

SUMMARY
Oligodendrocytes are specialized cells that insulate and support axons with their myelin membrane, allowing proper brain function. Here, we identify lamin A/C (LMNA/C) as essential for transcriptional and functional stability of myelinating oligodendrocytes. We show that LMNA/C levels increase with differentiation of progenitors and that loss of Lmna in differentiated oligodendrocytes profoundly alters their chromatin accessibility and transcriptional signature. Lmna deletion in myelinating glia is compatible with normal developmental myelination. However, altered chromatin accessibility is detected in fully differentiated oligodendrocytes together with increased expression of progenitor genes and decreased levels of lipid-related transcription factors and inner mitochondrial membrane transcripts. These changes are accompanied by altered brain metabolism, lower levels of myelin-related lipids, and altered mitochondrial structure in oligodendrocytes, thereby resulting in myelin thinning and the development of a progressively worsening motor phenotype. Overall, our data identify LMNA/C as essential for maintaining the transcriptional and functional stability of myelinating oligodendrocytes.
In brief
Nuclear lamina protein LMNA/C increases as progenitors differentiate. Pruvost et al. identify that it is essential for the transcriptional stability of myelinating oligodendrocytes. Its absence results in aberrant expression of progenitor genes in mature cells, altered lipid metabolism and mitochondrial structure, and onset of a progressively worsening clinical phenotype.
Graphical Abstract
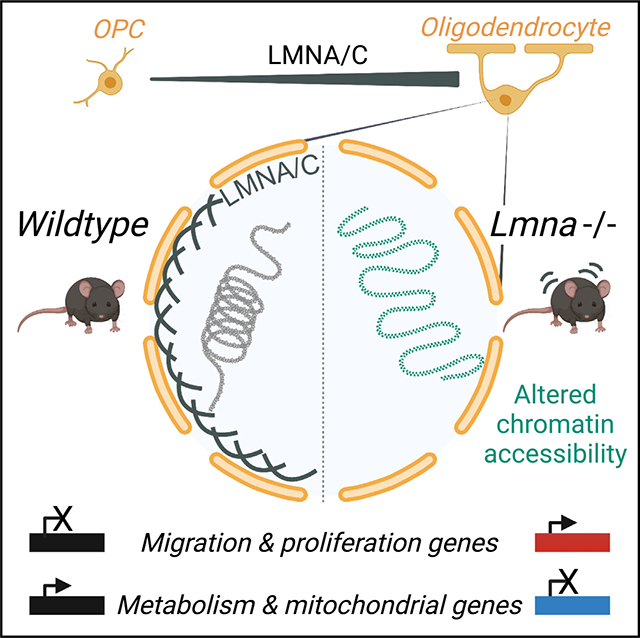
INTRODUCTION
The acquisition of cell identity is critical to ensure the unique function of specialized cells and their topological organization within different tissues.1,2 This process is clearly important during development, when cell identity is first established, but is also relevant in adulthood, when the functionality of each organ relies on functional specialization.3,4 Myelinating oligodendrocytes are post-mitotic, highly branched, stationary cells in the central nervous system (CNS), with each cell wrapping axons with their specialized membrane, called myelin, and providing insulation and metabolic support.5 In the developing brain, functional neuronal networks rely on the functional specialization of these cells, which provide insulation and metabolic support. Myelinating oligodendrocytes derive from bipolar, proliferating, highly migratory and electrically active progenitors, which exit from the cell cycle, stop migrating, lose synaptic contacts, and form the insulating membrane called myelin.5,6 At a molecular level, this transition is modulated by transcription factors and epigenetic components,7–10 including histone11–16 and DNA modifications,17–19 microRNAs,20–22 and non-coding RNAs,23,24 which alter chromatin structure and gene expression.25 Genes related to migration, transcriptional inhibition of myelin transcripts, cell division, and electrical excitability are downregulated in myelinating cells,12,26,27 while those related to myelin lipid synthesis and metabolism are upregulated.10,28–34
At an ultrastructural level, the process of differentiation is characterized by the transition from predominantly euchromatic nuclei in progenitors to electron-dense nuclei in myelinating oligodendrocytes, which are characterized by large masses of peripheral heterochromatin, in the proximity of the nuclear lamina.27,35 The nuclear lamina is the 30- to 100-nm-thick network36–38 providing an interface between chromatin and the nuclear envelope.39,40 It is formed by intermediate filaments composed of lamin A/C and B proteins, which are encoded by distinct genes.41 Lamin A and C are alternatively spliced products of the Lmna gene, and lamin B1 is encoded by the Lmnb1 gene.42 A-type lamins (lamins A and C) are expressed in differentiated cells while B-type lamins are expressed in almost all somatic undifferentiated cells at early stages of development.43–46 Alterations of the epigenomic landscape of oligodendrocytes have been reported in neurological conditions characterized by demyelination, such as multiple sclerosis47–49 or adrenoleukodystrophy,50 and in autism,51 as well as psychiatric conditions including major depression52–54 and schizophrenia.55 Altered expression of nuclear lamins has also been linked to alterations of white matter tracts. For instance, we and others have previously reported on the importance of decreasing lamin B1 levels during the differentiation of cultured oligodendrocyte progenitors,56 a process regulated in part by the levels of the microRNA miR-23.57 Duplications of the LMNB1 gene in humans,58–62 as well as large deletions resulting in re-localization of enhancers,63 were shown to result in altered myelin content in white matter tracts. Overexpression studies in oligodendrocyte-related cell lines56,57 and mice64–66 revealed disrupted lipid metabolism and late myelin alterations. For lamin A/C, however, mutations have been mostly associated with cardiomyopathies, lipodystrophies, and muscular dystrophies.67,68 The first evidence linking the LMNA locus (on chromosome 1) to a neurological phenotype was reported in 199969 and further refined to the 1q21–3 region.70 Although LMNA mutations have also been associated with autosomal recessive forms of peripheral neuropathies in North Africa and in the Middle East,71,72 it is important to note that the clinical phenotype has been characterized by wide variability, attributed to the strict cellular specificity of LMNA function, and prompted us to analyze its role in myelinating cells. Although the Lmna gene is expressed at low levels in the brain,73 we report here a role for lamin A/C (LMNA/C) in regulating metabolism and transcriptional stability of myelinating oligodendrocytes.
RESULTS
The nuclear lamina of myelinating oligodendrocytes is enriched in lamin A
As progenitors transition into myelinating oligodendrocytes during the period of developmental myelination, the levels of Lmnb1 transcripts in the mouse corpus callosum decrease (Figure 1A), while the levels of Lmna transcripts slowly increase from birth to post-natal day 21 (P21; Figure 1B). Protein levels follow a similar trend, with LMNB1 levels decreasing in the adult mouse brain and LMNA/C protein levels increasing over time (Figures 1C and 1D). Immunohistochemical analysis of murine brain sections (Figure 1E) further validated the presence of LMNA/C in the nuclei of differentiated oligodendrocytes, defined by the co-expression of the transcription factor OLIG2 and the myelin basic protein (MBP). The levels of LMNA/C were significantly higher in differentiated OLIG2+/MBP+ oligodendrocytes compared with OLIG2+/MBP− progenitors (Figure 1F). A similar reciprocal pattern of Lmna and Lmnb1 expression was observed in cultured progenitors and differentiated oligodendrocytes, both at the RNA (Figures 1G and 1H) and protein levels (Figure 1I). In addition, the LMNA/C protein levels were inversely proportional to the mitogen concentration in the culture medium (Figure 1J), suggesting that LMNA/C accumulation in the nuclei of differentiating oligodendrocytes was triggered by the absence of mitogenic stimulation. Immunohistochemical analysis of human brain sections stained with antibodies recognizing either human LMNAor LMNB1, and either the progenitor marker NG2 (Figure 1K) or the oligodendrocytic marker CC1 (Figure 1M), detected LMNB1 immunoreactivity predominantly in NG2+ cells (Figure 1L) and LMNA in mature CC1+ oligodendrocytes (Figure 1N). The detection of a reciprocal pattern of LMNB1 expression in progenitors and LMNA/C in mature oligodendrocytes underscored an important feature of nuclear lamins in myelinating glia of the CNS that was conserved across species. Interestingly, this reciprocal pattern was not detected in Schwann cells, the myelinating cells of the peripheral nervous system (PNS), which retain the ability to de-differentiate and re-enter the cell cycle. In Schwann cells, LMNB1 and LMNA/C levels remained constant over time and were not affected by the presence of mitogens or differentiation signals in the culture medium (Figure S1). Overall, these data are consistent with the notion that nuclear lamins play distinct roles in different cells including myelinating glia of the CNS or the PNS, which are characterized by distinct phenotypic stability (e.g., oligodendrocytes) and plasticity (e.g., Schwann cells).
Figure 1. The expression of Lmna transcripts and LMNA/C proteins increases during developmental myelination and oligodendrocyte progenitor differentiation.
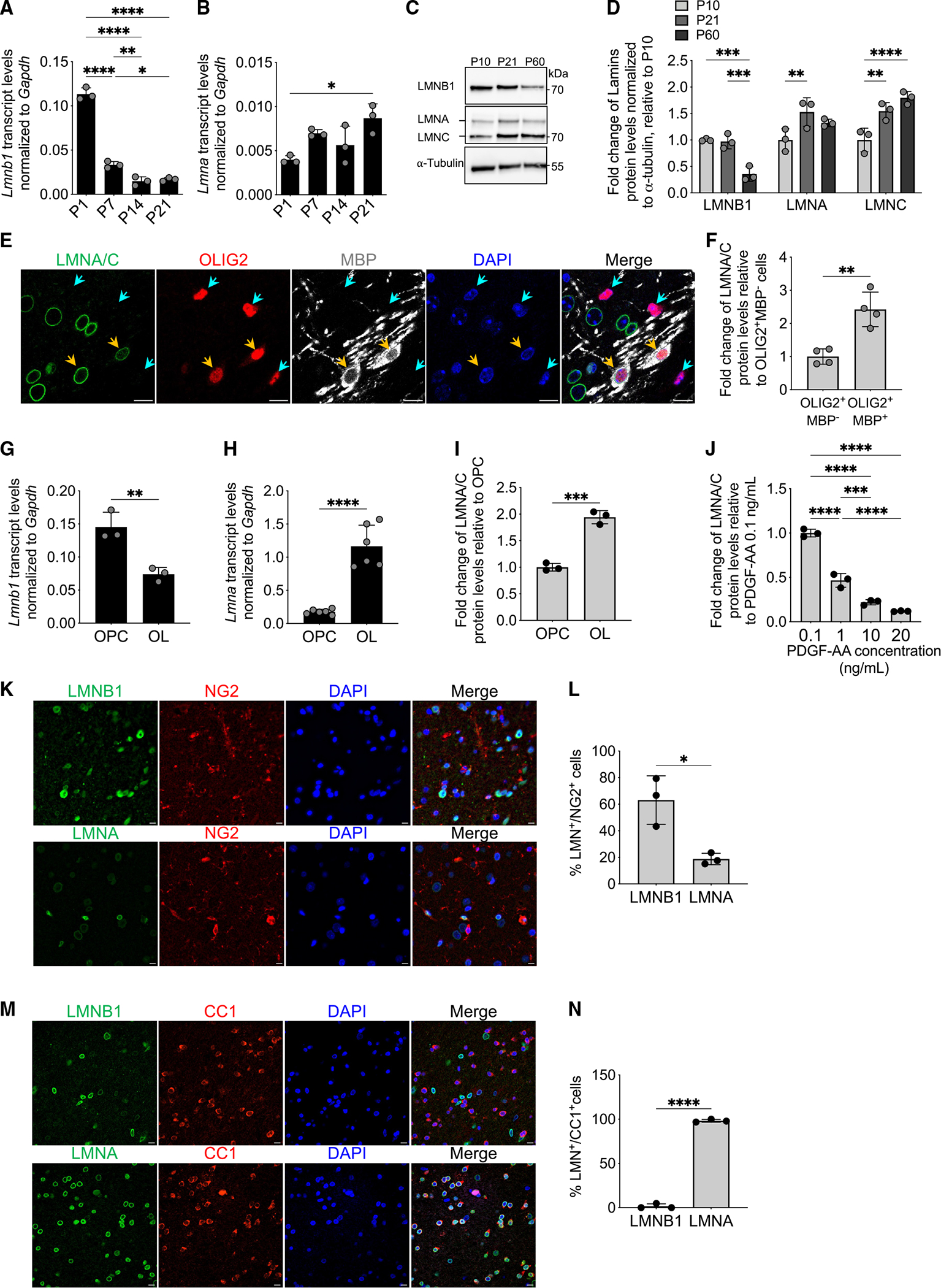
(A and B) Scatterplot of Lmnb1 (A) and Lmna (B) transcripts, normalized to Gapdh levels. Measurements by real-time qPCR of RNA samples extracted from mouse corpus callosum at postnatal day 1 (P1), P7, P14, and P21. Data are represented as mean ± SD, n = 3 mice per group (*p < 0.05, **p < 0.01, ****p < 0.0001, one-way ANOVA).
(C) Representative western blot of expression levels of LMNB1 and LMNA/C in the corpus callosum of mice at P10, P21, and P60. α-Tubulin used as a loading control. Molecular weights indicated on the right.
(D) Fold change of protein levels of LMNB1, LMNA, and LMNC in the corpus callosum of mice at P10, P21, and P60 relative to P10, measured by western blot and normalized to α-tubulin levels. Data are represented as mean ± SD, n = 3 mice per group (**p < 0.01, ***p < 0.001, ****p < 0.0001, two-way ANOVA).
(E) Representative confocal image of P7 mouse corpus callosum stained for LMNA/C (green) with OLIG2 (red) and MBP (white). DAPI (blue) was used as nuclear counterstain. Scale bar, 10 μm.
(F) Scatterplot of the fold change of LMNA/C protein levels measured by immunofluorescence in OLIG2+MBP− cells (identified by a turquoise arrowhead in E) and OLIG2+ MBP+ cells (identified by an orange arrowhead in E), relative to OLIG2+MBP− cells. Data are represented as mean ± SD, n = 4 mice per group (**p < 0.01, two-tailed Student’s t test).
(G and H) Scatterplots of the transcript levels for Lmnb1 (G) and Lmna (H), normalized to Gapdh, measured by real-time qPCR. RNA samples were extracted from proliferating progenitors oligodendrocyte progenitor cells (OPCs) or from cells differentiated into oligodendrocytes (OLs) following 48 h in the absence of mitogens and presence of T3. Data are represented as mean ± SD, n = 3 independent cultures for Lmnb (**p < 0.01, two-tailed Student’s t test) and n = 6 independent cultures for Lmna (****p < 0.0001, two-tailed Student’s t test).
(I) Scatterplot of the fold change of LMNA/C protein levels in OLs relative to OPCs and measured by immunofluorescence. Data are represented as mean ± SD, n = 3 independent cultures (***p < 0.001, two-tailed Student’s t test).
(J) LMNA/C protein levels measured by immunofluorescence in OPC cultured in the presence of the indicated concentrations of PDGF-AA. Data are presented as fold change relative to those in OPC cultured in 0.1 ng/mL PDGF-AA. Mean ± SD, n = 3 independent cultures (***p < 0.001, ****p < 0.0001, one-way ANOVA).
(K) Representative confocal images of adult human brain stained for the indicated lamins (LMNB1 or LMNA in green) and NG2 (in red). DAPI (blue) was used as nuclear counterstain. Scale bar, 10 μm.
(L) Scatterplot of the percentage of NG2+ cells that are either LMNB1+ or LMNA+. Data are represented as mean ± SD, n = 3 human brains (*p < 0.05, two-tailed Student’s t test).
(M) Representative confocal images of adult human brain stained for the indicated lamins (LMNB1 or LMNA in green) and CC1 (in red). DAPI (blue) was used as nuclear counterstain. Scale bar, 10 μm.
(N) Scatterplot of the percentage of CC1+ cells that are either LMNB1+ or LMNA+. Data are represented as mean ± SD, n = 3 human brains (****p < 0.0001, two-tailed Student’s t test).
Lineage-specific genetic ablation of Lmna gene in myelinating glia is compatible with normal developmental myelination
To genetically delete Lmna in myelinating glia, we crossed mice with loxP sites before exon 10 and in exon 12 of the Lmna gene (Lmnafl/fl) with a knockin Cre line driven by the Cnp1 promoter (Cnp-Cre) to target myelinating glia in the CNS and PNS74 (Figure 2A). Decreased Lmna transcript (Figure 2B) and LMNA/C protein levels (Figures 2C and 2D) were detected in oligodendrocytes and Schwann cells (Figure S2). No significant difference in the percentage of LMNA/C-expressing NeuN+ neurons (Figures 2E and 2F) or GFAP+ astrocytes (Figures 2G and 2H) was found between wild-type (WT; CnpCre/+;Lmnafl/fl) and mutant mice (CnpCre/+;Lmnafl/fl; conditional knockout [cKO]), thereby confirming that the ablation of LMNA/C was lineage specific and confined to myelinating glia.
