In (A-E), one-way ANOVA and Tukey’s post-hoc test: * p< 0.05, ** and ++ p<0.01, *** and +++ p<0.001, **** and ++++ p<0.0001. Data are expressed as mean ± SEM.
CAIs promote microglial and perivascular macrophage phagocytic activity
The triggering receptor expressed on myeloid-cells-2 (TREM2) is expressed by microglia and promotes microglial survival, proliferation and phagocytic activity84–86. In our TgSwDI mice, TREM2 levels appeared slightly lower (−19% in DG and −16% in cortex, not significant) compared to age-matched WT animals. However, ATZ and MTZ treatment significantly boosted TREM2 expression compared to untreated Tg mice in the DG, with a similar trend in the cortex (Fig. 6A). When compared to WT mice, ATZ also significantly enhanced TREM2 expression also in the CA1 hippocampal area (Supplementary Fig. 5A). The increase in TREM2 induced by CAIs suggests that these drugs promote microglial phagocytic activity and therefore Aβ clearance, in line with the observed reduction of total, vascular and glial Aβ deposits. To corroborate this hypothesis, we analyzed the presence of CD68, a microglial/perivascular macrophage (PVM) activation marker, located in the endosomal/lysosomal compartment, which plays a major role in the clearance of brain waste material87. IHC analysis revealed that TgSwDI mice exhibited increased CD68 levels in the DG (Fig. 6B, more than +2.5 F.C.), and even greater levels in the CA1 (+3.8 F.C.) (Supplementary Fig. 5B), in contrast to WT animals. Strikingly, in both areas, CAI treatment further increased CD68 expression compared to untreated Tg mice (in DG, ATZ- and MTZ-treated mice showed +1.65 and +1.44F.C., respectively, compared to Tg; in CA1, ATZ and MTZ groups had +1.7 and +1.26F.C., respectively, in contrast to untreated Tg). These data, in combination with the increase in TREM2, reinforce the hypothesis that CAIs may reduce Aβ deposition by promoting microglial/PVM phagocytic activity. We observed the presence of an elevated number of CD68+ cells that colocalize with vascular Aβ deposits in the perivascular spaces (Fig. 6B, DG, magnified images), suggesting that CAIs (particularly ATZ) may prompt PVM phagocytic activity, likely contributing to perivascular Aβ clearance. Indeed, the CAI-treated groups presented a reduced Aβ content within CD68+ cells (DG, Fig. 6B) compared to untreated Tg animals. These results may suggest an increased amount of active perivascular CD68+ cells migrating over the pathological deposits, accompanied by an increased degradation of Aβ, likely resulting in the observed reduction of vascular Aβ (Fig. 2A, 2B, and Supplementary Fig. 2A). In the cortex (Fig. 6C), MTZ treatment particularly boosted the total CD68 signal compared to Tg mice. The percentage of Aβ signal within CD68+ cells was slightly reduced in CAI-treated mice, although it did not reach statistical significance. Similarly to what observed in the DG, ATZ induced a higher CD68 signal over Aβ deposits (Fig. 6C), suggesting an increased trophism of CD68+ cells towards Aβ degradation. Immunoblot analysis of CD68 expression corroborated the IHC assessment, showing increased CD68 levels in TgSwDI animals compared to WT, with an even greater expression in CAI-diet fed mice (Fig. 6D). Overall, these results suggest that CA inhibition promotes Aβ removal by microglia/PVM, facilitating Aβ clearance.
Figure 6: CAI treatment increases TREM2 and CD68+ perivascular phagocytic cells.
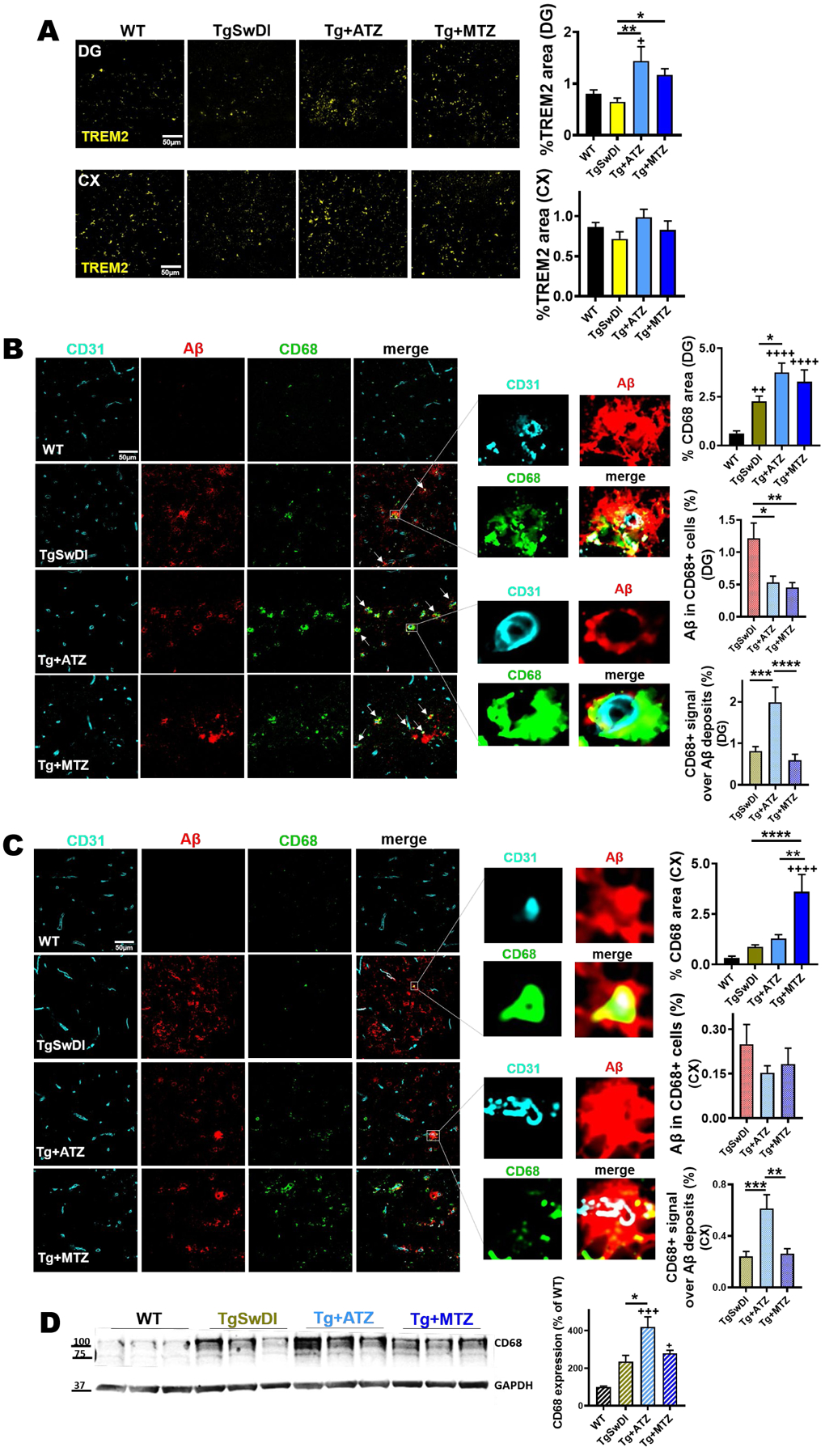
A) ATZ and MTZ significantly increase microglial TREM2 expression in DG in TgSwDI mice, and show a trend to increase in the cortex (cx). Relative plots on the right. For % TREM2 area, both in DG and cortex, WT, TgSwDI and MTZ: N=5, ATZ: N=3, n≥9 measurements acquired /group. * and + p<0.05 and **p<0.01, One-way ANOVA and Tukey’s post-hoc test. B, C) Expression of CD68 (green) around microvasculature (CD31, cyan), and co-localization with Aβ (red) in DG (B) and cortex (C). Original magnification, 60x. Scale bar, 50μm. CAI-fed animals present higher phagocytic activity marker (CD68), plotted as %CD68 area per acquisition field, compared to WT and Tg mice. TgSwDI: N=7, WT: N=6, ATZ: N=4, MTZ: N=5, n≥12 measurements acquired/group. * p<0.05, ++ p<0.01, ++++ p<0.0001, One-way ANOVA and Tukey’s post-hoc test. Arrows indicate vascular Aβ internalized by CD68+ perivascular macrophages (PVM), as shown in yellow in the magnified images. On the right, colocalization plots for both Aβ within CD68+ cells (Aβ/CD68) and CD68+ area over Aβ deposits (CD68/Aβ). TgSwDI: N=7, MTZ: N=5, ATZ: N=4, n≥11 measurements/group. For Aβ/CD68 colocalization, * p<0.05 and ** p<0.01. For CD68/Aβ colocalization, *** p<0.001 and **** p<0.0001, ++++P<0.0001. One-way ANOVA and Tukey’s post-hoc test. D) WB of total brain homogenates showing that CD68 expression is significantly increased by CAIs, particularly ATZ. N=3/group, * and + p<0.05, +++ p<0.001, One-way ANOVA and Tukey’s post-hoc test. Data are expressed as mean ± SEM.
