

Abstract
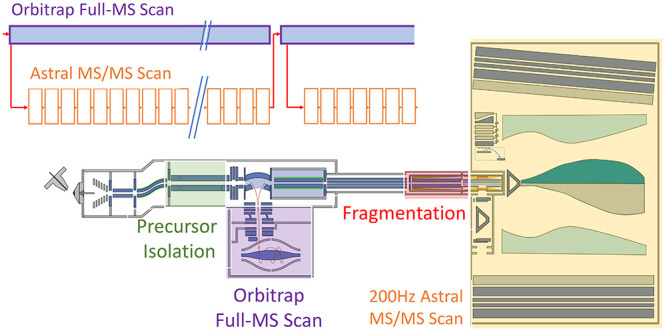
The growing trend toward high-throughput proteomics demands rapid liquid chromatography–mass spectrometry (LC–MS) cycles that limit the available time to gather the large numbers of MS/MS fragmentation spectra required for identification. Orbitrap analyzers scale performance with acquisition time and necessarily sacrifice sensitivity and resolving power to deliver higher acquisition rates. We developed a new mass spectrometer that combines a mass-resolving quadrupole, the Orbitrap, and the novel Asymmetric Track Lossless (Astral) analyzer. The new hybrid instrument enables faster acquisition of high-resolution accurate mass (HRAM) MS/MS spectra compared with state-of-the-art mass spectrometers. Accordingly, new proteomics methods were developed that leverage the strengths of each HRAM analyzer, whereby the Orbitrap analyzer performs full scans with a high dynamic range and resolution, synchronized with the Astral analyzer’s acquisition of fast and sensitive HRAM MS/MS scans. Substantial improvements are demonstrated over previous methods using current state-of-the-art mass spectrometers
Bottom-up proteomics workflows profile complex mixtures of proteins through the analysis of enzymatically digested peptides delivered to a mass spectrometer via chromatographic separation.1 In a typical workflow, peptide precursor ions are optionally detected within a panoramic MS scan, then isolated, fragmented, and the resulting MS/MS spectra analyzed by bioinformatic tools to identify parent peptides and infer predigest proteins.2−5 A typical sample may easily contain hundreds of thousands of peptide features,6 with the depth of coverage defined by the mass spectrometer’s ability to generate sufficient quality spectra across a high dynamic range in the available time.
For MS/MS spectra, sensitivity has historically been the most pressing challenge due to the extremely low abundance of many peptide features, caused by the enormous range of protein abundances.7,8 Thermo Scientific Orbitrap mass spectrometers evolved as important proteomics instruments due to their relatively high transmission and accurate precursor mass measurements with a high dynamic range.9−12 Pairing of an Orbitrap analyzer with a more sensitive, albeit lower resolving power, linear ion trap with single ion detection has long been an effective strategy, allowing the Orbitrap analyzer to carry out highly resolved full mass scans while the linear ion trap performed isolation and sensitive MS/MS acquisition.10 An important evolution of this strategy was the Tribrid instrument architecture combining an Orbitrap analyzer for precursor detection, a quadrupole mass filter for precursor isolation, and a dual-pressure linear ion trap for sensitive MS/MS.9 A parallelized acquisition method allows each analyzer to carry out fit-for-purpose acquisition and operate efficiently across the entire instrument.
For ultrahigh-throughput proteomics with chromatographic gradients compressed down to 5 or 8 min, large numbers of sample ions pass through the instrument within a short time frame, and the requisite number of MS/MS spectra to identify peptides need to be generated much more quickly. An analyzer acquiring 25 spectra per second (25 Hz) to target all detectable peptides in 100 min, as had been estimated,6 would have to operate above 300 Hz to manage the same MS/MS coverage in 8 min. Although there are ways to mitigate this fundamental burden, such as via parallel identification of multiple coisolated precursor ions within an individual MS/MS spectrum,13,14 there are multiple technological innovations required to reach such a challenging acquisition rate with sufficient sensitivity and dynamic range.
Orbitrap MS/MS acquisition has been limited to above 40 Hz,15 and this comes with a trade-off of sensitivity and resolution due to shorter ion detection transient. Thus, Orbitrap MS/MS methods must balance the repetition rate with resolution and sensitivity.16 Moderately faster acquisition rates of 70 Hz MS/MS are fundamentally possible with Orbitrap analyzers and demonstrate some benefit for high-throughput workflows.17
Time-of-flight (ToF) analyzers on the other hand have typically very short ion measurement periods, tens of microseconds to a few milliseconds for ions to traverse the flight path, depending on size and design, and may routinely generate spectra at >100 Hz.18,19 Such analyzers have historically suffered from poor sensitivity, due largely to low duty cycle and transmission losses within the orthogonal accelerators that introduce ions into the analyzer20 and on multiple grids along the ion path. Some of these losses have been reduced with ion trapping followed by mass-dependent extraction into an orthogonal accelerator21 or by replacement of the device with a direct-extraction ion trap22,23 in a similar manner to how a curved linear trap (C-trap) is used to inject ions into Orbitrap analyzers.24 However, severe temporal broadening of ion packets and a strong dependence on the numbers of ions combined with complex electronics and detector saturation precluded the widespread utilization of this linear trap approach with continuous ion beams.
Multireflection or multiturn time-of-flight analyzer designs with folded and grid-free flight paths offer far higher resolving power and consequently higher statistical mass accuracy than conventional linear or single-reflection designs.25 Numerous “MR-ToF” or “Open Electrostatic Trap” designs of this type have been introduced over decades.25−27 In spite of the potential of such analyzers, they are yet to achieve widespread adoption for liquid chromatography/mass spectrometry (LC/MS) applications as they have lacked an effective way of interfacing to continuous ion sources.
This work aspires to resolve the limitations of previous approaches by developing a novel Astral mass analyzer, implemented in a hybrid mass spectrometer in combination with a quadrupole mass filter and an Orbitrap analyzer. The properties of the instrument are described in detail, and parallelized methods of operation are shown that employ Orbitrap acquisition for full scans and the Astral analyzer for MS/MS measurements to leverage the strengths of each analyzer. Experiments have been performed in accordance with standard proteomics workflows and compared against the results of current state-of-the-art instrumentation.
Experimental Methods
Chromatography and Samples
Instrument characterization was carried out with an infused Pierce Flexmix calibration solution, which includes caffeine, the MRFA peptide, and Ultramark. LC/MS applications were performed with Pierce Hela digest ranging in amount from 250 pg to 2 μg and a 3-proteome mixture of human, E. coli, and yeast digests mixed at different ratios.
