

Summary
Anthrax toxin (AT), secreted by Bacillus anthracis, is a three-protein cocktail of lethal factor (LF, 90 kDa), edema factor (EF, 89 kDa), and the protective antigen (PA, 83 kDa). Steps in anthrax toxicity involve (1) binding of ligand (EF/LF) to a heptamer of PA63 (PA63h) generated after N-terminal proteolytic cleavage of PA and, (2) following endocytosis of the complex, translocation of the ligand into the cytosol by an as yet unknown mechanism. The PA63h.LF complex was directly visualized from analysis of images of specimens suspended in vitrified buffer by cryo-electron microscopy, which revealed that the LF molecule, localized to the nonmembrane-interacting face of the oligomer, interacts with four successive PA63 monomers and partially unravels the heptamer, thereby widening the central lumen. The observed structural reorganization in PA63h likely facilitates the passage of the large 90 kDa LF molecule through the lumen en route to its eventual delivery across the membrane bilayer.
Introduction
Anthrax toxin (AT) is the major virulence factor secreted by the Gram-positive pathogenic bacterium Bacillus anthracis (Leppla, 1999; Dixon et al., 1999). This pathogen has attracted worldwide attention because of the relevance to bio-terrorism. AT belongs to the family of so-called A-B toxins, in which the B moiety binds and inserts into the membrane of sensitive cells to activate translocation of the A moiety. AT is a cocktail of three nontoxic monomeric proteins, the lethal factor (LF, 90 kDa), the edema factor (EF, 89 kDa), and the protective antigen (PA, 83 kDa). LF and EF are the catalytic components, while PA is the receptor binding component. The process of intoxication first involves a binding of PA to a cell-surface receptor (Bradley et al., 2001), followed by cleavage of PA by furin at a single site to produce PA63 (63 kDa), which spontaneously oligomerizes into a heptamer (PA63h) (Milne et al., 1994). Binding of the catalytic components to the oligomer produces the toxic PA63h.LF or PA63h.EF binary complexes (Leppla, 1999), which are then endocytosed. The complex inserts into the endosomal membrane under acidic conditions, forming a transmembrane pore (Collier, 1999; Blaustein et al., 1989; Milne and Collier, 1993), and EF or LF is released into the cytosol by an as yet undetermined mechanism. LF, a Zn-dependent metalloprotease whose only known substrates are members of the mitogen-activated protein kinase kinase (MAPKK) family, causes inhibition of many signal transduction pathways, while EF, a calmodulin-dependent adenylate cyclase, elevates cellular levels of cAMP and causes local edema and impaired neutrophil function.
Understanding the molecular recognition processes involved in the formation of the toxic complex and the delivery of the lethal and edema factors is critical to gaining insight into the pathogenesis of anthrax, and eventually to rational design of agents to block toxin action (Young and Collier, 2002). X-ray crystal structures of full-length LF (Pannifer et al., 2001), the catalytic domain of EF (Drum et al., 2002), and PA and soluble PA63h (Petosa et al., 1997) have provided a basis for further studies of the intermolecular interactions in the binary complexes. The PA monomer is a flat (100 × 50 × 30 Å) four-domain molecule. Heptamerization to PA63h with minor attendant alterations in tertiary structure leads to a hollow ring (160 Å diameter and 85 Å high) that encloses a negatively charged lumen that is 20–35 Å in diameter. Residues in domain 3, as determined from mutagenesis studies (Mogridge et al., 2001), mediate the oligomerization process. In planar lipid-bilayer membranes, PA63h generates cation-selective, voltage-gated ion channels with a minimal pore diameter of ~12 Å that are blocked upon binding of LF or LFN, the recombinant N-terminal domain of LF (Zhao et al., 1995). Based on the PA63h X-ray structure, a model has been proposed that implicates an amphipathic, 23 amino acid long, chymotrypsin-sensitive, disordered segment of domain 2 in membrane insertion. In this model, analogous to that of staphylococcal α-haemolysin, each amphipathic segment contributes two β strands to form a 14-stranded, porin-like, β barrel channel (Petosa et al., 1997; Benson et al., 1998).
LF and EF bind to receptor bound or unbound PA63h with high affinity (nanomolar dissociation constant), as determined from measurements with purified proteins (Elliott et al., 2000; Mogridge et al., 2002a). It was demonstrated that the recombinant N-terminal domain of the ligand (homologous LFN or EFN) is sufficient for tight binding as well as translocation mediated by PA63h (Ballard et al., 1996; Goletz et al., 1997). Two groups have reported the stoichiometry of binding of LF and/or EF to PA63h. The earlier study by Singh et al. (1999) suggested that one LF molecule binds to each monomer (i.e., seven per heptamer). More recent studies by Collier and colleagues (Mogridge et al. 2002b) show that the ligand binding site is shared between two PA63 monomers, and that a maximum occupancy of three EF and/or LF molecules per heptamer is indicated (Mogridge et al. 2002a). Site-directed mutagenesis of a stretch of residues in domain 1 of PA63, which was postulated to harbor the ligand binding site from the 4.5 Å resolution X-ray structure of PA63h (Petosa et al., 1997), has demonstrated their role in ligand binding (Cunnigham et al., 2002). Similar mutagenesis studies, directed by the X-ray crystal structure of LF (Pannifer et al., 2001), have identified the cognate binding sites on lethal and edema factors (Lacy et al., 2002).
In this study, we report the direct visualization of the binding of LF to PA63h as determined by analysis of electron cryo-microscopic images of the complex while suspended in vitrified buffer. Comparison of the averaged three-dimensional (3D) views of the liganded and unliganded complexes showed that the PA63h molecule binds to one LF molecule, which is localized to the nonmembrane-interacting (called hereafter “top”) face of the heptamer. The observed binding of the ligand involves interaction with four successive PA63 monomers and elicits structural perturbations in the oligomer structure and in the central lumen. This could be important if the lumen, acting as a vestibule for the transmembrane pore, is the conduit for ligand translocation, as is generally thought.
