

Key Points
Question
How are variants of uncertain significance (VUSs) distributed across genetic disorders and what evidence contributes to their resolution?
Findings
In this cohort study of 1 689 845 individuals who predominantly underwent multigene panel testing, 41% had VUSs (mostly missense changes), most often among those who underwent testing for disorders with incomplete penetrance or were identified as Asian or Shephardic Jewish individuals. Among unique VUSs, 7.3% were reclassified, with most reclassified as benign, often using clinical and experimental evidence.
Meaning
These findings suggest that improving diversity in genomic databases, methods to evaluate missense variants, and use of clinical and experimental data would reduce uncertainty in genetic testing.
This cohort study describes the sources, gene distribution, and population-level attributes of variants of uncertain significance and evaluates the impact of the different types of evidence used to reclassify them.
Abstract
Importance
Variants of uncertain significance (VUSs) are rampant in clinical genetic testing, frustrating clinicians, patients, and laboratories because the uncertainty hinders diagnoses and clinical management. A comprehensive assessment of VUSs across many disease genes is needed to guide efforts to reduce uncertainty.
Objective
To describe the sources, gene distribution, and population-level attributes of VUSs and to evaluate the impact of the different types of evidence used to reclassify them.
Design, Setting, and Participants
This cohort study used germline DNA variant data from individuals referred by clinicians for diagnostic genetic testing for hereditary disorders. Participants included individuals for whom gene panel testing was conducted between September 9, 2014, and September 7, 2022. Data were analyzed from September 1, 2022, to April 1, 2023.
Main Outcomes and Measures
The outcomes of interest were VUS rates (stratified by age; clinician-reported race, ethnicity, and ancestry groups; types of gene panels; and variant attributes), percentage of VUSs reclassified as benign or likely benign vs pathogenic or likely pathogenic, and enrichment of evidence types used for reclassifying VUSs.
Results
The study cohort included 1 689 845 individuals ranging in age from 0 to 89 years at time of testing (median age, 50 years), with 1 203 210 (71.2%) female individuals. There were 39 150 Ashkenazi Jewish individuals (2.3%), 64 730 Asian individuals (3.8%), 126 739 Black individuals (7.5%), 5539 French Canadian individuals (0.3%), 169 714 Hispanic individuals (10.0%), 5058 Native American individuals (0.3%), 2696 Pacific Islander individuals (0.2%), 4842 Sephardic Jewish individuals (0.3%), and 974 383 White individuals (57.7%). Among all individuals tested, 692 227 (41.0%) had at least 1 VUS and 535 385 (31.7%) had only VUS results. The number of VUSs per individual increased as more genes were tested, and most VUSs were missense changes (86.6%). More VUSs were observed per sequenced gene in individuals who were not from a European White population, in middle-aged and older adults, and in individuals who underwent testing for disorders with incomplete penetrance. Of 37 699 unique VUSs that were reclassified, 30 239 (80.2%) were ultimately categorized as benign or likely benign. A mean (SD) of 30.7 (20.0) months elapsed for VUSs to be reclassified to benign or likely benign, and a mean (SD) of 22.4 (18.9) months elapsed for VUSs to be reclassified to pathogenic or likely pathogenic. Clinical evidence contributed most to reclassification.
Conclusions and Relevance
This cohort study of approximately 1.6 million individuals highlighted the need for better methods for interpreting missense variants, increased availability of clinical and experimental evidence for variant classification, and more diverse representation of race, ethnicity, and ancestry groups in genomic databases. Data from this study could provide a sound basis for understanding the sources and resolution of VUSs and navigating appropriate next steps in patient care.
Introduction
Genomic sequencing has led to the identification of many rare, novel DNA variants in diverse populations. To determine whether a variant is associated with disease, laboratory geneticists follow guidelines for weighing clinical information, population allele frequency, in silico projections of molecular effects, and other evidence.1 Classifying variants as pathogenic or likely pathogenic (P/LP) helps clinicians confirm diagnoses, provide prognoses, refine treatment strategies, and address risks for family members.2 However, as evident in the National Institutes of Health’s ClinVar database,3 many variants are classified as variants of uncertain significance (VUSs) because available evidence at the time of their discovery is not sufficient to establish their involvement in disease. VUSs can be reassessed when additional evidence emerges, sometimes leading to reclassification as P/LP or benign or likely benign (B/LB).
VUSs frustrate clinicians and patients and can be misinterpreted.4,5 Physicians are sometimes uncomfortable counseling patients about VUSs, and patients report a range of responses to VUSs, from quick dismissal to long-term anxiety.6,7,8 Exacerbating this challenge, VUSs are most commonly reported among Asian, Black, and Hispanic individuals, adding to the health disparities already affecting these populations.9,10,11 This is in part due to their underrepresentation in the medical literature and genomics projects.12,13
Most studies on VUSs have focused on a few genes, variant types, or medical specialties.14,15,16 We retrospectively reviewed VUSs in a clinical cohort of 1.6 million individuals to build a comprehensive picture of the prevalence and distribution of VUSs across hundreds of disease genes, the types of variants that constitute VUSs, and the evidence types that contribute to resolving them.
Methods
This cohort study was approved by the Western Independent Review Board, without the need for additional individual informed consent because data were deidentified. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline for cohort studies.
