

Abstract
Congenital idiopathic megaesophagus (CIM) is a gastrointestinal disorder of dogs wherein the esophagus is dilated and swallowing activity is reduced, causing regurgitation of ingesta. Affected individuals experience weight loss and malnourishment and are at risk for aspiration pneumonia, intussusception, and euthanasia. Great Danes have among the highest incidences of CIM across dog breeds, suggesting a genetic predisposition. We generated low-pass sequencing data for 83 Great Danes and used variant calls to impute missing whole genome single nucleotide variants (SNVs) for each individual based on haplotypes phased from 624 high-coverage dog genomes, including 21 Great Danes. We validated the utility of our imputed data set for genome-wide association studies (GWASs) by mapping loci known to underlie coat phenotypes with simple and complex inheritance patterns. We conducted a GWAS for CIM with 2,010,300 SNVs, identifying a novel locus on canine chromosome 1 (P-val = 2.76×10−10). Associated SNVs are intergenic or intronic and are found in two clusters across a 1.7 Mb region. Inspection of coding regions in high-coverage genomes from affected Great Danes did not reveal candidate causal variants, suggesting that regulatory variants underlie CIM. Further studies are necessary to assess the role of these non-coding variants.
Keywords: low-coverage sequence, Great Dane, imputation, gastrointestinal dysmotility, DOK6, CDH19
INTRODUCTION
Dysmotility is a central component of gastrointestinal disease and a comorbidity of disorders of the central nervous system, both of which are major concerns for human and canine health (Clark et al. 2023; Miron and Dumitrascu 2019; Niesler and Rappold 2021; Poirier et al. 2016; Sperber et al. 2021; Whitehead et al. 2016). Congenital idiopathic megaesophagus (CIM) is a hypomotility disorder of dogs characterized by reduced peristaltic activity and dilation of the esophagus that impede passage of food into the stomach (Richter 2014; Tams 2003). The congenital form of megaesophagus is usually idiopathic and diagnosed shortly after weaning onto solid food. The predominant clinical sign is regurgitation, which may occur only occasionally in mild cases, or several times a day in severe cases (Johnson et al. 2009, Palmer 1968; Tams 2003). Affected dogs suffer from weight loss and malnourishment and are at lifelong risk for aspiration pneumonia and intussusception (Graham et al. 1998; Grimes et al. 2020; Kogan et al. 2008). CIM is diagnosed via radiography, with or without barium contrast (Fig. 1) (Johnson et al. 2009; McBrearty et al. 2011). Although mortality is high in neonates, CIM cases can usually be managed with frequent, high-caloric, liquid meals fed in an elevated position (Guilford 1990).
Fig. 1.

Barium-contrast radiographs of affected and healthy Great Dane littermates. Radiographs were taken at five weeks of age following a barium meal. Barium is retained in the enlarged esophagus of the CIM-affected individual (left) and passes through to the stomach in the healthy puppy (right)
While CIM has been reported across breeds, German shepherd dogs (GSDs) and Great Danes have among the highest incidences (Cox et al. 1980; Guilford 1990; Haines et al. 2019, Harvey et al. 1974). In GSDs, we previously uncovered a sex bias wherein males are approximately twice as likely to be affected as females (Bell et al. 2022). Using a genome-wide association study (GWAS) and high-coverage whole genome sequence, we detected a major locus associated with CIM in GSDs and an allele of a variable number tandem repeat (VNTR) that confers risk (Bell et al. 2022).
The study of complex diseases such as CIM may be aided by the emerging method of low-pass whole genome sequencing, which is a cost-effective alternative to SNP arrays for the generation of genome-wide genetic profiles (Martin et al. 2021). Variant calls from low coverage sequence (usually an average of 1X) are used to impute missing whole genome data for each individual based on haplotypes phased from high-coverage genomes (Buckley et al. 2022; Li et al. 2011; Wasik et al. 2021). Unlike commercial arrays where markers were selected for informativeness in limited populations, low-pass whole genome sequencing is an unbiased alternative in which novel variants can be detected in specific populations (Martin et al. 2021). GWASs conducted with complete genome data have allowed for direct identification of the genetic basis for disease (Hayward et al. 2019; Mansour et al. 2018). Further, high-density genotype data may reduce post-GWAS efforts to refine candidate intervals and identify the best regional associations in genetically complex disorders, which are often attributed to non-coding regulatory variants (Hayward et al. 2019; Li et al. 2011; Plassais et al. 2019).
Here, we generated low-pass whole genome sequences for CIM-affected and unaffected dogs, imputed genotypes using a multi-breed reference panel, and conducted a GWAS to identify genomic regions associated with esophageal hypomotility in the Great Dane.
METHODS
Population
Whole blood, buccal cells, or skin tissues were obtained from purebred Great Danes with informed owner consent, and DNA was prepared using the Gentra Puregene DNA Isolation kit (Qiagen). Pedigrees, radiographs, and additional phenotypic data (e.g., coat color/pattern) were collected when available. Cases were diagnosed by a veterinarian via a standard or barium contrast radiograph, history of clinical signs from puppyhood, and exclusion of secondary causes (e.g., myasthenia gravis, persistent right aortic arch), with the exception of four cases diagnosed based on clinical signs alone. Controls were at least one year old, had no history of clinical signs consistent with CIM, and had no relatives known to be affected by CIM. A subset of 81 dogs with sufficient DNA were genotyped for the VNTR associated with CIM in GSDs, as previously described (Bell et al. 2022). A two-tailed Fisher’s exact probability test was performed to evaluate the association of CIM with the VNTR. A one-way chi square test was used to assess sex bias (http://vassarstats.net/).