CA-VB is a mediator of Aβ-driven neurovascular dysfunction
We have shown above that CAI treatment inhibits caspase-3 activation in ECs, astrocytes and microglia in Tg mice. We have previously demonstrated in cellular models that CAIs reduce caspase activation in cells composing the NVU by preventing Aβ-driven mitochondrial dysfunction and mitochondrial ROS production (particularly H2O2)16,32–35,43,44. Among the 15 known human CA isoforms, CA-VA and -VB are expressed solely in the mitochondria, and CA-II (a widespread cytosolic isoform abundant in the CNS) translocates to the mitochondria during aging and neurodegeneration88. Because mitochondrial mechanisms are responsible for the protective effects exerted by CAIs, we tested whether changes in mitochondrial CA isoforms were present in TgSwDI mice. Immunoblot analysis unveiled that, compared to WT, 16-month-old TgSwDI brains displayed a significant increased CA-VB expression (+1.75 F.O.C, p<0.01) (Fig. 7A), and, strikingly, CAI treatment restored CA-VB levels similar to WT brains (Fig. 7A). When we measured the cerebral levels of CA-VA and CA-II in our model, we did not observe significant alterations among the different groups (Fig. 7A), suggesting that specifically CA-VB overexpression may be pathologically induced by cerebral amyloidosis. Corroborating this result, we found a positive correlation between the expression of brain CA-VB and cerebral insoluble Aβ40, the most abundant Aβ species in this mouse model (Supplementary Fig. 6A). Moreover, we evaluated mitochondrial CA expression in CAA human cortices, and detected upregulated human CA-VB levels, with a strong trend to significance (127% increase, p=0.06; Fig. 7B), compared to the controls, and no changes in CA-VA and CA-II expression (Fig. 7B). Similarly, we found that CA-VB was overexpressed approaching significance (511% increase, p=0.06; Fig. 7C) also in human AD brains presenting CAA pathology (AD+CAA). Combining the human CAA and AD+CAA groups, we found a significant upregulation of CA-VB levels (Fig. 7D; 215% increase, p<0.05), suggesting that brain amyloidosis and CAA may trigger a specific CA-VB overexpression. To confirm this, and due to the importance of vascular dysfunction in our model, we evaluated the expression of CA-VB in human cerebral microvascular ECs challenged with 25μM Aβ40-Q22 (the vasculotropic Dutch mutant present in the TgSwDI mice) or 10μM Aβ42. In line with the expression pattern observed in the Tg animals and in the human subjects, we observed that Aβ, particularly Aβ40-Q22, induced an increase in CA-VB expression (Fig.7E), but not in that of CA-VA or CA-II (Supplementary Fig. 6B and 6C). Hence, to test the hypothesis that inhibition of endothelial CA-VB results in protective effects, we assessed microvascular EC apoptosis following Aβ challenge in the presence or absence of CA-VB silencing RNA (Fig. 7F and 7G), and compared its effects to CA-VA and CA-II silencing (Supplementary Fig. 6D and 6E). Silencing efficiency was confirmed via PCR after 48hrs (−80% CA-VB expression in siCA-VB conditions, compared to a scrambled siRNA control) (Fig. 7F). As expected, ECs treated with Aβ peptides at concentrations known to cause apoptosis in our models (10μM Aβ42 or 25μM Aβ40-Q22, for 24hrs after silencing)16,32,43 underwent apoptosis in presence of the scrambled siRNA control, while CA-VB silencing significantly rescued Aβ42- and Aβ40-Q22-induced EC apoptosis (Fig. 7G). In contrast, when CA-VA and CA-II were silenced, we did not observe any attenuation of Aβ-induced apoptosis (Supplementary Fig. 6D and 6E), pointing to CA-VB as a specific target for mitigating Aβ-induced EC stress, and thus the resultant neurovascular dysregulation, in AD and CAA. On the contrary, CA-VA downregulation, per se, induced apoptosis (Supplementary Fig. 6D), as expected, since CA-VA is considered an essential enzyme89,90. Interestingly, CA-VA was also decreased in ECs treated with Aβ40-Q22 (Supplementary Fig. 6B). Overall, this set of experiments suggests that the protective properties of CAIs observed in our cellular and animal models may be mediated, at least in part, through the mitigation of a maladaptive Aβ-induced overexpression of the mitochondrial CA-VB isoform. Importantly, upon CAI treatment, both Aβ deposition and CA-VB overexpression were reduced in Tg mice. This finding, together with the fact that CA-VB silencing reduces Aβ-induced EC apoptosis, indicates that Aβ-induced CA-VB overexpression may be a mediator of cerebrovascular stress and cell death.
Figure 7: CA-VB expression increases in Tg mice, AβQ22-treated ECs and CA-VB silencing prevents endothelial apoptosis.
