

Abstract
We previously reported the discovery and characterization of two novel proteins (ORF1 and ORF2) generated by the alternative splicing of the JC virus (JCV) late coding region. Here, we report the discovery and partial characterization of three additional novel ORFs from the same coding region, ORF3, ORF4 and ORF5, which potentially encode 70, 173 and 265 amino acid long proteins respectively. While ORF3 protein exhibits a uniform distribution pattern throughout the cells, we were unable to detect ORF5 expression. Surprisingly, ORF4 protein was determined to be the only JCV protein specifically targeting the promyelocytic leukemia nuclear bodies (PML-NBs) and inducing their reorganization in nucleus. Although ORF4 protein has a modest effect on JCV replication, it is implicated to play major roles during the JCV life cycle, perhaps by regulating the antiviral response of PML-NBs against JCV infections and thus facilitating the progression of the JCV-induced disease in infected individuals.
Keywords: Promyelocytic leukemia nuclear bodies; interferon; PML-NBs; progressive multifocal leukoencephalopathy; ORF, splicing; Polyomavirus; JCV, SV40; BKV; papillomavirus; merkel cell carcinoma virus; DNA replication; transcription; RNA splicing
INTRODUCTION
The nucleus of the mammalian cells harbors remarkable macromolecular assemblies including Cajal bodies, polycomb group proteins, nuclear stress bodies, speckles, paraspeckles, nucleoli, nucleolus and promyelocytic leukemia nuclear bodies (PML-NBs) (Cremer and Cremer, 2001; Misteli, 2007). Unlike the cytoplasmic organelles, they all lack the biological membranes which separate them from the surrounding nucleoplasm. They play specific roles in the function of the nucleus (Lamond and Sleeman, 2003; Lamond and Spector, 2003; Leung et al., 2003; Leung and Lamond, 2003; Mao et al., 2011; Misteli, 2010). Among those, PML-NBs were identified 27 years ago (Ascoli and Maul, 1991) and were shown to play critical roles in initiation of both intrinsic and interferon-mediated antiviral responses against DNA and RNA viral infections (Regad and Chelbi-Alix, 2001; Regad et al., 2001; Stepp et al., 2013). PML-NBs are relatively small protein assemblies and display a distinct and punctate morphology under microscope (Lallemand-Breitenbach and de The, 2018). Their numbers fluctuate during cell division depending on the cell cycle stage. In addition to their antiviral response, PML-NBs are also involved in several cellular processes, including apoptosis, DNA damage response, transcription, chromatin remodeling and oncogenesis (Bernardi and Pandolfi, 2007; Everett and Chelbi-Alix, 2007).
The major component of the PML-NBs is the promyelocytic leukemia protein (PML) itself, which was originally discovered as a fusion protein with retinoic acid receptor as a result of a chromosomal translocation found in patients with acute promyelocytic leukemia (de The et al., 1990; de The et al., 1991; Kakizuka et al., 1991). hDaxx Sp100 and ATRX transcription factors are the other three constitutive components of the PML-NBs. However, some other proteins can also be a temporary resident of these structures, including but not limited to MORC3 and SUMO under certain stress conditions, (Lallemand-Breitenbach and de The, 2018; Sloan et al., 2016; Stepp et al., 2013; Wang et al., 2018). Posttranslational modifications such as phosphorylation and sumoylation on PML proteins play critical roles in the assembly of PML-NBs (Hsu and Kao, 2018). Certain viruses encode PML-NB-targeting proteins with the aim of reducing or eliminating their antiviral activity [see for review (Scherer and Stamminger, 2016)]. Two prime examples of these anti-PML-NB viral proteins are the HSV-1 immediate early ICP0 protein (Maul et al., 1993), and the immediate early protein IE1 of human cytomegalovirus (hCMV) (Lee et al., 2004). ICP0 acts as SUMO-targeted ubiquitin ligase and leads to the proteasome-dependent degradation of sumoylated PML components to counteract the PML-mediated silencing of the HSV-1 gene expression. hCMV IE1 protein, on the other hand, inhibits the sumoylation of PML protein itself, through which it prevents the assembly of PML-NBs and thereby inhibits their antiviral effect.
Infection by several DNA viruses was previously reported to modulate the reorganization of the PML-NBs (Day et al., 1998; Florin et al., 2002; Jiang et al., 2011). For example, the late capsid protein, L2, of bovine (Day et al., 1998) and human (Florin et al., 2002) papillomaviruses were clearly shown to reorganize the PML-NBs in the nucleus. Similarly, a human polyomavirus, BK virus, (BKV), infection was also demonstrated to induce alterations in morphology and numbers of PML-NBs (Jiang et al., 2011), however, targeting PML protein by siRNA silencing did not result in a dramatic effect on the rate of the BKV DNA replication suggesting that PML-NBs do not directly target viral genome (Jul-Larsen et al., 2004). Similar infection studies with mouse polyomavirus and SV40 revealed the replication of SV40 DNA at the vicinity of PML-NB complexes (Erickson et al., 2012; Tang et al., 2000). Infection studies with another human polyomavirus, JC virus (JCV), revealed that JCV infection does not cause the degradation of PML-NB complexes and the silencing of PML expression by short hairpin RNA (shRNA) did not have a significant effect on JCV replication either (Gasparovic et al., 2009), further suggesting that PML complexes are not directly involved in JCV DNA replication. In another study by Shishido-Hara et al, the accumulation of JCV major (VP1) and minor (VP2, VP3) capsid proteins at the PML-NBs was detected not only in transfected cells but also in progressive multifocal leukoencephalopathy patient brain samples (Shishido-Hara et al., 2004). In addition, the same group also detected the viral-like particles at the discrete locations along the inner nuclear periphery and concluded that PML-NBs can serve as a scaffold for JCV viral assembly (Shishido-Hara et al., 2008; Shishido-Hara et al., 2004).
JCV is a neurotropic human polyomavirus that infects oligodendrocytes and astrocytes in the central nervous system (CNS); and causes a fatal brain disease known as progressive multifocal leukoencephalopathy in a subset of the immunocompromised individuals including AIDS and multiple sclerosis patients (Berger, 1992, 2000, 2007; Berger and Koralnik, 2005; Kleinschmidt-DeMasters and Tyler, 2005; Korman et al., 2009; Langer-Gould et al., 2005; Langer-Gould and Steinman, 2006). Like any other virus, JCV also heavily relies on the regulatory mechanisms provided by the host cells for the splicing of its pre-mRNA transcripts. JCV genome (a double stranded circular DNA, ~ 5kb) possesses two coding regions (early and late), each of which generates a single transcript. These transcripts then undergo an alternative splicing process to produce the splicing products that encode regulatory and structural proteins (Coric et al., 2014; Frisque et al., 1984; Saribas et al., 2013; Saribas et al., 2018; Saribas et al., 2014; Saylor and Venkatesan, 2016; Shishido-Hara et al., 2000). For example, the late coding region of JCV produces two primary splice products, resulting from the removal of an intron located between agnoprotein and VP2 coding regions; and a second intron located between agnoprotein and VP1 coding regions (Frisque et al., 1984; Saribas et al., 2013). Generation of additional splice products were also partially described by Shishido-Hara et al., for JCV using an in vitro tissue culture system and in vivo progressive multifocal leukoencephalopathy patient samples (Shishido-Hara et al., 2000) but no viral protein product associated with the individual splice products was reported by these investigators. Our lab also recently reported the discovery and characterization of two new open reading frames (ORF1 and ORF2) for JCV late transcripts (Saribas et al., 2018). These ORFs encode a 58 and a 72 aa long protein respectively and both share a common amino acid region with the N-terminus of VP1 protein but they also have a unique carboxy terminal region of their own (Saribas et al., 2018).
Over the years, some of the polyomavirus infection studies have led to a unifying conclusion that the morphology and number of the PML-NBs are significantly altered by the viral infection and yet, to date, none of these studies identified a single or a group of viral proteins directly linked to such alterations. In this study, for the first time, we report the discovery of such a protein encoded by one of the JCV late splice variants designated as the ORF4 protein (173 aa long). We demonstrated that ORF4 protein is the only viral protein of JCV that directly targets the PML-NBs and triggers their reorganization in the nucleus. We also report the discovery of two additional novel ORFs for JCV late coding region, designated as ORF3 and ORF5, each of which potentially encodes 70 and 265 aa-long proteins respectively. ORF4 protein has a modest effect on JCV replication but we believe that it may play major roles during the JCV life cycle, perhaps by regulating the antiviral response of PML-NBs against JCV infections and thus by facilitating the progression of the JCV-induced disease, progressive multifocal leukoencephalopathy, in the infected individuals.
MATERIALS AND METHODS
Cell lines
SVG-A is a human cell line established by transforming primary human fetal glial cells with an origin-defective SV40 mutant (49). These cells do not express the SV40 late proteins (VP1, VP2, VP3 and Agno) but they do express the early proteins including SV40 LT-Ag (Saribas et al., 2014; Sariyer et al., 2006). HeLa cells are a human epithelial cervical carcinoma cell line (ATCC, catalog no. ATCC CCL-2). Both cell lines were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) (Life Technologies, catalog no. 31600–034) supplemented with 10% heat inactivated fetal bovine serum (FBS, Gemini, catalog no 100–106) and antibiotics [penicillin-streptomycin (100 µg/ml) (Gemini, catalog no 400–1001), ciprofloxacin (MP biochemical, catalog no. 199020, 10 µg/ml)]. Primary human fetal astrocytes (PHFA) were obtained from the “Comprehensive NueroAIDS Tissue Culture Core Facility”, Temple University, and maintained in the primary astrocyte media (Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 Ham (DMEM F12 Ham) (Sigma, catalog no. D8900–1L), supplemented with GlutaMAX (Gibco, catalog no. 35050–0610), insulin (Sigma, catalog no. I0516–5ml, 10 µg/ml final), Fetal Bovine Serum (10 % final) (FBS) penicillin/streptomycin (100 µg/ml) and ciprofloxacin (10 µg/ml). Cells were maintained at 37°C in a humidified atmosphere and supplemented with 7 % CO2.
Plasmids
The coding sequences of the ORF3, ORF4 and ORF5 were subcloned into pCGT7 vector at XbaI/BamHI enzyme restriction sites and tagged with T7-tag sequence (MASMTGGQQMG) at their N-termini. The resulting expression plasmids were designated as pCGT7-JCV-ORF3, pCGT7-JCV-ORF4 and pCGT7-JCV ORF5. The integrity of each construct was validated by DNA sequencing. The nuclear localization signal sequence (NLS) (QRLKRRVKNP) of ORF4 was mutated into (QRLKAAVKNP) in the pCGT7-JCV-ORF4 and in the Bluescript KS+JCV Mad-1 viral plasmid by employing the Quik Change™ site–directed mutagenesis kit (Agilent Technologies, catalog no. 200524) and by using appropriate PCR primers. The resulting plasmids were designated as pCGT7-JCV-ORF4-NLS-Mut (RR105–106AA) and Bluescript KS+JCV-Mad-1-VP1/ORF4-Mut (RR286–287AA).
Transfections
SVG-A and HeLa cells were plated onto tissue culture plates (100 mm in diameter, Becton Dickinson, catalog no. 353002) (2 × 106 cells/plate) the day before transfection. Cells were washed with growth medium (DMEM), re-fed with fresh medium and grown for 3 h at 37°C. Each culture plate was then transfected with the following expression plasmids separately: pCGT7-JCV-ORF3, pCGT7-JCV-ORF4 and pCGT7-JCV-ORF5 (8 µg each) using lipofectamine™ 3000 transfection reagent according to the manufacturer’s recommendations (ThermoFisher, catalog no. L3000001). At 24h post-transfection, whole-cell extracts were prepared as described below. The description for the rest of the transfection assays were provided under corresponding figure legends.
Whole-cell extract preparation and Western blotting
Whole-cell extracts were prepared from cells (SVG-A and HeLa) transfected with pCGT7-JCV-ORF3 or pCGT7-JCV-ORF4 or pCGT7-JCV-ORF5 plasmids as described previously (Coric et al., 2014; Sariyer et al., 2011). Briefly, cells (transfected/untransfected) were lysed in RIPA lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1.0 % NP-40 and 0.1 SDS in the presence of protease inhibitors (Sigma, catalog no. P8340)] on a rocking platform at 4°C for 30 min (2 × 106 cells/1000 µl lysis buffer). The cell lysates were then cleared by centrifugation at 17,000 x g for 10 min at 4°C and stored at - 80°C until use. For Western blot analysis, 40 µg of protein was resolved on an SDS-15% PAGE, transferred onto a nitrocellulose membrane with 0.2 µm pore size (Bio Rad, catalog no. 162–0097) for 1h at 250 mA. Membranes were first blocked with 5 % bovine serum albumin prepared in TBST (50 mM Tris-HCL, pH 7.4, 150 mM NaCl, 0.01 % Tween 20) buffer for 30 min at room temperature. Membranes were then probed with the primary mouse monoclonal α-T7 (Novagen, catalog no. 69522) and secondary goat anti-mouse IRDye 680LT (LI-COR, catalog no. 926–68070) antibodies. Finally, membranes were washed twice with 1 x PBS and scanned using Odyssey® CLx Infrared Imaging System (LI-COR) to detect the protein of interest. Blots were also probed with α-GAPDH antibody (Abcam, catalog no. ab8245) as a loading control.
Indirect immunofluorescence microscopy
Indirect immunofluorescence microscopy studies were performed as previously described (Coric et al., 2014). Briefly, SVG-A and/or HeLa cells (2 × 106 cells/100 mm tissue culture plate) were transfected separately with the following plasmids: pCGT7-JCV-ORF3, pCGT7-JCV-ORF4 and pCGT7-JCV-ORF5 (8 µg each) using lipofectamine™ 3000 transfection reagent according to the manufacturer recommendations. The rest of the transfection of the specific plasmids are described under corresponding figure legends. At 24 h post-transfection, cells were trypsinized, reseeded at subconfluency on polylysine-coated glass chamber slides (Nunc, catalog no. 154461) and incubated for an additional 24h. The next day, the cells were washed twice with 1 x PBS, fixed in cold acetone and incubated with 5% bovine serum albumin in 1 x PBS for 2 h. Chamber slides were then incubated with either α-T7 monoclonal antibody (1:200 dilution) (Novagen, catalog no. 69522) or α-T7 polyclonal antibody (Millipore, catalog no. AB3790) alone or combinations of anti-T7 polyclonal and anti-PML monoclonal (Santa Cruz, catalog no sc-966) primary antibodies overnight. Cells were washed three times with TBST buffer for 10 min intervals and subsequently incubated either with a fluorescein isothiocyanate (FITC)-conjugated goat α-mouse (Novus, catalog no. NB720-F) or FITC-conjugated-goat α-rabbit (Abcam, catalog no. Ab6717) or combination of FITC or Rhodamine-conjugated goat α-mouse (Millipore, catalog no. AP124R) secondary antibodies for 45 min. Cells were then washed with TBST buffer three times for 10 min each, incubated with DAPI (ThermoFisher, 4’,6-Diamidino-2-Phenylindole, Dihydrochloride, catalog no. D1306) (300 ng/ml prepared in 1 x PBS) for 5 min. to stain the nucleus, mounted using “ProLong® Gold Αntifade” mounting medium (Life Technologies, catalog no. P36934) and dried overnight. Slides were then examined under a fluorescence microscope (Leica, DMI-6000B, objective: HCX PL APD 40x or 60x/1.25 oil, employing LAS AF operating software) for visualization of the protein of interest.
Transfection/infection of the PHFA and SVG-A cells
Transfection/infection of the primary human fetal astrocytes (PHFA) was carried out as previously described (Sariyer et al., 2011). Briefly, the plasmid constructs [Bluescript KS (+)-JCV-Mad-1-WT were digested with BamHI to liberate the viral genome from the vector, Bluescript KS (+). PHFA (2 × 106 cells/T75 cm2 flask), obtained from “Comprehensive NueroAIDS Tissue Culture Core”, Temple University, were transfected/infected with viral DNA (8 µg DNA) using lipofectamine™ 3000 transfection reagent. Note that, after transfection of the viral DNA into the cells, the viral infection cycle starts. After overnight incubation, cells were transferred into T175cm2 flasks and fed with complete (DMEM F12 Ham) media (35 ml) (Sigma, catalog no. D8900–1L). The content of the complete DMEM F12 Ham media is described under the “cell lines” section above. The half-volume of the media was replenished every three days post-transfection until the 15th day post-transfection/infection. At the 15th day post-transfection/infection, the samples were processed for immunocytochemistry and total RNA isolation as described below. In parallel, SVG-A cells were also subjected to transfection/infection as described for PHFG cells and as described previously (Saribas et al., 2018).
RNA isolation from the JCV-infected PHFA and progressive multifocal leukoencephalopathy brain tissue samples
The total RNA was isolated from PHFA (untransfected or transfected/infected cells). At the 15th day post-transfection/infection, the cells were collected by trypsinization and homogenized in 1 ml of Trizol reagent (ThermoFisher Scientific, catalog no. 15596018) by gentle pipetting. Subsequently, total RNA was isolated by using the RNeasy lipid tissue mini kit (Qiagen, catalog no. 74804) following the protocols provided by the manufacturer. In addition, total RNA was also isolated from progressive multifocal leukoencephalopathy positive and Non-progressive multifocal leukoencephalopathy brain tissue samples obtained from the National NeuroAIDS Tissue Consortium (NNTC, www.nntc.org). Each sample (50–100 mg) was homogenized in 3 ml of Trizol reagent using a glass-Teflon homogenizer. Subsequently, total RNA was isolated employing the RNeasy lipid tissue mini kit following the protocols provided by the manufacturer.