Clinical Cohort and DNA Sequencing
Next-generation sequencing data and clinical and demographic information were obtained for individuals who were affected by or at-risk of hereditary disease and were referred to a laboratory (Invitae), where clinician-ordered germline testing of at least 1 gene panel for Mendelian disorders was performed between September 9, 2014, and September 7, 2022, as described previously (eAppendix in Supplement 1).17,18,19 Panel sizes ranged from 1 to nearly 700 genes. Individuals aged 90 years or older at time of testing, those who requested data deletion or opted out of data use, and family members of probands were excluded from the study.
Variant Classification
Variants were classified as B/LB, P/LP, or VUSs using Sherloc, a validated system based on guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.1,20,21 Sherloc uses a semiquantitative point-based rubric for evaluating variant type, allele frequency, and clinical, functional, and computational evidence (including from publicly available software). Fixed values were assigned to each type of evidence on separate pathogenic and benign scales, and sums in both directions were assessed independently. Any variant with fewer than 4 cumulative pathogenic points or fewer than 3 cumulative benign points was classified as a VUS. Variants in genes with preliminary evidence of disease association were excluded, consistent with recommendations by ClinGen.22 Clinical reports were generated following professional guidelines and included details on variant classifications and the evidence used to justify those classifications.
Demographics and Clinical Areas
Information on patient age, sex, and race, ethnicity, and ancestry (REA) groups was provided by ordering clinicians. For analysis of age-related data, patients were grouped as 0 to 12 months, 13 months to 5 years, 6 to 12 years, 13 to 18 years, 19 to 40 years, 41 to 59 years, and 60 years and older. For REA groups, clinicians selected from preset groups on test order forms: Ashkenazi Jewish, Asian, Black, French Canadian, Hispanic, Native American, Pacific Islander, Sephardic Jewish, White (non-Jewish and non–French Canadian for the purposes of this study), or other. When other was selected, a free-text response could be added. If free-text responses matched a preset group, individuals were included in that group. Individuals with more than 1 reported REA background were grouped as multiple.
Clinical areas included cardiology, dermatology, hematology, hereditary cancer, immunology, inherited metabolic disorders, neurology, ophthalmology, and pediatric-onset rare disease. If genes were included in tests associated with 2 clinical areas (eg, NF1 in both hereditary cancer and neurology), both clinical areas were considered. Since some genes are involved in multiple hereditary diseases, we analyzed patient attributes based on the clinical area of the ordered panel (ie, test clinical area) and variant attributes based on primary gene-disease relationships (ie, gene clinical area).
Genetic Testing Results and VUS Counts
Findings were grouped into 5 categories based on variant classification and disease inheritance patterns: molecular diagnosis, potentially positive, carrier, VUS only, and negative. At the time data were analyzed for this study, all variants currently classified as VUSs (ie, excluding variants originally reported as VUSs but later reclassified as P/LP or B/LB) were counted as observed VUSs, with variants counted multiple times if observed in multiple individuals. The observed VUSs remaining after duplicates were excluded were counted as unique VUSs. Variants that were classified as VUSs at any time before data analyses (including those later reclassified) were also counted and duplicates were excluded; these were referred to as all-time unique VUSs.
Unique VUSs were stratified to illustrate the spectrum of variant types in this clinical cohort (eAppendix in Supplement 1). To analyze correlations between the types of disease genes tested and the prevalence of uncertain results, unique VUSs were also stratified by the inheritance patterns and penetrance of disorders associated with the VUS-harboring genes. Separately, observed VUSs were stratified by REA groups, age at testing, and test clinical area. Overall and across stratified groups, VUS rates were calculated and normalized to the number of genes tested to account for differences in panel sizes.
Reclassification
To identify new evidence that could help reclassify VUSs, we routinely monitored published literature, gathered information on additional patients with the same variants, and noted how other clinical laboratories were classifying them. On initial reporting of a VUS, clinicians were also given the opportunity to provide additional clinical information for the patient and to consider complimentary family testing.
To estimate the relative impact of different types of evidence on reclassification, we separated all-time unique VUSs according to whether or not they had been reclassified. We then determined which types of evidence were applied more frequently in reclassified VUSs than in nonreclassified VUSs.
Statistical Analysis
We used 1-way analysis of variance and the Bonferroni post hoc test to analyze differences among means. The 2-sample z-test for proportions and the Pearson χ2 test were used to compare categorical variables among groups when appropriate. Statistical analyses were performed using R software version 4.1.1. (R Project for Statistical Computing). All significance tests were 2-tailed, and P < .05 was considered statistically significant. Data were analyzed from September 1, 2022, to April 1, 2023.
Results
The study cohort included 1 689 845 individuals ranging in age from 0 to 89 years at time of testing (median age, 50 years), with 1 203 210 (71.2%) female participants. By REA group, there were 39 150 Ashkenazi Jewish individuals (2.3%), 64 730 Asian individuals (3.8%), 126 739 Black individuals (7.5%), 5539 French Canadian individuals (0.3%), 169 714 Hispanic individuals (10.0%), 5058 Native American individuals (0.3%), 2696 Pacific Islander individuals (0.2%), 4842 Sephardic Jewish individuals (0.3%), and 974 383 White individuals (57.7%) (eTable 1 in Supplement 1).