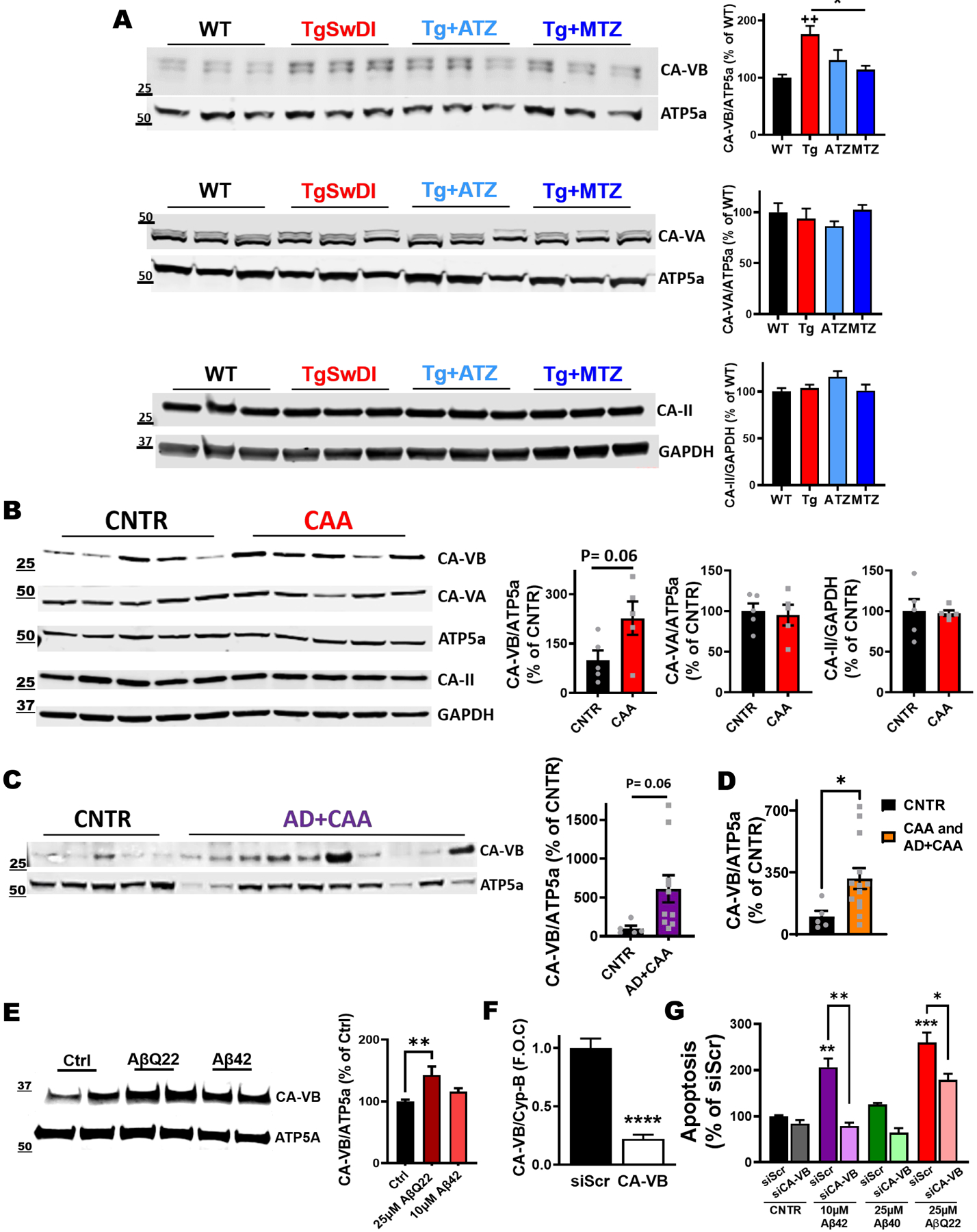
A) Immunoblot for CA-VA, -VB and -II in total brain lysates of 16-month-old mice. TgSwDI brains display a significant increase in the mitochondrial carbonic anhydrase-VB (CA-VB) compared to age-matched WT mice, while CAI-treated brains do not show the CA-VB increase (CA-VB normalized to the mitochondrial protein ATP5a). The expression of CA-VA (normalized to ATP5a) and of CA-II (normalized to GAPDH) does not change. N=3 animals/group, n=6 technical replicates/group. * p<0.05 and ++ p<0.01, One-way ANOVA and Tukey’s post-hoc test. B, C) Biochemical analysis of mitochondrial carbonic anhydrases in human cortices. CA-VB expression is upregulated in CAA (+127%, p=0.06) (B) and in AD+CAA subjects (+511%, p=0.06) (C), compared to healthy controls (CNTR). No significant alterations in CA-VA and CA-II levels, between CAA and CNTR brains (B). CNTR and CAA groups N=5, and AD+CAA group N=10. Two-tailed unpaired t test. D) The combination of CAA and AD+CAA groups shows significant upregulation of CA-VB levels compared to CNTR. CNTR N=5; CAA group combined with AD+CAA group N=15. *p<0.05. Two-tailed unpaired t test. E) Western Blot analysis of CA-VB in cerebral ECs after 24hr challenge with Aβ40-Q22 (25μM) and Aβ42 (10μM). CA-VB was normalized to the mitochondrial protein ATP5a. Quantification is represented on the right. The expression of CA-VB is significantly increased following 24hrs Aβ40-Q22 treatment. Data represents the combination of at least three experiments each with 2 replicates, graphed as mean + SEM. One-way ANOVA and Dunnett’s post-test. **p< 0.01 F) qRT-PCR for mRNA expression levels of CA-VB and Cyp-B (housekeeping control gene) in cerebral ECs 48hrs post-transfection with siRNA for CA-VB (siCA-VB) or with a scrambled siRNA sequence (siScr) as control. **** p<0.0001 vs. siScr, Unpaired two-tailed t-test. G) CA-VB silencing prevents apoptosis, measured as the formation of fragmented nucleosomes (by Cell death ELISAplus), after challenge with Aβ42 (10μM), Aβ40 (25μM) or Aβ40-Q22 (25μM) for 24hrs (starting after the 48hr silencing). The graph displays one representative experiment of at least N=3 experiments, each performed in duplicate (n=2). * p<0.05 and ** p<0.01 vs. siScr control, One-way ANOVA, and Tukey’s post-hoc test. Data are expressed as mean ± SEM.
Discussion
This study demonstrated, for the first time in an AD/ADRD mouse model, that a chronic treatment with CAIs results in a reduction of Aβ pathology in all brain areas analyzed. The CAIs showed no toxicity. TgSwDI animals treated with ATZ and MTZ showed reduced vascular, microglial and astrocytic Aβ accumulation and lower caspase-3 activation in endothelial and glial cells, together with an amelioration of microvascular abnormalities and glial reactivity phenotypes, with associated cognitive improvement.
CVD is one of the major causal players in AD onset and progression. Many clinical studies, including the Alzheimer’s Disease Neuroimaging Initiative (ADNI), suggest the involvement of cerebrovascular disease very early in AD, both preceding and as a result of Aβ deposition7,91. Recent transcriptomic studies also revealed selective vulnerability of vascular cell subpopulations associated with clinically diagnosed AD92.
ATZ and MTZ cross the BBB, are safe for chronic systemic administration in humans, and are used clinically against glaucoma, high-altitude sickness, high-altitude cerebral edema and other non-AD-related indications, including seizures36,37,39,40. We have shown that CAIs reduce Aβ-induced loss of mitochondrial membrane potential, mitochondrial H2O2 overproduction, caspase-9 and −3 activation and Aβ-mediated apoptosis in cerebral endothelial, neuronal and glial cells in vitro, and acutely in the mouse brain39,43,44.