Whole genome sequencing
Dogs were selected for low-pass whole genome sequencing such that known relatives within three generations were excluded, and sex and coat color were roughly balanced between cases and controls. Libraries were constructed for 83 dogs with the Illumina DNA Prep kit and low-pass whole genome sequencing was performed on an Illumina NovaSeq 6000, generating dual-index 150 bp paired-end reads. Total reads generated ranged from 18,453,514 to 157,596,075 per sample. Paired-end reads were aligned to CanFam3.1 using Burrows-Wheeler Aligner (Li and Durbin 2009) and variants were jointly called using BCFtools mpileup function (Li 2011). Variants with a phred-scaled quality score < 20 were removed, and biallelic SNVs were selected using the BCFtools view command. These variants comprised the imputation target panel.
Nine ancestrally diverse Great Danes were selected for high-coverage whole genome sequencing. We generated paired-end 100, 125, or 150 bp sequencing on Illumina HiSeq systems. For each sample, paired-end reads were trimmed and aligned to CanFam3.1.
Imputation reference panel
Publicly available and private high-coverage whole genome sequence data for 624 dogs representing 65 breeds were used to create a custom variant call format (VCF) file to serve as an imputation reference panel. The reference panel includes 21 genetically diverse Great Danes, including 16 private genomes and five public genomes sequenced herein (four controls and one CIM-affected dog). This VCF was created using a previously described bioinformatics pipeline (Friedenberg and Meurs 2016), with the following updates: GATK was updated to version 4.1 and dbSNP was updated to version 151. Variants were subset using BCFtools (Li et al. 2009; Li 2011) to include only bi-allelic SNVs (n=21,809,094) called in at least 98% of dogs and phased using Beagle 5.1 (Browning and Browning 2007) on a per chromosome basis with standard options.
Imputation
Prior to imputation, the target panel from low-pass sequencing was compared to the reference panel using conform-gt (Browning and Browning 2007) to exclude target variants without a corresponding reference panel variant and to adjust target variants to match the allele order and chromosome strand in the reference panel. The target panel was imputed to the reference panel on a chromosome-by-chromosome basis with Beagle 4.0 (Browning and Browning 2007) using the genotype likelihood method with the following flags: window=50000, overlap=20000, phase-its=12, burnin-its=12.
Imputation accuracy was assessed using four Great Danes who were not included in the imputation reference panel and from whom both high-coverage and low-pass whole genome sequences were generated. Genotype concordance was calculated by comparing imputed SNV genotypes having DR2 > 0.8 (n=8,227,616) with high-coverage genotypes from the same individual.
Genome-wide association studies
Linkage disequilibrium (LD) pruning was performed using SNP & Variation Suite v8 (SVS, Golden Helix) with the following parameters: window size = 50, window increment = 5, CHM r2 LD threshold = 0.99. All GWASs were performed in SVS using the filtered set of 2,010,300 SNVs. Validation GWASs for dilute and merle pigmentation phenotypes were conducted using a single locus mixed linear model (EMMAX), under recessive and dominant models, respectively. GWAS for CIM was performed using a single locus mixed linear model (EMMAX) under an additive model with sex as a covariate and correction for hemizygous males on the X chromosome. P-values were calculated using an additive genetic model.
Variant analyses
A regional genotype association test (additive model) was performed for all (unpruned) chromosome 1 SNVs. The top 20 lead variants were evaluated using Ensembl Genes 100, RefSeq Genes 105 (NCBI), and the UCSC Genome Browser to determine if variants were expressed and/or coding. Genotypes from 1,351 publicly available dog genomes (Bell et al. 2022) were used to determine the major allele for each variant and to identify alleles unique to the Great Dane.
Data Availability
Sample Read Archive accession numbers for 9 high-coverage and 83 low-pass Great Dane genomes are provided in Supplementary Table 1. The VCF, index file, and metadata are available through the Data Repository for University of Minnesota (https://doi.org/10.13020/GKXV-GT86).
RESULTS
Population
Among the total number of affected dogs collected (n=69), we observed a slight overrepresentation of males (27 females, 42 males, P-val = 0.09). Genotyping of the tri-allelic VNTR, previously associated with CIM in GSDs, in the reference panel and 81 study dogs (n=102) revealed that allele 2 is the major allele (68%) among Great Danes (Bell et al. 2022). Great Danes also possess GSD risk allele 1, but allele 3 was not observed herein. The VNTR was not associated with CIM (44 cases vs. 37 controls, P-val = 0.51; Supplementary Table 2).
Whole genome sequence & imputation
High-coverage whole genome sequencing data (13X to 25X coverage) were generated for four CIM-affected and five CIM-unaffected Great Danes. Low-pass whole genome sequences (1.1X to 9.5X coverage, average coverage = 2.9X) were generated for 83 dogs (45 females, 38 males). These numbers include four dogs for whom we generated both high- and low-coverage sequence data. A single VCF was created from the low-pass data containing variant calls for all 83 dogs, and the data were imputed using a reference panel of 624 high-coverage whole genome sequences, which includes 21 Great Danes from diverse backgrounds. A total of 21,809,094 SNVs were imputed, and among those, only SNVs having DR2 > 0.8 (n=8,227,616) were retained for downstream analysis. Imputation accuracy calculated for four dogs ranged from 96.4 to 98%.