Diagnostic single-gene or multigene panel testing for this cohort represented a combined equivalent of 132 902 170 single-gene tests. Only a single gene was tested in 29 409 individuals (1.7%), while a mean (SD) of 78.6 (100.0) genes were analyzed per person in the whole cohort (Figure 1A). By the end of the study, 692 227 individuals (41.0%) had at least 1 observed VUS (range, 1-63 VUSs). VUSs were the only finding in 535 385 individuals (31.7%). Overall, 1 368 493 observed VUSs corresponded to 475 284 unique VUSs (eTable 2 in Supplement 1); at least 1 unique VUS (range, 1-4756 VUSs) was identified in 99.4% of genes tested (eTable 3 and eTable 4 in Supplement 1). Although the diagnostic rate remained less than 20% regardless of how many genes were tested, the rate of VUS-only results increased as the panel size grew (Figure 1B). The number of observed VUSs per individual also increased as the number of genes in a panel expanded, with the slope of this positive correlation varying by REA group (Figure 1C).
Figure 1. Number of Genes Tested and Proportion of Variants of Uncertain Significance (VUSs).

A, Only 29 409 individuals (1.7%) were tested for a single gene; 16 448 individuals (1.0%) were tested for more than 400 genes and have been excluded from the x-axis. Of note, the 2 highest proportions of individuals tested, encompassing 47 genes and 84 genes, each, represent the 2 highest volume hereditary cancer panels. C, Slope of each line was calculated using a linear regression model, with the SE shown as shading around the line. Definitions of relevant terms are provided in the eAppendix in Supplement 1.
VUS Rates Across REA and Age Groups
The proportion of individuals with at least 1 observed VUS was highest among those with Asian or Sephardic Jewish backgrounds, while it was lowest among Ashkenazi Jewish and French Canadian individuals. Compared with White individuals, the proportion of Ashkenazi Jewish or French Canadian individuals with observed VUSs was significantly lower and the proportion of observed VUSs among all other populations was significantly higher (Figure 2A).
Figure 2. Variants of Uncertain Significance (VUSs) by Race, Ethnicity, and Ancestry (REA) Groups, Age, and by Clinical Areas.
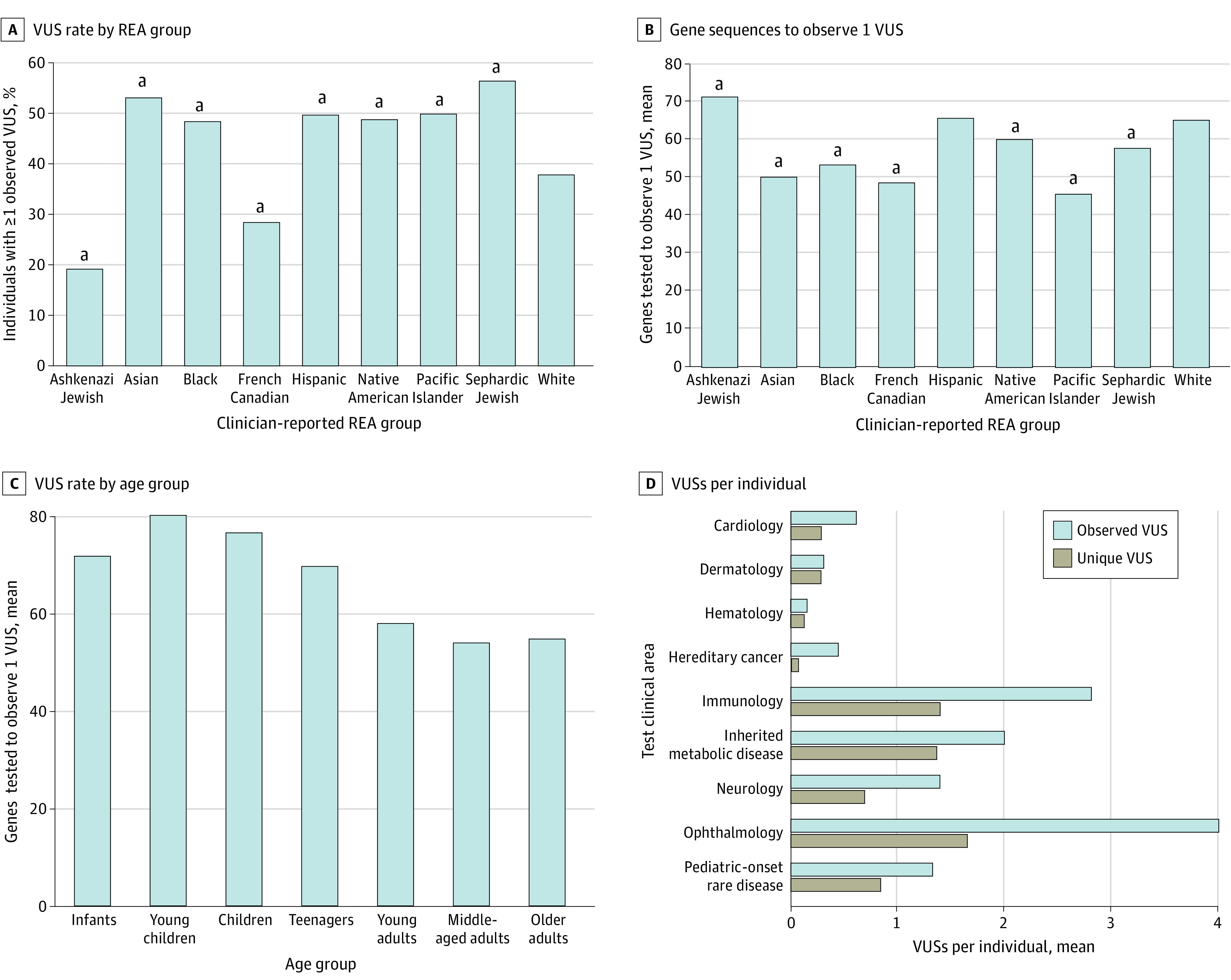
A and B, White individuals were used as the reference group for comparisons. B and C, Higher y-axis value reflects a lower VUS rate. C, Individuals tested at multiple ages (n = 8871) were treated as multiple individuals. Normalized VUS rates differed significantly among age groups (P < .001). Age group definitions are provided in the eAppendix in Supplement 1.