Results
In vitrified as well as negatively stained specimens, both PA63h and the PA63h.LF complex displayed preferential en face (view parallel to the lumen axis) orientation, with the en face fraction greater in the former. This is likely due to hydrophobic patches located at the “top” face of the oligomer that are revealed upon conversion of PA to PA63 by proteolysis and are involved in LF binding. Initially, using images of en face views that were within a tilt of ±15°, we calculated a projected difference map (PA63h.LF-PA63h) (Supplemental Figure S1; see the Supplemental Data available with this article online). This difference map revealed (1) a set of three density peaks contiguously located on one half of the projected view of the oligomer and (2) major density modulation in the lumen. This result indicated that the ligand bound the oligomer asymmetrically (Mogridge et al., 2002a) and that upon binding there is a large redistribution of protein mass along the lumen volume.
We found that sub-c.m.c. (critical micellar concentration) amounts of the detergent β-octyl glucopyranoside (OG), added just prior to vitrification, diminished the preferential orientation and yielded a range of views (Figure 1) that proved sufficient for 3D reconstruction described hereafter. Figure 2 shows raw images and class averages for three different views of the complex. Various initial models (see the Experimental Procedures) led to independently generated 3D reconstructions of the complex; however, these reconstructions appeared visually to be essentially similar with very similar final resolution (~18 Å ) calculated by Fourier shell correlation analysis (Frank, 1996) (Supplemental Figure S2). This analysis established the internal consistency of the data from the images, the lack of bias extended to the reconstructed, averaged 3D map of the complex, and therefore the confidence level for the inferences drawn from the calculated map. In all, 12,080 and 19,265 particle images were selected, out of which about 45% were finally used for PA63h and PA63h.LF reconstructions, respectively.
Figure 1. Cryo-EM Images Used in Reconstruction.

A micrograph showing a field of the PA63h.LF complex suspended in vitrified buffer over a hole in a carbon film and recorded at nominal defocus of 2.0 μm. The protein is displayed with a lighter density compared to the surrounding vitrified buffer, i.e., in reversed contrast. Both en face views and relatively fewer “side views” can be seen. Excess (unbound) LF molecules contribute to the background. (Bar = 1000 Å ).
Figure 2. Single-Particle Image Analysis.
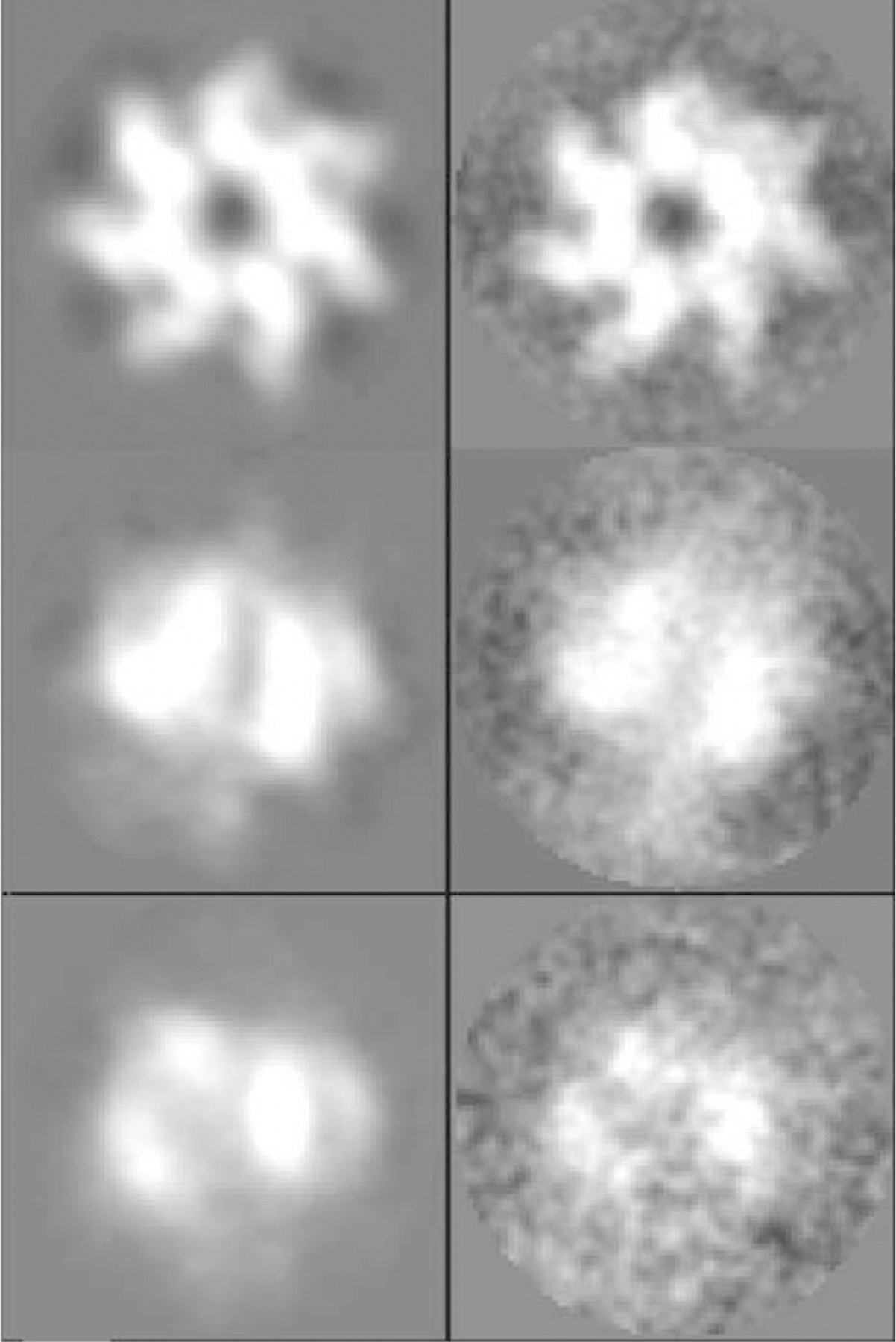
Class averages (left panel) and raw particle images (right panel) for three example views of the PA63h.LF complex that were in the set included in the 3D reconstruction.