Liquid chromatography was performed using a Vanquish Neo UHPLC system, operated in either a direct injection or trap-and-elute configuration. The sample was injected via an autosampler and separated on either an Easy-Spray PepMap Neo UHPLC 150 μm x 15 or 50 cm low load- or 110 cm μPACTM HPLC columns, respectively. The instrument was operated in Data-Independent Acquisition (DIA) mode; data were processed with Proteome Discoverer using the CHIMERYS search algorithm12 or Biognosys SpectronautTM 17.
Mass Spectrometry
Experiments were performed using a prototype Orbitrap Astral mass spectrometer, a schematic representation of which is shown in Figure 1. Electrosprayed ions are admitted into the first ∼4 mbar vacuum region through a heated steel transfer tube, captured by an ion funnel, and passed via quadrupole ion guides through a series of differentially pumped regions to the hyperbolic-rod quadrupole mass filter.28
Figure 1.
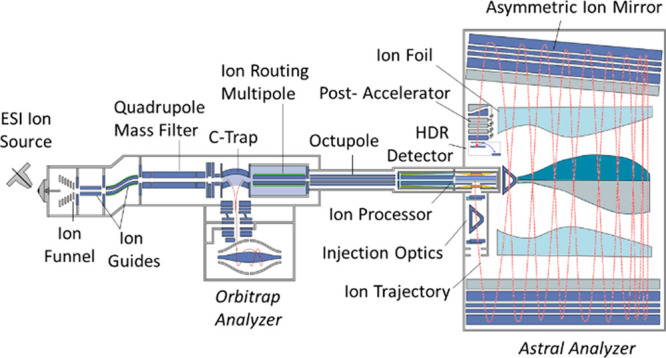
Schematic representation of the mass spectrometer.
For a full mass scan, a wide m/z range of ions passes through the mass filter into the C-Trap, with the admission controlled by a gate between them. In normal operation, the ions are not stored in the C-trap but pass further to the Ion Routing Multipole (IRM) for trapping within this gas-filled PCB-mounted quadrupole ion guide. Inside the IRM, excess ion kinetic energy is quenched in buffer gas (ca. 10–2 mbar nitrogen). A DC gradient along the IRM drives ions back to the C-Trap,28 where the ion packet is further collisionally cooled, compressed, and ejected into the Orbitrap analyzer.29−31
For Astral MS/MS spectra, a precursor isolation window is set by the quadrupole mass filter, either identified from the full mass scan (Data Dependent Acquisition, DDA) or predefined by the acquisition method (Data Independent Acquisition, DIA). Isolated ions pass through the C-Trap to the IRM. The IRM axial gradient is reversed to drive ions to the far end, where they are stopped by a trapping voltage on an exit aperture. Here, the ions are trapped and stored at the far end of the IRM for a set accumulation period, after which the trapping voltage is reduced to a transmitting level and the ions are released into the 350 mm-long octupole ion guide.
The octupole transmits ions to the ion processor, a dual-pressure linear quadrupole ion trap operated at 3.8 MHz RF and having the inscribed radius r0 = 2 mm. The first section of the ion processor is a high-pressure region fed with nitrogen by a PEEKSil capillary and operates at 10–2 mbar. Here, the ions are accelerated to a higher energy and undergo high-energy collisional dissociation (HCD).32 Wedge-shaped DC electrodes mounted between the RF electrodes create a DC gradient that drives the fragment ions to the far end of the high-pressure section, where they accumulate and thermalize. A rise of the DC offset of the high-pressure section pushes the ions to the following low-pressure section for subsequent orthogonal extraction to the Astral analyzer. The two sections are driven with phase-locked RF supplies, which minimize harmful fringe fields at the interface and allow ion transfer at moderate kinetic energy below about 1 eV per unit charge. The low-pressure section is separated by a gas conductance restriction which allows its operation with a pressure of 2–4 × 10–3 mbar, found to reach a good compromise between thermalization time and avoidance of unwanted collisions during the orthogonal extraction.
The low-pressure region of the ion processor contains one pair of RF rods that are split longitudinally, creating an equatorial space within which auxiliary DC electrodes are mounted, which defines an axial potential well to store and thermalize the ions in the middle of the low-pressure section in front of a slot for subsequent orthogonal ejection.33 The low-pressure region is lifted to 4 kV, RF is rapidly quenched, and a 500 V/mm DC gradient is applied across the trap to extract the ions through the slot into the Astral analyzer.
This accumulation process allows for a high utilization of the ion beam compared to the poor duty cycle and transmission losses typical of traditional orthogonal accelerators. Before an ion packet is ejected from the low-pressure section, the next ion packet is already prepared in the high-pressure section to ensure maximal instrument utilization.
The extracted ion packet is accelerated to 4 kV as it passes a grounded aperture before being shaped by a pair of lenses and an electrostatic prism comprising the Injection Optics. The tabletop-sized Astral analyzer is of a form previously described by Grinfeld and Makarov.26 It features a pair of elongated asymmetric ion mirrors, between which ions perform multiple oscillations while slowly drifting down the mirrors’ length due to the initial inclination of trajectories,.34−36 After the first reflection, the inclination angle of ion packets is adjusted by the second electrostatic prism to the optimal value of about two degrees.
The asymmetric ion mirrors are designed to be slightly converging toward each other, making the ion drift decelerate over the course of the first 12–13 oscillations toward the distant end of the mirrors. The ion foil electrodes are mounted between the mirrors, above and below the ion path, and are biased with a small tunable potential between 0 and −20 V. The precise shapes of the ion foil electrodes serve to compensate for the temporal aberration induced by the mirror asymmetry and improve the quality of the spatial focus at the detector, as well as to compensate for mechanical misalignments of the mirrors. The drift is eventually reversed by a returning electrostatic potential formed by mirror tilt as well as refraction on the ion foil, and over the following 12–13 oscillations, the ions drift back to the second electrostatic prism of the injection optics. While making a complete set of 24–26 oscillations over the >30m track, the ion packets are separated according to their mass-to-charge ratios and, being refocused spatially, arrive at a high dynamic range detector located at the proximal end next to the ion processor. In essence, this ion-optical solution makes ion motion isochronous both along and across the mirror system.
