

Abstract
TURBOMOLE is a highly optimized software suite for large-scale quantum-chemical and materials science simulations of molecules, clusters, extended systems, and periodic solids. TURBOMOLE uses Gaussian basis sets and has been designed with robust and fast quantum-chemical applications in mind, ranging from homogeneous and heterogeneous catalysis to inorganic and organic chemistry and various types of spectroscopy, light–matter interactions, and biochemistry. This Perspective briefly surveys TURBOMOLE’s functionality and highlights recent developments that have taken place between 2020 and 2023, comprising new electronic structure methods for molecules and solids, previously unavailable molecular properties, embedding, and molecular dynamics approaches. Select features under development are reviewed to illustrate the continuous growth of the program suite, including nuclear electronic orbital methods, Hartree–Fock-based adiabatic connection models, simplified time-dependent density functional theory, relativistic effects and magnetic properties, and multiscale modeling of optical properties.
1. Introduction
TURBOMOLE is a collaborative, multinational software development project aiming to provide highly efficient and stable computational tools for quantum-chemical simulations of molecules, clusters, periodic systems, and solutions. The software suite is optimized for widely available, inexpensive, and resource-efficient hardware, such as multicore workstations and medium-size compute clusters. TURBOMOLE specializes in electronic structure methods with an outstanding accuracy–cost ratio, such as density functional theory including the random phase approximation (RPA), GW-Bethe–Salpeter equation (BSE) methods, second-order Møller–Plesset (MP2) theory, and coupled-cluster (CC) methods. The code is based on Gaussian basis sets and has been pivotal for the development of many fast and low-scaling algorithms in the past three decades, such as integral-direct methods, the resolution-of-the-identity (RI) approximation, fast multipole methods, imaginary frequency integration, Laplace transform, and pair natural orbital methods.
The development of TURBOMOLE was started in the late 1980s by Reinhart Ahlrichs and his group. Integral-direct algorithms and non-Abelian point group symmetry were among the first distinctive capabilities of TURBOMOLE, which initially focused on Hartree–Fock (HF) methods, with subsequent extensions for second-order MP2 perturbation theory1−7 and time-dependent HF (TDHF) response properties.8 A major milestone was the relatively early adoption of density functional theory (DFT) using newly designed quadrature algorithms9 and that of time-dependent DFT (TDDFT) shortly afterward.10 With extensions to meta-GGA,11 RPA, and other fifth-rung methods,12−16 as well as current density17 and local hybrid functionals,18,19 critical performance improvements,20−24 and a plethora of available analytical derivatives of ground- and excited-state energies,25−39 TURBOMOLE has become one of the leading all-purpose molecular (TD)DFT codes. The development and implementation of the RI approximation for Coulomb (RI-J40,41) and exchange contributions, (RI-K42,43), as well as its generalization to post-HF theories such as MP2 and CC244−46 and its multipole-accelerated version for extended systems (MARI-J), were other critical innovations.47 The RI methods are still cornerstones in many modern implementations and outstanding features of the program suite.23,31,48−68 More recent additions include explicitly correlated wave function methods up to CCSD(T) and BCCD(T),68,69 efficient pair natural orbital (PNO) approaches,70−73 solvation models and embedding,74−83 two-component relativistic methods,84−86 GW-BSE type methods52,87,88 real-time (RT) TDDFT,89 and nonadiabatic molecular dynamics.90
To ensure continuity and coordinate the development, maintenance, and distribution independent of individual developers or groups, TURBOMOLE GmbH, a limited liability company located in Karlsruhe, Germany, was founded in 2007. TURBOMOLE GmbH has adopted an irrevocable bylaw preventing the distribution of dividends to ensure that all profits are reinvested into the project. TURBOMOLE GmbH distributes fee-based end-user licenses itself and through partners, as well as free developer licenses and access to the source code based on project proposals.91
Here we focus on recent developments and provide illustrative applications to chemistry and materials science. For an overview of existing features, as well as development, licensing, and distribution, the reader is referred to refs (92) and (93) and the TURBOMOLE Web site.91
2. Brief Feature Overview
The program suite consists of a series of modules with a broad range of methods from universal force field to fast semiempirical methods, state-of-the-art DFT and MP2, and coupled-cluster and post-HF methods for ground and excited states. For convenience, the use of modules is facilitated by various tools such as the scripts woelfling, raman, vcd, and genetic.py for reaction path optimization,103 vibrational Raman spectra,29 vibrational circular dichroism spectra (VCD),104 and genetic algorithms, respectively.105 Moreover, the graphical user interface TmoleX is of great help for running calculations and visualizing results.106
Almost all time-consuming parts are parallelized for multicore systems or clusters using OpenMP107 for shared-memory parallelization (SMP)46,62,95−97 and the message-passing interface108 (MPI) for parallelization across multiple nodes,98−102 as outlined in Table 1. The older Fork-SMP94 is available as a fallback for some modules. Starting with the latest release (V7.7), support for graphics processing units (GPUs) has become available.109
Table 1. Available Parallelizations of Various Modules Shown in Version 7.7a.
| module | functionality | Fork-SMP | OpenMP | MPI | OpenMP/MPI | GPU |
|---|---|---|---|---|---|---|
| dscf | HF/DFT energy | √ | √ | √ | √ | √ |
| grad | HF/DFT gradient | √ | √ | √ | √ | √ |
| ridft | RI-HF/RI-DFT energy | √ | √ | √ | X | √ |
| rdgrad | RI-HF/RI-DFT gradient | √ | √ | √ | X | √ |
| aoforce | HF/DFT Hessian | √ | √ | √ | √ | √ |
| escf | HF/DFT/GW-BSE excitation energies | √ | √ | √ | √ | √ |
| egrad | HF/DFT excited-state gradient | √ | √ | X | X | √ |
| mpshift | NMR/EPR parameters (HF/DFT/MP2) | X | √ | X | X | √ |
| evib | electron transport (HF/DFT) | √ | √ | √ | √ | X |
| odft | orbital-dependent DFT energies | X | √ | X | X | X |
| mpgrad | MP2 energy, gradient | √ | X | √ | X | X |
| ricc2 | RI-MP2, ADC(2), CC2 energies, gradients, spectra | X | √ | √ | √ | X |
| pnoccsd | PNO-MPPT and PNO–CC energies with F12 | X | √ | √ | √ | X |
| ccsdf12 | CCSD, CCSD(T) energies with F12 | X | √ | X | X | X |
| rirpa | RPA energy, gradient | X | √ | X | X | X |
| riper | periodic HF/DFT energy, gradient | X | √ | X | X | X |
The list of parallelized HF/DFT modules includes those for molecular self-consistent field (SCF) energy (dscf and ridft) and gradient calculations (grad and rdgrad), response properties such as vibrational frequencies (aoforce), NMR/EPR spectra (mpshift), excited-state properties (escf, egrad), and electron transport properties (evib). For these modules, the OpenMP version is recommended for most calculations due to its cost–benefit ratio in terms of computer hardware. Accordingly, post-Kohn–Sham methods (rirpa) and calculations with periodic boundary conditions at the HF/DFT level (riper), as well as the CCSD and CCSD(T) program ccsdf12, are only parallelized with OpenMP. In contrast, the implementation of approximate CC methods in ricc2 and pnoccsd widely supports the OpenMP and MPI standard and combinations thereof for large-scale calculations on multiple nodes.
3. Recent Developments
3.1. Local Hybrid Functionals for Strong Correlation and Range-Separated Local Hybrid Functionals
Local hybrid functionals (LHs)110,111 with a position-dependent exact-exchange (EXX) admixture governed by a local mixing function (LMF) have been part of TURBOMOLE since release V7.2 and have since been extended in various ways over the past years. Using seminumerical integration techniques, such LHs have been implemented in an efficient way in the code,18 with functionalities that exceed by far those available in any other quantum-chemistry package that contains LHs. Beyond ground-state SCF112 and nuclear gradients,33 this now includes linear-response TDDFT energies19,113 and excited-state gradients,35 frequency-dependent and frequency-independent polarizabilities,113,114 NMR chemical shifts115 and spin–spin coupling constants,60,86 EPR hyperfine couplings and g-tensors,116,117 the related NMR shieldings of paramagnetic systems,118 magnetizabilities,119 and quasiparticle states based on GW.119 From the variety of possible applications and evaluations, many of which have been touched upon in the 2020 overview of TURBOMOLE92 and in a 2019 comprehensive review of local hybrids,110 we highlight in particular the outstanding performance of LHs for mixed-valence systems120−122 and for phosphorescence spectra.123−125 We also point to further recent LH publications and to reviews by other authors.111,117,119,126−128
Here, we focus on two recent extensions. Essentially, the aims of these works have been to conserve the established advantages of LHs and improve other aspects. We start with the fundamental goal to escape the often invoked “zero-sum game”129,130 between reduced self-interaction errors and delocalization errors or “fractional-charge errors” (FCEs) on one side and minimizing static-correlation errors or “fractional-spin errors” (FSEs) on the other side.131 The enhanced EXX admixture usually helps minimize FCEs, and LHs have been shown to achieve this goal while retaining some of the important left–right correlation in bonding regions.110 On the other hand, larger EXX admixtures usually are detrimental in cases with large FSEs, such as dissociating or stretched bonds or many transition-metal systems with appreciable static correlation. Standard LHs so far have not been a way out of this dilemma, at least not to a larger extent. Relevant real-space approaches to reduced FSEs are Becke’s B13 functional132 and a modified approach by Kong and Proynov (KP16/B13),133 which have both been implemented self-consistently into a local developer’s version of TURBOMOLE.134
Circumventing the numerical difficulties and poor SCF convergence in many cases of the B13 and KP16/B13 functionals, the idea of a local strong-correlation factor has recently been transferred to the LH framework. Initial attempts were still based on relatively simple first-generation LHs but did already show that FSEs can be reduced when multiplying the LH term for nondynamical correlation (NDC) by a somewhat modified KP16/B13-type qAC factor.134 Most recently, the more advanced scLH22t functional has been constructed.135 It is based on the more recent and overall more accurate LH20t functional.120 Using a damping factor for smaller NDC contributions, an almost complete decoupling between the underlying LH20t and the added qAC factor could be obtained. That is, the optimized parameters of LH20t, as well as its excellent performance for weakly correlated situations (e.g., for GMTKN55 main-group energetics), are retained, but FSEs and the related spin-restricted dissociation curves of covalent bonds are dramatically improved.135 Notably, the qAC factor forms part of a new LMF (Figure 1).
Figure 1.

Spin-restricted dissociation curve of P2 with the original (LH20t) and sc-corrected t-LMF (scLH22t) along the bond axis for different distances on the dissociation curve (right). See ref (135) for related graphics for other molecules.
The second extension of LHs has been the implementation and construction of range-separated local hybrids (RSLHs), combining the ideas of local hybridization in real space and range-separated hybrids in interelectronic distance space.136,137 That is, instead of full-range semilocal exchange, short-range exchange is mixed in. This corresponds to the use of a long-range EXX admixture governed by a suitable range-separation parameter ω. Based on earlier work for locally range-separated hybrids,138 RSLHs have been implemented for ground-state SCF (modules dscf/ridft) and gradients (modules grad/rdgrad), as well as for linear-response TDDFT (module escf).139 Using this implementation, the ωLH22t RSLH has been constructed and optimized. It retains most of the good performance of LH20t for main-group and transition-metal energetics, as well as for core, Rydberg, and triplet valence excitations. At the same time, however, it decisively corrects errors for excitations with appreciable charge-transfer character and improves on the potential-energy curves of three-electron cations known to be affected strongly by self-interaction errors.139 Most recently, ωLH22t has been demonstrated to provide unprecedented accuracy in quasiparticle energies for applications in molecular electronics and organic photovoltaics, without the usual system-dependent tuning of range-separated hybrids.140
3.2. Inclusion of the Current Density in DFT
The kinetic energy density τ(r) is a commonly
used ingredient in many functionals to detect iso-orbital regions
or describe the inhomogeneity of the electron density ρ(r). For its extension τ(r,t), used in the time-dependent Kohn–Sham formalism (TDKS),
it has been shown17 that this quantity
is not invariant under a gauge transformation in the external potential.
Substitution of τ by its generalization141−143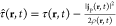 , where jP is the
paramagnetic current density, restores gauge invariance. This leads
to additional terms in the magnetic orbital rotation Hessian in linear-response
TDDFT calculations, accounting for the response of jP. While the original implementation of these terms in TURBOMOLE
dates back to 2012 (release V6.4), we highlight four recent important
updates.
, where jP is the
paramagnetic current density, restores gauge invariance. This leads
to additional terms in the magnetic orbital rotation Hessian in linear-response
TDDFT calculations, accounting for the response of jP. While the original implementation of these terms in TURBOMOLE
dates back to 2012 (release V6.4), we highlight four recent important
updates.
First, an incorrect prefactor 2 has been removed from the original implementation with V7.6.119,144 TDDFT calculations employing τ-dependent functionals performed with previous versions erroneously overcorrected for the effects of the current density response and should be reassessed, although average changes are on the order of 0.03 eV.144
Second, a recent investigation145 reveals that the effect restoring gauge invariance has on the final excitation energies can be significantly larger than previously assumed depending on the functional and type of excitation. In one particular investigation of d–d excitations in nickel(II) complexes, restoring gauge invariance shifts the excitation energies with the M06-2X functional by more than 0.4 eV closer to the experimental reference values, as shown in Figure 2.145
Figure 2.
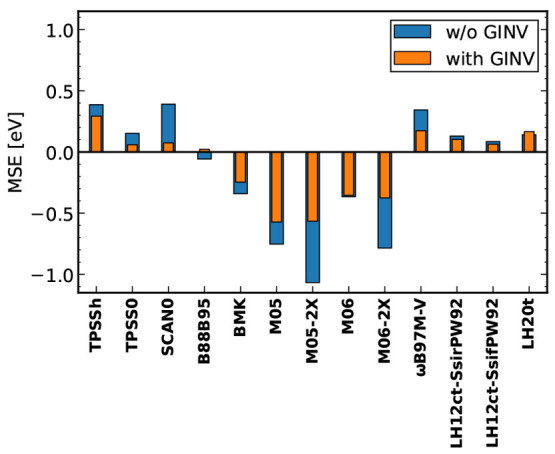
Effect of restoring gauge invariance (GINV) for various τ-dependent functionals on the average deviation of vertical TDDFT excitation energies from the experimental absorption maxima for five Ni(II) complexes.146 Reprinted from ref (145) with permission. Copyright 2022 AIP Publishing.
A broader analysis reveals that the importance of imposing gauge invariance can be linked to the derivative of the exchange energy integrand with respect to τ.145 Moreover, n → π* excitations are significantly more affected by restoring gauge invariance than most π → π* excitations with the exception of π → π*⊥ excitations, where the dominantly contributing molecular orbitals (MOs) are perpendicular (⊥) to each other.145 These findings suggest that a reassessment of previously reported TDDFT results obtained with τ-dependent functionals is warranted, particularly for cases that are potentially more sensitive due to the choice of the functional, the type of excitation, or both. Gauge invariance is restored by default with τ-dependent functionals at moderate (nonhybrid functionals) or negligible (hybrid functionals) additional computational cost.17 Recently, excited-state gradients and quadratic response properties for τ̂-dependent meta-generalized gradient approximations (mGGAs) have been implemented, enabling gauge invariant computations of excited-state equilibrium structures, relaxed dipole moments, (dynamic) hyperpolarizabilities, and two-photon absorption cross sections.147 These developments will be available in a future TURBOMOLE release.
Third, the inclusion of jP is required for gauge invariance of magnetic properties and implemented for magnetizabilities,119 NMR coupling constants,119 and NMR shifts of closed-shell119,148 and open-shell systems,118 as well as EPR hyperfine coupling constants149 and g-tensors.118 Recent findings indeed hint at the inclusion of the current density response also being crucial for NMR and EPR properties.118,119,148 Especially for open-shell systems, neglecting the current density leads to large deviations,118 as shown in Figure 3.
Figure 3.

Impact of the current-dependent generalization of τ for various density functional approximations on the isotropic Δg-shift of [MoNCl4]2–, [MoOF4]−, [MoOCl4]−, [MoOF5]2–, [MoOBr5]2–, [TcNF4]−, [TcNCl4]−, and [TcNBr4]−. Results with the well-known field-dependent generalization serve as reference.150 We list the mean signed percentwise deviation (MSPD), the mean absolute percentwise deviation (MAPD), and the root-mean-square deviation (RMSD). Reprinted from ref (118) under a CC BY license. Copyright 2022 the Authors.
Finally, it was shown that the inclusion of the paramagnetic current is also crucial in relativistic two-component calculations.151 Contrary to all previous cases, the modifications outlined above must already be taken into account for the ground state in the presence of spin–orbit coupling (SOC). SOC gives rise to an internal magnetic field, inducing a paramagnetic current that already has a nonvanishing contribution at the energy-level.151 Accordingly, if properties such as light–matter interactions are targeted in the presence of SOC, the interplay of the ground- and excited-state paramagnetic currents must be taken into account. This gives rise to highly nonlinear jP-dependent terms, which have a profound impact on many properties.151,152 Current density functional theory (CDFT) for τ-based functionals is available for ground-state energies (module ridft) and gradients (module rdgrad) and linear response properties (modules escf and mpshift).151 Given the profound impact of jP, we therefore strongly recommend the exclusive use of the current-dependent forms.
3.3. Methods for Finite Magnetic Fields
Quantum-chemical calculations are routinely carried out for various types of molecular properties in magnetic fields, including NMR, EPR, and magnetic circular dichroism (MCD) spectroscopy (see also sections 3.4, 3.5, 3.6, and 3.9.2 of this Review).153−155 For these applications, the magnetic field is usually treated perturbatively, as it is orders of magnitude smaller than the electronic interactions responsible for the formation of a chemical bond.
Other applications necessitate the use of a more general approach, particularly if the magnetic field becomes strong enough to compete with the electronic interactions within a molecule (>1000 T).156−163 Such conditions may be found in the vicinity of interstellar objects like magnetic white dwarfs and cannot be reproduced in a laboratory.164−167 Consequently, spectra obtained from such interstellar objects can only be interpreted using quantum-chemical calculations168−172 In such extreme conditions, entirely new types of chemical bonding, spin-phase transitions, and other exotic phenomena have been shown to occur.162,173,174 The effects of arbitrarily weak or strong magnetic fields on atoms or molecules may be computed using the finite magnetic field approach, which is implemented for Hartree–Fock,175 CDFT,152,176GW/BSE,177,178 RPA,179 and CC2.178 Through the calculation of electronic ground states, molecular gradients, and electronic excitations, a wide variety of applications for molecular spectroscopy in magnetic fields are now accessible.
Due to the efficiency of our implementation, systems containing dozens of atoms can be routinely computed in explicit magnetic fields.152,175,176,178 To highlight the capabilities of our approach, we calculated the MCD spectrum of ZnDiNTAP,176,180 a zinc tetraazaporphyrin with two fused naphthalene units, Figure 4a, using CAM-B3LYP and a mixed def2-TZVP (Zn)/def2-SVP (all other atoms) basis set. Similarly to the experiment,180Figure 4b, a magnetic field of 5 T was applied. The resulting spectrum is shown in Figure 4c. Minor differences can all be attributed to solvation effects and vibronic coupling, which were neglected in our calculation.176 Furthermore, the MCD spectrum of ZnDiNTAP in an explicit magnetic field of 1000 T is shown in Figure 4c. While some of the bands, particularly in the fingerprint region, are not affected by nonlinear effects induced via such a strong field, the two Q bands are significantly shifted (650 → 693 nm and 536 → 514 nm).
Figure 4.

MCD spectrum of ZnDiNTAP. (a) Molecular structure. (b) Experimental spectrum. Reprinted with permission from ref (180). Copyright 2007 RSC Publishing. (c) Spectra as calculated in finite magnetic fields of 5 and 1000 T. Adapted with permission from ref (176). Copyright 2022 the Authors.
Other applications of the finite magnetic field ansatz include higher-order properties such as magnetizabilites and hypermagnetizabilities through the use of numerical derivatives. Moreover, molecules that are naturally prone to exhibiting “magnetic” effects, such as aromatic compounds (see also section 3.7), may show a nonlinear response even to weak magnetic fields.152 This is not captured by a perturbative approach but can be routinely investigated using our implementation.
3.4. EPR Properties and Single-Molecule Magnets
Over the past decade, TURBOMOLE has pioneered the in silico study of f-element chemistry. To provide accurate descriptions of compounds containing these heavy elements, robust DFT routines are leveraged to deliver a balanced treatment of dynamic correlation, static correlation, solvation, and relativistic effects. More recently, these developments have enabled the discovery of new species with novel chemistry, subsequently necessitating new and improved computational methods capable of describing them.
For example, a series of recently discovered Ln-based single-molecule magnets (SMMs), [La(OAr*)3]−, [Lu(NR2)3]−, and [Lu(OAr*)3]− (OAr* = 2,6-Ad2-4-t-Bu-C6H2O, Ad = adamantyl, t-Bu= tert-butyl, R = SiMe3 with Me = methyl), were found to exhibit exotic EPR properties, with [Lu(OAr*)3]− producing hyperfine coupling (HFC) constants of unforeseen magnitude and furthermore demonstrating extended magnetic coherence facilitated by a hyperfine clock transition. A primary investigation of these species with nonrelativistic HFC operators attributed the large hyperfine coupling constants to a sizable Fermi contact contribution from the highest occupied MO (HOMO) of each system. However, the predictions of hyperfine coupling constants and g-tensor values themselves produced errors of roughly one order of magnitude, strongly suggesting the need for more rigorous methods.181
Such improved predictions of EPR spectra are possible with relativistic exact two-component (X2C) theory,38,39 including spin–orbit effects up to the noncollinear two-component (2c) DFT framework.38,39,117 For meta-generalized gradient approximations and local hybrid functionals, this also includes the paramagnetic current density in the ground state (cf. section 3.2).151 This rigorous formulation can be truncated to the scalar-relativistic limit63 or a perturbative ansatz149 to study the individual contributions of each term to the EPR parameters. Gauge origin invariance of the g-tensor calculations is ensured by the gauge including atomic orbitals,36,39,149 which are crucial for systems with a spatially distributed spin density. These methods were implemented, and their performance is shown here for the aforementioned SMM [Lu(OAr*)3]− shown in Figure 5. The all-electron relativistic methods lead to good agreement with the experiment, as shown in Table 2. The HFC constant of [Lu(OAr*)3]− is dominated by the scalar-relativistic contribution due to the localization of the spin-density in the 6s/d HOMO producing a large Fermi contact interaction. The importance of the paramagnetic spin–orbit contribution increases with the number of unpaired electrons and the scalar formulation, as well as the spin–orbit perturbation theory (SOPT) break down for systems such as [TbPc2]− with six unpaired electrons, Pc = bis(phthalocyaninato).38,39,117,151 The self-consistent 2c methods are thus pivotal.
Figure 5.

Molecular structure of single-molecule magnets [Lu(OAr*)3]− and [TbPc2]−. Reprinted with permission from ref (151) under a CC BY license. Copyright 2022 the Authors.
Table 2. Principal Components for the HFC A of [Lu(OAr*)3]− and Isotropic HFC A of [TbPc2]− at Various Levels of theory38,39,117,149 and Comparison with Experimental Results (expt.)181,182a.
| [Lu(OAr*)3]− |
[TbPc2]− | ||||
|---|---|---|---|---|---|
| method | Axx | Ayy | Azz | A | |
| TPSS | SR | 3192 | 3192 | 3148 | 190 |
| cTPSS | SO | 3190 | 3190 | 3153 | 375 |
| ωB97X-D | SR | 3489 | 3489 | 3456 | 136 |
| ωB97X-D | SO | 3464 | 3464 | 3448 | 488 |
| TMHF | SR | 3225 | 3225 | 3171 | 293 |
| cTMHF | SO | 3208 | 3208 | 3161 | 526 |
| Expt. | 3500 | 3500 | 3400 | 519 | |
HFC is given in MHz. SR and SO denote scalar and spin–orbit relativistic results (x2c-TZVPall-2c/x2c-SVPall-2c basis), respectively.
With the next release version, the EPR Euler transformations for the HFC, g-tensor, and electric-field gradient as well as the nuclear quadrupole interaction tensor will further become available for users.39
3.5. NMR Coupling Constants Across the Periodic Table of Elements
NMR spectroscopy is key to the analysis and structure determination not only for organic compounds but also for inorganic systems consisting of heavy elements. The NMR spin–spin coupling constant describes the splitting of the signals or peaks in the NMR spectra and is a characteristic property driven by the chemical environment. Formally, the coupling tensor is obtained as the mixed derivative of the energy with respect to the corresponding nuclear magnetic moments, which are introduced via the principle of minimal coupling. NMR couplings are available within a nonrelativistic60 scalar X2C183 and the spin–orbit X2C framework.86 All functional classes up to local hybrids are supported and include the current density for gauge invariance.61,119,151
For systems made up of light elements, the nonrelativistic treatment is sufficient. Here, the coupling constant is generally partitioned into the Fermi-contact (FC), spin–dipole (SD), paramagnetic spin–orbit (PSO), and diamagnetic spin–orbit (DSO) contributions. The FC, SD, and PSO terms necessitate the solution of the response equations, whereas the DSO term is computed with the ground-state density. Typically, the FC contribution is the leading term, and accurate coupling constants require large basis sets. Thus, a nuclear selection scheme and locally dense basis sets are often applied to large-scale calculations.
Systems containing heavy elements such as Sn, Pb, Pd, and Pt require the inclusion of relativistic effects,61,86,183,185−187i.e., methods based on the Dirac equation are introduced. For such methods, the FC, SD, and PSO terms are coupled due to spin–orbit interaction, and they come with drastically increased computational demands. Nevertheless, when using a local X2C ansatz,84−86 large-scale calculations are possible, as illustrated in Figure 6 for the Karplus relationship of Me3Sn–CH2–CHR–SnMe3, where R is different substituents (Me = CH3). The relativistic DFT approach reproduces the experimental findings with fairly good agreement. Improvements are possible with the correlation kernel augmented BSE (cBSE) based on the Green’s function GW ansatz. Here, the DFT response equations are replaced with their BSE counter parts.61,113
Figure 6.
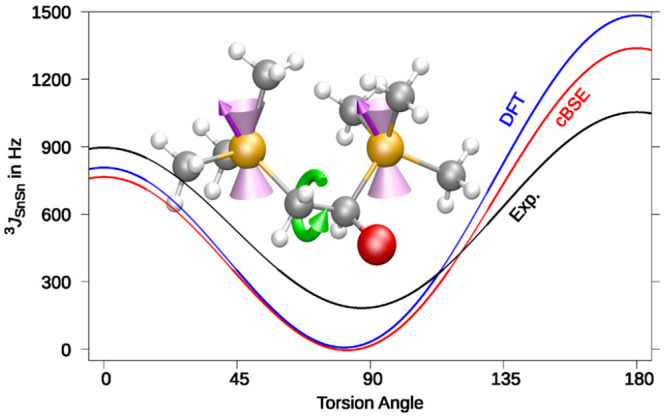
Fitted Karplus equation for Sn compounds: 3J = Acos(2ϕ) + Bcos(ϕ) + C. For each Sn–Sn torsion angle ϕ, the average of the 3JSnSn coupling constant over 13 compounds is computed. Blue, BH&HLYP/x2c-TZVPall-2c; red, GW-cBSE@BH&HLYP; and Exp., experimental findings.184 Adapted with permission from ref (61). Copyright 2022 the Authors.