The 3D reconstructed maps for the unliganded heptamer (PA63h) and the complex (PA63h.LF) are shown in Figure 3. Compared to the unliganded molecule, the most prominent alteration in density features characterizing the complex is observed on the nonmembrane binding (“top”) face of the PA63 heptamer, and this alteration is distributed over the region occupied by approximately one half (~4 monomers–C, D, E, and F) of the oligomers (Figure 3G). In order to ensure that the observed density modifications are not due to noise, we proceeded to estimate, as a measure of the noise level in our reconstruction, the departure from the exact 7-fold symmetry in the PA63h reconstruction where no symmetry was imposed (Figure 3, middle panel). For this purpose, we subdivided the unsymmetrized PA63h and PA63h.LF maps into seven equal segments (pie wedges of equal volume) and calculated, for both maps, pairwise chi-squared values between the segments (see the Supplemental Data available with this article online). The aim was to quantify separately, in terms of the distributions of the sets of 21 (= 7 × (7 – 1)/2) chi-squared differences, the variations amongst the density profiles of the monomers in the liganded and in the unliganded map. For the unliganded complex, the chi-squared values ranged from 2,760 to 6,316 (median = 3,969, mean = 4,235), while these ranged from 3,105 to 10,308 (median = 5,180, mean = 5,908) in the case of the complex (Supplemental Table S1). These calculated values elaborated distinct distribution profiles (Supplemental Figures S3A and S3B) and indicated density variations between many pairs of monomers in the complex that were significantly larger than those for unsymmetrized PA63h. Further analysis to demonstrate that the distributions are significantly different was carried out by using the nonparametric Kolmogorov-Smirnov statistical test (Conover, 1971). The calculated p-value of 0.01591 strongly indicated (usual cutoff = 0.05) that the two sets of pairwise chi-squared values do not belong to the same distribution.
Figure 3. 3D Reconstructed Images of Uncomplexed and Complexed Heptamer in Three Analogous Mutually Orthogonal Views.
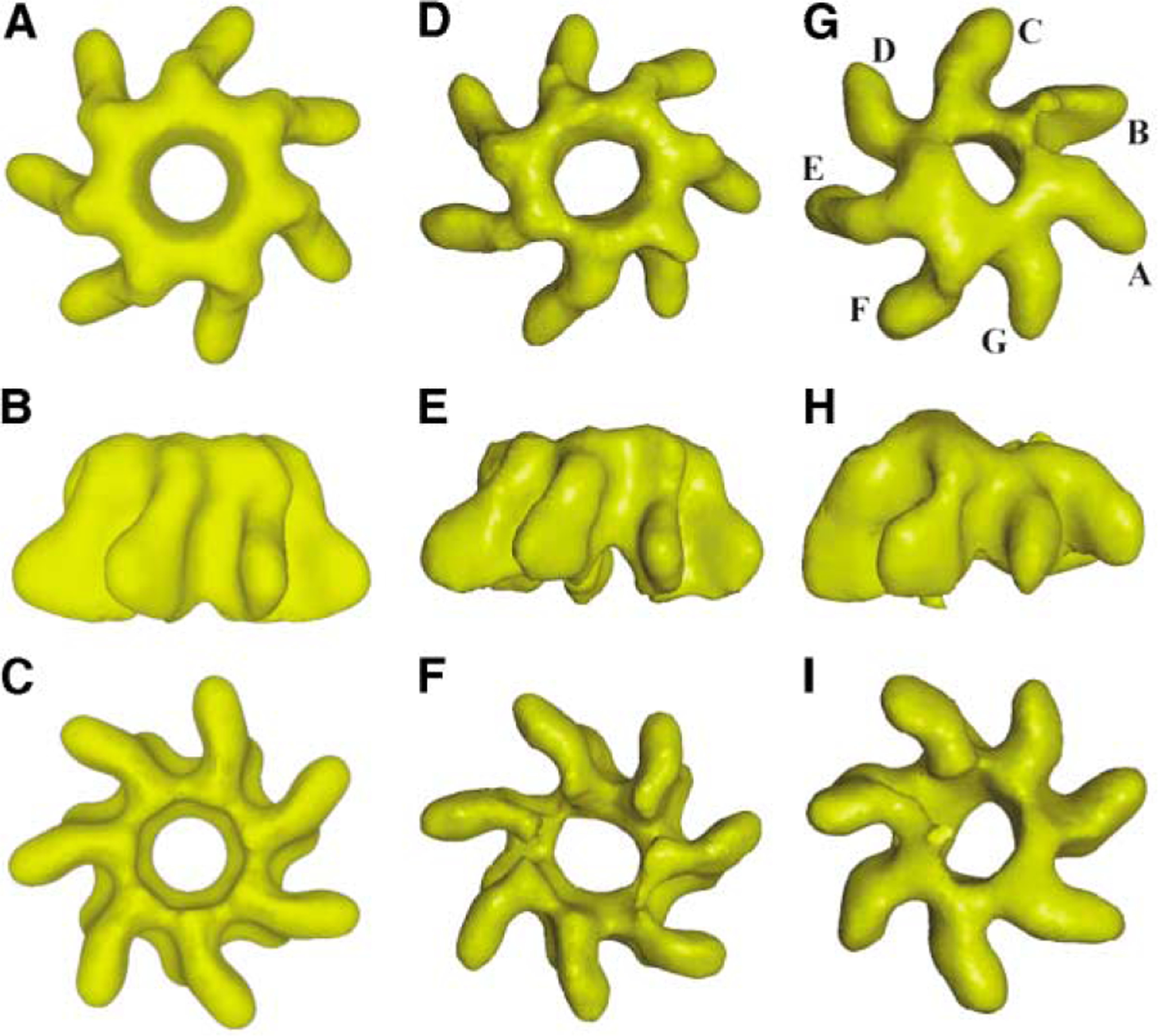
(A–I) The (A–C) left panel and the (D–F) middle panel correspond to the reconstruction of PA63h with and without using C7 symmetry in the calculation. The (G–I) right panel corresponds to the reconstruction of the PA63h.LF complex. The reconstructions shown in the middle and right panels were used to establish the statistical significance of the signal due to bound ligand in the complex (Supplemental Table S1). The nomenclature followed for identification of the monomers (seven, A–G) is indicated in (G). In each panel, the topmost view is from the nonmembrane-interacting face, the middle is a view from the side, and the bottom view is one looking down the membrane-interacting face. The maps were rendered by using the visual 5D software (http://www.ssec.wisc.edu/billh/vis5d.html).