The combination of gridless design with spatial focusing enables a high transmission of ions throughout the analyzer. The asymmetric geometry of the analyzer as well as its high transmission are reflected in the Astral abbreviation, which stands for ASymmetric TRAck Lossless analyzer. To reduce collisional ion losses with residual gas, the main chamber of the analyzer is maintained at a pressure below 10–8 mbar by a 6-port split-flow turbopump (Pfeiffer, Asslar, Germany).
The ion arrival is detected by a novel custom-designed high dynamic range (HDR) detector (El-Mul Technologies Ltd.), which incorporates a conversion dynode, a magnet for focusing secondary electrons, a scintillator, a light guide, and a photomultiplier tube (PMT). A postaccelerator, implemented as a stack of slit-shaped apertures, is mounted in front of the detector to deliver an extra 10 kV acceleration voltage to the ions before hitting the conversion dynode; the total impinge energy is thus as high as 14 keV per charge, maximizing the probability of successful detection of ions in a wide mass range. Secondary electrons are converted to photons by the scintillator optically coupled to the PMT. Thanks to optical coupling, the PMT is hermetically sealed, which excludes contamination of dynodes by residual gases and extends its lifetime. The PMT output signal is split into two channels with a 10x difference in gain, which further enhances the dynamic range.
The repetition rate is limited by numerous factors, but most importantly, the accumulation time (fill time) for a required number of ions to be accumulated for analysis, the time taken by power supplies to dynamically adjust during scan execution, and the time required for ions to traverse the mass spectrometer. A complex instrument with multiple sequential stages would bring the total operation cycle to well above 10 ms. For this reason, each stage of operation has been highly parallelized, with multiple ion packets simultaneously being handled on different stages, which elevates the repetition rate above 200 Hz.
Figure 2 presents the operational sequence for an Astral analyzer MS/MS scan, indicating the time required for each stage. The time for the ion source, including the interface ion guides, varies depending on the difference in m/z between the targeted ion packet and its predecessor. For the small mass steps common in DIA acquisition, there is little difference in transmission before and after voltage adjustment. Larger mass jumps, as in DDA experiments, incur an extra penalty for ions to travel from the capillary to the quadrupole. The accumulation of ions in the IRM, rather than directly to the ion processor, is added to maximize allowed ion accumulation/fill time through parallelization with the ion processor’s operation. No nonparallelizable series of stages exceeds >4.5 ms in DIA, allowing a 200 Hz repetition rate.
Figure 2.
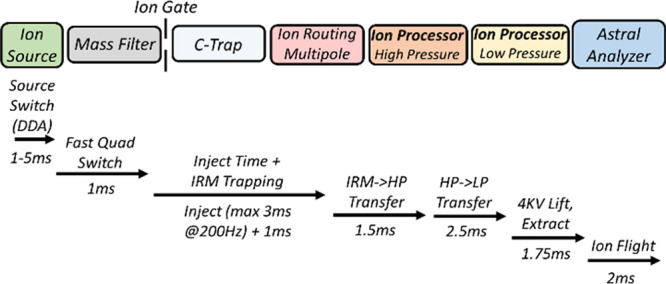
Timing sequence for 200 Hz parallelized ion transfer of ions from the source to Astral analyzer.
LC–MS Analysis Method
The hybrid acquisition cycle is drawn schematically in Figure 3. For both DIA and DDA acquisition modes, a full mass range of ions is first injected into the Orbitrap analyzer and then a series of MS/MS scans are performed. In this scheme, the Orbitrap and Astral analyzers operate simultaneously. Full mass scans are also performed in the Astral analyzer at regular intervals for automatic gain control (AGC37), so-called “fluxscans” to measure ion current and set appropriate accumulation times. The AGC target level was set to 100% for full mass scans, equivalent to approximately 50,000 charges for full mass scans and 10,000 charges for MS/MS scans (if within the 3 ms maximal injection time). Accumulation time for MS/MS scans varies from 0.03 to 3 ms but mostly reaches the upper limit. Orbitrap resolution was set to 240 K, equivalent to 2 Hz full-MS acquisition.
Figure 3.
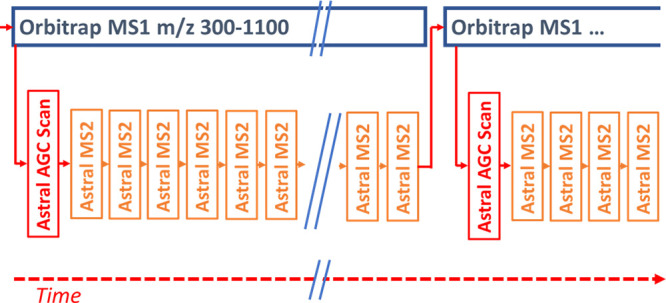
Representation of parallelized acquisition method, with Orbitrap full MS scans overlapped with Astral MS/MS scans.
Instrument Characterization
Various properties of the Astral analyzer were characterized with the Flexmix calibration solution. Figure 4 shows the resolution and mass deviation of averaged peaks across the 190–3000 m/z range, with 5000 and 50,000 total ions detected per shot. The number of ions was calibrated by measuring the signal response of single ions. Increased ion loads generate space charge effects within the ion processor, expanding the trapped ion cloud, while also causing additional expansion of extracted ion packets as they traverse the analyzer. Unsurprisingly, an average resolution was reduced from above 100 to ∼90 K at the higher ion load. More severe is the loss of resolution for individual very intense analyte species. The mass shift imposed at high ion load was also less than 1 ppm for all ions, with no applied correction to mass position for peak intensity.
Figure 4.
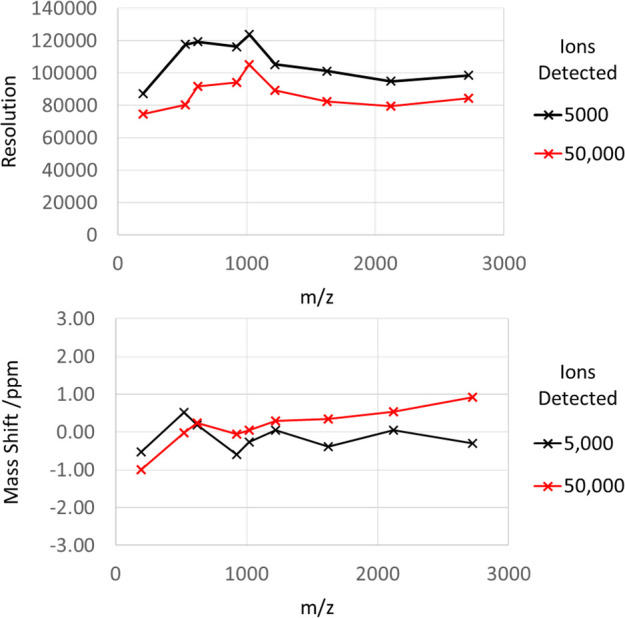
Resolution and m/z measurement shift of calibration mixture ions at 5000 and 50,000 ions detected per shot.