Genome-wide association studies
After LD pruning, 2,010,300 SNVs were used for GWAS. A principal component analysis (PCA) showed minimal underlying population substructure between CIM cases and controls (Supplementary Fig. 1). Genomic differences between coat phenotypes were apparent and consistent with common breeding practices that separate fawn and brindle lines from harlequin, merle, and mantle lines (Fig. 2A).
Fig. 2.

Principal component analysis and GWAS for coat phenotypes. A Principal component 1 (PC1) is plotted against seven common coat color and pattern combinations in Great Danes (n=69). A fawn individual homozygous for the dilute causal allele is marked by a blue arrow. B Manhattan plots illustrate results of GWASs conducted with 2,010,300 SNVs for dilute coat color (13 cases vs. 56 controls; top) and merle coat pattern (14 cases vs. 55 controls; bottom). Bonferroni significance is denoted by the black horizontal lines
To validate the utility of this data set for GWAS, we mapped two phenotypes for which the genetic bases are known: dilute coat color and merle coat pattern. Both test GWASs were performed using 69 dogs with owner-reported coat phenotypes (Fig. 2B). The lead SNV (chr25:48138845, P-val = 4.08×10−31) in the GWAS for recessive dilute coat color is located within the second intron of Melanophilin (MLPH, D locus). Dilute fur across breeds, including the Great Dane, is proposed to be caused by an exon 1 splice variant in MLPH, which yielded the second best association (chr25:48121642, P-val = 9.71×10−22; Drögemüller et al. 2007). Examination of the genotypes for the causal variant revealed that a fawn individual classified as a dilute control was homozygous for the causal allele. This individual was heterozygous at the lead GWAS variant. In a second GWAS that excluded this individual, the causal splice variant is the leading SNV (P-val =1.92×10−30).
The GWAS for merle coat patterning, a semidominant and incompletely penetrant trait, revealed a primary signal on chromosome 10 (chr10:643797, P-val = 4.08×10−20) and a secondary signal on chromosome 31 (chr31:15722, P-val = 6.96×10−16). The lead variant for merle is located downstream of Premelanosome Protein (PMEL, M locus), 351 kb telomeric to the causal short-interspersed element insertion (Clark et al. 2006; Murphy et al. 2018). The chromosome 31 signal represents a CanFam3.1 genome assembly error; these SNVs are in LD with the M locus on chromosome 10. The assembly has been corrected in CanFam4 (Wang et al. 2021), placing these SNVs 340 kb centromeric to PMEL. The results of these test GWASs validate the utility of our imputed data set for mapping simple Mendelian and complex traits.
We conducted a GWAS for CIM with sex as a covariate, using 45 cases (23 females, 22 males) and 38 controls (22 females, 16 males; Fig. 3A). A 2.3 Mb region of association on chromosome 1 includes 14 SNVs surpassing Bonferroni significance (Supplementary Table 3). Ten SNVs lie within an 800 kb region starting at 9 Mb, and four SNVs lie in a 93 kb window at 11 Mb.
Fig. 3.

Manhattan plots illustrating association results for CIM in 45 case and 38 control Great Danes. A GWAS conducted with sex as a covariate using 2,010,300 SNVs. Bonferroni significance is denoted by the black horizontal line. A Q-Q plot shows observed vs. expected −log10P-vals with the genomic inflation factor (λ) (inset). B Regional Fisher’s exact association tests for 394,308 chromosome 1 SNVs with genes 1Mb centromeric and telomeric to the two lead SNVs shown below (UCSC, CanFam3.1)
Variant analyses
To identify the best associated variants within the unpruned dataset, Fisher’s exact association tests were performed for all 394,308 chromosome 1 SNVs (Fig. 3B). The lead SNV was identical to the GWAS. The top 20 variants were annotated and none were within protein coding or splicing regions (Supplementary Table 4) or unique to the Great Dane breed. The lead variant (chr1:9814231) is intronic to a predicted lncRNA (ENSCAFT00000060437.1), and all other variants are intergenic. Using IGV, we manually scanned four high-coverage genomes (three affected, one unaffected) for exonic variants in the seven positional genes (Fig. 3B) or structural variants. We identified a single nonsynonymous variant in CCDC102B that fits the expected pattern of zygosity in the four genomes but is not associated with CIM (P-val = 0.25). No evidence of structural variation was observed.
The top two SNVs (chr1:9814231, P-val = 2.76×10−10; chr1:11437660, P-val = 6.41×10−10) are not in high LD (r2 = 0.59; Fig. 4A). In the intervening region, from approximately 10 to 11 Mb, we observed high levels of homozygosity across both cases and controls (Fig. 4B). Comparisons between canine reference genomes reveals no major rearrangements from 8–13 Mb, but highlights a genome misassembly at ~10.5 Mb in both CanFam3 and CanFam4. Approximately 40 kb of extra sequence has been removed in CanFam5 and CanFam6, corresponding to the region of lowest heterozygosity in Fig. 4B. The leading SNVs occur in regions where the controls have higher frequencies of the dog minor alleles, based on 1,351 dogs representing at least 233 breeds (Fig. 4C). We observed increased homozygosity (29–42%) for the dog minor allele among controls and lower levels in cases (2–4%).
Fig. 4.