The protein mass predicted by the 3D reconstruction of the complex was determined as follows. The starting contour level in the PA63h 3D map was adjusted until the volume of the 3D reconstruction agreed with the expected volume for the 441 kDa (63 × 7) protein. Next, the PA63h density map was overlaid on the PA63h.LF map. The starting contour in the latter map was adjusted such that the volume occupied by the monomer whose density features showed minimal alteration from those in the unliganded state (monomer A) best matched the monomer volume in the PA63h map scaled as above. Such a choice ensured that the deduced protein mass for the complex approximately reflected the maximal, observed number of bound ligand molecules. Based on this starting contour level, the volume occupied by the 3D map of the PA63h.LF complex corresponded to a species with a molecular weight of ~520 kDa, i.e., to a complex of one LF molecule and one PA63h oligomer.
Visually, by using the program O (Jones et al., 1991), the density envelope of the complex could be accounted for by the location of one LF molecule (Pannifer et al., 2001) together with seven PA63 monomers (Petosa et al., 1997), leading to a 1:1 (LF: PA63h) docked model (Figure 4). The calculated crosscorrelation coefficients for docking of the X-ray structure of the heptamer (Petosa et al. 1997) in the PA63h reconstruction with C7 symmetry imposed was 0.87, while this coefficient was 0.77 for the docked model in the density map of the complex. In the above calculation, density maps for the atomic coordinates were truncated to a limiting 18 Å resolution. In the docking exercise in the case of the PA63h.LF complex, the orientations of the two protein moieties (PA63 and LF) defined by their X-ray structures were varied only as rigid bodies. Given the modest (~18 Å) resolution of the 3D maps, no attempts were made to adjust domain orientations to fully locate all parts of the two molecules within the density envelope. In the docked model, the LF molecule drapes the nonmembrane-interacting “top” surfaces of four contiguous PA63 monomers, with the extent (~150 Å) and curvature (~70° between the N- and C-terminal domains) of the bound ligand, as viewed from the top, closely matching those for the four contiguous PA63 monomers. This results in an intimate overlap of the binding surfaces in the binary complex. This arrangement leaves three “non-interacting” PA63 monomers where the available space on top of the molecule is insufficient to bind another LF molecule. The N-terminal domain (LFN) interacts with the cognate PA63 monomer (F, Figure 4 and Supplemental Figure S4) through LF residues that were previously identified by mutagenesis (Cunningham et al., 2002; Chauhan and Bhatnagar, 2002). In addition, the deduced model indicates that part of the C-terminal region of LF (LFC) also interacts with the oligomer. In support of this deduction, we note that there exists internal sequence similarity between the segments 110–150 in LFN and the segment 680–725 of the LFC, at the level of ~40.0% (http://ca.expasy.org/), which is expected to reflect a significant similarity in structure (Marti-Renom et al., 2000; Sanchez et al., 2000). This result suggests the presence of binding motifs in LFC (Figure 4 and Supplemental Figure S4) similar to those identified (Lacy et al., 2002) in LFN. In fact, domain 4 in LF was recognized to have the same fold as domain 1, even though the overall sequence similarity is limited (Pannifer et al., 2001).
Figure 4. Docked Atomic Model of the Complex.
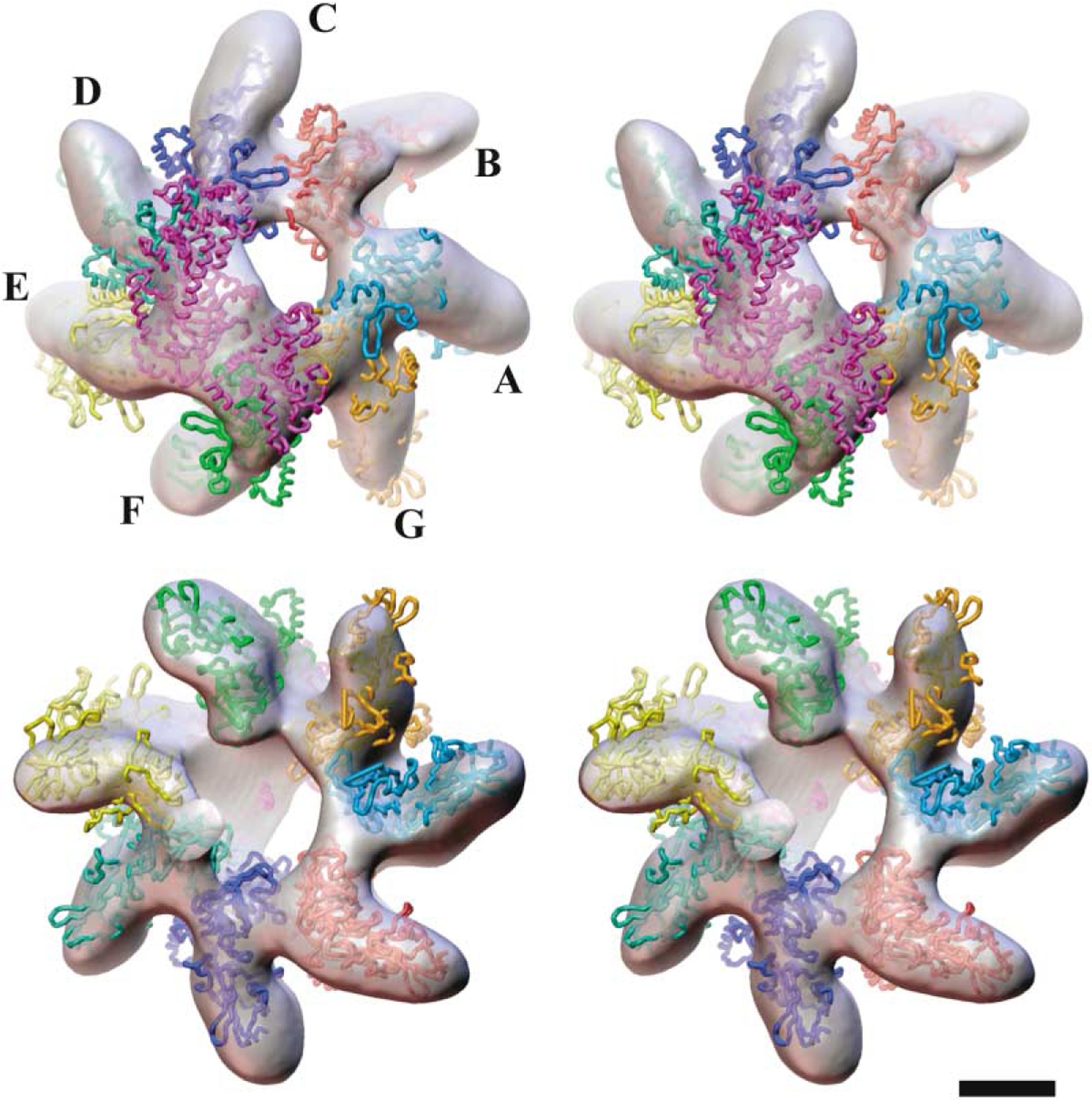
Stereo views of the atomic model of the complex of the heptamer (PA63h: A–G) and lethal factor (LF) docked into the 3D reconstructed density map of the PA63h.LF complex viewed from the nonmembrane-interacting (top pair) and the membrane-interacting (bottom pair) faces, respectively. The ligand molecule LF is shown in purple. This illustration was created by using AVS (Upson et al., 1989) software. (Bar = 50 Å ).