aP < .001.
When VUS rates were normalized to the number of genes tested, Pacific Islander individuals had the highest rate of VUSs and Ashkenazi Jewish individuals had the lowest rate, as approximately 46 genes had to be sequenced to observe a VUS in Pacific Islander individuals vs 71 in Ashkenazi Jewish individuals; White individuals had a significantly lower VUS rate than any other group except for the Ashkenazi Jewish and Hispanic groups (Figure 2B). Normalized VUS rates also differed significantly among age groups, with a lower rate (ie, more genes sequenced to find a VUS) in young children and a higher rate in adults (Figure 2C).
Clinical Distribution of VUSs
The highest proportions of observed VUSs occurred in the clinical areas of ophthalmology, immunology, neurology, pediatric onset rare disease, and inherited metabolic disorders (eTable 2 in Supplement 1). The mean number of observed VUSs per individual was also significantly higher in these test clinical areas than in others (Figure 2D). These results may be partly explained by the large panels (ie, more genes tested per patient) used in these areas as opposed to areas like hereditary cancer or cardiology, in which smaller panels with more well-characterized genes are used. Similarly, the mean number of unique VUSs per individual was higher in clinical areas with large panels (Figure 2D). These VUSs likely do not have sufficient evidence for reclassification, perhaps because they are rare in population databases or are in genes that have not been well characterized.
Distribution of VUS Types
Unique VUSs were most frequent in genes associated with both autosomal recessive (AR) and autosomal dominant (AD) disorders (Figure 3A) and in genes associated with disorders of varying penetrance (Figure 3B). Most unique VUSs were missense variants (Figure 3C). A small proportion of truncating sequence variants (including splicing variants), copy number variants, and other types of variants were also classified as VUSs when the variant’s expected molecular effect was incompatible with the disease mechanism or mode of disease inheritance. Copy number variants were typically intragenic duplications or other changes projected to preserve the transcript reading frame.
Figure 3. Distribution of Variants of Uncertain Significance (VUSs) by Genetic and Molecular Attributes.
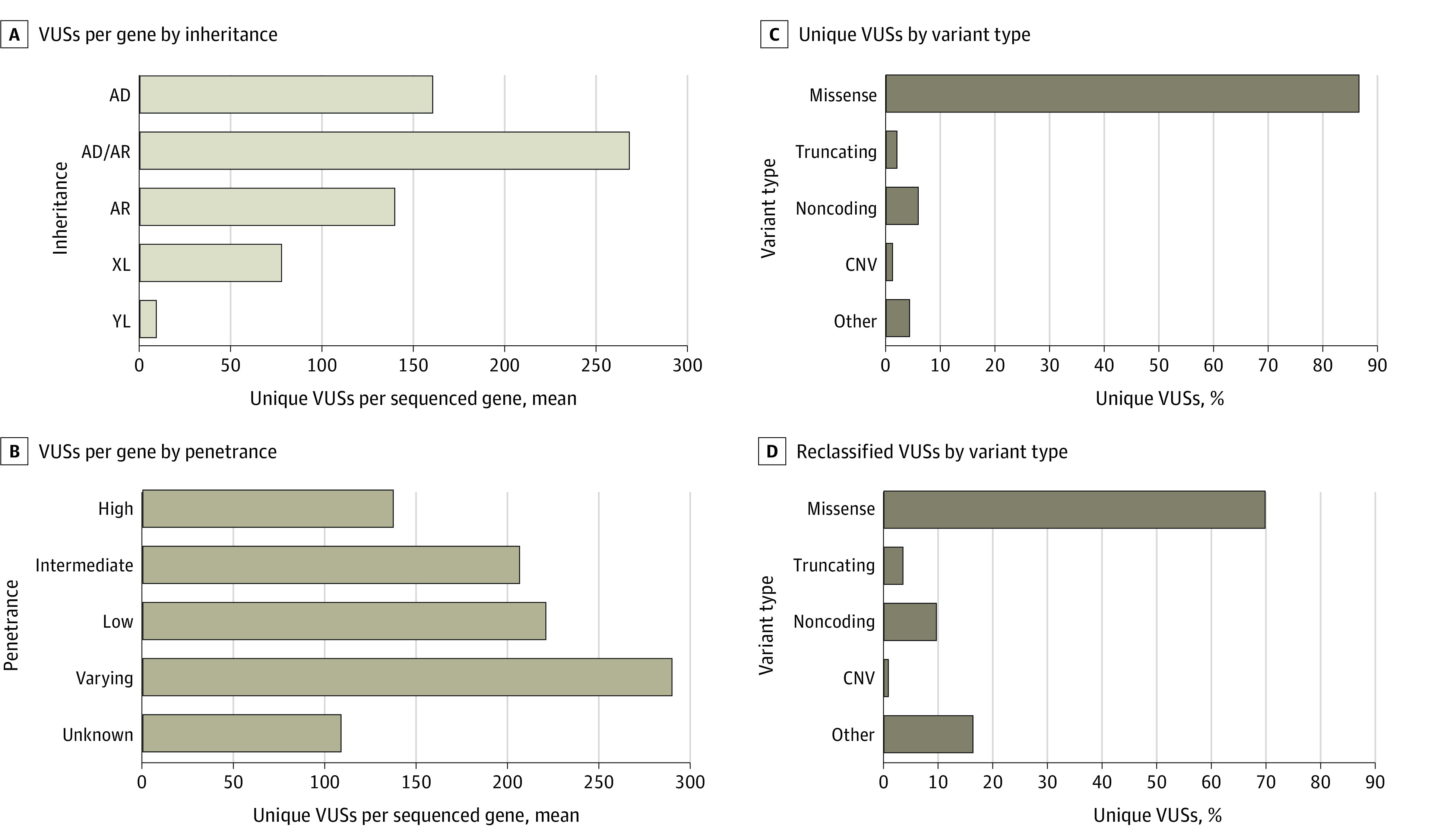
A, The total numbers of unique genes sequenced for each inheritance were 723 genes for autosomal dominant (AD), 1581 genes for autosomal recessive (AR), 467 genes for AD/AR, 157 genes for X-linked (XL), and 1 gene for Y-linked (YL). D, A total of 37 699 VUSs were reclassified. Variant type descriptions are provided in the eAppendix in Supplement 1.
VUSs in genes associated with AR diseases (ie, AR genes), which have unique implications for clinical follow-up, were observed in 1581 genes, accounting for 41.5% of all observed VUSs. Approximately 95% of individuals with at least 1 VUS in an AR gene (252 523 individuals) did not have a second VUS or P/LP variant in the same gene (eFigure 1 in Supplement 1). However, a small proportion of VUSs were present alongside other VUSs or P/LP variants within the same AR gene, warranting further follow-up if the patient’s phenotype was suggestive of an associated AR disorder.