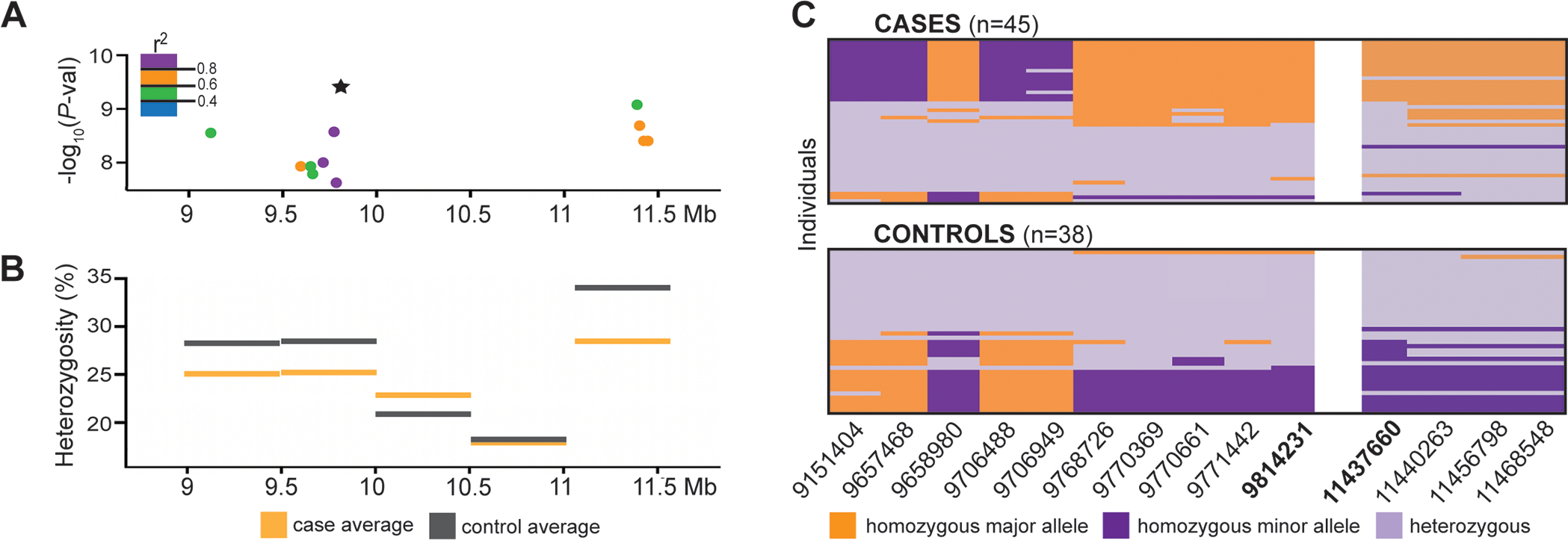
Analyses of the chromosome 1 CIM locus. A Pairwise LD (r2) for the 14 significant SNVs relative to the lead SNV (star). B Average case and control percent heterozygosity in 500 kb windows. C Genotypes for the 14 significant SNVs are color-coded relative to the major allele across dogs (based on 1,351 dogs of 233 pure and mixed breeds). Individuals are in rows, SNVs are in columns with chromosome 1 position at the bottom. White bar denotes the large interval with no associated or informative SNVs
DISCUSSION
We conducted one of the first GWASs in dogs with imputed low-pass whole genome sequence and identified a novel locus associated with a genetically complex gastrointestinal dysmotility in Great Danes (Morrill et al. 2022, Piras et al. 2020). A single region of association with CIM was detected on chromosome 1, tagged by two leading SNVs positioned 1.6 Mb apart and in modest LD. No SNVs in the intervening region reach significance, likely a consequence of the low levels of heterozygosity among both cases and controls. Genomic comparisons with the latest canine reference genomes reveal an assembly error in this region and highlight the need for future studies to understand its structure. All lead SNVs are intergenic or intronic and none were unique to the Great Dane breed, suggesting that the causal variants are regulatory polymorphisms. CIM cases possess the dog major alleles at these loci. The minor dog alleles are predominant among controls, suggesting that the identified locus may confer protection from CIM. Protective haplotypes have been previously identified in complex canine diseases (Bianchi et al. 2020; Karamatic et al. 2022; Tsai et al. 2013). Alternatively, the major alleles could uniquely confer risk in the Great Dane because of their genetic background (Abrams et al. 2020; Evans et al. 2017; Ivansson et al. 2016). Notably, none of the variants surpassing Bonferroni significance are represented on the Illumina CanineHD BeadChip array (containing 220,853 SNPs), thus our approach allowed us to detect an association that may have been missed using SNP array technology.
The lead SNV is intronic to a lncRNA. While lncRNAs have been implicated in diseases and other traits, the importance of intronic variants is largely unknown (Aznaourova et al. 2020; Plassais et al. 2020). The broad chromosome 1 signal spans seven protein coding genes. Docking Protein 6 (DOK6) and Cadherin-19 (CDH19) are intriguing candidates because they have both been associated with the gastrointestinal motility disorder, Hirschsprung disease (Gao et al. 2017; Huang et al. 2022; Lan et al. 2021). Also known as congenital aganglionic megacolon, Hirschsprung disease is a developmental disorder of the enteric nervous system characterized by lack of peristalsis in the colon (Diposarosa et al. 2021). DOK6 is an intracellular adapter molecule and a key promoter of neurite outgrowth (Li et al. 2010). CDH19 lies in the region of low variability between the two leading SNVs. A type II cadherin, CDH19 is a marker of Schwann cell lineage (originating from neural crest-derived cells) and may play a role in myelin formation (Kameneva et al. 2021; Stratton et al. 2017; Takahashi et al. 2005). Neural crest cell migration is important in the development of the enteric nervous system, which regulates many gastrointestinal activities, including motility (Huang et al. 2022). Our low-pass GWAS resulted in a prioritized list of non-coding variants for future studies to determine their impact on expression of DOK6, CDH19, and/or other positional genes.