The most striking effect of the ligand binding is on the shape and geometry of the lumen and the regular structure of the PA63 heptamer. There is significant enlargement of the lumen volume, which now assumes an irregular shape with an axis inclined by about 13° to the C7 axis of the unliganded molecule. This change in the lumen architecture is in large part due to a pronounced displacement between monomers E and F, which is apparent especially at the membrane-interacting face (Figures 3I and 4). The shape of the density at this face, which is adjacent to the location of monomer E (Figure 4), suggests that a rigid body movement of monomer E has occurred that is approximately defined by a translation of ~20 Å along the vertical (z axis) toward the bottom (membrane-interacting face), an ~180° rotation about this axis, and a tilt of ~35° around an axis perpendicular to the z axis. It may be that the movement of monomer E drives the displacement of monomers C, D, and F to various degrees, such as that observed on the membrane-interacting bottom face of the oligomer. Here, the separation between these monomers has increased by 5–10 Å . The cumulative result is that the regular heptamer structure is somewhat unraveled on this face of the oligomer. A likely consequence of the displacement of the monomers is that the chemical nature of the surface lining the vestibule through the PA63h oligomer is changed, an alteration that could facilitate the putative translocation of the ligand through the lumen.
Discussion
The Interaction between PA63 and LF
To our knowledge, our study, for the first time, directly visualizes the binding of full-length LF to PA63h, an essential step in anthrax toxin action. The envelope of the 3D reconstruction calculated at ~18 Å resolution indicates that in the PA63h.LF complex an equivalent of one LF molecule binds to one oligomer. As mentioned earlier, reported values for the stoichiometry of LF (or EF) and LFN binding to PA63h have varied. This was determined to be seven LF per heptamer from native gel electrophoresis (Singh et al., 1999). However, the recent results by Collier and colleagues (Mogridge et al., 2002a), who used recombinant LFN to deduce a maximum binding of three LFN molecules per heptamer, have led us to question the validity of the earlier data reported by one of us (S.H.L. in Singh et al. [1999]). In our cryo-electron microscopy (cryo-EM) experiment, the full-length LF was used in excess – a ratio of 10.5 LF molecules to 1 PA63h, which is expected to cover all available binding sites on the oligomer. The deduced 1:1 PA63h.LF complex visualized in our analysis is more in line with an earlier study by the Collier group (unpublished data referred to in Elliott et al. [2000]), in which the number of EF/LF molecules per PA63h was estimated by Surface Plasmon Resonance to be one or two. Also, recent studies by Zhang et al. (2004) have shown that the efficiency of the translocation of LFN mediated by PA63h heptamer is the same, irrespective of the amount (e.g., up to saturating levels) of LFN present, which would be easier to rationalize if the LF:PA63h stoichiometry were 1:1.
In the absence of additives, even though predominant, the fraction of en face views for the complex is less, ~70% compared to ~90%, for the unliganded molecules. This is consistent with our model for LF binding, in which the binding interface covering the “top” surface of four of the seven PA63 monomers hides some of the residual hydrophobic patches. Further, in line with such an argument is the effect of small amounts of detergent OG (0.05% final concentration), which induced the display of a fuller range of views for both unliganded and liganded specimens, but, again, more so for the complex. The addition of such sub-c.m.c. amounts of OG does not adversely affect the nanomolar binding of ligand to the oligomer.
The docking of one molecule of LF and the seven PA63 monomers in the density map of the complex was visually carried out based on the known X-ray structures (Petosa et al., 1997; Pannifer et al., 2001) and by using knowledge of the interacting regions of the two-polypeptide chains, as determined by site-directed mutagenesis studies. For PA63, these have been identified by Cunningham et al. (2002) and Chauhan and Bhatnagar (2002), and, for LF, they have been identified by Gupta et al. (2001) and Lacy et al. (2002). In our docked model (Figure 4), LFN residues in segments 40–50, 100–113, 143–147, 191–194, and 207–212 are proximal to the oligomer surface. In addition, several residues in LFC, e.g., in segments 300–340 and 710–756, are also proposed to interact with the PA63 monomer. This is consistent with the noted, as described above, sequence and hence structural similarity between regions in LFN (residues 110–150) harboring interaction motifs with PA63 and regions in LFC (residues 680–725). The cognate binding surface on PA63 includes residues in segment 205–221 (Chauhan and Bhatnagar, 2002) in monomer C, residues in segment 479–490 in monomer D, residues in segment 567–580 in monomer E, and residues in segments 465–470 and 185–225 (Cunningham et al., 2002) in monomer F (Supplemental Figure S5).