To demonstrate the efficiency, the calculation of the Sn–P coupling constants of [({SIDipp}P)2Sn] (SIDipp = 1,3-bis(2,6-di-isopropylphenyl)-imidazolidin-2-ylidene) with 137 atoms187 takes about 44 min (PBE) and 55 h (PBE0) using 12 OpenMP threads of an Intel Xeon Gold 6212U CPU (2.40 GHz).86 Notably, using a single NVIDIA A100 GPU, the PBE0 timing can be reduced to 3.5 h.
3.6. Paramagnetic NMR Shieldings and Shifts
NMR spectroscopy is also an important technique for the characterization of open-shell chemical compounds. NMR shielding tensors and chemical shifts describe the positions of the peaks in NMR spectra. In the closed-shell case, only the temperature-independent orbital contribution is relevant to the calculation of the shielding tensor. Both a nonrelativistic treatment3,62,188 and a scalar-relativistic treatment36,104 are available for the orbital contribution, including the response of the current density.118,119,148 The paramagnetic NMR (pNMR) shielding tensor σtotI for a nucleus I reads
| 1 |
with S denoting the spin, μe denoting the Bohr magneton, γI denoting the gyromagnetic ratio of nucleus I, kB denoting the Boltzmann constant, and T denoting the temperature. Here, the orbital contribution σorb is the straightforward open-shell generalization of the closed-shell limit.63,118 Additionally, a temperature-dependent contribution arises, which includes the HFC tensor AI of nucleus I and the g-tensor g already discussed in section 3.4. Both the HFC and the g-tensor depend on spin–orbit coupling. For the calculation of 1H/13C pNMR spectra of large molecules, a perturbative treatment of spin–orbit coupling is preferred over the 2c ansätze due to lower computational costs.149 The viability of the pertubative ansatz in the X2C framework is demonstrated for two negatively charged Ru(III) complexes in Figure 7, which depicts the good agreement between calculated results and the experimentally measured189 pNMR 1H and 13C shifts of the two compounds.
Figure 7.
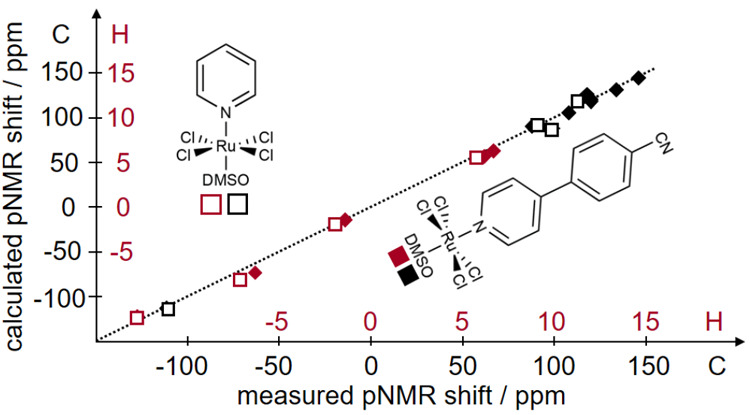
Computational results (ωB97X-D/x2c-TZVPall-s) and experimental findings for two negatively charged Ru(III) compounds. Adapted with permission from ref (149). Copyright 2022 the Authors.
The calculation of properties depending on the density in the vicinity of the nuclei requires additional tight basis functions. Thus, the pcJ,190 pcS,191 and pcH192 basis sets are recommended for nonrelativistic calculations. For relativistic calculations, the x2c-s basis sets were developed.193,194
The efficiency of the pNMR implementation is similar to that of the closed-shell case, as shown in Figure 8. Coulomb integrals can be calculated with the RI-J/MARI-J approximations for the Coulomb contribution62,63 and the seminumerical scheme for exchange integrals.18,109
Figure 8.
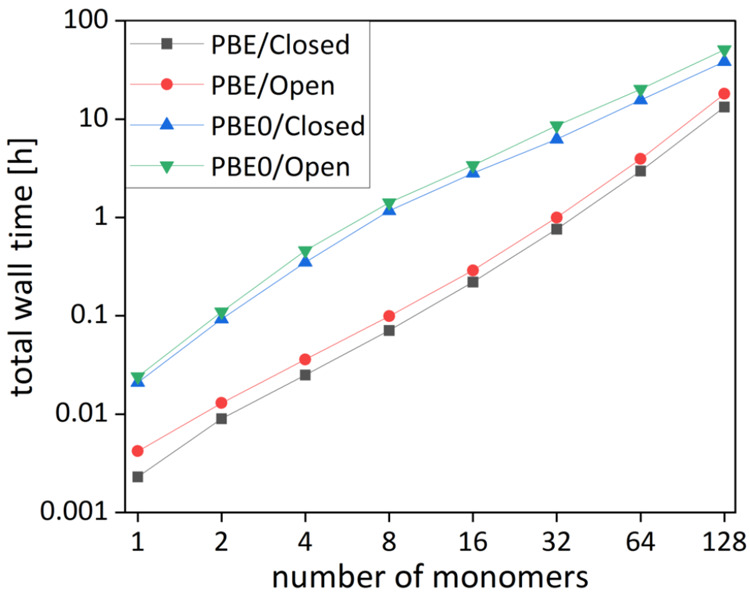
Total wall times for SCF and NMR calculations with respect to the number of glucose units (6-31G* basis). Wall times were measured on a single thread of an Intel Xeon Gold 6212U CPU (2.40 GHz). Reprinted with permission from ref (63). Copyright 2021 the Authors.
3.7. Ring Currents of Heavy-Metal Clusters
Aromatic compounds, such as benzene, show characteristic signals at 7 ppm in the 1H NMR spectra. This shift is a consequence of the cyclic electron delocalization associated with the π-orbitals, which deshield the nuclei due to an induced ring current.195 This magnetically induced current density may be calculated indirectly with the nucleus-independent chemical shift196 (NICS) or directly using TURBOMOLE’s interface to the GIMIC program, which was reworked for release V7.7 and now supports open-shell calculations.197−200 The latter approach is more flexible and also applicable to complicated multicyclic systems.201−204
The occurrence of ring currents and the concept of aromaticity are not restricted to cyclic conjugated hydrocarbons and related organic compounds. All-metal systems may also sustain a ring current, and these systems are therefore classified as all-metal aromatic compounds. For instance, the endohedral [Th@Bi12]4– cluster features a nonlocalizable π-orbital around the {Bi12} torus, which leads to a ring current.205Figure 9 shows a streamlined representation of this ring current, whose strength amounts to about 25 nA/T.119,205
Figure 9.
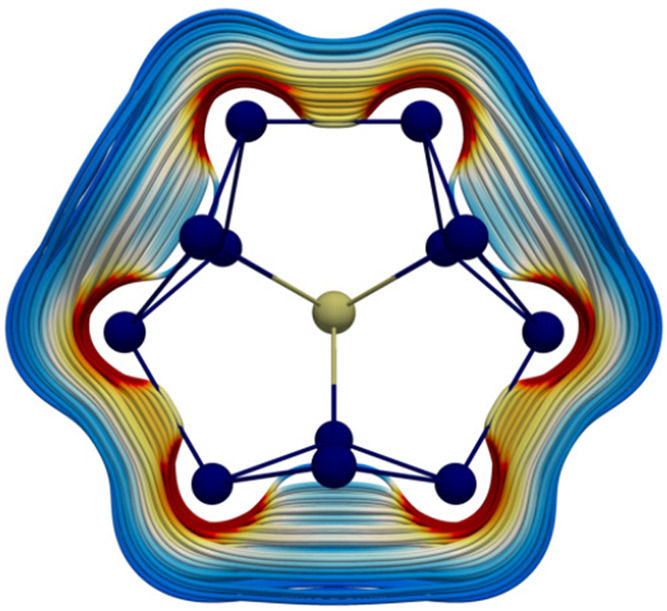
Aromatic ring current of [Th@Bi12]4–. The color scheme (red to blue) indicates strong to weak currents. Data from ref (205)
Despite featuring only two delocalized π-electrons, almost the same ring current strength as that in porphine is induced. Here, the thorium atom in the torus center is only needed for stability and the synthesis, but it does not take part in the ring current. Furthermore, the open-shell variant [U@Bi12]3– shows a strong ring current,63 as the same π-orbital is occupied by two electrons.205,206 Moreover, prismatic {Bi6}-based clusters such as [(CpRu)3Bi6]− show ring currents of more than 25 nA/T.207 Therefore, all-metal clusters help to push the frontiers of aromaticity.
3.8. Characterization of Novel Electronic Configurations in f-Block Elements by DFT and RPA Methods
Electronic structure calculations of large f-element complexes contain a great deal of complexity due to the competition between metal oxidation states, d- and f-shell occupations, spin coupling, and relativistic effects.208 Computational studies in these compounds have to scan a range of formal electronic occupations and apply a range of techniques to ensure convergence to the desired electronic configuration, including Fermi smearing with suitable parameters,209,210 and a combination of damping, level shifting,211 and direct inversion in the iterative subspace (DIIS) extrapolation.212 Moreover, the stability of the ground-state reference is a concern in calculations of molecular properties, for example, electronic absorption spectra.213
DFT results helped characterize the structures and properties of nontraditional Ln2+ complexes possessing the 4fn5d1 configuration in the [Ln(C5H4SiMe3)3]− (Ln = Ce–Nd, Gd–Er),214−217 [Ln{N(SiMe3)2}3]− (Ln = La, Gd),218,219 and [Ln(CpiPr5)2] (Ln = Tb, Dy) series.220 The preference for the 4fn5d1 configuration relative to the traditional 4fn+1 occupation of Ln2+ results from the stabilization of the Ln 5dz2 orbital by the trigonal ligand environment or extremely bulky ligands. Nontraditional Ln2+ complexes show a characteristic intense absorption band in the visible range due to excitations from the occupied Ln 5d orbital. The prediction of UV–vis spectra of low-valent lanthanide complexes, in particular those with a nontraditional configuration, is improved by including diffuse augmentation in lanthanide basis sets.221 DFT and RPA methods were employed to examine the strong ferromagnetic coupling between the Ln3+ centers in [(C5Me5)2Ln(μ-S)2Mo(μ-S)2Ln(C5Me5)2]− (Ln = Y, Gd, Tb, Dy) and the Mo→Ln electronic excitations in the near-infrared spectral region.222 Excited-state studies of [Ln(C5Me5)2(C5Me4H)] and [Ln(C5Me5)2(η3-C3H4)] complexes (Ln = Y, Dy, Lu) using TDDFT elucidated their unexpected photochemical activation, which was used to reduce dinitrogen and sulfur and to polymerize isoprene.223,224
Computational studies of neutral actinide complexes [An(CpiPr5)2] (An = Th, U, Pu, Am, Bk, No, Lr) (pentaisopropylcyclopentadienyl = CpiPr5) using DFT predicted ground states with a linear ligand arrangement of S10 symmetry and significant An 6d orbital occupation for An = Th, U, Lr.225 The calculations were carried out with the TPSS exchange–correlation functional,226 Stuttgart–Cologne scalar-relativistic small-core effective core potentials (ECPs),227 and the corresponding basis sets.228,229 Mixed 5f/6d occupation was predicted in the corresponding Pu complex, while the An = Am, Bk, No complexes were found to have 5fn+1 configurations. The Pu and Am complexes showed a slight deviation from the perfectly symmetric structure, with Cp–M–Cp bending angles of 11° and 12°, respectively. The simulated absorption spectra showed intense peaks in the UV–vis range due to the metal–ligand charge transfer excitations from the An 6d orbital shown in Figure 10. Comparisons with the previously experimentally known Ln analogs (Ln = Dy, Tb)220 suggested that the synthesis of the predicted actinocene complexes was thermodynamically feasible. The computational predictions received experimental confirmation for An = U while the results were still under review. Layfield and co-workers reported the synthesis of the linear S10-symmetric “second-generation” uranocene [U(CpiPr5)2].230 The U–Cp centroid distance was determined from crystallographic studies as 2.504 Å, in good agreement with the computational result of 2.483 Å. The measured UV–vis spectra showed broad and intense absorption, as predicted by TDDFT calculations.
Figure 10.
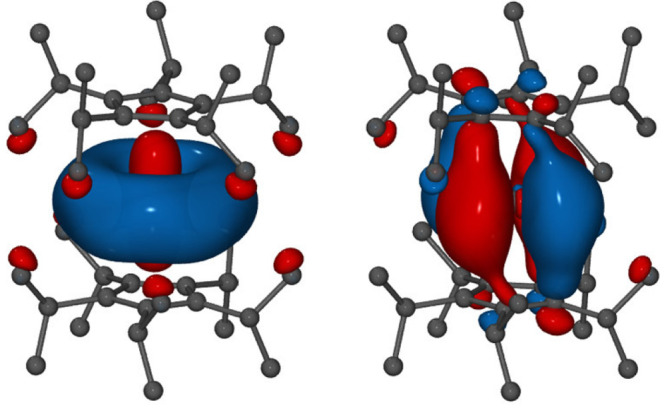
Contour plots of the HOMO (left) and LUMO (right) of [An(CpiPr5)2] determined with TPSS,226 Stuttgart–Cologne scalar relativistic ECPs,227 and the corresponding basis sets.228,229 Orbital isovalues of 0.03 were used. Reprinted with permission from ref (225). Copyright 2019 American Chemical Society.
3.9. Damped Response Theory
Assessing light–matter interactions in extended or exotic systems is an active field of development in TURBOMOLE.53,119,152,176,178,231,232 While root-by-root linear-response methods have been crucial for many applications, they are not suitable for spectrally dense systems or core excitation due to the high number of excited states (roots). Damped response theory,233,234 or the equivalent complex polarization propagator approach,235 provides a convenient framework to formulate resonance convergent response functions, circumventing these problems. It provides a convenient route to directly compute a variety of absorptive and dispersive effects in both UV–vis and X-ray frequency regions, which is particularly advantageous for large systems, and in frequency regions with high densities of states, as it does not require to solve eigenvalue equations for all contributing states and individual transitions matrix elements between them.236 It also allows to compute nonlinear transition properties in the vicinity of additional resonances, e.g., the resonant inelastic X-ray scattering (RIXS) transition amplitudes,237 as well as properties at imaginary frequencies, like the C6 dispersion coefficient.232,238−240
A number of implementations of damped response theory have been presented in the past two decades for time-dependent density functional theory and a variety of wavefunction methods.80,113,232,235−238,241−254 TURBOMOLE is, to the best of our knowledge, the only program to date that offers damped linear response functions at the RI-CC2 level of theory232,253 (available since release V7.7).
3.9.1. Damped Response for Multiscale Modeling
DFT-based damped response implementations in TURBOMOLE cover IR spectroscopy, VCD, and Raman spectroscopy, as well as absorption and electronic circular dichroism (ECD) in the visible and ultraviolet spectral range. Furthermore, this approach has recently been extended to the modern framework of the GW-BSE method, being especially useful for core excitations.113,255
At a given complex frequency ωex = ω + iγ, where ω and γ are the real and imaginary parts of the external field, respectively, the coupled perturbed equation254,256
| 2 |
is first solved. A and B are defined as
εp marks orbital or quasiparticle energies, vpq,rs is a Coulomb integral, fpq,rs is the exchange-correlation kernel, if present, and Kpq,rs is a exchange integral. The precise kinds of fpq,rs and Kpq,rs depend on the chosen method.254pν describes the external perturbation, e.g., an electric or magnetic field.254,256 The polarizability can then be calculated as
| 3 |
Damped polarizabilities and the related magnetizabilities have further emerged as the quantum-chemical cornerstone of the transition matrix (T-matrix) based approach for multiscale modeling of light–matter interactions.254,257,258 This way, the functionalization of molecular structures within optical devices is possible. In that regard, optical cavities filled with molecular materials or metasurfaces of cylinders consisting of molecular materials can be designed. These devices exhibit tailored optical properties for a variety of applications, such as enhancing the circular dichroism of a chiral molecule. Through the multiscale approach depicted in Figure 11, the properties of a molecular unit and the macroscopic sample can be distinguished and combined to achieve a specific effect. Combining TURBOMOLE and, for example, the multilayered periodic general Mie method (mpGMM),259 simulations and predictions of the light–matter interactions of layer-structured materials ranging from a few to hundreds of nanometers are now routinely possible.254,260 To target molecular materials of arbitrary shape, the T-matrix approach was furthermore coupled with state-of-the-art homogenization techniques.261 Combining classical electrodynamics and quantum mechanics has proven to be a worthwhile approach in the field of light–matter interactions, and TURBOMOLE will remain at the forefront of these developments.
Figure 11.

Scheme for T-matrix-based multiscale modeling of light–matter interactions using damped response polarizabilities as outlined in ref (254). Reprinted with permission from ref (254) under a CC BY-NC-ND license. Copyright 2022 the Authors.
3.9.2. Damped Response Theory for One-Photon Absorption and CD Spectra with RI-CC2
For RI-CC2, the equations for the damped response237,262 of the cluster amplitudes are recast in a form that only involves effective matrices in the space of single excitations, avoiding the storage of parameters for the double excitation space.
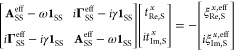 |
4 |
Subscripts Re and Im represent real and imaginary components, respectively. The effective matrices in the equations above are
| 5 |
| 6 |
where ASS, ASD, and ADS are, respectively, the singles–singles, singles-doubles, and doubles-singles blocks of the CC2 Jacobi matrix. Δijab = εa – εi + εb – εj – ω are frequency-shifted orbital energy differences, and ω and γ are again the real and imaginary parts of the frequency of the external field, respectively. The effective right-hand sides are
| 7 |
| 8 |
where ξxS and ξxD are the single and doubles parts of the right-hand sides in the nonpartitioned form, respectively.232,253 The partitioned formulation that avoids the need to store double excitation amplitudes and four-index integrals allows applications to system sizes otherwise not accessible at the CC2 level.
As an illustrative application, we computed the UV–vis absorption spectrum and electronic circular dichroism spectrum of a donor–acceptor cyclophane263 shown in Figure 12. The absorption spectrum was obtained from calculations of the imaginary damped dipole polarizability and the ECD spectrum from the imaginary part of the optical rotation tensor in the velocity gauge. The asymmetric form253 of the damped linear response function was used in the calculations (the symmetric form is also available).
Figure 12.
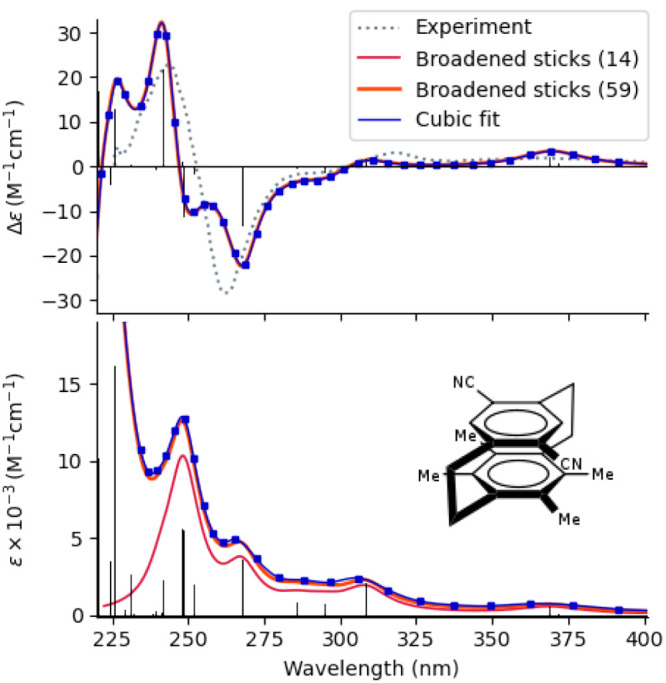
ECD (upper panel) and absorption spectra (lower panel) of (4Sp)-4,7-dicyano-12,13,15,16-tetramethyl[2.2]-paracyclophane computed at the CC2/aug-cc-pVDZ level. The MP2-optimized structure and the experimental CD spectrum have been taken from ref (263). Results from damped response theory are plotted as blue squares; the blue line is a cubic spline fit to these computed points. Stick spectra are the results from state-wise calculations, and the red and orange lines are obtained by convoluting these computed stick spectra with a Lorentzian broadening with a half-width-at-half-maximum of ≈1000 cm–1 including, respectively, the lowest 14 and 59 states.
Cyclophanes are well-studied examples of strained aromatic organic compounds (hydrocarbons) that exhibit planar chirality. The UV–vis spectrum is a typical example where the traditional state-wise approach converges only slowly with the number of states, as seen by the large difference between the results for 14 and 59 states that have been included for comparison. The problem does not appear in the damped response calculations.
Ongoing work is concerned with extending the implementation to induced and nonlinear spectra like MCD and RIXS.253
3.10. Nonadiabatic Molecular Dynamics Simulation for Spectroscopic Observables
Many photophysical and photochemical processes involve multiple electronic excited states coupled by radiative and nonradiative transitions. Efficient simulations of these processes by nonadiabatic molecular dynamics (NAMD) have recently become possible with transition dipole moments30 and nonadiabatic couplings between excited states32,264 computed within quadratic response TDDFT. The TURBOMOLE implementation of the multistate fewest-switches surface hopping (FSSH) algorithm enables simulations of molecular systems with 50–100 atoms and simulation times of >10 ps.265,266 In addition, time-resolved fluorescence (TRF) and transient absorption (TA)34 spectra can be simulated for comparison with experimental results.
The simultaneous treatment of multiple electronic excited states enables the examination of Kasha’s rule.267 According to Kasha’s rule, singlet excited states energetically located above S1 undergo ultrafast decay to the S1 state and thus are not directly involved in fluorescence or photoactivated reactions. However, exceptions to Kasha’s rule are well-known in molecules in the gas phase, such as azulene and pyrene.268
In our recent study, we used the NAMD implementation in TURBOMOLE to investigate the dynamics of several polycyclic aromatic hydrocarbons including pyrene, azulene, and bicyclo[6.2.0]decapentaene (BCDP, an isomer of azulene) at the PBE0/def2-SVP269,270 level. Azulene was found to exhibit non-Kasha behavior due to emission from the S2 state, in agreement with experiment.271 BCDP obeys Kasha’s rule and emits only from S1. Previous studies assigned the high-energy shoulder in the pyrene fluorescence spectrum to non-Kasha emission as a result of the reverse S1 → S2 internal conversion.268,272−275 Multistate NAMD simulations describe the non-Kasha behavior as a combination of S1 → S2 transitions and the change in the diabatic character of the S1 state from dark (Lb) to bright (La) at points of near degeneracy between the S1 and S2 states. The high-energy shoulder in the fluorescence spectrum of pyrene can be understood as originating from excited states with diabatic bright (La) character. The S2 lifetime in pyrene was computed by an exponential fit of the decay of the state population as 63 fs, in agreement with the experiment value of 85 fs in methanol.276 The S1 lifetime of azulene was computed to be 2.2 ps in comparison to the experimental result of 1.4 ps in cyclohexane.277 The computed lifetime of the S1 state of BCDP was found to be 0.8 ps.
The NAMD trajectories were also used to obtain the TA spectrum of pyrene as an ensemble average of the Gaussian-broadened excited-state absorption and emission spectra (Figure 13). The experimental TA spectrum of pyrene shows an intense band at 580 nm and the growth of a steady state signal at around 450 nm corresponding to S2 → Sn transient absorption that decays rapidly and a S1 → Sn transition (n > 4), respectively.276,278 NAMD simulations predict S2 → S4 and S1 → S4 bands at 1500 and 800 nm, respectively. The time evolution of the S1 and S2 states is in good agreement with experiment, while the absorption maxima (λ(S2 → S4) = 1500 nm and λ(S1 → S4) = 800 nm) are red-shifted due to truncation of the electronic excitation space.
Figure 13.

Computed (left) and experimental (right) TA spectra of pyrene in the gas phase. Band origins of S1 and S2 were shifted by −0.42 and −0.02 eV, respectively. Experimental spectra reprinted from ref (278). Copyright 1999 Hindawi Publishing Corporation. Distributed under a CC-BY license.
3.11. Generating Function Methods for Vibrationally Resolved Electronic Spectroscopy
The vibrational structure of electronic spectra gives detailed information about molecular structure279 and excited-state phenomena such as internal conversion and intersystem crossing.280,281 In the case of conformationally flexible molecules, molecular dynamics sampling often allows the shape of electronic spectra to be accounted for.282−287 However, this approach fails for rigid molecules with high vibrational frequencies.288 In this case, special care must be taken to include the quantum nature of nuclear vibrations. Efficient theoretical simulation of molecular vibrationally resolved electronic (i.e., vibronic) spectra has been advanced by switching from the time-independent approach,289 which requires a tedious sum-overstates evaluation of Franck–Condon overlaps, to the time-dependent formalism (generating function approach), where the spectrum is given by the Fourier transform of an appropriate time-correlation function.288,290−292 The radless module288,293,294 makes use of the generating function method to compute vibrationally resolved absorption and emission spectra, as well as photoelectron ionization spectra. Spectra can be computed within the global harmonic approximation, which only requires equilibrium geometries for initial and final structures as well as vibrational spectra of both structures. The method accounts for the full Duschinsky rotation,295 taking into account differences in initial and final state structures and vibrational modes. Due to its efficiency, the method can be applied to large molecules, such as polyaromatic hydrocarbons.123,124,293,296,297 An extension of the module further allows the computation of emission and absorption spectra arising from singly occupied vibrationally excited initial states, allowing the simulation of single vibronic level (SVL) fluorescence293 and vibrationally promoted electronic resonance (VIPER) spectra.298
In addition, the newly implemented semiclassical thawed Gaussian approximation (TGA)291,299 offers an efficient way of evaluating the time-correlation function without resorting to the global harmonic approximation. The relation between vibronic spectroscopy and semiclassical dynamics stems from the wavepacket autocorrelation picture of the dipole time correlation function, first popularized by Heller.290 In TGA, an initial Gaussian wave function is evolved under an effective local harmonic potential constructed at each step about the center of the wavepacket. As a result, its Gaussian form is conserved; the center of the wavepacket follows a classical trajectory, while its width is adjusted according to the instantaneous Hessian of the potential energy surface (PES). Whereas in the original ab initio TGA300−303 the Hessian of the potential energy is updated over time, in the single-Hessian version,304−306 implemented in TURBOMOLE as part of the radless module,294 the Hessian is kept constant throughout the dynamics. Therefore, the overall additional cost compared to the conventional harmonic approximation is that of a single ab initio trajectory in the final electronic state, which is simulated using the frog module. Since the trajectory experiences the true anharmonic PES, the method can account for anharmonicity at least approximately, although it cannot describe more subtle quantum dynamics, such as wavepacket splitting or tunneling. The TGA approach has proven to be especially useful in systems with a large displacement between the ground- and excited-state minima and in systems with a double-well-shaped PES along a low-frequency mode in the final electronic state. In such molecules, the harmonic approach typically fails because the global harmonic PES constructed around one of the wells is not adequate. Moreover, in contrast to the global harmonic methods, the TGA results often depend weakly on the choice of the Hessian, as illustrated in Figure 14. Overall, the implementation in TURBOMOLE combines these advantages of TGA with accurate and efficient excited-state electronic structures, such as ADC(2) and CC2 methods.