Figure 2. Effective and lineage-specific genetic ablation of Lmna in the central nervous system of mutant mice.
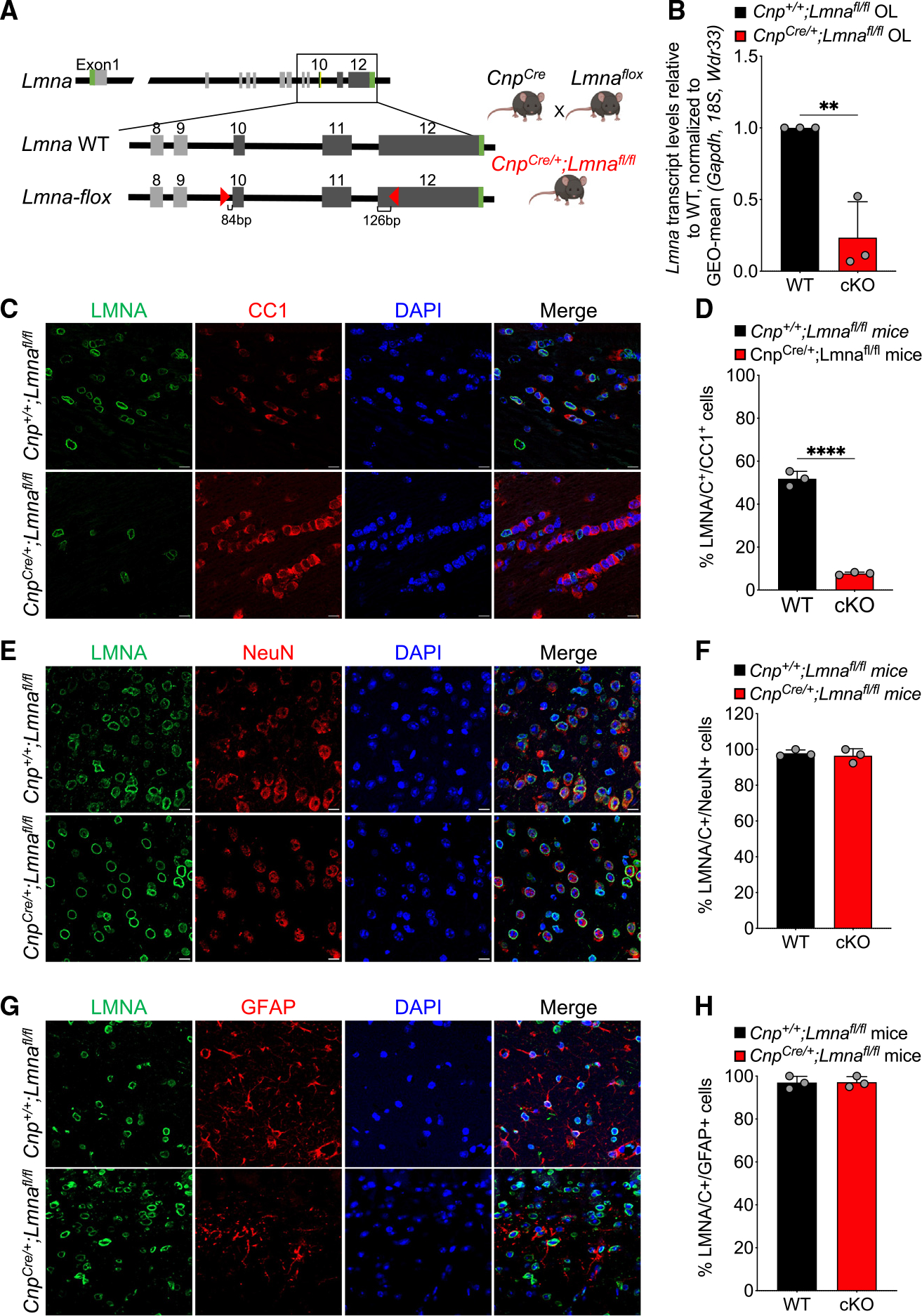
(A) Schematic of the breeding strategy used to obtain the conditional knockout targeting myelinating glia. Lmna wild-type allele (Lmna WT) and conditionally targeted allele of Lmna (Lmna-flox) with the indicated loxP sites (red triangles) are shown on the left side. The crossing between the Lmna-floxed and Cnp-Cre lines is shown on the right.
(B) Scatterplot of Lmna transcript levels measured by real-time qPCR in RNA samples extracted from cultured OPCs isolated from WT (Cnp+/+; Lmnafl/fl) and Lmna mutant mice (CnpCre/+;Lmnafl/fl) and differentiated in vitro. Data were normalized to the geometric mean (GEO-mean) of Gapdh, 18S, and Wdr33 transcript levels. Data are represented as mean ± SD, n = 3 independent cultures per group (**p < 0.01, two-tailed Student’s t test).
(C, E, and G) Representative confocal images of brain sections of control (Cnp+/+;Lmnafl/fl) and Lmna mutant mice (CnpCre/+;Lmnafl/fl), stained with LMNA/C (green) and CC1 (red, C), NeuN (red, E), or GFAP (red, G) antibodies. DAPI (blue) was used as nuclear counterstain. Scale bar, 20 μm.
(D, F, and H) Scatterplots representing the percentage of LMNA/C+/CC1+ cells (D), LMNA/C+/NeuN+ cells (F), and LMNA/C+/GFAP+ cells (H) in WT and Lmna mutant mice. Data are represented as mean ± SD.
Number of cells analyzed per genotype: WT, 560 (D), 93 (F), and 94 cells (H); cKO, 628 (D), 98 (F), and 92 cells (H); n = 3 mice per genotype (****p < 0.0001, D, two-tailed Student’s t test; p ≥ 0.05, F and H, two-tailed Student’s t test).
CnpCre/+;Lmnafl/fl mice were born at expected Mendelian ratios and were indistinguishable from control littermates until adulthood. At 8 weeks, Lmna mutant mice started to develop the gradual inability to hold and grip a grid upon inversion (Videos S1 and S2), although the phenotype was variable and not detected in all the mice (Figure 3A). Additional symptoms, including ataxia and tremors, became more consistent around 12 weeks, and by 26 weeks, all Lmna mutants displayed a severe motor phenotype (Figure 3A; Videos S3 and S4), which could be also quantified using the rotarod motor task (Figure 3B). Motor signs were not detected in mice with Lmna ablation in the neuronal lineage (Figure S3) and thereby were specific to Lmna mice with ablation in myelinating glia. In the absence of LMNA/C, developmental myelination proceeded normally, as myelin thickness in the corpus callosum of Lmna mutants and controls was virtually indistinguishable at P21 (Figures 3C–3E) and remained relatively unperturbed until adulthood (Figure S4). Oligodendrocyte cell counts in the corpus callosum (Figures 3F and 3G) and myelin protein levels in spinal cord extracts did not reveal any difference between mutant and WT mice at 8 weeks (Figure 3H). We conclude that the absence of LMNA/C is compatible with normal CNS developmental myelination.
Figure 3. Loss of Lmna in OLs does not interfere with the early stages of developmental myelination.
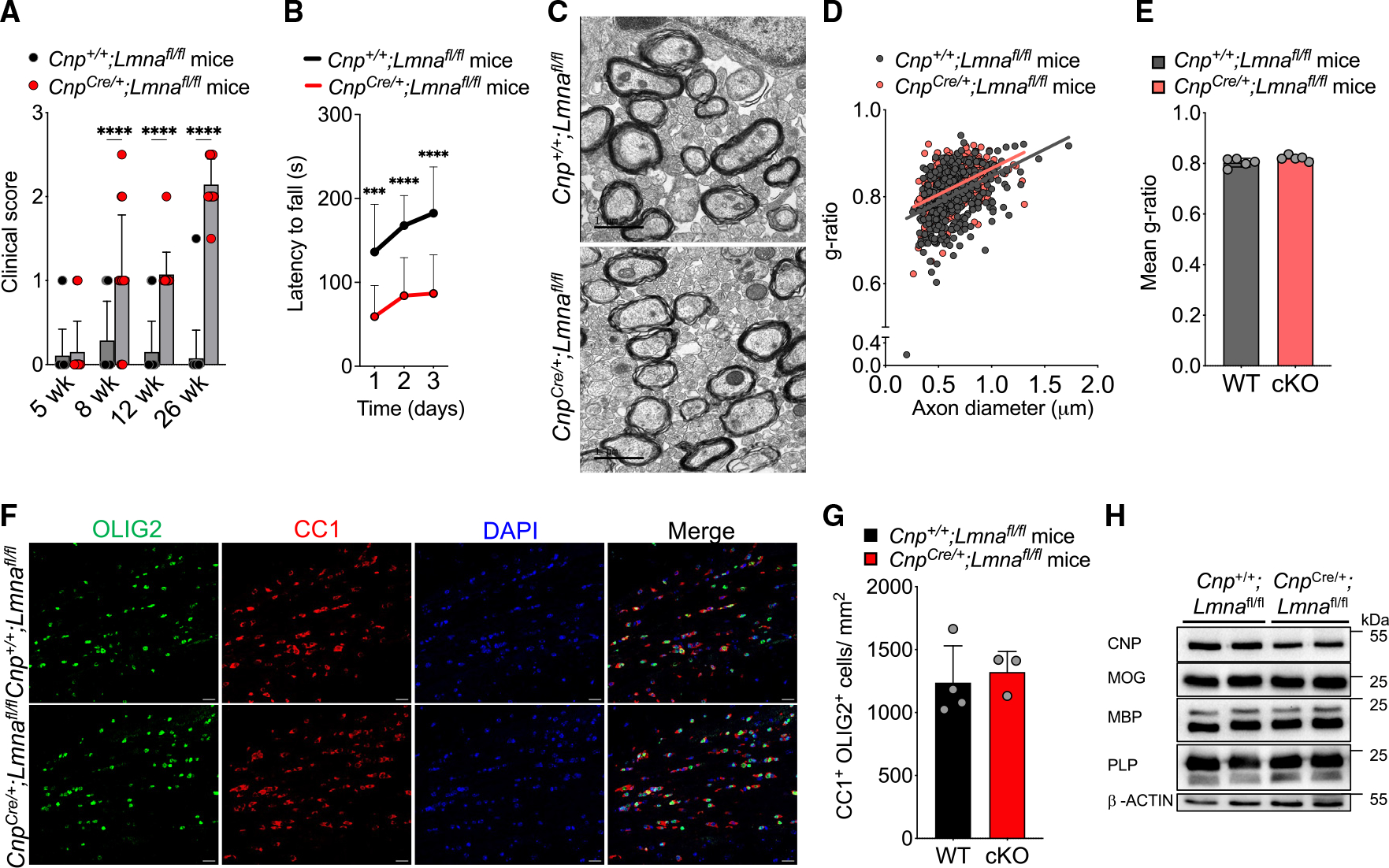
(A) Scatterplot of the clinical score observed in WT (Cnp+/+;Lmnafl/fl) and Lmna mutant (CnpCre/+;Lmnafl/fl) mice at 5, 8, 12, and 26 weeks. Data represented as mean + SD; WT mice: n = 19 at 5 weeks, 14 at 8 weeks, 20 at 12 weeks, and 20 at 26 weeks; Lmna mutant mice: n = 20 at 5 weeks, 14 at 8 weeks, 14 at 12 weeks, and 14 at 26 weeks (****p < 0.0001, two-way ANOVA).
(B) Latency to fall in the rotarod test at 26 weeks in WT (Cnp+/+;Lmnafl/fl; n = 18) and Lmna mutant mice (CnpCre/+;Lmnafl/fl; n = 13) tested on 3 consecutive days. Data represented as mean ± SD (***p < 0.001, ****p < 0.0001, two-way ANOVA).
(C) Representative electron microscopy images of the myelinated axons in the corpus callosum of P21 WT (Cnp+/+;Lmnafl/fl) and Lmna mutant mice (CnpCre/+; Lmnafl/fl). Scale bar, 1 μm.
(D) Scatterplot of g-ratios of myelinated axons presented as a function of axon diameter in WT (Cnp+/+;Lmnafl/fl) and Lmna mutant (CnpCre/+;Lmnafl/fl) P21 mice. Totals of n = 432 axons in 5 WT and 398 axons in 5 Lmna mutant mice were measured (WT, slope = 0.122 ± 0.014, intercept = 0.726 ± 0.009; cKO, slope = 0.120 ± 0.012, intercept = 0.745 ± 0.008).
(E) Scatterplot of the average ± SD g-ratio in the corpus callosum of WT and Lmna mutant P21 mice; n = 5 mice per genotype (p ≥ 0.05, two-tailed Student’s t test).
(F) Representative confocal images of the corpus callosum of WT (Cnp+/+;Lmnafl/fl) and Lmna mutant mice (CnpCre/+;Lmnafl/fl). Sections were stained with OLIG2 (green) and CC1 (red) antibodies. DAPI (blue) was used as counterstain. Scale bar, 20 μm.
(G) Scatterplot of the number of CC1+/OLIG2+ cells per mm2 corpus callosum in WT (Cnp+/+;Lmnafl/fl, n = 4) and Lmna mutant (CnpCre/+;Lmnafl/fl, n = 3) mice. Data are represented as average ± SD (p ≥ 0.05, two-tailed Student’s t test).
(H) Western blot of protein extracts from spinal cord lysates of WT (Cnp+/+;Lmnafl/fl, n = 2) and Lmna mutant mice (CnpCre/+;Lmnafl/fl, n = 2), probed for the myelin proteins indicated on the left. Molecular weights indicated on the right. β-Actin was used as loading control.
Altered chromatin accessibility and transcriptome in myelinating oligodendrocytes in the brain of 8-week-old mice lacking the Lmna gene
As nuclear lamins anchor chromatin to the nuclear periphery, we reasoned that the absence of LMNA/C may impact the pattern of heterochromatin distribution in the nuclei of myelinating oligodendrocytes. We therefore measured the distribution of heterochromatic areas in the nuclei of myelinating oligodendrocytes in Lmna mutants and controls (Figure S5). The detection of decreased peripheral heterochromatin in Lmna-null myelinating oligodendrocytes prompted us to identify the genomic regions with greater chromatin accessibility in these cells, using an assay for transposase-accessible chromatin using sequencing (ATAC-seq). Briefly, the reporter Ndrg1-EGFP mice, which express EGFP in the soma of myelinating oligodendrocytes,75 were bred with CnpCre/+;Lmnafl/fl mice in order to obtain triple mutants expressing EGFP from myelinating oligodendrocytes and either expressing or lacking Lmna. EGFP+ myelinating oligodendrocytes were then sorted from the 8-week-old control and Lmna mutant brains by fluorescence-activated cell sorting (FACS)75 and used to compare the pattern of chromatin accessibility in myelinating oligodendrocytes expressing or not expressing Lmna. The ATAC-seq assay identified a total of 90,497 peaks of open chromatin, of which 5,838 were differentially accessible (false discovery rate [FDR] < 0.05) between control (Ndrg1-EGFP;Cnp+/+; Lmnafl/fl) and Lmna mutant (Ndrg1-EGFP;Cnp-Cre/+;Lmnafl/fl) mice (Figure 4A). Among these peaks, we focused on the 3,914 peaks that were more accessible in the Lmna-null myelinating oligodendrocytes, with statistically significant differences between the two genotypes (FDR < 0.05, log2 fold change [FC] ≥ 0.25; Figure 4A). Among the genes with more accessible chromatin regions in the mutant cells, we detected the cell-surface glycoprotein Cd44, involved in migration and cell adhesion; the transcription factor Srebf1, which regulates several genes involved in myelin lipid biosynthesis; the metallopeptidase Adam11; the voltage-dependent calcium channel subunit Cacna1h; and several epigenomic modulators such as Cbx7, Setd2, and Dnmt3a (Figure 4B). The regions of higher chromatin accessibility in the mutant nuclei were mostly detected at promoter regions (77%), followed by regulatory distal intergenic and intronic regions (Figure 4C). Gene Ontology analysis of categories associated with genes with more accessible chromatin at promoters highlighted biological processes such as transcription, chromatin organization, and even cell cycle (Figure 4D). Accessible regions were also visualized using the WashU Epigenome Browser76 to show examples of differentially accessible genomic regions between mutants and controls (Figure 4E). The transcriptional consequences of LMNA/C loss in myelinating oligodendrocytes were then evaluated by RNA sequencing analysis of samples isolated from mature cells sorted from reporter mice either expressing (Ndrg1-EGFP;Cnp+/+;Lmnafl/fl) or not expressing Lmna (Ndrg1-EGFP;CnpCre/+;Lmnafl/fl). Differential expression analysis between myelinating oligodendrocytes from the two genotypes (p < 0.05) revealed 668 upregulated and 538 downregulated transcripts in the mutant oligodendrocytes compared with controls (Figure 5A). Genes characteristic of the progenitor state and involved in migration, the PDGFR signaling pathway, and modulation of synaptic transmission were found to be upregulated and expressed in mutant myelinating oligodendrocytes lacking Lmna (Figure 5B; Table S1). The transcript levels of genes with increased chromatin accessibility, like Cd44 (Figure 5C), were further validated by real-time qPCR (Figure 5D) as well as immunohistochemical analysis (Figures 5E and 5F). However, of the 2,626 genes with chromatin accessible at promoter regions, only 10% resulted in transcriptional changes (Figure S6A), thereby raising questions related to the remaining 2,365 genes. We first asked whether those genes overlapped with previous datasets of genes regulated by repressive histone methylation or DNA modifications in the oligodendrocyte lineage, which identified a subset of genes regulated by these epigenetic modifications (Figure S6B). We then conducted a chromatin immunoprecipitation enrichment analysis (ChEA) to define putative regulatory transcription factors in common among the 2,365 genes with accessible chromatin at the promoter and no transcriptional changes.