In this study we applied the drugs for the first time in a transgenic model of cerebral amyloidosis/CAA. The reduced Aβ deposition in hippocampus, cortex and hypothalamus appeared independent from APP processing. TgSwDI mice showed impairments in limb strength, motor learning or coordination, and, as expected, worse performance in the probe test of the Barnes maze (increased distance and errors), independent of observed changes in motor function, implicating deficits in learning and memory. However, transgenic mice treated with ATZ and MTZ were not significantly different from WT in grip strength (all limbs and forelimbs) and Barnes maze performance. These findings suggest that CAIs ameliorate peak limb force and spatial memory. Neither ATZ nor MTZ induced detectable changes on rotarod performance. CAIs may thus exert little to no impact on striatal and cerebellar function, but could act to improve cortical and/or hippocampal deficits associated with motor strength and mnemonic function.
Extracellular Aβ can be removed from the brain by multiple clearance systems4,26, such as the intramural periarterial drainage (IPAD) pathway4,25,93–95, or the astroglial-mediated glymphatic clearance96–98. The meningeal lymphatic system provides a complementary clearance route for cerebral waste products99,100. Glial cells such as astrocytes and microglia are essential for removing Aβ through phagocytosis and degradation, which, when efficient, prevent Aβ accumulation101–104 . Astrocytic end-feet wrap blood vessels, aiding endothelial junction stabilization and CBF regulation105. Astrocytic Aβ clearance is impaired in AD and CAA101,106,107, where reactive astrogliosis is an important pathogenic process108–110. Microglia, upon prolonged insults, prompt to pro-inflammatory phenotype111,112, hindering clearance and degradation of unwanted products. Hence, all clearance processes appear to be dependent on vascular fitness and functionality, glial cell health, or both4,26,113. In his study, we demonstrated that the treatment with CAIs improves both cerebrovascular and glial fitness.
Caspase activation, whether or not resulting in apoptosis, decreases the ability of vascular and glial cells to perform their metabolic functions114,115 and is linked to toxic signaling to neurons115–118. Interestingly, we showed caspase-3 activation in ECs, microglia and astrocytes of TgSwDI mice, which was robustly decreased by CAI treatment, likely improving vascular and glial physiological fitness.
Importantly, CAIs reduced the number of cerebral microhemorrhages, characterizing Dutch and Iowa CAA cases65–68, late onset AD and CAA brains119–121, and starting at 12 months in this animal model69. Additionally, vasoconstriction was reverted in CAI-treated mice. These results confirm that CAIs may regulate cerebral vessel tone74,122,123, ameliorating blood vessel dysfunction70. In an acute vasodilatory tests, 1000mg ATZ achieves a supramaximal dilative effect for 20’122. Of note, the dose we applied (20mg/kg), when translated to human doses (182mg/day for a 70kg person after allometric scaling), is many folds lower than the doses used clinically or in the acute ATZ challenge. Interestingly, we did not see significant effects in the maximum CBF or CBV response (or in their respective A.U.C.s) after functional activation in TgSwDI mice compared to WT animals. Tg mice fed with CAIs at our doses for 6 months did not differ significantly from untreated TgSwDI, indicating that chronic effects of low doses of CAIs on CBF and CBV appear different from acute effects of high doses of the drugs, used for the ATZ vasodilatory challenge.
Since CAIs prevent mitochondrial dysfunction, mitochondrial ROS production, caspase activation and cell death in multiple brain cell types, including endothelial and glial cells35,43,44,124,125, the reduction of endothelial and glial mitochondrial stress and caspase activation may be the primary mediator of the observed effects. It is clear that preventing microglial and astrocytic stress and over-reactivity can also promote vascular health, and vice versa113.
Indeed, CAIs decreased astrocytic and microglial caspase-3 activation, Aβ content and ameliorated glial reactivity markers in the Tg animals, suggesting that glial cells are healthier and can better degrade Aβ. Accordingly, insoluble Aβ40 species (or fibrillar Aβ40), the main component of CAA and the most abundant Aβ species in this mouse model (Fig 1D), were particularly reduced by CAIs treatment. Soluble Aβ40 was also partially reduced, albeit less dramatically. Oligomeric species measured in the soluble fraction did not change with treatment, confirming that CAIs mainly reduced deposition of fibrillar Aβ40 and likely higher molecular weight protofibrils within the soluble fraction.
The reduction of Aβ and caspase-3 activation observed in microglia of CAI-treated mice was associated with reduced IBA-1 and amoeboid shape, but with an augmented number of total microglia. Aβ-immunized AD subjects also have twice the number of microglia compared to AD and control groups, with an increased number of ramified microglia126, in line with the increased numbers and higher amounts of “bushy” microglia in our CAI-treated animals. Strikingly, treatment with CAIs, especially ATZ, increased TREM2 in Tg mice. TREM2 promotes microglial phagocytosis and Aβ clearance by fostering the survival of activated microglia86,127,128, and its variants are risk factors for late-onset AD129. TREM2 deficiency also reduces Aβ clearance130, and is associated with early stages of CAA110. The increase in TREM2 confirms that CAIs can facilitate microglial phagocytosis and, ultimately, Aβ clearance.
In addition, CAI-treated Tg mice showed elevated CD68. PVM and microglia express the phagocytic marker CD68131,132, which localizes at the late endosomes and lysosomes133, with roles in peptide transport/antigen processing134. PVM are associated with Aβ deposits in meningeal and cortical BVs and contribute to vascular health12,135,136. Notably, ATZ-treated mice show increased CD68+ cells over Aβ vascular deposits, indicating that CAIs may promote Aβ degradation by PVM.
It needs to be noted that ATZ, at high doses, can decrease CSF production137, impairing glymphatic clearance138. However, we believe this is not a detrimental factor for our therapeutic strategy, as supported by the observed reduction of Aβ deposits and improvement in cognition, without signs of toxicity, after long-term administration of the drug at the dose we propose. Confirming this, impaired glymphatic clearance is also present in normal pressure hydrocephalus (NPH)139, where treatment with low-dose ATZ (125–375mg/day) was shown to ameliorate periventricular white matter hyperintensities140, suggesting that the protective effects of low doses of ATZ may be uncoupled from its CSF/glymphatic effects.
Arterial pulsation, essential for multiple clearance mechanisms including IPAD25,141, may also be improved by CAIs40,74, by ameliorating vascular smooth muscle cell tone and function, which is currently the subject of further studies by our group.