High transmission and space-charge tolerance allow rapid spectra acquisition without spectral averaging, and every scan forms an individual spectrum with a sufficient signal-to-noise ratio. However, the mass accuracy is determined in this case by the shot-to-shot jitter. The jitter was assessed by gathering single-shot peak data over 60 s and calculating the standard deviation σ of peak centroids. 3σ jitter was observed (Figure 5) to be below 1.5 ppm (σ < 0.5 ppm), with the exception of low-intensity peaks m/z 1022 and 1922, for which the statistical error dominated over the instrumental jitter.
Figure 5.
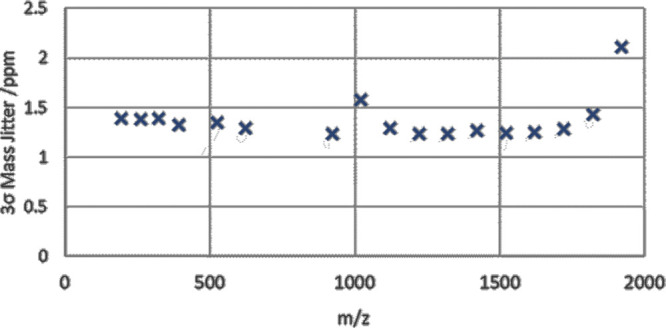
3-sigma level mass jitter for single shot repetitions.
Figure 6 shows recording of centroids of Ultramark 922 peaks over a 70 h period, where the 50× averaging was used to suppress shot-to-shot jitter. An uncorrected drift over a 2.5 ppm range was detected, mostly driven by changes of ambient temperature.
Figure 6.
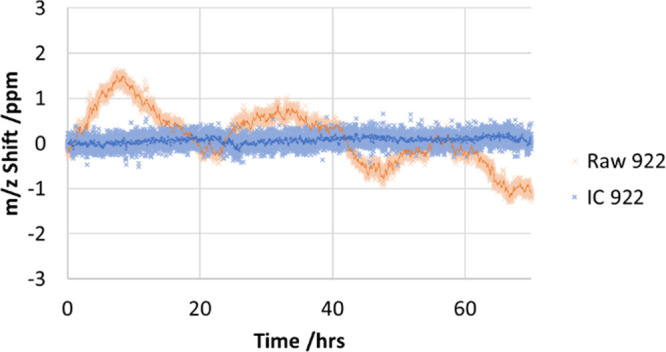
Mass drift of ultramark m/z 922 over 70 h, as uncorrected (raw) and with an intermittent internal calibrant (IC) correction.
To mitigate the mass measurement errors caused by drift of the analyzer mechanics or applied voltages, an intermittent internal calibration method was developed, whereby internal calibrant ions of fluoranthene +1 (m/z 202) are injected at regular intervals and provide a proportional calibration coefficient to measured ion flight times. A calibrant injection once every 10 s with a 10-point moving average was found to be sufficient for this purpose, although even less frequent injection such as every 5 min may also be used. While this has no benefit for control of jitter, as with conventional internal calibration applied to every mass spectrum, it instead has the advantage of having a negligible impact on the overall repetition rate. As Figure 6 shows, the use of an internal calibrant reduces the drift to 0.5 ppm (0.13 ppm standard deviation).
Combining the drift and jitter estimations, and with allowance for quality of calibration, implies that most peaks will be determined within 3 ppm mass accuracy.
Important considerations for fast LC–MS experiments are the Astral analyzer’s repetition rate and duty cycle. This latter property may be defined as the proportion of the scan interval over which the ion trap may spend accumulating ions from the source prior to pulsed extraction into the analyzer. A scan of ion accumulation time was performed, and the repetition rate was monitored and drawn graphically in Figure 7. Below 2.5 ms, the accumulation time has no impact on the repetition rate, but above this, the repetition rate falls precipitously as the accumulation time comes to dominate the operational cycle. At 200 Hz the maximum accumulation time was just over 3 ms, equating to a 60% duty cycle, although it may in the future be possible to increase this via preaccumulation of ions in the ion guide ahead of the quadrupole, as previously shown for the Orbitrap Exploris mass spectrometer.17 In a second experiment, the scan rate was recorded while the instrument was run in a DIA mode of operation, with a fixed 3 ms accumulation time. Here, the scan rate was recorded consistently above 200 Hz, as also shown in Figure 7. This is all very suitable for DIA experiments with small m/z transitions, however, precludes extra overheads for large transitions (DDA), that require an additional 2 ms to adjust the ion source.
Figure 7.
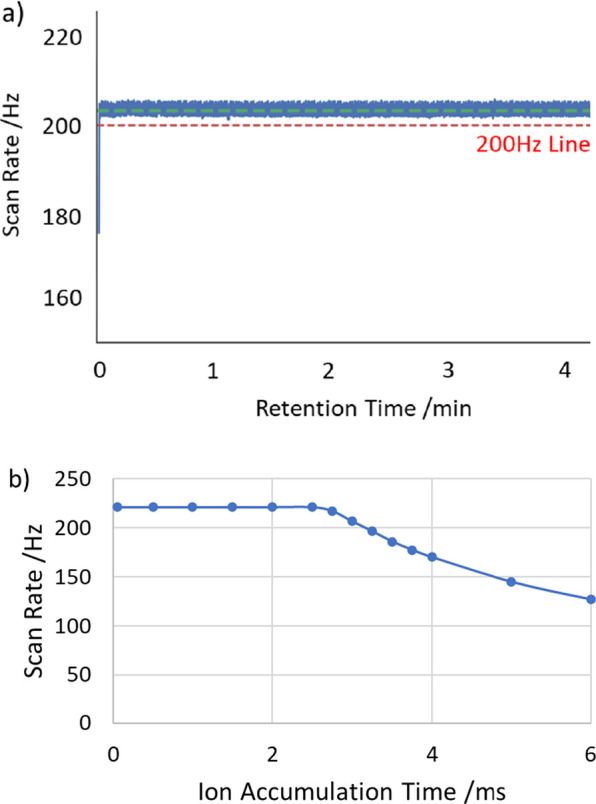
(a) Astral repetition rate with varying ion trap accumulation times, where acquisition drops below 200 Hz with >3 ms accumulation time. (b) Scan rate recorded while running the DIA method with fixed 3 ms ion accumulation time.