Figure 14.
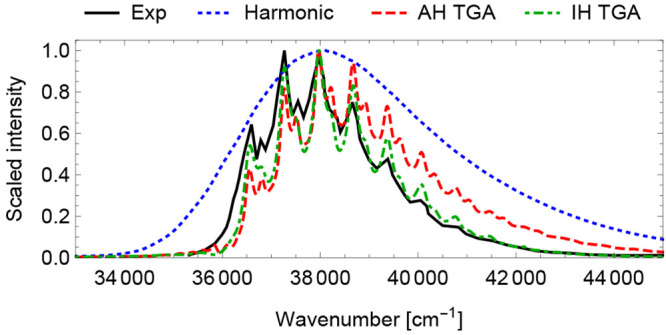
Experimental absorption spectrum of 1,2,4,5-tetrafluorobenzene (black) compared with the spectra simulated using the (adiabatic Hessian, AH) global harmonic method (“Harmonic”, blue dotted line) and single-Hessian TGA with either the adiabatic Hessian (AH, red dashed line, evaluated in the excited electronic state at its corresponding optimized geometry) or the initial Hessian (IH, green dash-dotted line, ground-state Hessian computed at the ground-state optimized geometry). All electronic structure calculations, including geometry optimization, energies, and forces for the ab initio dynamics and Hessians, were performed at the CC2/def2-TZVP level. Adapted with permission from ref (294). Copyright 2022 American Chemical Society.
3.12. Molecular Properties from Self-Consistent GKS-spRPA
The generalized Kohn–Sham semicanonical
projected random phase approximation (GKS-spRPA) provides a route
for obtaining one-particle energies at the RPA level of theory.16 These one-particle energies provide accurate
estimates of ionization potentials (IPs) and electron affinities due
to a correct description of orbital correlation and relaxation effects.
Its computational cost was reduced from 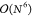 to
to 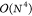 using well-known analytic continuation
(AC) techniques.307−309
using well-known analytic continuation
(AC) techniques.307−309
3.12.1. Applications to Nonvalence Anionic States and X-ray Emission Spectroscopy
The AC version of GKS-spRPA retains a high accuracy across energy scales from valence to core-ionization energies. The versatility of the AC GKS-spRPA was shown by its application to problems involving very weakly bound anionic states and very strongly bound core-hole ionization energies.
Nonvalence anionic states of molecules are weakly bound states where the excess electron occupies a diffuse orbital. The excess electron is bound by a combination of long-range electrostatic and correlation effects. When electrostatics are sufficient to bind an excess electron, the anionic states are referred to as nonvalence electrostatic-bound (NVEB), and when correlation effects are necessary the states are referred to as nonvalence correlation-bound (NVCB). Using a model water tetramer cluster, the GKS-spRPA approach was shown to provide electron affinities (EAs) from the lowest unoccupied molecular orbital (LUMO) energies that were within 10 meV of those provided by the EOM-CCSD(T)a* approach, see Figure 15. The high accuracy of the GKS-spRPA approach is due to the correct description of long-range correlation effects. The AC approach introduces errors of less than 5 meV for nonvalence states.
Figure 15.
EAs of a water tetramer cluster for different dimer separations (R) computed using GKS-spRPA, ΔHF, ΔCCSD(T), and EOM-CCSD(T)a* methods. The inset shows the arrangement of the tetramer with a 70% isosurface of the LUMO at R = 4.047 Å. aug-cc-pVDZ basis sets were used for O and H atoms, and a 7s7p set of basis functions located at the center of the cluster was used. The shaded (unshaded) area of the plot corresponds to NVCB (NVEB) anionic states. Reprinted with permission from ref (309). Copyright 2021 American Chemical Society.
The AC GKS-spRPA method can also be used for simulating nonresonant X-ray emission (XE) spectra using just the information from the one-particle eigenspectrum.310 XE energies, ΔE, computed by taking the difference between core and valence IPs and the oscillator strengths, fosc., are evaluated within a frozen orbital approximation
| 9 |
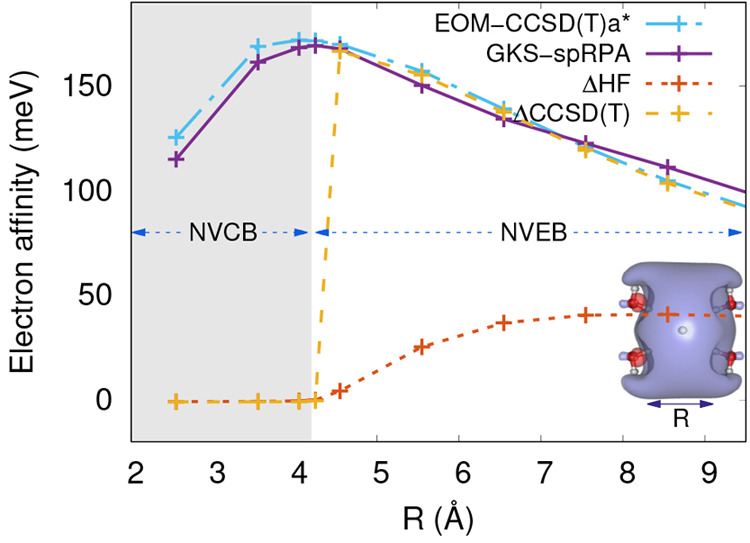
where ϕc and ϕv are the core orbital and valence GKS-spRPA orbitals involved in the XE process, respectively. The AC GKS-spRPA approach was used in conjunction with the scalar-relativistic (SR) X2C approach and uncontracted basis sets to estimate highly accurate XE spectra for molecules containing second- and third-period elements, for example, see Figure 16. Using uncontracted basis sets, the XE energies were found to have MAEs of 0.7 eV for both second and third period-based XE. The X2C-based AC GKS-spRPA approach thus enables the simulation of nonresonant X-ray emission within a simple one-particle picture while avoiding the use of empirical shifts or core-hole reference states. The latter is an appealing aspect, since issues related to variational instability, which are present in core–hole reference based methods, are avoided.
Figure 16.
XE spectra of S8 from experiments311,312 and the SR-GKS-spRPA method.310 The computed spectrum (in blue) was obtained by Gaussian broadening of the vertical transitions (red lines) using a width parameter of 1 eV. The vertical dashed lines denote the two intense peak positions for SR-GKS-spRPA. Reprinted and adapted with permission from ref (310). Copyright 2022 American Chemical Society.
3.12.2. Orbital Ordering in Quinacridone
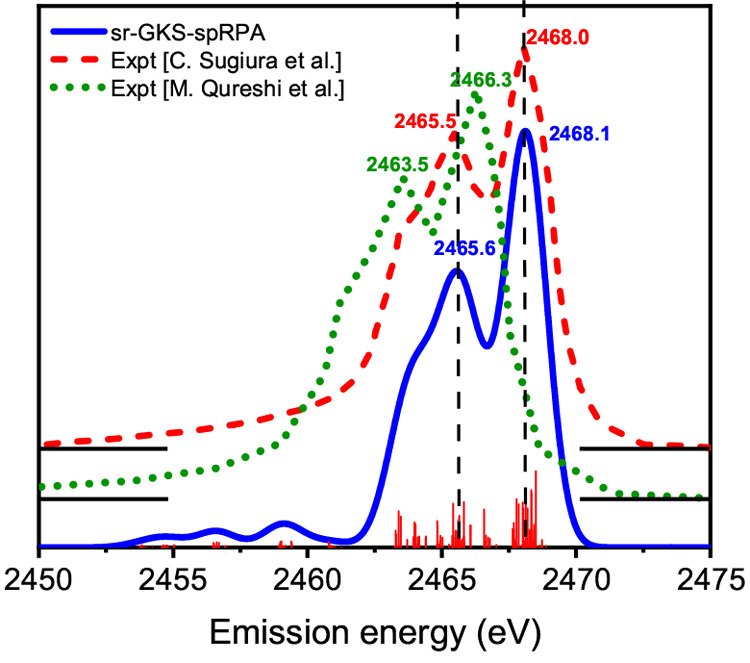
The accuracy of GKS-spRPA16 for ionization potentials (IPs) compared to nonselfconsistent RPA23 is due to partial satisfaction of functional self-consistency, which requires that the Kohn–Sham (KS) density equal the interacting density defined as the functional derivative of the ground-state energy with respect to the external potential.16,317 A comparison of the GKS-spRPA orbitals and orbital energies to experimental photoelectron spectra of quinacridone illustrates this point, see Figure 17. Within both the KS and the GKS approach, the HOMO energy equals the first IP and subsequent lower-lying orbital energies approximate higher principal IPs.318,319 However, this is often not true for semilocal density functional approximations. PBE313 predicts a HOMO(−1), HOMO(−2), and HOMO(−3) ordering inconsistent with the results of optimally tuned range-separated hybrid functional (OT-RSH) calculations, yielding an accurate description of experimental photoelectron spectra and G0W0 IPs.320,321 The canonical GKS-spRPA orbital ordering qualitatively agrees with the one OT-RSH one down to HOMO(−3), see Figure 17. Furthermore, the negative GKS-spRPA HOMO energy of 7.07 eV is close to the experimental IP of 7.23 eV,322 whereas the negative PBE HOMO energy of 4.92 eV is significantly too small.
Figure 17.
Plots of the five highest occupied orbitals at isovalues of ±0.02 shown for (a) PBE and (b) GKS-spRPA (with the PBE potential). The structure of quinacridone was optimized with PBE-D3313−315 and cc-pVTZ basis sets.316 Reproduced from ref (317) with the permission of AIP Publishing.
3.13. Subquadratic-Scaling PNO-CCSD(T) and PNO-CCSD(T)-F12 Methods
Since 2010, TURBOMOLE has supported accurate wave function methods for computing the ground-state energies of molecular configurations, and up to CCSD(T) and BCCD(T) are available, together with their explicitly correlated counterparts CCSD(T)(F12*) and BCCD(T)(F12*).68,69 When combined with a sufficiently large basis set, these methods deliver ground-state energy differences for reaction or interaction energies and potential energy surfaces to an accuracy that enables a quantative comparison with experimental results. The characteristics of the TURBOMOLE implementation are low memory and disk requirements and shared-memory parallelization, which make calculations on systems with ∼20 atoms routinely possible on modern machines.
To evaluate energies for larger molecules, the steep 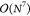 scaling of the costs with system size must
be overcome. Local approximations based on the short-ranged nature
of electron correlation in molecules provide a route to near-linear
scaling of costs with the system size. The PNO approach uses an approximate
MP2 pair density to construct the set of local virtual orbitals for
each pair that is best suited to describing the correlation of that
pair. This, in combination with screening and local density fitting
approximations, among others, reduces the scaling from
scaling of the costs with system size must
be overcome. Local approximations based on the short-ranged nature
of electron correlation in molecules provide a route to near-linear
scaling of costs with the system size. The PNO approach uses an approximate
MP2 pair density to construct the set of local virtual orbitals for
each pair that is best suited to describing the correlation of that
pair. This, in combination with screening and local density fitting
approximations, among others, reduces the scaling from 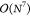 to
to 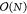 in the asymptotic limit. For ground-state
energies, PNO-MP2, PNO-CCSD, and PNO-CCSD(T) explicitly correlated
variants PNO-MP2-F12, PNO-CCSD(F12*), and PNO-CCSD(T)(F12*) have been
available since V7.6.70−73
in the asymptotic limit. For ground-state
energies, PNO-MP2, PNO-CCSD, and PNO-CCSD(T) explicitly correlated
variants PNO-MP2-F12, PNO-CCSD(F12*), and PNO-CCSD(T)(F12*) have been
available since V7.6.70−73
The efficiency of the implementation is greatly improved if the PNOs are expanded in projected atomic orbitals (PAOs) rather than directly in terms of atomic orbitals (AOs). This is known as domain-based local PNO theory (DLPNO). In TURBOMOLE, the PAO domains for each pair are determined also using the approximate MP2 density rather than by analyzing the MO coefficients and are consequently much more compact. The resulting domains are called principle domains.323 Very recently, the principle domain approach has been extended to F12 theory, where principle domains and PNOs are required for every subspace in the F12 strong orthogonality projector.324 In contrast to implementations in other software packages, we do not use the simplified “A” approximation95 for the MP2-F12 contributions. This has the benefit that the energies converging smoothly to both the canonical limit and the basis set limit can be extrapolated using PNO extrapolation techniques.325
To illustrate the characteristics of the TURBOMOLE implementation and what is now possible, we report timings of PNO-CCSD(T) and PNO-CCSD(T)(F12*) calculations on a sequence of alkane chains and rock salt crystal fragments in Figure 18. The default tight (10–7) PNO truncation threshold was used in all cases. The def2-TZVPP basis and def2-TZVP were used for CnH2n+2 and NanCln, respectively, and calculations were run on a 48 core Intel processor with 200 Gb of memory. For the linear systems, the observed scaling is subquadratic, with C128H256 taking 15 h to complete. For the globular systems, the observed scaling is subcubic, with Na50Cl50 taking 45 h to complete. The F12 calculations are 2–3× more costly than non-F12 calculations but provide energies close to the basis set limit without requiring basis sets with a large number of AOs per atom.
Figure 18.

Wall-clock timings of PNO-CCSD(T) and PNO-CCSD(T)(F12*) calculations for alkane chains (+) and NaCl clusters (cross).
3.14. Real-Time TDDFT for Molecules
While linear-response TDDFT can be used to study excitations under weak fields, a better understanding of nonlinear excited state dynamics at the femto- and sub-femtosecond time scales requires using RT-TDDFT.
RT-TDDFT for molecular systems has been implemented89 within the riper module55−58,327,328 using the Magnus propagator329 and the predictor-corrector scheme330 for time integration. The implementation utilizes density fitting and continuous fast multipole method (DF-CFMM) techniques56 to speed up the KS matrix evaluation and scales almost quadratically with system size. Previously, the implementation was used to simulate absorption spectra over a wide range of frequencies by perturbing the molecules with weak electric fields.89 Recently, the code has been extended to simulate high harmonic generation (HHG) spectra under intense laser pulses and was utilized to complement and analyze the pulse-induced electron dynamics in the organic semiconductor molecules tetraphenylporphyrin (TPP) and zinc-tetraphenylporphyrin (ZnTPP).326Figure 19 shows the good agreement of the experimental absorption and HHG spectra with the calculated ones. The difference in the first few harmonics is due to the fact that the experimental spectra contain contributions from the quartz substrate. Overall, the HHG and absorption spectra of both the porphyrins are quite similar due to the very similar electronic structure. Our simulations combined with experiments showed that π → π* excitation plays a major role in the harmonic generation process in porphyrins. It was also discovered that resonant excitation leads to an early onset of nonperturbative behavior for the fifth harmonic, and similar effects are expected in Brunel harmonic generation with other organic materials.326
Figure 19.

(a) Experimental linear absorption spectra and (b) HHG spectra of 100 nm thick TPP and ZnTPP samples. (c) Calculated linear absorption spectra and (d) HHG spectra of TPP and ZnTPP molecules.326
3.15. Developments of the DFT-Based Embedding Implementations
3.15.1. Frozen Density Embedding Implementation
Environment effects on molecular systems beyond static Coulomb potentials can be treated in TURBOMOLE with the conductor-like screening model (COSMO), polarizable embedding (PE), or frozen density embedding (FDE). Developments on COSMO and PE in TURBOMOLE have been presented in the previous review.92 In contrast to the latter approaches, FDE is an atomistic QM/QM embedding model, which compared to QM/MM schemes uses a purely quantum-mechanical description of the total system and does not require any system-specific parametrizations prior to a calculation.
The implementation of FDE in TURBOMOLE was until recently restricted to just two subsystems, limiting its applicability. It has now been extended to handle arbitrarily many subsystems and to an FDE variant free of intermediates evaluated on the supermolecular orbital basis. For the embedding potentials, the newly implemented approach uses only electron densities and electrostatic potentials of the subsystems, which are computed on an integration grid generated for the total system. It is available for HF and DFT within the dscf and ridft programs. In combination with the ricc2 program, it can be used to compute ground-state energies with MP2 and CC2 within the perturbation to the energy (PTE) or post-SCF reaction field coupling approaches. Excitation energies can be calculated within the frozen solvent approximations either with or without the kernel contributions. An important feature of the new implementation is the possibility to include pseudopotentials to improve the embedding potential. This allows for a more accurate description of the Pauli repulsion,83 which is particularly important for electronically excited states to prevent unphysical charge spill-out. Besides all of the new features in FDE, it should also be mentioned that the current implementation is still limited to closed-shell subsystems.
To demonstrate its capabilities, we computed the first eight excitation energies of acetone at the CC2/aug-cc-pVDZ level embedded in 237 water molecules within the frozen solvent approximation. The active subsystem also includes two water molecules beside acetone and was treated with the HF method during the freeze and thaw (FaT) cycles, while the remaining water molecules were treated as separate subsystems using DFT with the PBE exchange correlation functional313 and a gridsize of 3.9,331 For the embedding potential, the PBE exchange correlation and the LC94 kinetic energy functional332 with a gridsize of 3 were used. A comparison of the spectrum of solvated acetone with that of isolated acetone is shown in Figure 20.
Figure 20.
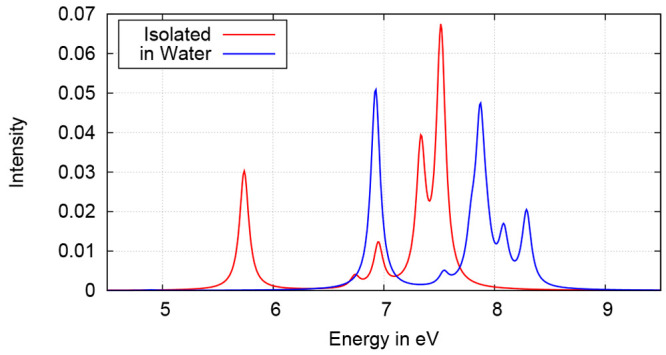
Computed spectra of acetone isolated (red) and surrounded by 237 water molecules (blue).
Recently, another update scheme of the subsystem densities was implemented, where the densities of all subsystems are updated simultaneously at the end of a FaT cycle instead of successive updates immediately after each subsystem calculation. Thus, the updated densities of each subsystem only contribute to the embedding potentials in the next FaT cycle, offering the advantage that interim results no longer depend on the enumeration of the subsystems; this allows the subsystem calculations to be performed simultaneously to improve efficiency in parallel runs. In Table 3, the wall times for both update schemes are compared for an reduced example of the above system containing only 78 water molecules. The timings show that this kind of parallelization leads to a decrease of the wall time in the case of many small subsystems where it is inefficient to parallelize single subsystem calculations over many CPU cores.
Table 3. Wall Times for an Acetone Molecule in a Water Cluster for the Two Different Update Schemesa.
| update scheme | successive | simultaneous |
|---|---|---|
| T(40), min | 122.8 | 71.4 |
| T(1), min | 369.6 | 586.2 |
| speedup | 3.0 | 8.2 |
| nFaT | 6 | 10 |
T(X) represents the computational wall time obtained using X CPU cores. nFaT gives the number of FaT iterations where it is necessary to reach convergence.
Future developments on the implementation of the FDE method aim to increase the applicability to systems containing large subsystems by combining it with PNO-based methods333 and to reduce further the computation time of the Coulomb contribution to the embedding potential.
3.15.2. FDE and Projection-Based Embedding for Molecules and Solids
The FDE implementation described above is suitable for molecular and weakly interacting subsystems, as it employs embedding potentials based on approximate kinetic energy density functionals (KEDFs).334 Recently, a DFT-based embedding scheme that treats both molecular and periodic systems on equal footing has been implemented within the riper module.82 This implementation supports both FDE and projection-based embedding335 (PbE) via a level-shift projection operator336 (LSPO). PbE along with FaT cycles can be used to perform exact DFT-in-DFT embedding for molecules and solids and reproduce the exact total DFT energies, even for strongly interacting subsystems. Similar to the implementation in section 3.15.1, the embedding scheme is also coupled with correlated wave function (CW) methods and additionally with RT-TDDFT, enabling CW-in-DFT and RT-TDDFT-in-DFT calculations, respectively, on a cluster embedded in a molecular/periodic environment. However, here the CW-in-DFT calculations are performed only by adding the DFT-based embedding potential as a static term to the HF core potential of the active subsystem and obtaining the converged HF reference orbitals for post-SCF calculations. RT-TDDFT-in-DFT, on the other hand, does support updating a portion of the embedding potential during the time evolution in select cases.
As an illustrative application of the work, Figure 21 and Table 4 show that the solvatochromic shift in the first excitation energy, calculated using the CC2-in-DFT method (molecule-in-periodic) for an acetone molecule (active) solvated with 113 water molecules (periodic environment) in a cubic box with 3D periodicity, is in remarkable agreement with the shifts calculated using standard CC246 for acetone + (H2O)20, acetone + (H2O)35, and acetone + (H2O)48 clusters at only a fraction of the computational cost. While beneficial, it is also important to acknowledge the limitations of the implementation as well, such as being restricted to only two closed-shell subsystems and using only LDA/GGA functionals for the embedding potential.
Figure 21.

Acetone + H2O in a cubic box of 15 Å containing 113 water molecules visualized using CrysX-3D Viewer.337
Table 4. Solvatochromic Shift ΔE of Acetone (Ac) S1 Excitation in Water Calculated via CC2 on Ac + (H2O)n Clusters and CC2-in-DFT (Periodic)a.
| CC2 |
CC2-in-DFT (periodic) | |||
|---|---|---|---|---|
| Ac + (H2O)20 | Ac + (H2O)35 | Ac + (H2O)48 | Ac in 3D H2O | |
| ΔE, eV | 0.19 | 0.19 | 0.21 | 0.20 |
| Nbf | 640 | 1000 | 1312 | 2872 |
| T, h | 0.88 | 8.36 | 37.33 | 4.10 |
T denotes the wall time of the CC2 solvatochromic shift calculations.
3.16. Periodic Hartree–Fock Exchange
In ref (328), we have presented a robust implementation of the periodic Hartree–Fock exchange in TURBOMOLE’s riper module.56,57 Without precautions, exchange matrix elements may be divergent, arising from an artificial periodicity of the density matrix. This periodicity of the density matrix is introduced in practical calculations by the discretization of wavevectors. The finite k-mesh determines in turn the size of the Born–von Kármán supercell. We have demonstrated328 that a minimum image convention338 removes the divergence for discrete k-meshes. While calculations with periodic HF exchange may be unstable for small supercells, stable SCF calculations and convergence of total energies are typically achieved for sufficiently large sizes of the supercells. The size of the supercell or k-mesh that is required for a reliable energy depends on the locality of the density matrix and hence both the electronic structure of the studied material and the chosen basis set. For selected insulators and semiconductors, we have demonstrated that HF total energies converge exponentially with the number of k-points,328 see Figure 22.
Figure 22.
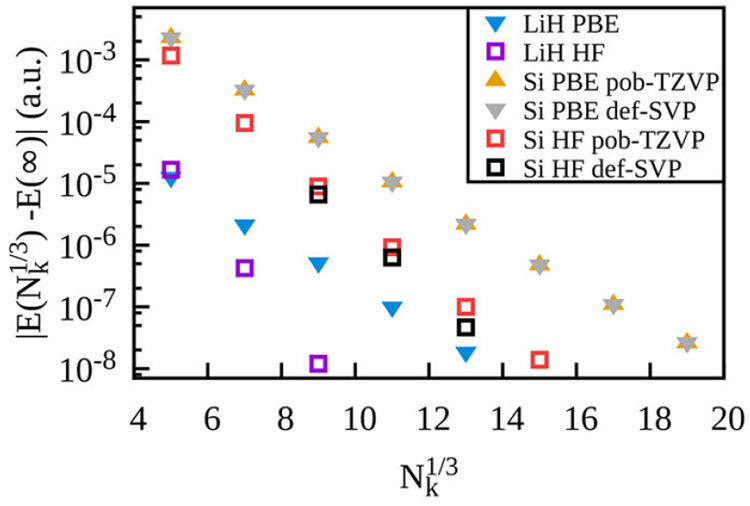
Self-consistent total energy differences |E(N1/3k) – E(∞)| per primitive cell for the PBE and HF methods. LiH is calculated in the rocksalt structure with a lattice constant of 4.084 Å339 and is described with the basis set from ref (340). For Si, we use the diamond structure with a lattice constant of 5.430 Å341 and the pob-TZVP342 and def-SVP343 basis sets. E(∞) is approximated by the energy obtained with a 31 × 31 × 31 k-mesh, since |E(25) – E(31)| < 4 × 10–10 a.u. for LiH and Si. Reprinted with permission from ref (328). Copyright 2018 American Chemical Society.
Through our implementation of periodic exchange, conventional Hartree–Fock calculations can be carried out with TURBOMOLE for periodic systems of any dimension. In addition, DFT calculations with hybrid functionals can now be performed routinely for semiconductors and insulators, and we showed successful applications of PBE0313,344 and HSE06345 hybrid and range-separated hybrid functionals.328 As the next important step, analytical gradients shall be added for structure optimization. Furthermore, the Hartree–Fock exchange infrastructure that is available may be used in the development of new electronic structure methods for periodic systems that require exchange.
4. Select Features under Development
4.1. Nuclear Electronic Orbital Method
Proton-coupled electron transfer (PCET) reactions are an important class of reactions that cannot be adequately described within the Born–Oppenheimer approximation.346 A remedy to this problem is the nuclear electronic orbital (NEO) methods, which treat not only electrons but also the protons of selected hydrogen atoms quantum mechanically.347,348 This is of particular importance for reactions that include proton transfer, such as, for example, acid–base reactions. The corresponding effects become especially important when the proton transfer is coupled to the electronic structure, such as in photoacids and photobases. In an initial proof-of-principle implementation, nuclear electronic orbitals were made available for the Hartree–Fock method (NEO-HF) and second-order Møller–Plesset perturbation theory (NEO-MP2) in a development version of TURBOMOLE. Furthermore, for the NEO-HF method, analytical gradients have since been implemented to allow for structure and basis set optimization.