Figure 4. Loss of Lmna in mature OLs increases chromatin accessibility in promoter regions of genes involved in transcription and chromatin organization.
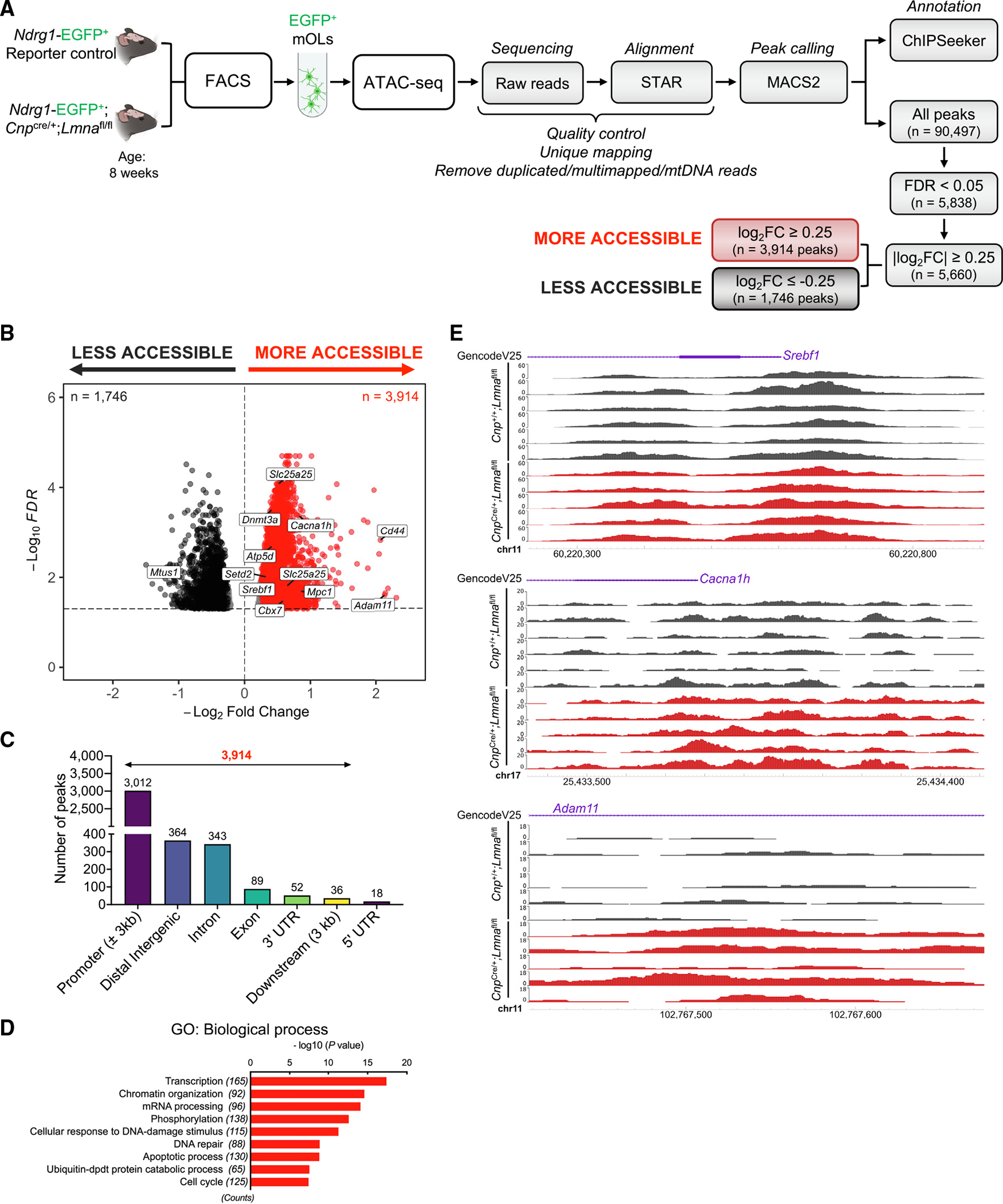
(A) Experimental design and analysis workflow for ATAC-seq on FACS myelinating OLs (mOLs) from the brain of 8-week-old Lmna mutant (Ndrg1-EGFP;CnpCre/+; Lmnafl/fl; n = 5) and control mice (Ndrg1-EGFP;Cnp+/+;Lmnafl/fl; n = 6).
(B) Volcano plot of differential ATAC-seq peaks (FDR < 0.05). Red dots represent more accessible peaks in mutant mOLs compared with controls. Black dots represent less accessible peaks in mutant cells (FDR < 0.05 and |log2 FC| ≥ 0.25), with the dotted line representing threshold for significance.
(C) Bar graph showing genomic distribution of more accessible chromatin peaks (n = 3,914; FDR < 0.05 and |log2 FC| ≥ 0.25) in Lmna mutant mOLs compared with control cells.
(D) Gene Ontology (GO) analysis of the unique genes (n = 2,626) with more accessible chromatin peaks at promoters in Lmna mutant OLs (n = 3,012; FDR < 0.05 and log2 FC ≥ 0.25).
(E) Visualization of ATAC-seq peaks in control mOLs (n = 6; black) and Lmna mutant mOLs (n = 5; red) at the indicated genomic regions. Data were visualized using the WashU Epigenome Browser. The scale on the y axis was selected for optimal visualization of peaks.
See also Figures S5 and S6.
Figure 5. Altered transcriptome in mOLs lacking Lmna.
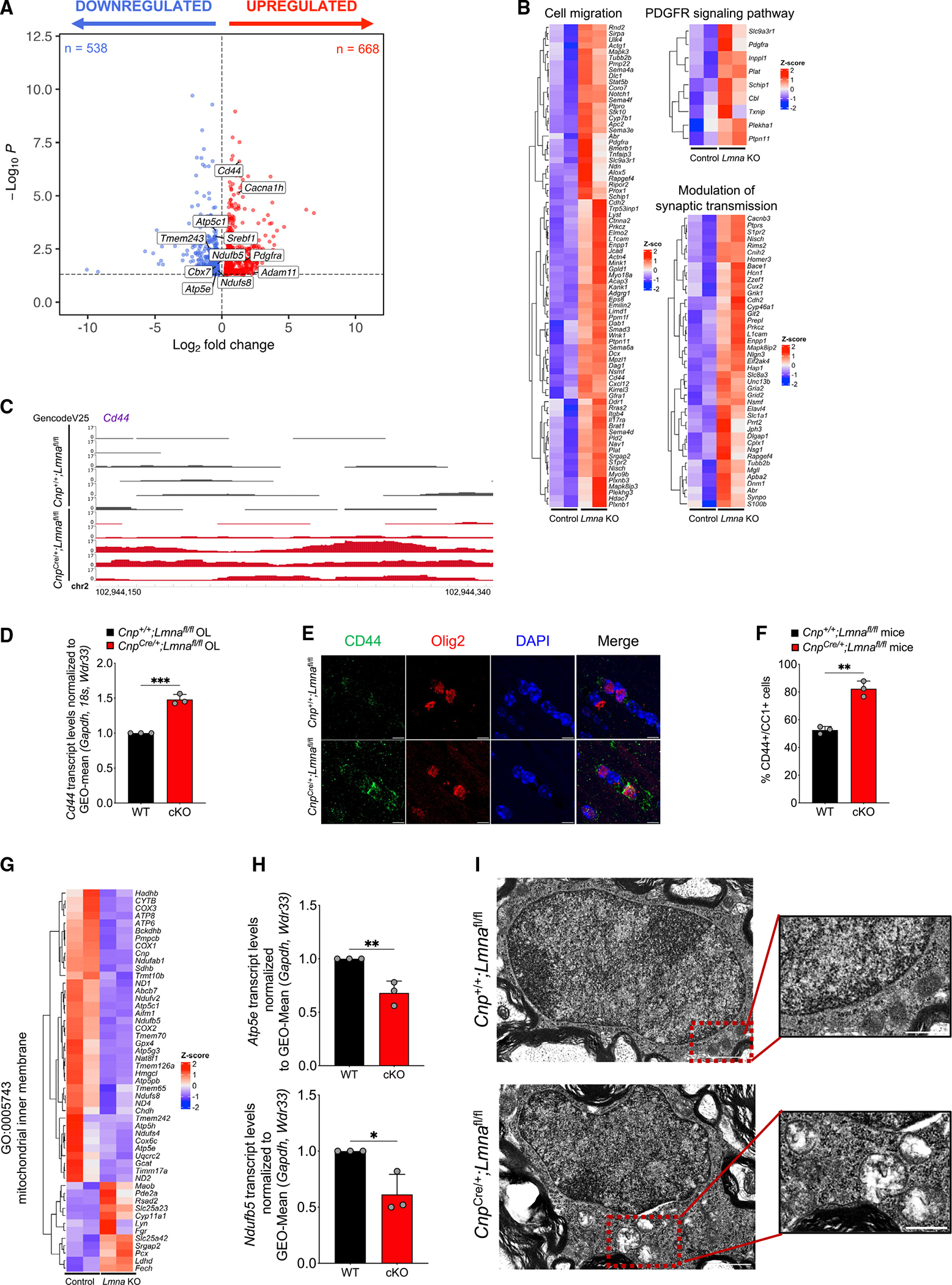
(B) Volcano plot of differentially expressed genes (p < 0.05) in FACS mOLs isolated from 8-week-old Lmna mutant mice (Ndrg1-EGFP;CnpCre/+;Lmnafl/fl, n = 3) and control mice (Ndrg1-EGFP;Cnp+/+;Lmnafl/fl, n = 3). After quality control, the following samples were used for differential expression analysis: n = 2 Lmna mutant and n = 2 control samples. Red dots represent significantly upregulated genes (n = 668), and blue dots represent significantly downregulated genes (n = 538).
(C) Gene expression heatmaps of upregulated biological process Gene Ontology (GO) terms in significantly upregulated (red) or downregulated (blue) genes in Lmna mutant mOLs (Ndrg1-EGFP;CnpCre/+;Lmnafl/fl) compared with control cells (Ndrg1-EGFP;Cnp+/+;Lmnafl/fl). Genes annotated for the GO terms “cell migration,” “PDGFR signaling pathway,” and “modulation of synaptic transmission” are shown.
(C) Visualization of ATAC-seq peaks in control (n = 6, black) and Lmna mutant (n = 5, red) OLs at the distal intergenic region of Cd44. Data were visualized using the WashU Epigenome Browser. The scale on the y axis was selected for optimal visualization of peaks.
(D) Scatterplot of Cd44 transcript levels measured by real-time qPCR in RNA samples extracted from cultured OPCs isolated from WT (Cnp+/+;Lmnafl/fl) and Lmna mutant mice (CnpCre/+;Lmnafl/fl) and differentiated in vitro. Data were normalized to the geometric (GEO)-mean of Gapdh, 18S, and Wdr33 transcript levels. Data represent average levels ± SD, n = 3 independent cultures per group (***p < 0.001, two-tailed Student’s t test).
(E) Representative confocal images of the corpus callosum of WT (Cnp+/+;Lmnafl/fl) and Lmna mutant 26-week-old mice (CnpCre/+;Lmnafl/fl). Sections were stained with CD44 (green) and OLIG2 (red). DAPI (blue) was used as counterstain. Scale bar, 5 μm.
(F) Scatterplot of the number of CD44+/CC1+ cells per mm2 corpus callosum in control mice (Cnp+/+;Lmnafl/fl, n = 3) and Lmna mutant mice (CnpCre/+;Lmnafl/fl, n = 3). Data represent average percentages ± SD (**p < 0.01, two-tailed Student’s t test).
(G) Heatmap of the transcript levels within the “mitochondrial inner membrane” GO category, showing downregulation (blue) in sorted mOLs lacking Lmna (Ndrg1-EGFP;CnpCre/+;Lmnafl/fl) compared with control cells (Ndrg1-EGFP;Cnp+/+;Lmnafl/fl).
(H) Scatterplot of the mitochondrial transcript Atp5e (C) and Ndufb5 (D) levels measured by real-time qPCR in RNA samples extracted from Lmna mutant (CnpCre/+; Lmnafl/fl) and control (Cnp+/+;Lmnafl/fl) OLs, differentiated in cultures from neonatal OPCs. Data were normalized to the geometric (GEO)-mean of Gapdh and Wdr33 transcript levels. Data are represented as mean ± SD, n = 3 independent cultures per group (*p < 0.05, **p < 0.01, two-tailed Student’s t test).
(I) Electron microscopy image of OLs in the ventral horn of the spinal cord of Lmna mutant (CnpCre/+;Lmnafl/fl) and control (Cnp+/+;Lmnafl/fl) mice. Scale bar, 1 μm. The insets represent higher-magnification images of the mitochondrial structure in OLs from mice of the two genotypes to highlight the loss of internal cristae definition in mOLs lacking LMNA. Scale bar, 0.5 μm.
Besides being targets for the oligodendrocyte lineage-specific OLIG2, we noted that most of the genes were also regulated by multiple transcription factors (Figure S6C) including MYC, TBX2, RARB, and HAND2, which are either not expressed or are expressed at very low levels in myelinating oligodendrocytes (Figure S6D) as shown in a published dataset.77 Therefore, it is conceivable that despite a higher chromatin accessibility at promoter regions in the absence of Lmna, associated genes may not be expressed due to the presence of repressive epigenetic marks or the absence of regulatory transcription factors. Collectively, these data identify changes in chromatin accessibility and increased transcript levels of stage-inappropriate genes in myelinating oligodendrocytes lacking LMNA/C.
Transcriptional and morphological alterations of inner mitochondrial membrane in Lmna mutant oligodendrocytes
Gene Ontology analysis of transcripts downregulated in Lmna mutant myelinating oligodendrocytes (directly sorted from the brain) highlighted mitochondrial inner membrane and myelin sheath as the top Gene Ontology categories related to cellular components (Figure 5G; Table S2). The downregulation of genes of the mitochondria inner membrane including Atp5e and Ndufb5 in mutant oligodendrocytes was further validated by real-time qPCR (Figure 5H) and prompted us to ask whether mitochondria integrity was affected in older mice. Electron microscopic analysis of myelinating oligodendrocytes in the ventral horn of the spinal cord of 26-week-old Lmna mutant mice compared with controls revealed the presence of severe morphological changes and disrupted mitochondria cristae (Figure 5I). Together with the transcriptional changes detected in myelinating oligodendrocytes lacking Lmna, the identified mitochondrial alterations prompted us to ask whether potential metabolic consequences could be related to the progressive motor phenotype detected in mutant mice. An untargeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) metabolomic analysis of 26-week-old Lmna mutant and control brains (Figure 6A) identified 99 metabolites with differential abundance in Lmna mutants (Figure 6B). Among the 56 upregulated ones, we detected several metabolites associated with the inner mitochondrial membrane, including those related to fatty acid β-oxidation (such as acetylcarnitine) and glutamine metabolism (e.g., D-glutamine and Nα-acetyl-L-glutamine) (Figure 6C). We further visualized the increased levels of acetylcarnitine and D-glutamine, and the lower levels of 5-aminovaleric acid betaine, using MALDI imaging (Figure 6D). Together, these data suggest that mitochondrial pathology and metabolic disruptions occur in vivo in myelinating cells that lack Lmna throughout the brain.