Structural Rearrangement in PA63h upon Ligand Binding
The inherently asymmetric nature of the complex is readily apparent in the orthogonal view (Figure 3H). This is due to the distortion brought about by structural rearrangement of the PA63h oligomer upon asymmetric ligand binding (one ligand molecule per heptamer as deduced above). Four of the seven monomers participating in LF binding have moved to different degrees with the result that the lumen, cylindrically symmetric in the unliganded state, assumes an irregular inclined shape with a significant increase in exposed volume (Figure 4). This is apparent from the observation that, for instance, if the LF molecule is removed in the docked model, the entrance to the lumen at the “top” face is enlarged; this new entrance is approximately elliptical (35 Å × 75 Å) rather than a circle of ~30 Å diameter that is seen in the unliganded state (Supplemental Figure S6). The overall features in the membrane binding face, especially when viewed along the direction of the lumen, however, appear rather similar in both the unliganded and liganded state. A number of ancillary experiments lend support to this observed distortion in the oligomer. On some occasions, crystals of PA63h were seen on carbon support films in negatively stained specimens. However, no crystalline order was visible in electron microscope (EM) grids prepared with complexes or when LF was diffused into PA63h suspension prior to staining on the EM grids. Helical crystals of PA63h (Wilson-Kubalek, E. et al., personal communication) generated at pH 8.0 and 7.0 on lipid tubules (Wilson-Kubalek, 2000) were dispersed upon incubation with LF, and lattices in 2D crystals of PA63h generated by lipid reconstitution at pH 6.0 were also destroyed when LF was added to these crystals. These observations taken together strongly suggest that the LF binding elicits large-scale conformational changes in the oligomer.
An intriguing aspect of the action of anthrax toxin, as well as for other toxins in the A-B family, such as diphtheria toxin (DT), is the molecular mechanism accompanying pore formation and ligand translocation. In analogy with the X-ray structure for S. aureus α-heamolysin (Song et al., 1996), the X-ray structure of PA63h (Petosa et al., 1997) and recent studies by Nassi et al. (2002) have led to the suggestion that the structure of the pore traversing the membrane bilayers is a β barrel. Nassi et al. (2002) indicate that the barrel extends beyond the bilayers involving residues that are otherwise buried in the PA monomer and suggest that a major rearrangement of domains in the “top” of the oligomer is required for membrane insertion of the β barrel stem. As indicated by our 3D density map of the liganded complex and the docked atomic model, flexibility and movement at the monomer interfaces appear also to be exploited to facilitate and/or accompany the ligand binding process. Studies with PA and the recombinant LFN-DTA chimera, in which DTA is the catalytic fragment of DT, have shown that delivery of the polypeptide into the cell was blocked when engineered DTA variants contained intramolecular disulfide links (Weshe et al., 1998). This supports the notion that the ligand in the A-B family of toxins very likely undergoes some degrees of unfolding to facilitate translocation. In the case of LF, this process may include flexing of domain 1 with respect to the rest of the molecule to suitably alter the resting dimension (100 Å tall and 70 Å wide) to ease passage across the membrane bilayer.
What is the situation accompanying the insertion of the toxic complex into the bilayers and its release? The enlargement of the lumen volume (Figures 3 and 4) may represent the first step needed to facilitate ligand translocation. This process may be further aided by partial unfolding of the ligand as mentioned above. Given the moderate ~18 Å resolution of the map, we had chosen to retain the LF and the PA63 at their respective X-ray structures in modeling the content of the density envelope. Therefore, no direct quantification of the likely unfolding of the ligand or alteration in the structure of the PA63 monomer(s) can be made from our maps. The inherent asymmetry brought about by ligand binding and the observed distortion in the oligomer structure suggests that the “regular” β barrel structure of the pore may also be compromised or replaced by a, as yet unknown, variant connected to the enlarged vestibular lumen. In any event, the insertion process in this case is also likely to be accompanied by a conformational rearrangement of domains in the “top” face of the oligomer. It is possible that such domain movements, which may be distinct from those in the unliganded state (Nassi et al., 2002), are utilized in the important step of releasing the bound ligand and its eventual translocation.
Experimental Procedures
Ligand Binding
Protein expression and purification were performed as described by Park and Leppla (Park and Leppla, 2000) and Klimpel et al. (1992). The molecular weights of the protein samples and their purity were checked by SDS-PAGE. Heptamers of protective antigen (PA63h) and the E697C variant of LF, which lacks catalytic activity but has normal Zn2+ binding, were stored at −80°C in 5 mM CHES, 5 mM NaCl (pH 9.4), and 10 mM Tris (pH 8.0). To generate the ligand heptamer complex, aliquots of freshly thawed PA63h and LF were mixed at a ratio of 10.5 ligand molecules to 1 PA63h heptamer (molar ratio of ligand to PA63 monomer = 1.5:1) and incubated at room temperature for 30 min with 1 mM DTT added. Specimen preparation for microscopy (described below) for the complex was carried out at 4°C or at room temperature, usually within 30 min of incubation.
EM Specimen Preparation and Data Collection
Negatively stained specimens were prepared by using 0.2% methylamine tungstate according to Kolodziej et al. (1997) and by using 1% uranyl acetate. Such specimens were tilted up to 55° in a Philips CM120 or CM100 microscope to capture tilted projections used to calculate low-resolution, initial 3D models from images recorded at under-focus of 3,000–8,000 Å . In order to examine the effect of additives to induce display of the full range of orientations in nominally untilted images of vitrified protein samples, detergents such as OG, Triton X-100, and 3-[3-Cholamidopropyl)-dimethylammonio]-1-propane sulfonate (CHAPS) at sub-c.m.c. concentrations and a variety of lipids (at low-millimolar concentration), such as E. coli phosphatidylcholine, di-myristoyl phosphatidylcholine, di-lauroyl phosphatidylcholine, di-oleoyl phosphatidylcholine, Palmitoyloleoyl phosphatidylcholine, E. coli phosphatidyl ethanolamine, and E. coli polar lipids, were added to protein samples just prior to vitrification. We found that OG (c.m.c. = 0.44%) at a final concentration of 0.05% was the most satisfactory additive for imaging frozen-hydrated specimens of both PA63h and PA63h.LF. Such specimens were prepared by plunge freezing (Dubochet et al., 1988) protein samples with 0.05% added OG and were suspended in buffer across holes in carbon-support film (Quantifoil, Germany). These were examined, nominally untilted, at −173°C. Minimal dose pairs of cryo-images, the first at close to focus (~0.7 μm under-focus) and the second at an underfocus of 1.6–3.5 μm, were recorded with a Philips CM200FEG operated at 120KV and at a nominal magnification of 50,000. Micrographs were digitized at 7 μm/pixel on a SCAI scanner (Zeiss) and then binned at 2 × 2 pixels so that each pixel corresponded to 2.8 Å on the specimen.