BRCA2, ATM, and APC had the highest number of unique VUS (eTable 3 in Supplement 1), while PLEC, AMPD1, and SYNE1 had the highest number of observed VUSs per patient tested in the cohort (eTable 5 in Supplement 1). The number of unique VUS per gene was correlated with gene size and with the number of times a gene was sequenced in the cohort (eTable 4 and eFigure 2 in Supplement 1). We also noted that observed VUSs for some genes declined over time, likely due to accumulated data for and experience with classifying variants in those genes.
Reclassification of VUSs
During the study, 37 699 unique VUSs (7.3%) of 512 983 all-time unique VUSs were reclassified, affecting 163 977 individuals. Approximately 80% were reclassified as B/LB, affecting 149 987 patients, and the remaining 20% were reclassified as P/LP, affecting 17 206 patients. The mean (SD) time between original classification of VUSs and reclassification was 30.7 (20.0) months for those reclassified as B/LB and 22.4 (18.9) months for those reclassified as P/LP.
Most reclassified VUSs (26 329 VUSs [69.8%]) were missense variants (Figure 3D), and most reclassified VUSs were in genes associated with AD disease (ie, AD genes) (eTable 6 in Supplement 1), likely reflecting a test-ordering bias toward AD genes. More than 20% of reclassified VUSs (8667 VUSs [23.0%]) were in AR genes (eTable 6 in Supplement 1).
The highest proportions of all-time unique VUSs that were reclassified were in pediatric rare disease, cardiology, neurology, and hereditary cancer genes and, in all gene clinical areas, most VUS reclassifications were downgrades (Figure 4A). Two types of evidence were enriched in reclassification of VUSs compared with VUSs that were not reclassified: clinical evidence (55.0-fold enrichment in upgraded VUSs and 18.2-fold in downgraded VUSs compared with VUSs that were not reclassified) and experimental studies (12.2-fold enrichment in upgraded VUSs and 3.1-fold in downgraded VUSs compared with VUSs that were not reclassified) (Figure 4B). Of the various clinical evidence subtypes (ie, clinical observations, clinical de novo, indirect evidence, variant segregation data, confirmation of a variant in trans with a P/LP variant), clinical de novo evidence was the most impactful: it was used during classification more than 1000 times more frequently for VUSs that were upgraded than for those that remained VUSs, and used in VUS upgrades more than 12 times more frequently than any other subtype (Figure 4C).
Figure 4. Reclassification of Variants of Uncertain Significance (VUSs).

A, Each percentage was calculated using the total number of all-time unique VUSs in that clinical area as the denominator. B and C, Fold change for current VUSs was set at 1×. Evidence descriptions are provided in the eAppendix in Supplement 1.
B/LB indicates benign or likely benign; P/LP, pathogenic or likely pathogenic; VUS, variant of uncertain significance.