The sensitivity of the Astral analyzer was compared to that of the Orbitrap analyzer by comparison of ion number counts within sequential injections to each analyzer of differing m/z Flexmix ions. Orbitrap ion numbers were estimated from signal-to-noise (S/N) measurement of multiple ion peaks, compared to earlier S/N measurements of multiply charged single ions.38 Highly charged ions, such as intact proteins, produce intense single ion peaks that the Orbitrap analyzer is capable of detecting,39 allowing estimation of the number of charges at the noise threshold; approximately 2.6 for an Orbitrap Exploris MS running a 256 ms transient (resolution 120 K) with eFT signal processing.
Astral analyzer peak intensities were also converted to ion numbers by comparison with the measurement of single ions of MRFA at m/z 524 with regard to m/z dependence of secondary ion conversion.40 The results, shown in Figure 8, indicate substantially greater transmission of ions into the Astral analyzer, more than double at higher m/z.
Figure 8.
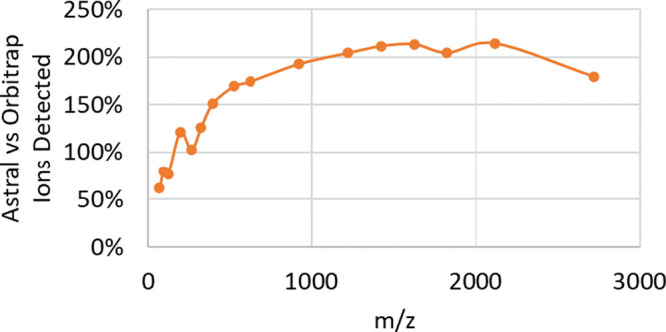
Relative number of ions detected in Astral vs Orbitrap analyzers.
Figure 9 reproduces the MS/MS spectrum of the MRFA (m/z = 524.26) peptide in both Astral and Orbitrap analyzers with 1000 ions detected. The fragment mass positions in the Astral spectrum matched that of the equivalent Orbitrap scan, but notably, the peak resolution was ∼100,000 except at m/z below ∼150, where resolution remained high but began to clearly drop off. An Orbitrap DDA or DIA method would typically run at resolution values of 7.5 or 15K at m/z 200. A major advantage of the Astral analyzer over the Orbitrap analyzer is the detection of single ions, rather than requiring >10 ions for a detectable peak in MS/MS acquisition. The advantage of the Astral spectrum is clear, with an order of magnitude higher signal-to-noise ratio and 132 peaks detected vs only 30 in the Orbitrap mass spectrum. For the identification of low-level analytes, this is expected to be highly advantageous and essential when cycling through precursors rapidly with limited ion accumulation time.
Figure 9.
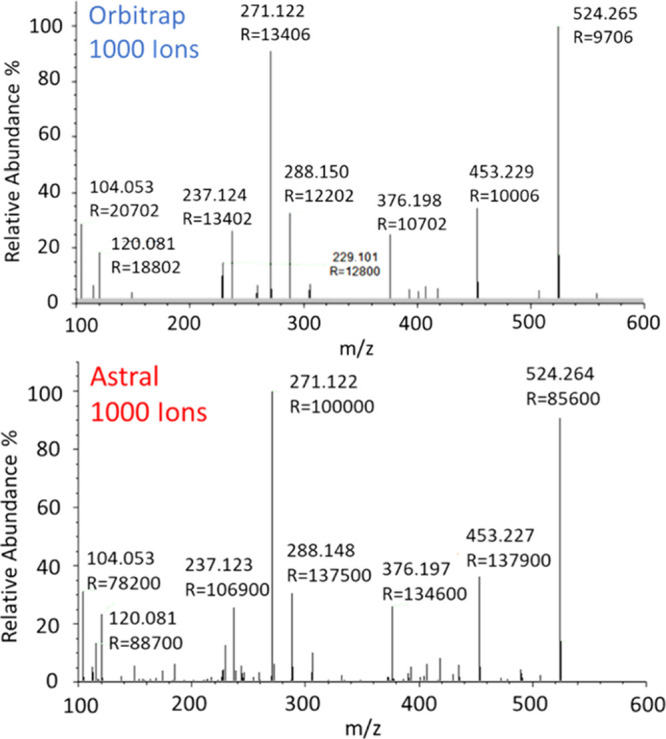
Comparison of MRFA MS/MS spectra in Astral and Orbitrap analyzers with only 1000 ions detected.
Assessment of Proteomics Application Performance
DIA Throughput and Depth of Analysis
Pierce HeLa digest (200 ng) was separated on a PepMap, 150 μm × 15 cm column with 300, 180, 100, and 60 samples per day (SPD) methods (3.6, 5.5, 11, and 20.1 min gradients corresponding to 4.8, 8, 14.4, and 24 min experimental cycles). A second experiment used a 1000 ng load over slower 30 SPD runs, separated on a μPAC Neo 110 cm column. The AGC target was set to 500%, the full-MS mass range was set to 380–980, and the MS/MS range was set to 150–2000 with 2 Th isolation windows. Figure 10 shows protein and peptide identifications reported with Chimerys using a match between runs for three technical replicates, filtered to <1% peptide FDR and ≤1% protein FDR.
Figure 10.
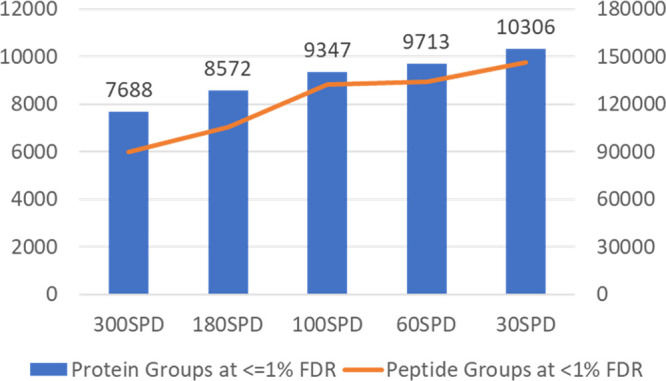
Single shot HeLa DIA protein groups and peptide groups IDs. Variation in the experiment length from 300 to 30 samples per day. 200 ng HeLa digest was injected for 300, 180, 100, and 60 SPD runs and 1000 ng HeLa digest for the 30 SPD runs.