Image Processing, 3D Reconstruction, and Docking of Atomic Models
Single particle image analysis and 3D reconstruction were carried out by using the EMAN program suite (Ludtke et al., 1999). Particle identification in a micrograph showing no visible drift and minimal astigmatism was done by using the BOXER program in EMAN. For cryo-image pairs, particles selected from the high-underfocus image were used to identify the same particles in the low-underfocus image by crossselection analysis; these particles were subsequently used for further analysis. The level of focus and astigmatism for a given micrograph was determined by the CTFit program in EMAN by using an 8-parameter fit. The centers of all the particles were aligned, and the box size was progressively reduced to 80 × 80 pixels.
After several rounds of rotational and translational refinement, aligned images were classified. The given initial 3D model or internally generated initial model (in the case of reference-free analysis) was sliced every 10° to generate 180 classes of projections. Particle images were classified based on correlation agreement with these 180 classes; a maximum of about 250 particle images were present in each class. The class-averaged projections were combined to generate a new model, and the process was iterated at least eight times, ending with calculations with 7° sliced projections. In each cycle of refinement, the particle center and the orientation of the projected image defined by the Euler angles were derived by cross-correlation with projections of the current 3D model. Classes containing spurious or persistently misaligned images were excluded from the data set for subsequent iterations of the refinement procedure. The resolution of the final reconstruction was estimated by Fourier shell correlation analysis of two independent reconstructions, each derived from a randomly chosen half of the raw images.
Docking of atomic models into the 3D map of the PA63h.LF complex was carried out by manually placing the atomic coordinates of the PA63 and LF X-ray structures (PA63: 1ACC; LF: 1J7N) into the electron density by using the program O (Jones et al., 1991) and orientating these in three dimensions to achieve the best fit within the density envelope.
Starting Models
Several initial models, either generated in the reference-free procedure or supplied externally, were used for calculating independent 3D reconstructions. These were (1) an X-ray structure-derived model of PA63h (Petosa et al., 1997) truncated to 50 Å resolution, (2) a 3D model generated by reference-free alignment of images from negatively stained samples of PA63h, and (3) a 3D model created by reference-free alignment of a low-pass filtered (25 Å ), high defocus (1.6–3.5 μm) member of cryo-image pairs and by using information only within the first CTF node. In the case of PA63h, calculations were done both with and without imposing the C7 symmetry, whereas in the case of the PA63h.LF complex, no C7 symmetry was imposed since the analysis in projection (see below) had indicated asymmetry induced by LF binding. Fourier shell correlation analysis was used to check the stability and convergence of the refinement process leading to the calculated averaged maps and was also used to assess the agreement between the maps determined independently by using different starting models.
Processing of Images of the En Face Nominally Untilted View
The en face views were analyzed as a pseudo 3D data set so as to account for small tilts. The initial model was the 3D X-ray structure of PA63h truncated to 50 Å resolution, and during this analysis, C7 symmetry was applied only in the case of PA63h. The refinement results showed that more than 90% of the en face images were within ~15° from the 7-fold axis direction. The averaged, nominally projected views for PA63h and PA63h.LF were generated from the pseudo 3D reconstructions. For calculating the PA63h.LF-PA63h difference map, the two maps were first normalized by using the mean and maximum density in each map. Next, the maps were rotated and aligned, and the difference map was calculated.
Supplementary Material
Acknowledgments
We thank Steven Ludtke and Wen Jiang for their help with the EMAN software; Hong Z. Zhou; and colleagues at the cryo-EM facility at Scripps, especially Elizabeth Wilson-Kubalek, for helpful suggestions and discussion. We thank Michael Pique for creating Figure 4 and, in particular, thank Chris Triggs for statistical analysis and Florence Tama for correlation calculations. We thank Anchi Cheng, Vijay Reddy, and Joerg Kistler for comments on the manuscript. This work was supported by a National Institutes of Health grant R01-GM52567 and an American Heart Association Established Investigator grant to A.K.M. There is no potential conflict of interest with regard to the contents of this manuscript.
Footnotes
Supplemental Data
Supplemental Data including Supplemental Table S1; a description of the chi-squared analysis calculation that was used to compute the entries in Supplemental Table S1; and additional figures, including the figures that describe statistical analysis based on the data in Supplemental Table S1, are available at http://www.structure.org/cgi/content/full/12/11/2059/DC1/.
References
- Ballard JD, Collier RJ, and Starnbach MN (1996). Anthrax toxin-mediated delivery of a cytotoxic T-cell epitope in vivo. Proc. Natl. Acad. Sci. USA 93, 12531–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson EL, Huynh PD, Finkelstein A, and Collier RJ (1998). Identification of residues lining the anthrax protective antigen channel. Biochemistry 37, 3941–3948. [DOI] [PubMed] [Google Scholar]
- Blaustein RO, Koehler TM, Collier RJ, and Finkelstein A (1989). Anthrax toxin: channel-forming activity of protective antigen in planar phospholipid bilayers. Proc. Natl. Acad. Sci. USA 86, 2209–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley KA, Mogridge J, Mourez M, Collier RJ, and Young JA (2001). Identification of the cellular receptor for anthrax toxin. Nature 414, 225–229. [DOI] [PubMed] [Google Scholar]
- Chauhan V, and Bhatnagar R (2002). Identification of amino acid residues of anthrax protective antigen involved in binding with lethal factor. Infect. Immun 70, 4477–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier RJ (1999). Mechanism of membrane translocation by anthrax toxin: insertion and pore formation by protective antigen. J. Appl. Microbiol 87, 283. [DOI] [PubMed] [Google Scholar]