Because mitochondrial mechanisms are responsible for the protective effects exerted by CAIs16,32–35,40,43,44,142–144, we asked if the mitochondrial CAs, CA-VA and CA-VB142,145, or CA-II, the most abundant CNS CA, which localizes at the mitochondria during aging and neurodegeneration88, mediate these effects. Interestingly, we found that the mitochondrial CA-VB is specifically overexpressed in TgSwDI brains, while CAI treatment reverts this effect. Importantly, we show that CA-VB expression is upregulated also in human subjects with CAA or AD+CAA, corroborating that cerebral Aβ deposition may induce CA-VB overexpression. In line with this hypothesis, we demonstrated that CA-VB is specifically increased in ECs after challenge with vasculotropic Aβ. Impressively, silencing of CA-VB, but not CA-VA or CA-II, prevents Aβ-induced endothelial apoptosis. This discovery points for the first time to CA-VB as a mediator of Aβ-induced cerebrovascular pathology. This finding also paves the way for future studies to clarify the specific impact of CA-VB and its modulation in CAA and AD, which are currently being developed in our laboratory.
Limitations of this study include the fact that this mouse model lacks tau pathology. Studies of the effects of CAIs in AD models containing both Aβ and P-tau (3xTg) are currently in progress in our laboratory. Imaging (MRI and two-photon microscopy) studies are also in progress to extend our understanding of the effects of these drugs on the cerebral microcirculation, oxygen diffusion, brain structures/volumes, blood flow and perivascular clearance pathways.
Conclusion
Overall, these results underscore the positive effects of CAIs on the cerebrovascular and neurovascular consequences of amyloidosis and provide a mechanistic understanding for the protective effects of these drugs, highlighting the high potential of repurposing CAIs in AD and CAA and paving the way to clinical trials.
Supplementary Material
Funding
This work was supported by NIH R01NS104127 and R01AG062572 grants, the Edward N. and Della L. Thome Memorial Foundation Awards Program in Alzheimer’s Disease Drug Discovery Research, the Alzheimer’s Association (AARG), the Pennsylvania Department of Health Collaborative Research on Alzheimer’s Disease (PA Cure) Grant, awarded to SF, and by the Karen Toffler Charitable Trust, and the Lemole Center for Integrated Lymphatics research. LD, BGL and AM are supported by P01AG060882 to TW. LD and TW are also supported by P30AG066512 to TW.
Footnotes
Consent Statement
All human samples were de-identified and obtained from brain banks, therefore, consent was not necessary for this study.
Conflicts of Interest. We declare no conflict of interest. SF in an inventor on US Patent 10780094 for the use of CAIs in Alzheimer’s disease and CAA. All other authors have nothing to disclose in relation to this study.
References
- 1.Zlokovic BV. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005;28(4):202–208. [DOI] [PubMed] [Google Scholar]
- 2.Ostergaard L, Aamand R, Gutierrez-Jimenez E, et al. The capillary dysfunction hypothesis of Alzheimer’s disease. Neurobiol Aging. 2013;34(4):1018–1031. [DOI] [PubMed] [Google Scholar]
- 3.Buee L, Hof PR, Delacourte A. Brain microvascular changes in Alzheimer’s disease and other dementias. Ann N Y Acad Sci. 1997;826:7–24. [DOI] [PubMed] [Google Scholar]
- 4.Carare RO, Aldea R, Agarwal N, et al. Clearance of interstitial fluid (ISF) and CSF (CLIC) group-part of Vascular Professional Interest Area (PIA): Cerebrovascular disease and the failure of elimination of Amyloid-beta from the brain and retina with age and Alzheimer’s disease-Opportunities for Therapy. Alzheimers Dement (Amst). 2020;12(1):e12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snyder HM, Corriveau RA, Craft S, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2015;11(6):710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alber J, Alladi S, Bae HJ, et al. White matter hyperintensities in vascular contributions to cognitive impairment and dementia (VCID): Knowledge gaps and opportunities. Alzheimers Dement (N Y). 2019;5:107–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sweeney MD, Montagne A, Sagare AP, et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2019;15(1):158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de la Torre JC. Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 2004;3(3):184–190. [DOI] [PubMed] [Google Scholar]
- 9.Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64(6):575–611. [DOI] [PubMed] [Google Scholar]
- 10.Kalaria RN. The role of cerebral ischemia in Alzheimer’s disease. Neurobiol Aging. 2000;21(2):321–330. [DOI] [PubMed] [Google Scholar]
- 11.Corriveau RA, Bosetti F, Emr M, et al. The Science of Vascular Contributions to Cognitive Impairment and Dementia (VCID): A Framework for Advancing Research Priorities in the Cerebrovascular Biology of Cognitive Decline. Cellular and molecular neurobiology. 2016;36(2):281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faraco G, Sugiyama Y, Lane D, et al. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J Clin Invest. 2016;126(12):4674–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. American journal of physiology Heart and circulatory physiology. 2002;283(1):H315–323. [DOI] [PubMed] [Google Scholar]
- 14.Ellis RJ, Olichney JM, Thal LJ, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, Part XV. Neurology. 1996;46(6):1592–1596. [DOI] [PubMed] [Google Scholar]
- 15.Ghiso J, Fossati S, Rostagno A. Amyloidosis associated with cerebral amyloid angiopathy: cell signaling pathways elicited in cerebral endothelial cells. J Alzheimers Dis. 2014;42(0):S167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fossati S, Cam J, Meyerson J, et al. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-beta variants in cells composing the cerebral vessel walls. FASEB J. 2010;24(1):229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghiso J, Frangione B. Cerebral amyloidosis, amyloid angiopathy, and their relationship to stroke and dementia. J Alzheimers Dis. 2001;3(1):65–73. [DOI] [PubMed] [Google Scholar]
- 18.Pantoni L Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9(7):689–701. [DOI] [PubMed] [Google Scholar]
- 19.Love S Contribution of cerebral amyloid angiopathy to Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004;75(1):1–4. [PMC free article] [PubMed] [Google Scholar]
- 20.Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke. 2011;42(9):2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grinberg LT, Thal DR. Vascular pathology in the aged human brain. Acta Neuropathol. 2010;119(3):277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boche D, Zotova E, Weller RO, et al. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain. 2008;131(Pt 12):3299–3310. [DOI] [PubMed] [Google Scholar]
- 23.Leurent C, Goodman JA, Zhang Y, et al. Immunotherapy with ponezumab for probable cerebral amyloid angiopathy. Ann Clin Transl Neurol. 2019;6(4):795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Leon MJ, Li Y, Okamura N, et al. Cerebrospinal Fluid Clearance in Alzheimer Disease Measured with Dynamic PET. J Nucl Med. 2017;58(9):1471–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aldea R, Weller RO, Wilcock DM, Carare RO, Richardson G. Cerebrovascular Smooth Muscle Cells as the Drivers of Intramural Periarterial Drainage of the Brain. Frontiers in aging neuroscience. 2019;11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarasoff-Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain-implications for Alzheimer disease. Nature reviews Neurology. 2015;11(8):457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roher AE, Kuo YM, Esh C, et al. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Mol Med. 2003;9(3–4):112–122. [PMC free article] [PubMed] [Google Scholar]