The depth of analysis is extremely high for a single shot method, evidenced by the 10,306 protein groups identified over a 48 min run. There remains a balance between throughput and analytical depth, but even the 4.8 min experiment (300 SPD) shows >7600 protein groups identified, rising to >9000 for the same quantity analyzed over 14.4 min (100 SPD). A rough comparison is that Orbitrap-only methods typically require at least 4× the LC separation time to produce a similar depth of analysis to the hybrid Orbitrap/Astral methods.
Label-Free Quantitation
Quantitative performance was assessed by analysis of two 3-proteome mixed samples containing E. coli, HeLa, and yeast digests at 2:3:1 (sample A) and 1:3:2 (sample B) ratio. Loads (500 ng) were separated on a 50 cm μPAC Neo column over a 24 min experiment. The mass spectrometer was operated in the same manner as the throughput experiments, but for a 3 ms Astral max inject time, and the output data was analyzed in Spectronaut 17 using the DirectDIA workflow.
Figure 11a shows the overall protein IDs and the proportion within 20 and 10% coefficients of variation CV. Of more than 13,000 identified proteins, the median CV was <4.7%, while over 72% had a CV < 10% and 87% a CV < 20%. Figure 11b shows average log2 ratios of intensities of the identified human, yeast, and E. coli proteins between the two 3-proteome mixtures, A and B. Theoretical A/B ratios were 1 for human, 0.5 for yeast, and 2 for E. coli proteomes. Experimentally obtained average ratios were 1.016 for humans, 0.490 for yeast, and 1.993 for E. coli. Run-to-run reproducibility of the quantitation was better than 1%. High quantitative performance was maintained throughout various loads and gradient lengths and remained unchanged, with minor MS acquisition method variations. Maintaining good quantitative performance at such an analytical depth is almost surprising, as greater sensitivity to smaller numbers of ions might be expected to result in greater statistical fluctuations of trace species.
Figure 11.
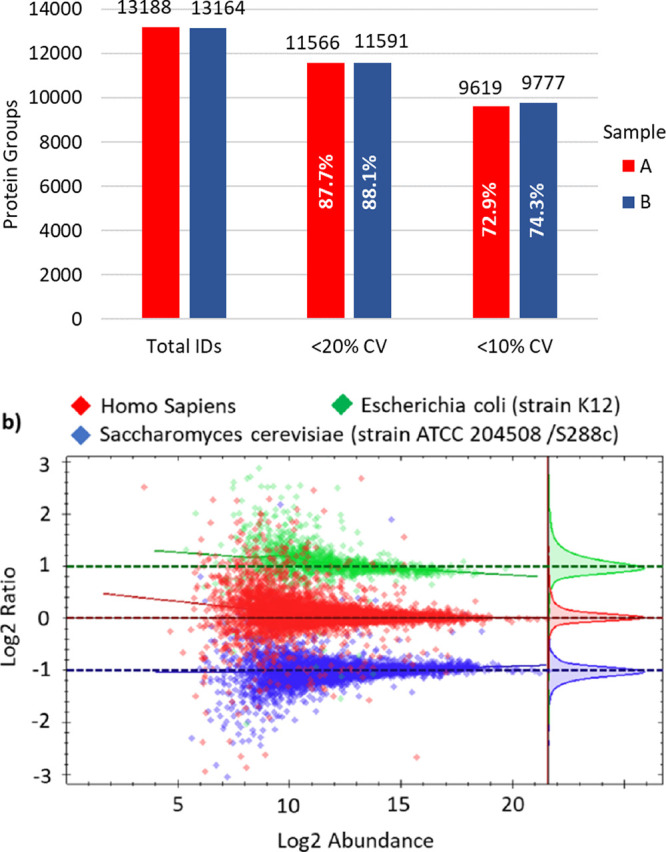
Comparative analysis of 500 ng 3-proteome mixtures (E. coli, HeLa, and yeast) at 2:3:1 and 1:3:2 ratios for samples A and B, respectively. (a) Protein identifications at various coefficients of variation between triplicate runs. (b) Average Log2 ratio of yeast and E. coli protein intensities between samples A and B.
Low Loads
With the explosion of the single-cell analysis field, high-throughput measurements of very low amounts of protein samples have become increasingly important. Small amounts of HeLa from 0.25 to 10 ng were separated over an 18 min, 80 SPD method on a 50 cm μPAC Neo low load column and measured by DIA with a full-MS mass range of 400–800. The isolation width and maximum injection time were decreased from 20 Th and 40 ms for 0.25 ng load to 5 Th and 10 ms for 10 ng load. The FAIMS Pro Duo interface15 was used with a compensation voltage of −50 V to remove singly charged background ions, which prevented the ion processor from becoming flooded by unwanted ions. Data were processed using Spectronaut 17, both with the library-free DirectDIA workflow and by using a spectral library generated from the series of runs.
Figure 12 shows the protein and peptide identifications over a series of runs. Even at 0.25 ng, 3900 protein groups may be identified with DirectDIA, increasing to 5700 with the aid of the generated library. The longer 40 ms accumulation times and wider DIA windows are required for such low ion loads, and the instrument operates at a lower repetition rate. The depth of identification may then be assumed to arise from the combination of high ion transmission and single ion sensitivity. Coefficients of variation remain relatively low, with 1 ng runs producing a median CV of 8.1% with MS1 quantitation and 8.7% with MS/MS quantitation. Protein IDs (87%) have a CV of <20%, and 70% have a CV <10%.
Figure 12.
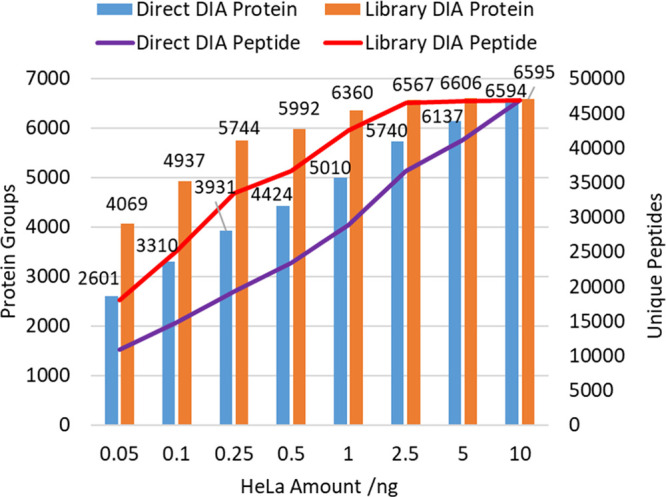
Low-load HeLa 80 samples per day DIA protein and peptide identifications with and without a generated library.