RT-PCR and DNA sequencing
RT-PCR amplification was carried out using SuperScript™ III one step RT-PCR system (Invitrogen, catalog no. 12574026) using 250 ng total RNA isolated from either PHFA or progressive multifocal leukoencephalopathy brains tissue samples, using appropriate primers (see figure legends) as previously described (Saribas et al., 2018). Each reaction was first incubated at 55°C for 30 min to synthesize cDNA and subsequently amplified by PCR using appropriate primers (see figure legends) as follows: Denaturation: 94°C for 2 min (1 cycle), amplification: 94°C for 15 sec., 55°C for 30 sec., 68°C for 1 min (40 cycles), extension cycle: 68°C for 5 min. (1 cycle), and maintenance: 4°C (1 cycle, 10 min). Specific PCR reactions were carried out as described under each figure legend. Specific RT-PCR products corresponding to the ORF3, ORF4 and ORF5 splicing fragments were subcloned into pcDNA3.1 (+) at XbaI and BamHI sites and sequenced commercially (Genewiz, http://www.genewiz.com) as indicated under corresponding figure legends. Note that RNA samples were treated with RNAse-free DNAse I (New England Biolabs, catalog no. M0303S) prior to use in RT-PCR reactions.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) analyses and data processing
Whole cell extracts (WCEs) prepared from SVG-A cells (uninfected and infected with JCV Mad1 strain) at 21st day post-infection using RIPA lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1.0 % NP-40 and 0.1 SDS in the presence of a cocktail of the protease inhibitors (Sigma, catalog no. P8340] were immunoprecipitated (1mg WCE /sample) by a newly raised anti-ORF4 antibody (#31) (10 µl/sample) in duplicate using protein G magnetic beads (cat. no. 101945, Active Motive) on a racking platform at 4°C overnight. The next day, samples were extensively washed with RIPA buffer and analyzed on an SDS-PAGE gradient gel (4–20%, cat. no. 5671094, Bio-Rad). The anti-ORF4 antibody was commercially raised in rabbits (Lampire Biologicals, Piperville, PA) against the following ORF4 peptide: C 52ATTVLLDEFGVGPLCKGDNLYLSAVDVCG80. A Cys residue was added to the ORF4 peptide at the first amino acid position to conjugate the “keyhole limpet hemocyanin (KLH)” to make the peptide more immunogenic in rabbits. The specificity of the anti-ORF4 was confirmed by Western blotting (WB) and immunoprecipitation (IP)/WB prior to the proteomics studies. LC-MS/MS analysis was performed by the Proteomics and Metabolomics Facility at the Wistar Institute, Philadelphia, PA, using a Q Executive Plus mass spectrometer (ThermoFisher Scientific) coupled with a Nano-ACQUITY UPLC system (Waters). Samples were digested in-gel with trypsin and injected onto a UPLC Symmetry trap column (180 μm i.d. x 2 cm packed with 5 μm C18 resin; Waters). Tryptic peptides were separated by reversed phase HPLC on a BEH C18 Nanocapillary analytical column (75 μm i.d. x 25 cm, 1.7 μm particle size; Waters) using a 95 min gradient formed by solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). A 30-min blank gradient was run between sample injections to minimize carryover. Eluted peptides were analyzed by the mass spectrometer set to repetitively scan m/z from 300 to 1800 in positive ion mode. The full MS scan was collected at 70,000 resolution followed by data-dependent MS/MS scans at 17,500 resolution on the 20 most abundant ions exceeding a minimum threshold of 10,000. The peptide match was set as preferred; exclude isotopes option and charge-state screening was enabled to reject unassigned and greater than 5 charged ions.
Peptide sequences were identified using MaxQuant 1.6.3.3 (Cox and Mann, 2008). (MS/MS spectra were searched against a UniProt human, JCV and common contaminant protein databases using full tryptic specificity with up to two missed cleavages, static carbamidomethylation of Cys, and variable Met oxidation, protein N-terminal acetylation and Asn deamidation. Consensus identification lists were generated with false discovery rates set at 1% for protein and peptide identifications. Proteins not consistently identified by more than one peptide are considered low confident identifications and were removed from further analysis.
Replication assay
Replication assays were carried out as previously described (Sariyer et al., 2011). Briefly, the plasmid constructs (Bluescript KS-JCV Mad-1 WT and Bluescript KS-JCV Mad-1 ORF4/VP1 M190I mutant) were digested with BamHI to liberate the inserts from the vector (Bluescript KS+). SVG-A cells (2×106 cells/75cm2 flask) were separately transfected/infected with each of the digested DNA (8µg each) using lipofectamine 3000 according to the manufacturer’s recommendations. The next day, cells were washed with 1 x PBS and fed with DMEM supplemented with 10% heat inactivated FBS and antibiotics [penicillin-streptomycin (100 µg/ml), ciprofloxacin (10 µg/ml)]. Half of the media was replenished every three days post-transfection. At the indicated time points, low-molecular-weight DNA containing both input and replicated viral DNA was isolated as follows: Cells were harvested by trypsinization, washed with 1 x PBS by centrifugation at 1000 xg for 3 min. Cells were then resuspended in Qiagen P1 buffer (250 µl, 50 mM Tris-HCl pH 8.0, 10 mM EDTA, 100 μg/ml RNaseA), lysed with Qiagen P2 buffer (250 µl, 200 mM NaOH, 1% SDS) and neutralized with Qiagen P3 buffer (250 µl, 3.0 M potassium acetate pH 5.5). The cell lysates were spun at 15000 xg for 4 min and the supernatant (650 µl) was transferred into a new Eppendorf tube without taking any cell debris. DNA was precipitated by isopropanol (650 µl) by spinning samples at 15000 xg for 5 min at room temperature. The DNA pellet was resuspended in 100 µl dH2O containing RNAase A (1 µg total/sample). Ten microliters of each sample were digested with BamHI and DpnI enzymes, resolved on 1% agarose gel, and transferred onto nitrocellulose membrane (catalog no. RPN 203N, Amersham). Southern blots were probed using radio-labeled JCV-specific probes as described previously (Sariyer et al., 2011).
RESULTS
Analysis of the splicing patterns of the JCV late coding region in JCV-infected cells as well as in progressive multifocal leukoencephalopathy brain patient samples
The transcripts generated by the JCV late coding region is known to produce two major alternatively spliced products, M1 and M2, (Bollag et al., 2006; Frisque et al., 1984) (Fig. 1A). These splice products encode primarily a regulatory protein, agnoprotein and three structural capsid proteins (VP1, VP2 and VP3) (Bollag et al., 2006; Frisque et al., 1984). We also recently reported the discovery of two novel open reading frames (ORF1 and ORF2) from the JCV late transcripts (Saribas et al., 2018) (Fig. 1A). These transcripts are detectable in cells infected with JCV in vitro and in progressive multifocal leukoencephalopathy patient brain tissue samples in vivo (Saribas et al., 2018). Based on these recent findings, we have reasoned that JCV late coding region most likely generates additional ORFs by alternative splicing. To this end, the bioinformatics analysis of the JCV late coding region, employing a “splice site predictor” web site, (http://spliceport.cbcb.umd.edu), revealed the presence of the additional splice donor and acceptor sites. They are as follows: 5’-splice donor sites: nucleotides positioned at: 315, 492, 1585 and 1643; and 3’- splice acceptor sites: nucleotides positioned at 521, 1426, 1635 and 1953 (Table 1).
Fig 1. Analysis of the splicing patterns of the JCV late coding region by RT-PCR.
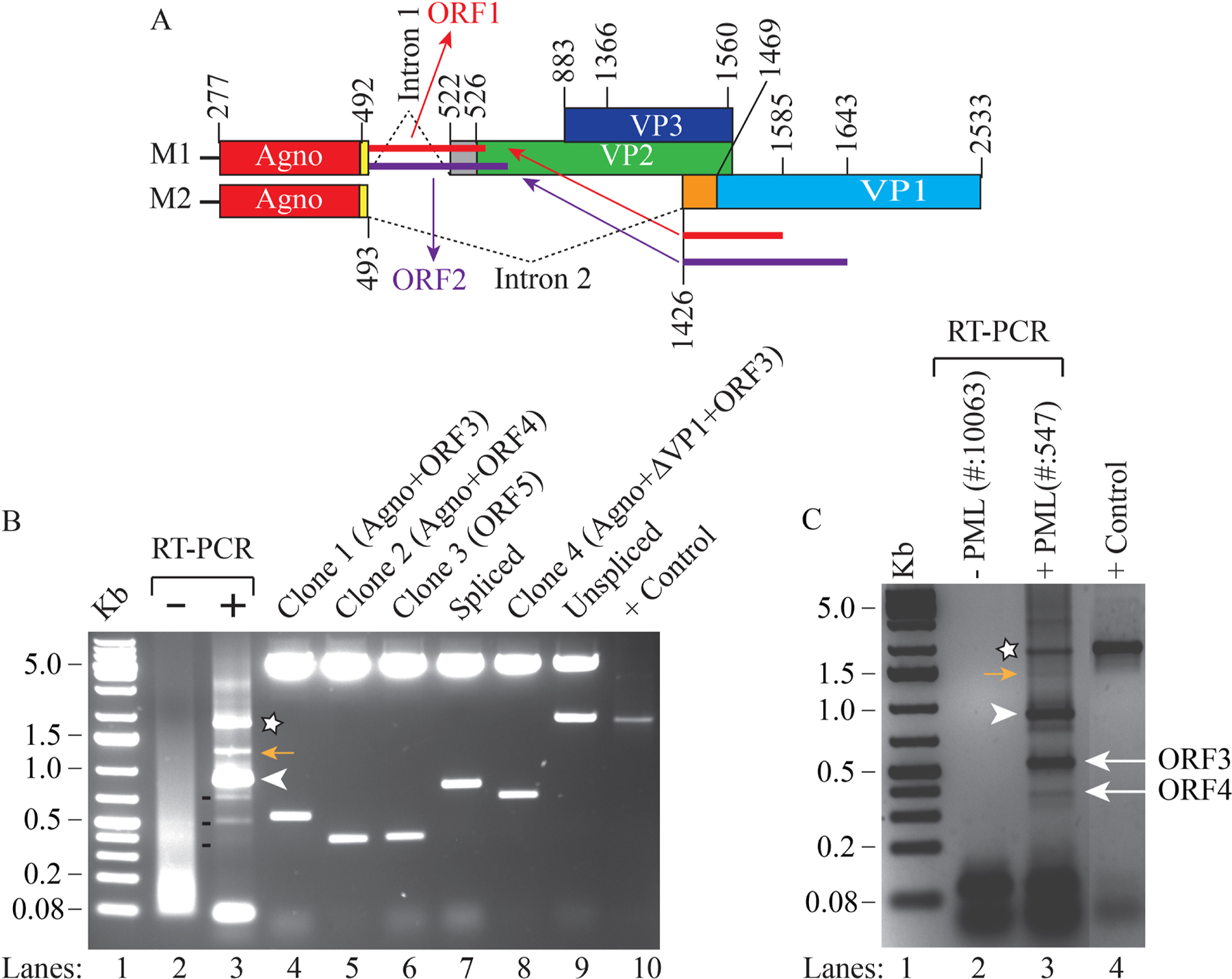
(A) A schematic representation of the splicing pattern of the JCV late coding region. The JCV late coding region primarily generates two major splice products, M1 and M2, by removing the intron 1 and intron 2. Both M1 and M2 splice products are produced by alternative cis-splicing. The JCV late coding region was also shown to produce two additional novel open reading frames (ORF1 and ORF2) by trans-splicing (Saribas et al., 2018). (B) Discovery of additional splice products from the JCV late coding region. Total RNA was isolated from PHFA infected with JCV Mad-1 strain [uninfected (lane 2) or infected (lane 3)] at 15th day post-infection and was subjected to RT-PCR using 5’-primer (JCV Mad-1 nt 277–294) and 3’-primer (JCV Mad-1 nt 2533–2511) as described in materials and methods. The RT-PCR products were then resolved on a 2% agarose gel. The band labeled with a star or an arrowhead or 3 dashed lines were subcloned into the pcDNA 3.1 vector at HindIII/Kpn1 sites, sequenced (clone 1, 2, 3 and 4) and analyzed by bioinformatics to determine the presence of the splice donor/acceptor sites and any open reading frame. The inserts in lane 7 and lane 9 represent the spliced and unspliced form of the M2 transcript respectively (panel A). In lane 10, DNA from the late coding region of the JCV Mad-1 strain was PCR-amplified using the same primers that were used for RT-PCR reaction above and loaded on the agarose gel as a positive control (+ control). Analysis of the sequencing data revealed that Clones 1, 2 and 3 contain open reading frames, designated as ORF3, ORF4 and ORF5 respectively. In addition, Clone 4, contains combinations of several open reading frames, including for the full-length of agnoprotein, ORF3, and a short coding sequence for the N-terminus of VP1. (C) Analysis of the total RNA obtained from a non-progressive multifocal leukoencephalopathy and progressive multifocal leukoencephalopathy brain tissue samples by RT-PCR to determine the splicing pattern of the JCV late coding region. The total RNA was isolated from a non-progressive multifocal leukoencephalopathy and progressive multifocal leukoencephalopathy brain in vivo tissue samples (Saribas et al., 2018), subjected to RT-PCR using the same PCR primer set that is used for panel B and RT-PCR products were resolved on a 2% agarose gel. The bands indicated by an asterisk, a yellow-colored arrow and an arrowhead correspond to the same bands indicated on panel B. The bands indicated by the labeled arrows on panel C were determined to be corresponding to the ORF3- and ORF4-containing RT-PCR products obtained from in vitro infected cells (panel B) after DNA sequencing.
Table 1.
Summary of the reported splice donor and acceptor sites for JCV late coding region.
| Nucleotide position | Donor site | Acceptor site | Donor/Acceptor site sequence | References |
|---|---|---|---|---|
| nt 315 | + | − | tgaaggttagta | Current work |
| nt 492 | + | − | cataggtaagta | Frisque et al., 1984 |
| nt521 | − | + | ttttcaggttca | Frisque et al., 1984 |
| nt 1426 | − | + | cttttagggttg | Frisque et al., 1984 |
| nt 1585 | + | − | cagaggtagaat | Shishido-Hara et al., 2000, Saribas et al., 2018 and current work |
| nt 1635 | − | + | atcttaggggtt | Saribas et al., 2018 |
| nt 1643 | + | − | ttttagtaagtc | Saribas et al., 2018 |
| nt 1953 | − | + | attacagaacaa | Shishido-Hara et al., 2000, and current work |
Splice site predictions: http://spliceport.cbcb.umd.edu
Note: Nucleotide numbering is according to the JCV Mad-1 strain (Accession number: 02226.1)
To determine whether the JCV late coding region generates additional novel ORFs, for the first step, the total RNA was prepared from the primary human fetal astrocytes (PHFA) (uninfected or infected with JCV) and RT-PCR was performed on the isolated RNA samples using specific JCV primers (indicated under legend for Fig. 1) and analyzed on an agarose gel (Fig. 1B, lanes 2 and 3). The RT-PCR products from the infected cells showed a distinct banding pattern on the gel compared to the control (Fig. 1B, compare lane 3 with lane 2). The two major bands designated by an asterisk and an arrowhead were determined to be unspliced and spliced products of the M2 transcript respectively (Fig. 1A) determined by a direct DNA sequencing of the RT- PCR products. The remaining bands indicated by three dash lines were subcloned into a vector, analyzed on an agarose gel following a restriction enzyme digestion and sequenced (Fig. 1B, lanes 4–8). Note that we also attempted to subclone the band indicated by a yellow arrow on the panel. Sequencing data about this band showed a combination of several splice products, including ORF3 and ORF4.
In parallel, the splicing pattern of the JCV late coding region was also investigated in progressive multifocal leukoencephalopathy patient brain samples. The total RNA was isolated from the non-progressive multifocal leukoencephalopathy and progressive multifocal leukoencephalopathy brain samples obtained from National NeuroAIDS Tissue Consortium (NNTC, www.nntc.org) and was subjected to RT-PCR, followed by agarose gel analysis. The bands indicated by an asterisk, a yellow-colored arrow and an arrowhead on the RT-PCR product were the same as those detected in the infected cell samples (Fig. 1B). In addition, the bands indicated by the labeled arrows on the progressive multifocal leukoencephalopathy patient brain samples correspond to ORF3- and ORF4-containing RT-PCR products as those detected for the tissue culture infected cells. The identity of those bands was confirmed by a direct DNA sequencing of the RT-PCR products.
Discovery of the novel open reading frames (ORFs) for the JCV late coding region
Next, following the DNA sequencing of the clones and RT-PCR products, we determined the slicing patterns and the protein coding capacity of the JCV late coding region by bioinformatics. As illustrated in Fig. 2B, for Clone 1, the following two introns were removed from the transcripts: (i) The one located between agnoprotein and VP1coding regions, corresponding to the nucleotides 493 and 1425. (ii) The other one is an internal intron within VP1 located between nucleotides 1585 and 1954 (Fig. 2B). The remaining exons are joined together and created two open reading frames: 1) One for the full-length agnoprotein and the other for the partial VP1 coding region, which is designated as ORF3. ORF3 potentially encodes a 70 aa long protein and harbors two regions: 1) a 41 amino acid region identical to the N-terminus of VP1 (aa 1–41), 2) a 29 aa long C-terminus region unique to ORF3, created due to a frame shift occurred at the exon junctions within VP1 coding region as illustrated in Fig. 2B.
Fig. 2. The splicing products of the JCV late transcripts revealed novel open reading frames.