- 28.Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxidants & redox signaling. 2011;15(5):1167–1178. [DOI] [PubMed] [Google Scholar]
- 29.Erickson MA, Banks WA. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab. 2013;33(10):1500–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009;118(1):103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fossati S, Ghiso J, Rostagno A. Insights into Caspase-Mediated Apoptotic Pathways Induced by Amyloid-beta in Cerebral Microvascular Endothelial Cells. Neurodegenerative Diseases. 2012;10(1–4):324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fossati S, Ghiso J, Rostagno A. TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer’s Abeta. Cell Death Dis. 2012;3:e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fossati S, Todd K, Sotolongo K, Ghiso J, Rostagno A. Differential contribution of isoaspartate post-translational modifications to the fibrillization and toxic properties of amyloid-beta and the asparagine 23 Iowa mutation. Biochem J. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parodi-Rullan R, Sone JY, Fossati S. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J Alzheimers Dis. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thiry A, Dogne JM, Supuran CT, Masereel B. Carbonic anhydrase inhibitors as anticonvulsant agents. Curr Top Med Chem. 2007;7(9):855–864. [DOI] [PubMed] [Google Scholar]
- 37.Wright A, Brearey S, Imray C. High hopes at high altitudes: pharmacotherapy for acute mountain sickness and high-altitude cerebral and pulmonary oedema. Expert Opin Pharmacother. 2008;9(1):119–127. [DOI] [PubMed] [Google Scholar]
- 38.de Leval X, Ilies M, Casini A, et al. Carbonic anhydrase inhibitors: synthesis and topical intraocular pressure lowering effects of fluorine-containing inhibitors devoid of enhanced reactivity. Journal of medicinal chemistry. 2004;47(11):2796–2804. [DOI] [PubMed] [Google Scholar]
- 39.Provensi G, Carta F, Nocentini A, et al. A New Kid on the Block? Carbonic Anhydrases as Possible New Targets in Alzheimer’s Disease. Int J Mol Sci. 2019;20(19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemon N, Canepa E, Ilies MA, Fossati S. Carbonic Anhydrases as Potential Targets Against Neurovascular Unit Dysfunction in Alzheimer’s Disease and Stroke. Front Aging Neurosci. 2021;13:772278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aamand R, Skewes J, Moller A, Fago A, Roepstorff A. Enhancing effects of acetazolamide on neuronal activity correlate with enhanced visual processing ability in humans. Neuropharmacology. 2011;61(5–6):900–908. [DOI] [PubMed] [Google Scholar]
- 42.Okazawa H, Yamauchi H, Sugimoto K, Toyoda H, Kishibe Y, Takahashi M. Effects of acetazolamide on cerebral blood flow, blood volume, and oxygen metabolism: a positron emission tomography study with healthy volunteers. J Cereb Blood Flow Metab. 2001;21(12):1472–1479. [DOI] [PubMed] [Google Scholar]
- 43.Solesio ME, Peixoto PM, Debure L, et al. Carbonic anhydrase inhibition selectively prevents amyloid beta neurovascular mitochondrial toxicity. Aging Cell. 2018:e12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fossati S, Giannoni P, Solesio ME, et al. The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol Dis. 2016;86:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scholtzova H, Do E, Dhakal S, et al. Innate Immunity Stimulation via Toll-Like Receptor 9 Ameliorates Vascular Amyloid Pathology in Tg-SwDI Mice with Associated Cognitive Benefits. J Neurosci. 2017;37(4):936–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scholtzova H, Kascsak RJ, Bates KA, et al. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci. 2009;29(6):1846–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goni F, Herline K, Peyser D, et al. Immunomodulation targeting of both Abeta and tau pathological conformers ameliorates Alzheimer’s disease pathology in TgSwDI and 3xTg mouse models. J Neuroinflammation. 2013;10:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zinchuk V, Zinchuk O, Okada T. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: pushing pixels to explore biological phenomena. Acta Histochem Cytochem. 2007;40(4):101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu S, Park S, Allington G, et al. Targeting Apolipoprotein E/Amyloid beta Binding by Peptoid CPO_Abeta17–21 P Ameliorates Alzheimer’s Disease Related Pathology and Cognitive Decline. Scientific reports. 2017;7(1):8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lo P, Crouzet C, Vasilevko V, Choi B. Corrigendum to “Visualization of microbleeds with optical histology in mouse model of cerebral amyloid angiopathy” [105, May 2016, 109–113]. Microvasc Res. 2016;106:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, Ji Y, Mehta P, Bates KA, Sun Y, Wisniewski T. Blocking the apolipoprotein E/amyloid-beta interaction reduces fibrillar vascular amyloid deposition and cerebral microhemorrhages in TgSwDI mice. J Alzheimers Dis. 2011;24(2):269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park L, Koizumi K, El Jamal S, et al. Age-dependent neurovascular dysfunction and damage in a mouse model of cerebral amyloid angiopathy. Stroke. 2014;45(6):1815–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desjardins M, Kilic K, Thunemann M, et al. Awake Mouse Imaging: From Two-Photon Microscopy to Blood Oxygen Level-Dependent Functional Magnetic Resonance Imaging. Biol Psychiatry Cogn Neurosci Neuroimaging. 2019;4(6):533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Devor A, Sakadzic S, Srinivasan VJ, et al. Frontiers in optical imaging of cerebral blood flow and metabolism. J Cereb Blood Flow Metab. 2012;32(7):1259–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kura S, Xie H, Fu B, Ayata C, Boas DA, Sakadzic S. Intrinsic optical signal imaging of the blood volume changes is sufficient for mapping the resting state functional connectivity in the rodent cortex. J Neural Eng. 2018;15(3):035003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crews FT, Vetreno RP. Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology (Berl). 2016;233(9):1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beynon SB, Walker FR. Microglial activation in the injured and healthy brain: what are we really talking about? Practical and theoretical issues associated with the measurement of changes in microglial morphology. Neuroscience. 2012;225:162–171. [DOI] [PubMed] [Google Scholar]
- 58.Fernandez-Arjona MDM, Grondona JM, Fernandez-Llebrez P, Lopez-Avalos MD. Microglial Morphometric Parameters Correlate With the Expression Level of IL-1beta, and Allow Identifying Different Activated Morphotypes. Front Cell Neurosci. 2019;13:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gomez-Nicola D, Perry VH. Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist. 2015;21(2):169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Damisah EC, Hill RA, Rai A, et al. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci Adv. 2020;6(26):eaba3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bell-Temin H, Culver-Cochran AE, Chaput D, et al. Novel Molecular Insights into Classical and Alternative Activation States of Microglia as Revealed by Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)-based Proteomics. Mol Cell Proteomics. 2015;14(12):3173–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bandeira PT, Vernal J, Matos GM, et al. A Type IIa crustin from the pink shrimp Farfantepenaeus paulensis (crusFpau) is constitutively synthesized and stored by specific granule-containing hemocyte subpopulations. Fish Shellfish Immunol. 2020;97:294–299. [DOI] [PubMed] [Google Scholar]