Conclusions
The Orbitrap/Astral dual analyzer mass spectrometer was characterized via LC–MS shotgun proteomics methods with a parallel hybrid mode of acquisition. From low to high sample loads, it was found to produce depth of analysis at higher throughputs than those that have hitherto been accessible. Analytical performance was thought to be enabled by the high scan rate, transmission, and single ion detection of the Astral analyzer, which allows both a high rate of acquisition to cover rapidly eluting peptides and sensitivity to measure low-abundance analyte species. It is anticipated that this new technology will make an important contribution to the field of biological mass spectrometry.
Acknowledgments
The authors would like to acknowledge the advice and support of colleagues throughout Thermo Fisher Scientific, without whom we would be lost, as well as academic and industrial collaborators who have been so vital to both building the next-generation mass analyzers and getting the best out of them.41,43
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.3c02856.
Additional details of vacuum chamber dimensions and pumping; instrument timing diagrams and scan parallelization; further characterization of Astral jitter, drift and dynamic range; summary table for method details; and additional application results covering plasma proteomics, tandem mass tags, and instrument robustness over a large cohort study (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): All authors are employees of Thermo Fisher Scientific, the manufacturer of instrumentation used in this research.
Supplementary Material
References
- Zhang Z.; Wu S.; Stenoien D. L.; Paša-Tolić L. High-throughput proteomics. Annu. Rev. Anal. Chem. 2014, 7, 427–454. 10.1146/annurev-anchem-071213-020216. [DOI] [PubMed] [Google Scholar]
- Eng J. K.; McCormack A. L.; Yates J. R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5 (11), 976–989. 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J.; Creasy D. M.; Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20 (18), 3551–3567. . [DOI] [PubMed] [Google Scholar]
- Demichev V.; Messner C. B.; Vernardis S. I.; Lilley K. S.; Ralser M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17 (1), 41–44. 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolg D. P.; Gessulat S.; Paschke C.; Graber M.; Rathke-Kuhnert M.; Seefried F.; Frejno M. INFERYS rescoring: Boosting peptide identifications and scoring confidence of database search results. Rapid Commun. Mass Spectrom. 2021, e9128 10.1002/rcm.9128. [DOI] [PubMed] [Google Scholar]
- Michalski A.; Cox J.; Mann M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC– MS/MS. J. Proteome Res. 2011, 10 (4), 1785–1793. 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- Kim M. S.; Pinto S. M.; Getnet D.; Nirujogi R. S.; Manda S. S.; Chaerkady R.; Pandey A. A draft map of the human proteome. Nature 2014, 509 (7502), 575–581. 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M.; Schlegl J.; Hahne H.; Gholami A. M.; Lieberenz M.; Savitski M. M.; Kuster B. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509 (7502), 582–587. 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- Senko M. W.; Remes P. M.; Canterbury J. D.; Mathur R.; Song Q.; Eliuk S. M.; Zabrouskov V. Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal. Chem. 2013, 85 (24), 11710–11714. 10.1021/ac403115c. [DOI] [PubMed] [Google Scholar]
- Eliuk S.; Makarov A. Evolution of Orbitrap mass spectrometry instrumentation. Annu. Rev. Anal. Chem. 2015, 8 (1), 61–80. 10.1146/annurev-anchem-071114-040325. [DOI] [PubMed] [Google Scholar]
- Scheltema R. A.; Hauschild J. P.; Lange O.; Hornburg D.; Denisov E.; Damoc E.; Mann M. The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteomics 2014, 13 (12), 3698–3708. 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski A.; Damoc E.; Hauschild J. P.; Lange O.; Wieghaus A.; Makarov A.; Horning S. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteomics 2011, 10 (9), 1–12. 10.1074/mcp.M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]; M111.011015
- Frejno M.; Zolg D. P.; Schmidt T.; Gessulat S.; Graber M.; Seefried F.; Rathke-Kuhnert M.; Wilhelm M.. CHIMERYS: An AI-Driven Leap Forward in Peptide Identification. In Proceedings of the 69th ASMS Conference on Mass Spectrometry and Allied Topic; Philadeplhia PA, 2021. [Google Scholar]
- Dorfer V.; Maltsev S.; Winkler S.; Mechtler K. CharmeRT: boosting peptide identifications by chimeric spectra identification and retention time prediction. J. Proteome Res. 2018, 17 (8), 2581–2589. 10.1021/acs.jproteome.7b00836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker-Jensen D. B.; Martínez-Val A.; Steigerwald S.; Rüther P.; Fort K. L.; Arrey T. N.; Olsen J. V. A compact quadrupole-orbitrap mass spectrometer with FAIMS interface improves proteome coverage in short LC gradients. Mol. Cell. Proteomics 2020, 19 (4), 716–729. 10.1074/mcp.TIR119.001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelstrup C. D.; Jersie-Christensen R. R.; Batth T. S.; Arrey T. N.; Kuehn A.; Kellmann M.; Olsen J. V. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field Orbitrap mass spectrometer. J. Proteome Res. 2014, 13 (12), 6187–6195. 10.1021/pr500985w. [DOI] [PubMed] [Google Scholar]
- Arrey T.; Stewart H.; Harder A.. Ion Pre-Accumulation for High Speed Orbitrap Exploris Operation. In Proceedings of the 70th ASMS Conference on Mass Spectrometry and Allied Topic; Minneapolis MN, 2022. [Google Scholar]
- Meier F.; Brunner A. D.; Koch S.; Koch H.; Lubeck M.; Krause M.; Mann M. Online parallel accumulation–serial fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer. Mol. Cell. Proteomics 2018, 17 (12), 2534–2545. 10.1074/mcp.TIR118.000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews G. L.; Simons B. L.; Young J. B.; Hawkridge A. M.; Muddiman D. C. Performance characteristics of a new hybrid quadrupole time-of-flight tandem mass spectrometer (TripleTOF 5600). Anal. Chem. 2011, 83 (13), 5442–5446. 10.1021/ac200812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernushevich I. V.; Loboda A. V.; Thomson B. A. An introduction to quadrupole–time-of-flight mass spectrometry. J. Mass. Spectrom. 2001, 36 (8), 849–865. 10.1002/jms.207. [DOI] [PubMed] [Google Scholar]
- Chernushevich I. V.; Merenbloom S. I.; Liu S.; Bloomfield N. A W-Geometry ortho-TOF MS with high resolution and up to 100% duty cycle for MS/MS. J. Am. Soc. Mass Spectrom. 2017, 28 (10), 2143–2150. 10.1007/s13361-017-1742-8. [DOI] [PubMed] [Google Scholar]
- Andrien B. A.; Whitehouse C.; Sansone M. A. Proceedings of the 46th ASMS Conference on Mass Spectrometry and Allied Topics. May 31–June 4; Orlando, FL, 1998; pp 889–890. [Google Scholar]
- Franzen J.Method and device for orthogonal ion injection into a time-of-flight mass spectrometer, US Patent US5763878A, 1998.