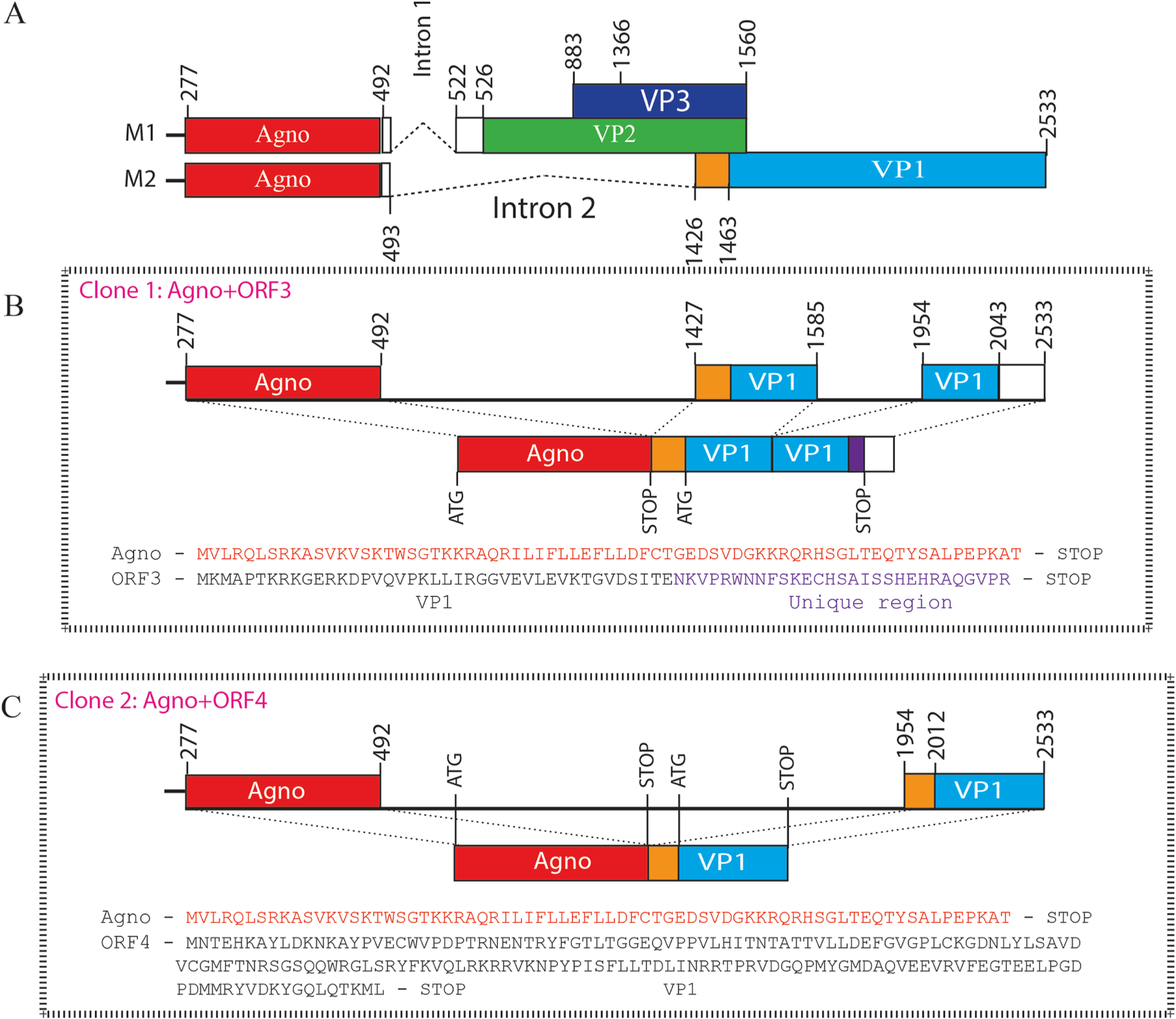
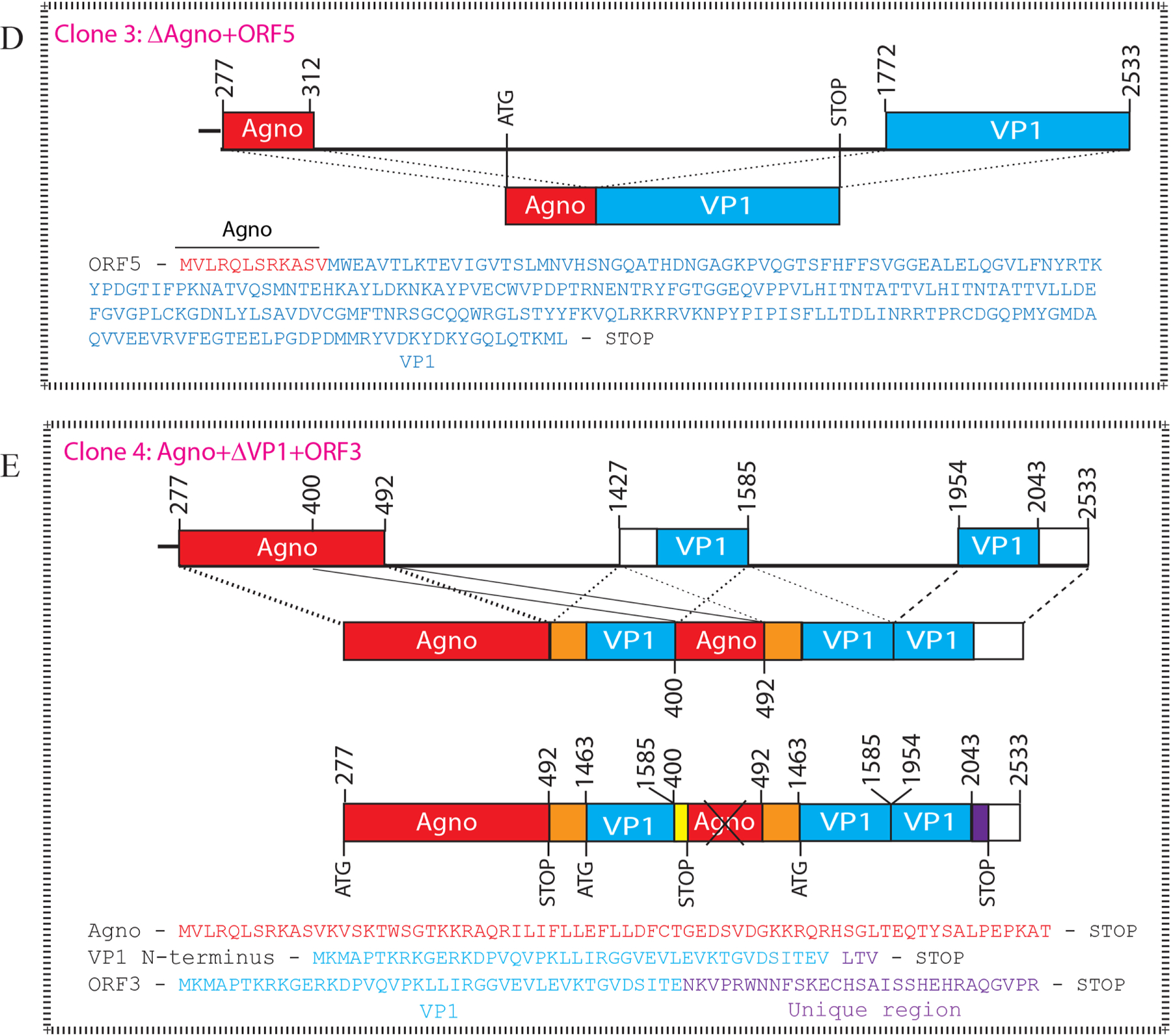
(A) Graphical representation of two major splicing patterns of the JCV late transcripts (M1 and M2). (B) The splicing pattern in Clone 1 produces a novel open reading frame, designated as ORF3, which is created by the removal of an internal intron and rejoining the exons within the VP1 coding region followed by a frame shift occurring at the splice junction. The N-terminus 41 aa region is identical to the VP1 coding sequences, but its C-terminus 29 aa region is unique. (C) The splicing pattern in Clone 2 also produces a unique ORF called ORF4. ORF4 is a truncated version of VP1 and encodes a 173 aa long protein (Table 2). (D) The splicing pattern in Clone 3 created a fusion protein (ORF5) after joining the N-terminus of agnoprotein (14 aa long) with the C-terminus coding region of VP1 (251 aa long). ORF5 can encode a 265 aa long fusion protein (Table 2). (E) The splicing pattern in Clone 4 creates multiple ORFs, where a short N-terminus of VP1 is duplicated as indicated. The C-terminus of agnoprotein is spliced into between the duplicated short N-terminus of VP1. Resulting ORFs are as follows: (i) Full-length agnoprotein coding region. (ii) An ORF designated as “VP1 N-terminus” which encodes a 45 aa long protein, 42 aa region of which is identical with VP1 and 3 aa are unique. (iii) This splicing pattern also creates an ORF identical to the ORF3 observed for the Clone 1. Note that partially duplicated agnoprotein coding region does not create any ORF.
The second clone (Clone 2) results from an interesting splicing pattern of the JCV late transcripts, where a splice donor site located at the end of agnoprotein coding region, corresponding to the nucleotide 492, is joined to a splice-accepter site located at the nucleotide position 1954, removing a 1462 nt intron fragment. This splicing pattern created two open reading frames: (i) One for the full-length agnoprotein and (ii) the other for a partial VP1 coding region, which is designated as ORF4 (Fig. 2C). ORF4 potentially encodes a 173 aa long protein, which is entirely identical to the C-terminal coding region of VP1 (Fig. 2C, Table 2).
Table 2.
Biochemical features of the putative ORF proteins produced by the translation of the alternatively spliced viral late transcripts.
| ORFs | Number of amino acids | Predicted molecular weigth | Splice type |
|---|---|---|---|
| ORF1 | 58 | 6.4 | Trans-splicing |
| ORF2 | 72 | 8.1 | Trans-splicing |
| ORF3 | 70 | 7.9 | Cis-splicing |
| ORF4 | 173 | 19.8 | Cis-splicing |
| ORF5 | 265 | 29.8 | Cis-splicing |
In Clone 3, a more distinct splicing pattern has also occurred, where a slice-donor site, located towards N-terminus coding region of agnoprotein positioned at nt 312 is spliced into a splice-accepter site located within VP1 positioned at nt 1772, removing a 1460 nt long intron. This splicing created a fusion protein between the N-terminus of agnoprotein and C-terminus of VP1. This open reading frame is designated as ORF5, which contains a 12 aa long fragment from the N-terminus of agnoprotein and a 253 aa long fragment from the C-terminus of VP1 (Fig. 2D). ORF5 can potentially encode a 265 aa long protein (Table 2).
The splicing pattern in Clone 4 resulted from a more complex splicing event, creating an (i) Agno open reading frame, (ii) a short VP1 N-terminus open reading frame and (iii) another ORF3 open reading frame (Fig 2E). Additionally, a direct DNA sequencing analysis of the RT-PCR products from the progressive multifocal leukoencephalopathy patient samples (Fig. 1C, lane 3) revealed identical splicing patterns as described for Clone 1 and Clone 2. They also contain ORFs for ORF3 and ORF4 respectively. Taken together, the RT-PCR analysis of the JCV late transcripts obtained from the JCV-infected cells and the progressive multifocal leukoencephalopathy patient brain samples revealed similar splicing patterns. Particularly, the splicing pattern of the ORF3 and ORF4 are clearly detectable in the JCV-infected cells in vitro and in the progressive multifocal leukoencephalopathy patient brain samples in vivo. Here, it should be emphasized that the detection of the novel splicing products for JCV late transcripts is not associated with any possible RT-PCR artifacts, because such RT-PCR amplifications were repeated several times using different RNA preparations from the infected cells to eliminate such a possibility. Additionally, the alternatively spliced product for ORF4 was also reported by Shishido-Hara et al., (Shishido-Hara et al., 2000) both from the transfected cells and progressive multifocal leukoencephalopathy brain tissue samples which are in agreement with our results. However, the protein coding capacity of the ORF4 was not investigated during that time as compared to our current study.
OFR3 and ORF4 display distinct subcellular distribution patterns
Next, the subcellular distribution patterns of each ORF were first examined in glial (SVG-A) and non-glial (HeLa) cells by immunocytochemistry (ICC). As shown in Fig. 3AB, the distribution pattern of ORF3 in glial and non-glial cells is not different from one another. That is, ORF3 showed a relatively uniform cytoplasmic and nuclear distribution pattern in both cell lines (Fig. 3AB). In contrast, ORF4 showed an exclusively nuclear and punctate distribution pattern in both cell types (Fig. 3AB), suggesting that ORF4 may co-localize with a particular or a number of known specific macromolecular protein assemblies in the nucleus including, polycomb-group proteins, nuclear stress bodies, Cajal bodies, speckles, paraspeckles, nucleoli, nucleolus and/or promyelocytic leukemia nuclear bodies (PML-NBs) (Cremer and Cremer, 2001; Misteli, 2007).
Fig. 3. Analysis of the expression profiles of ORFs in SVG-A and HeLa cells by immunocytochemistry (ICC) and Western blotting.
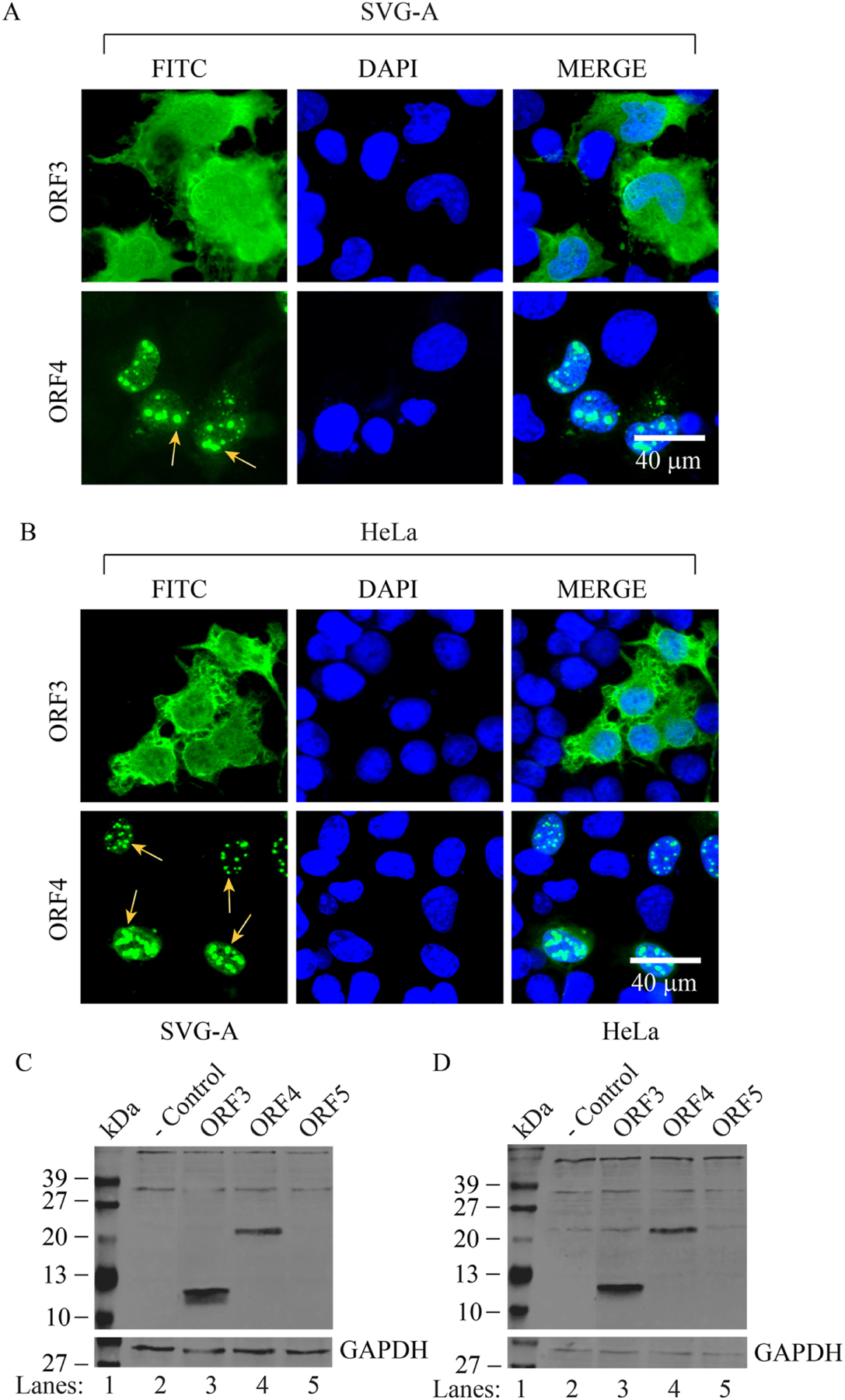
(A) Analysis of the subcellular distribution patterns of ORF3 and ORF4 in SVG-A cells. pCGT7-ORF3 and pCGT7-ORF4 expression plasmids were separately transfected into SVG-A cells by lipofectamine 3000 as described in materials and methods; and at 24h post-transfection, cells were processed for ICC using a primary mouse α-T7 monoclonal antibody and the secondary FITC-conjugated goat α-mouse antibody. Samples were then stained with DAPI and examined under a fluorescence microscope. Arrows point to a distinct punctate nuclear localization of ORF4 in the nucleus. (B) Analysis of the subcellular distribution patterns of ORF3 and ORF4 in HeLa cells. In parallel to the ICC experiments described for panel A, HeLa cells were also transfected separately with the same expression plasmids and processed for ICC as described for panel A. Scale bar: 40 µm in both panels. Immunocytochemistry experiments were repeated more than three times in different cell lines. Without exception, all ORF4-transfected cells showed a similar punctate distribution of ORF4 protein in the nucleus. (C and D) Analysis of the ORF3, ORF4 and ORF5 expression by Western blotting. In parallel to the ICC experiments described for panel A and B, whole-cell extracts were also prepared from SVG-A (C) and HeLa (D) cells and analyzed by Western blotting using α-T7 monoclonal antibody. GAPDH was used as a gel loading control.