Traditionally, GWASs in dogs are performed using commercial genotyping arrays (Ostrander et al. 2019a). At about 50% of the cost of array technologies in dogs, low-pass sequencing is emerging as a feasible alternative that yields considerably more data. Whereas only a fraction of SNPs on a genotyping array are informative in a given breed due to ascertainment bias, low-pass sequencing captures intrabreed variation within the study population. The advantage of this approach for complex disease where regulatory variants are involved is a comprehensive list of candidate causal variants as a GWAS outcome. Here, after pruning for markers in perfect LD, we were able to perform a dense GWAS with over 2 million variants informative in the Great Dane breed. For comparison, published GWASs in the Great Dane included about 100,000 SNPs (Metzger et al. 2015; Sarviaho et al. 2020).
Design of the reference panel for imputation presents study limitations. We excluded variants that are difficult to genotype accurately from next generation sequencing data (e.g., insertions, deletions, structural changes, multi-allelic variants), thus causal variants may not have been included in the GWAS. Our imputed data were sufficient to map directly to the causal SNV for dilute coat color (Drögemüller et al. 2007), a simple Mendelian trait. However, we could not directly identify the large insertion causing merle patterning. Inclusion of small insertions and deletions in the reference panel would maximize the utility of this approach, although the latter example would still necessitate post-GWAS analysis of high-coverage genomes.
While imputation accuracy across breeds improves with breed representation (Hayward et al. 2019), accuracy within a given breed requires inclusion of diverse individuals to capture breed-specific haplotypes. This necessity may hinder studies in breeds from whom high-coverage genomes are unavailable or limited. Five Great Danes in a multi-breed reference panel was previously insufficient for highly accurate imputation (86.8% average genotype concordance; Friedenberg and Meurs 2016). Here, genomes from 21 Great Danes were included in a larger multi-breed reference panel, resulting in greater accuracy (97% average genotype concordance). The number of individuals necessary to capture the haplotypes present within a breed will vary based on population structure, size, etc. Still, as more high-coverage genomes become publicly available, through sequencing efforts such Dog10k (Ostrander et al. 2019b), low-pass sequencing and imputation becomes increasingly feasible across dog breeds.
Supplementary Material
Supplementary Figure 1 Principal component analysis for the CIM GWAS cohort. Principal components (PC) 1 and 2 are plotted for 83 Great Danes
ACKNOWLEDGEMENTS
The authors wish to thank the dog owners who contributed to this study and the Upright Canine Brigade for help recruiting participants.
FUNDING
This project was supported by the American Kennel Club Canine Health Foundation (#02709 to L.A.C.). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the views of the Foundation. This work was also supported by the Great Dane Club of America Charitable Trust (L.A.C.).
Footnotes
STATEMENTS AND DECLARATIONS
The authors declare that they have no competing interests.
References
- Abrams SR, Hawks AL, Evans JM, Famula TR, Mahaffey M, Johnson GS, Mason JM, Clark LA, 2020. Variants in FtsJ RNA 2’-O-Methyltransferase 3 and Growth Hormone 1 are associated with small body size and a dental anomaly in dogs. Proc Natl Acad Sci U S A 117, 24929–24935. 10.1073/pnas.2009500117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aznaourova M, Schmerer N, Schmeck B, Schulte LN, 2020. Disease-Causing Mutations and Rearrangements in Long Non-coding RNA Gene Loci. Front Genet 11, 527484. 10.3389/fgene.2020.527484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SM, Evans JM, Evans KM, Tsai KL, Noorai RE, Famula TR, Holle DM, Clark LA, 2022. Congenital idiopathic megaesophagus in the German shepherd dog is a sex-differentiated trait and is associated with an intronic variable number tandem repeat in Melanin-Concentrating Hormone Receptor 2. PLoS Genet 18, e1010044. 10.1371/journal.pgen.1010044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi M, Rafati N, Karlsson Å, Murén E, Rubin C-J, Sundberg K, Andersson G, Kämpe O, Hedhammar Å, Lindblad-Toh K, Rosengren Pielberg G, 2020. Whole-genome genotyping and resequencing reveal the association of a deletion in the complex interferon alpha gene cluster with hypothyroidism in dogs. BMC Genomics 21, 307. 10.1186/s12864-020-6700-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL, 2007. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies By Use of Localized Haplotype Clustering. The American Journal of Human Genetics 81, 1084–1097. 10.1086/521987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley RM, Harris AC, Wang G-D, Whitaker DT, Zhang Y-P, Ostrander EA, 2022. Best practices for analyzing imputed genotypes from low-pass sequencing in dogs. Mamm Genome 33, 213–229. 