Figure 6. Unbiased metabolomic analysis of Lmna mutant brains reveals several metabolic alterations compared with controls.
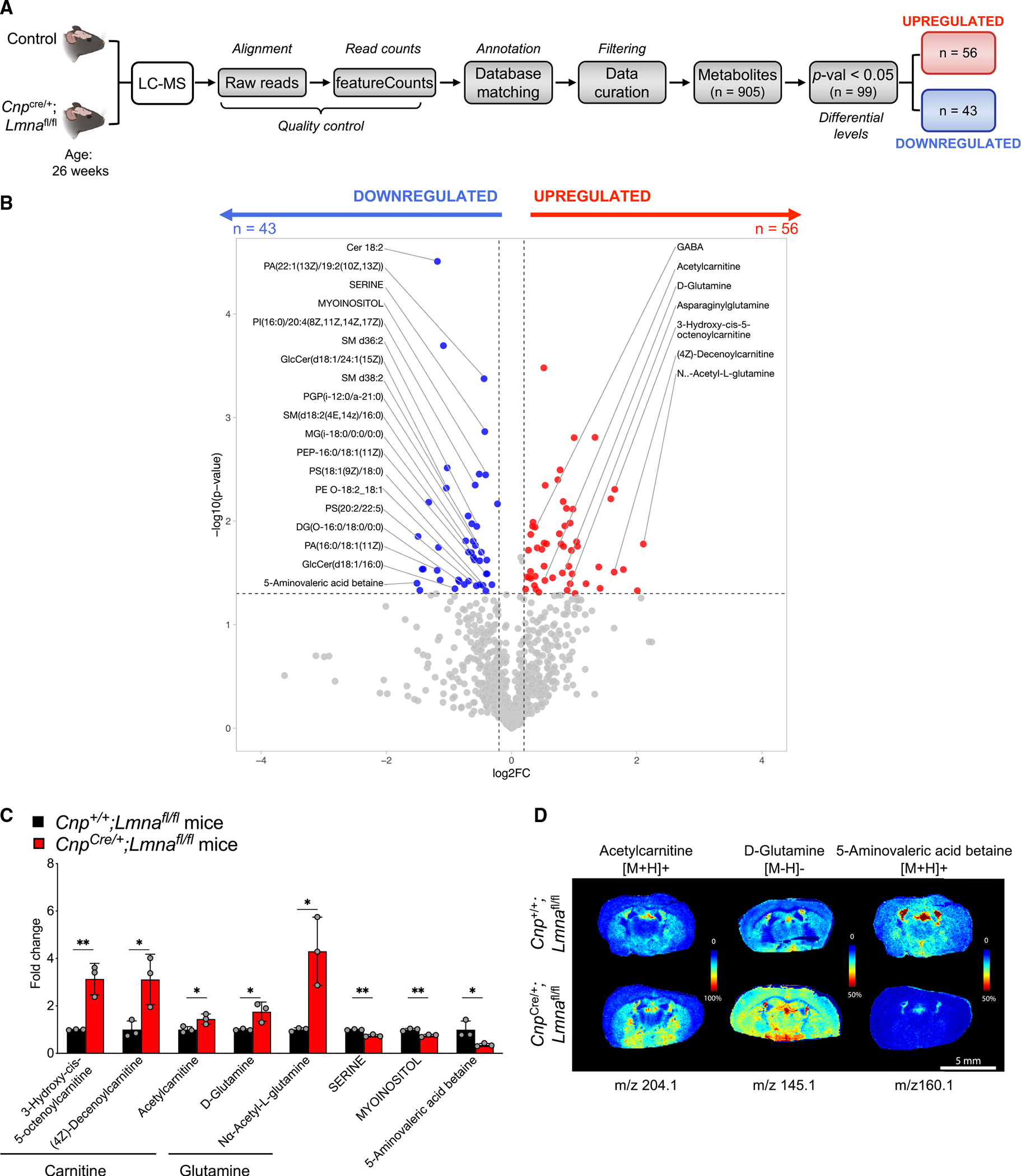
(A) Experimental design and analysis workflow for LC-MS on the brain of Lmna mutant mice (CnpCre/+;Lmnafl/fl; n = 3) compared with the brain of control mice (n = 3).
(B) Volcano plot of metabolites with differential levels in Lmna mutant brains compared with controls (p < 0.05). Red dots represent significantly upregulated metabolites (n = 56), Blue dots represent significantly downregulated metabolites (n = 46).
(C) Scatterplot of the fold change of selected metabolite levels in Lmna mutant (CnpCre/+;Lmnafl/fl) brains relative to the levels detected in control brains. Data represented as mean ± SD (n = 3 mice per group; *p < 0.05, **p < 0.01, Student’s t test).
(D) Representative MALDI imaging of selected metabolites with the indicated m/z in Lmna mutant (CnpCre/+;Lmnafl/fl) and control brains. Scale bar, 5 mm.
Lipid dysregulation and myelin thinning in older mice with Lmna ablation in myelinating oligodendrocytes
Among the 43 downregulated metabolites, we detected several lipid species, a finding that was consistent with the increased chromatin accessibility and decreased transcript levels of the sterol transcription factor Srebf1 in Lmna-null myelinating oligodendrocytes (Figure 7A). The lipids with significantly lower abundance in the Lmna mutant mice compared with controls included several sphingomyelins, ceramides, phosphatidylserines, and phosphatidic acids (Figure 7B). Decreased lipid content throughout the brain was also validated using MALDI imaging (Figure 7C). Based on these findings, it was not surprising to notice that, despite the similar percentage of myelinated axons (Figure S7), only in the Lmna mutants could we detect axons with thinner myelin both in the corpus callosum (Figures 7D–7F) and the spinal cord (Figures 7G–7I). Importantly, at the same time point, when the motor clinical phenotype was quite pronounced and we detected myelin alterations in the CNS, we did not detect significant changes in the peripheral sciatic nerve or in the skeletal muscle (Figure S7). Overall, these data suggest that the absence of LMNA/C in myelinating glia profoundly impacts the transcriptional and structural stability of myelinating oligodendrocytes, which show aberrant mitochondria ultrastructure, disrupted lipid metabolism, thinning of myelin, and associated motor deficits.
Figure 7. Altered lipid profile and late dysmyelination in Lmna mutants.
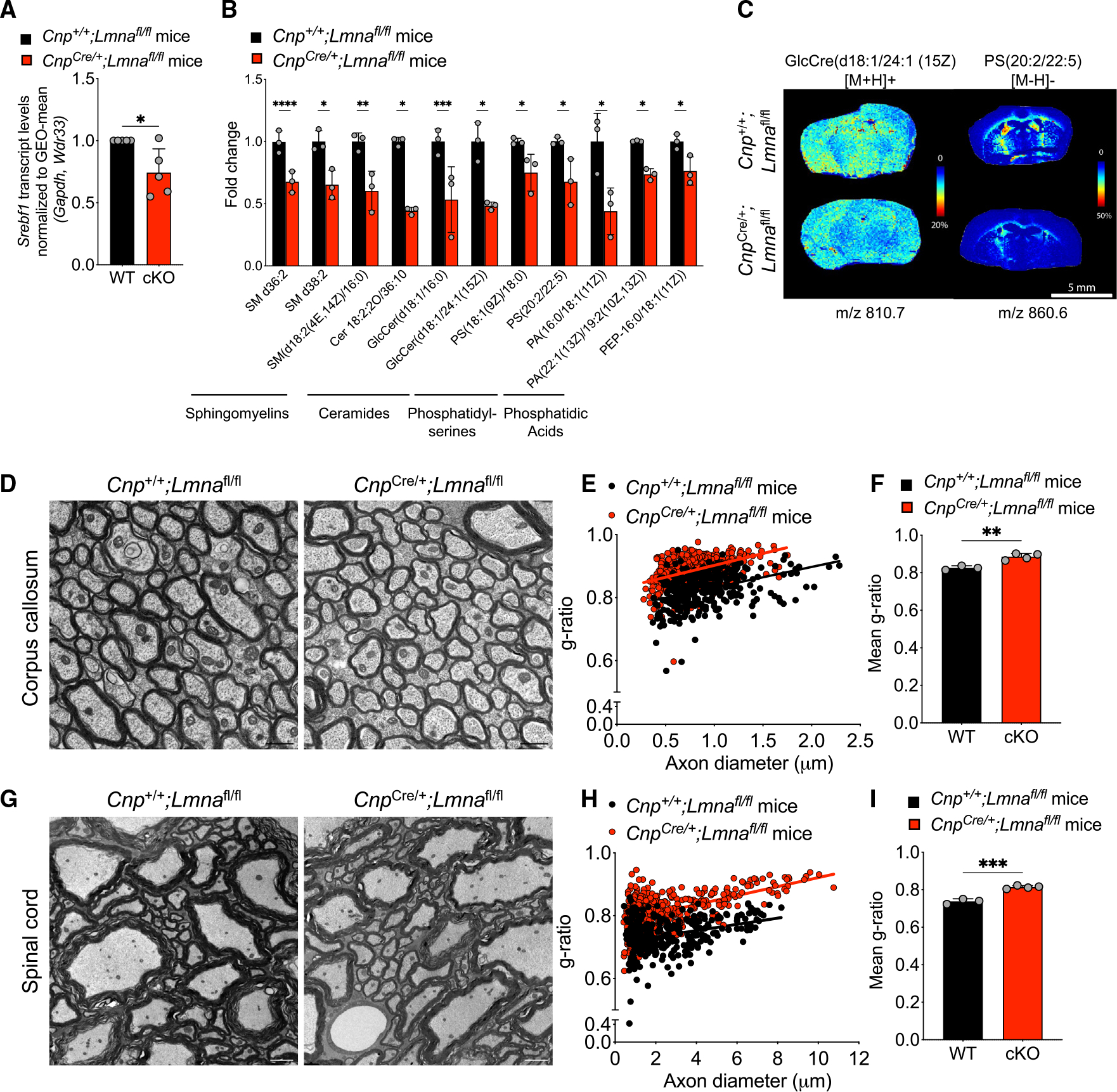
(A) Scatterplot of Srebf1 transcript levels measured by real-time qPCR in RNA samples extracted from cultured OL progenitors isolated from WT (Cnp+/+;Lmnafl/fl) and Lmna mutant mice (CnpCre/+;Lmnafl/fl) and differentiated in vitro, relative to WT. Data were normalized to the GEO-mean of Gapdh and Wdr33 transcript levels. Data represent the average ± SD, n = 5 independent cultures per group (*p < 0.05, two-tailed Student’s t test).
(B) Scatterplot of the fold change differences in selected lipids in the brain of Lmna mutants (CnpCre/+;Lmnafl/fl) relative to their levels in control brains, as resulting from the LC-MS analysis. Data represented as mean ± SD (n = 3 mice per group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001, Student’s t test).
(C) Representative MALDI imaging of Lmna mutant and control brain sections. Shown in pseudocolor is the relative abundance of selected lipids detected with 2,5-dihydroxybenzoic acid (DHB) matrix (GlcCre(d18:1/24:1 (15Z)) and N-(1-naphthyl) ethylenediamine dihydrochloride (NEDC) matrix (PS(20:2(11Z,14Z)/22:5(4Z,7Z,10Z,13Z,16Z)), abbreviated PS(20:2/22:5)), at the indicated m/z.
(D) Representative electron microscopy images of the corpus callosum of 26-week-old WT and Lmna mutant mice. Scale bar, 1 μm.
(E) Scatterplot of g-ratios relative to the diameter of myelinated axons in the corpus callosum of WT and Lmna mutant mice. Totals of n = 338 axons in 3 WT mice and 462 axons were measured in 4 Lmna mutant mice (WT, slope = 0.065 ± 0.008, intercept = 0.767 ± 0.008; cKO, slope = 0.075 ± 0.007, intercept = 0.827 ± 0.006).
(F) Scatterplot of the average g-ratio ± SD of myelinated axons in the corpus callosum of WT (n = 3) and Lmna mutant mice (n = 4). (**p < 0.01, two-tailed Student’s t test).
(G) Representative electron microscopy images of the spinal cord of 26-week-old WT and Lmna mutant mice. Scale bar, 2 μm.
(H) Scatterplot of g-ratios relative to the diameter of myelinated axons in the spinal cord of WT and Lmna mutant mice. Totals of n = 320 axons in 3 WT mice and 421 axons were measured in 4 Lmna mutant mice (WT, slope = 0.010 ± 0.002, intercept = 0.713 ± 0.005; cKO, slope = 0.014 ± 0.001, intercept = 0.778 ± 0.004).
(I) Scatterplot of the average g-ratio ± SD of myelinated axons in the spinal cord of WT (n = 3) and Lmna mutant mice (n = 4). Data are represented as mean +SD (***p < 0.001, two-tailed Student’s t test).
See also Figure S7.
DISCUSSION
Myelinating oligodendrocytes are highly specialized cells whose role is to guarantee metabolic support of neurons and electrical insulation of axons5 in order to allow rapid and accurate transmission of neural impulses. These cells differentiate from proliferating and migratory progenitors via a complex series of events, regulated by the interaction between transcription factors and epigenomic modulators6–9,78 and the formation of heterochromatin and its recruitment to the nuclear periphery. The nuclear periphery comprises the nuclear envelope and the nuclear lamina at the interface with heterochromatin. The lamina is composed of a meshwork of intermediate filament proteins, including LMNA/C, and LMNB.37,39 We show here that LMNB1 expression is high in oligodendroglial proliferating progenitors, characterized by nuclear euchromatin, and declines with differentiation. In contrast, LMNA/C levels are low in progenitors and increase as cells differentiate into myelinating oligodendrocytes, which are characterized by repressive heterochromatin at the nuclear periphery and progressively stiffer and more rigid nuclei.79 This expression pattern is conserved across species43–46 and is functionally important for myelinating glia of the CNS, as altered levels of LMNB1 have been previously linked to myelin disorders in humans58–62,80 and mice.64–66 Duplications of the Lmnb1 gene or alterations of its regulatory elements in human subjects result in a late-onset neurological disorder characterized by severe myelin damage and called autosomal dominant leukodystrophy.58–62,80 An altered ratio between nuclear lamins, such as in transgenic mice overexpressing Lmnb1,64–66 also results in a late-onset demyelinating pathology, characterized by vacuolar degeneration of myelin and decreased transcripts for myelin proteins64 and enzymes regulating lipid metabolism.66
We therefore hypothesized that the expression of the correct proportion of the two lamins at distinct stages of differentiation is important for the function of myelinating cells of the CNS. In this article, we tested the consequences of decreasing the levels of LMNA/C in myelinating oligodendrocytes using the Cnp-Cre line, which targets myelinating glia in both the CNS and the PNS and deletes Lmna in both oligodendrocytes and Schwann cells.
Starting at 8–12 weeks of age, mice with Lmna deletion in myelinating glia developed a mild motor phenotype characterized by the inability to grip and hold on to a grid, followed by developing onset of tremors and ataxia, which progressively worsened with age.
We cannot formally exclude the possibility that the age-dependent severity of the phenotype could be in part consequent to the single copy of Cnp in mutants due to the use of the knockin Cnp-Cre line.74 However, we note that the motor phenotype of the Lmna mutants was quite distinct from the catatonic phenotype previously described for the aged Cnp-Cre heterozygous mice, which were characterized by normal grip strength and prolonged hold to a rod, in the absence of motor signs.81 Importantly, we did not detect significant alterations in peripheral myelin. This led to the conclusion that loss of Lmna differentially impacts central (i.e., oligodendrocytes) and peripheral (i.e., Schwann cells) myelinating glia. These differences could be attributed to the unique functional features of the two myelinating cell types, with oligodendrocytes being post-mitotic stationary cells5 with a very stable transcriptome and the ability to wrap multiple axons with their myelin membrane and Schwann cells being more plastic, wrapping a single axon and retaining the ability to dedifferentiate and migrate in response to injury.82,83 Differences could also be related to the distinct expression patterns of the nuclear lamins detected in the two cell types. Although both oligodendrocytes and Schwann cells express LMNA/C, only central myelinating glia display a characteristic inverse relationship with LMNB1 levels, while Schwann cells have constant levels of both lamins regardless of whether they are proliferating or differentiating. Based on these considerations and the clinical phenotype suggestive of myelinating glia dysfunction (e.g., tremors, ataxia), we focused the study on a thorough characterization of the role of LMNA/C in regulating the epigenome and transcriptome of mature oligodendrocytes.