In parallel, a stable expression profiles of ORF3, ORF4 and ORF5 was also examined by Western blotting using whole-cell extracts prepared from transfected SVG-A and HeLa cells. As observed for ICC assays (Fig. 3AB), the expression of ORF3 and ORF4 were also readily detectable by Western blotting (Fig. 3C and Fig. 3D) in two different cell types. Note that, under similar expression conditions, we were unable to detect the expression of ORF5 by either Western blotting or ICC assays, even though the lysis buffer was supplemented with sufficient amount of protease inhibitors. We repeated both assays for ORF5 for several times with a different set of the transfection assays but did not obtain any better detectable signal for this protein, reinforcing the idea that ORF5 is an unstable protein (Fig. 3C and 3D).
ORF4 co-localizes with PML-NBs and alters their organization pattern in the nucleus
Immunocytochemistry studies demonstrated that ORF4 protein exhibits a punctate distribution pattern in the nucleus (Fig. 3AB). We then reasoned that this could be as a result of the co-localization of ORF4 with one or more of the nuclear macromolecular protein assemblies such as PML-NBs, nucleolus, Cajal bodies, and paraspeckles (Cremer and Cremer, 2001; Misteli, 2007). We addressed this question by immunocytochemistry by expressing ORF4 protein in the primary human fetal astrocytes (PHFA) and SVG-A cells using various antibodies that specifically detect PML-NBs, nucleolus, Cajal bodies, or paraspeckles. Among those, it was surprising to observe that ORF4 only co-localizes with PML-NBs and alters their reorganization not only in primary (PHFA) but also in established cells (SVG-A) (Fig. 4) suggesting that such an observation is not cell-type specific. Of note, every single ORF4 protein-positive cell shows a similar PML-NB targeting pattern in the nucleus regardless of the expression level of ORF4 protein in cells. In other words, we did not observe ORF4 protein co-localizing with other protein complexes in the nucleus as it does with PML-NBs. To further demonstrate this case, we provide the data, for example, for the “nucleolus plus ORF4” colocalization, where our data showed no significant colocalization of ORF4 with nucleolus (Supplement 11 and Supplement 13). In addition, with respect to PML-NB targeting by ORF4 protein, our findings also suggested that ORF4 protein could be the only or one of the JCV proteins responsible for the reorganization of PML-NBs. Similar ICC expression studies with other JCV proteins including, large T antigen, small t antigen, agnoprotein, ORF1, ORF2 and ORF3 were also performed, but none of these proteins induced the reorganization of PML-NBs (Supplements 1 and 2), supporting the idea that ORF4 is the only JCV protein responsible for such a phenomenon. In addition, we tested whether the putative BKV ORF4 protein co-localizes with PML-NBs as strongly as that observed for the JCV ORF4 by ICC studies. In contrast to JCV ORF4, BKV ORF4 protein did not show a relatively restricted nuclear localization pattern. Instead, it exhibited a uniform distribution throughout the cells (nuclear and cytoplasmic) (Supplement 3). However, BKV ORF4 also exhibits the ability to reorganize PML-NBs in the nucleus as observed for JCV ORF4.
Fig. 4. ORF4 co-localizes with PML-NBs in PHFA and SVG-A cells.
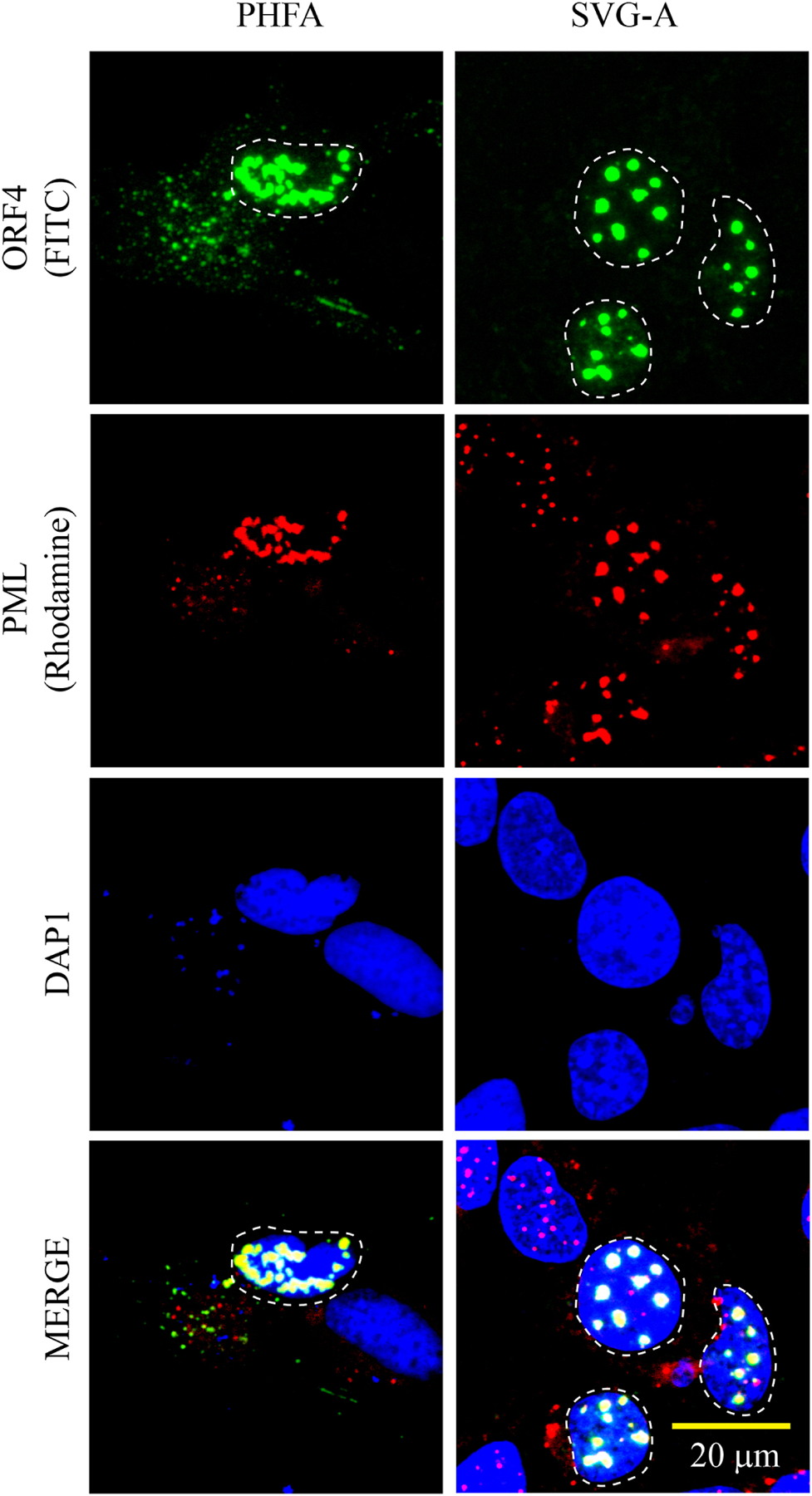
Co-localization of ORF4 protein with PML-NBs is demonstrated by ICC. pCGT7-ORF4 plasmid was transfected into PHFG and SVG-A cells and at 24h post-transfection, cells were fixed with cold acetone and processed for ICC as follows: The sample slides were incubated with the primary (α-T7 polyclonal and α-PML monoclonal) and the secondary (FITC-conjugated goat α-rabbit and Rhodamine-conjugated goat α-mouse) antibodies. Finally, the cells were stained with DAPI and examined under a florescence microscope as described under materials and methods. The cells with a punctate nuclear expression of ORF4 is demarcated by the dashed circles. These ICC experiments were repeated more than three times with the same consistent results. A representative group of cells is shown. Scale bar: 20 µm
ORF4 protein also co-localizes with other permanent members of the PML-NB members, including hDaxx and ATRX but not Sp100.
PML-NBs are implicated in regulation of transcription, growth suppression, apoptosis (Florin et al., 2002) ; and intrinsic and innate antiviral responses (Scherer and Stamminger, 2016). They also play important regulatory roles in type I and type II interferon immune responses as co-regulatory protein assemblies (Regad and Chelbi-Alix, 2001; Regad et al., 2001; Ulbricht et al., 2012). PML-NBs are known to composed of several permanent and transient members. The PML protein, for example, is a major contributor to the PML-NBs. The other permanent members of these remarkable protein assemblies include Sp100 (an interferon-inducible protein), hDaxx (a transcriptional co-repressor) and ATRX (a chromatin remodeling protein, containing an ATPase/helicase domain and belongs to the SWI/SNF family). In order to be a structural component of PML-NBs, any viral or host protein should be either sumoylated by SUMO-1 or SUMO-2 enzymes or have a SUMO-interacting domain (SIM) in their structure (Bund et al., 2014; Meinecke et al., 2007; Zhong et al., 2000).
Since we identified ORF4 protein as the PML-NB targeting protein, we then sought to determine whether ORF4 protein also alters the molecular dynamics of several other permanent members of the PML-NBs by employing ICC in SVG-A cells. As shown in Fig. 5A, in addition to PML, ORF4 protein was also found to co-localize and the reorganize the other two permanent members of PML-NBs, hDaxx and ATRX. In contrast, ORF4 failed to induce such a reorganization for Sp100, which is also a permanent member of PML-NBs. This observation perhaps represents a reminiscent case observed for HPV minor capsid protein L2 (Becker et al., 2003). That is, ORF4 either leads to the degradation of Sp100 or merely releases it from the PML-NB complexes into the nucleoplasm. These two possibilities need to be sorted out. Taken together, JCV ORF4 protein clearly modulates the molecular dynamics of the permanent members of the PML-NBs including PML, hDaxx, ATRX and Sp100 but the functional consequences of such a regulation are yet to be determined. Of note, the quantification studies related to the co-localization of ORF4 with PML-NBs and reorganization of PML-NBs by ORF4 showed the following results: Whenever ORF4 is expressed in cells, we observe the co-localization and reorganization of PML-NBs (PML, hDaxx, and ATRX) with ORF4 close to 100% of cases as shown in Fig. 5A, Fig. 5B and Supplement 12. More than 30 cells for each case were examined for this purpose with more than three different transfection cases and presented in the form of Pie graphs (Fig. 5B). However, as explained above, Sp100 does not significantly colocalize with ORF4 in colocalizations studies (Fig. 5A and Fig. 5B, Supplement 12).
Fig. 5. Analysis of the effect of ORF4 on the PML-NB members.
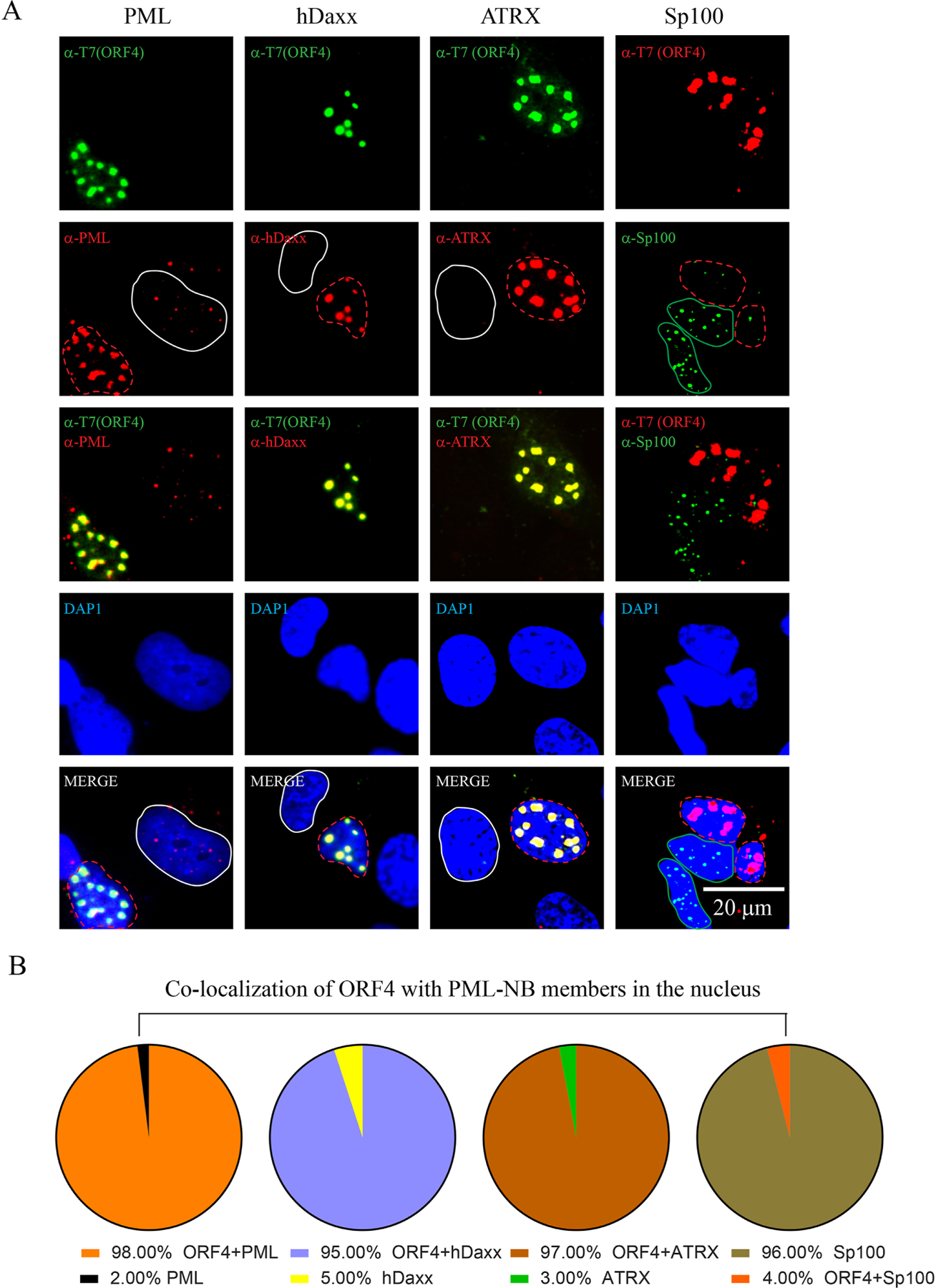
(A) SVG-A cells were transfected with a T7-tagged ORF4 expression plasmid (pCGT7-ORF4) and the next day, samples were processed for ICC. T7-ORF4, PML, hDaxx, ATRX proteins were detected by using α-T7 (polyclonal, catalog no. AB3790, Millipore), α-PML (monoclonal, catalog no. sc-966, Santa Cruz), α-hDaxx (monoclonal, catalog no. sc-8043, Santa Cruz), α-ATRX (monoclonal, catalog no. sc-55584, Santa Cruz) antibodies. In Sp100 column, T7-ORF4 and Sp100 were detected using α-T7 (monoclonal, catalog no. 69522, Novagen) and α-Sp100 (polyclonal, catalog no. NBP1–89457, Novus) antibodies. The white and red color-labeled circles in PML, hDaxx and ATRX columns indicate the ORF4-negative and -positive cells respectively. The red and green color-labeled circles in the Sp100 column indicate ORF4-positive and –negative cells respectively. These ICC experiments were repeated more than three times. A representative group of cells is shown here. Scale bar: 20 µm. (B) Presentation of the quantification of the results in the form of Pie graph.
ORF4 protein contains its own putative nuclear localization signal shared by VP1
Another peculiar observation about the nuclear localization of ORF4 protein is that even though ORF4 does not harbor any previously known nuclear localization signal of its own, it still strongly localizes to the nucleus. It is also important to note that the previously reported nuclear localization signal (NLS) of JCV VP1 is known to localize to the N-terminus of the protein (Ishii et al., 1996; Shishido-Hara et al., 2000) (Fig. 6). In addition, the amino acid alignment of VP1 proteins for JCV, BKV and SV40 showed a high sequence similarity among these proteins (Fig. 6). However, the N-terminus of JCV VP1, where the NLS sequence of each protein was reported to localize, significantly diverges from those of BKV and SV40, indicating that the previously reported JCV VP1 NLS sequence is most likely a weak NLS signal, and may have additional functional NLS sequences somewhere within its sequence. Indeed, bioinformatic predictions revealed a putative secondary NLS sequence for not only JCV VP1 but also BKV and SV40 VP1, located towards the C-terminus of each protein. It is now designated as NLS-2 (QLRKRRVKNP) of VP1 (Fig. 6). More importantly, the predicted secondary NLS-2 of VP1 completely overlaps with that ORF4 protein encompassing amino acids 282–291 (Fig. 6). Collectively, these findings suggest that NLS-2 could be a functional NLS for both VP1 and ORF4 proteins.
Fig. 6. JCV ORF4 protein contains a novel nuclear localization signal (NLS)-like motif.