10.1007/s00335-021-09914-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Amador A, Wang A, 2023. Convergence of human and veterinary medicine: leveraging canine naturally occurring neurological disorders to develop regenerative treatments. Neural Regen Res 18, 541. 10.4103/1673-5374.350195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark LA, Wahl JM, Rees CA, Murphy KE, 2006. Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc Natl Acad Sci U S A 103, 1376–1381. 10.1073/pnas.0506940103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox VS, Wallace LJ, Anderson VE, Rushmer RA, 1980. Hereditary esophageal dysfunction in the Miniature Schnauzer dog. Am J Vet Res 41, 326–330. [PubMed] [Google Scholar]
- Diposarosa R, Bustam NA, Sahiratmadja E, Susanto PS, Sribudiani Y, 2021. Literature review: enteric nervous system development, genetic and epigenetic regulation in the etiology of Hirschsprung’s disease. Heliyon 7, e07308. 10.1016/j.heliyon.2021.e07308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drögemüller C, Philipp U, Haase B, Günzel-Apel A-R, Leeb T, 2007. A noncoding melanophilin gene (MLPH) SNP at the splice donor of exon 1 represents a candidate causal mutation for coat color dilution in dogs. J Hered 98, 468–473. 10.1093/jhered/esm021 [DOI] [PubMed] [Google Scholar]
- Evans JM, Noorai RE, Tsai KL, Starr-Moss AN, Hill CM, Anderson KJ, Famula TR, Clark LA, 2017. Beyond the MHC: A canine model of dermatomyositis shows a complex pattern of genetic risk involving novel loci. PLoS Genet 13, e1006604. 10.1371/journal.pgen.1006604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedenberg SG, Meurs KM, 2016. Genotype imputation in the domestic dog. Mamm Genome 27, 485–494. 10.1007/s00335-016-9636-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z-G, Chen Q-J, Shao M, Qian Y-Z, Zhang L-F, Zhang Y-B, Xiong Q-X, 2017. Preliminary identification of key miRNAs, signaling pathways, and genes associated with Hirschsprung’s disease by analysis of tissue microRNA expression profiles. World J Pediatr 13, 489–495. 10.1007/s12519-017-0064-z [DOI] [PubMed] [Google Scholar]
- Graham KL, Buss MS, Dhein CR, Barbee DD, Seitz SE, 1998. Gastroesophageal intussusception in a Labrador retriever. Can Vet J 39, 709–711. [PMC free article] [PubMed] [Google Scholar]
- Grimes JA, Fleming JT, Singh A, Campbell BG, Hedlund CS, Tobias KM, Arai S, Ham KM, Repellin R, Schroeder R, Sumner JP, Abrams B, Boudreau B, Lewis B, Wallace ML, 2020. Characteristics and long-term outcomes of dogs with gastroesophageal intussusception. J Am Vet Med Assoc 256, 914–920. 10.2460/javma.256.8.914 [DOI] [PubMed] [Google Scholar]
- Guilford WG, 1990. Megaesophagus in the dog and cat. Semin Vet Med Surg Small Anim 5, 37–45. [PubMed] [Google Scholar]
- Haines JM, Khoo A, Brinkman E, Thomason JM, Mackin AJ, 2019. Technique for Evaluation of Gravity-Assisted Esophageal Transit Characteristics in Dogs with Megaesophagus. J Am Anim Hosp Assoc 55, 167–177. 10.5326/JAAHA-MS-6711 [DOI] [PubMed] [Google Scholar]
- Harvey CE, O’Brien JA, Durie VR, Miller DJ, Veenema R, 1974. Megaesophagus in the dog: a clinical survey of 79 cases. J Am Vet Med Assoc 165, 443–446. [PubMed] [Google Scholar]
- Hayward JJ, White ME, Boyle M, Shannon LM, Casal ML, Castelhano MG, Center SA, Meyers-Wallen VN, Simpson KW, Sutter NB, Todhunter RJ, Boyko AR, 2019. Imputation of canine genotype array data using 365 whole-genome sequences improves power of genome-wide association studies. PLoS Genet 15, e1008003. 10.1371/journal.pgen.1008003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Hou Y, Wang X, Wang L, Yi C, Wang C, Sun X, Tam PKH, Ngai SM, Sham MH, Burns AJ, Chan WY, 2022. Direct Interaction of Sox10 With Cadherin-19 Mediates Early Sacral Neural Crest Cell Migration: Implications for Enteric Nervous System Development Defects. Gastroenterology 162, 179–192.e11. 10.1053/j.gastro.2021.08.029 [DOI] [PubMed] [Google Scholar]
- Ivansson EL, Megquier K, Kozyrev SV, Murén E, Körberg IB, Swofford R, Koltookian M, Tonomura N, Zeng R, Kolicheski AL, Hansen L, Katz ML, Johnson GC, Johnson GS, Coates JR, Lindblad-Toh K, 2016. Variants within the SP110 nuclear body protein modify risk of canine degenerative myelopathy. Proc Natl Acad Sci U S A 113, E3091–3100. 10.1073/pnas.1600084113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BM, 2009. Canine Megaesophagus. Kirk’s Current Veterinary Therapy 486–492. [Google Scholar]
- Kameneva P, Kastriti ME, Adameyko I, 2021. Neuronal lineages derived from the nerve-associated Schwann cell precursors. Cell Mol Life Sci 78, 513–529. 10.1007/s00018-020-03609-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamatic S, Goode R, Bageswaran N, Willet CE, Samaha G, Ferguson R, Mazrier H, Wade CM, 2022. Genome-Wide Association Analysis for Chronic Superficial Keratitis in the Australian Racing Greyhound. Genes (Basel) 13, 1328. 10.3390/genes13081328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogan DA, Johnson LR, Sturges BK, Jandrey KE, Pollard RE, 2008. Etiology and clinical outcome in dogs with aspiration pneumonia: 88 cases (2004–2006). J Am Vet Med Assoc 233, 1748–1755. 10.2460/javma.233.11.1748 [DOI] [PubMed] [Google Scholar]