The extent to which each type of evidence was applied to the classification of upgraded and downgraded VUSs differed by clinical area (eFigure 3 in Supplement 1). For example, indirect evidence was applied to more downgraded VUSs than any other type of evidence (ie, ≥75% of VUSs reclassified to B/LB) in genes related to ophthalmology, immunology, hematology, inherited metabolic disorders, neurology, and dermatology. Evidence from internally developed computational tools was applied in at least 50% of upgraded VUSs in genes related to hereditary cancer, ophthalmology, and hematology. Clinical evidence had an outsized impact during classification of upgraded VUSs, and population data had an outsized impact during classification of downgraded VUSs in cardiology compared with other clinical areas. In all areas but cardiology and pediatric disorders, variant type evidence contributed substantially more to upgraded VUSs than to downgraded VUSs.
Discussion
The findings of this cohort study using expansive data across genes, clinical areas, modes of disease inheritance, REA groups, and patterns of reclassification offer insights that can inform the management of VUSs in laboratory and clinical genetics practice broadly. VUSs (most of which were missense changes) were found in 41% of 1.6 million individuals tested. In general, VUS rates were highest in individuals who were not of European White descent (eg, Asian, Black, Hispanic, Native American, Pacific Islander, and Sephardic Jewish) and in genes associated with disorders of incomplete penetrance. Although clinical evidence contributed more than any other evidence type to VUS reclassifications overall, we observed substantial variation in the impact of evidence type on upgrades vs downgrades.
Population data were applied at similar rates during classification of unique VUSs that remained as such and of all-time unique VUSs that were reclassified to B/LB. Although this seems counterintuitive because population evidence frequently contributes to B/LB classifications and therefore is expected to be enriched in reclassified VUSs, note that population data are readily available during initial classification of many variants and can be used as a standalone criterion to classify a variant as B/LB. Parsing data from each clinical area makes it evident that population data contribute more frequently to VUS downgrades than to upgrades.
In contrast to population data, clinical evidence (especially de novo evidence) was highly enriched (>1000×) in VUSs that had been upgraded to P/LP than in those that remained VUSs, highlighting the importance of parental testing for diagnosing certain conditions.23,24 We also found strong enrichment of clinical segregation data and in trans phase evidence. Lastly, enrichment of clinical observations suggesting pathogenicity during reclassifications of VUSs to P/LP extended findings from a 2022 study21 showing the benefits of systematically using such data to resolve VUSs. These observations emphasize the critical importance of the relationship between testing laboratories and clinical geneticists and genetic counselors, who can provide clinical information, facilitate family testing, and communicate the complexities of VUS resolution to patients.
Historically, genomic data have been aggregated largely from individuals of European White descent, and underrepresented populations have encountered disproportionate barriers to research participation and clinical genetic testing access.25,26 Consequently, databases, such as gnomAD and ClinVar, have accumulated variant allele frequency information with substantial gaps for individuals who are not from White European REA groups, leading to higher VUS rates in these groups. Our study found that VUS rates were in fact higher among Asian, Black, Native American, Pacific Islander, and Sephardic Jewish individuals than among those identified as White. Other studies have also reported on disparities in the population-specific utility of genetic testing.27,28 These disparities are likely to be mitigated over time as genomes from more individuals from different REA groups are sequenced through large-scale projects (eg, AllofUs) and as laboratories continue to share information through ClinVar.29
Population-agnostic approaches to generating evidence for variant classification could also help reduce VUS rates. Most VUSs in our study (and in ClinVar) were missense changes. Although in silico methods for evaluating the effects of missense variants have improved, there is still considerable room for improving their prognostic power (eg, by using gene-specific training data sets).30,31,32 Integration of clinical information and better experimental evidence will also resolve more missense VUSs; recent innovations, such as multiplex assays of variant effects have already enabled systematic use of experimental evidence for variant classification.33,34
Limitations
This study has some limitations. Because our data are from an unselected clinical cohort referred for genetic testing, the clinical areas and genes tested may be unevenly distributed, which could introduce bias and influence the number of observed VUSs. For example, the genes most commonly sequenced in children were different from genes most commonly sequenced in adults. Second, we included and analyzed clinical information wherever available, but, as is typical of clinical genetics laboratories, we did not routinely and consistently receive detailed clinical information for patients referred to our laboratory for genetic testing. Third, because information on REA was clinician-reported, it may be inaccurate or lacking in some individuals.
Conclusions
This cohort study of approximately 1.6 million individuals highlighted the need for better methods for interpreting missense variants, increased availability of clinical and experimental evidence for variant classification, and more diverse representation of REA groups in genomic databases. Experts have suggested what clinicians should consider when evaluating genetic testing results in their diagnostic approach.35 The VUS rate in a gene is influenced by several factors, including the variant spectrum, length of the coding regions, penetrance of associated disorders, severity of phenotypes as related to fitness, amount and type of natural variation, and what we know about the gene through published research. Given this complexity, a close partnership between clinicians and laboratory geneticists is essential for optimizing how genetic testing results are reported and next steps are implemented.