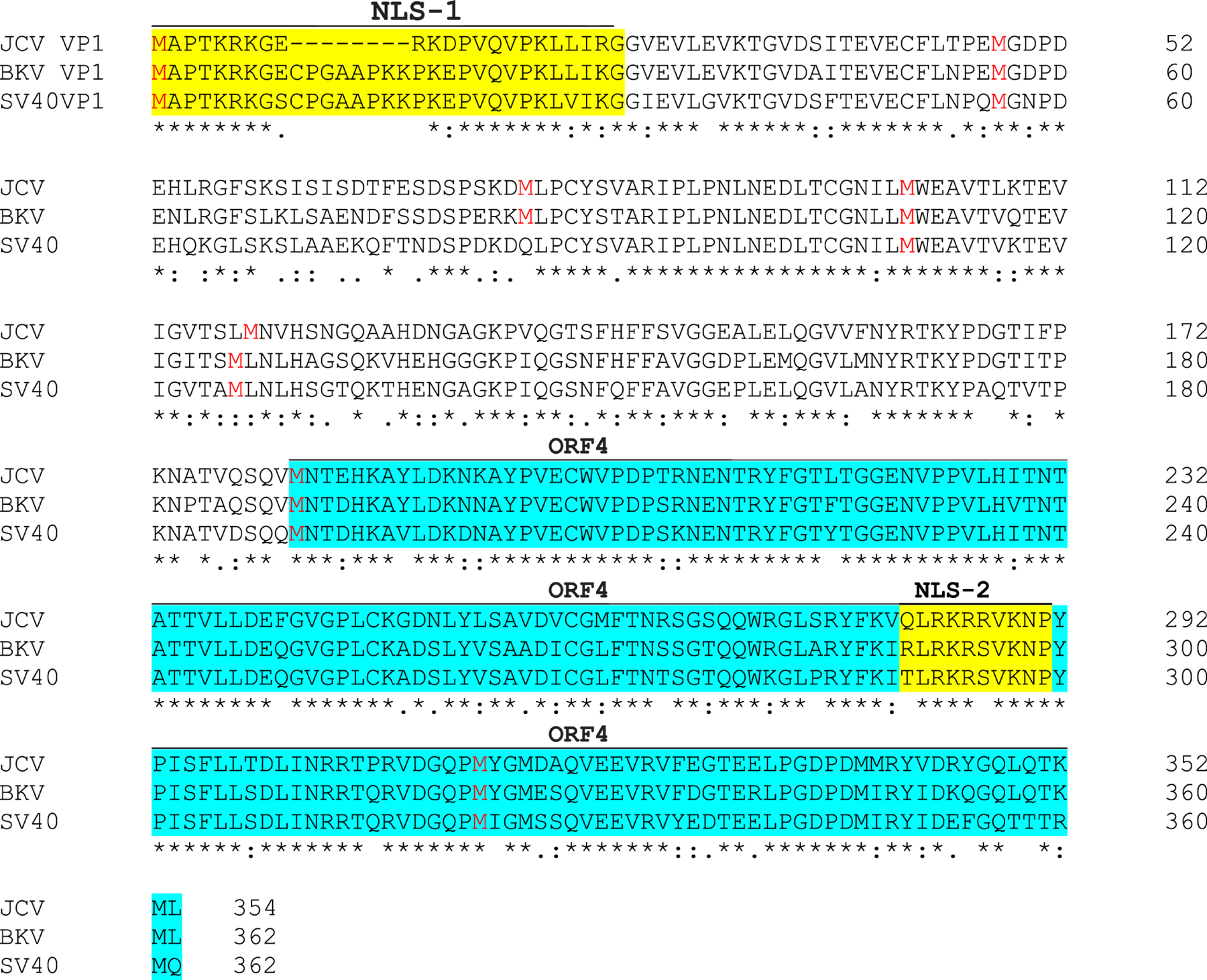
(A) Sequence alignment of JCV, BKV and SV40 VP1 proteins. VP1 protein sequence of JCV, BKV and SV40 were aligned with each other using the “Clustal Omega” program (https://www.ebi.ac.uk/Tools/msa/clustalo). Each protein contains two putative NLS designated as NLS-1 and NLS-2 as indicated in yellow color. Methionine residues in each VP1 protein sequence are highlighted in red color. ORF4 coding region is highlighted in light blue. The putative ORF4 protein of JCV, BKV and SV40 contains a predicted NLS sequence (NLS-2) of their own, which was predicted by using the following website: http://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi.
CLUSTAL O (1.2.4) multiple sequence alignment for JCV, BKV and SV40 VP1 proteins
ORF4 protein lost its predominant nuclear localization property when its NLS-2 sequence is mutated
Next, we tested whether the putative nuclear localization signal of ORF4 is indeed responsible for driving the ORF4 protein into the nucleus. To demonstrate this, an expression plasmid of ORF4 WT and that of ORF4 harboring NLS-2 mutant (QLRKAAVKNP) (Fig. 7A) were separately transfected into SVG-A cells and the transfected cells were examined by ICC at 24h post-transfection. As consistently observed, ORF4 WT protein predominantly localized to the nucleus and exhibited a punctate distribution pattern but the ORF4 NLS mutant protein partially lost its predominant nuclear localization feature and therefore localized to both the nuclear and cytoplasmic compartments of the cells (Fig. 7B). This finding demonstrates that the putative NLS-2 indeed functions as an authentic NLS signal of the ORF4 protein. Additionally, we tested co-localization of ORF4 NLS mutant protein with PML by ICC. ORF4 protein lost its overwhelming co-localization capacity with PML-NBs as observed for the ORF4 WT protein. Instead, a partial co-localization is observed in the nucleus (Supplement 4).
Fig. 7. ORF4 lost its predominant nuclear localization feature when its NLS is altered (RR105–106AA).
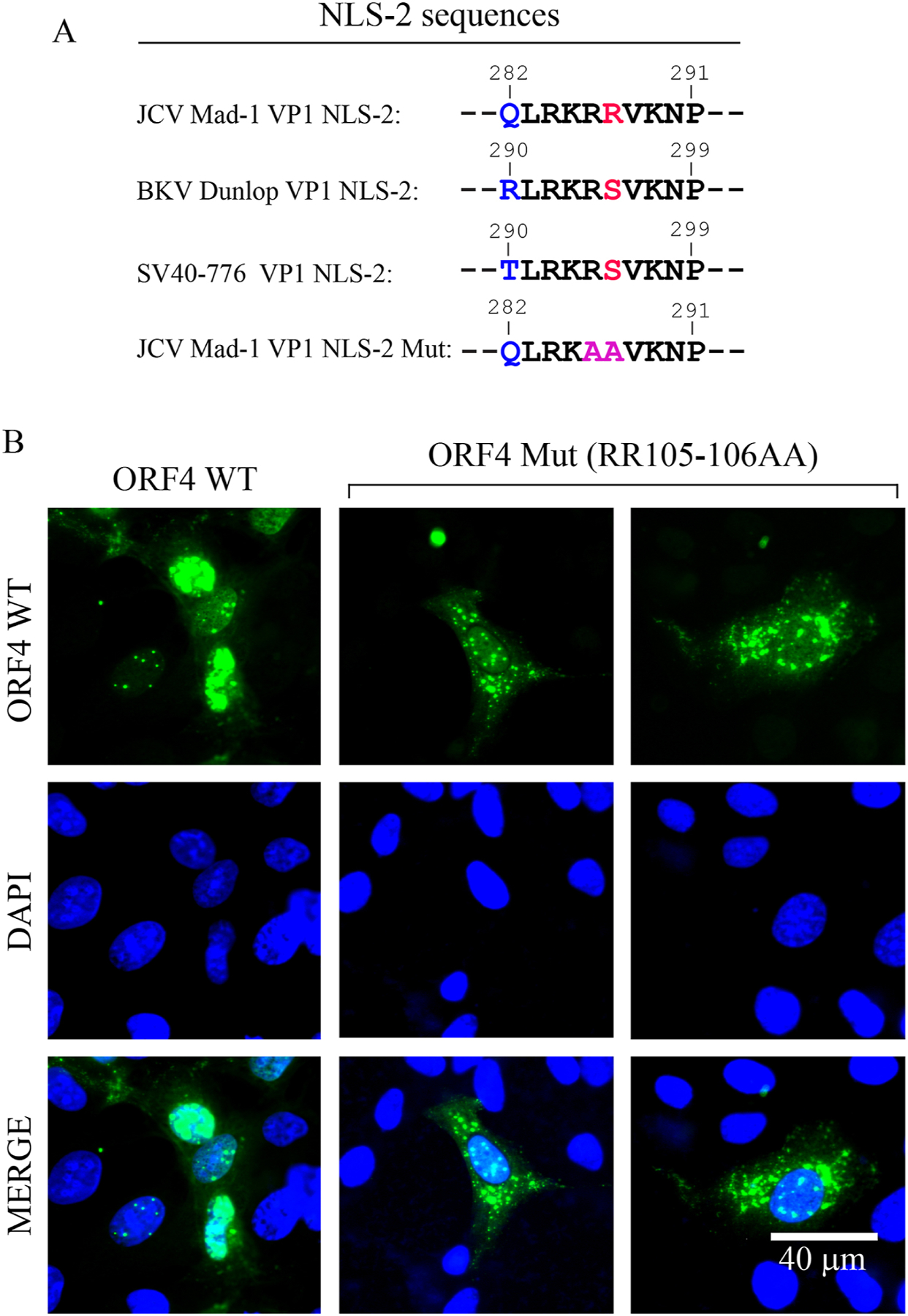
(A) Comparison of the NLS-2 sequences of JCV, BKV and SV40. (B) ORF4 protein containing the mutant NLS-2 sequence lost its predominant nuclear localization feature. The ORF4 (pCGT7-ORF4) and its NLS-2 mutant (pCGT7-ORF4-NLS-2 mut) expression plasmids were separately transfected into SVG-A cell and at 24h post-transfection, cells were processed for ICC using a primary α-T7 polyclonal and a secondary α-rabbit FITC conjugated antibodies and examined under a fluorescence microscope. Scale bar: 40 µm.
Next, we tested whether the NLS-2 motif also plays a major role in the nuclear localization of the full-length VP1 protein in the absence of the viral infection cycle. NLS-1 sequence of VP1 was always considered as a weak NLS signal of JCV VP1 protein compared to those of BKV and SV40 (Ishii et al., 1996; Saribas et al., 2018; Shishido-Hara et al., 2000; Wychowski et al., 1986). We then reasoned that, in addition to being a novel NLS signal for ORF4, NLS-2 motif could also be a novel NLS motif for the JCV VP1 protein. This idea was tested using an NLS-2 mutant of VP1 in transfected cells by ICC (Supplement 5). As expected, JCV VP1 WT protein is exclusively localized to the nucleus. However, the VP1 protein with mutations within its NLS-2 sequence largely localizes to both compartments of the transfected cells, demonstrating that NLS-2 signal also behaves as a strong NLS signal for the VP1 protein (Supplement 5).
Additionally, we also tested whether NLS-2 sequence of VP1 functions as NLS signal for the full-length VP1 protein in infected cells by using an NLS-2 mutant virus of JCV (VP1 RR286–287AA) in the infection assays (Supplement 6). As shown in Supplement 6, as expected, WT VP1 strongly localizes to nucleus but its strong nuclear distribution pattern significantly altered when its NLS-2 motif is mutated. That is, it lost its exclusively nuclear localization pattern and became almost evenly distributed to both cytoplasmic and nuclear compartments of the infected cells, corroborating with our findings from the VP1 NLS-2 expression studies in the transfected cells (Supplement 5). Agnoprotein (Agno) expression was used as a positive control in ICC for the infected cells. In summary, the results from the ICC studies demonstrate that the NLS-2 motif of VP1 functions as a NLS signal for both ORF4 and VP1 proteins.
Analysis of the reorganization of the PML-NBs in JCV-infected cells
We sought to further emphasize the relevance of all our findings by analyzing the impact of JCV WT and JCV ORF4 mutant viruses on the reorganization of PML-NBs by ICC. To achieve this, JCV Mad-1 WT and JCV Mad-1 ORF4 mutant (VP1M182I) viral genomes were transfected/infected into SVG-A cells and the infected cells were then processed for ICC at the 5th day post-transfection/infection. Note that the infection cycle of JCV starts once the viral DNA is transfected into the cells. The reorganization of PML-NB complexes in infected cells were compared to those uninfected cells and to those infected with JCV ORF4 mutant virus (VP1 M182I) (Fig. 8). As opposed to the infected cells, the reorganization of PML-NBs in the ORF4 mutant-infected cells was similar to the control (uninfected) cells, suggesting that JCV infection plays a role in reorganization of the PML-NBs. In particular, ORF4 protein is most likely involved in this reorganization process.
Fig. 8. Analysis of the ORF4 mutant of JCV in reorganization of PML-NBs.
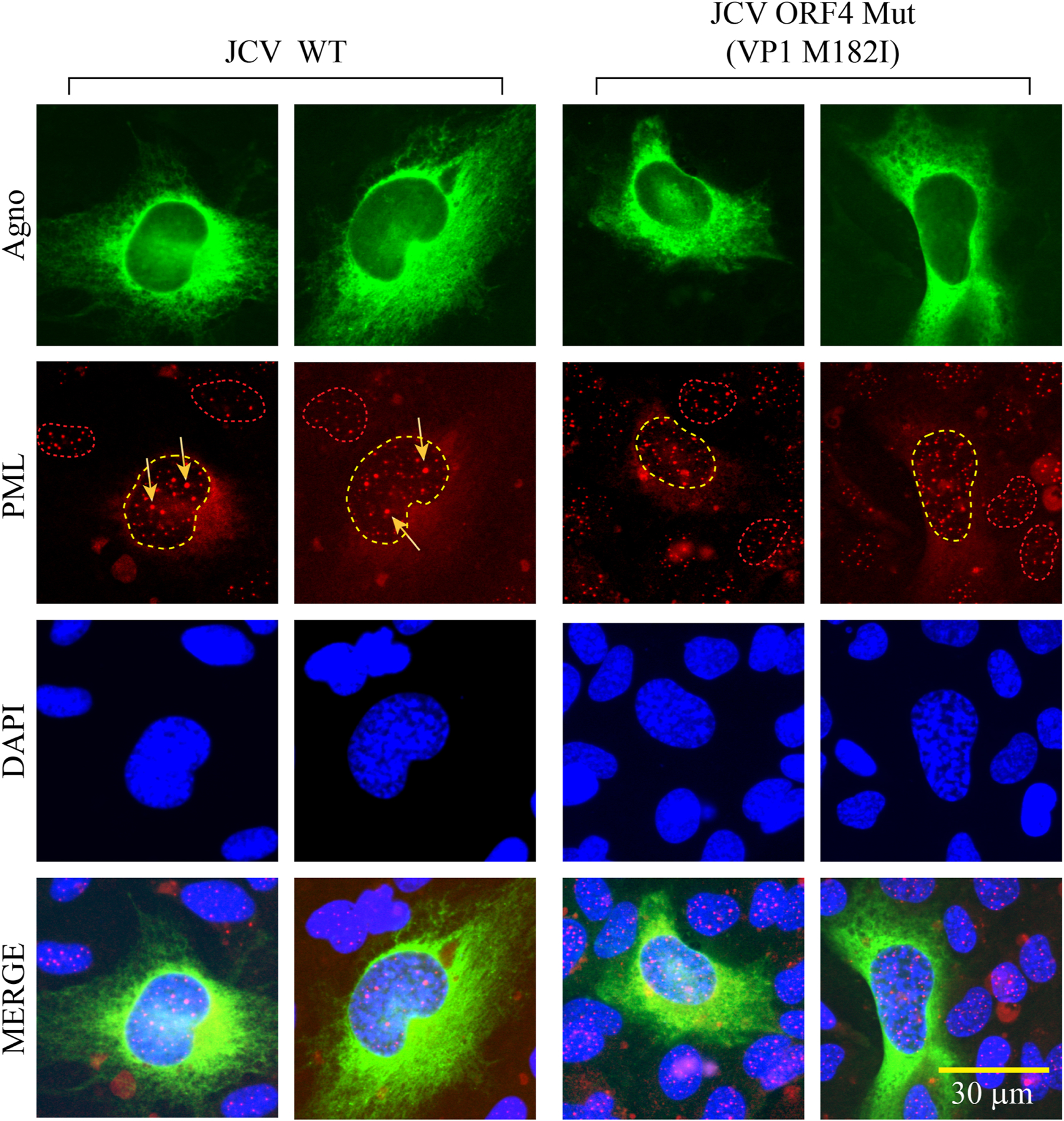
The JCV WT (Bluescript KS+JCV Mad-1) and its mutant [Bluescript KS+JCV Mad-1 VP1 Mut (M182I)] plasmids were digested with the BamHI restriction enzyme to release the viral DNA from the vector and transfected into SVG-A cells. On the 5th day post-transfection/infection, cells were processed for ICC to detect the expression of PML, using the primary (α-PML, monoclonal and anti-Agno, polyclonal) and secondary (rhodamine-conjugated goat α-mouse and FITC-conjugated goat α-rabbit) antibodies. Note that agnoprotein detection was used as a positive control to pinpoint the infected cells. Cells were then examined under a florescence microscope after staining the nucleus with DAPI. Note that the nucleus of the cells infected with JCV WT and those infected with JCV harboring ORF4 mutant (M182I) sequences are demarcated with yellow dashed-line circles to demonstrate the differential distribution pattern of PML protein in WT versus mutant virus-infected cells. The nuclei of the several uninfected cells are demarcated with red dashed-line circles. Arrows point to the reorganized PML-NBs. Scale bar: 30 µm.
Analysis of the ORF4 protein expression by proteomics approaches
To demonstrate the expression of the ORF4 protein during the infection cycle, SVG-A cells were infected with JCV Mad-1 strain of the virus. First, the infection status of the cells was evaluated by both Western blotting (Fig. 9A) and immunocytochemistry (Fig. 9B). Next, we tested whether the newly raised anti-ORF4 antibody can detect ORF4 protein by Western blotting (Fig. 9C) and immunoprecipitation (IP/WB) (Fig. 9D) assays. Indeed, the newly raised antibody can detect this protein by both Western blotting (Fig. 9C, lane 3) and immunoprecipitation (Fig. 9D, lane 2) assays. However, analysis of whole-cell extracts (WCEs) prepared from the infected cells by both assays (Fig. 9CD) did not yield the detection of ORF4 protein, suggesting the following possibilities: (i) Either ORF4 protein is not expressed during the viral infection cycle in intro tissue culture infection cases or (ii) expressed at low levels that are not detectable by both assays due to their low sensitivity issues. To differentiate between these two possibilities, we have employed a highly sensitive proteomics approach to detect ORF4 protein. To this end, WCEs from both uninfected and infected cells were immunoprecipitated using anti-ORF4 antibody and separated on a 4–20% gradient gel (Fig. 10A). The bands corresponding to the expected size of ORF4 (~19–21 kDa) were cut out and subjected to proteomics analysis. These proteomics studies indeed confirmed the expression of ORF4 protein during the viral infection cycle (Fig. 10B, Supplements, 7A, 8 and 9).