- 63.Weksler B, Romero IA, Couraud PO. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS. 2013;10(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cabrera E, Mathews P, Mezhericher E, et al. Abeta truncated species: Implications for brain clearance mechanisms and amyloid plaque deposition. Biochim Biophys Acta Mol Basis Dis. 2018;1864(1):208–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Greenberg SM, Shin Y, Grabowski TJ, et al. Hemorrhagic stroke associated with the Iowa amyloid precursor protein mutation. Neurology. 2003;60(6):1020–1022. [DOI] [PubMed] [Google Scholar]
- 66.Herzig MC, Winkler DT, Burgermeister P, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004;7(9):954–960. [DOI] [PubMed] [Google Scholar]
- 67.Levy E, Carman MD, Fernandez-Madrid IJ, et al. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248(4959):1124–1126. [DOI] [PubMed] [Google Scholar]
- 68.Tomidokoro Y, Rostagno A, Neubert TA, et al. Iowa variant of familial Alzheimer’s disease: accumulation of posttranslationally modified AbetaD23N in parenchymal and cerebrovascular amyloid deposits. Am J Pathol. 2010;176(4):1841–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis J, Xu F, Deane R, et al. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279(19):20296–20306. [DOI] [PubMed] [Google Scholar]
- 70.Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol. 1992;84(2):117–127. [DOI] [PubMed] [Google Scholar]
- 71.Giannoni P, Arango-Lievano M, Neves ID, et al. Cerebrovascular pathology during the progression of experimental Alzheimer’s disease. Neurobiol Dis. 2016;88:107–117. [DOI] [PubMed] [Google Scholar]
- 72.Kelly P, Denver P, Satchell SC, Ackermann M, Konerding MA, Mitchell CA. Microvascular ultrastructural changes precede cognitive impairment in the murine APPswe/PS1dE9 model of Alzheimer’s disease. Angiogenesis. 2017;20(4):567–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kelly P, McClean PL, Ackermann M, Konerding MA, Holscher C, Mitchell CA. Restoration of cerebral and systemic microvascular architecture in APP/PS1 transgenic mice following treatment with Liraglutide. Microcirculation. 2015;22(2):133–145. [DOI] [PubMed] [Google Scholar]
- 74.Kiss B, Dallinger S, Findl O, Rainer G, Eichler HG, Schmetterer L. Acetazolamide-induced cerebral and ocular vasodilation in humans is independent of nitric oxide. Am J Physiol. 1999;276(6):R1661–1667. [DOI] [PubMed] [Google Scholar]
- 75.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. [DOI] [PubMed] [Google Scholar]
- 76.Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468(7321):232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gordon GR, Howarth C, MacVicar BA. Bidirectional control of arteriole diameter by astrocytes. Exp Physiol. 2011;96(4):393–399. [DOI] [PubMed] [Google Scholar]
- 78.Filiano AJ, Gadani SP, Kipnis J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 2015;1617:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Davis EJ, Foster TD, Thomas WE. Cellular forms and functions of brain microglia. Brain Res Bull. 1994;34(1):73–78. [DOI] [PubMed] [Google Scholar]
- 80.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. [DOI] [PubMed] [Google Scholar]
- 81.Zhang J, Xie X, Tang M, et al. Salvianolic acid B promotes microglial M2-polarization and rescues neurogenesis in stress-exposed mice. Brain Behav Immun. 2017;66:111–124. [DOI] [PubMed] [Google Scholar]
- 82.Zhang JQ, Wu XH, Feng Y, et al. Salvianolic acid B ameliorates depressive-like behaviors in chronic mild stress-treated mice: involvement of the neuroinflammatory pathway. Acta Pharmacol Sin. 2016;37(9):1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Taylor SE, Morganti-Kossmann C, Lifshitz J, Ziebell JM. Rod microglia: a morphological definition. PLoS One. 2014;9(5):e97096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Konishi H, Kiyama H. Microglial TREM2/DAP12 Signaling: A Double-Edged Sword in Neural Diseases. Front Cell Neurosci. 2018;12:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zheng H, Jia L, Liu CC, et al. TREM2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J Neurosci. 2017;37(7):1772–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim SM, Mun BR, Lee SJ, et al. TREM2 promotes Abeta phagocytosis by upregulating C/EBPalpha-dependent CD36 expression in microglia. Scientific reports. 2017;7(1):11118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zotova E, Bharambe V, Cheaveau M, et al. Inflammatory components in human Alzheimer’s disease and after active amyloid-beta42 immunization. Brain. 2013;136(Pt 9):2677–2696. [DOI] [PubMed] [Google Scholar]
- 88.Pollard A, Shephard F, Freed J, Liddell S, Chakrabarti L. Mitochondrial proteomic profiling reveals increased carbonic anhydrase II in aging and neurodegeneration. Aging (Albany NY). 2016;8(10):2425–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baertling F, Wagner M, Brunet T, et al. Fatal metabolic decompensation in carbonic anhydrase VA deficiency despite early treatment and control of hyperammonemia. Genet Med. 2020;22(3):654–655. [DOI] [PubMed] [Google Scholar]
- 90.Diez-Fernandez C, Rufenacht V, Santra S, et al. Defective hepatic bicarbonate production due to carbonic anhydrase VA deficiency leads to early-onset life-threatening metabolic crisis. Genet Med. 2016;18(10):991–1000. [DOI] [PubMed] [Google Scholar]
- 91.Veitch DP, Weiner MW, Aisen PS, et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 2019;15(1):106–152. [DOI] [PubMed] [Google Scholar]
- 92.Yang AC, Vest RT, Kern F, et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature. 2022;603(7903):885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim SH, Ahn JH, Yang H, Lee P, Koh GY, Jeong Y. Cerebral amyloid angiopathy aggravates perivascular clearance impairment in an Alzheimer’s disease mouse model. Acta Neuropathol Commun. 2020;8(1):181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Albargothy NJ, Johnston DA, MacGregor-Sharp M, et al. Convective influx/glymphatic system: tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018;136(1):139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Veluw SJ, Hou SS, Calvo-Rodriguez M, et al. Vasomotion as a Driving Force for Paravascular Clearance in the Awake Mouse Brain. Neuron. 2020;105(3):549–561 e545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17(11):1016–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nedergaard M, Goldman SA. Glymphatic failure as a final common pathway to dementia. Science. 2020;370(6512):50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Reeves BC, Karimy JK, Kundishora AJ, et al. Glymphatic System Impairment in Alzheimer’s Disease and Idiopathic Normal Pressure Hydrocephalus. Trends Mol Med. 2020;26(3):285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Da Mesquita S, Papadopoulos Z, Dykstra T, et al. Meningeal lymphatics affect microglia responses and anti-Abeta immunotherapy. Nature. 2021;593(7858):255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Da Mesquita S, Louveau A, Vaccari A, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. 2018;560(7717):185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wyss-Coray T, Loike JD, Brionne TC, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9(4):453–457. [DOI] [PubMed] [Google Scholar]
- 102.Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14(7):463–477. [DOI] [PubMed] [Google Scholar]
- 103.Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm (Vienna). 2010;117(8):949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xiao Q, Yan P, Ma X, et al. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 2014;34(29):9607–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10(11):1369–1376. [DOI] [PubMed] [Google Scholar]