Figure 23a shows the nuclear orbitals of the trans-Zundel isomer H9O4+.349 The nuclear orbital energies, as calculated by the NEO-HF method, give an estimate of the binding energies of the protons. While the four outer ones have energies from −450 to −436 kcal/mol, the central one is the least stable with an energy of −417 kcal/mol. If the outer two water molecules are removed, the energies change to between −400.5 and −399.6 kcal/mol for the four outer protons and −371 kcal/mol for the central one. This hints at the ionic cluster being stabilized by the outer water molecules.
Figure 23.
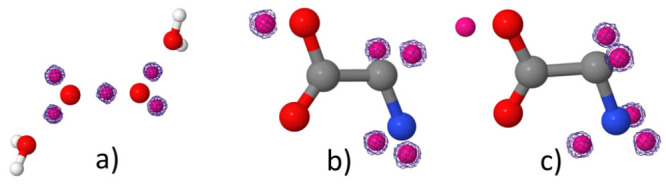
Nuclear orbitals as isosurfaces of the total densities with a cutoff of 0.0001 calculated with NEO-HF in the dscf program using the def2-TZVP basis for electrons and DZSPDN348 for protons. White, classical proton; pink, center for basis set of a quantum proton; red, oxygen; and blue, nitrogen. (a) trans-Zundel isomer of H9O4+, (b) neutral glycine, and (c) zwitterionic glycine.
The protons of the neutral glycine molecule in Figure 23b have orbital energies between −633 and −546 kcal/mol, with the least stable one being the one at the carboxyl group. The zwitterionic glycine structure in Figure 23c has been optimized as a positively charged system with six classical protons. A NEO-HF calculation with five protons resulted in proton orbital energies between −663 and −558 kcal/mol. This demonstrates that protonation and deprotonation at certain sites can be elegantly investigated by the ab initio occupation of nuclear orbitals at the respective sites instead of placing classical nuclei according to intuition.
Implementing these methods in TURBOMOLE allows for the use of highly efficient schemes that already exist for purely electronic methods. The NEO methods can also use various existing programs to analyze the results. In future developments, NEO–DFT350 will be implemented for molecular and periodic351 systems.
4.2. Hartree–Fock-Based Adiabatic Connection Models
MP2352 is one of the most used approaches for wavefunction-based correlation energies, as well as being used in the double-hybrid (DH) DFT.353,354 Nevertheless, the MP2 method shows several limitations, e.g., it overestimates the correlation energy in large systems355 and diverges for systems with a vanishing gap.356 For this reason, several regularized and/or scaled MP2 methods have been developed.356−358
Another more recent path in this direction is the Møller–Plesset or Hartree–Fock adiabatic connection (HFAC) method.359 In the HFAC approach, the correlation energy Ec is given as a nonlinear function of EMP2, EHFx (the HF exchange energy), and two semilocal functionals of the HF density (Wc = Wc[ρHF] and W′c = W′c[ρHF]). The latter are derived from the strong-correlation regime,360,361 and we have
| 10 |
The HFAC method de facto includes an infinite-order resummation of the MP correlation series thanks to the interpolation with the strong-correlation limit, as in the more conventional AC based on DFT.362 The nonlinear function F can be approximated by modeling the HFAC curve at various coupling strengths359 using known exact asymptotic conditions.361,363 Consequently, F satisfies two important limits:
| 11 |
where G is a nonlinear function whose form depends on the choice of F. For well-behaving approximations of F, the condition in eq 11b yields a finite energy whenever EMP2 → – ∞, thus removing one main limitation of MP2 and DH functionals for systems with a vanishing gap. The condition (eq 11a) is an exact condition,359 which is violated in all the regularized MP2 methods.356,357 Thus, the HFAC method allows us to overcome the main drawbacks of the MP2 approach within a well-defined theoretical framework at the small extra cost of a post-HF semilocal DFT calculation.
Some working approximations of F have been proposed (e.g., ISI,362,364 RevISI,365 and MPACF1363), and they have been implemented in TURBOMOLE together with the currently available DFT approximations for Wc and W′c.360,366 Note that eq 10 is not size-consistent for systems composed of different species of fragments (as F is a nonlinear function). However, a size-consistent correction (SCC)367 can be readily computed with TURBOMOLE at no additional costs, allowing the calculation of dissociation curves.
Two examples of applications where MP2 and DH functionals fail whereas the HFAC implementation can be readily used are displayed in Figure 24.
Figure 24.
Dissociation energy curves for (a) a coronene dimer and (b) H2, as computed with different methods. For the coronene dimer, reference data are taken from the literature,368,369 and the equilibrium distance is R0 = 3.458 Å. All calculations were performed with the hfacm script. The coronene dimer results are based on a complete basis set extrapolation from cc-pVQZ results. H2 calculations employed the aug-cc-pVQZ basis set. The strong-correlation functional used in the HFAC results is the hPC model.366
Figure 24a shows the dissociation curve for the coronene dimer, a prototype for a wide class of problems that are hardly tractable with high-level correlated wave function methods but are poorly described by MP2 because of the lack of high-order correlation contributions.355 In constrast, HFACM methods are very close to the available reference data at the equilibrium geometry. The MPACF1 functional has been tuned on dispersion complexes and on average exhibits an error 2–5× smaller than MP2.363
Figure 24b shows the H2 dissociation in a restricted formalism, a prototype of a strongly correlated system.129−131,357,364 All methods work well close to the equilibrium geometry. However, for a larger separation where the energy-gap closes, MP2 and DH functionals rapidly diverge. HFAC methods, however, remain well behaved, yielding a finite interaction energy, see eq 11b. The exact result is not reproduced, as the available HFAC functionals are approximated and do not take into account the recent theory developments.361 Further development and testing are thus required, and the HFAC implementation in TURBOMOLE represents an efficient platform to this end.
4.3. Approximate TDDFT Approaches
Time-dependent density functional theory is still the most-used approach for the calculation of excitation energies of molecular and extended systems, thanks to its favorable accuracy/computational-cost ratio. Nevertheless, the computational cost of first-principles TDDFT calculations limits routine calculations to systems with a few hundreds of atoms, depending on the choice of the exchange-correlation (XC) functional. Different methods and algorithms have been developed in order to increase the TDDFT efficiency,370−373 most prominently the RI-J technique.374−377 Another efficient, though approximated, path is to perform a semiempirical tight-binding linear-response (TBLR) approximation,378−380 using first-principles KS orbitals and eigenvalues. TBLR methods speed up the TDDFT calculation by about two orders of magnitude. TBLR is accompanied by a loss in accuracy of about 0.1–0.2 eV, which is comparable to the overall TDDFT accuracy.381,382 More recently, it has been shown that, for semilocal XC functionals, the TBLR approaches can be considered as an approximation of the RI-TDDFT scheme with only one s-type Gaussian basis function per atom in the RI auxiliary basis set (TDDFT-as)383 and with the three index RI integrals replaced with a Löwdin approximation.378−380 Instead, in the TDDFT-as method, the latter approximation is not employed and, moreover, the calculation of the semilocal XC kernel contribution on the grid is not required,383 as it can be modeled/approximated by the same exponent α of the s-type Gaussian auxiliary basis function. However, the exponent α needs to be optimized for each atom type separately.
Here, we shortly report on results for two computationally expensive cases using the PBE313 functional. First, a 120 atom silver nanoparticle, Ag120, with Td symmetry calculated using the def-SVP41 basis set. Second, a fullerene with C1 symmetry calculated using the def2-TZVP270 basis set. Both systems contain only a single type of atom. We optimized α by minimizing the root-mean square (RMS) averaged excitation energy error Eavg, avoiding state flipping,383 and considering 400 t2 (600 a) excited states for the silver nanoparticle (fullerene). The optimization yields αAg = 0.036 with Eavg = 5 meV and αC = 0.18 with Eavg = 12 meV for Ag120 and C60, respectively, as reported in Table 5. The amazing accuracy is also retained when the maximum error on all excitation energies Emax, including many optically dark states in C60, is considered. This is demonstrated in Table 5 and Figure 25, where we report the absorption spectra of the two systems considered. The TDDFT-as absorption spectra in Figure 25, reported on a log scale to also highlight states with low oscillator strengths, can be hardly distinguished from the reference TDDFT results in a wide energy range for both systems investigated. Compared to TBLR approaches,383 the accuracy of TDDFT-as is therefore increased by an order of magnitude, showing that it is a highly competitive approach.
Table 5. Optimized Exponent of the s-Type Auxiliary Basis Function, RMS-Averaged (Eavg) and Maximum (Emax) Errors on Excitation Energies, and Computational Cost (on a Single-Core Intel Xeon Gold 6132) for the First Davidson Step for the RI-J and XC Part for the Two Systems Considereda.
| Ag120 | C60 | |
|---|---|---|
| α | 0.036 | 0.18 |
| Eavg, meV | 5 | 12 |
| Emax, meV | 11 | 39 |
| TDDFT RI-J, s | 5719 | 7148 |
| TDDFT-as RI-J, s | 116 | 104 |
| TDDFT XC, s | 22363 | 29845 |
| TDDFT-as XC, s | 0 | 0 |
In the TDDFT(-as) calculations, the 1s core orbital and the 4s4p orbitals were kept frozen in C60 and Ag120, respectively.
Figure 25.
TDDFT and TDDFT-as absorption spectra (in log scale) for (a) Ag120 and (b) fullerene determined using the PBE XC functional and a Lorentzian broadening of 20 meV.
The computational speed up of the TDDFT-as method is shown in Table 5. The XC part is completely neglected, and the RI-Coulomb part is reduced by a factor of 50–70 due to the strongly reduced dimension of the auxiliary basis set.
More recently, TDDFT-as was extended to hybrid functionals and defined across the periodic table, forming the TDDFT-ris model.384 Using the PBE0 hybrid functional344 on a test set comprising small-to-large organic molecules, TDDFT-ris has an average error of Eavg = 60 meV384 compared to an average error of Eavg = 240 meV for sTDDFT.378,379 Thus, the TDDFT-as and TDDFT-ris methods, both fully available in the next release, are efficient and accurate approximations of standard TDDFT, providing a significantly less empirical alternative to TBLR approaches. Thanks to the flexible and efficient implementation, accurate simulations of the absorption spectra of large nanoparticles and organic molecules are available at a fraction of the computational cost of standard TDDFT.
4.4. Multiscale Modeling Extensions for the Nonlinear Optical Response of Molecular Materials
Further ongoing work concerning multiscale modeling of optical molecular materials is envisaged in the direction of integrating the automatic construction of T-matrices, as used in the multiscale approach in section 3.9.1. These T-matrices or effective material parameters derived thereof are often used in scattering codes and finite-element method-based programs. Examples for such codes are the multilayered periodic general Mie method code (mpGMM), JCMsuite, and COMSOL Multiphysics, which describe light–matter interactions of complex optical devices made form novel materials. In the foreseeable future, the current developments and implementations will be converted into a fully fledged workflow for optical material simulations. This will make the ab initio-based T-matrix approach available to a broader scientific community interested in a bottom-up approach of simulating complex artificial molecular materials and photonic devices. Future work will be dedicated to nonlinear optical properties, as this topic is currently seeing increasing interest in the scientific community. The change in the molecular dipole moment Δμi (polarization) upon exposure to the oscillating external electric field Ei at the excitation frequencies is often expressed as a power series of the incident field E.
| 12 |
In eq 12, αij denotes the polarizability, βijk denotes the first hyperpolarizability, γijkl denotes the second hyperpolarizability and so on. Currently, optical multiscale studies are limited to linear response, taking into account αij.254,261 To take into account nonlinear effects, i.e., βijk and/or γijkl, the additionally arising quadratic (and/or cubic) response terms of eq 12 need to be taken into account. While TURBOMOLE already allows calculation of the first hyperpolarizabilities βijk for real frequencies, ongoing work is dedicated to expanding this toward general complex frequencies. Ultimately, this will allow not only studies of the nonlinear light–matter interactions on the individual molecular level but also the construction of “hyper-T-matrices”. The latter can be used to investigate for example second-harmonic generation (SHG) efficiencies, macroscopic second-order susceptibilities, and two-photon absorption of photonic devices made from molecular materials.
4.5. Relativistic Effects and Magnetic Properties of Periodic Systems
As is evident from the many sections in this Review focusing on molecules, TURBOMOLE was initially developed to study finite molecular systems. However, the code infrastructure was extended to support calculations with periodic boundary conditions almost 15 years ago,55 and developments for molecular systems can be transferred to the periodic code. Recently, a two-component DFT framework was implemented for ground-state calculations, supporting energies and various plotting options for bands and the electron density on a grid.385 The reason to use such a framework for periodic systems is twofold. First, it allows for the inclusion of spin–orbit coupling in a variational ansatz for relativistic effects. Second, the 2c formalism is necessary to study magnetic properties and arbitrary spin alignments, i.e., ferromagnetic, antiferromagnetic, and noncollinear spin textures.
A pilot application to the band gaps of AgI is shown in Table 6. Here, relativistic effects substantially affect the band energies. That is, the nonrelativistic approach shows a large deviation toward the four-component (4c) ansatz directly based on the Dirac equation. In contrast, the scalar 1c approach yields substantial improvement. The spin–orbit 2c approach, employing ECPs, further improves upon these results and is in good agreement with the four-component reference. This shows that the 2c framework serves as an excellent approximation with drastically reduced computational demands. Here, a Kramers-restricted (time-reversal symmetric) approach is available for closed-shell systems, whereas a Kramers-unrestricted approach (breakdown of time-reversal symmetry) is used for open-shell systems. For the latter, a noncollinear treatment of the electron spin is applied.
Table 6. Energy Band Gaps (in eV) of Three-Dimensional AgI (Lattice Constant 6.169 Å, Rocksalt Structure386) Obtained for Various k-Points with the PBE Functionala.
| method | L–L | Γ–Γ | X–X | L–X |
|---|---|---|---|---|
| NR | 3.89 | 3.42 | 3.71 | 1.48 |
| 1c ECP | 3.49 | 2.16 | 2.98 | 0.65 |
| 2c ECP | 3.25 | 1.82 | 2.69 | 0.41 |
| 4c DKS | 3.25 | 1.88 | 2.74 | 0.49 |
Nonrelativistic calculations (NR) are performed with the TZVPalls2/TZVPall basis set,387 whereas the ECP-based 1c and 2c calculations use the dhf-SVP(-2c) bases.388 Results taken from ref (385). Four-component Dirac–Kohn–Sham (DKS) reference values are taken from ref (389), employing the uncontracted Dyall-VDZ basis.390,391.
Current endeavors cover the extension of the 2c formalism to energy gradients56 and the stress tensor,59 as well as the inclusion of the current density,151 see section 3.2. This will allow us to perform structure optimizations and sophisticated studies with τ-dependent functionals, as the current density is of crucial importance for materials such as Weyl semimetals392 and magnetic Hopfions.393 Another strong point of the 1c implementation for periodic systems is the availability of HF exchange, which can be applied at a reasonable cost as localized basis functions are used.328 This allows for the use of generally all available global and range-separated hybrid functionals in a stable and convergent framework. Extension of this feature to the 2c framework will allow for a more precise description of band gaps and other properties. For example, for magnetic properties it was shown that the amount of HF exchange incorporated is crucial.119 Therefore, a robust implementation of 2c HF exchange in the periodic framework will be useful in determining related quantities also for materials in the solid state and nanostructures.
5. Outlook
The quality of a code strongly correlates with the health and functioning of its developers’ community.394 TURBOMOLE developers are organized in small units pursuing their own scientific agendas, as illustrated by this Review. While this is a typical and, to a degree, necessary modus operandi for large scientific coding projects, the need to secure original authorship and demonstrate scientific independence often conflicts with sharing plans and code, taking collective responsibility, and avoiding “technical debt”. As a result, TURBOMOLE has historically not been particularly easy to use, contribute to, or interface with other codes. TURBOMOLE GmbH was founded precisely to address these issues and has provided a framework to advance common goals and improve code quality. Nevertheless, incentives to collaborate and adopt sustainable coding practices remain few and far between. The future of the TURBOMOLE project will vitally depend on whether the conditions set by the environment, i.e., academic institutions, funding agencies, reviewers, the developers, and not least the users, foster a thriving and collaborative community, which incentivizes continued investment in the code base.
Acknowledgments
All past and present developers’ contributions to the TURBOMOLE project are gratefully acknowledged. A list of TURBOMOLE contributors is available on the TURBOMOLE website.91 Y. J. Franzke was supported by fellowships from Fonds der Chemischen Industrie (FCI, German Chemical Industry Fonds), Deutscher Akademischer Austauschdienst (DAAD, German Academic Exchange Service), and TURBOMOLE GmbH. C. Holzer and M. Krstić gratefully acknowledge funding by Volkswagen Stiftung. T. Begušić and E. Tapavicza acknowledge scientific support from J. Vaníček in the development and application of the TGA method. F. Della Sala acknowledges the financial support from ICSC–Centro Nazionale di Ricerca in High Performance Computing, Big Data and Quantum Computing, funded by European Union–NextGenerationEU–PNRR. D. A. Fedotov and S. Coriani acknowledge support from the European Unions Horizon 2020 research and innovation program under the Marie Skłodowska-Curie European Training Network COSINE (grant agreement no. 765739). J. H. Andersen and S. Coriani acknowledge financial support from the Independent Research Fund Denmark-DFF-FNU RP2 (grant no. 7014-00258B). S. Gillhuber is supported by a fellowship from Fonds der Chemischen Industrie (FCI no. 110160). R. Grotjahn acknowledges support via a Walter-Benjamin postdoctoral fellowship funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), no. 501114520. C. Hättig acknowledges support by the Deutsche Forschungsgemeneinschaft (DFG) via project Ha 2588/10-1. The Kaupp group has been supported by the Deutsche Forschungsgemeinschaft (DFG) via projects KA1187/14-1 and KA1187/14-2. M. Kehry acknowledges financial support by the DFG through the Transregional CRC 88 “Cooperative Effects in Homo- and Heterometallic Complexes” (project C1). F. Mack acknowledges support from TURBOMOLE GmbH and from the DFG through the CRC 1176 (Project Q5). The material in section 3.8 and parts of the material in section 3.12 is based upon work supported by the US National Science Foundation under CHE-2102568. The material in section 3.2 and in section 3.10 is based upon work supported by the US Department of Energy, Office of Basic Energy Sciences, under award number DE-SC0018352. A. Pausch was supported by a fellowship from Fonds der Chemischen Industrie and Studienstiftung des deutschen Volkes (German Academic Scholarship Foundation). E. Perlt and T. Schrader acknowledge support from the Carl Zeiss Foundation within the CZS Breakthroughs Program. M. Sierka and M. Sharma gratefully acknowledge financial support from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the CRC 1375 NOA, project A4, and from the Carl Zeiss Foundation within the CZS Breakthroughs Program. B. Samal and V. K. Voora were supported by the Department of Atomic Energy, Government of India, under project no. RTI2001. F. Weigend acknowledges support from the DFG through the Collaborative Research Centre (CRC) 1573 (Project Q). J.M. Yu acknowledges support from the US National Science Foundation under Grant DGE-1839285. B. Zerulla acknowledges support by the KIT through the “Virtual Materials Design” (VIRTMAT) project.
Author Contributions
‡ Y. J. Franzke and C. Holzer contributed equally to this work. Y. J. Franzke contributed to the conception and implementation of one-component current density ansätze for pNMR shifts, NMR couplings, EPR, and magnetizabilities as well as the two-component generalization for spin–orbit coupling (SCF, NMR, EPR, TDDFT). Further, Y. J. Franzke contributed to the conception and execution of research on relativistic EPR and NMR properties detailed in sections 3.4, 3.5, 3.6, and 3.7. This includes the implementation of integrals as well as the X2C/DLU-X2C response and picture-change routines. He designed the implementation of all relativistic EPR and (paramagnetic) NMR methods. Furthermore, the 2c damped response and static 2c CPKS framework were designed and implemented together with C. Holzer and M. Kehry. Y. J. Franzke contributed to the two-component DFT approach for periodic systems. He co-organized the collaboration for this paper and wrote and revised parts of the manuscript. C. Holzer contributed to the conception and implementation of the current density framework for one-component and two-component ansätze (finite magnetic field, SCF, NMR, EPR, and TDDFT). Additionally, C. Holzer implemented the initial graphics processing (GPU) unit support in the modules listed in Table 1. C. Holzer further contributed to the conception and implementation of the (2c) GW-BSE method in general and the related solvers. Furthermore, the 2c damped response and static 2c CPKS framework were designed and implemented together with Y. J. Franzke and M. Kehry. Additionally, the implementation and interfacing for the T-matrix based multiscale modeling of optical properties was codesigned by C. Holzer. He co-organized the collaboration for this paper and wrote and revised parts of the manuscript. Y. J. Franzke and C. Holzer have rewritten the local hybrid code from 2020–2022, i.e., increased efficiency, the just in time (JIT) framework, corrected memory handling, and so on. They implemented the general two-component version (Kramers-unrestricted LMFs and calibration function, XC kernel, and CDFT). T. Begušić and E. Tapavicza implemented the TGA method for vibronic spectra. F. Bruder added spin–orbit perturbation theory for EPR and pNMR using the X2C routines developed above. He wrote and revised parts of the manuscript. F. Della Sala implemented the HFACM approach and developed the TDDFT-as/TDDFT-ris methods. E. Fabiano developed and implemented the HFACM approach. S. Fürst carried out the implementation, optimization, and evaluation of range-separated local hybrid functionals, including the new ωLH22t functional. S. Gillhuber implemented the nonrelativistic EPR and pNMR extension. Y. J. Franzke added the scalar X2C part. S. Gillhuber contributed to the research of section 3.7. He wrote and revised parts of the manuscript. R. Grotjahn performed investigations on the importance of the current-density correction in TDDFT and implemented the excited-state gradients and quadratic response properties for current-dependent MGGAs. Furthermore, R. Grotjahn contributed to the development of the ωLH22t functional. M. Kaupp supervised the work of S. Fürst and A. Wodyński as well as part of the work of R. Grotjahn and supported work on local hybrid functionals, including range-separated local hybrids, strong-correlation corrections, and aspects pertaining to current dependence. M. Kehry was involved in the design and programming steps of the underlying (2c) damped response modules for GW-BSE and 2c TDDFT. He wrote and revised parts of the manuscript. J. H. Andersen and D. A. Fedotov contributed to the development of the damped-response RI-CC2 functionalities. J. H. Andersen and C. Hättig carried out the illustrative damped RI-CC2 calculation here reported. C. Hättig and S. Coriani conceived, conceptualized, and supervised the damped-response RI-CC2 project and wrote the text in section 3.9.2. M. Krstić and B. Zerulla devised the multiscale modeling approach and designed and programmed the interface to external Maxwell-solver based programs. They wrote and revised parts of the manuscript. F. Mack contributed to the implementation of the NMR coupling constants (nonrelativistic and 2c) at the DFT and GW-BSE levels. S. Majumdar contributed to the research discussed in section 3.10 and was involved in conceptualization, execution, and writing. G. S. Phun contributed to writing section 3.10. A. Rajabi and D. Rappoport were involved in both the conceptualization and execution of the research discussed in section 3.8. S. M. Parker developed and supervised the TDDFT-ris method. F. Pauly and his group contributed to method development on periodic Hartree–Fock exchange and relativistic effects in periodic systems and the corresponding sections of this review. He wrote and revised parts of the manuscript. A. Pausch supported Y. J. Franzke and C. Holzer in the implementation of the two-component CDFT framework. A. Pausch and C. Holzer developed the methods for finite magnetic fields. He wrote and revised parts of the manuscript. E. Perlt performed implementations of the NEO methods and contributed to writing of section 4.1. T. Schrader performed NEO calculations and wrote section 4.1. M. Sharma, under the supervision of M. Sierka, contributed to the RT-TDDFT code and extended it to perform HHG simulations and also wrote the corresponding section 3.14. Additionally, M. Sharma implemented the molecular and periodic DFT-based embedding coupled with RT-TDDFT and wave function methods and wrote section 3.15.2. B. Samal and B. D. Nguyen contributed to writing section 3.12. V. K. Voora developed the AC version of the GKS-spRPA method and contributed to the writing of section 3.12. A. Wodyński carried out the implementation, optimization, and evaluation of strong-correlation-corrected local hybrid functionals. J. M. Yu contributed to the conception and execution of the research detailed in section 3.4. R. Treß developed the frozen density embedding implementation and carried out the calculation reported in section 3.15.1, and C. Hättig supervised the project. F. Furche conceived, oversaw, and participated in the development of the material in sections 3.8 and 3.10 and parts of the material in sections 3.2, 3.12, 3.4, and 4.1. He also initiated and organized the collaboration for this paper and wrote and revised parts of the manuscript. M. Sierka supervised the work of M. Sharma described in sections 3.14 and 3.15.2, as well as conceived, oversaw, and participated in the development of the RT-TDDFT code (section 3.14). He co-organized the collaboration for this paper and wrote and revised parts of the manuscript. F. Weigend supervised the work of F. Bruder, S. Gillhuber, and F. Mack and supported the EPR, pNMR shift, NMR coupling, and ring current studies. He co-organized the collaboration for this paper and wrote and revised parts of the manuscript.
The authors declare the following competing financial interest(s): Principal Investigator Filipp Furche has an equity interest in TURBOMOLE GmbH. The terms of this arrangement have been reviewed and approved by the University of California, Irvine, in accordance with its conflict of interest policies. Christof Hattig and David P. Tew have an equity interest in TURBOMOLE GmbH. Marek Sierka and Florian Weigend have an equity interest in TURBOMOLE GmbH and serve as its chief executive officers.