- Lan C, Wu Y, Wang N, Luo Y, Zhao J, Zheng Y, Zhang Y, Huang L, Zhu Y, Lu L, Zhong W, Zeng J, Xia H, 2021. Association between ABHD1 and DOK6 polymorphisms and susceptibility to Hirschsprung disease in Southern Chinese children. J Cell Mol Med 25, 9609–9616. 10.1111/jcmm.16905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. 10.1093/bioinformatics/btr509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R, 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup, 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. qi, Shi L, You Y. gang, Gong Y. hua, Yin B, Yuan J. gang, Peng X. zhong, 2010. Downstream of tyrosine kinase/docking protein 6, as a novel substrate of tropomyosin-related kinase C receptor, is involved in neurotrophin 3-mediated neurite outgrowth in mouse cortex neurons. BMC Biol 8, 86. 10.1186/1741-7007-8-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sidore C, Kang HM, Boehnke M, Abecasis GR, 2011. Low-coverage sequencing: Implications for design of complex trait association studies. Genome Res. 21, 940–951. 10.1101/gr.117259.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour TA, Lucot K, Konopelski SE, Dickinson PJ, Sturges BK, Vernau KL, Choi S, Stern JA, Thomasy SM, Döring S, Verstraete FJM, Johnson EG, York D, Rebhun RB, Ho H-YH, Brown CT, Bannasch DL, 2018. Whole genome variant association across 100 dogs identifies a frame shift mutation in DISHEVELLED 2 which contributes to Robinow-like syndrome in Bulldogs and related screw tail dog breeds. PLoS Genet 14, e1007850. 10.1371/journal.pgen.1007850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AR, Atkinson EG, Chapman SB, Stevenson A, Stroud RE, Abebe T, Akena D, Alemayehu M, Ashaba FK, Atwoli L, Bowers T, Chibnik LB, Daly MJ, DeSmet T, Dodge S, Fekadu A, Ferriera S, Gelaye B, Gichuru S, Injera WE, James R, Kariuki SM, Kigen G, Koenen KC, Kwobah E, Kyebuzibwa J, Majara L, Musinguzi H, Mwema RM, Neale BM, Newman CP, Newton CRJC, Pickrell JK, Ramesar R, Shiferaw W, Stein DJ, Teferra S, van der Merwe C, Zingela Z, NeuroGAP-Psychosis Study Team, 2021. Low-coverage sequencing cost-effectively detects known and novel variation in underrepresented populations. Am J Hum Genet 108, 656–668. 10.1016/j.ajhg.2021.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBrearty AR, Ramsey IK, Courcier EA, Mellor DJ, Bell R, 2011. Clinical factors associated with death before discharge and overall survival time in dogs with generalized megaesophagus. J Am Vet Med Assoc 238, 1622–1628. 10.2460/javma.238.12.1622 [DOI] [PubMed] [Google Scholar]
- Metzger J, Wöhlke A, Mischke R, Hoffmann A, Hewicker-Trautwein M, Küch E-M, Naim HY, Distl O, 2015. A Novel SLC27A4 Splice Acceptor Site Mutation in Great Danes with Ichthyosis. PLoS One 10, e0141514. 10.1371/journal.pone.0141514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron I, Dumitrascu DL, 2019. Gastrointestinal Motility Disorders in Obesity. Acta Endo (Buc) 15, 497–504. 10.4183/aeb.2019.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrill K, Hekman J, Li X, McClure J, Logan B, Goodman L, Gao M, Dong Y, Alonso M, Carmichael E, Snyder-Mackler N, Alonso J, Noh HJ, Johnson J, Koltookian M, Lieu C, Megquier K, Swofford R, Turner-Maier J, White ME, Weng Z, Colubri A, Genereux DP, Lord KA, Karlsson EK, 2022. Ancestry-inclusive dog genomics challenges popular breed stereotypes. Science 376, eabk0639. 10.1126/science.abk0639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SC, Evans JM, Tsai KL, Clark LA, 2018. Length variations within the Merle retrotransposon of canine PMEL: correlating genotype with phenotype. Mob DNA 9, 26. 10.1186/s13100-018-0131-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesler B, Rappold GA, 2021. Emerging evidence for gene mutations driving both brain and gut dysfunction in autism spectrum disorder. Mol Psychiatry 26, 1442–1444. 10.1038/s41380-020-0778-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrander Elaine A., Dreger DL, Evans JM, 2019a. Canine Cancer Genomics: Lessons for Canine and Human Health. Annu. Rev. Anim. Biosci. 7, 449–472. 10.1146/annurev-animal-030117-014523 [DOI] [PubMed] [Google Scholar]
- Ostrander Elaine A, Wang G-D, Larson G, vonHoldt BM, Davis BW, Jagannathan V, Hitte C, Wayne RK, Zhang Y-P, Dog10K Consortium, André C, Axelsson E, Boyko A, Davis BW, Forman O, Frantz L, Hitte C, Jagannathan V, Karlsson E, Kidd J, Larson G, Leeb T, Lindblad-Toh K, Lohi H, Lohmueller KE, Marques-Bonet T, Mellersh C, Ostrander Elaine A, Savolainen P, Schnabel R, vonHoldt BM, Wang Guo-Dong, Wayne Robert K, Yang Z, Zhai W, Zhang Ya-Ping, 2019b. Dog10K: an international sequencing effort to advance studies of canine domestication, phenotypes and health. National Science Review 6, 810–824. 10.1093/nsr/nwz049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, 1968. Achalasia or cardiospasm in Great Dane puppies. Vet Med Small Anim Clin 63, 574–576. [PubMed] [Google Scholar]