- Hardman M.; Makarov A. A. Interfacing the Orbitrap mass analyzer to an electrospray ion source. Anal. Chem. 2003, 75 (7), 1699–1705. 10.1021/ac0258047. [DOI] [PubMed] [Google Scholar]
- Brais C. J.; Ibañez J. O.; Schwartz A. J.; Ray S. J. Recent advances in instrumental approaches to time-of-flight mass spectrometry. Mass. Spectrom. Rev. 2021, 40 (5), 647–669. 10.1002/mas.21650. [DOI] [PubMed] [Google Scholar]
- Grinfeld D.; Makarov A.. Multi-reflection mass spectrometer, US Patent US9136102B2, 2013. 10.1111/bjh.12591. [DOI]
- Stewart H., Grinfeld D., Makarov A.. Multi-reflection mass spectrometers, US Patent US10964520B2, 2019.
- Hauschild J. P., Peterson A. C., Couzijn E., Denisov E., Chernyshev D., Hock C., Makarov A.A Novel Family of Quadrupole-Orbitrap Mass Spectrometers for a Broad Range of Analytical Applications. Preprints 2020–06–04, 2020060111. 10.20944/preprints202006.0111.v1. [DOI] [Google Scholar]
- Bekker-Jensen D. B.; Martínez-Val A.; Steigerwald S.; Rüther P.; Fort K. L.; Arrey T. N.; Olsen J. V. A compact quadrupole-orbitrap mass spectrometer with FAIMS interface improves proteome coverage in short LC gradients. Mol. Cell. Proteomics 2020, 19 (4), 716–729. 10.1074/mcp.TIR119.001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange O.; Damoc E.; Wieghaus A.; Makarov A.. Enhanced FT for Orbitrap Mass Spectrometry. Proceedings of the 59th ASMS Conference on Mass Spectrometry and Allied Topics, Denver, CO, 2011, 10.5172/conu.2011.40.1.15. [DOI] [Google Scholar]
- Lange O.; Damoc E.; Wieghaus A.; Makarov A. Enhanced Fourier transform for Orbitrap mass spectrometry. Int. J. Mass Spectrom. 2014, 369, 16–22. 10.1016/j.ijms.2014.05.019. [DOI] [Google Scholar]
- Olsen J. V.; Macek B.; Lange O.; Makarov A.; Horning S.; Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4 (9), 709–712. 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- Stewart H., Hock C., Giannakopulos A., Grinfeld D., Heming R.; Makarov A., A rectilinear pulsed-extraction ion trap with auxiliary axial DC trapping electrodes. In Proceedings of the 66th ASMS Conference on Mass Spectrometry and Allied Topics; San Antonio, TX, 2018. [Google Scholar]
- Sudakov M.; Kumashiro S. TOF systems with two-directional isochronous motion. Nucl. Instrum. Methods Phys. Res., Sect. A 2011, 645 (1), 210–215. 10.1016/j.nima.2010.12.154. [DOI] [Google Scholar]
- Verentchikov A. N.; Yavor M. I.; Hasin Y. I.; Gavrik M. A. Multireflection planar time-of-flight mass analyzer. I: An analyzer for a parallel tandem spectrometer. Technol. Phys. 2005, 50 (1), 73–81. 10.1134/1.1854827. [DOI] [Google Scholar]
- Nazarenko L. M.; Sekunova L. M.; Yakushev E. M.. Time-of-flight mass spectrometer with multiple reflections, Soviet Patent No. SU1725289, 1989.
- Schwartz J. C.; Xaio-Guang Z.; Bier M. E.. Method and apparatus of increasing dynamic range and sensitivity of a mass spectrometer, United States Patent No. US5572022A, 1995.
- Denisov E.; Damoc E.; Makarov A. Exploring frontiers of Orbitrap performance for long transients. Int. J. Mass. Spectrom. 2021, 466, 116607 10.1016/j.ijms.2021.116607. [DOI] [Google Scholar]
- Rose R. J.; Damoc E.; Denisov E.; Makarov A.; Heck A. J. High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat. Meth. 2012, 9 (11), 1084–1086. 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- Liu R.; Li Q.; Smith L. M. Detection of large ions in time-of-flight mass spectrometry: effects of ion mass and acceleration voltage on microchannel plate detector response. J. Am. Soc. Mass Spectrom. 2014, 25 (8), 1374–1383. 10.1007/s13361-014-0903-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil L. R., Damoc N. E., Arrey T. N., Pashkova A., Denisov E., Petzoldt J., MacCoss M. J.Evaluating the performance of the Astral mass analyzer for quantitative proteomics using data independent acquisition. bioRxiv 2023, 10.1021/acs.jproteome.3c00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosius V., Aragon-Fernandez P., Arrey T. N., Uresin N., Furtwangler B., Stewart H., Schoof E. M.Evaluating the capabilities of the Astral mass analyzer for single-cell proteomics. bioRxiv 2023, 10.1038/s41467-023-41602-1. [DOI] [Google Scholar]
- Guzman U. H., Martinez Del Val A.; Ye Z.; Damoc E.; Arrey T. N.; Pashkova A., Olsen J. V.. Narrow-window DIA: Ultra-fast quantitative analysis of comprehensive proteomes with high sequencing depth. bioRxiv 2023, 10.1101/2023.06.02.543374. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.