- 106.Nielsen HM, Veerhuis R, Holmqvist B, Janciauskiene S. Binding and uptake of A beta1–42 by primary human astrocytes in vitro. Glia. 2009;57(9):978–988. [DOI] [PubMed] [Google Scholar]
- 107.Escartin C, Galea E, Lakatos A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24(3):312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schipper HM, Bennett DA, Liberman A, et al. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging. 2006;27(2):252–261. [DOI] [PubMed] [Google Scholar]
- 109.Carter SF, Scholl M, Almkvist O, et al. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med. 2012;53(1):37–46. [DOI] [PubMed] [Google Scholar]
- 110.Taylor X, Cisternas P, You Y, et al. A1 reactive astrocytes and a loss of TREM2 are associated with an early stage of pathology in a mouse model of cerebral amyloid angiopathy. J Neuroinflammation. 2020;17(1):223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Streit WJ, Conde JR, Fendrick SE, Flanary BE, Mariani CL. Role of microglia in the central nervous system’s immune response. Neurol Res. 2005;27(7):685–691. [DOI] [PubMed] [Google Scholar]
- 112.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–1543. [DOI] [PubMed] [Google Scholar]
- 113.Taylor X, Cisternas P, Jury N, et al. Activated endothelial cells induce a distinct type of astrocytic reactivity. Commun Biol. 2022;5(1):282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang XX, Zhang B, Xia R, Jia QY. Inflammation, apoptosis and autophagy as critical players in vascular dementia. Eur Rev Med Pharmacol Sci. 2020;24(18):9601–9614. [DOI] [PubMed] [Google Scholar]
- 115.Burguillos MA, Deierborg T, Kavanagh E, et al. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472(7343):319–324. [DOI] [PubMed] [Google Scholar]
- 116.de Calignon A, Fox LM, Pitstick R, et al. Caspase activation precedes and leads to tangles. Nature. 2010;464(7292):1201–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Venero JL, Burguillos MA, Brundin P, Joseph B. The executioners sing a new song: killer caspases activate microglia. Cell Death Differ. 2011;18(11):1679–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Canepa E, Fossati S. Impact of Tau on Neurovascular Pathology in Alzheimer’s Disease. Front Neurol. 2020;11:573324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vinters HV, Secor DL, Read SL, et al. Microvasculature in Brain Biopsy Specimens from Patients with Alzheimers-Disease - an Immunohistochemical and Ultrastructural-Study. Ultrastruct Pathol. 1994;18(3):333–348. [DOI] [PubMed] [Google Scholar]
- 120.Grabowski TJ, Cho HS, Vonsattel JP, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49(6):697–705. [DOI] [PubMed] [Google Scholar]
- 121.Vinters HV, Natte R, Maat-Schieman ML, et al. Secondary microvascular degeneration in amyloid angiopathy of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D). Acta Neuropathol. 1998;95(3):235–244. [DOI] [PubMed] [Google Scholar]
- 122.Settakis G, Molnar C, Kerenyi L, et al. Acetazolamide as a vasodilatory stimulus in cerebrovascular diseases and in conditions affecting the cerebral vasculature. Eur J Neurol. 2003;10(6):609–620. [DOI] [PubMed] [Google Scholar]
- 123.Stoppe G, Schutze R, Kogler A, et al. Cerebrovascular reactivity to acetazolamide in (senile) dementia of Alzheimer’s type: relationship to disease severity. Dementia. 1995;6(2):73–82. [DOI] [PubMed] [Google Scholar]
- 124.Lochhead JJ, McCaffrey G, Quigley CE, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30(9):1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Parodi-Rullan R, Ghiso J, Cabrera E, Rostagno A, Fossati S. Alzheimer’s amyloid beta heterogeneous species differentially affect brain endothelial cell viability, blood-brain barrier integrity, and angiogenesis. Aging Cell. 2020;19(11):e13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Franco-Bocanegra DK, Gourari Y, McAuley C, et al. Microglial morphology in Alzheimer’s disease and after Abeta immunotherapy. Sci Rep. 2021;11(1):15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kleinberger G, Yamanishi Y, Suarez-Calvet M, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6(243):243ra286. [DOI] [PubMed] [Google Scholar]
- 128.Wang Y, Cella M, Mallinson K, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160(6):1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xiang X, Werner G, Bohrmann B, et al. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med. 2016;8(9):992–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chinnery HR, Ruitenberg MJ, McMenamin PG. Novel characterization of monocyte-derived cell populations in the meninges and choroid plexus and their rates of replenishment in bone marrow chimeric mice. J Neuropathol Exp Neurol. 2010;69(9):896–909. [DOI] [PubMed] [Google Scholar]
- 132.Fabriek BO, Van Haastert ES, Galea I, et al. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia. 2005;51(4):297–305. [DOI] [PubMed] [Google Scholar]
- 133.Marsh SE, Abud EM, Lakatos A, et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A. 2016;113(9):E1316–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chistiakov DA, Killingsworth MC, Myasoedova VA, Orekhov AN, Bobryshev YV. CD68/macrosialin: not just a histochemical marker. Lab Invest. 2017;97(1):4–13. [DOI] [PubMed] [Google Scholar]
- 135.Thanopoulou K, Fragkouli A, Stylianopoulou F, Georgopoulos S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc Natl Acad Sci U S A. 2010;107(48):20816–20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2009;106(4):1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Faraci FM, Mayhan WG, Heistad DD. Vascular effects of acetazolamide on the choroid plexus. J Pharmacol Exp Ther. 1990;254(1):23–27. [PubMed] [Google Scholar]
- 138.Lundgaard I, Lu ML, Yang E, et al. Glymphatic clearance controls state-dependent changes in brain lactate concentration. J Cereb Blood Flow Metab. 2017;37(6):2112–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bae YJ, Choi BS, Kim JM, Choi JH, Cho SJ, Kim JH. Altered glymphatic system in idiopathic normal pressure hydrocephalus. Parkinsonism Relat Disord. 2021;82:56–60. [DOI] [PubMed] [Google Scholar]
- 140.Alperin N, Oliu CJ, Bagci AM, et al. Low-dose acetazolamide reverses periventricular white matter hyperintensities in iNPH. Neurology. 2014;82(15):1347–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li M, Kitamura A, Beverley J, et al. Impaired Glymphatic Function and Pulsation Alterations in a Mouse Model of Vascular Cognitive Impairment. Front Aging Neurosci. 2021;13:788519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nishimori I, Vullo D, Innocenti A, Scozzafava A, Mastrolorenzo A, Supuran CT. Carbonic anhydrase inhibitors. The mitochondrial isozyme VB as a new target for sulfonamide and sulfamate inhibitors. J Med Chem. 2005;48(24):7860–7866. [DOI] [PubMed] [Google Scholar]
- 143.Shah GN, Morofuji Y, Banks WA, Price TO. High glucose-induced mitochondrial respiration and reactive oxygen species in mouse cerebral pericytes is reversed by pharmacological inhibition of mitochondrial carbonic anhydrases: Implications for cerebral microvascular disease in diabetes. Biochem Biophys Res Commun. 2013;440(2):354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shah GN, Price TO, Banks WA, et al. Pharmacological inhibition of mitochondrial carbonic anhydrases protects mouse cerebral pericytes from high glucose-induced oxidative stress and apoptosis. J Pharmacol Exp Ther. 2013;344(3):637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Boriack-Sjodin PA, Heck RW, Laipis PJ, Silverman DN, Christianson DW. Structure determination of murine mitochondrial carbonic anhydrase V at 2.45-A resolution: implications for catalytic proton transfer and inhibitor design. Proc Natl Acad Sci U S A. 1995;92(24):10949–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.