Fig. 9. The newly raised anti-ORF4 antibody immunoprecipitates and detects ORF4 protein.
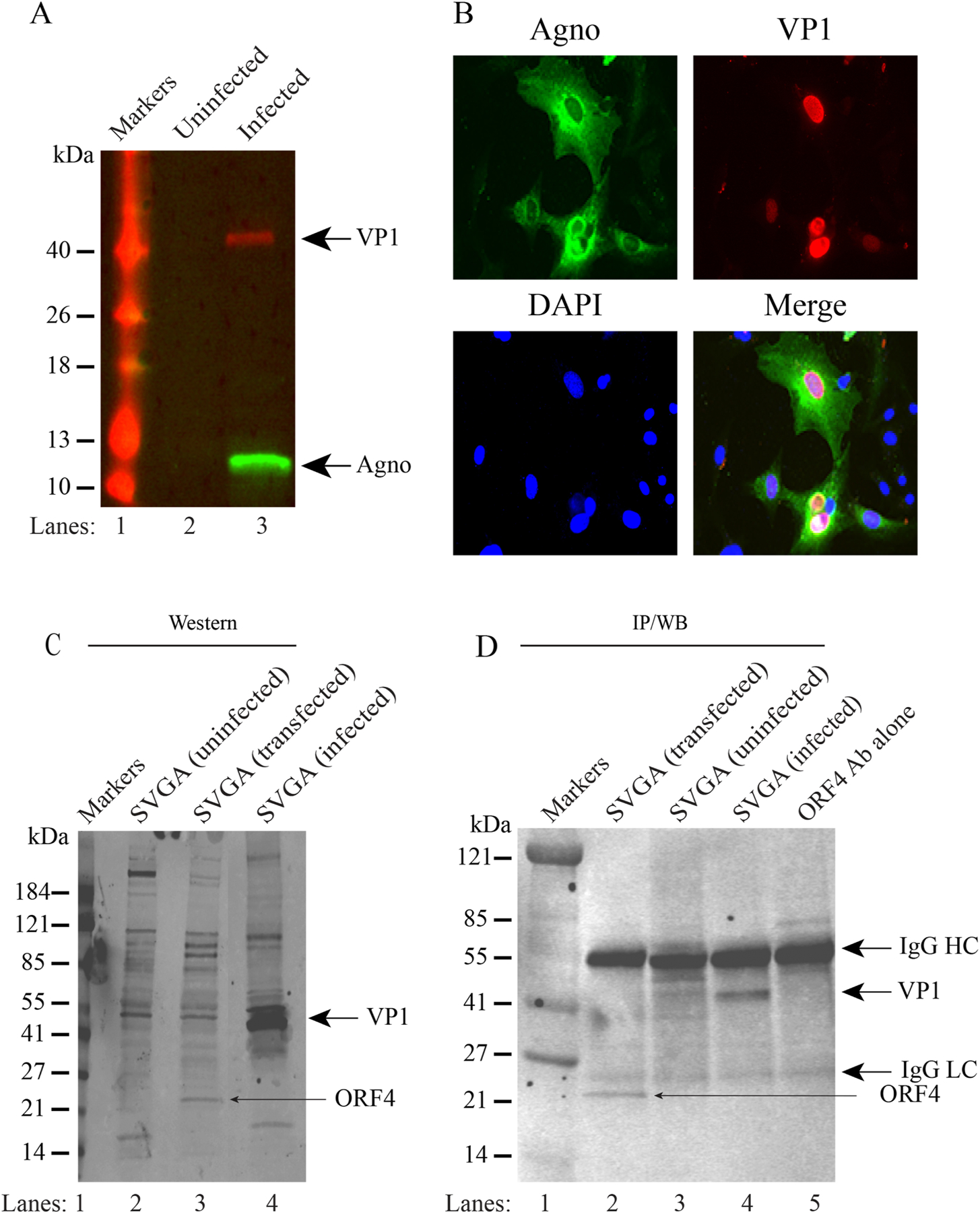
(A) Detection of JCV Agnoprotein and VP1 by Western blotting in extracts prepared from the JCV-infected SVG-A cells using anti-Agno (rabbit polyclonal (Del Valle et al., 2005)) and anti-VP1 (monoclonal, PAB597, a gift from Dr. Walter Atwood, Brown University, USA) antibodies. (B) Detection of Agnoprotein and JCV VP1 protein by immunocytochemistry using the same anti-Agno and anti-VP1 antibodies mentioned for panel A in cells infected with JCV. (C) Detection of ORF4 and VP1 proteins by the newly raised anti-ORF4 antibody. Whole-cell extracts prepared from the uninfected, ORF4-transfected, and JCV-infected SVG-A cells were analyzed by Western blotting using the newly raised anti-ORF4 antibody (rabbit polyclonal, #31 antibody). Anti-ORF4 antibody detects ORF4 and VP1 proteins. (D) Anti-ORF4 antibody immunoprecipitates ORF4 and VP1. Whole cell extracts (200 µg) prepared from the ORF4-transfected, untransfected and JCV-infected cells were subjected to immunoprecipitation by anti-ORF4 antibody (10 µl/sample) and immunoprecipitants were analyzed by immunoblotting using the anti-ORF4 antibody (1:200 dilution).
Fig. 10. Proteomics studies confirm the expression of ORF4 protein in JCV-infected cells.
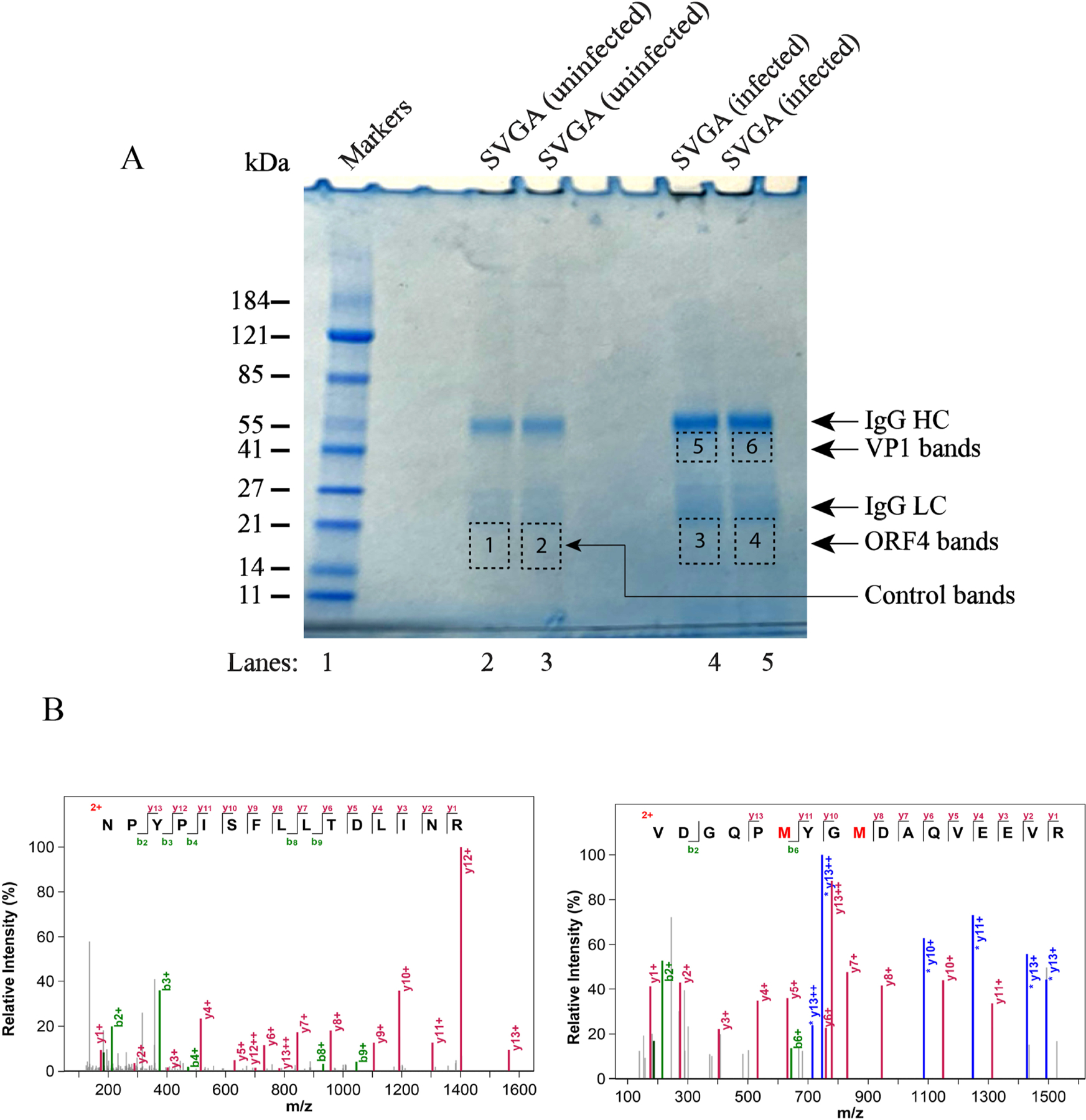
(A) Analysis of the immunoprecipitated ORF4 protein on an SDS-PAGE gradient gel (4–20%). Whole-cell extracts (1 mg) prepared from the SVG-A cells (untransfected or JCV-infected) were subjected to immunoprecipitation in duplicate with the newly raised anti-ORF4 antibody (10 µl/sample) using protein G magnetic beads (catalog no. 101945, Active Motive) and analyzed on a gradient gel (catalog no. 5671094, Bio-Rad). Gel was stained with “gel code blue safe protein stain” (catalog no. 1860957, ThermoScientific) to reveal the immunoprepitated proteins; and antibody heavy (HC) and light (LC) chains. The labeled bands on the gel were marked with the dashed rectangles (bands 1 and 2 for uninfected cells, bands 3 and 4 for infected cells where ORF4 is supposed to migrate, bands 5 and 6 for infected cells, where VP1 is supposed to migrate). These labeled bands were individually cut out, subjected to in-gel digestion with trypsin, and analyzed by proteomics (LC-MS/MS) to detect the ORF4 and VP1 proteins.
Effect of ORF4 mutant on the viral DNA replication
We next investigated the effect of a null mutant of ORF4 on JCV replication (Fig. 10A–D). To achieve this, the translation initiation codon (Met) of ORF4 was mutated to Ile (VP1M182I) to eliminate the ORF4 expression from the viral background and functional consequences of this mutation were then assessed by Southern blotting. In this mutation, we made a conservative substitution (Met to Ile), because the side chains of Ile mimic the bulky side chain of Met and thus may have a minimal effect on the structure of JCV VP1. Note that the infection cycle of JCV is relatively longer compared to those of BKV and SV40. While BKV and SV40 require 2–3 days to complete their first round of the infection cycle, it usually takes 6–7 days for JCV in a cell culture system. Additionally, it is also our experience working with the JCV infection cycle that the scale of the effect of a mutation on the viral replication can be better assessed when, at least, the first two rounds of the viral infection cycle are examined. To investigate this effect, SVG-A cells were separately transfected/infected either with JCV WT or its ORF4 mutant [VP1(M182I)] and the replicated DNA was isolated at the indicated time points and analyzed by Southern blotting. As expected, the wild-type virus replicated efficiently during the first two rounds of infection cycle. That is, the level of the replication of WT virus significantly increased by 15th day post-transfection/infection compared to that of 5th day (~ 5-fold increase, Fig. 11D). The ORF4 mutant virus, on the other hand, replicated modestly less efficiently compared to WT during the first two rounds of infection cycle (Fig. 11CD), suggesting that although ORF4 protein has a modest impact on JCV replication in in vitro cell culture replication assays, there would be a different scenario in in vivo patient infection cases. That is, ORF4 protein would be expressed relatively in large quantities and thus inhibit PML-NB-induced antiviral responses more efficiently in vivo, where this would then lead to a more rapid brain tissue destruction in infected individuals. Note that, JCV is known to spread more rapidly in the brains of the infected individuals.
Fig. 11. Analysis of the replication efficiency of a ORF4 mutant by Southern blotting.
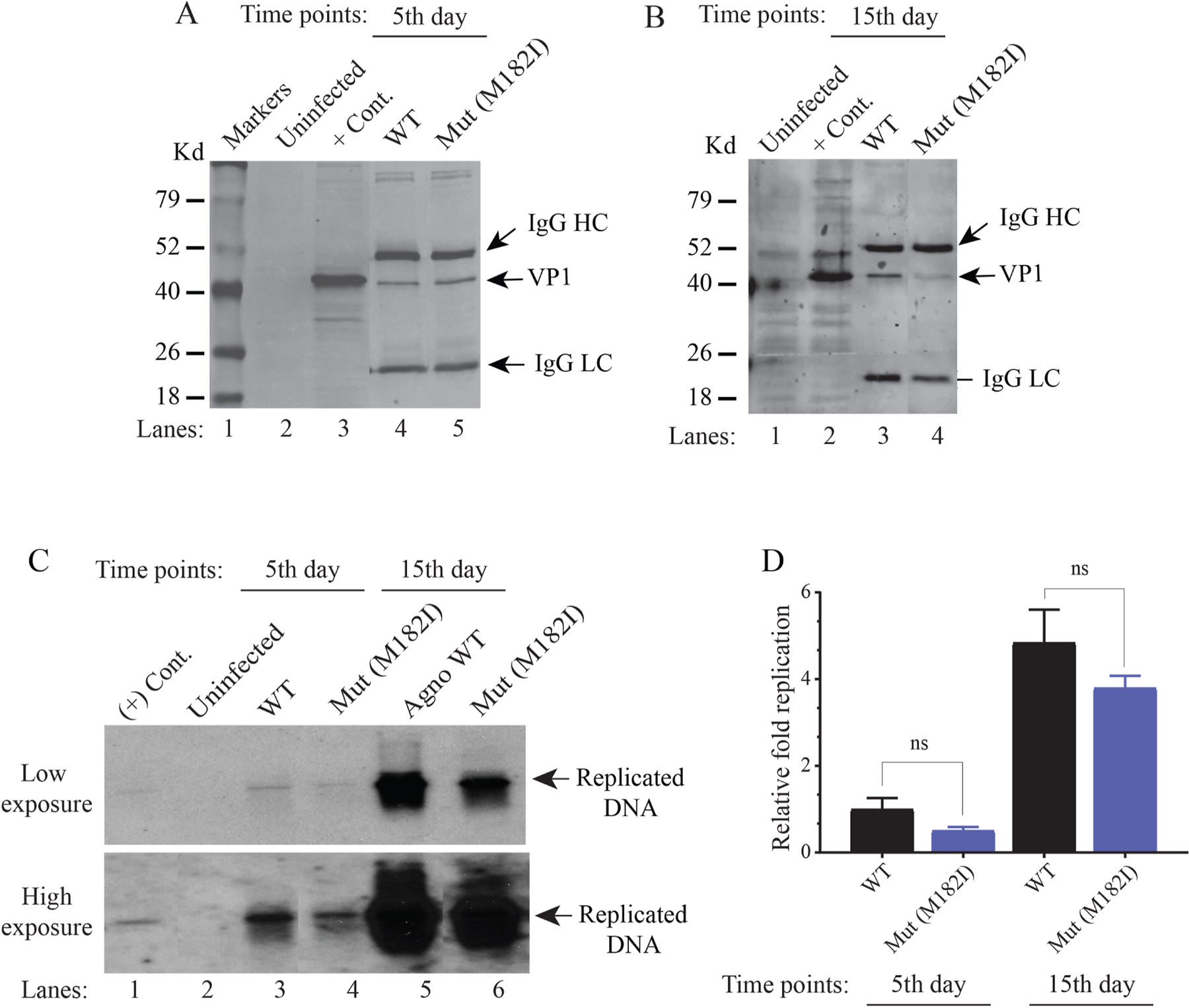
(A and B) Western blot analysis of JCV VP1 expression. The plasmid constructs [Bluescript KS-JCV Mad-1 WT and its mutant (Bluescript KS-JCV Mad-1 VP1 M182I) were separately transfected/infected into SVG-A cells. At the indicated time points, whole-cell extracts were prepared from SVG-A cells (uninfected and transfected/infected), immunoprecipitated (150 µg protein/sample) using an α-VP1 (PAB597) antibody (2 µl/sample) and analyzed by Western blotting by using the same antibody. (C) In parallel, the low-molecular-weight DNA containing both input and replicated viral DNA was isolated from the uninfected and transfected/infected cells, digested with BamHI and DpnI restriction enzymes. Digested DNA was resolved on a 1% agarose gel, transferred onto a nitrocellulose membrane (Bio-Rad, catalog no, 1620094) and probed for detection of the newly replicated DNA using a probe prepared from the JCV Mad-1 WT as a template. In lane 1, 2 ng of JCV Mad-1 WT linearized by BamHI digestion was loaded as positive control (+ Cont.). Replication assays were repeated twice in duplicate, and a representative data is shown here. (D) Quantification analysis of Southern blots by a semi-quantitative densitometry method (using the NIH Image J program) and presentation of the results in arbitrary units. Results were statistically analyzed by the GraphPad program using One-way ANOVA and data columns were compared by Tukey’s multiple comparison test.