References
- Ahlrichs R.; Bär M.; Häser M.; Horn H.; Kölmel C. Electronic structure calculations on workstation computers: The program system TURBOMOLE. Chem. Phys. Lett. 1989, 162, 165–169. 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]
- Häser M.; Ahlrichs R. Improvements on the direct SCF method. J. Comput. Chem. 1989, 10, 104–111. 10.1002/jcc.540100111. [DOI] [Google Scholar]
- Häser M.; Ahlrichs R.; Baron H. P.; Weis P.; Horn H. Direct computation of second-order SCF properties of large molecules on workstation computers with an application to large carbon clusters. Theor. Chim. Acta 1992, 83, 455–470. 10.1007/BF01113068. [DOI] [Google Scholar]
- Haase F.; Ahlrichs R. Semidirect MP2 gradient evaluation on workstation computers: The MPGRAD program. J. Comput. Chem. 1993, 14, 907–912. 10.1002/jcc.540140805. [DOI] [Google Scholar]
- Brode S.; Horn H.; Ehrig M.; Moldrup D.; Rice J. E.; Ahlrichs R. Parallel Direct SCF and Gradient Program For Workstation Clusters. J. Comput. Chem. 1993, 14, 1142–1148. 10.1002/jcc.540141004. [DOI] [Google Scholar]
- Kollwitz M.; Gauss J. A direct implementation of the GIAO-MBPT(2) method for calculating NMR chemical shifts. Application to the naphthalenium and anthracenium ions. Chem. Phys. Lett. 1996, 260, 639–646. 10.1016/0009-2614(96)00897-4. [DOI] [Google Scholar]
- Kollwitz M.; Häser M.; Gauss J. Non-Abelian point group symmetry in direct second-order many-body perturbation theory calculations of NMR chemical shifts. J. Chem. Phys. 1998, 108, 8295–8301. 10.1063/1.476258. [DOI] [Google Scholar]
- Weiss H.; Ahlrichs R.; Häser M. A direct algorithm for self-consistent-field linear-response theory and application to C60-excitation-energies, oscillator-strengths, and frequency-dependent polarizabilities. J. Chem. Phys. 1993, 99, 1262–1270. 10.1063/1.465370. [DOI] [Google Scholar]
- Treutler O.; Ahlrichs R. Efficient molecular numerical integration schemes. J. Chem. Phys. 1995, 102, 346–354. 10.1063/1.469408. [DOI] [Google Scholar]
- Bauernschmitt R.; Ahlrichs R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. 10.1016/0009-2614(96)00440-X. [DOI] [Google Scholar]
- Furche F.; Perdew J. P. The performance of semi-local and hybrid density functionals in 3d transition metal chemistry. J. Chem. Phys. 2006, 124, 044103. 10.1063/1.2162161. [DOI] [PubMed] [Google Scholar]; also published in Virtual J. Biol. Phys. Res. 2006, 11.
- Furche F. Molecular tests of the random phase approximation to the exchange-correlation energy functional. Phys. Rev. B 2001, 64, 195120. 10.1103/PhysRevB.64.195120. [DOI] [Google Scholar]
- Bates J. E.; Furche F. Communication: Random phase approximation renormalized many-body perturbation theory. J. Chem. Phys. 2013, 139, 171103. 10.1063/1.4827254. [DOI] [PubMed] [Google Scholar]
- Chen G. P.; Voora V. K.; Agee M. M.; Balasubramani S. G.; Furche F. Random-phase approximation methods. Annu. Rev. Phys. Chem. 2017, 68, 421–445. 10.1146/annurev-physchem-040215-112308. [DOI] [PubMed] [Google Scholar]
- Chen G. P.; Agee M. M.; Furche F. Performance and scope of perturbative corrections to random-phase approximation energies. J. Chem. Theory Comput. 2018, 14, 5701–5714. 10.1021/acs.jctc.8b00777. [DOI] [PubMed] [Google Scholar]
- Voora V. K.; Balasubramani S. G.; Furche F. Variational generalized Kohn-Sham approach combining the random-phase-approximation and Green’s-function methods. Phys. Rev. A 2019, 99, 012518. 10.1103/PhysRevA.99.012518. [DOI] [Google Scholar]
- Bates J. E.; Furche F. Harnessing the meta-generalized gradient approximation for time-dependent density functional theory. J. Chem. Phys. 2012, 137, 164105. 10.1063/1.4759080. [DOI] [PubMed] [Google Scholar]
- Plessow P.; Weigend F. Seminumerical calculation of the Hartree-Fock exchange matrix: Application to two-component procedures and efficient evaluation of local hybrid density functionals. J. Comput. Chem. 2012, 33, 810–816. 10.1002/jcc.22901. [DOI] [PubMed] [Google Scholar]
- Maier T. M.; Bahmann H.; Kaupp M. Efficient Semi-numerical Implementation of Global and Local Hybrid Functionals for Time-Dependent Density Functional Theory. J. Chem. Theory Comput. 2015, 11, 4226–4237. 10.1021/acs.jctc.5b00624. [DOI] [PubMed] [Google Scholar]
- Furche F.; Ahlrichs R.; Wachsmann C.; Weber E.; Sobanski A.; Vögtle F.; Grimme S. Circular dichroism of helicenes investigated by time-dependent density functional theory. J. Am. Chem. Soc. 2000, 122, 1717–1724. 10.1021/ja991960s. [DOI] [Google Scholar]
- Furche F.; Rappoport D.. Density functional methods for excited states: equilibrium structure and electronic spectra In Computational Photochemistry; Olivucci M., Ed.; Computational and Theoretical Chemistry, Vol. 16; Elsevier: Amsterdam, The Netherlands, 2005; pp 93–128. [Google Scholar]
- Kattannek M.Entwicklung und Implementierung optimierter Algorithmen für molekulare Hartree-Fock- und Dichtefunktional-Rechnungen. Ph.D. Thesis, Universität Karlsruhe (TH), Karlsruhe, Germany, 2006. [Google Scholar]
- Eshuis H.; Yarkony J.; Furche F. Fast computation of molecular random phase approximation correlation energies using resolution of the identity and imaginary frequency integration. J. Chem. Phys. 2010, 132, 234114. 10.1063/1.3442749. [DOI] [PubMed] [Google Scholar]
- Furche F.; Krull B. T.; Nguyen B. D.; Kwon J. Accelerating molecular property calculations with nonorthonormal Krylov space methods. J. Chem. Phys. 2016, 144, 174105. 10.1063/1.4947245. [DOI] [PubMed] [Google Scholar]
- Deglmann P.; Furche F.; Ahlrichs R. An efficient implementation of second analytical derivatives for density functional methods. Chem. Phys. Lett. 2002, 362, 511–518. 10.1016/S0009-2614(02)01084-9. [DOI] [Google Scholar]
- Grimme S.; Furche F.; Ahlrichs R. An improved method for density functional calculations of the frequency-dependent optical rotation. Chem. Phys. Lett. 2002, 361, 321–328. 10.1016/S0009-2614(02)00975-2. [DOI] [Google Scholar]
- Deglmann P.; May K.; Furche F.; Ahlrichs R. Nuclear second analytical derivative calculations using auxiliary basis set expansions. Chem. Phys. Lett. 2004, 384, 103–107. 10.1016/j.cplett.2003.11.080. [DOI] [Google Scholar]
- Rappoport D.; Furche F. Analytical time-dependent density functional derivative methods within the RI-J approximation, an approach to excited states of large molecules. J. Chem. Phys. 2005, 122, 064105. 10.1063/1.1844492. [DOI] [PubMed] [Google Scholar]
- Rappoport D.; Furche F. Lagrangian approach to molecular vibrational Raman intensities using time-dependent hybrid density functional theory. J. Chem. Phys. 2007, 126, 201104. 10.1063/1.2744026. [DOI] [PubMed] [Google Scholar]
- Send R.; Furche F. First-order nonadiabatic couplings from time-dependent hybrid density functional response theory: Consistent formalism, implementation, and performance. J. Chem. Phys. 2010, 132, 044107. 10.1063/1.3292571. [DOI] [PubMed] [Google Scholar]
- Burow A. M.; Bates J. E.; Furche F.; Eshuis H. Analytical first-order molecular properties and forces within the adiabatic connection random phase approximation. J. Chem. Theory Comput. 2014, 10, 180–194. 10.1021/ct4008553. [DOI] [PubMed] [Google Scholar]
- Ou Q.; Bellchambers G. D.; Furche F.; Subotnik J. E. First-order derivative couplings between excited states from adiabatic TDDFT response theory. J. Chem. Phys. 2015, 142, 064114. 10.1063/1.4906941. [DOI] [PubMed] [Google Scholar]
- Klawohn S.; Bahmann H.; Kaupp M. Implementation of Molecular Gradients for Local Hybrid Density Functionals Using Seminumerical Integration Techniques. J. Chem. Theory Comput. 2016, 12, 4254–4262. 10.1021/acs.jctc.6b00486. [DOI] [PubMed] [Google Scholar]
- Parker S. M.; Rappoport D.; Furche F. Quadratic Response Properties from TDDFT: Trials and Tribulations. J. Chem. Theory Comput. 2018, 14, 807–819. 10.1021/acs.jctc.7b01008. [DOI] [PubMed] [Google Scholar]
- Grotjahn R.; Furche F.; Kaupp M. Development and Implementation of Excited-State Gradients for Local Hybrid Functionals. J. Chem. Theory Comput. 2019, 15, 5508–5522. 10.1021/acs.jctc.9b00659. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Weigend F. NMR Shielding Tensors and Chemical Shifts in Scalar-Relativistic Local Exact Two-Component Theory. J. Chem. Theory Comput. 2019, 15, 1028–1043. 10.1021/acs.jctc.8b01084. [DOI] [PubMed] [Google Scholar]
- Wodyński A.; Kaupp M. Density Functional Calculations of EPR g- and Hyperfine-Coupling Tensors Using the Exact Two-Component (X2C) Transformation and Efficient Approximations to the Two-Electron Spin-Orbit Terms. J. Phys. Chem. A 2019, 123, 5660–5672. 10.1021/acs.jpca.9b03979. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Yu J. M. Hyperfine Coupling Constants in Local Exact Two-Component Theory. J. Chem. Theory Comput. 2022, 18, 323–343. 10.1021/acs.jctc.1c01027. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Yu J. M. Quasi-Relativistic Calculation of EPR g Tensors with Derivatives of the Decoupling Transformation, Gauge-Including Atomic Orbitals, and Magnetic Balance. J. Chem. Theory Comput. 2022, 18, 2246–2266. 10.1021/acs.jctc.1c01175. [DOI] [PubMed] [Google Scholar]
- Eichkorn K.; Treutler O.; Öhm H.; Häser M.; Ahlrichs R. Auxiliary Basis Sets to Approximate Coulomb Potentials. Chem. Phys. Lett. 1995, 240, 283–290. 10.1016/0009-2614(95)00621-A. [DOI] [Google Scholar]
- Eichkorn K.; Weigend F.; Treutler O.; Ahlrichs R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. 10.1007/s002140050244. [DOI] [Google Scholar]
- Weigend F. A fully direct RI-HF algorithm: Implementation, optimized auxiliary basis sets, demonstration of accuracy and efficiency. Phys. Chem. Chem. Phys. 2002, 4, 4285–4291. 10.1039/b204199p. [DOI] [Google Scholar]
- Weigend F. Hartree–Fock exchange fitting basis sets for H to Rn. J. Comput. Chem. 2008, 29, 167–175. 10.1002/jcc.20702. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Häser M.; Patzelt H.; Ahlrichs R. RI-MP2: optimized auxiliary basis sets and demonstration of efficiency. Chem. Phys. Lett. 1998, 294, 143–152. 10.1016/S0009-2614(98)00862-8. [DOI] [Google Scholar]
- Weigend F.; Häser M. RI-MP2: first derivatives and global consistency. Theor. Chem. Acc. 1997, 97, 331–340. 10.1007/s002140050269. [DOI] [Google Scholar]
- Hättig C.; Weigend F. CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. J. Chem. Phys. 2000, 113, 5154–5161. 10.1063/1.1290013. [DOI] [Google Scholar]
- Sierka M.; Hogekamp A.; Ahlrichs R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. 10.1063/1.1567253. [DOI] [Google Scholar]
- Armbruster M. K.; Weigend F.; van Wüllen C.; Klopper W. Self-consistent treatment of spin-orbit interactions with efficient Hartree-Fock and density functional methods. Phys. Chem. Chem. Phys. 2008, 10, 1748–1756. 10.1039/b717719d. [DOI] [PubMed] [Google Scholar]
- Baldes A.; Weigend F. Efficient two-component self-consistent field procedures and gradients: implementation in TURBOMOLE and application to Au20–. Mol. Phys. 2013, 111, 2617–2624. 10.1080/00268976.2013.802037. [DOI] [Google Scholar]
- Kühn M. Correlation Energies from the Two-Component Random Phase Approximation. J. Chem. Theory Comput. 2014, 10, 623–633. 10.1021/ct400994x. [DOI] [PubMed] [Google Scholar]
- Krause K.; Klopper W. Implementation of the Bethe-Salpeter equation in the TURBOMOLE program. J. Comput. Chem. 2017, 38, 383–388. 10.1002/jcc.24688. [DOI] [PubMed] [Google Scholar]
- Gui X.; Holzer C.; Klopper W. Accuracy Assessment of GW Starting Points for Calculating Molecular Excitation Energies Using the Bethe-Salpeter Formalism. J. Chem. Theory Comput. 2018, 14, 2127–2136. 10.1021/acs.jctc.8b00014. [DOI] [PubMed] [Google Scholar]
- Holzer C.; Klopper W. Ionized, electron-attached, and excited states of molecular systems with spin–orbit coupling: Two-component GW and Bethe–Salpeter implementations. J. Chem. Phys. 2019, 150, 204116. 10.1063/1.5094244. [DOI] [PubMed] [Google Scholar]
- Kühn M.; Weigend F. Two-component hybrid time-dependent density functional theory within the Tamm-Dancoff approximation. J. Chem. Phys. 2015, 142, 034116. 10.1063/1.4905829. [DOI] [PubMed] [Google Scholar]
- Burow A. M.; Sierka M.; Mohamed F. Resolution of identity approximation for the Coulomb term in molecular and periodic systems. J. Chem. Phys. 2009, 131, 214101–1–214101–6. 10.1063/1.3267858. [DOI] [PubMed] [Google Scholar]
- Łazarski R.; Burow A. M.; Sierka M. Density functional theory for molecular and periodic systems using density fitting and continuous fast multipole methods. J. Chem. Theory Comput. 2015, 11, 3029–3041. 10.1021/acs.jctc.5b00252. [DOI] [PubMed] [Google Scholar]
- Łazarski R.; Burow A. M.; Grajciar L.; Sierka M. Density functional theory for molecular and periodic systems using density fitting and continuous fast multipole method: Analytical gradients. J. Comput. Chem. 2016, 37, 2518–2526. 10.1002/jcc.24477. [DOI] [PubMed] [Google Scholar]
- Grajciar L. Low-memory Iterative Density Fitting. J. Comput. Chem. 2015, 36, 1521–1535. 10.1002/jcc.23961. [DOI] [PubMed] [Google Scholar]
- Becker M.; Sierka M. Density functional theory for molecular and periodic systems using density fitting and continuous fast multipole method: Stress tensor. J. Comput. Chem. 2019, 40, 2563–2570. 10.1002/jcc.26033. [DOI] [PubMed] [Google Scholar]
- Mack F.; Schattenberg C. J.; Kaupp M.; Weigend F. Nuclear Spin–Spin Couplings: Efficient Evaluation of Exact Exchange and Extension to Local Hybrid Functionals. J. Phys. Chem. A 2020, 124, 8529–8539. 10.1021/acs.jpca.0c06897. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Holzer C.; Mack F. NMR Coupling Constants Based on the Bethe–Salpeter Equation in the GW Approximation. J. Chem. Theory Comput. 2022, 18, 1030–1045. 10.1021/acs.jctc.1c00999. [DOI] [PubMed] [Google Scholar]
- Reiter K.; Mack F.; Weigend F. Calculation of Magnetic Shielding Constants with meta-GGA Functionals Employing the Multipole-Accelerated Resolution of the Identity: Implementation and Assessment of Accuracy and Efficiency. J. Chem. Theory Comput. 2018, 14, 191–197. 10.1021/acs.jctc.7b01115. [DOI] [PubMed] [Google Scholar]
- Gillhuber S.; Franzke Y. J.; Weigend F. Paramagnetic NMR Shielding Tensors and Ring Currents: Efficient Implementation and Application to Heavy Element Compounds. J. Phys. Chem. A 2021, 125, 9707–9723. 10.1021/acs.jpca.1c07793. [DOI] [PubMed] [Google Scholar]
- Friese D. H.; Hättig C.; Ruud K. Calculation of two-photon absorption strengths with the approximate coupled cluster singles and doubles model CC2 using the resolution-of-identity approximation. Phys. Chem. Chem. Phys. 2012, 14, 1175–1184. 10.1039/C1CP23045J. [DOI] [PubMed] [Google Scholar]
- Friese D. H.; Hättig C.; Rizzo A. Origin-independent two-photon circular dichroism calculations in coupled cluster theory. Phys. Chem. Chem. Phys. 2016, 18, 13683–13692. 10.1039/C6CP01653G. [DOI] [PubMed] [Google Scholar]
- Winter N. O. C.; Hättig C. Scaled opposite-spin CC2 for ground and excited states with fourth order scaling computational costs. J. Chem. Phys. 2011, 134, 184101. 10.1063/1.3584177. [DOI] [PubMed] [Google Scholar]
- Winter N. O. C.; Hättig C. Quartic scaling analytical gradients of scaled opposite-spin CC2. Chem. Phys. 2012, 401, 217–227. 10.1016/j.chemphys.2011.10.002. [DOI] [Google Scholar]
- Hättig C.; Tew D. P.; Köhn A. Communications: Accurate and efficient approximations to explicitly correlated coupled-cluster singles and doubles, CCSD-F12. J. Chem. Phys. 2010, 132, 231102. 10.1063/1.3442368. [DOI] [PubMed] [Google Scholar]
- Tew D. P. Explicitly correlated coupled-cluster theory with Brueckner orbitals. J. Chem. Phys. 2016, 145, 074103. 10.1063/1.4960655. [DOI] [PubMed] [Google Scholar]
- Schmitz G.; Hättig C.; Tew D. P. Explicitly Correlated PNO-MP2 and PNO-CCSD and their Application to the S66 Set and Large Molecular Systems. Phys. Chem. Chem. Phys. 2014, 16, 22167–22178. 10.1039/C4CP03502J. [DOI] [PubMed] [Google Scholar]
- Schmitz G.; Hättig C. Perturbative triples correction for local pair natural orbital based explicitly correlated CCSD(F12*) using Laplace transformation techniques. J. Chem. Phys. 2016, 145, 234107. 10.1063/1.4972001. [DOI] [PubMed] [Google Scholar]
- Frank M. S.; Schmitz G.; Hättig C. The PNO–MP2 gradient and its application to molecular geometry optimizations. Mol. Phys. 2017, 115, 343–356. 10.1080/00268976.2016.1263762. [DOI] [Google Scholar]
- Schmitz G.; Hättig C. Accuracy of Explicitly Correlated Local PNO-CCSD(T). J. Chem. Theory Comput. 2017, 13, 2623–2633. 10.1021/acs.jctc.7b00180. [DOI] [PubMed] [Google Scholar]
- Laricchia S.; Fabiano E.; Della Sala F. Frozen density embedding with hybrid functionals. J. Chem. Phys. 2010, 133, 164111. 10.1063/1.3494537. [DOI] [PubMed] [Google Scholar]
- Bachorz R. A.; Bischoff F. A.; Glöß A.; Hättig C.; Höfener S.; Klopper W.; Tew D. P. The MP2-F12 method in the TURBOMOLE program package. J. Comput. Chem. 2011, 32, 2492–2513. 10.1002/jcc.21825. [DOI] [PubMed] [Google Scholar]
- Schwabe T.; Sneskov K.; Haugaard Olsen J. M.; Kongsted J.; Christiansen O.; Hättig C. PERI-CC2: A Polarizable Embedded RI-CC2 Method. J. Chem. Theory Comput. 2012, 8, 3274–3283. 10.1021/ct3003749. [DOI] [PubMed] [Google Scholar]
- Höfener S. Coupled-cluster frozen-density embedding using resolution of the identity methods. J. Comput. Chem. 2014, 35, 1716–1724. 10.1002/jcc.23679. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Diedenhofen M. Calculation of Solvation Free Energies with DCOSMO-RS. J. Phys. Chem. A 2015, 119, 5439–5445. 10.1021/jp511158y. [DOI] [PubMed] [Google Scholar]
- Hršak D.; Marefat Khah A.; Christiansen O.; Hättig C. Polarizable Embedded RI-CC2 Method for Two-Photon Absorption Calculations. J. Chem. Theory Comput. 2015, 11, 3669–3678. 10.1021/acs.jctc.5b00496. [DOI] [PubMed] [Google Scholar]
- Reinholdt P.; Nørby M. S.; Kongsted J. Modeling of Magnetic Circular Dichroism and UV/Vis Absorption Spectra Using Fluctuating Charges or Polarizable Embedding within a Resonant-Convergent Response Theory Formalism. J. Chem. Theory Comput. 2018, 14, 6391–6404. 10.1021/acs.jctc.8b00660. [DOI] [PubMed] [Google Scholar]
- Marefat Khah A.; Karbalaei Khani S.; Hättig C. Analytic Excited State Gradients for the QM/MM Polarizable Embedded Second-Order Algebraic Diagrammatic Construction for the Polarization Propagator PE-ADC(2). J. Chem. Theory Comput. 2018, 14, 4640–4650. 10.1021/acs.jctc.8b00396. [DOI] [PubMed] [Google Scholar]
- Sharma M.; Sierka M. Efficient Implementation of Density Functional Theory Based Embedding for Molecular and Periodic Systems Using Gaussian Basis Functions. J. Chem. Theory Comput. 2022, 18, 6892–6904. 10.1021/acs.jctc.2c00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treß R. S.; Hättig C.; Höfener S. Employing Pseudopotentials to Tackle Excited-State Electron Spill-Out in Frozen Density Embedding Calculations. J. Chem. Theory Comput. 2022, 18, 1737–1747. 10.1021/acs.jctc.1c00732. [DOI] [PubMed] [Google Scholar]
- Peng D.; Middendorf N.; Weigend F.; Reiher M. An efficient implementation of two-component relativistic exact-decoupling methods for large molecules. J. Chem. Phys. 2013, 138, 184105. 10.1063/1.4803693. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Middendorf N.; Weigend F. Efficient implementation of one- and two-component analytical energy gradients in exact two-component theory. J. Chem. Phys. 2018, 148, 104110. 10.1063/1.5022153. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Mack F.; Weigend F. NMR Indirect Spin-Spin Coupling Constants in a Modern Quasirelativistic Density Functional Framework. J. Chem. Theory Comput. 2021, 17, 3974–3994. 10.1021/acs.jctc.1c00167. [DOI] [PubMed] [Google Scholar]
- van Setten M. J.; Weigend F.; Evers F. The GW-Method for Quantum Chemistry Applications: Theory and Implementation. J. Chem. Theory Comput. 2013, 9, 232–246. 10.1021/ct300648t. [DOI] [PubMed] [Google Scholar]
- Kaplan F.; Harding M. E.; Seiler C.; Weigend F.; Evers F.; van Setten M. J. Quasi-Particle Self-Consistent GW for Molecules. J. Chem. Theory Comput. 2016, 12, 2528–2541. 10.1021/acs.jctc.5b01238. [DOI] [PubMed] [Google Scholar]
- Müller C.; Sharma M.; Sierka M. Real-time time-dependent density functional theory using density fitting and the continuous fast multipole method. J. Comput. Chem. 2020, 41, 2573–2582. 10.1002/jcc.26412. [DOI] [PubMed] [Google Scholar]
- Tapavicza E.; Bellchambers G.; Vincent J. C.; Furche F. Ab initio non-adiabatic dynamics. Phys. Chem. Chem. Phys. 2013, 15, 18336–18348. 10.1039/c3cp51514a. [DOI] [PubMed] [Google Scholar]
- TURBOMOLE. https://www.turbomole.org (accessed 2022-12-01).
- Balasubramani S. G.; Chen G. P.; Coriani S.; Diedenhofen M.; Frank M. S.; Franzke Y. J.; Furche F.; Grotjahn R.; Harding M. E.; Hättig C.; Hellweg A.; Helmich-Paris B.; Holzer C.; Huniar U.; Kaupp M.; Marefat Khah A.; Karbalaei Khani S.; Müller T.; Mack F.; Nguyen B. D.; Parker S. M.; Perlt E.; Rappoport D.; Reiter K.; Roy S.; Rückert M.; Schmitz G.; Sierka M.; Tapavicza E.; Tew D. P.; van Wüllen C.; Voora V. K.; Weigend F.; Wodyński A.; Yu J. M. TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations. J. Chem. Phys. 2020, 152, 184107. 10.1063/5.0004635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furche F.; Ahlrichs R.; Hättig C.; Klopper W.; Sierka M.; Weigend F. Turbomole. WIREs Comput. Mol. Sci. 2014, 4, 91–100. 10.1002/wcms.1162. [DOI] [Google Scholar]
- van Wüllen C. Shared-memory parallelization of the TURBOMOLE programs AOFORCE, ESCF, and EGRAD: How to quickly parallelize legacy code. J. Comput. Chem. 2011, 32, 1195–1201. 10.1002/jcc.21692. [DOI] [PubMed] [Google Scholar]
- Bachorz R. A.; Bischoff F. A.; Glöß A.; Hättig C.; Höfener S.; Klopper W.; Tew D. P. The MP2-F12 Method in the TURBOMOLE Program Package. J. Comput. Chem. 2011, 32, 2492–2513. 10.1002/jcc.21825. [DOI] [PubMed] [Google Scholar]
- Almaraz E. P.; Furche F.. Initial OpenMP version of aoforce and escf, released with TURBOMOLE V6.3; TURBOMOLE GmbH: Karlsruhe, Germany, 2011.
- Holzer C.; Franzke Y. J.. OpenMP version of ridft, rdgrad, and egrad with contributions to mpshift, dscf, and grad; improved OpenMP version of aoforce and escf, released with TURBOMOLE V7.4 and further improved in TURBOMOLE V7.5; TURBOMOLE GmbH: Karlsruhe, Germany, 2020.
- Hättig C.; Hellweg A.; Köhn A. Distributed memory parallel implementation of energies and gradients for second-order Møller-Plesset perturbation theory with the resolution-of-the-identity approximation. Phys. Chem. Chem. Phys. 2006, 8, 1159–1169. 10.1039/b515355g. [DOI] [PubMed] [Google Scholar]
- Müller T.Global Array toolkit based distributed shared memory version of ridft and rdgrad, released with TURBOMOLE V6.0; TURBOMOLE GmbH: Karlsruhe, Germany, 2011.
- Müller T.Parallel DFT in Turbomole, Linear Algebra. In High Performance Computing in Chemistry; Grotendorst J., Ed.; NIC Series, Vol. 25; John von Neumann Institute for Computing: Jülich, Germany, 2005; pp 83–107. [Google Scholar]
- Müller T.MPI based version of ridft and rdgrad with native distributed shared memory support, released with TURBOMOLE V7.2; TURBOMOLE GmbH: Karlsruhe, Germany, 2017.
- van Wüllen C.Hybrid OpenMP/MPI parallelization of dscf, grad, and aoforce, released with TURBOMOLE V7.2; TURBOMOLE GmbH: Karlsruhe, Germany, 2017.