At a molecular level, we attribute the phenotype to the progressive thinning of the myelin sheath detected in Lmna mutants over time. We propose that decreased lipid abundance, consequent to transcriptional changes, and altered use of energy in myelinating oligodendrocyte, due to misexpression of progenitor-stage-related genes, as the consequence of increased chromatin accessibility, may account for the progressively worsening dysfunction of these cells. In other words, we propose that the transcriptional control of cell identity resulting in highly specialized functional states is challenged by the loss of LMNA/C in mature oligodendrocytes. We further postulate that the heterochronic expression of progenitor-related genes in myelinating oligodendrocytes lacking Lmna may induce mutant cells to redirect energy demands toward transcription and synthesis of proteins that are not needed for the differentiated state. For instance, the expression of the migration-related genes and proteins (e.g., CD4484) in myelinating oligodendrocytes is not compatible with their stationary state and functional specialization. At the same time, decreased LMNA/C levels in myelinating oligodendrocytes result in increased chromatin accessibility and decreased expression of genes related to the inner mitochondrial membrane85 as well as key sterol-binding transcription factors responsible for lipid synthesis.86,87 As a result, the myelinating oligodendrocytes may respond to the increased energy demands (needed for the synthesis of molecules unrelated to their functionally specialized state) by utilizing lipids as an energy source. The decreased expression of sterol-binding factor Srebf1 further impairs synthesis of myelin lipids, as detected by the metabolomic analysis, and eventually leads to myelin thinning, especially evident in large-caliber motor axons, which are also the ones with higher energy demands.
This mechanism is distinct from the one previously described for LMNB1 overexpression in oligodendrocytes in which genomic regions encoding for essential cholesterol metabolism enzymes remained silenced and attached to the periphery and then were released as progenitors differentiated.56 Importantly, in the Lmna mutant oligodendrocytes, not all chromatin regions that become accessible also became transcriptionally active. In part, this discrepancy between chromatin accessibility and transcriptional output could be due to the fact that the transcription factors responsible for activation of the regions of increased accessibility (e.g., MYC) are no longer expressed in the differentiated state. However, it is also conceivable that the absence of LMNA/C may impact the transcriptional activity of associated LAP2-emerin-MAN1 (LEM) domain proteins. It has been previously reported that besides chromatin tethering to the nuclear periphery,88 LEM proteins also serve an important role in transcriptional activation.89,90 As such, it will be important, in the future, to determine how these LEM proteins affect the transcriptome of oligodendrocytes.
In summary, this study characterized the effects of Lmna gene deletion in myelinating glia. It reveals that the absence of LMNA/C is compatible with normal development and differentiation of oligodendrocytes and Schwann cells, likely because the transcriptional networks responsible for differentiation and the related epigenomic landscape responsible for heterochromatin formation are not affected. However, as the animals age and reach adulthood, tethering of heterochromatin to the nuclear lamina is compromised. In the absence of an anchoring point for repressed genomic regions and altered association with binding proteins, the stability of the transcriptome of myelinating oligodendrocytes is jeopardized. Genes characteristic of the progenitor stage start being re-expressed, and those related to the inner mitochondrial membrane and to the synthesis of lipid components decline over time. This leads to an increased energy demand for the synthesis of molecules incompatible with the terminally differentiated specialized phenotype and decreased lipids, resulting in myelin thinning and clinical signs late in life.
Overall, our findings are consistent with the notion that the role of LMNA/C is cell specific. In myelinating oligodendrocytes, the balanced reciprocal expression of LMNB1 in progenitors and LMNA/C in fully specialized myelinating cells is responsible for the transcriptional and functional stability of these cells. The distinct phenotype detected in the CNS and the PNS also opens important questions regarding the specificity of LMNA/C’s role in central and peripheral myelinating glia. Future studies will be needed to clarify these aspects and define the role of LEM proteins in modulating the terminally differentiated phenotype.
Limitations of the study
A potential limitation of this study is that we used the Cnp-Cre mouse line in which Lmna is deleted in the myelinating cells of both the PNS and the CNS and in which KO cells contain a single copy of Cnp, thereby potentially contributing to the severity of the phenotype. Since we detect a different pattern of lamin expression of LMNA/C and LMNB1 between the CNS and the PNS, it would be interesting to determine what genes are regulated by LMNA/C in the Schwann cells and if they relate to the differences in plasticity of the two cell types since only Schwann cells have the ability to dedifferentiate and proliferate. A second consideration is that the loss of LMNA/C may impact the function of lamina-associated transcriptional regulators, such as LEM proteins, which could be the subject of important follow-up studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Patrizia Casaccia (pcasaccia@gc.cuny.edu).
Materials availability
Mouse lines used in this study are subject to MTA from the original investigator.
Data and code availability
RNA-Seq and ATAC-Seq data have been deposited at NCBI’s Gene Expression Omnibus (GEO)91 and are accessible through GEO: GSE222574,GSE190404 respectively. Metabolomics data have been deposited at NIH Common Fund’s National Metabolomics Data Repository (NMDR) Website, the Metabolomics Workbench,114 https://www.metabolomicsworkbench.org where it has been assigned Study ID ST002739. The data can be accessed directly via its Project DOI: 10.21228/M8FM85. RNA-seq, ATAC-seq and Metabolomics data are publicly available as of the date of publication. All data reported in this study will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All breeding and experiments were performed according to approved protocols by the Institutional Animal Care and Use Committee (IACUC) at Mount Sinai Medical Center and at the Advanced Science Research Center (ASRC) of the Graduate Center of The City University of New York (CUNY). Animals were housed in a temperature- and humidity-controlled facility on a 12-h light/dark cycle with ad libitum access to food and water. Animals from either sex were used. Non-transgenic mice (C57BL/6J, RRID:IMSR_JAX:000664) were purchased from the Jackson Laboratory. To generate conditional knockout mouse lines, we crossed the Lmna-flox line (Gift from Dr. Colin L. Stewart44) with the Cnp1-Cre line (MGI:3051635, Gift from Dr. Klaus-Armin Nave74) or with the CamK2a-Cre line (RRID:IMSR_JAX:005359115). To isolate mature oligodendrocytes from the control and Lmna mutant (CnpCre/+;Lmnafl/fl) mice, mice were crossed with the reporter mouse line Ndrg1-EGFP mouse line, described in Marechal et al.75 Mice were used at different ages: postnatal day 1 (P1), P7, P14, P21, P60 and at 8 weeks and 26 weeks (26–30 weeks). Sample size (n) is indicated in each figure legend.
Human
Normal human brain specimens were obtained from the areas surrounding the surgical resection for intractable epilepsy. Surgical tissue was acquired from the Bartoli brain tumor bank at Columbia University Medical Center. All diagnoses were rendered by board-certified neuropathologists. Study protocols were approved by Columbia University Irving Medical Center Institutional Review Board. All clinical samples were de-identified prior to analysis. Analyses were carried out in alignment with the principles outlined in the WMA Declaration of Helsinki and the Department of Health and Human services Belmont Report. Informed written consents were provided by all patients. The demographics of the cases used are provided in Table S3.
Primary oligodendrocyte progenitor cultures
Oligodendrocyte progenitor cells were isolated from the brain of C57BL/6J, CnpCre/+;Lmnafl/fl and Lmnafl/fl mice at postnatal day 7. Both male and female pups were used. Briefly, forebrains brains were digested in papain buffer to obtain single cell suspension. Two negative immunopanning selections were performed using BSL1 (2.5 μg/mL, Vector Labs, L-1100) to deplete microglia. To isolate oligodendrocyte progenitors, a positive immunopanning selection was then performed using a rat anti-mouse CD140a antibody (0.75 μg/mL, BD Biosciences, 558774), binding to the Platelet-Derived Growth Factor Receptor alpha chain (PDGFR-α). Adherent cells were trypsinized and seeded on Poly-D-lysine (0.1 mg/mL, Sigma) coated dishes. Cells were cultured in tissue-culture incubator (37°C, 5% CO2) in SATO medium (Dulbecco’s modified Eagle’s medium, DMEM), 0.1 mg/mL bovine serum albumin (BSA, Sigma), 0.1 mg/mL apo-Transferrin (Sigma), 1.6 mg/mL putrescine (Sigma), 62 ng/mL progesterone (Sigma), 4 μg/mL sodium selenite (Sigma), 5 mg/mL insulin human (Sigma), 1 mM sodium pyruvate (Gibco, Thermo Fisher Scientific), 1% GlutaMAX Supplement (Gibco, Thermo Fisher Scientific), B27 supplement (Gibco, Thermo Fisher Scientific), 5 μg/mL N-acetyl-cysteine (Sigma), Trace Element B (Corning, Fisher), 10 μg/mL biotin (Sigma), 5 μM forskolin (Sigma) supplemented with the growth factors PDGF-AA (10 ng/mL, PeproTech, 100–13A) and basic fibroblast growth factor (bFGF; 20 ng/mL, Peprotech, 100–18B). To differentiate OPC, growth factors were removed and 3,3′,5-Triiodo-L-thyronine sodium salt (T3, 60 nM, Sigma, T5516) was added for 48 or 72 h.
Schwann cell cultures
Primary mouse Schwann cells were obtained using the method by Shan et al.116 with modifications. Sciatic nerves were dissected from CnpCre/+;Lmnafl/fl and Lmnafl/fl mice at postnatal day 7 (P7; male and female pups) then dissociated by enzymatic digestion (0.05% collagenase/dispase (Sigma) and 0.25% trypsin-EDTA (Gibco, Thermo Fisher Scientific)) and trituration. Cells were cultured on Poly-D-lysine (0.1 mg/mL, Sigma)/laminin (10 μg/mL, Sigma) coated dishes in a tissue-culture incubator (37°C, 5% CO2) with Schwann cell culture medium (SCCM: DMEM containing 10% Fetal Bovine Serum (VWR), 2 μM forskolin (Sigma), 10 ng/mL EGF domain of rhNRG-1-β1 (R&D systems) and 1% penicillin/streptomycin (Gibco, Thermo Fisher Scientific)). For Schwann cell enrichment, cells were treated with cytosine arabinoside (AraC, 10 mM, Sigma) for 48 h, then cultured in fresh SCCM for further cell growth.
Rat Schwann cells were obtained from sciatic nerves at postnatal day 2 (P2). Cultures were then expanded in vitro for 3 weeks on Poly-D-lysine (0.1 mg/mL, Sigma) coated dishes, in a tissue-culture incubator (37°C, 5% CO2) using M+ media (Minimum Essential Medium containing 10% Fetal Bovine Serum (VWR), 1% GlutaMAX Supplement (Gibco, Thermo Fisher Scientific), 4 μM forskolin (Sigma), 5 ng/mL EGF domain of rhNRG-1-β1 (R&D systems) and 1% penicillin/streptomycin) with fresh medium change every 2–3 days. Cells for Western blot and immunocytochemistry then seeded on Poly-D-lysine (0.1 mg/mL, Sigma)/laminin (10 μg/mL, Sigma) coated dishes or chamber slides. For differentiation, cells were cultured in M+ media supplemented with 1 mM dibutyryl-cAMP (db-cAMP) (Stemcell Technologies) for 72 h.
METHOD DETAILS
Immunohistochemistry (mouse tissues)
For immunohistochemistry, mice were deeply anesthetized with ketamine/xylazine (150 mg/kg and 15 mg/kg) and perfused with 4% paraformaldehyde (PFA). After tissue processing and paraffin embedding, 5–7 μm sections were cut using a microtome. Sections were de-paraffinized, immersed in 10 mM citrate buffer, pH 6.0, for 10 min in a microwave at 650 W. Sections were permeabilized and blocked in PGBA (0.1 M Phosphate buffer pH 7.4, 0.1% Gelatin porcine type A (Sigma), 1% BSA, and 0.002% Sodium Azide, 10% normal goat or donkey serum and 0.1% Triton X-100) at room temperature (RT) for 1 h. Sections were then incubated overnight at 4°C with primary antibodies (CC1, 1:50, Millipore MABC200; CD44, 1:250, Abcam ab157107; GFAP, 1:500, Millipore NE1015; LMNA/C, 1:200, Abcam ab169532; NeuN, 1:200, Millipore MAB377 OLIG2, 1:200, Millipore AB9610; OLIG2, 1:200, Millipore MABN50). After PBS washes, sections were incubated with Alexa Fluor secondary antibodies (1:500) for 2 h at RT then washed in PBS. Stained slides were mounted with DAPI Fluoromount G mounting medium (Thermo Fisher Scientific). Primary and secondary antibodies used are listed in the key resources table. Confocal images were captured using the Zeiss LSM800 Fluorescence Microscope.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-α-Tubulin (WB) | Sigma | Cat#CP06; RRID:AB_2617116 |
| Mouse monoclonal anti-APC, clone CC1 (IHC Human) | Millipore | Cat#OP80; RRID:AB_2057371 |
| Mouse monoclonal anti-APC, clone CC1 (IHC) | Millipore | Cat#MABC200; RRID:AB_11203645 |
| Mouse monoclonal anti-β-Actin (WB) | Sigma | Cat#A4700; RRID:AB_476730 |
| Rat monoclonal anti-mouse CD140a (Cells) | BD Biosciences | Cat#558774; RRID:AB_397117 |
| Rabbit polyclonal anti-CD44 (IHC) | Abcam | Cat#ab157107; RRID:AB_2847859 |
| Mouse monoclonal anti-GAPDH (WB) | Abcam | Cat#ab8245; RRID:AB_2107448 |
| Mouse monoclonal anti-GFAP (IHC) | Millipore | Cat# NE1015; RRID:AB_10679796 |
| Rabbit polyclonal anti-Lamin A (IHC Human) | Abcam | Cat#ab226198; RRID:AB_2927652 |
| Mouse monoclonal anti-Lamin A/C (WB) | Active Motif | Cat#39287; RRID:AB_2793218 |
| Goat polyclonal anti-Lamin A/C (ICC) | Santa Cruz | Cat#sc-6215; RRID:AB_648152 |
| Rabbit polyclonal anti-Lamin A (ICC) | Abcam | Cat#ab26300; RRID:AB_775965 |
| Rabbit monoclonal anti-Lamin A/C (IHC) | Abcam | Cat#ab169532; RRID:AB_2927696 |
| Rabbit polyclonal anti-Lamin B1 (IHC, WB) | Abcam | Cat#ab16048; RRID:AB_443298 |
| Mouse monoclonal anti-Lamin B1 (ICC) | Santa Cruz | Cat#sc-374015; RRID:AB_10947408 |
| Rabbit polyclonal anti-MBP (WB) | Abcam | Cat#ab40390; RRID:AB_1141521 |
| Rat monoclonal anti-MBP (IHC) | Millipore | Cat#MAB386; RRID:AB_94975 |
| Mouse monoclonal anti-MOG clone Z12 (WB) | Gift from Prof. Richard Reynolds | N/A |
| Mouse monoclonal anti-NeuN (IHC) | Millipore | Cat#MAB377; RRID:AB_2298772 |
| Mouse monoclonal anti-NG2 (IHC Human) | Millipore | Cat#MAB5384; RRID:AB_177646 |
| Mouse monoclonal anti-Olig2 (ICC, IHC) | Millipore | Cat#MABN50; RRID:AB_10807410 |
| Rabbit polyclonal anti-Olig2 (IHC) | Millipore | Cat#AB9610; RRID:AB_570666 |
| Rabbit polyclonal antibody PLP/DM20 A431 (WB) | Gift from Dr. Klaus-Armin Nave | N/A |
| Rabbit monoclonal anti-SOX10 (ICC) | Cell Signaling | Cat#89356; RRID:AB_2792980 |
| Goat anti-mouse IgG (H + L; WB) | Jackson ImmunoResearch | Cat#115–035-166; RRID:AB_2338511 |
| Goat anti-rabbit IgG (H + L; WB) | Jackson ImmunoResearch | Cat#111–035-144; RRID:AB_2307391 |
| Goat anti-rat Alexa Fluor 647(ICC, IHC) | Thermo Fisher Scientific | Cat#A-21247; RRID:AB_141778 |
| Goat anti-mouse Alexa Fluor 546 (ICC, IHC) | Thermo Fisher Scientific | Cat#A-11030; RRID:AB_2534089 |
| Goat anti-rabbit Alexa Fluor 488 (ICC, IHC) | Thermo Fisher Scientific | Cat#A-11034; RRID:AB_2576217 |
| Donkey anti-goat Alexa Fluor 488 (ICC) | Thermo Fisher Scientific | Cat# A32814; RRID:AB_2762838 |
| Donkey anti-mouse Alexa Fluor 488 (ICC) | Thermo Fisher Scientific | Cat# A-21202; RRID:AB_141607 |
| Donkey anti-rabbit Alexa Fluor 555 (ICC) | Thermo Fisher Scientific | Cat# A31572; RRID:AB_162543 |
|
| ||
| Biological samples | ||
|
| ||
| Human brain tissue | Bartoli brain tumor bank | Columbia University Medical Center |
|
| ||
| Critical commercial assays | ||
|
| ||
| Nextera index kit | Illumina | FC-121–1011 |
| RNeasy Mini Kit | Qiagen | 74106 |
| Ultra-Low-RNA-seq RNA Sample Prep Kit | Illumina | N/A |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq data | This paper | GEO: GSE222574 |
| ATAC-seq data | This paper | GEO: GSE190404 |
| Metabolomics data | This paper | NMDR: Study ID ST002739; Project http://dx.doi.org/10.21228/M8FM85 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: Cnp1-Cre (Cnptm1(cre)Kan) | Gift from Dr. Klaus A Nave; (Lappe-Siefke et al.)74 | MGI:3051635 |
| Mouse: Lmna-flox | Gift from Dr. Colin L. Stewart; (Solovei et al.)44 | N/A |
| Mouse: Tg(Ndrg1-EGFP)EQ125Gsat/Mmucd | (Marechal et al.)75 | RRID:MMRRC_011851-UCD |
| Mouse: B6.Cg-Tg(Camk2a-cre)T29–1Stl/J | Jackson Laboratory; (Tsien et al.)91 | RRID:IMSR_JAX:005359 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S4 for RT-qPCR primers | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ-Fiji | NIH Image | RRID:SCR_003070; RRID:SCR_002285 |
| GraphPad Prism 9 | GraphPad Software | RRID:SCR_002798 |
| R Project for Statistical Computing | R Software | RRID:SCR_001905 |
| ataqv | (Orchard et al.)92 | RRID:SCR_023112 |
| BEDtools | (Quinlan & Hall)93 | RRID:SCR_006646 |
| ChIPSeeker | (Yu et al.)94 | RRID:SCR_021322 |
| ComplexHeatmap | (Gu et al.)95 | RRID:SCR_017270 |
| DESeq2 | (Anders & Huber)96 | RRID:SCR_015687 |
| EnhancedVolcano | (Bilghe et al.)97 | RRID:SCR_018931 |
| fastp | (Chen et al.)98 | RRID:SCR_016962 |
| FASTQC | (Andrews)99 | RRID:SCR_014583 |
| HISAT2 | (Kim et al.)100 | RRID:SCR_015530 |
| MACS2 | (Zhang et al.)101 | RRID:SCR_013291 |
| Picard | Broad Institute | RRID:SCR_006525;http://broadinstitute.github.io/picard |
| Rsubread | (Liao et al.)102 | RRID:SCR_016945 |
| Samtools | (Li et al.)103 | RRID:SCR_002105 |
| STAR | (Dobin et al.)104 | RRID:SCR_004463 |
| ChEA_2022 Library | (Keenan et al.)105 | https://maayanlab.cloud/chea3/ |
| Enrichr | (Chen et al.; Kuleshov et al.; Xie et al.)106–108 | RRID:SCR_001575; https://maayanlab.cloud/Enrichr/ |
| DAVID | (Huang et al.; Sherman et al.)109,110 | RRID:SCR_001881; https://david.ncifcrf.gov |
| VolcaNoseR | (Goedhart & Luijsterburg)111 | https://huygens.science.uva.nl/VolcaNoseR/ |
| FlexImaging v3.0 | Bruker Daltonics | N/A |
| SCiLS (2015b) | Bruker Daltonics | RRID:SCR_014426 |
| MetaboScape | Bruker Daltonics | N/A |
| MetaboBASE® Personal Library 3.0 | Bruker Daltonics | N/A |
| HMDB | (Wishart et al.)112 | RRID:SCR_007712; http://www.hmdb.ca |
| XCMSPlus | (Domingo-Almenara et al.)113 | N/A |
Immunohistochemistry (human tissues)
Human paraffin embedded formalin fixed sections (7 μm) were deparaffinized then subjected to antigen retrieval (20 min in pressure cooker in Trilogy pretreatment buffer, Sigma, 920P-05). Sections were blocked in 10% goat-serum buffer, for 1 h at RT. Primary antibodies for CC1 (1:500, Millipore OP80), LMNA (1:1,000, Abcam, ab226198), LMNB1 (1:1,000, Abcam, ab16048), NG2 (1:500, Sigma, MAB5384-I), were incubated for 4 h at RT. After washes, sections were incubated with Alexa fluor conjugated goat secondary antibodies (1:500) for 1 h at RT. After washes, sections were incubated with DAPI (1:1,000) for 5 min. Primary and secondary antibodies used are listed in the key resources table. Slides were mounted with fluorogold mounting medium and imaged at 20x and/or 40x on Zeiss Axio Vert with Apotome 3.