A 2020 commentary by Appelbaum et al36 suggest that the responsibility for tracking VUSs and gathering evidence to reclassify them falls in large part on laboratories. As genetic testing increases, tracking VUSs across millions of individuals will require advanced computational methods, including those incorporating machine learning. Algorithms for evaluating variant effects with high prognostic values and quantitative approaches for scoring evidence types can enhance accuracy in classifying variants.37 Laboratories could also develop methods to highlight which additional evidence types might resolve VUSs and thereby guide clinicians on follow-up. Together, these approaches would accelerate the adoption of a quantitative system for variant classification,38 enabling laboratories and clinicians to explain genetic testing results better to patients and resolve VUSs sooner.
eAppendix. Additional Definitions
eTable 1. Characteristics of Clinical Cohort
eTable 2. Observed VUS Across Test Order Clinical Areas
eTable 3. Percentage of Unique Variants That are VUS in Disease Genes
eTable 4. Genes With Most Unique VUS Normalized to Coding Sequence (CDS) Length
eTable 5. Genes With Most Observed VUS per Patient
eTable 6. Reclassification by Variant Type and Inheritance
eFigure 1. Results Categories Among Individuals With at Least 1 VUS in a Gene Associated With Autosomal Recessive Disease
eFigure 2. Coding Region Length or the Number of Times a Gene is Sequenced and Their Correlation With the Number of Unique VUS per Gene
eFigure 3. Proportion of Unique VUS That Underwent Reclassification With Different Evidence Types Applied, Shown by Clinical Areas
eReferences.
Data Sharing Statement
References
- 1.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.ACMG Board of Directors . Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2015;17(6):505-507. doi: 10.1038/gim.2015.41 [DOI] [PubMed] [Google Scholar]
- 3.National Library of Medicine . ClinVar. Accessed September 19, 2023. https://www.ncbi.nlm.nih.gov/clinvar/
- 4.Gould D, Walker R, Makari-Judson G, Seven M. Experiences of individuals with a variant of uncertain significance on genetic testing for hereditary cancer risks: a mixed method systematic review. J Community Genet. 2022;13(4):371-379. doi: 10.1007/s12687-022-00600-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reuter C, Chun N, Pariani M, Hanson-Kahn A. Understanding variants of uncertain significance in the era of multigene panels: through the eyes of the patient. J Genet Couns. 2019;28(4):878-886. doi: 10.1002/jgc4.1130 [DOI] [PubMed] [Google Scholar]
- 6.Macklin SK, Jackson JL, Atwal PS, Hines SL. Physician interpretation of variants of uncertain significance. Fam Cancer. 2019;18(1):121-126. doi: 10.1007/s10689-018-0086-2 [DOI] [PubMed] [Google Scholar]
- 7.Clift K, Macklin S, Halverson C, McCormick JB, Abu Dabrh AM, Hines S. Patients’ views on variants of uncertain significance across indications. J Community Genet. 2020;11(2):139-145. doi: 10.1007/s12687-019-00434-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amano Y, Raz A, Timmermans S, Shkedi-Rafid S. Cancer patients’ understandings of genetic variants of uncertain significance in clinical care. J Community Genet. 2022;13(4):381-388. doi: 10.1007/s12687-022-00594-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giri VN, Hartman R, Pritzlaff M, Horton C, Keith SW. Germline variant spectrum among African American men undergoing prostate cancer germline testing: need for equity in genetic testing. JCO Precis Oncol. 2022;6(1):e2200234. doi: 10.1200/PO.22.00234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caswell-Jin JL, Gupta T, Hall E, et al. Racial/ethnic differences in multiple-gene sequencing results for hereditary cancer risk. Genet Med. 2018;20(2):234-239. doi: 10.1038/gim.2017.96 [DOI] [PubMed] [Google Scholar]
- 11.Tatineni S, Tarockoff M, Abdallah N, et al. Racial and ethnic variation in multigene panel testing in a cohort of BRCA1/2-negative individuals who had genetic testing in a large urban comprehensive cancer center. Cancer Med. 2022;11(6):1465-1473. doi: 10.1002/cam4.4541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sirugo G, Williams SM, Tishkoff SA. The missing diversity in human genetic studies. Cell. 2019;177(1):26-31. doi: 10.1016/j.cell.2019.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Popejoy AB, Ritter DI, Crooks K, et al. ; Clinical Genome Resource (ClinGen) Ancestry and Diversity Working Group (ADWG) . The clinical imperative for inclusivity: race, ethnicity, and ancestry (REA) in genomics. Hum Mutat. 2018;39(11):1713-1720. doi: 10.1002/humu.23644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Landry LG, Rehm HL. Association of racial/ethnic categories with the ability of genetic tests to detect a cause of cardiomyopathy. JAMA Cardiol. 2018;3(4):341-345. doi: 10.1001/jamacardio.2017.5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burke W, Parens E, Chung WK, Berger SM, Appelbaum PS. The challenge of genetic variants of uncertain clinical significance: a narrative review. Ann Intern Med. 2022;175(7):994-1000. doi: 10.7326/M21-4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caputo SM, Golmard L, Léone M, et al. Classification of 101 BRCA1 and BRCA2 variants of uncertain significance by cosegregation study: a powerful approach. Am J Hum Genet. 2021;108(10):1907-1923. doi: 10.1016/j.ajhg.2021.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lincoln SE, Kobayashi Y, Anderson MJ, et al. A Systematic comparison of traditional and multigene panel testing for hereditary breast and ovarian cancer genes in more than 1000 patients. J Mol Diagn. 2015;17(5):533-544. doi: 10.1016/j.jmoldx.2015.04.009 [DOI] [PubMed] [Google Scholar]