- Plessow P. Reaction Path Optimization without NEB Springs or Interpolation Algorithms. J. Chem. Theory Comput. 2013, 9, 1305–1310. 10.1021/ct300951j. [DOI] [PubMed] [Google Scholar]
- Reiter K.; Kühn M.; Weigend F. Vibrational circular dichroism spectra for large molecules and molecules with heavy elements. J. Chem. Phys. 2017, 146, 054102. 10.1063/1.4974897. [DOI] [PubMed] [Google Scholar]
- Sierka M.; Döbler J.; Sauer J.; Santambrogio G.; Brümmer M.; Wöste L.; Janssens E.; Meijer G.; Asmis K. Unexpected Structures of Aluminum Oxide Clusters in the Gas Phase. Angew. Chem., Int. Ed. 2007, 46, 3372–3375. 10.1002/anie.200604823. [DOI] [PubMed] [Google Scholar]
- Steffen C.; Thomas K.; Huniar U.; Hellweg A.; Rubner O.; Schroer A. TmoleX – A graphical user interface for TURBOMOLE. J. Comput. Chem. 2010, 31, 2967–2970. 10.1002/jcc.21576. [DOI] [PubMed] [Google Scholar]
- OpenMP. https://www.openmp.org (accessed 2022-12-05).
- MPI Forum. https://www.mpi-forum.org (accessed 2022-12-05).
- Holzer C. An improved seminumerical Coulomb and exchange algorithm for properties and excited states in modern density functional theory. J. Chem. Phys. 2020, 153, 184115. 10.1063/5.0022755. [DOI] [PubMed] [Google Scholar]
- Maier T. M.; Arbuznikov A. V.; Kaupp M. Local hybrid functionals: Theory, implementation, and performance of an emerging new tool in quantum chemistry and beyond. WIREs Comput. Mol. Sci. 2019, 9, e1378. 10.1002/wcms.1378. [DOI] [Google Scholar]
- Janesko B. G. Replacing hybrid density functional theory: motivation and recent advances. Chem. Soc. Rev. 2021, 50, 8470–8495. 10.1039/D0CS01074J. [DOI] [PubMed] [Google Scholar]
- Bahmann H.; Kaupp M. Efficient Self-Consistent Implementation of Local Hybrid Functionals. J. Chem. Theory Comput. 2015, 11, 1540–1548. 10.1021/ct501137x. [DOI] [PubMed] [Google Scholar]
- Kehry M.; Franzke Y. J.; Holzer C.; Klopper W. Quasirelativistic two-component core excitations and polarisabilities from a damped-response formulation of the Bethe-Salpeter equation. Mol. Phys. 2020, 118, e1755064. 10.1080/00268976.2020.1755064. [DOI] [Google Scholar]
- Grotjahn R.; Lauter G. J.; Haasler M.; Kaupp M. Evaluation of Local Hybrid Functionals for Electric Properties: Dipole Moments and Static and Dynamic Polarizabilities. J. Phys. Chem. A 2020, 124, 8346–8358. 10.1021/acs.jpca.0c06939. [DOI] [PubMed] [Google Scholar]
- Schattenberg C. J.; Reiter K.; Weigend F.; Kaupp M. An Efficient Coupled-Perturbed Kohn-Sham Implementation of NMR Chemical Shift Computations with Local Hybrid Functionals and Gauge-Including Atomic Orbitals. J. Chem. Theory Comput. 2020, 16, 931–943. 10.1021/acs.jctc.9b00944. [DOI] [PubMed] [Google Scholar]
- Wodyński A.; Kaupp M. Noncollinear Relativistic Two-Component X2C Calculations of Hyperfine Couplings Using Local Hybrid Functionals. Importance of the High-Density Coordinate Scaling Limit. J. Chem. Theory Comput. 2020, 16, 314–325. 10.1021/acs.jctc.9b00911. [DOI] [PubMed] [Google Scholar]
- Holzer C.; Franzke Y. J. A Local Hybrid Exchange Functional Approximation from First Principles. J. Chem. Phys. 2022, 157, 034108. 10.1063/5.0100439. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Holzer C. Impact of the current density on paramagnetic NMR properties. J. Chem. Phys. 2022, 157, 031102. 10.1063/5.0103898. [DOI] [PubMed] [Google Scholar]
- Holzer C.; Franzke Y. J.; Kehry M. Assessing the Accuracy of Local Hybrid Density Functional Approximations for Molecular Response Properties. J. Chem. Theory Comput. 2021, 17, 2928–2947. 10.1021/acs.jctc.1c00203. [DOI] [PubMed] [Google Scholar]
- Haasler M.; Maier T. M.; Grotjahn R.; Gückel S.; Arbuznikov A. V.; Kaupp M. A Local Hybrid Functional with Wide Applicability and Good Balance between (De)Localization and Left–Right Correlation. J. Chem. Theory Comput. 2020, 16, 5645–5657. 10.1021/acs.jctc.0c00498. [DOI] [PubMed] [Google Scholar]
- Harrison D. P.; Grotjahn R.; Naher M.; Ghazvini S. M. B. H.; Mazzucato D. M.; Korb M.; Moggach S. A.; Lambert C.; Kaupp M.; Low P. J. Quantum Interference in Mixed-Valence Complexes: Tuning Electronic Coupling Through Substituent Effects. Angew. Chem., Int. Ed. 2022, 61, e202211000. 10.1002/anie.202211000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gückel S.; Safari P.; Bagher Hosseini Ghazvini S. M.; Hall M. R.; Gluyas J. B. G.; Kaupp M.; Low P. J. Iron Versus Ruthenium: Evidence for the Distinct Differences in the Electronic Structures of Hexa-1,3,5-triyn-1,6-diyl-bridged Complexes [Cp* (dppe){M}{μ-(C≡C)3}{M(dppe)Cp*}]+ (M = Fe, Ru). Organometallics 2021, 40, 346–357. 10.1021/acs.organomet.0c00681. [DOI] [Google Scholar]
- Grotjahn R.; Kaupp M. Reliable TDDFT Protocol Based on a Local Hybrid Functional for the Prediction of Vibronic Phosphorescence Spectra Applied to Tris(2,2′-bipyridine)-Metal Complexes. J. Phys. Chem. A 2021, 125, 7099–7110. 10.1021/acs.jpca.1c05101. [DOI] [PubMed] [Google Scholar]
- Grotjahn R.; Kaupp M. Validation of Local Hybrid Functionals for Excited States: Structures, Fluorescence, Phosphorescence, and Vibronic Spectra. J. Chem. Theory Comput. 2020, 16, 5821–5834. 10.1021/acs.jctc.0c00520. [DOI] [PubMed] [Google Scholar]
- Grotjahn R.; Kaupp M. Assessment of hybrid functionals for singlet and triplet excitations: Why do some local hybrid functionals perform so well for triplet excitation energies?. J. Chem. Phys. 2021, 155, 124108. 10.1063/5.0063751. [DOI] [PubMed] [Google Scholar]
- Grotjahn R.; Kaupp M. A Look at Real-World Transition-Metal Thermochemistry and Kinetics with Local Hybrid Functionals. Isr. J. Chem. 2022, e202200021. 10.1002/ijch.202200021. [DOI] [Google Scholar]
- Schattenberg C. J.; Kaupp M. Extended Benchmark Set of Main-Group Nuclear Shielding Constants and NMR Chemical Shifts and Its Use to Evaluate Modern DFT Methods. J. Chem. Theory Comput. 2021, 17, 7602–7621. 10.1021/acs.jctc.1c00919. [DOI] [PubMed] [Google Scholar]
- Schattenberg C. J.; Lehmann M.; Bühl M.; Kaupp M. Systematic Evaluation of Modern Density Functional Methods for the Computation of NMR Shifts of 3d Transition-Metal Nuclei. J. Chem. Theory Comput. 2022, 18, 273–292. 10.1021/acs.jctc.1c00964. [DOI] [PubMed] [Google Scholar]
- Mori-Sánchez P.; Cohen A. J. The derivative discontinuity of the exchange–correlation functional. Phys. Chem. Chem. Phys. 2014, 16, 14378–14387. 10.1039/C4CP01170H. [DOI] [PubMed] [Google Scholar]
- Janesko B. G.; Proynov E.; Kong J.; Scalmani G.; Frisch M. J. Practical Density Functionals beyond the Overdelocalization–Underbinding Zero-Sum Game. J. Phys. Chem. Lett. 2017, 8, 4314–4318. 10.1021/acs.jpclett.7b02023. [DOI] [PubMed] [Google Scholar]
- Mori-Sánchez P.; Cohen A. J.; Yang W. Discontinuous Nature of the Exchange-Correlation Functional in Strongly Correlated Systems. Phys. Rev. Lett. 2009, 102, 066403. 10.1103/PhysRevLett.102.066403. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density functionals for static, dynamical, and strong correlation. J. Chem. Phys. 2013, 138, 074109. 10.1063/1.4790598. [DOI] [PubMed] [Google Scholar]
- Kong J.; Proynov E. Density Functional Model for Nondynamic and Strong Correlation. J. Chem. Theory Comput. 2016, 12, 133–143. 10.1021/acs.jctc.5b00801. [DOI] [PubMed] [Google Scholar]
- Wodyński A.; Arbuznikov A. V.; Kaupp M. Local hybrid functionals augmented by a strong-correlation model. J. Chem. Phys. 2021, 155, 144101. 10.1063/5.0058917. [DOI] [PubMed] [Google Scholar]
- Wodyński A.; Kaupp M. Local Hybrid Functional Applicable to Weakly and Strongly Correlated Systems. J. Chem. Theory Comput. 2022, 18, 6111–6123. 10.1021/acs.jctc.2c00795. [DOI] [PubMed] [Google Scholar]
- Haunschild R.; Scuseria G. E. Range-separated local hybrids. J. Chem. Phys. 2010, 132, 224106. 10.1063/1.3451078. [DOI] [PubMed] [Google Scholar]
- Janesko B. G.; Krukau A. V.; Scuseria G. E. Self-consistent generalized Kohn-Sham local hybrid functionals of screened exchange: Combining local and range-separated hybridization. J. Chem. Phys. 2008, 129, 124110. 10.1063/1.2980056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klawohn S.; Bahmann H. Self-Consistent Implementation of Hybrid Functionals with Local Range Separation. J. Chem. Theory Comput. 2020, 16, 953–963. 10.1021/acs.jctc.9b01015. [DOI] [PubMed] [Google Scholar]
- Fürst S.; Haasler M.; Grotjahn R.; Kaupp M. Full Implementation, Optimization, and Evaluation of a Range-Separated Local Hybrid Functional with Wide Accuracy for Ground and Excited States. J. Chem. Theory Comput. 2023, 19, 488–502. 10.1021/acs.jctc.2c00782. [DOI] [PubMed] [Google Scholar]
- Fürst S.; Kaupp M. Accurate Ionization Potentials, Electron Affinities, and Band Gaps from the ωLH22t Range-Separated Local Hybrid Functional: No Tuning Required. J. Chem. Theory Comput. 2023, 19 (11), 3146–3158. 10.1021/acs.jctc.3c00173. [DOI] [PubMed] [Google Scholar]
- Dobson J. F. Alternative expressions for the Fermi hole curvature. J. Chem. Phys. 1993, 98, 8870–8872. 10.1063/1.464444. [DOI] [Google Scholar]
- Becke A. D. Current density in exchange-correlation functionals: Application to atomic states. J. Chem. Phys. 2002, 117, 6935–6938. 10.1063/1.1503772. [DOI] [Google Scholar]
- Tao J. Explicit inclusion of paramagnetic current density in the exchange-correlation functionals of current-density functional theory. Phys. Rev. B 2005, 71, 205107. 10.1103/PhysRevB.71.205107. [DOI] [Google Scholar]
- Bates J. E.; Heiche M. C.; Liang J.; Furche F. Erratum: “Harnessing the meta-generalized gradient approximation for time-dependent density functional theory” [J. Chem. Phys. 137, 164105 (2012)]. J. Chem. Phys. 2022, 156, 159902. 10.1063/5.0088123. [DOI] [PubMed] [Google Scholar]
- Grotjahn R.; Furche F.; Kaupp M. Importance of imposing gauge invariance in time-dependent density functional theory calculations with meta-generalized gradient approximations. J. Chem. Phys. 2022, 157, 111102. 10.1063/5.0113083. [DOI] [PubMed] [Google Scholar]
- Sciortino G.; Lihi N.; Czine T.; Maréchal J.-D.; Lledós A.; Garribba E. Accurate prediction of vertical electronic transitions of Ni(II) coordination compounds via time dependent density functional theory. Int. J. Quantum Chem. 2018, 118, e25655. 10.1002/qua.25655. [DOI] [Google Scholar]
- Grotjahn R.; Furche F. Gauge-Invariant Excited-State Linear and Quadratic Response Properties within the Meta-Generalized Gradient Approximation. J. Chem. Theory Comput. 2023, 10.1021/acs.jctc.3c00259. [DOI] [PubMed] [Google Scholar]
- Schattenberg C. J.; Kaupp M. Effect of the current dependence of tau-dependent exchange-correlation functionals on nuclear shielding calculations. J. Chem. Theory Comput. 2021, 17, 1469–1479. 10.1021/acs.jctc.0c01223. [DOI] [PubMed] [Google Scholar]
- Bruder F.; Franzke Y. J.; Weigend F. Paramagnetic NMR Shielding Tensors Based on Scalar Exact Two-Component and Spin-Orbit Perturbation Theory. J. Phys. Chem. A 2022, 126, 5050–5069. 10.1021/acs.jpca.2c03579. [DOI] [PubMed] [Google Scholar]
- Maximoff S. N.; Scuseria G. E. Nuclear magnetic resonance shielding tensors calculated with kinetic energy density-dependent exchange-correlation functionals. Chem. Phys. Lett. 2004, 390, 408–412. 10.1016/j.cplett.2004.04.049. [DOI] [Google Scholar]
- Holzer C.; Franzke Y. J.; Pausch A. Current density functional framework for spin–orbit coupling. J. Chem. Phys. 2022, 157, 204102. 10.1063/5.0122394. [DOI] [PubMed] [Google Scholar]
- Pausch A.; Holzer C. Linear Response of Current-Dependent Density Functional Approximations in Magnetic Fields. J. Phys. Chem. Lett. 2022, 13, 4335–4341. 10.1021/acs.jpclett.2c01082. [DOI] [PubMed] [Google Scholar]
- Helgaker T.; Jaszuński M.; Ruud K. Ab initio methods for the calculation of NMR shielding and indirect spin-spin coupling constants. Chem. Rev. 1999, 99, 293–352. 10.1021/cr960017t. [DOI] [PubMed] [Google Scholar]
- Vaara J. Theory and computation of nuclear magnetic resonance parameters. Phys. Chem. Chem. Phys. 2007, 9, 5399–5148. 10.1039/b706135h. [DOI] [PubMed] [Google Scholar]
- Kjærgaard T.; Coriani S.; Ruud K. Ab initio calculation of magnetic circular dichroism. WIREs Comput. Mol. Sci. 2012, 2, 443–455. 10.1002/wcms.1091. [DOI] [Google Scholar]
- Tellgren E. I.; Soncini A.; Helgaker T. Nonperturbative ab initio calculations in strong magnetic fields using London orbitals. J. Chem. Phys. 2008, 129, 154114. 10.1063/1.2996525. [DOI] [PubMed] [Google Scholar]
- Stopkowicz S.; Gauss J.; Lange K. K.; Tellgren E. I.; Helgaker T. Coupled-cluster theory for atoms and molecules in strong magnetic fields. J. Chem. Phys. 2015, 143, 074110. 10.1063/1.4928056. [DOI] [PubMed] [Google Scholar]
- Irons T. J. P.; Zemen J.; Teale A. M. Efficient Calculation of Molecular Integrals over London Atomic Orbitals. J. Chem. Theory Comput. 2017, 13, 3636–3649. 10.1021/acs.jctc.7b00540. [DOI] [PubMed] [Google Scholar]
- Sun S.; Williams-Young D. B.; Stetina T. F.; Li X. Generalized Hartree-Fock with Nonperturbative Treatment of Strong Magnetic Fields: Application to Molecular Spin Phase Transitions. J. Chem. Theory Comput. 2019, 15, 348–356. 10.1021/acs.jctc.8b01140. [DOI] [PubMed] [Google Scholar]
- Pausch A.; Gebele M.; Klopper W. Molecular point groups and symmetry in external magnetic fields. J. Chem. Phys. 2021, 155, 201101. 10.1063/5.0069859. [DOI] [PubMed] [Google Scholar]
- David G.; Irons T. J. P.; Fouda A. E. A.; Furness J. W.; Teale A. M. Self-Consistent Field Methods for Excited States in Strong Magnetic Fields: a Comparison between Energy- and Variance-Based Approaches. J. Chem. Theory Comput. 2021, 17, 5492–5508. 10.1021/acs.jctc.1c00236. [DOI] [PubMed] [Google Scholar]
- Stetina T. F.; Sun S.; Williams-Young D. B.; Li X. Modeling Magneto-Photoabsorption Using Time-Dependent Complex Generalized Hartree-Fock. ChemPhotoChem. 2019, 3, 739–746. 10.1002/cptc.201900161. [DOI] [Google Scholar]
- Monzel L.; Pausch A.; Peters L. D. M.; Tellgren E. I.; Helgaker T.; Klopper W. Molecular Dynamics of Linear Molecules in Strong Magnetic Fields. J. Chem. Phys. 2022, 157, 054106. 10.1063/5.0097800. [DOI] [PubMed] [Google Scholar]
- Ferrario L.; de Martino D.; Gänsicke B. T. Magnetic white dwarfs. Space Sci. Rev. 2015, 191, 111–169. 10.1007/s11214-015-0152-0. [DOI] [Google Scholar]
- Mereghetti S.; Pons J. A.; Melatos A. Magnetars: properties, origin and evolution. Space Sci. Rev. 2015, 191, 315–338. 10.1007/s11214-015-0146-y. [DOI] [Google Scholar]
- Liebert J.; Ferrario L.; Wickramasinghe D. T.; Smith P. S. Enigmas from the Sloan Digital Sky Survey DR7 Kleinman White Dwarf Catalog. Astrophys. J. 2015, 804, 93. 10.1088/0004-637X/804/2/93. [DOI] [Google Scholar]
- Ferrario L.; Wickramasinghe D.; Kawka A. Magnetic fields in isolated and interacting white dwarfs. Adv. Space Res. 2020, 66, 1025–1056. 10.1016/j.asr.2019.11.012. [DOI] [Google Scholar]
- Stopkowicz S. Perspective: Coupled cluster theory for atoms and molecules in strong magnetic fields. Int. J. Quantum Chem. 2018, 118, e25391. 10.1002/qua.25391. [DOI] [PubMed] [Google Scholar]
- Hall P. B.; Maxwell A. J. C2 in Peculiar DQ White Dwarfs. Astrophys. J. 2008, 678, 1292. 10.1086/586889. [DOI] [Google Scholar]
- Kowalski P. M. The origin of peculiar molecular bands in cool DQ white dwarfs. Astron. Astrophys. 2010, 519, L8. 10.1051/0004-6361/201015238. [DOI] [Google Scholar]
- Berdyugina S. V.; Berdyugin A. V.; Piirola V. Molecular Magnetic Dichroism in Spectra of White Dwarfs. Phys. Rev. Lett. 2007, 99, 091101. 10.1103/PhysRevLett.99.091101. [DOI] [PubMed] [Google Scholar]
- Vornanen T.; Berdyugina S. V.; Berdyugin A. V.; Piirola V. GJ 841B—The Second DQ White Dwarf With Polarized CH Molecular Bands. Astrophys. J. 2010, 720, L52–L55. 10.1088/2041-8205/720/1/L52. [DOI] [Google Scholar]
- Lange K. K.; Tellgren E. I.; Hoffmann M.; Helgaker T. A paramagnetic bonding mechanism for diatomics in strong magnetic fields. Science 2012, 337, 327–331. 10.1126/science.1219703. [DOI] [PubMed] [Google Scholar]
- Tellgren E. I.; Fliegl H. Non-perturbative treatment of molecules in linear magnetic fields: Calculation of anapole susceptibilities. J. Chem. Phys. 2013, 139, 164118. 10.1063/1.4826578. [DOI] [PubMed] [Google Scholar]
- Pausch A.; Klopper W. Efficient evaluation of three-centre two-electron integrals over London orbitals. Mol. Phys. 2020, 118, e1736675. 10.1080/00268976.2020.1736675. [DOI] [Google Scholar]
- Pausch A.; Holzer C.; Klopper W. Efficient Calculation of Magnetic Circular Dichroism Spectra Using Spin-Noncollinear Linear-Response Time-Dependent Density Functional Theory in Finite Magnetic Fields. J. Chem. Theory Comput. 2022, 18, 3747–3758. 10.1021/acs.jctc.2c00232. [DOI] [PubMed] [Google Scholar]
- Holzer C.; Teale A. M.; Hampe F.; Stopkowicz S.; Helgaker T.; Klopper W. GW quasiparticle energies of atoms in strong magnetic fields. J. Chem. Phys. 2019, 150, 214112. 10.1063/1.5093396. [DOI] [PubMed] [Google Scholar]
- Holzer C.; Pausch A.; Klopper W. The GW/BSE Method in Magnetic Fields. Front. Chem. 2021, 9, 746162 10.3389/fchem.2021.746162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer C. Practical Post-Kohn–Sham Methods for Time-Reversal Symmetry Breaking References. J. Chem. Theory Comput. 2023, 9 (11), 3131–3145. 10.1021/acs.jctc.3c00156. [DOI] [PubMed] [Google Scholar]
- Kobayashi N.; Nakai K. Applications of magnetic circular dichroism spectroscopy to porphyrins and phthalocyanines. Chem. Commun. 2007, 4077–4092. 10.1039/b704991a. [DOI] [PubMed] [Google Scholar]
- Kundu K.; White J. R. K.; Moehring S. A.; Yu J. M.; Ziller J. W.; Furche F.; Evans W. J.; Hill S. A 9.2-GHz clock transition in a Lu(II) molecular spin qubit arising from a 3,467-MHz hyperfine interaction. Nat. Chem. 2022, 14, 392–397. 10.1038/s41557-022-00894-4. [DOI] [PubMed] [Google Scholar]
- Ishikawa N.; Sugita M.; Wernsdorfer W. Quantum Tunneling of Magnetization in Lanthanide Single-Molecule Magnets: Bis(phthalocyaninato)terbium and Bis(phthalocyaninato)dysprosium Anions. Angew. Chem., Int. Ed. 2005, 44, 2931–2935. 10.1002/anie.200462638. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J. Reducing Exact Two-Component Theory for NMR Couplings to a One-Component Approach: Efficiency and Accuracy. J. Chem. Theory Comput. 2023, 19, 2010–2028. 10.1021/acs.jctc.2c01248. [DOI] [PubMed] [Google Scholar]
- Mitchell T. N.; Kowall B. Karplus-type dihedral angle dependence for the coupling constants 3J(119Sn--C--C--119Sn) and 3J(119Sn--C--C--29Si). Magn. Reson. Chem. 1995, 33, 325–328. 10.1002/mrc.1260330502. [DOI] [Google Scholar]
- Krätschmer F.; Sun X.; Gillhuber S.; Kucher H.; Franzke Y. J.; Weigend F.; Roesky P. Fully tin coated coinage metal ions: A pincer type bis-stannylene ligand for exclusive tetrahedral complexation. Chem. Eur. J. 2023, 29, e202203583. 10.1002/chem.202203583. [DOI] [PubMed] [Google Scholar]
- Peters B.; Stuhrmann G.; Mack F.; Weigend F.; Dehnen S. Highly Soluble Supertetrahedra upon Selective Partial Butylation of Chalcogenido Metalate Clusters in Ionic Liquids. Angew. Chem., Int. Ed. 2021, 60, 17622–17628. 10.1002/anie.202104867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer M.; Franzke Y. J.; Weigend F.; von Hänisch C. Low valent group 14 phosphinidenide complexes [(SIDippP)2M] exhibit P-M pπ-pπ interaction (M = Ge, Sn, Pb). Chem. Eur. J. 2020, 26, 192–197. 10.1002/chem.201905061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huniar U.Berechnung der chemischen Verschiebung der NMR mit Methoden der Dichtefunktionaltheorie (DFT) Diploma Thesis, University of Karlsruhe (TH), Karlsruhe, Germany, 1999. [Google Scholar]
- Novotný J.; Sojka M.; Komorovsky S.; Nečas M.; Marek R. Interpreting the Paramagnetic NMR Spectra of Potential Ru(III) Metallodrugs: Synergy between Experiment and Relativistic DFT Calculations. J. Am. Chem. Soc. 2016, 138, 8432–8445. 10.1021/jacs.6b02749. [DOI] [PubMed] [Google Scholar]
- Jensen F. The Basis Set Convergence of Spin–Spin Coupling Constants Calculated by Density Functional Methods. J. Chem. Theory Comput. 2006, 2, 1360–1369. 10.1021/ct600166u. [DOI] [PubMed] [Google Scholar]
- Jensen F. Basis Set Convergence of Nuclear Magnetic Shielding Constants Calculated by Density Functional Methods. J. Chem. Theory Comput. 2008, 4, 719–727. 10.1021/ct800013z. [DOI] [PubMed] [Google Scholar]
- Jakobsen P.; Jensen F. Probing basis set requirements for calculating hyperfine coupling constants. J. Chem. Phys. 2019, 151, 174107. 10.1063/1.5128286. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Treß R.; Pazdera T. M.; Weigend F. Error-consistent segmented contracted all-electron relativistic basis sets of double- and triple-zeta quality for NMR shielding constants. Phys. Chem. Chem. Phys. 2019, 21, 16658–16664. 10.1039/C9CP02382H. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Spiske L.; Pollak P.; Weigend F. Segmented Contracted Error-Consistent Basis Sets of Quadruple-ζ Valence Quality for One- and Two-Component Relativistic All-Electron Calculations. J. Chem. Theory Comput. 2020, 16, 5658–5674. 10.1021/acs.jctc.0c00546. [DOI] [PubMed] [Google Scholar]
- Sundholm D.; Fliegl H.; Berger R. J. F. Calculations of magnetically induced current densities: Theory and applications. WIREs Comput. Mol. Sci. 2016, 6, 639–678. 10.1002/wcms.1270. [DOI] [Google Scholar]
- von Ragué Schleyer P.; Maerker C.; Dransfeld A.; Jiao H.; van Eikema Hommes N. J. R. Nucleus-independent chemical shifts: A simple and efficient aromaticity probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. 10.1021/ja960582d. [DOI] [PubMed] [Google Scholar]
- GIMIC, ver. 2.1.4 (merge 4bce2c9). GitHub, 2022. https://github.com/qmcurrents/gimic (accessed 2022-11-23).