H&E staining
For Hematoxylin and eosin (H&E) staining, mice were euthanized by cervical dislocation. Soleus muscles were dissected from the mouse in prone position on a dissection board,117 then embedded in OCT and rapidly frozen in isopentane cooled with liquid nitrogen for approximately 10–20 s.118 Hematoxylin Gill No.1 (GHS116, Sigma) and Eosin Y (HT110116, Sigma) were used to perform the staining as per manufacturer’s instructions. Sections were imaged using a slide scanner (Leica Aperio CS2 slide scanner, objective 20×/0.75 NA Plan Apo).
Immunocytochemistry
Cells for immunocytochemistry were seeded in 8-well chamber slides (Thermo Fisher Scientific, 154941PK) and fixed with 4% PFA for 15 min at RT. Fixed cells were incubated in blocking buffer (PGBA, 5% normal goat or donkey serum, 0.1% Triton X-100) at RT for 1 h, then with primary antibodies (LMNA/C, 1:500, Abcam ab26300; LMNA/C, 1:800, Santa Cruz sc-6215; LMNB1, 1:500, Santa cruz sc-374015; OLIG2, 1:500, Millipore MABN50; MBP, 1:500, Millipore MAB386, SOX10, 1:1,000, Cell Signaling 89356) diluted in blocking buffer overnight at 4°C. After washes, appropriate Alexa Fluor secondary antibodies were applied for 1 h at RT and slides were mounted using DAPI Fluoromount-G mounting medium (Thermo Fisher Scientific). Primary and secondary antibodies used are listed in the key resources table.
Protein extraction and Western blot
For Western blot on mouse samples, mice were euthanized by cervical dislocation and spinal cord or corpus callosum was dissected. Protein lysates were obtained from tissues and cells using RIPA buffer (50mM Tris, 150mM NaCl, 1mM EDTA, 1% Triton X-100, 0.5% Sodium Deoxycholate) supplemented with protease inhibitors and dithiothreitol (DTT, 1mM, Thermo Fisher Scientific). Protein concentrations were measured using DC Protein assay (Biorad) and 10 to 20 μg of protein lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and transferred onto a Polyvinylidene fluoride (PVDF) membrane (25mM Tris base, 192mM Glycine, 20% Methanol, pH 8.3). Membranes were blocked in TBS (150mM NaCl, 20mM Tris-HCl, pH 7.5) containing 0.1% Tween 20, 5% non-fat milk, for 1 h at RT, then incubated overnight at 4°C with primary antibodies (α-Tubulin, 1:5,000, Calbiochem CP06; β-Actin, 1:2,500, Sigma A4700; GAPDH, 1:3,000, Abcam ab8245; LMNA/C, 1:1,000, Active Motif 39287; LMNB1, 1:10,000, Abcam ab16048; MBP, 1:1,000 Abcam ab40390; MOG, 1:100, Clone Z12, kindly provided by Prof. R. Reynolds, Imperial College, London, UK; PLP/DM20, 1:1,000, A431, kindly provided by Dr. Klaus-Armin Nave, Max Planck Institute for Multidisciplinary Sciences, Göttingen, Germany). Following 0.1%-Tween-TBS washes, secondary HRP-coupled antibodies (1:10,000) were incubated for 2 h at RT followed by washes. Immunoreactivity of target protein was detected using the ECL Prime Western Blotting System (Sigma) with ChemiDoc Imaging System (Bio-Rad). α-Tubulin, β-Actin or GAPDH were used as loading control for normalization. Primary and secondary antibodies used are listed in the key resources table.
RNA extraction, RT-qPCR and RNA-sequencing
RNA was extracted from cells and tissues (corpus callosum and sciatic nerve) using TRIzol (Invitrogen) and isopropanol precipitation. RNA samples were resuspended in Nuclease-free H2O and further purified using the RNeasy Mini Kit (Qiagen, 74106) with on-column DNase treatment. RNA purity was tested by measuring the A260/A280 ratio with a NanoDrop. For real-time quantitative PCR (RT-qPCR), RNA was reverse-transcribed with qScript XLT cDNA SuperMix (Quantabio, 95161). RT-qPCR reactions were run in triplicates using PerfeCTa SYBR GREEN FastMix, ROX reagent (Quantabio, 95072) at the ASRC Epigenetic Core facility. After normalizing each sample to indicated housekeeping genes, CT values of technical triplicates were averaged for each biological replicate. Sequences of primers used are listed in Table S4. For RNA-sequencing, approximately 52 ng of total RNA per sample (each sample pooled from 2 to 3 sorted brains) was used for library construction with the Ultra-Low-RNA-seq RNA Sample Prep Kit (Illumina) and sequenced using the Illumina HiSeq 2500 instrument by the Epigenomics Core of Weill Cornell Medical College according to the manufacturer’s instructions for 50 bp single-end read runs. Samples were run in triplicates and one sample per group was excluded after failing quality controls.
Electron microscopy
Mice were deeply anesthetized with ketamine/xylazine (150 mg/kg and 15 mg/kg) and perfused with 4% PFA, 5% glutaraldehyde, 0.1 M sucrose in 0.1 M PB, pH 7.4. Corpus callosum, spinal cord and sciatic nerves and were collected. Tissues embedded in EMbed 812 were sectioned on a Leica UCT ultramicrotome (1 μm) and stained with a solution of 1% toluidine blue. For transmission electronic microscopy (TEM), 90 nm sections were placed on copper slotted grids or occasionally on mesh grids (Virginia Commonwealth University). Sections were stained with 3% uranyl acetate in 50% methanol for 20 min followed by lead citrate for 5 min. Grids were viewed on a Philips CM12 Tungsten Emission TEM at 120 kV and imaged with a GATAN 4k × 2.7k digital camera. 8-week-old samples were imaged on Talos 120C TEM (New York University). g-ratios were calculated by dividing the axon diameter by the total diameter of myelinated fiber. The percentage of heterochromatin area at the periphery over the total area of heterochromatin was measured by calculating the area of electron-dense signal directly bound to the nuclear periphery over the total area of electron-dense signal in the nuclei.
Clinical score
Mice were monitored from birth and their behavior was evaluated according to the following clinical score: Score 0, normal; Score 0.5, mild reduction of hindlimb mobility, occasional clasping when held by the tail; Score 1, mouse slips on grid; Score 1,5, tremors, mouse hardly able to walk on grid; Score 2, mouse is unable to hold on inverted grid and walking deficit; Score 2.5, weakness of hindlimbs but preserved ability to hold on upright grid with forelimbs, presence of tremors and walking deficit; Score 3, tremors and progressive ataxia.
Rotarod
Mice were evaluated on the rotarod test at 26 weeks. During 5 min trial sessions, we monitored the time until the mice fell (latency to fall) from the rotating rod in accelerating speed mode (5–45 r.p.m.). Mice were tested 3 consecutive days, 3 trials per day with 15 min inter-trial interval.
FACS sorting
Mice were euthanized at 8 weeks by cervical dislocation to isolate brains and perform fluorescence-activated cell sorting (FACS).75 Brains were dissected in HBSS 1X (HBSS 10X (Invitrogen), 0.01 M HEPES buffer, 0.75% sodium bicarbonate (Invitrogen), 1% penicillin/streptomycin) and mechanically dissociated. After enzymatic dissociation using papain (30 μg/mL in DMEM-Glutamax, with 0.24 μg/mL L-cysteine and 40 μg/mL DNase I), samples were layered over a pre-formed Percoll density gradient and then centrifuged for 15 min at 22,000 × g. Cells were then collected and stained with propidium iodide (PI) for 2 min at RT to exclude dead cells. Subsequently, GFP-positive and PI-negative cells were sorted by FACS (Aria, Becton Dickinson) and collected in pure fetal bovine serum.
Generation of ATAC-seq libraries
ATAC-seq reactions were performed using an established protocol.119 Briefly, each sample consisted of 65,000 oligodendrocytes, sorted from the brain of Ndrg1-EGFP;Cnp-Cre;Lmna controls and mutants. Following cells lysis, nuclei were pelleted at 500 × g for 10 min at 4°C. To each nuclei pellet, 25 μL of TD buffer (2X reaction buffer from Nextera Kit), 2.5 μL of TDE1 (Nextera TN5 Transposase from Nextera Kit) and 22.5 μL of Nuclease-free H2O were added. Samples were kept at 37°C in gentle mixing for 30 min. DNA was eluted using Qiagen mini-elution kit. To each 10 μL of transposed DNA, 10 μL of Nuclease-free H2O, 2.5 μL of 25 μM PCR Primer 1, 2.5 μL of 25 μM Barcoded PCR Primer 2 and 25 μL of NEB Next High Fidelity 2x PCR Master mix, were added. Libraries were amplified using the Nextera index kit (Illumina, FC-121-1011), under the following cycling conditions: 72°C for 5 min, 98°C for 30 s, followed by thermocycling at 98°C for 10 s, 63°C for 30 s and 72°C for 1 min for a total of 5 cycles. To avoid saturation due to over-amplification, a 10% aliquot of this reaction was then removed and subjected to qPCR for 20 cycles to calculate the optimal number of cycles needed for the remaining 90%. The additional number of cycles was determined as follows: (1) Plot linear Rn vs. Cycle (2) Calculate the # of cycles that corresponded to 1/4 of maximum fluorescent intensity. In general, we found adding 4 cycles to this estimate yielded optimal ATAC-seq libraries, as determined by analysis on Agilent D5000 screen tapes (Agilent technologies, 5067–5588 and 5067–5589). Libraries were amplified for a total of 13–15 cycles. Following PCR, ATAC-seq libraries were resolved on 2% agarose gels and fragments ranging in size from 100 bp - 1.5 kb were excised and purified (Qiagen Minelute Gel Extraction Kit – Qiagen, 28604). Libraries were quantified by quantitative PCR (KAPA Biosystems, KK4873) prior to sequencing. Libraries were sequenced on Hi-Seq2500 (Illumina) obtaining 2 × 50 paired-end reads.
MALDI-TOF MS imaging and LC-MS/MS
The Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry (MS) imaging was carried out in MALDI MS Imaging Joint Facility, and the Liquid chromatography-mass-spectrometry (LC-MS/MS) experiment was carried out in Mass Spectrometry Core Facility at Advanced Science Research Center of City University of New York Graduate Center. High purity grade N-(1-Naphthyl) Ethylenediamine Dihydrochloride (NEDC) and 2,5-dihydroxybenzoic acid (DHB) matrices, Phosphorus (red), amino acid standards and SeQuant ZIC-HILIC liquid chromatography 3.5 μm, 100 × 2.1 mm column were purchased from Millipore Sigma-Aldrich. Optima UHPLC/MS-grade acetonitrile, ammonium acetate, methanol, isopropanol and water were purchased from Fisher Scientific.
Murine brains were harvested at 26 weeks immediately after cervical dislocation and were snap-frozen for 5 min on an aluminum boat floating on liquid nitrogen.99,120 The frozen tissue was cryosectioned (10 μm thickness) using CryoStar NX50 (Thermo Scientific) at −15°C (specimen head and chamber). Collected brain cryosections at −0.88 mm to −1.335mm from bregma were gently transferred onto the pre-cooled conductive side of indium tin oxide (ITO)-coated glass slides (Bruker Daltonics) for MALDI imaging. Mounted cryosections were desiccated in vacuum for 45 min at RT, followed by matrix deposition using HTX M5 sprayer (HTX Technologies, LLC). Both NEDC (10 mg/mL in isopropanol/water (70/30, v/v); flow rate of 0.05 mL/min, nozzle temperature 75°C for 30 cycles, 5s drying between each cycle) and DHB (40 mg/mL in methanol/water (70/30, v/v), flow rate of 0.05 mL/min and a nozzle temperature of 85°C for 8 cycles) were used to detect metabolites and lipids. The following parameters were used for both matrices: spray velocity of 1300 mm/min, track spacing of 2 mm, N2 gas pressure of 10 psi and flow rate of 3 L/min and nozzle height of 40 mm. MALDI mass spectra were acquired in negative ion mode (NEDC) or positive ion mode (DHB) acquired by MALDI-TOF MS Autoflex (Bruker Daltonics). MS spectra were calibrated using red phosphorus as the standard for all experiments. Acquisition raster width set at 120 μm with laser smartbeam parameter at “Medium”. The imaging data for each array position were summed up by 500 shots at a laser repetition rate of 500 Hz. To minimize broadening of ion peaks, all the experiments were ran by setting laser power to its lowest value while allowing to accumulate ion spectra with appropriate S/N ratio. Spectra were acquired in the mass range from m/z 50 to 1000 with a low mass gate at 50 Da.