DISCUSSION
We have previously reported the discovery and characterization of two novel ORFs (ORF1 and ORF2) generated by the alternative splicing of the JCV late coding region, each of which encodes a 58 and 72 aa long protein respectively (Saribas et al., 2018). The aim of this study was to further explore the possibility of whether JCV late coding region generates additional protein-coding ORFs by alternative splicing. Indeed, we have now identified three additional ORFs, named ORF3, ORF4 and ORF5, each of which encodes a 70, 173 and 265 aa long protein respectively. Expression studies revealed that ORF3 exhibits a relatively uniform distribution pattern both in the nucleus and cytoplasm, while ORF5 was undetectable either because it is a highly unstable protein, or its transcript is untranslatable. Initial characterization of these ORFs with respect to their amino acid composition, cellular distribution pattern and cellular targeting in cells led to new discoveries. For instance, ORF3 contains an interesting amino acid composition with two distinct domains: While the N-terminal region of the protein (41 aa long) completely overlaps with that of VP1, the rest of the protein (a 29 amino acid C-terminus region) is unique. This uniqueness results from a frame shift occurred at the splice junction, allowing the protein to encode an additional 29 amino acid stretch, and ending up with a stop codon (Fig. 2B). The expression of ORF3 is detectable by RT-PCR not only in JCV-infected cells in vitro (Fig. 1B) but also progressive multifocal leukoencephalopathy patient brain samples in vivo (Fig. 1C). It is uniformly distributed both in the nucleus and cytoplasm, but the importance of its expression in JCV-infected cells is yet to be further investigated. The amino acid sequence of ORF4, on the other hand, exhibits a complete overlap with the C-terminus region of the VP1 protein with no unique sequence of its own. Motif prediction and expression studies confirmed that ORF4 protein contains a novel nuclear localization signal (NLS) of its own (QLRKRRVKNP) localizing towards the middle portion of the protein (Fig. 6). This novel NLS sequence is also structurally and functionally shared with the JCV VP1 protein (Fig. 6). This NLS also behaves as the main NLS signal for JCV VP1 and ORF4 protein; and is a stronger NLS signal than that was previously reported for JCV VP1 (Ishii et al., 1996). The previously reported NLS of VP1 is localized towards the N-terminus of the protein (Ishii et al., 1996). Furthermore, mutational analysis revealed that ORF4 protein has a modest effect on the viral DNA replication. ORF4 protein primarily targets PML-NBs in the nucleus and induces their reorganization. Current studies additionally revealed the co-accumulation of ORF4 with the other two permanent members of the PML-NBs including hDaxx and ATRX (Fig. 5A and Supplement 12). However, the third permanent member of the PML-NBs, Sp100, was released from PML-NBs complexes either by degradation or merely dispersed into the nucleoplasm by ORF4, which is a reminiscent case observed for human papillomavirus L2 protein (Florin et al., 2002) (Fig. 5A and Supplement 12). These findings suggest that ORF4 has a differential effect on the assembly of the permanent members of the PML-NBs and thus most likely plays critical roles during the viral life cycle perhaps by modulating the antiviral effects of PML-NBs against JCV infection.
We provide evidence that ORF4 is expressed during the JCV infection cycle in vitro (Fig. 1B) and in vivo at the RNA level (Fig. 1C). The expression of ORF4 at the RNA level was also reported by Shishido-Hara et al., in vitro and in vivo, (Shishido-Hara et al., 2000) which is consistent with our results. Shisido-Hara et al also demonstrated the increased level of PML-NBs expression at the protein level in “progressive multifocal leukoencephalopathy patient tissue samples” by employing immunohistochemistry and electron microscopy (Shishido-Hara et al., 2012; Shishido-Hara et al., 2014) techniques. Their findings also correlate with our current findings and yet which protein of JCV is responsible for such a reorganization was unknown during that time.
In addition to the RNA based evidence, we also demonstrated the expression of ORF4 protein during the viral infection cycle by proteomics studies (Fig. 10AB). The proteomic analysis of the ORF4 bands (bands 3 and 4) also detected a trace amount of two contaminant peptides overlapping with the VP1 N-terminal region, (Supplement 7A). The detection of such contaminant peptides could be explained as follows: (i) One possible explanation is that some of the known or unknown spliced products of VP1 most likely strongly interacted with ORF4 protein, pulled down during the co-immunoprecipitation process, closely migrated with ORF4 protein on SDS-PAGE and thus contributed to the co-detection along with ORF4 during the proteomics analysis. (ii) Another explanation is that a trace amount of the full-length VP1 (which runs around 39 kDa area on the gel), (Fig. 10A) somehow closely migrated along with ORF4 protein and thus contributed to the low-level detection of VP1 N-terminus peptides (Supplement 7AB).
VP1 is known to form monomeric, heterodimeric and oligomeric structures (Chen et al., 1998; Stehle and Harrison, 1997; Suzuki et al., 2012). Since ORF4 protein sequence completely overlaps with the C-terminus of the VP1 protein, it is reasonable to expect that ORF4 protein may also establish similar kinds of interactions with its partners. Indeed, we recently reported the discovery of two short proteins called ORF1 and ORF2 (Saribas et al., 2018), each of which shares overlapping sequences with the N-terminus of VP1 as well as with the “contaminant peptide sequences” detected by proteomics (Supplement 7A). Another feature of these two proteins is that they closely run with ORF4 bands on SDS-PAGE. These two proteins may also contribute to the detection of the “contaminant peptides” in proteomics if and only if, they interact with ORF4 protein. Thus, we believe that “the contaminant peptides” originate from such known and unknown proteins which have overlapping sequences with the N-terminus of VP1 and from those that have interacting domains with ORF4 and VP1 (Supplement 7A), but not from the contamination of the full-length VP1 originating from the VP1 bands (Fig. 10A).
To further confirm that the detection of ORF4 in bands 3 and 4 is not simply due to contribution by the contamination of VP1, we carried out additional proteomics studies by analyzing the full-length VP1 bands (bands 5 and 6) (Fig. 10A, Supplement 10). We compared the total intensity of peptides belonging to the VP1 N-terminus (aa 1–181) with those obtained for the C-terminus (aa 182–354). This comparison was made for VP1 and ORF4 bands as shown in Supplement 7B. The results showed that the total intensity of the N-terminus peptides for full-length VP1 bands (bands 5 and 6) was 2-fold higher than that of the C-terminus peptides (Supplement 7B, panel on the right). However, when a similar analysis was made for the ORF4 bands (bands 3 and 4), we observed an inverse trend in that the total intensity of the C-terminus peptides was approximately 20-fold higher than that of N-terminus peptides (Supplement 7B, panel on the left). These results strongly support the idea that the detection of ORF4 in ORF4 bands is, in fact, due to its own expression during the viral infection cycle, not merely from the contamination of the full-length VP1. In other words, if VP1 protein itself contaminated ORF4 bands, we would expect to obtain a similar ratio with respect to the peptide intensity analysis between ORF4 and VP1 bands.
An important novelty in our current findings is that ORF4 protein specifically targets and co-localizes with one of the remarkable macromolecular assemblies in the nucleus, called PML-NBs; and induces their reorganization (Fig. 4 and 5). Although the significance of such targeting is yet to be further investigated for the JCV life cycle, PML-NBs were previously shown to play critical roles in initiation of both intrinsic and interferon-mediated antiviral responses against DNA and RNA viral infections (Regad and Chelbi-Alix, 2001; Regad et al., 2001; Stepp et al., 2013). In addition to their antiviral response, PML-NBs were also reported to be involved in apoptosis, DNA damage response, transcription, chromatin remodeling and oncogenesis (Bernardi and Pandolfi, 2007; Everett and Chelbi-Alix, 2007). The induction of the reorganization of the PML-NBs during the polyomavirus infections was previously reported at least for BKV (Jiang et al., 2011) and yet, to date, no specific or a group of polyomavirus proteins directly involved in such reorganization was reported. Our data clearly demonstrate that ORF4 protein of JCV is the only viral protein responsible for the direct targeting and reorganization of PML-NBs (Fig. 4 and 5), because the expression of several other JCV proteins including agnoprotein, large T antigen, small T antigen, ORF1, ORF2 and ORF3 did not show such an effect (Supplement 1 and 2).
There are a number of viral proteins that were reported to target PML-NBs with the aim of reducing or eliminating the antiviral effect of these nuclear structures [see for review (Scherer and Stamminger, 2016)]. One of the well-studied examples of these proteins is the HSV-1 immediate early ICP0 protein, which acts as a SUMO-targeted ubiquitin ligase and leads to the proteasome-dependent degradation of sumoylated PML component of PML-NBs to prevent the PML-mediated silencing of the HSV-1 gene expression (Maul et al., 1993). Another prominent example of such a viral protein is the immediate early protein of the human cytomegalovirus (hCMV IE1) which prevents the assembly of PML complexes by an alternative pathway through the inhibition of the sumoylation of PML protein itself (Lee et al., 2004). There are numerous additional examples from the literature indicating that viruses use various alternative strategies to antagonize the antiviral effects of PML-NBs. For example, the tegument protein, pp71 produced by CMV displaces the chromatin-associated factor, ATRX from PML and leads to its degradation in a proteasome-dependent manner (Tavalai and Stamminger, 2009). Several other subfamily members of gamma-herpesviruses also encode viral proteins that target PML-NBs to either modify their functions or lead to their degradation. For instance, murine gamma-herpes virus 68 protein (MHV-68) induces the proteasomal degradation of PML (Ling et al., 2008). In contrast, Epstein-Barr virus (EBV) BNRF1 protein prevents the interaction of hDaxx with ATRX (Tsai et al., 2011) and the herpesvirus saimiri ORF3 protein initiates the degradation of Sp100 (Full et al., 2012). Another viral protein termed, tegument protein ORF75, encoded by Kaposi-sarcoma associated herpesvirus leads to the degradation of ATRX (Full et al., 2014). Adenovirus E4-ORF3 protein, which forms linear and oligomeric structures inhibits the function of PML-NBs through the disruption of their formation (Ou et al., 2012). It is important to note that not all the viral infections lead to the degradation of the PML-NBs (Jiang et al., 2011; Jul-Larsen et al., 2004; Tang et al., 2000). The viral infection by the polyomavirus family, including JCV, BKV, SV40 and mouse polyomavirus do seem to follow this pathway (Jiang et al., 2011; Jul-Larsen et al., 2004; Tang et al., 2000). Even the elimination of PML protein by different strategies including siRNA silencing (Jul-Larsen et al., 2004) and short hairpin RNA (shRNA) (Gasparovic et al., 2009) did not have a significant effect on either BKV (Jul-Larsen et al., 2004) or JCV (Gasparovic et al., 2009) replication, indicating that PML complexes are not directly involved in viral DNA replication of polyomaviruses. However, Shishido-Hara et al., suggested a role for PML-NBs during the viral infection cycle. That is, they can serve as a scaffold for the virus assembly (Shishido-Hara et al., 2008; Shishido-Hara et al., 2004). Our current finding also demonstrates that JCV ORF4 protein induce not only the reorganization of PML but also that of two other permanent members of PML-NBs including hDaxx and ATRX (Fig. 5A). In contrast, the level of another permanent member of PML-NBs, Sp100, significantly decreased (Fig. 5A) suggesting that ORF4 protein appears to differentially regulate the stability of the PML-NB members.
Sequence alignment and splice prediction studies also revealed that, like JCV, both BKV and SV40 genomes could also encode an ORF4-like protein as a result of alternative splicing of their late transcripts, because both viruses contain potential splice donor/acceptor sites in their late coding regions to create an ORF4-like protein. To further support this assumption, we carried out a partial characterization of the putative BKV ORF4-like protein by expression studies, where we observed not only a strong co-localization of BKV-ORF4 with PML-NBs but also their reorganization as observed for JCV ORF4 (Supplement 3). However, the targeting of PML-NBs by BKV ORF4 protein was not as strong as the one that was observed for JCV ORF4 protein (Fig. 3). Instead, in addition to targeting PML-NBs in the nucleus, BKV ORF4 protein also showed a cytoplasmic distribution pattern. Such behavior may result from the fact that the NLS sequence of BKV ORF4 protein (RLRKRSVKNP) contains a slightly different amino acid composition than those of JCV ORF4 NLS (QLRKRRVKNP) (Fig. 7). These differences appear to influence the differential distribution pattern of both proteins in cells.
In this study, we also discovered that ORF4 contains a novel NLS motif within its coding sequence, which is also shared by JCV VP1 (Fig. 6). This NLS sequence plays a primary role in the nuclear localization of both ORF4 and VP1 (Fig. 7B, and Supplements 4 and 5). Specific mutations made within this sequence disrupted the nuclear localization of both proteins (Fig. 7B, and Supplement 5), supporting the idea that this newly identified NLS motif could be designated as the primary NLS sequence of both VP1 and ORF4 proteins. The strength of this new NLS was further tested by linking it to a heterologous protein. It was shown to be sufficient to mediate the transport a green fluorescent protein, GFP (a heterologous protein) into the nucleus as strongly as a well-described NLS sequence of SV40 large T antigen, which was used as a positive control in this assay (data not shown). It is also important to note here that the previously described NLS sequence of JCV VP1 (MAPT K5R6K7 GEK8K9D) (Fig. 6) was always considered as a weak NLS compared to those described for BKV and SV40 VP1 (MADTKRKGSCPGAAPKKPKE) (Fig. 6) (Ishii et al., 1996; Saribas et al., 2018; Shishido-Hara et al., 2000; Wychowski et al., 1986). Indeed, the sequence alignment among the VP1 proteins of JCV, BKV and SV40 supports this conclusion (Fig. 6). There are clear differences between JCV VP1 NLS and with those of BKV and SV40, which could explain the reasons why it is considered such a weak NLS sequence.
The functional importance of why ORF4 protein targets PML-NBs is currently unknown. However, we partially addressed this fundamental question by mutating ORF4 sequence and thus preventing its expression at the protein level during the viral life cycle. Note that, this mutation leaves the alternative splicing capacity of ORF4 intact at the RNA level. We have made a conservative amino acid substitution at the translational initiation codon of ORF4 (VP1 M182I) and the effect of this mutation on viral DNA replication was then assayed. Met substitution with Ile exhibited a modest negative effect on both first and second round of the viral replication cycle and suggested that the ORF4 protein has little effect on viral replication cycle in in vitro replication assay conditions. We believe that ORF4 protein would have more significant impact on the JCV replication in in vivo patient infection cases, perhaps by modulating the antiviral responses of PML-NBs against JCV infections in vivo. This would then lead to a more rapid viral spread and brain destruction in the infected individuals. The mechanism by which ORF4 mediates the reorganization of PML-NBs is currently unknown. One of the possible mechanisms in this respect is that ORF4 promotes the sumoylation of the major components of the PML-NBs, including PML isoforms, hDaxx, ATRX, because it is known that the sumoylation is a prerequisite for the assembly of PML-NBs. Note that ORF4 protein itself also contains a potential sumoylation site at Lys125 (VK125NP) with high probability by predictions suggesting that it would also be sumoylated prior to the targeting PML-NBs. Another possibility is that ORF4 promotes fusion of the scattered individual PML-NBs so that their apparent concentration is increased. Further studies are required to differentiate between such possibilities. Implications of the PML-NB targeting by JCV ORF4 is currently unknown. However, it is reasonable to suggest that JCV uses ORF4 protein to reduce or eliminate the “intrinsic and innate immunity” mediated by PML-NBs by directly targeting these protein complexes and inhibiting their antiviral responses. PML-NBs are known to restrict the viral gene expression through either “epigenetic silencing” and/or physical entrapping the viral particles within the NBs [see for review (Scherer and Stamminger, 2016). In addition, there are a number of studies describing a direct and positive regulation of innate immune signaling at the level of IFN induction and cytokine production (Chen et al., 2015; Kim and Ahn, 2015; Scherer et al., 2016; Ulbricht et al., 2012) by PML-NBs. Type I interferons (IFN-α and IFN-β) and type II interferon (IFN-γ) are upregulated by PML-NBs. These cytokines then stimulate “interferon-stimulated genes” (ISGs) in order to restrict viral propagation (Chen et al., 2015; Kim and Ahn, 2015; Scherer et al., 2016; Ulbricht et al., 2012). However, the molecular mechanisms by which PML-NBs stimulate the IFN pathways are not fully understood. Some reports indicate that PML isoforms play a critical role in this process. For instance, PML II isoform appears to bind and recruit the MHC class II trans-activator CIITA to PML-NBs thereby prolongs its activation (Ulbricht et al., 2012). PML II protein also binds to STAT1 and interferon regulatory factor 3 (IRF3) and promotes their recruitment onto the ISG and IFN-β promoters respectively (Chen et al., 2015). PML IV protein was also shown to enhance IRF3 activity by preventing the Pin1-mediated IRF3 degradation during vesicular stomatitis virus (VSV) infection (Chen et al., 2015). PML-NBs were also shown to modulate the expression of a number of cytokines including interleukin 1β (IL-1β) and IL-6 to regulate the acute inflammatory immune responses (Lo et al., 2013; Lunardi et al., 2011). In summary, PML NBs appear to not only act as powerful intrinsic repressors to silence viral gene expression but also serve as co-regulators of IFNs and interferon-stimulated genes (ISGs) to regulate the innate immune response against viral infections. Then, the next question is whether or not ORF4 plays a role as an antagonist during the JCV infection to overcome the intrinsic and IFN-mediated antiviral activity imposed by PML-NBs. If it does, what is the mechanism? This is another fundamental question that requires further investigation. Polyomaviruses contain a small DNA genome and are known to encode a limited number of structural and regulatory proteins. However, a more recent detailed analysis of some of their splicing patterns revealed that they utilize cis- and trans-alternative splicing mechanisms to diversify their genomic coding capacity (Frisque et al., 1984; Saribas et al., 2018).
Some of these alternative splicing variants are associated with the generation of highly interesting ORFs with the capacity of producing novel viral proteins, some of which appear to play important regulatory roles for the viral life cycle. A great detail about their identity and functions has been recently reported by Saribas et al [see for review (Saribas et al., 2019)]. Taken together, despite having a small genome size, polyomaviruses generate a number of novel open reading frames as a result of alternative splicing and produce unexpected but functionally important proteins in order to successfully complete their replication cycle. Further studies are required to better understand the functional importance of these novel proteins in order to design effective strategies against polyomavirus-induced diseases, including progressive multifocal leukoencephalopathy.