- Conover WJ (1971). Practical Nonparametric Statistics (New York: John Wiley & Sons; ). [Google Scholar]
- Cunningham K, Lacy DB, Mogridge J, and Collier RJ (2002). Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc. Natl. Acad. Sci. USA 99, 7049–7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon TC, Meselson M, Guillemin J, and Hanna PC (1999). Anthrax. N. Engl. J. Med 341, 815–862. [DOI] [PubMed] [Google Scholar]
- Drum CL, Yan SZ, Bard J, Shen YQ, and Lu D (2002). Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 415, 396–402. [DOI] [PubMed] [Google Scholar]
- Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, Mc-Dowell AW, and Schultz P (1988). Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys 21, 129–228. [DOI] [PubMed] [Google Scholar]
- Elliott JL, Mogridge J, and Collier RJ (2000). A quantitative study of the interactions of Bacillus anthracis edema factor and lethal factor with activated protective antigen. Biochemistry 39, 6706–6713. [DOI] [PubMed] [Google Scholar]
- Frank J (1996). Three-dimensional Electron Microscopy of Macromolecular Assemblies (San Diego: Academic Press; ). [Google Scholar]
- Goletz TJ, Klimpel KR, Arora N, Leppla SH, Keith JM, and Berzofsky JA (1997). Targeting HIV proteins to the major histocompatibility complex class I processing pathway with a novel gp120-anthrax toxin fusion protein. Proc. Natl. Acad. Sci. USA 94, 12059–12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Singh A, Chauhan V, and Bhatnagar R (2001). Involvement of residues 147VYYEIGK153 in binding of lethal factor to protective antigen of Bacillus anthracis. Biochem. Biophys. Res. Commun 280, 158–163. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowans SW, and Kjeldgaard M (1991). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Klimpel KR, Molloy SS, Thomas G, and Leppla SH (1992). Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad. Sci. USA 89, 10277–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziej SJ, Penczek PA, and Stoops JK (1997). Utility of Butvar support film and methylamine tungstate stain in three-dimensional electron microscopy: agreement between stain and frozen-hydrated reconstructions. J. Struct. Biol 120, 158–167. [DOI] [PubMed] [Google Scholar]
- Lacy DB, Mourez M, Fouassier A, and Collier RJ (2002). Mapping the anthrax protective antigen binding site on the lethal and edema factors. J. Biol. Chem 277, 3006–3010. [DOI] [PubMed] [Google Scholar]
- Leppla SH (1999). The bifactorial Bacillus anthracis lethal and oedema toxins. In Comprehensive Sourcebook of Bacterial Protein Toxins, Second Edition, Alouf JA, and Freer J, eds. (London: Academic Press; ), pp. 243–263. [Google Scholar]
- Ludtke SJ, Baldwin PR, and Chiu W (1999). EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol 128, 82–97. [DOI] [PubMed] [Google Scholar]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, and Sali A (2000). Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct 29, 291–325. [DOI] [PubMed] [Google Scholar]
- Milne JC, and Collier RJ (1993). pH-dependent permeabilization of the plasma membrane of mammalian cells by anthrax protective antigen Mol. Microbiol 10, 647–653. [DOI] [PubMed] [Google Scholar]
- Milne JC, Furlong D, Hanna PC, Wall JS, and Collier RJ Anthrax protective antigen forms oligomers during intoxication of mammalian cells. (1994). J. Biol Chem 269, 20607–20612. [PubMed] [Google Scholar]
- Mogridge J, Mourez M, and Collier RJ (2001). Involvement of domain 3 in oligomerization by the protective antigen moiety of anthrax toxin. J. Bacteriol 183, 2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogridge J, Cunningham K, and Collier RJ (2002a). Stoichiometry of anthrax toxin complexes. Biochemistry 41, 1079–1082. [DOI] [PubMed] [Google Scholar]
- Mogridge J, Cunningham K, Lacy DB, Mourez M, and Collier RJ (2002b). The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc. Natl. Acad. Sci. USA 99, 7045–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassi S, Collier RJ, and Finkelstein A (2002). PA63 channel of anthrax toxin: an extended beta-barrel. Biochemistry 41, 1445–1450. [DOI] [PubMed] [Google Scholar]
- Pannifer AD, Wong TY, Schwarzenbacher R, Renatus M, Petosa C, Bienkowska J, Lacy DB, Collier RJ, Park S, Leppla SH, et al. (2001). Crystal structure of the anthrax lethal factor. Nature 414, 229–233. [DOI] [PubMed] [Google Scholar]
- Park S, and Leppla SH (2000). Optimized production and purification of Bacillus anthracis lethal factor. Protein Expr. Purif 18, 293–302. [DOI] [PubMed] [Google Scholar]
- Petosa C, Collier RJ, Klimpel KR, Leppla SH, and Liddington RC (1997). Crystal structure of the anthrax toxin protective antigen. Nature 385, 833–838. [DOI] [PubMed] [Google Scholar]
- Sanchez R, Pieper U, Melo F, Eswar N, Marti-Renom MA, Madhusudhan MS, Mirkovic N, and Sali A (2000). Protein structure modeling for structural genomics. Nat. Struct. Biol 7 (Suppl), 986–990. [DOI] [PubMed] [Google Scholar]
- Singh Y, Klimpel K, Goel S, Swain P, and Leppla SH (1999). Oligomerization of anthrax toxin protective antigen and binding of lethal factor during endocytic uptake into mammalian cells. Infect. Immun 67, 1853–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, and Gouaux JE (1996). Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274, 1859–1866. [DOI] [PubMed] [Google Scholar]
- Upson C, Faulheber T Jr., Kamins D, Laidlaw D, Sclegel D, Vroom J, Gurwitz R, and Van Dam A (1989). The application visualization system: a computational environment for scientific visualization. Comput. Graph. Appl 9, 30–42. [Google Scholar]
- Wesche J, Elliott JL, Falnes PO, Olsnes S, and Collier RJ (1998). Characterization of membrane translocation by anthrax protective antigen. Biochemistry 37, 15737–15746. [DOI] [PubMed] [Google Scholar]
- Wilson-Kubalek EM (2000). Preparation of functionalized lipid tubules for electron crystallography of macromolecules. Methods Enzymol 312, 515–519. [DOI] [PubMed] [Google Scholar]
- Young JA, and Collier RJ (2002). Attacking Anthrax. Sci. Amer 286, 48–50, 54–59. [DOI] [PubMed] [Google Scholar]
- Zhang S, Cunningham K, and Collier RJ (2004). Anthrax protective antigen: efficiency of translocation is independent of the number of ligands bound to prepore. Biochemistry 43, 6339–6343. [DOI] [PubMed] [Google Scholar]
- Zhao J, Milne JC, and Collier RJ (1995). Effect of anthrax toxin’s lethal factor on ion channels formed by the protective antigen. J. Biol. Chem 270, 18626–18630. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.