For LC-MS/MS experiment, cold methanol method modified from previous study was used to extract metabolites.98 Briefly, a slice of brain (approximate 15 mg) was cut off the plane of MALDI imaging brain samples and homogenized in cold Methanol/Water (80/20, v/v) to a final concentration of 30 mg/mL. Following 20 min of gentle sonication in Bioruptor (30 s on, 30 s off, 20 cycles) at 4°C samples were centrifuged for 10 min of 10,000 × g at 4°C. 10 μL injection volume was used for LC-MS/MS experiments. LC-MS/MS global metabolic profiling was carried on all sample tissues using ZIC-HILIC chromatography (based on Acetonitrile/Water/7mM Ammonium acetate solvent systems) and high-resolution mass spectrometry. We employed maXis-II-ETD UHR-ESI-Qq-TOF mass-spectrometer (Bruker Daltonics) equipped with Dionex Ultimate-3000 liquid chromatography system ZIC-HILIC column operated at mildly acidic pH provided good performance on the metabolites of interest. Each LC-MS/MS experiment was performed in 3 biological replicates and 4 technical replicates per genotype.
QUANTIFICATION AND DATA ANALYSIS
RNA-seq analysis
Initial quality analyses were performed on FastQ files before trimming using FASTQC.100 To trim and filter low quality reads, fastp103 processor was used. High-quality reads were aligned to the mouse reference genome (mm10) by splice-aware aligner HISAT2.102 Obtained BAM files of mapped single-end reads sorted by genomic coordinates using samtools (v1.4.1).96 Raw counts were generated with the NCBI mouse RefSeq annotation (build 38), using uniquely aligned reads to exons by the featureCounts tool (Rsubread v2.12.0).104 Original experiment was performed on n = 3 samples per genotype; one sample per group was excluded following quality controls. The raw read counts were input into DESeq2 (v1.38.1),93 for normalizing the signal for each transcript and to ascertain differential gene expression with associated p values. Cut-off of p value < 0.05 was used to identify differentially expressed genes. Additional filtering step was applied to retain only the known protein-coding genes. Data deposited in GEO: GSE222574.
ATAC-seq data processing and analysis
Raw sequencing paired-end files were aligned by STAR (v2.7.0.e)92 to the mm10 reference genome using the following parameters: –alignIntronMax 1 –out FilterMismatchNmax 100 –alignEndsType EndToEnd –out FilterScoreMinOverLread 0.3 –out FilterMatchNminOverLread 0.3. This yielded for each sample a BAM file of mapped paired-end reads sorted by genomic coordinates. From these BAM-files, we removed reads that were mapped to multiple loci or to the mitochondrial genome using samtools (v0.1.19)96 as well as the duplicated reads using PICARD (v2.24.0). The read coverage files (bedGraph) were generated by genomeCoverageBed from BEDTools (v2.25.0)121 and converted to the indexed binary format files (bigWig) using bedGraphToBigWig. TSS (+/− 3kb) enrichment was calculated by ataqv package101 using TSS positions of 2,816 housekeeping genes. The calculated enrichment for this dataset (mean = 12.8) was comparable to other brain-related mouse datasets (mean = 13.3 for94). MACS2 package109 was used for generating signal tracks and peak calling. Peaks were annotated to different genomic regions by ChIPseeker package110 using the annotatePeak function. Data deposited in GEO: GSE190404.
Gene Ontology analysis
Enrichment analysis was performed with the Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics.105,106 Gene Ontology categories were sorted by lowest p value, then by higher of percentage of DAVID genes in the list associated with a particular annotation term. Selected top Gene Ontology categories were represented.
Comparative analysis of sequencing data
To compare our gene list of interest with previously published databases, we reported the percentage of genes overlapping with gene lists previously reported to be characterized by H3K27me3 or H3K9me3 (ChIP-seq12) occupancy, or regulation by DNA methylation (ERRBS17) or DNA hydroxymethylation (RRHP18).
Transcription factor analysis
Enrichment analysis of transcription factors that potentially regulate the gene list of interest was done using the ChIP enrichment analysis ChEA_2022 library107 on the Enrichr platform.108,112,113
MALDI-TOF MS imaging and LC-MS/MS analyses
MALDI imaging data were recorded and processed using FlexImaging v3.0, and further analyzed using SCiLS (2015b). Ion images were generated with root-mean-square (RMS) normalization and a bin width of ±0.10 Da. The spectra were interpreted manually, and analyte assignment was achieved by comparing with LC-MS experiment results.
Compound identification and descriptive statistical analysis of the LC-MS/MS data were performed through MetaboScape and XCMSPlus software.111 Bruker MetaboBase Personal 3.0, MoNA, MSDIAL, METLIN, and HMDB97 metabolomic libraries were used in compound identification. Ultimately, both accurate mass-measurements (with less than 5 pmm accuracy) and fragmentation spectra (or simply MS/MS spectra) were used for confident identification of metabolites and lipids. In total, 905 metabolites were identified in both wild type and mutant animals and were plotted in volcano plot. p value between wild type and mutant animals were analyzed by Student’s t-test using three animals per group. The level change of metabolites with p < 0.05 is considered as significant.
Image analysis and data visualization
Microscope images were analyzed using Fiji-ImageJ (RRID:SCR_003070). Volcano-plots and heatmaps were generated using VolcaNoseR95 and EnhancedVolcano (v1.16.0)122 and ComplexHeatmap (v2.14.0)123 package of R environment (v4.2.2).124 WashU Epigenome Browser (v53.8.0)76 was used to visualize ATAC-seq peaks on the genome.
Statistical analysis
GraphPad Prism (GraphPad Software, Inc, RRID:SCR_002798; v9.4.1) was used to generate all other graphs and to perform the statistical analyses indicated in each figure legend. When p value (p) < 0.05, statistical significance levels are indicated as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All data are represented as average ±SD.
Supplementary Material
Highlights.
LMNA/C levels increase as oligodendrocyte progenitors differentiate
Lmna deletion in oligodendrocytes impairs their transcriptional signature
Lmna deletion alters brain metabolism, myelin lipids, and mitochondrial structure
Lmna deletion in myelinating glia induces a progressive clinical motor phenotype
ACKNOWLEDGMENTS
The authors thank Dr. Colin Stewart for the Lmna floxed mice and Dr. Klaus-Armin Nave for the Cnp1-Cre mice used in this study. They would also like to acknowledge the initial work conducted by M.H. on Lmna systemic mutants with Dr. Lindsay Shopland, which has not been included in the current article. We thank the Epigenomics Core at Weill Cornell for sequencing, the NYU EM Core for electron microscopy on 8-week-old tissues, the ASRC-MALDI MS Imaging Joint Facility and Mass Spectrometry core (Dr. Rinat Abzalimov) for the untargeted metabolomic analysis, and the ASRC-Epigenetics core (Dr. Jia Liu) for the use of qPCR and cell-sorting instruments. The authors are grateful to Fatima Khan, Benjamin Inbar, Rebecca Frawley, and Jennifer Chou for technical help. The graphical abstract was created using BioRender.com. This work was performed thanks to grants from the National Institutes of Health (R35-NS111604) to P.C. and fellowships from the National Multiple Sclerosis Society to M.P. (FG-1907-34565) and J.P. (FG-1804-30631). This study was also supported by the National Institutes of Health (NIH), Bethesda, MD, under award numbers R01MH125246, R01AG065582, R01AG067025, R01AG050986, U01MH116442, and R01MH109677. The NMDR metabolomics repository is supported by NIH grant U2C-DK119886 and an OT2-OD030544 grant.
INCLUSION AND DIVERSITY
We support inclusive, diverse and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112848.
REFERENCES
- 1.Bitman-Lotan E, and Orian A (2021). Nuclear organization and regulation of the differentiated state. Cell. Mol. Life Sci. 78, 3141–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holmberg J, and Perlmann T (2012). Maintaining differentiated cellular identity. Nat. Rev. Genet. 13, 429–439. [DOI] [PubMed] [Google Scholar]
- 3.Natoli G (2010). Maintaining cell identity through global control of genomic organization. Immunity 33, 12–24. [DOI] [PubMed] [Google Scholar]
- 4.Pope SD, and Medzhitov R (2018). Emerging principles of gene expression programs and their regulation. Mol. Cell 71, 389–397. [DOI] [PubMed] [Google Scholar]
- 5.Baumann N, and Pham-Dinh D (2001). Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 81, 871–927. [DOI] [PubMed] [Google Scholar]
- 6.Dansu DK, Sauma S, and Casaccia P (2021). Oligodendrocyte progenitors as environmental biosensors. Semin. Cell Dev. Biol. 116, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pruvost M, and Moyon S (2021). Oligodendroglial epigenetics, from lineage specification to activity-dependent myelination. Life 11, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hernandez M, and Casaccia P (2015). Interplay between transcriptional control and chromatin regulation in the oligodendrocyte lineage. Glia 63, 1357–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selcen I, Prentice E, and Casaccia P (2023). The epigenetic landscape of oligodendrocyte lineage cells. Ann. N. Y. Acad. Sci. 1522, 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu Y, Chen Y, Kim B, Wang H, Zhao C, He X, Liu L, Liu W, Wu LMN, Mao M, et al. (2013). Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell 152, 248–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falcão AM, Meijer M, Scaglione A, Rinwa P, Agirre E, Liang J, Larsen SC, Heskol A, Frawley R, Klingener M, et al. (2019). PAD2-mediated citrullination contributes to efficient oligodendrocyte differentiation and myelination. Cell Rep. 27, 1090–1102.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Magri L, Zhang F, Marsh NO, Albrecht S, Huynh JL, Kaur J, Kuhlmann T, Zhang W, Slesinger PA, et al. (2015). Chromatin landscape defined by repressive histone methylation during oligodendrocyte differentiation. J. Neurosci. 35, 352–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marin-Husstege M, Muggironi M, Liu A, and Casaccia-Bonnefil P (2002). Histone deacetylase activity is necessary for oligodendrocyte lineage progression. J. Neurosci. 22, 10333–10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scaglione A, Patzig J, Liang J, Frawley R, Bok J, Mela A, Yattah C, Zhang J, Teo SX, Zhou T, et al. (2018). PRMT5-mediated regulation of developmental myelination. Nat. Commun. 9, 2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen S, Li J, and Casaccia-Bonnefil P (2005). Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J. Cell Biol. 169, 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJM, and Casaccia-Bonnefil P (2008). Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat. Neurosci. 11, 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moyon S, Huynh JL, Dutta D, Zhang F, Ma D, Yoo S, Lawrence R, Wegner M, John GR, Emery B, et al. (2016). Functional characterization of DNA methylation in the oligodendrocyte lineage. Cell Rep. 15, 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moyon S, Frawley R, Marechal D, Huang D, Marshall-Phelps KLH, Kegel L, Bøstrand SMK, Sadowski B, Jiang Y-H, Lyons DA, et al. (2021). TET1-mediated DNA hydroxymethylation regulates adult remyelination in mice. Nat. Commun. 12, 3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang M, Wang J, Zhang K, Lu G, Liu Y, Ren K, Wang W, Xin D, Xu L, Mao H, et al. (2021). Ten-eleven translocation 1 mediated-DNA hydroxymethylation is required for myelination and remyelination in the mouse brain. Nat. Commun. 12, 5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dugas JC, Cuellar TL, Scholze A, Ason B, Ibrahim A, Emery B, Zamanian JL, Foo LC, McManus MT, and Barres BA (2010). Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination. Neuron 65, 597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tripathi A, Volsko C, Garcia JP, Agirre E, Allan KC, Tesar PJ, Trapp BD, Castelo-Branco G, Sim FJ, and Dutta R (2019). Oligodendrocyte intrinsic miR-27a controls myelination and remyelination. Cell Rep. 29, 904–919.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao X, He X, Han X, Yu Y, Ye F, Chen Y, Hoang T, Xu X, Mi Q-S, Xin M, et al. (2010). MicroRNA-mediated control of oligodendrocyte differentiation. Neuron 65, 612–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He D, Wang J, Lu Y, Deng Y, Zhao C, Xu L, Chen Y, Hu Y-C, Zhou W, and Lu QR (2017). lncRNA functional networks in oligodendrocytes reveal stage-specific myelination control by an lncOL1/Suz12 complex in the CNS. Neuron 93, 362–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katsel P, Roussos P, Fam P, Khan S, Tan W, Hirose T, Nakagawa S, Pletnikov MV, and Haroutunian V (2019). The expression of long noncoding RNA NEAT1 is reduced in schizophrenia and modulates oligodendrocytes transcription. NPJ Schizophr. 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenzie AT, Wang M, Hauberg ME, Fullard JF, Kozlenkov A, Keenan A, Hurd YL, Dracheva S, Casaccia P, Roussos P, et al. (2018). Brain cell type specific gene expression and co-expression network architectures. Sci. Rep. 8, 8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boshans LL, Factor DC, Singh V, Liu J, Zhao C, Mandoiu I, Lu QR, Casaccia P, Tesar PJ, and Nishiyama A (2019). The chromatin environment around interneuron genes in oligodendrocyte precursor cells and their potential for interneuron reprograming. Front. Neurosci. 13, 829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Magri L, Swiss VA, Jablonska B, Lei L, Pedre X, Walsh M, Zhang W, Gallo V, Canoll P, and Casaccia P (2014). E2F1 coregulates cell cycle genes and chromatin components during the transition of oligodendrocyte progenitors from proliferation to differentiation. J. Neurosci. 34, 1481–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao C, Dong C, Frah M, Deng Y, Marie C, Zhang F, Xu L, Ma Z, Dong X, Lin Y, et al. (2018). Dual requirement of CHD8 for chromatin landscape establishment and histone methyltransferase recruitment to promote CNS myelination and repair. Dev. Cell 45, 753–768.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swiss VA, Nguyen T, Dugas J, Ibrahim A, Barres B, Androulakis IP, and Casaccia P (2011). Identification of a gene regulatory network necessary for the initiation of oligodendrocyte differentiation. PLoS One 6, e18088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernhardt C, Sock E, Fröb F, Hillgärtner S, Nemer M, and Wegner M (2022). KLF9 and KLF13 transcription factors boost myelin gene expression in oligodendrocytes as partners of SOX10 and MYRF. Nucleic Acids Res. 50, 11509–11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH, and Barres BA (2009). Myelin gene regulatory factor is a critical transcriptional regulator required for cns myelination. Cell 138, 172–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howng SYB, Avila RL, Emery B, Traka M, Lin W, Watkins T, Cook S, Bronson R, Davisson M, Barres BA, et al. (2010). ZFP191 is required by oligodendrocytes for CNS myelination. Genes Dev. 24, 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stolt CC, Rehberg S, Ader M, Lommes P, Riethmacher D, Schachner M, Bartsch U, and Wegner M (2002). Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 16, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turnescu T, Arter J, Reiprich S, Tamm ER, Waisman A, and Wegner M (2018). Sox8 and Sox10 jointly maintain myelin gene expression in oligodendrocytes. Glia 66, 279–294. [DOI] [PubMed] [Google Scholar]
- 35.Mori S, and Leblond CP (1970). Electron microscopic identification of three classes of oligodendrocytes and a preliminary study of their proliferative activity in the corpus callosum of young rats. J. Comp. Neurol. 139, 1–28. [DOI] [PubMed] [Google Scholar]
- 36.Cabianca DS, and Gasser SM (2016). Spatial segregation of heterochromatin: uncovering functionality in a multicellular organism. Nucleus 7, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sapra KT, Qin Z, Dubrovsky-Gaupp A, Aebi U, Müller DJ, Buehler MJ, and Medalia O (2020). Nonlinear mechanics of lamin filaments and the meshwork topology build an emergent nuclear lamina. Nat. Commun. 11, 6205–6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Briand N, and Collas P (2020). Lamina-associated domains: peripheral matters and internal affairs. Genome Biol. 21, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shevelyov YY, and Ulianov SV (2019). The nuclear lamina as an organizer of chromosome architecture. Cells 8, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aaronson RP, and Blobel G (1975). Isolation of nuclear pore complexes in association with a lamina. Proc. Natl. Acad. Sci. USA 72, 1007–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fisher DZ, Chaudhary N, and Blobel G (1986). cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. USA 83, 6450–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin F, and Worman HJ (1995). Structural organization of the human gene (LMNB1) encoding nuclear Lamin B1. Genomics 27, 230–236. [DOI] [PubMed] [Google Scholar]