- Piras IS, Perdigones N, Zismann V, Briones N, Facista S, Rivera JL, Rozanski E, London CA, Hendricks WPD, 2020. Identification of Genetic Susceptibility Factors Associated with Canine Gastric Dilatation-Volvulus. Genes 11, 1313. 10.3390/genes11111313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassais J, Kim J, Davis BW, Karyadi DM, Hogan AN, Harris AC, Decker B, Parker HG, Ostrander EA, 2019. Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology. Nat Commun 10, 1489. 10.1038/s41467-019-09373-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassais J, vonHoldt BM, Parker HG, Carmagnini A, Dubos N, Papa I, Bevant K, Derrien T, Hennelly LM, Whitaker DT, Harris AC, Hogan AN, Huson HJ, Zaibert VF, Linderholm A, Haile J, Fest T, Habib B, Sacks BN, Benecke N, Outram AK, Sablin MV, Germonpré M, Larson G, Frantz L, Ostrander EA, 2022. Natural and human-driven selection of a single non-coding body size variant in ancient and modern canids. Current Biology 32, 889–897.e9. 10.1016/j.cub.2021.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier A-A, Aubé B, Côté M, Morin N, Di Paolo T, Soulet D, 2016. Gastrointestinal Dysfunctions in Parkinson’s Disease: Symptoms and Treatments. Parkinson’s Disease 2016, 1–23. 10.1155/2016/6762528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JE, 2014. High-Resolution Manometry in Diagnosis and Treatment of Achalasia: Help or Hype. Curr Gastroenterol Rep 16, 0. 10.1007/s11894-014-0420-2 [DOI] [PubMed] [Google Scholar]
- Sarviaho R, Hakosalo O, Tiira K, Sulkama S, Niskanen JE, Hytönen MK, Sillanpää MJ, Lohi H, 2020. A novel genomic region on chromosome 11 associated with fearfulness in dogs. Transl Psychiatry 10, 169. 10.1038/s41398-020-0849-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperber AD, Bangdiwala SI, Drossman DA, Ghoshal UC, Simren M, Tack J, Whitehead WE, Dumitrascu DL, Fang X, Fukudo S, Kellow J, Okeke E, Quigley EMM, Schmulson M, Whorwell P, Archampong T, Adibi P, Andresen V, Benninga MA, Bonaz B, Bor S, Fernandez LB, Choi SC, Corazziari ES, Francisconi C, Hani A, Lazebnik L, Lee YY, Mulak A, Rahman MM, Santos J, Setshedi M, Syam AF, Vanner S, Wong RK, Lopez-Colombo A, Costa V, Dickman R, Kanazawa M, Keshteli AH, Khatun R, Maleki I, Poitras P, Pratap N, Stefanyuk O, Thomson S, Zeevenhooven J, Palsson OS, 2021. Worldwide Prevalence and Burden of Functional Gastrointestinal Disorders, Results of Rome Foundation Global Study. Gastroenterology 160, 99–114.e3. 10.1053/j.gastro.2020.04.014 [DOI] [PubMed] [Google Scholar]
- Stratton JA, Kumar R, Sinha S, Shah P, Stykel M, Shapira Y, Midha R, Biernaskie J, 2017. Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. eNeuro 4, ENEURO.0307–16.2017. 10.1523/ENEURO.0307-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Osumi N, 2005. Identification of a novel type II classical cadherin: rat cadherin19 is expressed in the cranial ganglia and Schwann cell precursors during development. Dev Dyn 232, 200–208. 10.1002/dvdy.20209 [DOI] [PubMed] [Google Scholar]
- Tams TR, 2003. Handbook of Small Animal Gastroenterology. Elsevier Health Sciences. [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP, 2013. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 14, 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KL, Starr-Moss AN, Venkataraman GM, Robinson C, Kennedy LJ, Steiner JM, Clark LA, 2013. Alleles of the major histocompatibility complex play a role in the pathogenesis of pancreatic acinar atrophy in dogs. Immunogenetics 65, 501–509. 10.1007/s00251-013-0704-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wallerman O, Arendt M-L, Sundström E, Karlsson Å, Nordin J, Mäkeläinen S, Pielberg GR, Hanson J, Ohlsson Å, Saellström S, Rönnberg H, Ljungvall I, Häggström J, Bergström TF, Hedhammar Å, Meadows JRS, Lindblad-Toh K, 2021. A novel canine reference genome resolves genomic architecture and uncovers transcript complexity. Commun Biol 4, 185. 10.1038/s42003-021-01698-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasik K, Berisa T, Pickrell JK, Li JH, Fraser DJ, King K, Cox C, 2021. Comparing low-pass sequencing and genotyping for trait mapping in pharmacogenetics. BMC Genomics 22, 197. 10.1186/s12864-021-07508-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead K, Cortes Y, Eirmann L, 2016. Gastrointestinal dysmotility disorders in critically ill dogs and cats: Gastrointestinal dysmotility disorders in small animals. Journal of Veterinary Emergency and Critical Care 26, 234–253. 10.1111/vec.12449 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Principal component analysis for the CIM GWAS cohort. Principal components (PC) 1 and 2 are plotted for 83 Great Danes
Data Availability Statement
Sample Read Archive accession numbers for 9 high-coverage and 83 low-pass Great Dane genomes are provided in Supplementary Table 1. The VCF, index file, and metadata are available through the Data Repository for University of Minnesota (https://doi.org/10.13020/GKXV-GT86).