CONCLUSION
The human neurotropic polyomavirus, JCV, causes a fatal disease of the central nervous system, known as progressive multifocal leukoencephalopathy. Its genome is known to encode a limited number of regulatory and structural proteins. However, recent studies demonstrate that its genome in fact generate additional open reading frames (ORFs) as a result of alternative splicing, some of which indeed encode additional regulatory proteins. We have previously reported the generation of such novel proteins including ORF1 and ORF2 (Saribas et al., 2018). Here, we also report the discovery and partial characterization of three additional ORF-encoded proteins, ORF3, ORF4 and ORF5. In this study, we demonstrated for the first time that the ORF4 protein is the only JCV protein that specifically targets PML-NBs and induces their reorganization. PML-NBs are known to participate in induction of innate immunity including IFN production. Our novel findings suggest important regulatory roles for the ORF4 protein during the JCV replication cycle, perhaps by regulating the antiviral response of PML-NBs against JCV infections, and thus promoting the rate of infection in progressive multifocal leukoencephalopathy patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Research Highlights:
JC virus is the causative agent of a fatal brain disease, progressive multifocal leukoencephalopathy.
ORF4 protein of JCV is the only viral protein that causes the reorganization of PML-NBs.
ACKNOWLEDGEMENTS
This work was made possible by a grant awarded to M. Safak. [(RO1NS09049) by the NIH. Primary human fetal astrocytes (PHFA) were obtained from the Temple University Comprehensive NeuroAIDS Center. progressive multifocal leukoencephalopathy and non-progressive multifocal leukoencephalopathy patient brain tissue samples were provided by the National NeuroAIDS Tissue Consortium (NNTC, www.nntc.org). We specifically thank Dr. Susan Morgello (Manhattan HIV Brain Bank) for helping us to obtain the brain tissue samples from NNTC. We also thank Dr. Krzysztof Reiss, Louisiana State University, for providing several antibodies to detect several nuclear protein complexes including PML-NBs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
Authors declare no conflict of interest.
REFERENCES
- Ascoli CA, Maul GG, 1991. Identification of a Novel Nuclear Domain. Journal of Cell Biology 112, 785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KA, Florin L, Sapp C, Sapp M, 2003. Dissection of human papillomavirus type 33 L2 domains involved in nuclear domains (ND) 10 homing and reorganization. Virology 314, 161–167. [DOI] [PubMed] [Google Scholar]
- Berger JR, 1992. PML in AIDS. Neurology 42, 1845–1846. [DOI] [PubMed] [Google Scholar]
- Berger JR, 2000. Progressive Multifocal Leukoencephalopathy. Curr Treat Options Neurol 2, 361–368. [DOI] [PubMed] [Google Scholar]
- Berger JR, 2007. Progressive multifocal leukoencephalopathy. Curr Neurol Neurosci Rep 7, 461–469. [DOI] [PubMed] [Google Scholar]
- Berger JR, Koralnik IJ, 2005. Progressive multifocal leukoencephalopathy and natalizumab--unforeseen consequences. N Engl J Med 353, 414–416. [DOI] [PubMed] [Google Scholar]
- Bernardi R, Pandolfi PP, 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8, 1006–1016. [DOI] [PubMed] [Google Scholar]
- Bollag B, Kilpatrick LH, Tyagarajan SK, Tevethia MJ, Frisque RJ, 2006. JC virus T’135, T’136 and T’165 proteins interact with cellular p107 and p130 in vivo and influence viral transformation potential. J Neurovirol 12, 428–442. [DOI] [PubMed] [Google Scholar]
- Bund T, Spoden GA, Koynov K, Hellmann N, Boukhallouk F, Arnold P, Hinderberger D, Florin L, 2014. An L2 SUMO interacting motif is important for PML localization and infection of human papillomavirus type 16. Cell Microbiol 16, 1179–1200. [DOI] [PubMed] [Google Scholar]
- Chen XS, Stehle T, Harrison SC, 1998. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. Embo J 17, 3233–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wright J, Meng X, Leppard KN, 2015. Promyelocytic Leukemia Protein Isoform II Promotes Transcription Factor Recruitment To Activate Interferon Beta and Interferon-Responsive Gene Expression. Mol Cell Biol 35, 1660–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coric P, Saribas AS, Abou-Gharbia M, Childers W, White MK, Bouaziz S, Safak M, 2014. Nuclear magnetic resonance structure revealed that the human polyomavirus JC virus agnoprotein contains an α-helix encompassing the Leu/Ile/Phe-rich domain. J Virol 88, 6556–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M, 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cremer T, Cremer C, 2001. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet 2, 292–301. [DOI] [PubMed] [Google Scholar]
- Day PM, Roden RB, Lowy DR, Schiller JT, 1998. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J Virol 72, 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de The H, Chomienne C, Lanotte M, Degos L, Dejean A, 1990. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 347, 558–561. [DOI] [PubMed] [Google Scholar]
- de The H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A, 1991. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell 66, 675–684. [DOI] [PubMed] [Google Scholar]
- Del Valle L, White MK, Enam S, Pina Oviedo S, Bromer MQ, Thomas RM, Parkman HP, Khalili K, 2005. Detection of JC virus DNA sequences and expression of viral T antigen and agnoprotein in esophageal carcinoma. Cancer 103, 516–527. [DOI] [PubMed] [Google Scholar]
- Erickson KD, Bouchet-Marquis C, Heiser K, Szomolanyi-Tsuda E, Mishra R, Lamothe B, Hoenger A, Garcea RL, 2012. Virion assembly factories in the nucleus of polyomavirus-infected cells. PLoS Pathog 8, e1002630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Chelbi-Alix MK, 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89, 819–830. [DOI] [PubMed] [Google Scholar]
- Florin L, Schafer F, Sotlar K, Streeck RE, Sapp M, 2002. Reorganization of nuclear domain 10 induced by papillomavirus capsid protein l2. Virology 295, 97–107. [DOI] [PubMed] [Google Scholar]
- Frisque RJ, Bream GL, Cannella MT, 1984. Human polyomavirus JC virus genome. J Virol 51, 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Full F, Jungnickl D, Reuter N, Bogner E, Brulois K, Scholz B, Sturzl M, Myoung J, Jung JU, Stamminger T, Ensser A, 2014. Kaposi’s sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLoS Pathog 10, e1003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Full F, Reuter N, Zielke K, Stamminger T, Ensser A, 2012. Herpesvirus saimiri antagonizes nuclear domain 10-instituted intrinsic immunity via an ORF3-mediated selective degradation of cellular protein Sp100. J Virol 86, 3541–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparovic ML, Maginnis MS, O’Hara BA, Dugan AS, Atwood WJ, 2009. Modulation of PML protein expression regulates JCV infection. Virology 390, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu KS, Kao HY, 2018. PML: Regulation and multifaceted function beyond tumor suppression. Cell Biosci 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Minami N, Chen EY, Medina AL, Chico MM, Kasamatsu H, 1996. Analysis of a nuclear localization signal of simian virus 40 major capsid protein Vp1. J Virol 70, 1317–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Entezami P, Gamez M, Stamminger T, Imperiale MJ, 2011. Functional Reorganization of Promyelocytic Leukemia Nuclear Bodies during BK Virus Infection. MBio 2 [DOI] [PMC free article] [PubMed]
- Jul-Larsen A, Visted T, Karlsen BO, Rinaldo CH, Bjerkvig R, Lonning PE, Boe SO, 2004. PML-nuclear bodies accumulate DNA in response to polyomavirus BK and simian virus 40 replication. Exp Cell Res 298, 58–73. [DOI] [PubMed] [Google Scholar]
- Kakizuka A, Miller WH Jr., Umesono K, Warrell RP Jr., Frankel SR, Murty VV, Dmitrovsky E, Evans RM, 1991. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 66, 663–674. [DOI] [PubMed] [Google Scholar]
- Kim YE, Ahn JH, 2015. Positive role of promyelocytic leukemia protein in type I interferon response and its regulation by human cytomegalovirus. PLoS Pathog 11, e1004785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt-DeMasters BK, Tyler KL, 2005. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med 353, 369–374. [DOI] [PubMed] [Google Scholar]
- Korman BD, Tyler KL, Korman NJ, 2009. Progressive multifocal leukoencephalopathy, efalizumab, and immunosuppression: a cautionary tale for dermatologists. Arch Dermatol 145, 937–942. [DOI] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V, de The H, 2018. PML nuclear bodies: from architecture to function. Curr Opin Cell Biol 52, 154–161. [DOI] [PubMed] [Google Scholar]
- Lamond AI, Sleeman JE, 2003. Nuclear substructure and dynamics. Curr Biol 13, R825–828. [DOI] [PubMed] [Google Scholar]
- Lamond AI, Spector DL, 2003. Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol 4, 605–612. [DOI] [PubMed] [Google Scholar]
- Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D, 2005. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med 353, 375–381. [DOI] [PubMed] [Google Scholar]
- Langer-Gould A, Steinman L, 2006. Progressive multifocal leukoencephalopathy and multiple sclerosis: lessons from natalizumab. Curr Neurol Neurosci Rep 6, 253–258. [DOI] [PubMed] [Google Scholar]
- Lee HR, Kim DJ, Lee JM, Choi CY, Ahn BY, Hayward GS, Ahn JH, 2004. Ability of the human cytomegalovirus IE1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. J Virol 78, 6527–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Andersen JS, Mann M, Lamond AI, 2003. Bioinformatic analysis of the nucleolus. Biochem J 376, 553–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AK, Lamond AI, 2003. The dynamics of the nucleolus. Crit Rev Eukaryot Gene Expr 13, 39–54. [DOI] [PubMed] [Google Scholar]
- Ling PD, Tan J, Sewatanon J, Peng R, 2008. Murine gammaherpesvirus 68 open reading frame 75c tegument protein induces the degradation of PML and is essential for production of infectious virus. J Virol 82, 8000–8012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo YH, Huang YW, Wu YH, Tsai CS, Lin YC, Mo ST, Kuo WC, Chuang YT, Jiang ST, Shih HM, Lai MZ, 2013. Selective inhibition of the NLRP3 inflammasome by targeting to promyelocytic leukemia protein in mouse and human. Blood 121, 3185–3194. [DOI] [PubMed] [Google Scholar]
- Lunardi A, Gaboli M, Giorgio M, Rivi R, Bygrave A, Antoniou M, Drabek D, Dzierzak E, Fagioli M, Salmena L, Botto M, Cordon-Cardo C, Luzzatto L, Pelicci PG, Grosveld F, Pandolfi PP, 2011. A Role for PML in Innate Immunity. Genes Cancer 2, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao YS, Zhang B, Spector DL, 2011. Biogenesis and function of nuclear bodies. Trends Genet 27, 295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul GG, Guldner HH, Spivack JG, 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J Gen Virol 74 ( Pt 12), 2679–2690. [DOI] [PubMed] [Google Scholar]
- Meinecke I, Cinski A, Baier A, Peters MA, Dankbar B, Wille A, Drynda A, Mendoza H, Gay RE, Hay RT, Ink B, Gay S, Pap T, 2007. Modification of nuclear PML protein by SUMO-1 regulates Fas-induced apoptosis in rheumatoid arthritis synovial fibroblasts. Proc Natl Acad Sci U S A 104, 5073–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli T, 2007. Beyond the sequence: Cellular organization of genome function. Cell 128, 787–800. [DOI] [PubMed] [Google Scholar]
- Misteli T, 2010. Higher-order genome organization in human disease. Cold Spring Harb Perspect Biol 2, a000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou HD, Kwiatkowski W, Deerinck TJ, Noske A, Blain KY, Land HS, Soria C, Powers CJ, May AP, Shu X, Tsien RY, Fitzpatrick JA, Long JA, Ellisman MH, Choe S, O’Shea CC, 2012. A Structural Basis for the Assembly and Functions of a Viral Polymer that Inactivates Multiple Tumor Suppressors. Cell 151, 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regad T, Chelbi-Alix MK, 2001. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 20, 7274–7286. [DOI] [PubMed] [Google Scholar]
- Regad T, Saib A, Lallemand-Breitenbach V, Pandolfi PP, de The H, Chelbi-Alix MK, 2001. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. Embo J 20, 3495–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas A, Abou-Gharbia M, Childers W, Sariyer IK, White MK, Safak M, 2013. Essential roles of Leu/Ile/Phe-rich domain of JC virus agnoprotein in dimer/oligomer formation, protein stability and splicing of viral transcripts. Virology 443, 161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Coric P, Bouaziz S, Safak M, 2019. Expression of novel proteins by polyomaviruses and recent advances in the structural and functional features of agnoprotein of JC virus, BK virus, and simian virus 40. J Cell Physiol 234, 8295–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, DeVoto J, Golla A, Wollebo HS, White MK, Safak M, 2018. Discovery and characterization of novel trans-spliced products of human polyoma JC virus late transcripts from PML patients. J Cell Physiol 233, 4137–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Mun S, Johnson J, El-Hajmoussa M, White MK, Safak M, 2014. Human polyoma JC virus minor capsid proteins, VP2 and VP3, enhance large T antigen binding to the origin of viral DNA replication: evidence for their involvement in regulation of the viral DNA replication. Virology 449, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Akan I, Palermo V, Gordon J, Khalili K, Safak M, 2006. Phosphorylation mutants of JC virus agnoprotein are unable to sustain the viral infection cycle. J Virol 80, 3893–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Saribas AS, White MK, Safak M, 2011. Infection by agnoprotein-negative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol J 8, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saylor D, Venkatesan A, 2016. Progressive Multifocal Leukoencephalopathy in HIV-Uninfected Individuals. Current infectious disease reports 18, 33. [DOI] [PubMed] [Google Scholar]
- Scherer M, Otto V, Stump JD, Klingl S, Muller R, Reuter N, Muller YA, Sticht H, Stamminger T, 2016. Characterization of Recombinant Human Cytomegaloviruses Encoding IE1 Mutants L174P and 1–382 Reveals that Viral Targeting of PML Bodies Perturbs both Intrinsic and Innate Immune Responses. J Virol 90, 1190–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer M, Stamminger T, 2016. Emerging Role of PML Nuclear Bodies in Innate Immune Signaling. J Virol 90, 5850–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido-Hara Y, Hara Y, Larson T, Yasui K, Nagashima K, Stoner GL, 2000. Analysis of capsid formation of human polyomavirus JC (Tokyo-1 strain) by a eukaryotic expression system: splicing of late RNAs, translation and nuclear transport of major capsid protein VP1, and capsid assembly. J Virol 74, 1840–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido-Hara Y, Higuchi K, Ohara S, Duyckaerts C, Hauw JJ, Uchihara T, 2008. Promyelocytic leukemia nuclear bodies provide a scaffold for human polyomavirus JC replication and are disrupted after development of viral inclusions in progressive multifocal leukoencephalopathy. J Neuropathol Exp Neurol 67, 299–308. [DOI] [PubMed] [Google Scholar]
- Shishido-Hara Y, Ichinose S, Higuchi K, Hara Y, Yasui K, 2004. Major and minor capsid proteins of human polyomavirus JC cooperatively accumulate to nuclear domain 10 for assembly into virions. J Virol 78, 9890–9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishido-Hara Y, Ichinose S, Uchihara T, 2012. JC virus intranuclear inclusions associated with PML-NBs: analysis by electron microscopy and structured illumination microscopy. Am J Pathol 180, 1095–1106. [DOI] [PubMed] [Google Scholar]
- Shishido-Hara Y, Yazawa T, Nagane M, Higuchi K, Abe-Suzuki S, Kurata M, Kitagawa M, Kamma H, Uchihara T, 2014. JC virus inclusions in progressive multifocal leukoencephalopathy: scaffolding promyelocytic leukemia nuclear bodies grow with cell cycle transition through an S-to-G2-like state in enlarging oligodendrocyte nuclei. J Neuropathol Exp Neurol 73, 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan E, Orr A, Everett RD, 2016. MORC3, a Component of PML Nuclear Bodies, Has a Role in Restricting Herpes Simplex Virus 1 and Human Cytomegalovirus. J Virol 90, 8621–8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle T, Harrison SC, 1997. High-resolution structure of a polyomavirus VP1-oligosaccharide complex: implications for assembly and receptor binding. Embo J 16, 5139–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepp WH, Meyers JM, McBride AA, 2013. Sp100 provides intrinsic immunity against human papillomavirus infection. MBio 4, e00845–00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Semba S, Sunden Y, Orba Y, Kobayashi S, Nagashima K, Kimura T, Hasegawa H, Sawa H, 2012. Role of JC virus agnoprotein in virion formation. Microbiol Immunol 56, 639–646. [DOI] [PubMed] [Google Scholar]
- Tang Q, Bell P, Tegtmeyer P, Maul GG, 2000. Replication but not transcription of simian virus 40 DNA is dependent on nuclear domain 10. J Virol 74, 9694–9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalai N, Stamminger T, 2009. Interplay between Herpesvirus Infection and Host Defense by PML Nuclear Bodies. Viruses 1, 1240–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K, Thikmyanova N, Wojcechowskyj JA, Delecluse HJ, Lieberman PM, 2011. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog 7, e1002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbricht T, Alzrigat M, Horch A, Reuter N, von Mikecz A, Steimle V, Schmitt E, Kramer OH, Stamminger T, Hemmerich P, 2012. PML promotes MHC class II gene expression by stabilizing the class II transactivator. J Cell Biol 199, 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Benhenda S, Wu H, Lallemand-Breitenbach V, Zhen T, Jollivet F, Peres L, Li Y, Chen SJ, Chen Z, de The H, Meng G, 2018. Publisher Correction: RING tetramerization is required for nuclear body biogenesis and PML sumoylation. Nat Commun 9, 1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wychowski C, Benichou D, Girard M, 1986. A domain of SV40 capsid polypeptide VP1 that specifies migration into the cell nucleus. Embo J 5, 2569–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP, 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 95, 2748–2752. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.