- Jusélius J.; Sundholm D.; Gauss J. Calculation of current densities using gauge-including atomic orbitals. J. Chem. Phys. 2004, 121, 3952–3963. 10.1063/1.1773136. [DOI] [PubMed] [Google Scholar]
- Taubert S.; Sundholm D.; Jusélius J. Calculation of spin-current densities using gauge-including atomic orbitals. J. Chem. Phys. 2011, 134, 054123. 10.1063/1.3549567. [DOI] [PubMed] [Google Scholar]
- Sundholm D.; Dimitrova M.; Berger R. J. F. Current density and molecular magnetic properties. Chem. Commun. 2021, 57, 12362–12378. 10.1039/D1CC03350F. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Sundholm D.; Weigend F. Calculations of current densities and aromatic pathways in cyclic porphyrin and isoporphyrin arrays. Phys. Chem. Chem. Phys. 2017, 19, 12794–12803. 10.1039/C7CP00624A. [DOI] [PubMed] [Google Scholar]
- Reiter K.; Weigend F.; Wirz L. N.; Dimitrova M.; Sundholm D. Magnetically Induced Current Densities in Toroidal Carbon Nanotubes. J. Phys. Chem. C 2019, 123, 15354–15365. 10.1021/acs.jpcc.9b03769. [DOI] [Google Scholar]
- Eulenstein A. R.; Franzke Y. J.; Bügel P.; Massa W.; Weigend F.; Dehnen S. Stabilizing a metalloid {Zn12} unit within a polymetallide environment in [K2Zn20Bi16]6–. Nat. Commun. 2020, 11, 5122. 10.1038/s41467-020-18799-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood A.; Dimitrova M.; Wirz L. N.; Sundholm D. Magnetically Induced Current Densities in π-Conjugated Porphyrin Nanoballs. J. Phys. Chem. A 2022, 126, 7864–7873. 10.1021/acs.jpca.2c04856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulenstein A. R.; Franzke Y. J.; Lichtenberger N.; Wilson R. J.; Deubner H. L.; Kraus F.; Clérac R.; Weigend F.; Dehnen S. Substantial π-aromaticity of the anionic heavy-metal cluster [Th@Bi12]4–. Nat. Chem. 2021, 13, 149–155. 10.1038/s41557-020-00592-z. [DOI] [PubMed] [Google Scholar]
- Lichtenberger N.; Wilson R. J.; Eulenstein A. R.; Massa W.; Clérac R.; Weigend F.; Dehnen S. Main Group Metal–Actinide Magnetic Coupling and Structural Response Upon U4+ Inclusion Into Bi, Tl/Bi, or Pb/Bi Cages. J. Am. Chem. Soc. 2016, 138, 9033–9036. 10.1021/jacs.6b04363. [DOI] [PubMed] [Google Scholar]
- Peerless B.; Schmidt A.; Franzke Y. J.; Dehnen S. φ-Aromaticity in prismatic {Bi6}-based clusters. Nat. Chem. 2023, 15, 347–356. 10.1038/s41557-022-01099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Computational Methods in Lanthanide and Actinide Chemistry; Dolg M., Ed.; John Wiley & Sons: Chichester, England, 2015. [Google Scholar]
- Rabuck A. D.; Scuseria G. E. Improving self-consistent field convergence by varying occupation numbers. J. Chem. Phys. 1999, 110, 695–700. 10.1063/1.478177. [DOI] [Google Scholar]
- Nava P.; Sierka M.; Ahlrichs R. Density functional study of palladium clusters. Phys. Chem. Chem. Phys. 2003, 5, 3372–3381. 10.1039/B303347C. [DOI] [Google Scholar]
- Saunders V.; Hillier I. A “Level–Shifting” method for converging closed shell Hartree–Fock wave functions. Int. J. Quantum Chem. 1973, 7, 699–705. 10.1002/qua.560070407. [DOI] [Google Scholar]
- Pulay P. Convergence acceleration of iterative sequences. The case of SCF iteration. Chem. Phys. Lett. 1980, 73, 393–398. 10.1016/0009-2614(80)80396-4. [DOI] [Google Scholar]
- Chung A. B.; Rappoport D.; Ziller J. W.; Cramer R. E.; Furche F.; Evans W. J. Solid-State End-On to Side-On Isomerization of (N = N)2– in {[(R2N)3Nd]2N2}2– (R = SiMe3) Connects In Situ LnIII(NR2)3/K and Isolated [LnII(NR2)3]1– Dinitrogen Reduction. J. Am. Chem. Soc. 2022, 144, 17064–17074. 10.1021/jacs.2c06716. [DOI] [PubMed] [Google Scholar]
- MacDonald M. R.; Bates J. E.; Fieser M. E.; Ziller J. W.; Furche F.; Evans W. J. Expanding rare-earth oxidation state chemistry to molecular complexes of holmium (II) and erbium (II). J. Am. Chem. Soc. 2012, 134, 8420–8423. 10.1021/ja303357w. [DOI] [PubMed] [Google Scholar]
- MacDonald M. R.; Bates J. E.; Ziller J. W.; Furche F.; Evans W. J. Completing the series of + 2 ions for the lanthanide elements: synthesis of molecular complexes of Pr2+, Gd2+, Tb2+, and Lu2+. J. Am. Chem. Soc. 2013, 135, 9857–9868. 10.1021/ja403753j. [DOI] [PubMed] [Google Scholar]
- Fieser M. E.; MacDonald M. R.; Krull B. T.; Bates J. E.; Ziller J. W.; Furche F.; Evans W. J. Structural, spectroscopic, and theoretical comparison of traditional vs recently discovered Ln2+ ions in the [K(2.2.2-cryptand)][(C5H4SiMe3)3Ln] complexes: the variable nature of Dy2+ and Nd2+. J. Am. Chem. Soc. 2015, 137, 369–382. 10.1021/ja510831n. [DOI] [PubMed] [Google Scholar]
- Woen D. H.; Evans W. J.. Expanding the + 2 Oxidation State of the Rare-Earth Metals, Uranium, and Thorium in Molecular Complexes In Handbook on the Physics and Chemistry of Rare Earths, Vol. 50; Bünzli J.-C. G., Pecharsky V. K., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; pp 337–394. [Google Scholar]
- Ryan A. J.; Darago L. E.; Balasubramani S. G.; Chen G. P.; Ziller J. W.; Furche F.; Long J. R.; Evans W. J. Synthesis, structure, and magnetism of tris(amide)[Ln{N(SiMe3)2}3]1– complexes of the non-traditional + 2 lanthanide ions. Chem.: Eur. J. 2018, 24, 7702–7709. 10.1002/chem.201800610. [DOI] [PubMed] [Google Scholar]
- Moore W. N.; White J. R.; Wedal J. C.; Furche F.; Evans W. J. Reduction of Rare-Earth Metal Complexes Induced by γ Irradiation. Inorg. Chem. 2022, 61, 17713–17718. 10.1021/acs.inorgchem.2c02857. [DOI] [PubMed] [Google Scholar]
- Gould C. A.; McClain K. R.; Yu J. M.; Groshens T. J.; Furche F.; Harvey B. G.; Long J. R. Synthesis and Magnetism of Neutral, Linear Metallocene Complexes of Terbium(II) and Dysprosium(II). J. Am. Chem. Soc. 2019, 141, 12967–12973. 10.1021/jacs.9b05816. [DOI] [PubMed] [Google Scholar]
- Rappoport D. Property-optimized Gaussian basis sets for lanthanides. J. Chem. Phys. 2021, 155, 124102. 10.1063/5.0065611. [DOI] [PubMed] [Google Scholar]
- Darago L. E.; Boshart M. D.; Nguyen B. D.; Perlt E.; Ziller J. W.; Lukens W. W.; Furche F.; Evans W. J.; Long J. R. Strong Ferromagnetic Exchange Coupling and Single-Molecule Magnetism in MoS43––Bridged Dilanthanide Complexes. J. Am. Chem. Soc. 2021, 143, 8465–8475. 10.1021/jacs.1c03098. [DOI] [PubMed] [Google Scholar]
- Fieser M. E.; Bates J. E.; Ziller J. W.; Furche F.; Evans W. J. Dinitrogen reduction via photochemical activation of heteroleptic tris (cyclopentadienyl) rare-earth complexes. J. Am. Chem. Soc. 2013, 135, 3804–3807. 10.1021/ja400664s. [DOI] [PubMed] [Google Scholar]
- Fieser M. E.; Johnson C. W.; Bates J. E.; Ziller J. W.; Furche F.; Evans W. J. Dinitrogen reduction, sulfur reduction, and isoprene polymerization via photochemical activation of trivalent bis (cyclopentadienyl) rare-earth-metal allyl complexes. Organometallics 2015, 34, 4387–4393. 10.1021/acs.organomet.5b00613. [DOI] [Google Scholar]
- Yu J. M.; Furche F. Theoretical Study of Divalent Bis(Pentaisopropylcyclopentadienyl) Actinocenes. Inorg. Chem. 2019, 58, 16004–16010. 10.1021/acs.inorgchem.9b02505. [DOI] [PubMed] [Google Scholar]
- Tao J.; Perdew J. P.; Staroverov V. N.; Scuseria G. E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. 10.1103/PhysRevLett.91.146401. [DOI] [PubMed] [Google Scholar]
- Dolg M.; Cao X. Accurate Relativistic Small-Core Pseudopotentials for Actinides. Energy Adjustment for Uranium and First Applications to Uranium Hydride. J. Phys. Chem. A 2009, 113, 12573–12581. 10.1021/jp9044594. [DOI] [PubMed] [Google Scholar]
- Cao X.; Dolg M.; Stoll H. Valence basis sets for relativistic energy-consistent small-core actinide pseudopotentials. J. Chem. Phys. 2003, 118, 487–496. 10.1063/1.1521431. [DOI] [Google Scholar]
- Cao X.; Dolg M. Segmented contraction scheme for small-core actinide pseudopotential basis sets. J. Mol. Struc. (THEOCHEM) 2004, 673, 203–209. 10.1016/j.theochem.2003.12.015. [DOI] [Google Scholar]
- Guo F.-S.; Tsoureas N.; Huang G.-Z.; Tong M.-L.; Mansikkamäki A.; Layfield R. A. Isolation of a Perfectly Linear Uranium(II) Metallocene. Angew. Chem., Int. Ed. 2020, 59, 2299–2303. 10.1002/anie.201912663. [DOI] [PubMed] [Google Scholar]
- Holzer C.; Klopper W. Communication: A hybrid Bethe-Salpeter/time-dependent density-functional-theory approach for excitation energies. J. Chem. Phys. 2018, 149, 101101. 10.1063/1.5051028. [DOI] [PubMed] [Google Scholar]
- Fedotov D. A.; Coriani S.; Hättig C. Damped (linear) response theory within the resolution-of-identity coupled cluster singles and approximate doubles (RI-CC2) method. J. Chem. Phys. 2021, 154, 124110. 10.1063/5.0042759. [DOI] [PubMed] [Google Scholar]
- Kristensen K.; Kauczor J.; Kjaergaard T.; Jørgensen P. Quasienergy formulation of damped response theory. J. Chem. Phys. 2009, 131, 044112. 10.1063/1.3173828. [DOI] [PubMed] [Google Scholar]
- Helgaker T.; Coriani S.; Jørgensen P.; Kristensen K.; Olsen J.; Ruud K. Recent Advances in Wave Function-Based Methods of Molecular Property Calculations. Chem. Rev. 2012, 112, 543–631. 10.1021/cr2002239. [DOI] [PubMed] [Google Scholar]
- Norman P. A perspective on nonresonant and resonant electronic response theory for time-dependent molecular properties. Phys. Chem. Chem. Phys. 2011, 13, 20519–20535. 10.1039/c1cp21951k. [DOI] [PubMed] [Google Scholar]
- Kauczor J.; Norman P. Efficient Calculations of Molecular Linear Response Properties for Spectral Regions. J. Chem. Theory Comput. 2014, 10, 2449–2455. 10.1021/ct500114m. [DOI] [PubMed] [Google Scholar]
- Faber R.; Coriani S. Resonant Inelastic X-ray Scattering and Nonesonant X-ray Emission Spectra from Coupled-Cluster (Damped) Response Theory. J. Chem. Theory Comput. 2019, 15, 520–528. 10.1021/acs.jctc.8b01020. [DOI] [PubMed] [Google Scholar]
- Hättig C.; Heß B. A. Correlated Frequency-dependent Polarizabilities and Dispersion Coefficients in the Time-dependent Second-order Møller-Plesset Approximation. Chem. Phys. Lett. 1995, 233, 359–370. 10.1016/0009-2614(94)01487-G. [DOI] [Google Scholar]
- Hättig C.; Heß B. A. TDMP2 calculation of dynamic multipole polarizabilities and dispersion coefficients for the halogen anions F–, Cl–, Br– and I–. J. Chem. Phys. 1998, 108, 3863–3870. 10.1063/1.475789. [DOI] [Google Scholar]
- Kauczor J.; Norman P.; Saidi W. A. Non-additivity of polarizabilities and van der Waals C6 coefficients of fullerenes. J. Chem. Phys. 2013, 138, 114107. 10.1063/1.4795158. [DOI] [PubMed] [Google Scholar]
- Norman P.; Bishop D. M.; Jensen H. J. A.; Oddershede J. Near-resonant absorption in the time-dependent self-consistent field and multiconfigurational self-consistent field approximations. J. Chem. Phys. 2001, 115, 10323–10334. 10.1063/1.1415081. [DOI] [Google Scholar]
- Norman P.; Bishop D. M.; Jensen H. J. A.; Oddershede J. Nonlinear response theory with relaxation: The first-order hyperpolarizability. J. Chem. Phys. 2005, 123, 194103. 10.1063/1.2107627. [DOI] [PubMed] [Google Scholar]
- Jensen L.; Autschbach J.; Schatz G. C. Finite lifetime effects on the polarizability within time-dependent density-functional theory. J. Chem. Phys. 2005, 122, 224115. 10.1063/1.1929740. [DOI] [PubMed] [Google Scholar]
- Ekström U.; Norman P.; Carravetta V.; Ågren H. Polarization Propagator for X-Ray Spectra. Phys. Rev. Lett. 2006, 97, 143001. 10.1103/PhysRevLett.97.143001. [DOI] [PubMed] [Google Scholar]
- Ekström U.; Norman P. X-ray absorption spectra from the resonant-convergent first-order polarization propagator approach. Phys. Rev. A 2006, 74, 042722. 10.1103/PhysRevA.74.042722. [DOI] [Google Scholar]
- Fahleson T.; Ågren H.; Norman P. A Polarization Propagator for Nonlinear X-ray Spectroscopies. J. Phys. Chem. Lett. 2016, 7, 1991–1995. 10.1021/acs.jpclett.6b00750. [DOI] [PubMed] [Google Scholar]
- Cukras J.; Kauczor J.; Norman P.; Rizzo A.; Rikken G. L. J. A.; Coriani S. A complex-polarization-propagator protocol for magneto-chiral axial dichroism and birefringence dispersion. Phys. Chem. Chem. Phys. 2016, 18, 13267–13279. 10.1039/C6CP01465H. [DOI] [PubMed] [Google Scholar]
- Coriani S.; Fransson T.; Christiansen O.; Norman P. Asymmetric-Lanczos-Chain-Driven Implementation of Electronic Resonance Convergent Coupled-Cluster Linear Response Theory. J. Chem. Theory Comput. 2012, 8, 1616–1628. 10.1021/ct200919e. [DOI] [PubMed] [Google Scholar]
- Coriani S.; Christiansen O.; Fransson T.; Norman P. Coupled-cluster response theory for near-edge x-ray-absorption fine structure of atoms and molecules. Phys. Rev. A 2012, 85, 022507. 10.1103/PhysRevA.85.022507. [DOI] [Google Scholar]
- Rehn D. R.; Dreuw A.; Norman P. Resonant Inelastic X-ray Scattering Amplitudes and Cross Sections in the Algebraic Diagrammatic Construction/Intermediate State Representation (ADC/ISR) Approach. J. Chem. Theory Comput. 2017, 13, 5552–5559. 10.1021/acs.jctc.7b00636. [DOI] [PubMed] [Google Scholar]
- Nørby M.; Coriani S.; Kongsted J. Modeling magnetic circular dichroism within the polarizable embedding approach. Theor. Chem. Acta. 2018, 137, 49. 10.1007/s00214-018-2220-5. [DOI] [Google Scholar]
- Vaara J.; Rizzo A.; Kauczor J.; Norman P.; Coriani S. Nuclear spin circular dichroism. J. Chem. Phys. 2014, 140, 134103. 10.1063/1.4869849. [DOI] [PubMed] [Google Scholar]
- Andersen J. H.; Coriani S.; Hättig C. Damped quadratic response within the resolution-of-identity coupled cluster singles and approximate doubles (RI-CC2) method. ChemRxiv 2023, 10.26434/chemrxiv-2023-dgbdz. [DOI] [PubMed] [Google Scholar]
- Zerulla B.; Krstić M.; Beutel D.; Holzer C.; Wöll C.; Rockstuhl C.; Fernandez-Corbaton I. A Multi-Scale Approach for Modeling the Optical Response of Molecular Materials Inside Cavities. Adv. Mater. 2022, 34, 2200350. 10.1002/adma.202200350. [DOI] [PubMed] [Google Scholar]
- Feuerstein W.; Holzer C.; Gui X.; Neumeier L.; Klopper W.; Breher F. Synthesis of New Donor-Substituted Biphenyls: Pre-ligands for Highly Luminescent (ĈĈD) Gold(III) Pincer Complexes. Eur. J. Chem. 2020, 26, 17156–17164. 10.1002/chem.202003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S. M.; Roy S.; Furche F. Unphysical divergences in response theory. J. Chem. Phys. 2016, 145, 134105. 10.1063/1.4963749. [DOI] [PubMed] [Google Scholar]
- Müller M. M.; Perdana N.; Rockstuhl C.; Holzer C. Modeling and measuring plasmonic excitations in hollow spherical gold nanoparticles. J. Chem. Phys. 2022, 156, 094103. 10.1063/5.0078230. [DOI] [PubMed] [Google Scholar]
- Perdana N.; Holzer C.; Rockstuhl C. Multiscale Modeling of Broadband Perfect Absorbers Based on Gold Metallic Molecules. ACS Omega 2022, 7, 19337–19346. 10.1021/acsomega.2c00911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutel D.; Groner A.; Rockstuhl C.; Fernandez-Corbaton I. Efficient simulation of biperiodic, layered structures based on the T-matrix method. J. Opt. Soc. Am. B 2021, 38, 1782–1791. 10.1364/JOSAB.419645. [DOI] [Google Scholar]
- Zerulla B.; Li C.; Beutel D.; Oßwald S.; Holzer C.; Bürck J.; Bräse S.; Wöll C.; Fernandez-Corbaton I.; Heinke L.; Rockstuhl C.; Krstić M. Exploring Functional Photonic Devices made from a Chiral MetalOrganic Framework Material by a Multiscale Computational Method. Adv. Funct. Mater. 2023, 2301093. 10.1002/adfm.202301093. [DOI] [Google Scholar]
- Zerulla B.; Venkitakrishnan R.; Beutel D.; Krstić M.; Holzer C.; Rockstuhl C.; Fernandez-Corbaton I. A T-Matrix Based Approach to Homogenize Artificial Materials. Adv. Opt. Mater. 2023, 11, 2201564. 10.1002/adom.202201564. [DOI] [Google Scholar]
- Kauczor J.; Norman P.; Christiansen O.; Coriani S. Communication: A reduced-space algorithm for the solution of the complex linear response equations used in coupled cluster damped response theory. J. Chem. Phys. 2013, 139, 211102. 10.1063/1.4840275. [DOI] [PubMed] [Google Scholar]
- Mori T.; Inoue Y.; Grimme S. Quantum Chemical Study on the Circular Dichroism Spectra and Specific Rotation of Donor-Acceptor Cyclophanes. J. Phys. Chem. A 2007, 111, 7995–8006. 10.1021/jp073596m. [DOI] [PubMed] [Google Scholar]
- Li Z.; Liu W. First-order nonadiabatic coupling matrix elements between excited states: A Lagrangian formulation at the CIS, RPA, TD-HF, and TD-DFT levels. J. Chem. Phys. 2014, 141, 014110. 10.1063/1.4885817. [DOI] [PubMed] [Google Scholar]
- Parker S. M.; Roy S.; Furche F. Multistate hybrid time-dependent density functional theory with surface hopping accurately captures ultrafast thymine photodeactivation. Phys. Chem. Chem. Phys. 2019, 21, 18999–19010. 10.1039/C9CP03127H. [DOI] [PubMed] [Google Scholar]
- Roy S.; Furche F.. Quinonile Photobasicity is Mediated by Hole Injection. In Bulletin of the American Physical Society, APS March Meeting 2018, Vol. 63, Los Angeles, CA, March 5–9, 2018; American Physical Society, 2018; Paper V03.00007
- Kasha M. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 1950, 9, 14–19. 10.1039/df9500900014. [DOI] [Google Scholar]
- Itoh T. Fluorescence and Phosphorescence from Higher Excited States of Organic Molecules. Chem. Rev. 2012, 112, 4541–4568. 10.1021/cr200166m. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Ernzerhof M.; Burke K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. 10.1063/1.472933. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Viswanath G.; Kasha M. Confirmation of the anomalous fluorescence of azulene. J. Chem. Phys. 1956, 24, 574–577. 10.1063/1.1742548. [DOI] [Google Scholar]
- Geldof P.; Rettschnick R.; Hoytink G. Fluorescence from the second excited singlets of pyrene and 3,4-benzpyrene. Chem. Phys. Lett. 1969, 4, 59–61. 10.1016/0009-2614(69)85066-9. [DOI] [Google Scholar]
- Baba H.; Nakajima A.; Aoi M.; Chihara K. Fluorescence from the second excited singlet state and radiationless processes in pyrene vapor. J. Chem. Phys. 1971, 55, 2433–2438. 10.1063/1.1676429. [DOI] [Google Scholar]
- Chihara K.; Baba H. Effects of foreign gases on dual fluorescences of pyrene vapor. Bull. Chem. Soc. Jpn. 1975, 48, 3093–3100. 10.1246/bcsj.48.3093. [DOI] [Google Scholar]
- Braun G.; Borges I. Jr; Aquino A. J.; Lischka H.; Plasser F.; Do Monte S. A.; Ventura E.; Mukherjee S.; Barbatti M. Non-Kasha fluorescence of pyrene emerges from a dynamic equilibrium between excited states. J. Chem. Phys. 2022, 157, 154305. 10.1063/5.0113908. [DOI] [PubMed] [Google Scholar]
- Borrego-Varillas R.; Ganzer L.; Cerullo G.; Manzoni C. Ultraviolet transient absorption spectrometer with Sub-20-fs time resolution. Appl. Sci. 2018, 8, 989. 10.3390/app8060989. [DOI] [Google Scholar]
- Foggi P.; Neuwahl F. V.; Moroni L.; Salvi P. R. Absorption of Azulene: Femtosecond Transient Spectra and Excited State Calculations. J. Phys. Chem. A 2003, 107, 1689–1696. 10.1021/jp021313e. [DOI] [Google Scholar]
- Neuwahl F. V.; Foggi P. Direct observation of s2-s1 internal conversion in pyrene by femtosecond transient absorption. Laser Chem. 1999, 19, 375–379. 10.1155/1999/37692. [DOI] [Google Scholar]
- Hollas J. M. Determination of molecular conformation from large amplitude vibrations in electronic spectra of organic molecules in a supersonic jet. Chem. Soc. Rev. 1993, 22, 371–382. 10.1039/cs9932200371. [DOI] [Google Scholar]
- Villa E.; Amirav A.; Lim E. C. Single-vibronic-level and excitation-energy dependence of radiative and nonradiative transitions in jet-cooled S1 pyridine. J. Phys. Chem. 1988, 92, 5393–5397. 10.1021/j100330a015. [DOI] [Google Scholar]
- Diau E. W.-G.; De Feyter S.; Zewail A. H. Direct observation of the femtosecond nonradiative dynamics of azulene in a molecular beam: The anomalous behavior in the isolated molecule. J. Chem. Phys. 1999, 110, 9785. 10.1063/1.478031. [DOI] [Google Scholar]
- Cisneros C.; Thompson T.; Baluyot N.; Smith A. C.; Tapavicza E. The role of tachysterol in vitamin D photosynthesis - a non-adiabatic molecular dynamics study. Phys. Chem. Chem. Phys. 2017, 19, 5763–5777. 10.1039/C6CP08064B. [DOI] [PubMed] [Google Scholar]
- Tapavicza E.; Thompson T.; Redd K.; Kim D. Tuning the photoreactivity of Z-hexatriene photoswitches by substituents - a non-adiabatic molecular dynamics study. Phys. Chem. Chem. Phys. 2018, 20, 24807–24820. 10.1039/C8CP05181J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson T.; Tapavicza E. First-Principles Prediction of Wavelength-Dependent Product Quantum Yields. J. Phys. Chem. Lett. 2018, 9, 4758–4764. 10.1021/acs.jpclett.8b02048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grathwol C. W.; Wössner N.; Swyter S.; Smith A. C.; Tapavicza E.; Hofstetter R. K.; Bodtke A.; Jung M.; Link A. Azologization and repurposing of a hetero-stilbene-based kinase inhibitor: towards the design of photoswitchable sirtuin inhibitors. Beilstein J. Org. Chem. 2019, 15, 2170–2183. 10.3762/bjoc.15.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofferman D. L.; Konar A.; Mastron J. N.; Spears K. G.; Cisneros C.; Smith A. C.; Tapavicza E.; Sension R. J. Probing the Formation and Conformational Relaxation of Previtamin D3 and Analogues in Solution and in Lipid Bilayers. J. Phys. Chem. B 2021, 125, 10085–10096. 10.1021/acs.jpcb.1c04376. [DOI] [PubMed] [Google Scholar]
- Tapavicza E.Conformationally controlled photochemistry studied by trajectory surface hopping In Time-Dependent Density Functional Theory; Zhu C., Ed.; Jenny Stanford Publishing: Singapore, China, 2022; pp 141–197. [Google Scholar]
- Tapavicza E.; Furche F.; Sundholm D. Importance of vibronic effects in the UV-Vis spectrum of the 7, 7, 8, 8-tetracyanoquinodimethane anion. J. Chem. Theory Comput. 2016, 12, 5058–5066. 10.1021/acs.jctc.6b00720. [DOI] [PubMed] [Google Scholar]
- Santoro F.; Improta R.; Lami A.; Bloino J.; Barone V. Effective method to compute Franck-Condon integrals for optical spectra of large molecules in solution. J. Chem. Phys. 2007, 126, 084509. 10.1063/1.2437197. [DOI] [PubMed] [Google Scholar]
- Heller E. J. The semiclassical way to molecular spectroscopy. Acc. Chem. Res. 1981, 14, 368–375. 10.1021/ar00072a002. [DOI] [Google Scholar]
- Heller E. J.The semiclassical way to dynamics and spectroscopy; Princeton University Press: Princeton, NJ, 2018. [Google Scholar]
- Baiardi A.; Bloino J.; Barone V. General time dependent approach to vibronic spectroscopy including Franck-Condon, Herzberg–Teller, and Duschinsky effects. J. Chem. Theory Comput. 2013, 9, 4097–4115. 10.1021/ct400450k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapavicza E. Generating Function Approach to Single Vibronic Level Fluorescence Spectra. J. Phys. Chem. Lett. 2019, 10, 6003–6009. 10.1021/acs.jpclett.9b02273. [DOI] [PubMed] [Google Scholar]