- 18.Lincoln SE, Truty R, Lin CF, et al. A rigorous interlaboratory examination of the need to confirm next-generation sequencing-detected variants with an orthogonal method in clinical genetic testing. J Mol Diagn. 2019;21(2):318-329. doi: 10.1016/j.jmoldx.2018.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Truty R, Paul J, Kennemer M, et al. Prevalence and properties of intragenic copy-number variation in Mendelian disease genes. Genet Med. 2019;21(1):114-123. doi: 10.1038/s41436-018-0033-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nykamp K, Anderson M, Powers M, et al. ; Invitae Clinical Genomics Group . Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19(10):1105-1117. doi: 10.1038/gim.2017.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson B, Ouyang K, Frank L, et al. Systematic use of phenotype evidence in clinical genetic testing reduces the frequency of variants of uncertain significance. Am J Med Genet A. 2022;188(9):2642-2651. doi: 10.1002/ajmg.a.62779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Strande NT, Riggs ER, Buchanan AH, et al. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the Clinical Genome Resource. Am J Hum Genet. 2017;100(6):895-906. doi: 10.1016/j.ajhg.2017.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ku CS, Polychronakos C, Tan EK, et al. A new paradigm emerges from the study of de novo mutations in the context of neurodevelopmental disease. Mol Psychiatry. 2013;18(2):141-153. doi: 10.1038/mp.2012.58 [DOI] [PubMed] [Google Scholar]
- 24.Wilfert AB, Sulovari A, Turner TN, Coe BP, Eichler EE. Recurrent de novo mutations in neurodevelopmental disorders: properties and clinical implications. Genome Med. 2017;9(1):101. doi: 10.1186/s13073-017-0498-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu YL, Maio A, Kemel Y, et al. Disparities in cancer genetics care by race/ethnicity among pan-cancer patients with pathogenic germline variants. Cancer. 2022;128(21):3870-3879. doi: 10.1002/cncr.34434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saylor KW, Klein WM, Calancie L, et al. Genetic testing and other healthcare use by Black and White individuals in a genomic sequencing study. Public Health Genomics. 2023;26(1):90-102. doi: 10.1159/000533356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Florentine MM, Rouse SL, Stephans J, et al. Racial and ethnic disparities in diagnostic efficacy of comprehensive genetic testing for sensorineural hearing loss. Hum Genet. 2022;141(3-4):495-504. doi: 10.1007/s00439-021-02338-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Appelbaum PS, Burke W, Parens E, et al. Is there a way to reduce the inequity in variant interpretation on the basis of ancestry? Am J Hum Genet. 2022;109(6):981-988. doi: 10.1016/j.ajhg.2022.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Denny JC, Rutter JL, Goldstein DB, et al. ; All of Us Research Program Investigators . The “All of Us” research program. N Engl J Med. 2019;381(7):668-676. doi: 10.1056/NEJMsr1809937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pejaver V, Byrne AB, Feng BJ, et al. ; ClinGen Sequence Variant Interpretation Working Group . Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. 2022;109(12):2163-2177. doi: 10.1016/j.ajhg.2022.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghosh R, Oak N, Plon SE. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017;18(1):225. doi: 10.1186/s13059-017-1353-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877-885. doi: 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livesey BJ, Marsh JA. Interpreting protein variant effects with computational predictors and deep mutational scanning. Dis Model Mech. 2022;15(6):dmm049510. doi: 10.1242/dmm.049510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gelman H, Dines JN, Berg J, et al. ; Brotman Baty Institute Mutational Scanning Working Group . Recommendations for the collection and use of multiplexed functional data for clinical variant interpretation. Genome Med. 2019;11(1):85. doi: 10.1186/s13073-019-0698-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biesecker LG, Nussbaum RL, Rehm HL. Distinguishing variant pathogenicity from genetic diagnosis: how to know whether a variant causes a condition. JAMA. 2018;320(18):1929-1930. doi: 10.1001/jama.2018.14900 [DOI] [PubMed] [Google Scholar]
- 36.Appelbaum PS, Parens E, Berger SM, Chung WK, Burke W. Is there a duty to reinterpret genetic data: the ethical dimensions. Genet Med. 2020;22(3):633-639. doi: 10.1038/s41436-019-0679-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aradhya S, Facio FM, Metz H, et al. Applications of artificial intelligence in clinical laboratory genomics. Am J Med Genet C Semin Med Genet. Published online July 28, 2023. doi: 10.1002/ajmg.c.32057 [DOI] [PubMed] [Google Scholar]
- 38.Tavtigian SV, Greenblatt MS, Harrison SM, et al. ; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) . Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20(9):1054-1060. doi: 10.1038/gim.2017.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix. Additional Definitions
eTable 1. Characteristics of Clinical Cohort
eTable 2. Observed VUS Across Test Order Clinical Areas
eTable 3. Percentage of Unique Variants That are VUS in Disease Genes
eTable 4. Genes With Most Unique VUS Normalized to Coding Sequence (CDS) Length
eTable 5. Genes With Most Observed VUS per Patient
eTable 6. Reclassification by Variant Type and Inheritance
eFigure 1. Results Categories Among Individuals With at Least 1 VUS in a Gene Associated With Autosomal Recessive Disease
eFigure 2. Coding Region Length or the Number of Times a Gene is Sequenced and Their Correlation With the Number of Unique VUS per Gene
eFigure 3. Proportion of Unique VUS That Underwent Reclassification With Different Evidence Types Applied, Shown by Clinical Areas
eReferences.
Data Sharing Statement