- 43.Röber RA, Weber K, and Osborn M (1989). Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 105, 365–378. [DOI] [PubMed] [Google Scholar]
- 44.Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et al. (2013). LBR and Lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152, 584–598. [DOI] [PubMed] [Google Scholar]
- 45.Stewart C, and Burke B (1987). Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell 51, 383–392. [DOI] [PubMed] [Google Scholar]
- 46.Young SG, Jung H-J, Coffinier C, and Fong LG (2012). Understanding the roles of nuclear A- and B-type Lamins in brain development. J. Biol. Chem. 287, 16103–16110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huynh JL, Garg P, Thin TH, Yoo S, Dutta R, Trapp BD, Haroutunian V, Zhu J, Donovan MJ, Sharp AJ, et al. (2014). Epigenome-wide differences in pathology-free regions of multiple sclerosis–affected brains. Nat. Neurosci. 17, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huynh JL, and Casaccia P (2010). Defining the chromatin landscape in demyelinating disorders. Neurobiol. Dis. 39, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huynh JL, and Casaccia P (2013). Epigenetic mechanisms in multiple sclerosis: implications for pathogenesis and treatment. Lancet Neurol. 12, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schlüter A, Sandoval J, Fourcade S, Díaz-Lagares A, Ruiz M, Casaccia P, Esteller M, and Pujol A (2018). Epigenomic signature of adrenoleukodystrophy predicts compromised oligodendrocyte differentiation: Epigenomic signature of adrenoleukodystrophy. Brain Pathol. 28, 902–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ginsberg MR, Rubin RA, Falcone T, Ting AH, and Natowicz MR (2012). Brain transcriptional and epigenetic associations with Autism. PLoS One 7, e44736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu J, Dietz K, DeLoyht JM, Pedre X, Kelkar D, Kaur J, Vialou V, Lobo MK, Dietz DM, Nestler EJ, et al. (2012). Impaired adult myelination in the prefrontal cortex of socially isolated mice. Nat. Neurosci. 15, 1621–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagy C, Maitra M, Tanti A, Suderman M, Théroux JF, Davoli MA, Perlman K, Yerko V, Wang YC, Tripathy SJ, et al. (2020). Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat. Neurosci. 23, 771–781. [DOI] [PubMed] [Google Scholar]
- 54.Lutz P-E, Tanti A, Gasecka A, Barnett-Burns S, Kim JJ, Zhou Y, Chen GG, Wakid M, Shaw M, Almeida D, et al. (2017). Association of a history of child abuse with impaired myelination in the anterior cingulate cortex: convergent epigenetic, transcriptional, and morphological evidence. Aust. J. Pharm. 174, 1185–1194. [DOI] [PubMed] [Google Scholar]
- 55.Li M, Xiao L, and Chen X (2022). Histone acetylation and methylation underlie oligodendroglial and myelin susceptibility in schizophrenia. Front. Cell. Neurosci. 16, 823708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yattah C, Hernandez M, Huang D, Park H, Liao W, and Casaccia P (2020). Dynamic Lamin B1-gene association during oligodendrocyte progenitor differentiation. Neurochem. Res. 45, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin S-T, and Fu Y-H (2009). miR-23 regulation of lamin B1 is crucial for oligodendrocyte development and myelination. Dis. Model. Mech. 2, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptácek LJ, and Fu Y-H (2006). Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet. 38, 1114–1123. [DOI] [PubMed] [Google Scholar]
- 59.Meijer IA, Simoes-Lopes AA, Laurent S, Katz T, St-Onge J, Verlaan DJ, Dupré N, Thibault M, Mathurin J, Bouchard J-P, et al. (2008). A novel duplication confirms the involvement of 5q23.2 in autosomal dominant leukodystrophy. Arch. Neurol. 65, 1496–1501. [DOI] [PubMed] [Google Scholar]
- 60.Brussino A, Vaula G, Cagnoli C, Mauro A, Pradotto L, Daniele D, Di Gregorio E, Barberis M, Arduino C, Squadrone S, et al. (2009). A novel family with Lamin B1 duplication associated with adult-onset leucoencephalopathy. J. Neurol. Neurosurg. Psychiatry 80, 237–240. [DOI] [PubMed] [Google Scholar]
- 61.Padiath QS, and Fu Y-H (2010). Autosomal dominant leukodystrophy caused by Lamin B1 duplications: a clinical and molecular case study of altered nuclear function and disease. In Methods in Cell Biology (Elsevier; ), pp. 337–357. 10.1016/S0091-679X(10)98014-X. [DOI] [PubMed] [Google Scholar]
- 62.Schuster J, Sundblom J, Thuresson A-C, Hassin-Baer S, Klopstock T, Dichgans M, Cohen OS, Raininko R, Melberg A, and Dahl N (2011). Genomic duplications mediate overexpression of lamin B1 in adult-onset autosomal dominant leukodystrophy (ADLD) with autonomic symptoms. Neurogenetics 12, 65–72. [DOI] [PubMed] [Google Scholar]
- 63.Giorgio E, Robyr D, Spielmann M, Ferrero E, Di Gregorio E, Imperiale D, Vaula G, Stamoulis G, Santoni F, Atzori C, et al. (2015). A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD). Hum. Mol. Genet. 24, 3143–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heng MY, Lin S-T, Verret L, Huang Y, Kamiya S, Padiath QS, Tong Y, Palop JJ, Huang EJ, Ptáček LJ, et al. (2013). Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J. Clin. Invest. 123, 2719–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lo Martire V, Alvente S, Bastianini S, Berteotti C, Bombardi C, Calandra-Buonaura G, Capellari S, Cohen G, Cortelli P, Gasparini L, et al. (2018). Mice overexpressing lamin B1 in oligodendrocytes recapitulate the age-dependent motor signs, but not the early autonomic cardiovascular dysfunction of autosomal-dominant leukodystrophy (ADLD). Exp. Neurol. 301, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rolyan H, Tyurina YY, Hernandez M, Amoscato AA, Sparvero LJ, Nmezi BC, Lu Y, Estécio MRH, Lin K, Chen J, et al. (2015). Defects of lipid synthesis are linked to the age-dependent demyelination caused by Lamin B1 overexpression. J. Neurosci. 35, 12002–12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Janin A, Bauer D, Ratti F, Millat G, and Méjat A (2017). Nuclear envelopathies: a complex LINC between nuclear envelope and pathology. Orphanet J. Rare Dis. 12, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Malashicheva A, and Perepelina K (2021). Diversity of nuclear Lamin A/C action as a key to tissue-specific regulation of cellular identity in health and disease. Front. Cell Dev. Biol. 9, 761469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bouhouche A, Benomar A, Birouk N, Mularoni A, Meggouh F, Tassin J, Grid D, Vandenberghe A, Yahyaoui M, Chkili T, et al. (1999). A locus for an axonal form of autosomal recessive Charcot-Marie-Tooth disease maps to chromosome 1q21.2-q21.3. Am. J. Hum. Genet. 65, 722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat J-M, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, et al. (2002). Homozygous defects in LMNA, encoding Lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth Disorder Type 2) and mouse. Am. J. Hum. Genet. 70, 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chaouch M, Allal Y, De Sandre-Giovannoli A, Vallat JM, Amer-el-Khedoud A, Kassouri N, Chaouch A, Sindou P, Hammadouche T, Tazir M, et al. (2003). The phenotypic manifestations of autosomal recessive axonal Charcot–Marie–Tooth due to a mutation in Lamin A/C gene. Neuromuscul. Disord. 13, 60–67. [DOI] [PubMed] [Google Scholar]
- 72.Tazir M, Azzedine H, Assami S, Sindou P, Nouioua S, Zemmouri R, Hamadouche T, Chaouch M, Feingold J, Vallat JM, et al. (2004). Phenotypic variability in autosomal recessive axonal Charcot-Marie-Tooth disease due to the R298C mutation in lamin A/C. Brain 127, 154–163. [DOI] [PubMed] [Google Scholar]
- 73.Jung H-J, Coffinier C, Choe Y, Beigneux AP, Davies BSJ, Yang SH, Barnes RH, Hong J, Sun T, Pleasure SJ, et al. (2012). Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc. Natl. Acad. Sci. USA 109, E423–E431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, and Nave K-A (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 33, 366–374. [DOI] [PubMed] [Google Scholar]
- 75.Marechal D, Dansu DK, Castro K, Patzig J, Magri L, Inbar B, Gacias M, Moyon S, and Casaccia P (2022). N-myc downstream regulated family member 1 (NDRG1) is enriched in myelinating oligodendrocytes and impacts myelin degradation in response to demyelination. Glia 70, 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li D, Purushotham D, Harrison JK, Hsu S, Zhuo X, Fan C, Liu S, Xu V, Chen S, Xu J, et al. (2022). WashU Epigenome Browser update 2022. Nucleic Acids Res. 50, W774–W781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marques S, Zeisel A, Codeluppi S, Van Bruggen D, Mendanha Falcão A, Xiao L, Li H, Häring M, Hochgerner H, Romanov RA, et al. (2016). Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu J, Moyon S, Hernandez M, and Casaccia P (2016). Epigenetic control of oligodendrocyte development: adding new players to old keepers. Curr. Opin. Neurobiol. 39, 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Makhija E, Jagielska A, Zhu L, Bost AC, Ong W, Chew SY, Shivashankar GV, and Van Vliet KJ (2018). Mechanical strain alters cellular and nuclear dynamics at early stages of oligodendrocyte differentiation. Front. Cell. Neurosci. 12, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Padiath QS (2016). Lamin B1 mediated demyelination: linking lamins, lipids and leukodystrophies. Nucleus 7, 547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hagemeyer N, Goebbels S, Papiol S, Kästner A, Hofer S, Begemann M, Gerwig UC, Boretius S, Wieser GL, Ronnenberg A, et al. (2012). A myelin gene causative of a catatonia-depression syndrome upon aging. EMBO Mol. Med. 4, 528–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cristobal CD, and Lee HK (2022). Development of myelinating glia: an overview. Glia 70, 2237–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mirsky R, Woodhoo A, Parkinson DB, Arthur-Farraj P, Bhaskaran A, and Jessen KR (2008). Novel signals controlling embryonic Schwann cell development, myelination and dedifferentiation. J. Peripher. Nerv. Syst. 13, 122–135. [DOI] [PubMed] [Google Scholar]
- 84.Piao J-H, Wang Y, and Duncan ID (2013). CD44 is required for the migration of transplanted oligodendrocyte progenitor cells to focal inflammatory demyelinating lesions in the spinal cord. Glia 61, 361–367. [DOI] [PubMed] [Google Scholar]
- 85.Signes A, and Fernandez-Vizarra E (2018). Assembly of mammalian oxidative phosphorylation complexes I–V and supercomplexes. Essays Biochem. 62, 255–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Monnerie H, Romer M, Jensen BK, Millar JS, Jordan-Sciutto KL, Kim SF, and Grinspan JB (2017). Reduced sterol regulatory element-binding protein (SREBP) processing through site-1 protease (S1P) inhibition alters oligodendrocyte differentiation in vitro. J. Neurochem. 140, 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, Goldstein JL, and Brown MS (1993). SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 75, 187–197. [PubMed] [Google Scholar]
- 88.Barton LJ, Soshnev AA, and Geyer PK (2015). Networking in the nucleus: a spotlight on LEM-domain proteins. Curr. Opin. Cell Biol. 34, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bourgeois B, Gilquin B, Tellier-Lebègue C, Östlund C, Wu W, Pérez J, El Hage P, Lallemand F, Worman HJ, and Zinn-Justin S (2013). Inhibition of TGF-β Signaling at the Nuclear Envelope: Characterization of Interactions Between MAN1, Smad2 and Smad3, and PPM1A. Sci. Signal. 6, ra49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dedeic Z, Cetera M, Cohen TV, and Holaska JM (2011). Emerin inhibits Lmo7 binding to the Pax3 and MyoD promoters and expression of myoblast proliferation genes. J. Cell Sci. 124, 1691–1702. [DOI] [PubMed] [Google Scholar]
- 91.Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, and Song H (2017). Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat. Neurosci. 20, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goedhart J, and Luijsterburg MS (2020). VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 10, 20560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wishart DS, Guo A, Oler E, Wang F, Anjum A, Peters H, Dizon R, Sayeeda Z, Tian S, Lee BL, et al. (2022). HMDB 5.0: the human metabolome database for 2022. Nucleic Acids Res. 50, D622–D631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen X, Momin A, Wanggou S, Wang X, Min H-K, Dou W, Gong Z, Chan J, Dong W, Fan JJ, et al. (2023). Mechanosensitive brain tumor cells construct blood-tumor barrier to mask chemosensitivity. Neuron 111, 30–48.e14. [DOI] [PubMed] [Google Scholar]
- 99.Chen YX, Veerasammy K, Yin J, Choetso T, Zhong T, Choudhury MA, Weng C, Xu E, Hein MA, Abzalimov R, et al. (2022). Sample preparation for rapid lipid analysis in Drosophila brain using matrix-assisted laser desorption/ionization mass spectrometry imaging. J. Vis. Exp. 10.3791/63930. [DOI] [PubMed] [Google Scholar]
- 100.Andrews S (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. [Google Scholar]
- 101.Orchard P, Kyono Y, Hensley J, Kitzman JO, and Parker SCJ (2020). Quantification, dynamic visualization, and validation of bias in ATAC-seq data with ataqv. Cell Syst. 10, 298–306.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- 105.Huang DW, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. [DOI] [PubMed] [Google Scholar]
- 106.Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC, Imamichi T, and Chang W (2022). DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Keenan AB, Torre D, Lachmann A, Leong AK, Wojciechowicz ML, Utti V, Jagodnik KM, Kropiwnicki E, Wang Z, and Ma’ayan A (2019). ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 47, W212–W224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yu G, Wang L-G, and He Q-Y (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- 111.Domingo-Almenara X, Montenegro-Burke JR, Ivanisevic J, Thomas A, Sidibé J, Teav T, Guijas C, Aisporna AE, Rinehart D, Hoang L, et al. (2018). XCMS-MRM and METLIN-MRM: a cloud library and public resource for targeted analysis of small molecules. Nat. Methods 15, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene set knowledge discovery with Enrichr. Curr. Protoc. 1, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sud M, Fahy E, Cotter D, Azam K, Vadivelu I, Burant C, Edison A, Fiehn O, Higashi R, Nair KS, et al. (2016). Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 44, D463–D470. 10.1093/nar/gkv1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, and Tonegawa S (1996). Subregion- and cell type–restricted gene knockout in mouse brain. Cell 87, 1317–1326. [DOI] [PubMed] [Google Scholar]
- 116.Shan F, Ji Q, Song Y, Chen Y, Hao T, Li R, Zhang N, and Wang Y (2022). A fast and efficient method for isolating Schwann cells from sciatic nerves of neonatal mice. J. Neurosci. Methods 366, 109404. [DOI] [PubMed] [Google Scholar]
- 117.Wang C, Yue F, and Kuang S (2017). Muscle histology characterization using H&E staining and muscle fiber type classification using immunofluorescence staining. Bio Protoc. 7, e2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Meng H, Janssen PML, Grange RW, Yang L, Beggs AH, Swanson LC, Cossette SA, Frase A, Childers MK, Granzier H, et al. (2014). Tissue Triage and Freezing for Models of Skeletal Muscle Disease. J. Vis. Exp, 51586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Veerasammy K, Chen YX, Sauma S, Pruvost M, Dansu DK, Choetso T, Zhong T, Marechal D, Casaccia P, Abzalimov R, et al. (2020). Sample preparation for metabolic profiling using MALDI mass spectrometry imaging. J. Vis. Exp, 62008. [DOI] [PubMed] [Google Scholar]
- 121.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bilghe K, Rana S, and Lewis M (2018). EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. [Google Scholar]
- 123.Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- 124.R Core Team (2022). R: A Language and Environment for Statistical Computing. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq and ATAC-Seq data have been deposited at NCBI’s Gene Expression Omnibus (GEO)91 and are accessible through GEO: GSE222574,GSE190404 respectively. Metabolomics data have been deposited at NIH Common Fund’s National Metabolomics Data Repository (NMDR) Website, the Metabolomics Workbench,114 https://www.metabolomicsworkbench.org where it has been assigned Study ID ST002739. The data can be accessed directly via its Project DOI: 10.21228/M8FM85. RNA-seq, ATAC-seq and Metabolomics data are publicly available as of the date of publication. All data reported in this study will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this study is available from the lead contact upon request.