- Begušić T.; Tapavicza E.; Vaníček J. Applicability of the Thawed Gaussian Wavepacket Dynamics to the Calculation of Vibronic Spectra of Molecules with Double-Well Potential Energy Surfaces. J. Chem. Theory Comput. 2022, 18, 3065–3074. 10.1021/acs.jctc.2c00030. [DOI] [PubMed] [Google Scholar]
- Duschinsky F. The importance of the electron spectrum in multi atomic molecules. Concerning the Franck-Condon principle. Acta Physicochim. URSS 1937, 7, 551–566. [Google Scholar]
- Benkyi I.; Tapavicza E.; Fliegl H.; Sundholm D. Calculation of vibrationally resolved absorption spectra of acenes and pyrene. Phys. Chem. Chem. Phys. 2019, 21, 21094–21103. 10.1039/C9CP04178H. [DOI] [PubMed] [Google Scholar]
- De Queiroz T. B.; De Figueroa E. R.; Coutinho-Neto M. D.; Maciel C. D.; Tapavicza E.; Hashemi Z.; Leppert L. First principles theoretical spectroscopy of methylene blue: Between limitations of time-dependent density functional theory approximations and its realistic description in the solvent. J. Chem. Phys. 2021, 154, 044106. 10.1063/5.0029727. [DOI] [PubMed] [Google Scholar]
- von Cosel J.; Cerezo J.; Kern-Michler D.; Neumann C.; van Wilderen L. J.; Bredenbeck J.; Santoro F.; Burghardt I. Vibrationally resolved electronic spectra including vibrational pre-excitation: Theory and application to VIPER spectroscopy. J. Chem. Phys. 2017, 147, 164116. 10.1063/1.4999455. [DOI] [PubMed] [Google Scholar]
- Heller E. J. Time-dependent approach to semiclassical dynamics. J. Chem. Phys. 1975, 62, 1544–1555. 10.1063/1.430620. [DOI] [Google Scholar]
- Wehrle M.; Šulc M.; Vaníček J. On-the-fly Ab Initio Semiclassical Dynamics: Identifying Degrees of Freedom Essential for Emission Spectra of Oligothiophenes. J. Chem. Phys. 2014, 140, 244114. 10.1063/1.4884718. [DOI] [PubMed] [Google Scholar]
- Wehrle M.; Oberli S.; Vaníček J. On-the-fly ab initio semiclassical dynamics of floppy molecules: Absorption and photoelectron spectra of ammonia. J. Phys. Chem. A 2015, 119, 5685. 10.1021/acs.jpca.5b03907. [DOI] [PubMed] [Google Scholar]
- Begušić T.; Patoz A.; Šulc M.; Vaníček J. On-the-fly ab initio three thawed Gaussians approximation: a semiclassical approach to Herzberg-Teller spectra. Chem. Phys. 2018, 515, 152–163. 10.1016/j.chemphys.2018.08.003. [DOI] [Google Scholar]
- Vaníček J.; Begušić T.. Ab Initio Semiclassical Evaluation of Vibrationally Resolved Electronic Spectra With Thawed Gaussians. In Molecular Spectroscopy and Quantum Dynamics; Marquardt R., Quack M., Eds.; Elsevier: St. Louis, MO, 2021; pp 199–229. [Google Scholar]
- Begušić T.; Cordova M.; Vaníček J. Single-Hessian thawed Gaussian approximation. J. Chem. Phys. 2019, 150, 154117. 10.1063/1.5090122. [DOI] [PubMed] [Google Scholar]
- Prlj A.; Begušić T.; Zhang Z. T.; Fish G. C.; Wehrle M.; Zimmermann T.; Choi S.; Roulet J.; Moser J.-E.; Vaníček J. Semiclassical Approach to Photophysics Beyond Kasha’s Rule and Vibronic Spectroscopy Beyond the Condon Approximation. The Case of Azulene. J. Chem. Theory Comput. 2020, 16, 2617–2626. 10.1021/acs.jctc.0c00079. [DOI] [PubMed] [Google Scholar]
- Begušić T.; Vaníček J. Efficient semiclassical dynamics for vibronic spectroscopy beyond harmonic, Condon, and zero-temperature approximations. Chimia 2021, 75, 261. 10.2533/chimia.2021.261. [DOI] [PubMed] [Google Scholar]
- Vidberg H. J.; Serene J. W. Solving the Eliashberg equations by means of N-point Padé approximants. J. Low Temp. Phys. 1977, 29, 179–192. 10.1007/BF00655090. [DOI] [Google Scholar]
- Rojas H. N.; Godby R. W.; Needs R. J. Space-Time Method for Ab Initio Calculations of Self-Energies and Dielectric Response Functions of Solids. Phys. Rev. Lett. 1995, 74, 1827–1830. 10.1103/PhysRevLett.74.1827. [DOI] [PubMed] [Google Scholar]
- Voora V. K. Molecular Electron Affinities Using the Generalized Kohn-Sham Semicanonical Projected Random Phase Approximation. J. Phys. Chem. Lett. 2021, 12, 433–439. 10.1021/acs.jpclett.0c03362. [DOI] [PubMed] [Google Scholar]
- Samal B.; Voora V. K. Modeling Nonresonant X-ray Emission of Second- and Third-Period Elements without Core-Hole Reference States and Empirical Parameters. J. Chem. Theory Comput. 2022, 18, 7272–7285. 10.1021/acs.jctc.2c00647. [DOI] [PubMed] [Google Scholar]
- Sugiura C. Kβ X-Ray Emission and K X-Ray Absorption Spectra of Sulfur in Sulfate Compounds. Jpn. J. Appl. Phys. 1993, 32, 3509–3514. 10.1143/JJAP.32.3509. [DOI] [Google Scholar]
- Qureshi M.; Nowak S. H.; Vogt L. I.; Cotelesage J. J. H.; Dolgova N. V.; Sharifi S.; Kroll T.; Nordlund D.; Alonso-Mori R.; Weng T.-C.; Pickering I. J.; George G. N.; Sokaras D. Sulfur Kβ X-ray emission spectroscopy: comparison with sulfur K-edge X-ray absorption spectroscopy for speciation of organosulfur compounds. Phys. Chem. Chem. Phys. 2021, 23, 4500–4508. 10.1039/D0CP05323F. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Dunning T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Yu J. M.; Nguyen B. D.; Tsai J.; Hernandez D. J.; Furche F. Selfconsistent random phase approximation methods. J. Chem. Phys. 2021, 155, 040902. 10.1063/5.0056565. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Yang W.; Burke K.; Yang Z.; Gross E. K. U.; Scheffler M.; Scuseria G. E.; Henderson T. M.; Zhang I. Y.; Ruzsinszky A.; Peng H.; Sun J.; Trushin E.; Görling A. Understanding band gaps of solids in generalized Kohn–Sham theory. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 2801–2806. 10.1073/pnas.1621352114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerends E. J.; Gritsenko O. V.; van Meer R. The Kohn-Sham gap, the fundamental gap and the optical gap: the physical meaning of occupied and virtual Kohn-Sham orbital energies. Phys. Chem. Chem. Phys. 2013, 15, 16408–16425. 10.1039/c3cp52547c. [DOI] [PubMed] [Google Scholar]
- Kronik L.; Stein T.; Refaely-Abramson S.; Baer R. Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2012, 8, 1515–1531. 10.1021/ct2009363. [DOI] [PubMed] [Google Scholar]
- Lüftner D.; Refaely-Abramson S.; Pachler M.; Resel R.; Ramsey M. G.; Kronik L.; Puschnig P. Experimental and theoretical electronic structure of quinacridone. Phys. Rev. B 2014, 90, 075204. 10.1103/PhysRevB.90.075204. [DOI] [Google Scholar]
- Slattery D. K.; Linkous C. A.; Gruhn N. E.; Baum J. Semiempirical MO and voltammetric estimation of ionization potentials of organic pigments. Comparison to gas phase ultraviolet photoelectron spectroscopy. Dyes Pgm. 2001, 49, 21–27. 10.1016/S0143-7208(01)00009-2. [DOI] [Google Scholar]
- Tew D. P. Principal Domains in Local Correlation Theory. J. Chem. Theory Comput. 2019, 15, 6597–6606. 10.1021/acs.jctc.9b00619. [DOI] [PubMed] [Google Scholar]
- Tew D. P.Principal domains in F12 explicitly correlated theory. In New Electron Correlation Methods and their Applications, and Use of Atomic Orbitals with Exponential Asymptotes; Musial M., Hoggan P. E., Eds.; Advances in Quantum Chemistry, Vol. 83; Academic Press, 2021; pp 83–106. [Google Scholar]
- Sorathia K.; Tew D. P. Basis set extrapolation in pair natural orbital theories. J. Chem. Phys. 2020, 153, 174112. 10.1063/5.0022077. [DOI] [PubMed] [Google Scholar]
- Li W.; Saleh A.; Sharma M.; Hunecke C.; Sierka M.; Neuhaus M.; Hedewig L.; Bergues B.; Alharbi M.; ALQahtani H.; Azzeer A. M.; Gräfe S.; Kling M. F.; Alharbi A. F.; Wang Z. Resonance effects in Brunel harmonic generation in thin film organic semiconductors. Adv. Opt. Mater. 2023, 2203070 10.1002/adom.202203070. [DOI] [Google Scholar]
- Burow A. M.; Sierka M. Linear scaling hierarchical integration scheme for the exchange-correlation term in molecular and periodic systems. J. Chem. Theory Comput. 2011, 7, 3097–3104. 10.1021/ct200412r. [DOI] [PubMed] [Google Scholar]
- Irmler A.; Burow A. M.; Pauly F. Robust Periodic Fock Exchange with Atom-Centered Gaussian Basis Sets. J. Chem. Theory Comput. 2018, 14, 4567–4580. 10.1021/acs.jctc.8b00122. [DOI] [PubMed] [Google Scholar]
- Blanes S.; Casas F.; Oteo J.; Ros J. The Magnus expansion and some of its applications. Phys. Rep 2009, 470, 151–238. 10.1016/j.physrep.2008.11.001. [DOI] [Google Scholar]
- Cheng C.-L.; Evans J. S.; Van Voorhis T. Simulating molecular conductance using real-time density functional theory. Phys. Rev. B 2006, 74, 155112. 10.1103/PhysRevB.74.155112. [DOI] [Google Scholar]
- Treutler O.Entwicklung und Anwendung von Dichtefunktionalmethoden. Ph.D. Dissertation, University of Karlsruhe (TH), Karlsruhe, Germany, 1995. [Google Scholar]
- Lembarki A.; Chermette H. Obtaining a gradient-corrected kinetic-energy functional from the Perdew-Wang exchange functional. Phys. Rev. A 1994, 50, 5328–5331. 10.1103/PhysRevA.50.5328. [DOI] [PubMed] [Google Scholar]
- Treß R. S.; Liu J.; Hättig C.; Höfener S. Pushing the limits: efficient wavefunction methods for excited states in complex systems using frozen-density embedding. J. Chem. Phys. 2022, 157, 204101. 10.1063/5.0100393. [DOI] [PubMed] [Google Scholar]
- Wesolowski T. A.; Warshel A. Frozen density functional approach for ab initio calculations of solvated molecules. J. Chem. Phys. 1993, 97, 8050–8053. 10.1021/j100132a040. [DOI] [Google Scholar]
- Lee S. J. R.; Welborn M.; Manby F. R.; Miller T. F. I. Projection-Based Wavefunction-in-DFT Embedding. Acc. Chem. Res. 2019, 52, 1359–1368. 10.1021/acs.accounts.8b00672. [DOI] [PubMed] [Google Scholar]
- Manby F. R.; Stella M.; Goodpaster J. D.; Miller T. F. I. A Simple, Exact Density-Functional-Theory Embedding Scheme. J. Chem. Theory Comput. 2012, 8, 2564–2568. 10.1021/ct300544e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M.; Mishra D. CrysX: crystallographic tools for the Android platform. J. Appl. Crystallogr. 2019, 52, 1449–1454. 10.1107/S1600576719013682. [DOI] [Google Scholar]
- Tymczak C. J.; Weber V. T.; Schwegler E.; Challacombe M. Linear scaling computation of the Fock matrix. VIII. Periodic boundaries for exact exchange at the Γ point. J. Chem. Phys. 2005, 122, 124105. 10.1063/1.1869470. [DOI] [PubMed] [Google Scholar]
- Guidon M.; Hutter J.; VandeVondele J. Robust Periodic Hartree–Fock Exchange for Large-Scale Simulations Using Gaussian Basis Sets. J. Chem. Theory Comput. 2009, 5, 3010–3021. 10.1021/ct900494g. [DOI] [PubMed] [Google Scholar]
- Paier J.; Diaconu C. V.; Scuseria G. E.; Guidon M.; VandeVondele J.; Hutter J. Accurate Hartree-Fock energy of extended systems using large Gaussian basis sets. Phys. Rev. B 2009, 80, 174114. 10.1103/PhysRevB.80.174114. [DOI] [Google Scholar]
- Paier J.; Marsman M.; Hummer K.; Kresse G.; Gerber I. C.; Ángyán J. G. Screened hybrid density functionals applied to solids. J. Chem. Phys. 2006, 124, 154709. 10.1063/1.2187006. [DOI] [PubMed] [Google Scholar]
- Peintinger M. F.; Oliveira D. V.; Bredow T. Consistent Gaussian basis sets of triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 2013, 34, 451–459. 10.1002/jcc.23153. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Horn H.; Ahlrichs R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. 10.1063/1.478522. [DOI] [Google Scholar]
- Krukau A. V.; Vydrov O. A.; Izmaylov A. F.; Scuseria G. E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. 10.1063/1.2404663. [DOI] [PubMed] [Google Scholar]
- Warburton R. E.; Soudackov A. V.; Hammes-Schiffer S. Theoretical Modeling of Electrochemical Proton-Coupled Electron Transfer. Chem. Rev. 2022, 122, 10599–10650. 10.1021/acs.chemrev.1c00929. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Nuclear–electronic orbital methods: Foundations and prospects. J. Chem. Phys. 2021, 155, 030901. 10.1063/5.0053576. [DOI] [PubMed] [Google Scholar]
- Webb S. P.; Iordanov T.; Hammes-Schiffer S. Multiconfigurational nuclear-electronic orbital approach: Incorporation of nuclear quantum effects in electronic structure calculations. J. Chem. Phys. 2002, 117, 4106–4118. 10.1063/1.1494980. [DOI] [Google Scholar]
- Zundel G. Hydration Structure and Intermolecular Interaction in Polyelectrolytes. Angew. Chem., Int. Ed. Engl. 1969, 8, 499–509. 10.1002/anie.196904991. [DOI] [Google Scholar]
- Sirjoosingh A.; Pak M. V.; Hammes-Schiffer S. Multicomponent density functional theory study of the interplay between electron-electron and electron-proton correlation. J. Chem. Phys. 2012, 136, 174114. 10.1063/1.4709609. [DOI] [PubMed] [Google Scholar]
- Xu J.; Zhou R.; Tao Z.; Malbon C.; Blum V.; Hammes-Schiffer S.; Kanai Y. Nuclear–electronic orbital approach to quantization of protons in periodic electronic structure calculations. J. Chem. Phys. 2022, 156, 224111. 10.1063/5.0088427. [DOI] [PubMed] [Google Scholar]
- Cremer D. Møller–Plesset perturbation theory: from small molecule methods to methods for thousands of atoms. WIREs Comput. Mol. Sci. 2011, 1, 509–530. 10.1002/wcms.58. [DOI] [Google Scholar]
- Grimme S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. 10.1063/1.2148954. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Grimme S. Double-hybrid density functionals. WIREs Comput. Mol. Sci. 2014, 4, 576–600. 10.1002/wcms.1193. [DOI] [Google Scholar]
- Nguyen B. D.; Chen G. P.; Agee M. M.; Burow A. M.; Tang M. P.; Furche F. Divergence of Many-Body Perturbation Theory for Noncovalent Interactions of Large Molecules. J. Chem. Theory Comput. 2020, 16, 2258–2273. 10.1021/acs.jctc.9b01176. [DOI] [PubMed] [Google Scholar]
- Shee J.; Loipersberger M.; Rettig A.; Lee J.; Head-Gordon M. Regularized Second-Order Møller-Plesset Theory: A More Accurate Alternative to Conventional MP2 for Noncovalent Interactions and Transition Metal Thermochemistry for the Same Computational Cost. J. Phys. Chem. Lett. 2021, 12, 12084–12097. 10.1021/acs.jpclett.1c03468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A. J.; Mori-Sánchez P.; Yang W. Second-Order Perturbation Theory with Fractional Charges and Fractional Spins. J. Chem. Theory Comput. 2009, 5, 786–792. 10.1021/ct8005419. [DOI] [PubMed] [Google Scholar]
- Grimme S. Improved second-order Møller–Plesset perturbation theory by separate scaling of parallel- and antiparallel-spin pair correlation energies. J. Chem. Phys. 2003, 118, 9095–9102. 10.1063/1.1569242. [DOI] [Google Scholar]
- Seidl M.; Giarrusso S.; Vuckovic S.; Fabiano E.; Gori-Giorgi P. Communication: Strong-interaction limit of an adiabatic connection in Hartree-Fock theory. J. Chem. Phys. 2018, 149, 241101. 10.1063/1.5078565. [DOI] [PubMed] [Google Scholar]
- Seidl M.; Perdew J. P.; Kurth S. Density functionals for the strong-interaction limit. Phys. Rev. A 2000, 62, 012502. 10.1103/PhysRevA.62.012502. [DOI] [PubMed] [Google Scholar]
- Daas T. J.; Kooi D. P.; Grooteman A. J. A. F.; Seidl M.; Gori-Giorgi P. Gradient Expansions for the Large-Coupling Strength Limit of the Møller-Plesset Adiabatic Connection. J. Chem. Theory Comput. 2022, 18, 1584–1594. 10.1021/acs.jctc.1c01206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl M.; Perdew J. P.; Kurth S. Simulation of All-Order Density-Functional Perturbation Theory, Using the Second Order and the Strong-Correlation Limit. Phys. Rev. Lett. 2000, 84, 5070–5073. 10.1103/PhysRevLett.84.5070. [DOI] [PubMed] [Google Scholar]
- Daas T. J.; Fabiano E.; Della Sala F.; Gori-Giorgi P.; Vuckovic S. Noncovalent Interactions from Models for the Møller–Plesset Adiabatic Connection. J. Phys. Chem. Lett. 2021, 12, 4867–4875. 10.1021/acs.jpclett.1c01157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiano E.; Gori-Giorgi P.; Seidl M.; Della Sala F. Interaction-Strength Interpolation Method for Main-Group Chemistry: Benchmarking, Limitations, and Perspectives. J. Chem. Theory Comput. 2016, 12, 4885–4896. 10.1021/acs.jctc.6b00713. [DOI] [PubMed] [Google Scholar]
- Gori-Giorgi P.; Vignale G.; Seidl M. Electronic Zero-Point Oscillations in the Strong-Interaction Limit of Density Functional Theory. J. Chem. Theory Comput. 2009, 5, 743–753. 10.1021/ct8005248. [DOI] [PubMed] [Google Scholar]
- Śmiga S.; Della Sala F.; Gori-Giorgi P.; Fabiano E. Self-Consistent Implementation of Kohn-Sham Adiabatic Connection Models with Improved Treatment of the Strong-Interaction Limit. J. Chem. Theory Comput. 2022, 18, 5936–5947. 10.1021/acs.jctc.2c00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuckovic S.; Gori-Giorgi P.; Della Sala F.; Fabiano E. Restoring Size Consistency of Approximate Functionals Constructed from the Adiabatic Connection. J. Phys. Chem. Lett. 2018, 9, 3137–3142. 10.1021/acs.jpclett.8b01054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlak R.; Janowski T.; Pitoňák M.; Řezáč J.; Pulay P.; Hobza P. Accuracy of Quantum Chemical Methods for Large Noncovalent Complexes. J. Chem. Theory Comput. 2013, 9, 3364–3374. 10.1021/ct400036b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros F.; Dunivan S.; Lao K. U. Coupled cluster benchmarks of large noncovalent complexes: The L7 dataset as well as DNA-ellipticine and buckycatcher-fullerene. J. Chem. Phys. 2021, 154, 154104. 10.1063/5.0042906. [DOI] [PubMed] [Google Scholar]
- Rocca D.; Gebauer R.; Saad Y.; Baroni S. Turbo charging time-dependent density-functional theory with Lanczos chains. J. Chem. Phys. 2008, 128, 154105. 10.1063/1.2899649. [DOI] [PubMed] [Google Scholar]
- Zuehlsdorff T. J.; Hine N. D. M.; Spencer J. S.; Harrison N. M.; Riley D. J.; Haynes P. D. Linear-scaling time-dependent density-functional theory in the linear response formalism. J. Chem. Phys. 2013, 139, 064104. 10.1063/1.4817330. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Neuhauser D.; Baer R.; Rabani E. Sublinear scaling for time-dependent stochastic density functional theory. J. Chem. Phys. 2015, 142, 034106. 10.1063/1.4905568. [DOI] [PubMed] [Google Scholar]
- Baseggio O.; Fronzoni G.; Stener M. A new time dependent density functional algorithm for large systems and plasmons in metal clusters. J. Chem. Phys. 2015, 143, 024106. 10.1063/1.4923368. [DOI] [PubMed] [Google Scholar]
- Bauernschmitt R.; Häser M.; Treutler O.; Ahlrichs R. Calculation of excitation energies within time-dependent density functional theory using auxiliary basis set expansions. Chem. Phys. Lett. 1997, 264, 573–578. 10.1016/S0009-2614(96)01343-7. [DOI] [Google Scholar]
- Heinze H. H.; Görling A.; Rösch N. An efficient method for calculating molecular excitation energies by time-dependent density-functional theory. J. Chem. Phys. 2000, 113, 2088. 10.1063/1.482020. [DOI] [Google Scholar]
- Weigend F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Kattannek M.; Ahlrichs R. Approximated electron repulsion integrals: Cholesky decomposition versus resolution of the identity methods. J. Chem. Phys. 2009, 130, 164106. 10.1063/1.3116103. [DOI] [PubMed] [Google Scholar]
- Grimme S. A Simplified Tamm-Dancoff Density Functional Approach for the Electronic Excitation Spectra of Very Large Molecules. J. Chem. Phys. 2013, 138, 244104. 10.1063/1.4811331. [DOI] [PubMed] [Google Scholar]
- Bannwarth C.; Grimme S. A simplified time-dependent density functional theory approach for electronic ultraviolet and circular dichroism spectra of very large molecules. Comput. Theoret. Chem. 2014, 1040–1041, 45. 10.1016/j.comptc.2014.02.023. [DOI] [Google Scholar]
- Rüger R.; van Lenthe E.; Heine T.; Visscher L. Tight-binding approximations to time-dependent density functional theory A fast approach for the calculation of electronically excited states. J. Chem. Phys. 2016, 144, 184103. 10.1063/1.4948647. [DOI] [PubMed] [Google Scholar]
- Jacquemin D.; Wathelet V.; Perpète E. A.; Adamo C. Extensive TD-DFT Benchmark: Singlet-Excited States of Organic Molecules. J. Chem. Theory Comput. 2009, 5, 2420–2435. 10.1021/ct900298e. [DOI] [PubMed] [Google Scholar]
- Liang J.; Feng X.; Hait D.; Head-Gordon M. Revisiting the Performance of Time-Dependent Density Functional Theory for Electronic Excitations: Assessment of 43 Popular and Recently Developed Functionals from Rungs One to Four. J. Chem. Theory Comput. 2022, 18, 3460–3473. 10.1021/acs.jctc.2c00160. [DOI] [PubMed] [Google Scholar]
- Giannone G.; Della Sala F. Minimal Auxiliary Basis Set for Time-Dependent Density Functional Theory and Comparison with Tight-Binding Approximations: Application to Silver Nanoparticles. J. Chem. Phys. 2020, 153, 084110. 10.1063/5.0020545. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Della Sala F.; Parker S. M. Minimal auxiliary basis set approach for the electronic excitation spectra of organic molecules. J. Phys. Chem. Lett. 2023, 14, 1968–1976. 10.1021/acs.jpclett.2c03698. [DOI] [PubMed] [Google Scholar]
- Franzke Y. J.; Schosser W. M.; Pauly F.. Self-consistent and efficient treatment of relativistic effects with periodic density functional methods: Energies, Gradients, and Stress Tensors arXiv (Physics.Chemical Physics), May 20, 2023, 2305.03817, ver. 2. 10.48550/arXiv.2305.03817. [DOI]
- Zhao R.; Zhang Y.; Xiao Y.; Liu W. Exact two-component relativistic energy band theory and application. J. Chem. Phys. 2016, 144, 044105. 10.1063/1.4940140. [DOI] [PubMed] [Google Scholar]
- Ahlrichs R.; May K. Contracted all-electron Gaussian basis sets for atoms Rb to Xe. Phys. Chem. Chem. Phys. 2000, 2, 943–945. 10.1039/a908859h. [DOI] [Google Scholar]
- Weigend F.; Baldes A. Segmented contracted basis sets for one- and two-component Dirac-Fock effective core potentials. J. Chem. Phys. 2010, 133, 174102. 10.1063/1.3495681. [DOI] [PubMed] [Google Scholar]
- Kadek M.; Repisky M.; Ruud K. All-electron fully relativistic Kohn-Sham theory for solids based on the Dirac-Coulomb Hamiltonian and Gaussian-type functions. Phys. Rev. B 2019, 99, 205103. 10.1103/PhysRevB.99.205103. [DOI] [Google Scholar]
- Dyall K. G. Relativistic Quadruple-Zeta Triple-Zeta and Revised and Double-Zeta Basis Sets for the 4p, 5p, and 6p Elements. Theor. Chem. Acc. 2006, 115, 441–447. 10.1007/s00214-006-0126-0. [DOI] [Google Scholar]
- Dyall K. G. Relativistic double-zeta, triple-zeta, and quadruple-zeta basis sets for the 4d elements Y-Cd. Theor. Chem. Acc. 2007, 117, 483–489. 10.1007/s00214-006-0174-5. [DOI] [Google Scholar]
- Bodo F.; Desmarais J. K.; Erba A. Spin current density functional theory of Weyl semimetals. Phys. Rev. B 2022, 105, 125108. 10.1103/PhysRevB.105.125108. [DOI] [Google Scholar]
- Desmarais J. K.; Flament J.-P.; Erba A. Spin-orbit coupling in periodic systems with broken time-reversal symmetry: Formal and computational aspects. Phys. Rev. B 2020, 101, 235142. 10.1103/PhysRevB.101.235142. [DOI] [Google Scholar]
- Lyman I.Code quality: a concern for businesses, bottom lines, and empathetic programmers. The Overflow, October 18, 2021. https://stackoverflow.blog/2021/10/18/code-quality-a-concern-for-businesses-bottom-lines-and-empathetic-programmers/ (accessed 2023-03-20).